Note: Descriptions are shown in the official language in which they were submitted.
':~,r~
..
HOECHST AKTIEIdGESELLSCHAf'T HOE 89/F 266 Dr.DA/sch
Description
Process for the preparation of a polyolefin
The invention relates to an olefin polymer of high
molecular weight and low residual catalyst content.
Isotactic PP is prepared with the aid of ethylene-bis-
(4,5,6,7)-tetrahydro-1-indenyl)-zirconium dichloride
together with an alumino~ane in a suspension polymeriza-
tion reaction (cf. EP-A-185,918). The polymer has a
narrow molecular weight distribution (z~/I~, 1.6 to 2.6).
It has been possible to achieve a considerable increase
in the activity of the catalyst system by a specific :,
preactivation method (cf. DE-3,726,067). The particle
morphology of the polymer has likewise been improved by
this preactivation method.
The molecular weights of the polymers obtained in accor-
dance with these two applications are still too low far
industrial use.
There was thus the object of discovering a process for
the preparatioxi of a high molecular weight olefin polymer
which can be carried out in an industrially interesting
temperature range with a high catalyst activity.
It has been found that the object can be achieved by
polymerization of olefins in the presenee of certain
metallocene catalysts.
The invention thus relates to a process for the prepara-
tion of a polyolefin by polymerization of an olefin of
the formula R11-CH=CH-R12, in which R11 and R12 are identi-
cal or different and are a hydrogen atom or a C~-C1w-alkyl
radical, or R11 and Rla, together with the carbon atom
joining them, form a ring having ~ to 28 carbon atoms, at
~.~ 3' ~a ~-
- 2 -
a temperature of OnC to 150°C, under a pressure of 0>5 to
100 bar, in solution, in suspension or in the gas phase
and in the presence of a catalyst which consists of a
metallocene and an aluminoxane of the formula (II)
R10 R30 R10
j ~1 - o ~1- o ~ ~ ~ a. 0 ( I I )
R10 n R
for the linear type, and/or of the f~renula (III)
R10
Al - O (III)
n-~ a
for the cyclic type, in which, in the formulae (II) and
( III ) , R1° is a C1-Cs-alkyl group and n is an integer from
2 to 50, wherein the metallocene is at least one compound
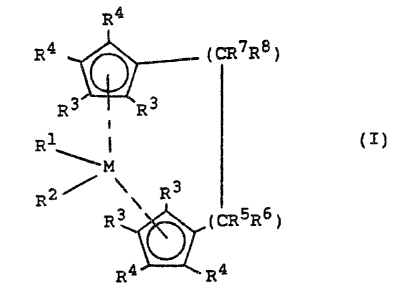
of the formula (I)
R~
R~ (CR7R~)
R~ ~R~
R 1~.~°., i ( I )
M
R2 ~ \ R3
R9 (OR'R6)
R4 ~ R~
in which
7M is zirconium or hafnium,
R1 and R2 are identical or differewt and are a hydrogen
atom, a C1-C1°-alkyl group, a C1-C1°-alkoa~y gxoup, a C6-
C1°-aryl group, a C6-Clo-aryloxy group, a C2-C1o-alkenyl
group, a C~-C4°-arylalkyl group, a C~-Coo-alkylaryl
group, a C8-C4°--arylalkenyl group or a halogen atom,
a0 R3 and R4 are identical or different and are a hydrogen
r
r~ ~lW~ r~ .'~?
-
atom, a halogen atom, a C1-Clo-alkyl group or
a -PIRi, -SR°, -QR°, -OSiR3, -~iR3 Or -PRi radical, ~.n
which R° is a C1-Clo-alkyl group, a C6-Clo-aryl group
or, in the case of radicals containing Si or P, also
a halogen atom,
or in each case two adjacent radicals R' or R'', together
with the carbon atoms joining them, form a ring and
R5, R6, R' and Ra are identical or different and are a
hydrogen atom, a halogen atom, a Cl-C3o-alkyl group, a
ZO C1-Clo-fluoroalkyl group, a Cs-Clo-aryl group, a C6-Czo
fluoroaryl group, a Cl-Clo-alkoxy group, a CZ-Cao-alkenyl
group, a C~-Cao-arylalkyl group, a C8-Cao-arylalkenyl
group, a -SiMe3 group, an -oSiMe3 group or a C7-C4o
alkylaryl group, or R5 and R6 or R' and R°, in each case
toga~ther with the atoms joining them, form a ring.
The catalyst to be used fax the process according to the
invention consists of an aluminoxane and at least one
metallocene of the formula I
R ( ~~RB )
R R
~I)
Rl~ I
M
R2.r~ ~, R3
R3 (CRSR6)
R4 R~
in which
M is hafnium or zirconium, preferably zirconium,
R1 and RZ are identical or different and are a hydrogen
atom, a C1-Clo-, preferably C1-C~-alkyl group, a C1~Clo--.
preferably C1-C3-alkoxy group, a Cs-C1o-, preferably CB-CB-
aryl group, a CB-Clo-. Preferably C6-CB-aryloxy group, a
Cz-Clo-~ Preferably C~-C4-alkenyl group, a C~-C4o-, prefer-
ably C,-Clo-arylalkyl group, a C~-Coo-, preferably CT-Clz-
alkylaryl group, a CB-Coo-, preferably C8-C12-arylalkenyl
R~
3 ('- 3
s ~r4 ~'r s~3 !"' i".' ~ 1
~L) ~d ..~.,°f ,"~ ~,9 f J
,.. Lk ..
group or a halogen atom, preferably chlorine.
R3 and R4 are identical or different and are a hydrogen
atom, a halogen atom, preferably a fluorine, chlorine or
bromine atom, a C1-Clo-, preferably C1-C3-alkyl group or a
-~lRi, -SR9, -ORg, -OSiR3, -SiR3 ox -PRz radical, in which
R9 is a Cl-Clo-, preferably Cl-C3-alkyl group or C~-Cxo-.
preferably C6-CB-aryl group, or in the case of radicals
containing Si or P also a halogen atom, preferably a
chlorine atom, or two adjacent radicals R3 ar R~~ together
with the carbon atoms joining them, form a ring. Particu-
larly preferred ligands are indenyl, fluorenyl and
cyclopentadienyl.
R5, R6, R' and R8 are identical or different and are a
hydrogen atom, a halogen atom, a Cl-C3o-, preferably C1-C4_
alkyl group, in particular a methyl group or ethyl group,
a C1-Clo-fluoroalkyl group, preferably a CF3 group, a C6-
Clo-fluoroaryl group, preferably a pentafluorophenyl
group, a C6-Ci9-, preferably Cs-C8-aryl group, in parti-
cular -CHz-C6H5 or -C6H5, a C1-Clo°, preferably C1-Cu-alkoxy
group, in particular a methoxy group, a Cz-Clo-, prefer-
ably Cz-C4-alkenyl group, a C7-Coo-, preferably C~-Clo-
arylalkyl group, a CB-Coo-, preferably Cg-C1z-arylalkenyl
group or a C?-C~,o'. Preferably C~-Czz-alkylaryl group, or R5
and R6 or R' and R8, in each case together with the atoms
2S joining them, form a ring.
Especially preferably, R5, Re and R' are a hydrogen atom
and RB is a phenyl, benzyl, methyl, ethyl, ~trifluoromethyl
or methoxy group, or RS and R' are a hydrogen atom and R6
and RB are a phenyl, benzyl, ethyl, methyl, trifluoro
methyl or methoxy group.
The metallocenes described above can be prepared in
accordance with the following general equations
A ~ r ~ f... ax..
Nj!.~ n,~
Hz R ~~ + But y 1 L i -----~--~ HR aL i
X° (CR~RB)m_ (CRSR~)n_X
BZRb + ButylLi --~ HRbLi
HRa- (CR~R$)m_ (CR5R6)~_Rbg 2 B~-7
LiRa-(CR~RB)m_(CR5R6)n_RbLi MC1~
---
7 8 s
(cR R ~~_R (oR7R~, -R~
C1 m i 1
M ~ g R~Li
C1
(CRSRf )n_Rb ~ C1 "°'°
(CR'R6) _Rb
n
(CR7R8)m-Rs
i ~ RI.
M
j ~ R~
(CR5R6)n-Rb R3 3
R
R3 R~
(X = C1, Br, J, O-Tosyl, HRa = ~ , HRH' = ) .
gar R~ R~
''rhe aoca~,alys~ is an aluminoxane of the foxznula IT
g10 R7.0 R20
~'''°- Al - o Al - Al'°/ ( T I )
Rio -,''~ ~ °"~. yo
for the linear type, and/or of the foranula (TTT)
Rlo
d~11 - ~ ( T T T )
n~~
for the cyclic type. In these formulae, R'~° is a Cl~Cs
a13cy1 group, preferably me~.hyl, ethyl or isobu~,yl, in
~~ ~ ~ q3 ~ ~r~ J
- ~ -
particular methyl, and n is an integer from 2 to 50,
preferably 5 to 40. However, the exact structure of the
aluminoxane is not known.
The aluminoxane can be prepared in various ways.
One possibility is careful addition of water to a dilute
solution of an aluminum trialkyl by introducing the
solution of the aluminum trialkyl, preferably aluminum
trimethyl, and the water, in each ease in small portions,
into a larger amount of an inert solvent initially
introduced into the vessel, and awaiting the end of the
evalutian of gas between each addition.
Tn another process, finely powdered copper sulfate
pentahydrate is suspended in toluene and, in a glass
flask under an inert gas at about -20°C, aluminum tri-
alkyl is added in an amount so that about 1 mol of
CuSOk ~ 5Hz0 is available for eve~.y 4 A1 atoms . After slaw
hydrolysis, alkane being split off, the reaction mixture
is left at room temperature for 24 to 4~ hours, during
which it must be cooled if appropriate, s~ that the
temperature does not rise above 30°C. The aluminoxane
dissolved in the toluene is then filtered off from the
copper sulfate and the solutian is concentrated in vacuo.
Tt is assumed that :Ln this preparation process the low
molecular weight aluminoxanes coedense to form higher
oligomers, aluminum trialkyl being split off.
Aluminoxanes are furthermore obtained when aluminum tri-
alkyl, preferably aluminum trimethyl, dissolved in an
inert aliphatic or aromatic solvent, preferably heptane
or toluene, is reacted with aluminum salts containing
water of crystallization, preferably aluminum sulfate, at
a temperature of -20 to 100°C. In this procedure, the
volume ratio between the solvent and the aluminum tri-
alkyl used is 1:1 to 50x1 - preferably 5:1 - and the
reaction time, which can be monitored by the splitting
off of the alkane, is 1 to 200 hours - preferably 10 to
~: ~. ~ ~ ;~. r.
a t:a~ -.~ ,. i c.l
-
40 hours.
Of the aluminum salts which contain water of crystalliza-
tion, those which have a high content of water of crys-
tallization are used in particular. Aluminum sulfate
hydrate, especial ly the compounds A1z ( S04 ) 3 ~ 16Ha0 and
A12 ( S04 ) 3 ~ 18HZ0 with the particularly high water of
crystallization content of 16 and, respectively, 18 mot
of HZfJ/mol of AlZ ( SQ4 ) 3, is particularly preferred .
Another variant for the preparation of aluminoxanes
comprises dissolving an aluminum trialkyl, preferably
aluminum trimethyl, in the suspending agent which has
been initially introduced into the polymerization kettle,
preferably in the liquid monomer flr in heptane or tolu-
ene, and then reacting the aluminum compound with water.
In addition to the processes described above for the
preparation of aluminoxanes, there are others which can
be used.
Regardless of the nature of the preparation, all the
aluminoxane solutions have a common feature of a varying
content of unreacted aluminum trialkyl which .is present
in the free form or as an adduct.
It is possible to preact3va~t~ the metallocene with an
ahuninoxane of the formula (TI) andlor (IIT) before use
in the polymerization reaction. The polymerization
activity is in this way significantly increased and the
particle morphology is improved.
The preactivatian of the transition metal compound is
carried out in solution. Preferably, in this procedure,
the metallocene is dissolved in a solution of the alumin-
oxane in an inert hydrocarbon. An aliphatic or aromatic
hydrocarbon is suitable as the inert hydrocarbon. Toluene
is preferably used.
The concentration of the aluminoxane in the solution is
~~ ~~ f~ ,.~ p
Vie! 9~,J ,,y' ~..i , ;1
g -
in the range from about 1~ by weight to the saturation
limit, preferably 5 to 30~ by weight, in each case based
on the total solution. The metallocene can be used in the
same concentration, but it is preferably used in an
amount of 10-4 - 1 mol per mol of aluminoxane. The pre-
activation time is 5 minutes to 60 hours, preferably 5 to
50 minutes. The preactivation is carried out at a tem-
perature of -78°C to 100°C, preferably 0 to 70°C.
The polymerization is carried out in a known manner in
solution, in suspension or in the gas phase, continuously
or discontinuously, in one or more stages at a tempera-
ture of 0 to 150°C, preferably 30 to 80°C. Olefins of the
formula R11-CH=CH-R1z are polymerized. In this formula, Rll
and R12 are identical or different and are a hydrogen atom
or an alkyl radical having 1 to 28 carbon atoms. However,
R11 and R12, together with the carbon atoms joining them,
can also form a ring having 4 to 28 carbon atoms.
Examples of such olefins are ethylene, propylene, 1-
butene, 1-hexane, ~-methyl-1-pentane, ~.-octane, norborn-
ene, norbornadiene, pentane, hexane or octane. Propylene
is polymerized in particular.
Hydrogen is added as a molecular weight regulator if
necessary. The total pressure in the polymerization
system is 0.5 to 100 bar. The polymerization is prefer-
ably carried out in the pressure range from 5 to 64 bar,
which is of particular interest industrially.
The metallocene compound is used in the polymerization in
a concentration, based on the transition metal, of 10-3 to
10-', preferably 10-4 to 106 mol of transition metal per
dm3 of solvent or per dm3 of reactor volume. The aluminox-
ane is used in a concentration of 10-5 to 10-~ mol, prefer-
ably 10-4 to 10-Z mol per dm3 of solvent or per dm3 of
reactor volume. However, in principle higher concentra-
tions are also possible. ~t least one compound of the
formula I is used as the metallocene. Mixtures of several
compounds of the formula I or mixtures of isomers are
~% ~e a.,3 fg ~,,1 ~1
g
also possible.
Tf the polymerization is carried out as suspension or
solution polymerization, an inert solvent which is
customary for the Ziegler low pressure process is used.
For example, the polymerization is carried out in an
aliphatic or cycloaliphatic hydrocarbon; examples of
these which may be mentioned are butane, pentane, hexane,
heptane, isooctane, cyclohexane and methylcyclohexane.
A benzine or hydrogenated diesel oil fraction can
furthermore be used. Toluene can also be used. The
polymerization is preferably carried out in the liquid
monomer.
If inert solvents are used, the monomers are metered in
as a gas or in liquid form.
If only one monomer is used as the suspending agent, the
comonomer or the comonomers is or are metered in as a gas
or in liquid form.
It is furthermore possible to carry out the polymeriza
tion in a mixture of different monomers as the suspending
agent; another monomer can then be metered in as a liquid
or in gaseous form. Tf ethylene is used, it is advantage-
ous for some of 'the ethylene to be initially iwtroduced
and for the remainder to be metered in during the poly-
merization.
The duration of the polymerization can be as desired,
since the catalyst system to Sae used according to the
invention shows only a slight time-related drop in
polymerization activity.
The process according to the invention is distinguished
by the fact that the metallocenes used are very heat-
stable, so that they can be used with high activity even
at temperatures up to 90°G. Tpie aluminoxanes used as
cocatalysts can moreover be added in lower concentrations
than previously. Finally, it is now possible to prepare
random copolymers at temperatures of industrial interest.
- 10 -
The metallocenes or metallocene mixtures to be used
according to the invention contain compounds which can
polymerize propylene to give polymers having a molecular
weight of more than 150,000 g/mol, preferably
200,000 g/mol. This is confirmed by the molecular weight
distribution, which has a high M~,/M~, ratio (> 2). The
molecular weight distribution is sometimes multimodal.
Synthesis of 1,2-bis(1-indenyl)-1,2-bis(phenyl)ethane
(diastereomer mixture)
A mixture of 68.64 g (336 mmol) of methylphenyl-benzo-
fulvene, 4.92 cm~ (61 mmol) of CC14 and 100 cm3 of tetra-
hydrofuran was added to 8.17 g (336 mmol) of magnesium
filings in the course of 0.5 hour. The reaction mixture,
which was warm because of the reaction which occurred,
was then stirred overnight. The resulting Grignard
mixture was added to ethereal HC1, and water was then
added. The organic phase was separated off, dried over
PIaZSO4, filtered and evaporated.
The crude product was purified by column chromatography
(50 x 250 mm; 60 ~., 70 - 200 ~sm, starting with pure n--
hexane with an increasing HZCC12 content).
Yield 3.1 g ( 7 .55 mmol, 4 . 5~ ) , rF ~ 0. 26 ( 3 volumes of
hexane/1 volume of H~CC12), melting point 225 - 230°C.
~x~ample
$(~5-1-Indenyl)-CHZ*CHEt-(r~s-1-indenyl)~~rClz (1)
56 cm3 of a 1.6 N (89.6 mmol) solution of butyllithium in
hexane were added dropwise to 12.2 g (42.6 mmol) of the
ligand (racemate) in 200 cm3 of tetrahydrofuran at room
temperature in the course of 1 hour and the mixture was
stirred at 60°C for 0.5 hour after the end of the
evolution of butane.
The resulting dilithium salt solution was added dropwise,
simultaneously with a solu~ta.on of 16.6 g (44 mmol) of
c;3 4~ 63 ~') ~.. ;;' h l
11
ZrCl4(tetrahydrofuran)Z in 300 cm3 of tetrahydrofuran, to
50 cm3 of tetrahydrofuran at room temperature in the
course of 2 hours. The mixture was then stirred at room
temperature for 1.5 hours, and 4 cm3 of a 1.0 N {4 mmol)
ethereal solution of HC1 were added. After the dark
reaction mixture had spontaneously brightened, an orange
color persisted. The mixture was now concentrated to
200 cm3 and the inorganic salts were precipitated by
addition of toluene and filtered off.
The filtrate was concentrated further. When the volume
was still 100 cm3, a yellow-orange solid was obtained. To
bring the precipitation to completion, the mixture was
placed in a deep-freeze (-3.5°C) overnight and then
filtered and the precipitate was washed with a little
cold toluene and then with n-pentane and dried in vacuo.
Yield I: 2.6 g (5.82 mmol - 13.70 ; 1~MR showed a 1:1
mixture of two complexes.
The precipitate which separates out on further concentra
tion of the filtrate was treated as described above.
Yield Its 3.3 g (7.39 mmol = 17.40 ; NMR showed a mixture
of the complexes from I.
The filtrate was evaporated to dryness. Yield III: 0.2 g.
Fraction II was 'taken up in ether and the mixture was
concentrated slowly. The first precipitate ~ obtained
shaved an -. 1;3 mixture according to IdMR, the complex
which appeared in the five-membered ring proton range in
the higher field predominating. When the procedure was
repeated with .A, an enrichment to better than 1:4 was
obtained (precipitate B, 1.2 g (2.59 mmol = 6.3~).
The filtrate of B evaporated to dryness contained the
comgound in resonance in the lower field enriched to more
than 4:1 (yield 0.8 g (1.79 mmol = 4.2~)).
Fraction II showed a correct CH analysis: found 58.5
{calculated 59.18) ~ C; 4.6 (4.51)$ H.
Example 2
7.9~ g (27.79 mmol) of ligand (racemate) in 100 cm3 of
tetrahydrofuran and 36 cm3 of a 1. 6 N ( 57 . 6 mmol ) solution
of butyllithium in hexane were reacted as described in
~.~i~.>,?v
- 12 -
Example 1 to give the d:ilithium salt. 10.78 g
(28.58 mmol) of ZrClu (tetrahydrofuran)2 in 2fJ0 cm~ of
tetrahydrofuran and the dilithium salt solution were
added dropwise to 50 cm3 of tetrahydrofuran at room
temperature. The procedure followed here was such that a
larger amount of zirconium compound was always initially
introduced, i.e. the zirconium solution was added in the
course of 3 hours and the dilithium salt solution was
added in the course of 8.5 hours. .~.fter the mixture had
been stirred at room temperature for two hours, 2.5 cm3 of
1. 0 N ( 2 . 5 mmol ) ethereal HCl were added - the mixture
brightened immediately - and the mixture was concentrated
to about 100 cm3. 150 cm3 of toluene were then added and
the mixture was left to stand overnight. The precipitate
obtained was separated off and the filtrate was concent-
rated. A small amount of oil which occurred during this
operation was decanted off and the liquid was evaporated
to dryness. The crude product was analyzed by NMR spec-
troscopy. The species known from Example 1 was found as
the main component, together with at least one other
compound. The mixture was extracted with 2 x 120 cm~ of
Et2p and the filtrate was evaporated . Tts NMR spectrum
shows, in addition to the signals known from Example 1,
further resonances in the five-membered ring proton
range. The further ether extracts obtained (~ 1 cm~) were
evaporated to dryness. 10.5 g of yellow solid were
obtained (23.5 mmol = 8~.6~; PdMR: two complexes).
Example 3
~ ( q5-1-Indenyl ) -C~Ia*CHP~ie- ( r~ 5-1-indenyl ) } ZrClz ( 2 )
~1 cm3 of a 1.6 N (65.6 mmol) solution of butyllithium in
hexane were added dropwise to 7.7 g (30.53 mmol) of
ligand (racemate) in 100 cm3 of tetrahydrofuran at room
temperature in the course of 1 hour and the mixture was
stirred at 60°C for 0.5 hour after the evolution of
butane had ended.
11.7 g (31.01 mmol) of ZrCl4(tetrahydrofuran)Z in 250 cm3
of tetrahydrofuran were added, simultaneously with the
6' F !~ r~ r. ~
~; ~~ ~ «~ r~ '..a P~J
- 13
dilithiurn salt solution, to 50 cm3 of tetrahydrofuran at
roam temperature in the course of 5 hours. After the
mixture had been stirred at room temperature for two
days, ~ cm3 (4 mmol) of ethereal HC1 were added. The now
clear yellow-orange reaction mixture was concentrated to
dryness. The orange-red evaporation residue was extracted
with 6 x 80 cm3 of toluene and the mixture was filtered
and concentrated. An oil which was obtained during
concentration to 200 cm3 was decanted off and the filtrate
was concentrated further to 80 cm~. The precipitate A
obtained by this procedure was filtered off, washed with
a little n-pentane and dried in vacuo. Yield A: 1.85 g
(4.28 mmol = 14~); ldP~R shows two complexes.
The filtrate was concentrated further to 30 cm3 and placed
at a low temperature (-35°C) overnight, decanted off from
precipitate B and evaporated to dryness (0.1 g; I~7MR
showed starting materials i.e. ligand and ZxCI~,(tetra-
hydrofuran)2, toluene and probably four complexes).
Precipitate B was washed with pentane and dried in vacuo.
Yield B: 0.7 g (1.62 mmol = 5.3~); IdMR showed the pre-
sence of a four-component mixture.
Elemental analysis gave the following result:
found 57.9 (calculated 58.32) C, 4.2~ (4.4) H~
Example 4
~ ( r~5-1-Indenyl ) -CHz*CH ( benzyl ) - ( r~5-1-indenyl ) }ZrCl2
(3)
17.25 cm3 of a 1.6 13 (27.6 mmol) solution of butyllithium
in hexane were added dropwise to 4.53 g (13 mmol) of
ligand in 75 crn3 of tetrahydrofuran and the mixture was
then stirred at 55°C for 0.5 hour after the evolu-
tion of gas had ended.
This dilithium salt solution was added, simultaneously
with 4.9 g (12.99 mmol) of ZrCl4(tetrahydrofuranjz in
100 cm3 of tetrahydrofuran, to 30 cm3 of tetrahydrofuran
at room temperature in the course of 6 hours, 'the mixture
- 14 -
was evaporated to dryness, the residue was taken up in
toluene and the mixture was filtered. After the solvent
had been stripped off, the residue was digested several
times with n-pentane, the pentane being decanted off . The
residue now obtained was dried. Yield: 2.28 g (about
4.47 mmol - 34.40; NMR confirmed the presence of at
least three complexes, contaminated with tetrahydrofuran
and pentane.
Example 5
.((~5-1-Indenyl)_*CHPh*CHPh-(q5-1-indenyi)~ZxCl2 (4)
6.25 cm3 of a 1.6 N (10 mmol) solution of butyllithium in
hexane were added dropwise to 2.04 g (4.97 mmol) of
ligand (diastereomer mixture) in 40 cm3 of tetrahydrofuran
at room temperature. During this procedure, a white-green
precipitate separated out after abo~.at half the solution
had been added. After the mixture had been stirred at
60°C fox two hours, the dilithium salt obtained was added
in portions to 1.88 g (4.98 mmol) of ~xCl,,(tetrahydro-
furan)2, dissolved in 40 cm3 of te~trahydrofuran, at 0°C.
Thereafter, the mixture was stirred at room temperature
for 4 hours and concentrated 'to dryness, the residue was
extracted with a warm (about 40°C) n-pentane/toluene
mixture (2:1 by volume), the mixture was fia.tered and the
.clear yellow solution obtained was concentrated. A yellow
precipitate A obtained by this procedure was filtered
off, washed with a little cold Et20 and dried in vacuo.
Yield A: 0 . 45 g ( about 0 . 86 mmoi = 17 . 70 ; PIMR showed, in
addition to solvents ( EtaO, tetrahydrofuran, n-pentane,
toluene), signals having cleavage patterns of at least
two complex compounds, which patterns are characteristic
of indenyl complex five-membered ring protons. 25 cm3 of
n-pentane were added to the filtrate and precipitate B
which had separated out was filtered off, washed with a
little cold Et20 and dried in vacuo. Yield B: 1. 08 g ( 1.57
mmol = 31.5~j; NPY~t showed, in addition to the signals of
toluene and pentane, the probable presence of a single
complex species which was not present in A.
,.~ r
~,.e sJ . ~ =.~a
- 15 -
The fil~tra~te of H was evaporated to dryness. :Cts I~MR
showed, in addition to Et20, pentane and toluene, probably
signals of the complex from precipitate B and the ligand.
Rxample 6
~ ( ~~-1-Indenyl )-CHZ*CHMe-( r~5°1-indenyl ) ~HfCl2 ( 6 )
34 cm3 of a 1.6 P7 (54.4 mmol) solution of butyllithium in
hexane were added dropwise to 7.2 g (26.95 mmol) of
ligand (racemate) in 200 cm3 of tetrahydrofuran at room
temperature in the course of one hour and the mixture was
then stirred at 60°C for 1 hour. The dilithium salt was
then added dropwise, simultaneously together with 13 g
(28 mmol) of HfCl4(tetrahydrofuran)2 in 200 cm3 of tetra-
hydrofuran, to 50 cm3 of tetrahydrofuran, the procedure
being such that an Hf excess was present in the reactor
vessel. After 2/3 of the reaction partners had been
added, the mixture was left to stand overnight and the
remainder of the reaction partners was added in the
course of 4 hours. The mixture was then stirred overnight
and 4 cm3 of 1.0 N (4.0 mmol) ethereal HC1 were added, a
brightening in color being observed.
The yellow-brown evaporation residue was extracted with
toluene and the mixture was filtered and evaporated to
dryness. The resulting residue T was digested with n-
pentane, the filtered soleent was stripped off and the
remainder of about 25 cm~ was separated of:~ from the
precipitate obtained.
Yield A: 0.61 g (1.14 mmol - 4.2~); ~7MR showed, in
addition to the signals of the ligand, resonances in the
indenyl five-membered ring proton range, which demon-
strata the presence of three complex compounds, two being
present as main components.
Residue 2 was further extracted with pentane/Et20 (1:2 by
volume), the mixture was filtered and the filtrate was
concentrated. Yield Bs 0.26 g (0.49 mmol - 1.8~); I~lR
demonstrated the presence of a complex mixture, evidently of four
compound: (faur methyl group triplets of the ethyl group
in the range from 1.25 to 0.85 ppm).
~J ~ a;~ ~) ~a 1
- 16 -
'.the following elemental analysis was found: calculated
49.5 (found 50.9) ~ C; 3.78 (~.1)~ H.
Example 7
~ ( r~ $-1-Indenyl ) -CHZCHEt- ( ,~ 5-1-indenyl ) } Z n ( CH3 ) 2 ( 5 )
4.1 cm~ of a 1.6 N (6.56 mmol) solution of methyllithium
in ether were added dropwise to 1.45 g (3.25 mmol) of a
suspension of two complex compounds in 50 cm3 of EtzO at
-40°C. when a relatively large proportion of the compon-
ents had dissolved and a dirty white precipitate had
formed, the mixture was evaporated to dryness. after
extraction with 200 em3 of n-pentane and filtration, the
solvent was stripped off and the residue was dried in
vacuo.
3~ield: 1.15 g (about 2.83 mnnol = 87.00; NMR showed, in
addition to a few impurities, probably monomethylated
compounds, two main components with the resonances
typical of CH3Zr species in the high field range, i.e. as
expected two resonances fox the syn-form, at -0.76 and
-0.88 ppm, and two signals at an almost identical shift
of -1.02 ppm for the anti-form, which no longer has CZ
symmetry because of the unsymmetric C2 bridge.
Examuples 7 to 10 and Comparison Example A
A dry 7.6 dm3 kettle was flushed with nitrogen and filled
with 10 drn3 of a benzine (boiling range 100-120°C) at
20°C. The gas space in the kettle was then flushed free
from nitrogen by forcing in 2 bar of ethylene and letting
down S times. Thereafter, 30 cm3 of a toluene solution of
methylaluminoxane (10.5 by weight of methylaluminoxane,
molecular weight according to cryoscopic determination:
750 g/mol ) were added. The contents of the kettle were
heated up to 60°C in the course of 15 minutes, while
stirring. The total pressure was then adjusted to 5 bar
by feeding in ethylene, while stirring at 250 revolutions
per minute. In parallel with this, 3.1 mg of metallocene
were dissolved in 20 cm~ of a toluene solution of methyl-
aluminoxane (concentration and quality as above) and were
";,~ ~ ')
- 17 -
preactivated by being left to stand for 15 minutes. The
solution was then introduced into the kettle. The poly-
merization system was brought to a temperature of 65°C
and then kept at this temperature for 1 hour by approp-
riate cooling. During this period, the total pressure was
kept at 5 bar by appropriately feeding in ethylene. The
amounts shown in Table 1 were obtained.
The following abbreviations have been used in the tables:
~i'~I = viscosity number in cma/g,
Mw = weight-average molecular weight in g/mol
Mw/Mn - polydispersity determined by gel permeation
chromatography (GPC)
II = isotaxy, determined by 13C-NMR spectroscopy,
BD = bulk density, T8 = glass transition temperature.
Examples 11 and 12 and Coaa~para.son Example 1B
A clean, dry 1.5 dm3 polymerization reactor with a stirrer
was flushed with nitrogen and then with ethylene and fil-
led with a solution of norbornene in 750 cm3 of toluene.
The reactor was then brought to a temperature of 20°C,
while stirring, and 1 bar of ethylene was forced in. 20
cm~ of a toluene solution of methylaluminoxane (10.1 by
weight of methylaluminoxane of molecular we,igla~t 1300
g/mol, cryoscopic determination) were then metered into
the reactox and tha mixture was stirred at 20°C for 15
minutes, the ethylene pressure being kept at 1 bar by
topping up (saturation of the toluene with ethylene). In
parallel with this, the metallocene was dissolved in
10 cm3 of a toluene solution of methylaluminoxane (con-
centration and quality see above) and was preactivated by
being left to stand for 15 minutes. The solution of the
complex was then metered into 'the reactor. Polymerization
was subsequently carxied out at 20°C for 1 hour, while
stirring (750 revolutions per minute), the ethylene
pressure being kept under 1 bar by topping up. The
contents of the reactor were then drained rapidly into a
stirred vessel into which 100 cm3 of isopropanol had been
~~~~'~)'~''~~~~
K~ ~ 1 7i) TJt '.~) i_
m ~~ -
initially introduced. 2 dm3 of acetone were added to this
mixture, 'the mixture was stirred for 10 minutes and the
suspended polymeric solid was then filtered off, The
polymer filtered off was then added to 600 cm3 of a
mixture of two parts of 3 normal hydrochloric acid and
one part of ethanol and this suspension was stirred for
2 hours. The polymer was then filtered off again, washed
neutral with water and dried at 80°C under 0.2 bar for 15
hours.
The results of the experiments are summarized in Table 2.
J~xamples 13 to 15 and Comparison Example C
A clean, dry 1.5 dm3 polymerization reactor with a stirrer
was flushed with nitrogen and then with propylene and
filled with a solution of 30 g of norbornene in 750 cm3 of
toluene. The reactor was then brought to a temperature of
20°C, while stirring, and 1 bar of propylene was forced
in. 20 cm3 of a toluene solution of methylaluminoxane
(10.1 by weight of methylaluminoxane having a molecular
weight of 1300 g/mol) were then metered into the reactor
and the mixture was stirred at 20°C for 15 minutes, the
propylene pressure being kept at 1 bar by topping up
(saturation of the toluene with propylene). In parallel
with this the metallocene was dissolved in 10 cm' of a
toluene solution of methylaluminoxane (concentration and
guality see above) and preactivated by being left to
stand fox 15 minutes. The solution of the complex was
then metered into the reactor. Polymerization was sub-
sequently carried out at 20°C for 3 hours, while stirring
(750 revolutions per minute), the propylene pressure
being kept at 1 bar by topping up. The contents of the
reactor were then drained rapidly into a stirred vessel
into which 100 cm3 of isopropanol had been initially
introduced. 2 dm3 of acetone were added to this mixture,
the mixture was stirred for 30 minutes and the suspended
polymeric solid was filtered off. The polymer filtered
off was then added to 600 cm3 of a mixture of two parts of
3 normal hydrochloric acid and one part of ethanol and
I
~~~3~~
- 19 -
this suspension was stirred for 2 hours. 200 cm3 of
toluene were then added to the mixture and, after the
mixture had been stirred for a further 5 minutes, the
toluene phase was separated off and 1 dm3 of acetone was
added. The polymer which had been dissolved in toluene by
extraction of the hydrochloric acid mixture was precipi-
tated during this procedure. The polymeric solid precipi-
tated was filtered off and dried at 80°C under 0.2 bar
for 15 hours.
The experimental results are summarized in Table 3.
Examples 16 to ~7
A dry 16 dm~ kettle was flushed with nitrogen and then
filled with 10 dm3 of liquid propylene. Two thirds of the
amount of methylaluminoxane stated in Table ~ for the
particular experiment were then added as a solution in
toluene and the mixture was stirred at 30°C for 15
minutes. In para11e1 with this a solution of the metallo-
cene in one third of the amount of methylaluminoxane
stated in Table 4 was prepared and was preactivated by
being left to stand for 15 minutes. The metallocene
(Examples 1 to '7) were used as mixtures of compounds or
as isomer mixtures, without further purification.
This solution was then introduced into the kettle. The
polymerization system was brought to th~a appropriate
polymerization temperature and the polymerization was
started. After 60 minutes, the polymerization was stopped
by cooling the reactor and letting down. The resulting
polymer yield and the analytical data determined can be
seen from Table 4.
r-i
ri
0
.-.
0
'
o a o 0 0
~ cV
N ~ N ~ c
c1 n
U
.-,
O o o to 0
L~ U N tt7 M 0~ u1
I:f.1 w .-i .-i
'p W
r-1 W ~-. o O o o p
N tn M wf o ~o
' N N
O .iaO
~ N ~ rn ,-I t~ o-I
r- tn ;
I
a ~ a ~ ~r ~, an
~
N 1
I ~ O ~ i
F;
N U N U
U
r P- ~~
i J f~
~ I ~ ~
~ ~ ~ ~
I .~i ~ ff
~; - H 7
U ~ ~ Ial r
r p
~, O
i r
-II
Qi QI~.i >-I ~ .--Irl sl1
e-i ~C..'
U ,...1 o~Y r-! 'a~
.-I 3Y r'"~Y i
U
N ~ r9
~
.N N ,I-> ~.,~ p
r-1 C~ 'Py tt1
QI N N
N a1 ~ p ~ ~ ,fit
~ ~ '~ ~! ;h
~ "d 1
. 0
~i ~ ~ '~ ~
0 "~ O O
p p
~ ~ L r-I .-
.r ~.. I
1 r-1
as c~ v a ~ 9ro zi
.~ I .~ U
.~ ,~
U U U tp U .~.:i~
I t~ U i.d
U U
tit ft1 iL1 (i~ .-i
PI rI rl .i.)
rl rl rl r-I
ii i-I 1-I a-I
'-~'p .R ,.4 O
'b 'CJ N
A!
1
t0
~ t-. ao rn
U
~
Image
E,~ 1'" !,..' 9
~1~~~.~~.~e
-- ZZ
0 0
v ~ .-ia;~;
o,
a
~ N
U
b
fU ~ ~ Q N
'"'6t1 00M L7
N N
d
O
t~
W
O O
O
M M M M
H
O
4a
O .!~O
~ N A'~~-I
t~ r~ Q1~ M N M N
t ~i
U
N
A4
U V 'k
~N
O N ~N N
r'I
p.j W rWw n
,
r~ r~r~r~
d w
.r ar
N N N N
O
td~
M O ya
r-! (d
y
W ~ ~ n-1r1ri
~~~ ~wr
-- 2 3 -.
~t tC t0 CO IS7 eP CO N t,~ et C>7 CO ~ et SD O)
6p M ~9 h N C ip 90 M to O ~ tr
~
cr M M C' et M 6t! ~ M M ~~t CO St N N N N
~ ~ct Ln N Id7 6D (C N c~'
N ~ ~ 6t7
r~ O O O O O O O O O O O O O O O O O O O O
O O O O O O O O C
O
Q O O O O O O O O O O O O O O C? O O O O O
O O O O O O O O G
O
O O O O O lt7 Lt9 O LA O O O O O O tJ O
O O O O O !17 O O O C
O 6l! O
6t7 CO N 00 N 68 tJt M cf~ t0 ip 6D C~ 6~ O
'~' iwa h ,-~ 6L7 h M 60 ~ O ~ O Q.
~ VI
a ~ lff et CL7 N N M 00 dp ~ ~ h M OD CO h c> t0 (~
h O h 6L7 t<7 M tp a? is
Lff ~ c1
M ~ ~ O CJ O taa N iD h O N Off W~ rr CO
O N O O M W M h P tt7 ttf
M N Or
6 b' h h ~ h i8 ~' ~ O h . On u7
ITi ~ tD 00 tp
uT 6n c~ t~;
U
da
m
AO N 6f7 O O Ld7 C,7 tD ~ d' t7 O O
e-t O Ll7 '~ ~t (D
O 1
ri N ~tD h tA N O (~ O C9 OTf . ~ ct
IA CO M M O M N h h
f, h
N ~ N vb r-a N w N M ~ N rr .-s
M w in M ~
et
b ..r
O ra ~-1
~ 00 O OOD n OD 000 CO CO AO CO Op0 OpD 00 Op0 CO I~~, n ~ ~ OoD CO 000 ~ h ~
~ ~
W
~ Wit'.~ P cC '
~ Iw t17
. . M ct h Ln O M lp h O M O c0 C9 M
4-i ~ N Pw M !t CO ~t O cfi
00 1 N
M
a a
~1 CD C97 d' P. r4 Pa, ~ In N 6D h O .~nM.-~
Q f N O M f\ M t17 Cn N ld7
~ a CD ep N ip 00
L67 tp
PI h-11_ O '~ N M tp 1.c7 r-t N M l11
CT ~w ~ M (C) On
Cf~\ 4 O M ~' w N
(O
.u . a ,. .~' ''
x r .~
\
.
i
,Q
U
~ 00 O Y h W 617 N M O ~ f~ N O (~ O O Ll7
QI h IW .-H N LLy~ O G7 Rf' O 07
CT ' ' O O v-9
GD .-~ tn .-i 00 ~. cD CO 6c7 t0 O 6d7 Ca C17
tn M 0 .-~ Cn et et (Ot CO h.. .-4
CO n 1 6d7 .-~ (C1 6I7
d N
h h h r-a h O p In N l0 -1 M N M ~&t .-4
' OD (~ N N C7tQ tD On er 05 N ~t
M t ~w CO
pit r1 .-1 N .-~ n1 r-1 r1 v-9
.-f r-1 N .-1
N
W
'rat.-.
O CC~ 0 0 0 Cl O O t6'f0 6.C1Ca 0 O O 0 0 0
a C~ 0 6n M O 0 0 0 0 0
M ~Y 0
0
.. ., ., ., .. ., ., ., ., ., ., .. ., .,
O ., tt7 (O ., ., ., .. ., ., .,
d ., 6~ Qp ., 6d7 O .. ., Ln tc1 O to
O Lff ., !l7 C7 O O 6Y! O G7
M N O M O O O 00
~ tp O
!L'7
6Cf
'
r~ .~ .~ ~-1 .-1 7 r-i N M N lL1 M .-1 tn 667
U .-d .-i M '-1 N 117 .-f tl7
.-i N .-1 N !tJ .-r u7
N r-1 66'7
I,C
1
oC
N
~
O G7 O O O O O CI ~ G1 O C9 O O O ~ O
f O O O O O O t~ O 00 C9 O O
60 Lt l O O
' tD O
' '
W ha l L67 ~ 1~ ~ l~ ~' 6l9 0.'p' h Il~
7 e? 7 ~f (~ l0 h ~D IO lt7 (D
I h. f'w CU PW P'~.
w ~
Q
H
ro
ro ~ ~ ~
d-1 ~ N r w r qvy
a -a
N N N
U
~1
~
N N N N .
c
a
w w ia ~ ~
u ,, a:
,6 L
U N ~ ~ ~ ~ + ~
. e .
Q 6-~ sa
v kJ 6tJ 6L ud UJ 6JJ 16.i eJJ
N N N N N N N N N
r, ~ rr sr /~. v~1 a1 /9 n a~a
6B1 '~ '~ '~ a 'C9 't7 '~ 'li7 'O
C C C C C C Sr C C
F-1 H C.~I i~1 7~i 1..1 IW YW hf
ss v v v
v v w ,.n an
r'~e L L L L L L L L L
N N N N PJ N N hl N
Q
i"1rl
6D h 00 .-~ N tl7 C17 O M ~' 40 h O r4 ~ !l7
C~ M et LO wl N Lf9 Ct1 N M CD
O h, C77 I~
Cp
,.-~~ .-~ N N N N N M M M M M M ct 'et ~~ eY
.-~ N N M M M M eY ~ et
N N et
N