Note: Descriptions are shown in the official language in which they were submitted.
- 2028313
;
--1--
AROMATIC DIAMINE COMPOUNDS, BISMALEIMIDE COMPOUND~
THERMOSETTING RESIN FORMING COMPOSITIONS THEREFROM,
RESINS THEREFROM, AND METHODS FOR THEIR PREPARAITON
BACKGROUND OF THE INVENTION
The present invention relates to aromatic
diamine compounds which can be used as raw materials
for polyimide, polyamide, bismaleimide or epoxy
resins. The compounds can also be employed as curing
agents. The invention also relates to a method for
preparing the compounds.
Furthermore, the present invention relates to
bismaleimide compounds which are useful as raw
materials for heat-resistant resins, and which can be
obtained from the aforementioned aromatic diamine
compounds. The invention also relates to a method for
preparing the compounds.
In addition, the present invention relates to
thermosetting resin forming compositions, and
ultimately the resins, which are obtained from the
aforementioned aromatic diamine compounds and
bismaleimide compounds, as well as a method for
preparing the composition.
In recent years, raw materials for
heat-resistant resins have been required to provide a
combination of good thermal and mechanical properties,
as well as certain other characteristics which
composites should exhibit such as flexibility and
moldability/workability.
In this regard, polyimide resins have been
used. While providing some excellent properties, they
typically exhibit poor moldability/workability.
An example of such a polyimide is an
aromatic polyimide made by Du Pont and marketed under
2028313
--2--
the tradename "Vespel". This polyimide can be prepared
from 4,4'-diaminodiphenyl ether and pyromellitic
anhydride. It is insoluble and unmeltable. Thus, when
it is molded, a special procedure such as powder sinter
molding must be used. Unfortunately, this molding
technique cannot be easily used for the preparation of
articles having complex shapes. During manufacturing
of complex shaped articles, additional operations such
as cutting are necessary. As a result the polyimide is
difficult to mold and costs increasé.
To address those drawbacks associated with
polyimide resins, a variety of strategies have been
formulated, primarily focusing on improving the diamine
component of the raw materials. For example, there
have been attempts to introduce an ether linkage group
or an isopropylidene group into the molecule and to
increase the molecular chain. Unfortunately, these
techniques have not effectively provided the
flexibility and moldability/workability characteristics
necessary for composite materials.
A typical example of a bismaleimide is
N,N'-(methylene-di-p-phenylene)bismaleimide disclosed
in Japanese Patent Laid-open Nos. 47-8644 and
47-11500. However, this bismaleimide compound is
substantially insoluble in common organic solvents such
as ketone and petroleum solvents. And, when a
polyimide resin is prepared using this compound, the
occupation ratio of the polyimido group in the polymer
structure is high. Therefore, the polymer is hard,
brittle and provides extremely poor flexibility and
high hygroscopicity. The effective application of the
bismaleimide compounds is accordingly limited
considerably.
2028313
--3--
Recently, in an effort to eliminate such
disadvantages, much attention has been given to the
development of long-chain bismaleimide compounds. For
example, Japanese Patent Laid-open No. 63-500866
discloses a bismaleimide compound having three benzene
rings and having the following structure represented by
formula (X):
O O
Il 11
C~ / ~ ~ C~3 / ~ I (X)
In addition, Japanese Patent Laid-open No. 63-264566
discloses a bismaleimide compound having four benzene
rings and having the following structure represented by
the formula (XI):
O O
Il 11
~ C CH - CH2 C ~ 0 ~ N ¦¦ (XI)
Il 11
O O
However, even with these bismaleimide
compounds and polymers prepared from the compounds, an
effective balanced combination of heat resistance,
flexibility, adhesion to a metal and inorganic
materials, and workability is not provided.
2028313
.
--4--
Thermosetting resins having an imido
structure have been effectively used to prepare molded
articles having excellent electrical insulating proper-
ties, heat resistance and dimensional stability.
Therefore, this type of resin has been utilized in many
industrial fields.
However, thermosetting resins obtained by
subjecting aromatic bismaleimides alone to heat
polymerization are very brittle and provide poor
flexibility, although they do provide excellent heat
resistance. To address this drawback, attempts have
been made to develop thermosetting resin forming
compositions comprising an aromatic bismaleimide and an
aromatic diamine. For example, a polyaminobismaleimide
resin made by Rhone Poulenc, and marketed under the
tradename "Kelimide" comprising N,N'-4,4'-
diphenylmethane-bismaleimide and 4,4'-diaminodiphenyl-
methane has been widely utilized for impregnating
varnishes, laminates and molded articles (See Japanese
Patent Publication No. 46-23250). However, this type
of thermosetting resin is still unsatisfactory in terms
of impact resistance and flexibility. Furthermore,
when these thermosetting resins are used as base
materials for electrical and electronic parts, they
exhibit poor moldability/workability and high
hygroscopicity, which adversely affect electrical
properties.
SUMMARY OF THE INVENTION
Therefore, an object of the present invention
is to provide a diamine compound which is useful as a
raw material for various resins and which can provide
excellent flexibility and moldability/workability. It
2028313
-- 5
ls likewise an ob~ect to provide a method for preparing such
a dlamine compound.
Another ob~ect of the present invention ls to
provlde a blsmalelmide compound which ls readlly soluble ln
common solvents and can be used to prepare a polymer havlng
improved properties. It is likewise an ob~ect to provide a
method for preparing such a bismaleimide compound.
Still another ob~ect of the present invention is to
provide a thermosettlng resln formlng composition, as well as
a resin, whlch can provlde excellent mechanlcal strength,
partlcularly ln terms of lmpact resistance, and whlch has low
hygroscoplclty. It ls also an ob~ect to provlde a method for
preparlng such a composition and resin.
Accordingly, the present invention is directed to a
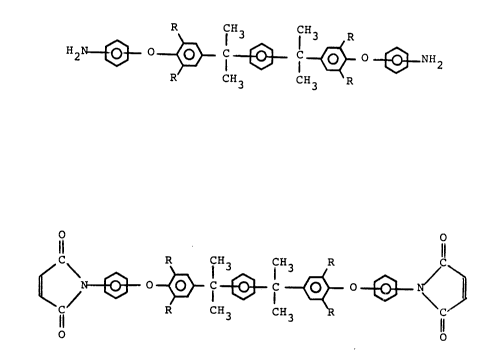
novel aromatlc diamine compound represented by formula (I):
H~ ~ ~ NH
R R
wherein R is H or CH3, with the proviso that:
(1) the amino group on the ring B is attached at the meta-
or para-position wlth respect to O, and
(2) when the amlno group on the ring B is attached at the
para-positlon wlth respect to O, then the two groups
represented by the formula:
27981-36
2028313
-- 6
H2N~O~ I--
are bonded to the ring A at the meta-posltion with respect to
each other and R ls H.
When the amino group on the ring B ls attached at
the meta-posltlon wlth respect to 0, the formula (I) ls
represented by the formula:
H~ ~O~NH3 (1--)
When the amino group on the ring B is attached at
the para-posltlon with respect to 0, the formula (I) is
represented by the formula:
H2N~O~CH\[~CH3 NH2
The dlamlne compound of the inventlon can be
obtalned by reacting a compound represented by the formula
(II)
27981-36
2028313
- 6a -
R R
HO~C [~OH ~)
R R
wlth a nltrobenzene compound such as nltrochlorobenzene or
dlnltrobenzene ln the presence of a base ln an aprotlc polar
solvent to obtaln a compound havlng the formula (III):
o2N~o--\~3, U `~N02 (m)
whereln R ls as deflned above and the provlso, relatlng to
the attachlng posltlons of the formula (I) ls also applled.
The compound of formula (III) ls then reduced to
obtaln the dlamlne compound of the lnventlon.
When R ls hydrogen and the two groups are attached
to rlng A at the meta-posltlon wlth respect to each other,
the formula (II) ls represented by the formula (II-a):
HO~C~3 ~CH3~--OH ~-a)
When the nltro group on the rlng B ls attached at
the metal-posltion with respect to 0, the formula (III) is
represented by the formula (III-a):
27981-36
2028313
- 6b -
02N~O~IC~O~NO~
When the nltro group on the rlng B ls attached at
the para-posltlon with respect to O and the two groups are
attached to the ring A at the meta-position wlth respect to
each other, the formula (III) ls represented by the formula
(III-b):
0~ ~ 0 ~ ~ IC ~ O ~ NO2 o~-b)
The present lnventlon provldes a relatlvely easy
technlque for provldlng the novel dlamlne, the dlamlne belng
useful as a raw materlal for reslns and whlch provldes
excellent flexlbllity and moldablllty/workability, or as a
curing agent.
Furthermore, the present lnvention provides a novel
bismaleimide compound whlch comprlses a skeleton having flve
benzene rlngs and ls represented by the formula (IV):
~b~ C~3 ~'
o R R o
whereln R ls H or CH3.
The blsmalelmlde compound represented by the
27981-36
2028313
- 6c -
formula (IV) ls advantageously soluble ln common solvents
such as toluene or xylene. A polylmlde prepared uslng thls
type of blsmalelmlde has excellent
27981-36
2028313
. . .
heat resistance and flexibility, as well as low
hygroscopicity.
The bismaleimide of the present invention can
be prepared by a dehydration/condensation reaction
between a diamine component and maleic anhydride. This
dehydration/condensation reaction can be carried out by
dissolving or dispersing the diamine of the invention,
maleic anhydride and an acid catalyst in a
predetermined solvent, and then heating the resulting
solution or dispersion under reflux. More
specifically, a diamine compound of formula (I) can be
reacted with maleic anhydride in the presence of an
acid catalyst under heating/reflux of an organic
solvent, while any water which is produced is removed
from the system.
The bismaleimide compound of the present
invention can be used as a raw material to prepare the
novel addition-type polyimide of the invention. Since
the occupation ratio of the imido group in the molecule
of the bismaleimide compound is low, the polyimide
resin made from this compound can provide improved
flexibility and low hygroscopicity. Therefore, when
using the bismaleimide compound of the present
invention, blending properties and performance of the
thermosetting-type polyimide resin can be considerably
improved.
Moreover, by the present invention there is
also provided a thermosetting resin forming composition
comprising the diamine compound of formula (I) and the
bismaleimide compound of formula (IV). The
thermosetting resin forming composition of the present
invention gives a thermosetting resin which has
excellent heat resistance, impact resistance and
toughness, as well as low hygroscopicity. Thus, it can
2028313
-- 8
be expected that the compositlon will be effectively applied
to electrlcal and electronic parts, various structural parts,
sllding parts and many other uses. The composition of the
present case is lndustrlally useful and effective.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1 is a mass spectrum of the bismaleimide
compound of Example 2A;
Fig. 2 is an IR spectrum of the bismaleimide
compound of Fig. l; and
Figs. 3 and 4 are IR spectra of the bismaleimide
compounds of Examples 2C and 2D, respectively.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS OF
THE INVENTION
Accordlng to the present lnventlon, the amlno
groups of the aminophenoxy groups are bonded at the meta- or
the para positions with respect to O, and preferred are those
compounds of formula (I) whereln the two amlnophenoxy-(methyl
or dimethyl)-a,a-dlmethylbenzyl groups are bonded at the
meta- or the para posltlons of the central benzene rlng.
Examples of preferable diamlne compounds lnclude
(1) 1,3-bls[4-(4-amlnophenoxy)-a,a-dlmethylbenzyl~benzene,
(11) 1,4-bls[4-(3-amlnophenoxy)-a,a-dlmethylbenzyl]benzene,
(iil) 1,3-bis[4-(3-aminophenoxy)-a,a-dlmethylbenzyl]benzene,
(lv) 1,4-bls[4-(3-amlnophenoxy)-3,5-dlmethyl-a,a-
dimethylbenzyl]benzene and (v) 1,3-bls[4-(3-amlnophenoxy)-
3,5-dlmethyl-a,a-dlmethylbenzyl]benzene (hereafter for
convenience, "1,4- or 1,3-" will be referred to as "1,4
(1,3)- ).
~4 27g81-36
2028313
g
Typlcally, lt has been found that compounds (11)-
(v) wlll provlde even better flexlbillty and
moldablllty/workablllty than the flrst compound (1).
There are known technlques for reduclng a bls(4-
nltrophenoxy) compound obtalned by reactlng a blsphenol wlth
4-chloronltrobenzene ln order to prepare a correspondlng
dlamlne. For example, bls(4-amlnophenoxy)blphenyl has been
manufactured by reduclng bls(4-nltrophenoxy)blphenyl obtalned
from 4,4'-blphenol and 4-chloronltrobenzene [28th Natlonal
Sampe Symposlum, Aprll 12-14, p. 728-736 (1983)].
Furthermore, 2,2'-bls(4-amlnophenoxy)propane has been
manufactured from 2,2'-bls(4-hydroxyphenyl)propane [Rocznlkl
Chem., 48, p. 1459 (1974)]. However, a dlamlne compound
havlng a structure of formula (I) accordlng to the present
lnventlon has not been so produced.
In order to obtaln a dlamlne compound of formula
(I), it ls necessary to pass through two steps of
condensatlon and reductlon. For example, when produclng 1,3-
bls[4-(4-amlnophenoxy)-a,a-dlmethylbenzyl]benzene, 1,3-bls(4-
hydroxy-a,a-dlmethylbenzyl]benzene), whlch has a structure
represented by formula (II-a), ls reacted wlth 4-
chloronltrobenzene ln the presence of a base ln an aprotlc
polar solvent ln order to prepare 1,3-bls[4-(4-nltrophenoxy)-
a,a-dimethylbenzyl]-benzene, whlch has a structure of formula
(III-b).
27981-36
2028313
--10--
The amount of 4-chloronitrobenzene is
preferably two to three, more preferably 2.05 to 2.5
times more per mole of 1,3-bis(4-hydroxy~
dimethylbenzyl]benzene.
Examples of suitable bases include
carbonates, hydrogencarbonates, hydroxides and
alkoxides of alkaline metals. Typical examples of these
bases include potassium carbonate, sodium carbonate,
potassium hydrogencarbonate, sodium hydrogencarbonate,
lo potas~um hydroxide, sodium hydroxide, lithium
hydroxide, sodium methox~de and potassium isopropoxide.
~mong these, potassium carbonate, sodium hydroxide and
potassium hydroxide are most preferably used and are
also preferred from an economical viewpoint. The
amount of the base i5 preferably not less than 2
equivalents, more preferably 2.2 to 3 equivalents when
1,3-bis(4-hydroxy~ dimethylbenzyl)benzene is
the raw material.
As the solvent used in the above described
condensation reaction, aprotic polar solvent~ are
suitable. Typical examples of these solvents include
dimethyl sulfoxide, N,N-dimethylformamide, N,N'-
dimethylacetamide, N-methylpyrrolidone, 1,3-dimethyl-2-
-imidazolidinone, hexamethylphosphortriamide and
2s sulfolane. The amount of the solvent is preferably 1
to 1 to 15 times, more preferably 2 to 5 times the
total weight of raw materials.
The reaction temperature may range from 50 to
220-C, and is preferably ln the range of 80 to 180'C.
The end point of the reactlon can be
determined by obser~ng a decrea~e of raw materials and
unreacted intermediate through thin-layer
chromatography or high-speed liquid chromatography.
27981-36
2028313
ThlC exemplified condensation reaction may be
carrled out by placing predetermined amounts of 1,~
(1,3)-bi~(4-hydroxy-Q,~-dlmethylbenzyl)benzene, the
base and the solvent in a reactor and forming an
alkaline metal salt of 1,4(1,3)-bis(4-hydroxy-
~dimethylbenzyl)benzene. Then, 4-chloronitrobenzene can
be added thereto. Alternatively, all the raw
materials, including 4-chloronitrobenzene can be
initially placed in the reactor. However, the reaction
is not limited to these procedures, and can be carried
out using other su~table procedures.
After completion of the reaction to obtain
the intermediate represented by the formula (III),
(e.g., 1,3-bis(4-(4-nitrophenoxy)~
dlmethylbenzyl)benzene), optional steps can be
performed. For example, the concentration of the
solvent can be modified or the reaction solution can be
discharged into water.
The intermedlate of formula (III) is then
reduced to prepare the desired compound represented by
the formula (I) (e.g., 1,3-bis[4-(4-aminophenoxy~-
dimethylbenzyl]benzene). The reduction can be
achieved by standard procedures such as those which
involve reducing a nitro group to an am~no group.
But, in an industrial context catalytic reduction or
hydrazine reduction is preferred.
In the case of catalytic reduction, the
reduction react~on is carrled out u~lng a reduction
cataly~t such as a metallic cataly~t or a carr~er
catalyst of nlckel, palladium or platinum or a Raney
catalyst of nickel or copper in an amount of o.Ol to
10% (by weight) in terms of the metal with respect to
the intermediate, e.g., l,3-bis[4 (4 nitro
phenoxy)-~,Q-dimethylbenzyllbenzene.
27981-36
2028313
-12-
In the reductlon reaction a solvent is used.
Preferable examples of the solvent include methanol,
ethanol, isopropyl alcohol, methyl cellosolve and N,N-
dimethylformamide which a~e inactive in the reaction.
s There is no particular limitation on the reaction
temperature, but it may be in the range of from 20 to
200 c, preferably 20 to loo C. The reaction pressure
maybe from atmospheric to about S0 atm.
In the case of hydrazine reduction, the
lo reduction reaction is carrled out uslng hydrazine in
amount which exceeds the theoretical amount by 1.1 to 5
tlmes.
The catalyst can be those metallic catalyst
usually employed with catalytlc reduct~on. The
particularly preferable catalyst, however, i~
palladium/carbon, platinum/carbon or that which is
preparad by allowing active carbon to adsorb ferric
chloride. The amount of the catalyst may range from
0.001 to 30~ (by weight) in terms of a metal with
respect to the intermediate, e.g.,
l,3-bis[4-(4-nitrophenoxy)
-d~methylbenzyl~-benzene.
As a reaction solvent which can be used in
this process, those same solvents can be used which are
2s useful with catalytic reduction. There is no
particular restriction on reaction temperature. But it
may usually be in the range of from 20 to 150-C,
preferably 40 to 120'C.
After completlon of the reaction, the
reaction solution is hot-filtrated and can be
sub~ected to optional treatments such as cooling,
concentration, dilution and crystallization in order to
obtain desired crystals of the product (e.g.,
1,3-bis~4-aminophenoxy)-
27981-36
'~ -
2028313
- 13 -
a,a-dimethylbenzyl]benzene). The crystals can be purifled by
sultable technlques such as washlng and recrystallizatlon.
The novel dlamlne accordlng to the lnventlon (e.g.,
1,3-bls[4-(4-amlnophenoxy)-a,a-dlmethylbenzyl]benzene can be
used to prepare polylmlde reslns, polyamide reslns,
bismalelmlde reslns or an epoxy resin uslng known technlques.
Furthermore, 1,4(1,3)-bls[4-(3-amlnophenoxy)-a,a-
dlmethylbenzyl]benzene can be prepared by the above-mentloned
procedure except that 4-chloronltrobenzene ls replaced wlth
m-dlnltrobenzene. In other words, varlous dlamlne compounds
of the present lnventlon can be obtalned by varylng the
comblnatlon of the compound of formula (II) and the
nltrobenzene compound.
The blsmalelmlde compound of the present lnventlon
can be prepared uslng a dehydratlon/condensatlon reactlon
between a dlamlne component represented by formula (I) and
malelc anhydrlde. As a result of thls reaction, amlno groups
present at the opposlte ends of formula (I) are replaced wlth
malelmlde groups.
The dehydratlon/condensation reactlon lnvolves
dlssolving or dispersing the dlamlne, malelc anhydrlde and an
acid catalyst in a predetermlned
27981-36
A
2.028313
solvent. Then, the resulting solution is heated under
reflux.
Preferable examples of the acid catalyst
include mineral acids such as sulfuric acid,
hydrochloric acid and phosphoric acid; heteropolyacids
such as phosphotungstic acid and phosphomolybdic acid;
organic sulfonic acids such as p-toluenesulfonic acid
and methanesulfonic acid; halogenated carboxylic acids
such as trichloroacetic acid and trifluoroacetic acid;
solid acids such as silica-alumina; and cation exchange
type ion exchange resins. In particular, sulfuric
acid, phosphoric acid and p-tolunenesulfonic acid are
preferred. In addition, the acids may be in the form
of salts with diamines.
The amount of acid catalyst depends upon its
type. But it may usually bé in the range of from 0.1
to 10% by weight with respect to the total weight of
maleic anhydride and the diamine. When the amount of
the catalyst is less than 0.1% by weight, the expected
effect of the catalyst will not always be achieved.
When the amount is more than 10% by weight, no
additional effect should be obtained from the catalyst,
which is uneconomical. In addition, it is
inconvenient and difficult to remove the excess
catalyst.
Examples of suitable solvents to be used in
the condensation reaction include aliphatic and
alicyclic hydrocarbons such as hexane, heptane, decane
and cyclohexane; aromatic hydrocarbons such as benzene,
toluene and xylene; halides of these aliphatic and
aromatic hydrocarbons; oxygen-containing,
nitrogen-containing and sulfur-containing polar
solvents such as dimethylformamide,
N-methylpyrrolidone, acetonitrile, dimethylacetamide,
2028313
.
-15-
dimethyl sulfoxide, sulfolane, anisole and n-butyl
ether; and mixtures thereof. The amount of the
solvent is preferably 1 to 20 times, particularly
preferably 3 to 10 times as much as the total weight of
the diamine component and maleic anhydride.
The reaction temperature under heating and
reflux depends slightly upon the type of solvent.
However, it may usually be in the range of from 80 to
190C, preferably from 100 to 160C. High pressure,
atmospheric pressure or a reduced pressure can all be
used, and will typically be selected to be compatible
with the type of solvent and reaction temperature.
The reaction time may usually be in the range
of from 2 to 10 hours, preferably 5 to 6 hours.
With regard to the amounts of maleic
anhydride and diamine component, it is desirable that
the amount of maleic anhydride is slightly in excess
relative to that of the diamine component. In general,
the molar ratio of maleic anhydride to diamine is
preferably 2.05 to 3Ø
After completion of the condensation
reaction, the reaction mixture is washed with water to
remove any remaining catalyst and unreacted maleic
anhydride therefrom. The solvent is then distilled
off, thereby obtaining a concentrate. Afterward,
alcohol is added to the concentrate, followed by
stirring, so that a crystal powder is obtained. The
resulting crude crystals can then be purified by
recrystallization.
In order to obtain the thermosetting resin
forming composition, and ultimately the resin, of the
present invention from the bismaleimide compound and
the diamine compound, it is preferred that a
bismaleimide compound represented by formula (I~) be
~028313
-16-
mixed with an aromatic diamine compound represented by
formula (I) in the absence of any solvent or in the
presence of an organic solvent. The mixture can then
be subjected to heat treatment at a temperature of from
70 to 220C to carry out prepolymerization; the mixture
and the prepolymer both being "resin forming
compositions". Thereafter, the resin can be produced.
The following procedures are preferably
employed. (1) The bismaleimide compound and the
aromatic diamine compound can be mixed and then ground
to a solid-solid state, wherein the ground mixture is
used. Alternatively, the mixture can be further
subjected to heat treatment to form a prepolymer. The
prepolymer can then be ground to pellets or powder. In
this case, heating conditions wherein the material is
partially cured to the level of the prepolymer are
preferable. In general, it is appropriate that the
heat treatment is effected at a temperature of from 70
to 220C for 5 to 240 minutes, preferably at 80 to
200C for 10 to 180 minutes.
Or, (2) The bismaleimide compound and the
aromatic diamine compound can be dissolved in an
organic solvent. Then, the solution can be discharged
into a poor solvent. The resulting precipitate can be
collected by filtration, dried and then formed into
pellets or powder. Alternatively, the bismaleimide
compound and the aromatic diamine compound can be
dissolved in an organic solvent, and then partially
cured up to the level of a prepolymer by heat
treatment. Afterward, the cured material can be
discharged into a poor solvent. The resulting
precipitate can be collected by filtration, dried and
then formed into pellets or powder. Conditions for the
heat treatment depend slightly upon the type of organic
202g313
-17-
solvent, but they are generally consistent with those
used for heat treatment according to procedure (1).
The organic solvent should not substantially
react with both components, but should be a good
s solvent for both components. Examples of suitable
solvents include halogenated hydrocarbons such as
methylene chloride, dichloroethane and
trichloroethylene; ketones such as acetone, methyl
ethyl ketone, cyclo-hexanone and di-isopropyl ketone;
ethers such as tetra-hydrofuran, dioxane and methyl
cellosolve; aromatic compounds such as benzene, toluene
and chlorobenzene; and aprotic polar solvents such as
acetonitrile, N,N-dimethylformamide, N,N-dimethyl-
acetamide, dimethyl sulfoxide, N-methyl-2-pyrrolidone
and 1,3-dimethyl-2-imidazolidinone.
The ratio between the bismaleimide compound
and the diamine compound is from 0.1 to 1.2 moles,
preferably 0.2 to 0.8 mole of diamine-compound per mole
of the bismaleimide compound. When the ratio is too
small, the resulting cured product will not have good
impact resistance or flexibility. When it is too
large, the cured product is adversely affected in terms
of heat resistance.
Additional components can be added to the
thermosetting resin forming composition of the present
invention, so long as the objects of the present
invention are not impaired. For example, a curing
accelerator can be added. Suitable accelerators
include radical polymerization initiators such as azo
com~ou..ds or organic peroxides, or ionic catalysts such
as tertiary amines, quaternary ammonium salts,
imidazoles or boron trifluoride-amine salts.
Powdery reinforcing material and fillers can
also be used. Examples include metal oxides such as
-18- 2028313
aluminum oxide or magnesium oxide; metal hydroxides
such as aluminum hydroxide; metal carbonates such as
calcium carbonate or magnesium carbonate; diatomaceous
earth powders; basic magnesium silicates; calcined
clays; finely powdered silica; melted silica;
crystalline silica, carbon black; kaolin; finely
powdered mica; quartz powder; graphite; asbestos;
molybdenum disulfide; or antimony trioxide.
Also, fibrous reinforcing materials and
fillers can be added. Examples include inorganic
fibers such as glass fiber, rock wool, ceramic fiber
alumina fiber, potassium titanate fiber or carbon
fiber; or organic fibers such as an aromatic polyamide.
For improving properties of the resin in a
final product such as a coating film, adhesive layer or
molded article, a synthetic resin can be added and
blended therewith. This type of synthetic resin is,
for example, a thermosetting resin such as a phenol
resin, an epoxy resin, a melamine resin or a silicone
resin; polyamide; polycarbonate; polysulfone; polyether
sulfone; polyether ether ketone; modified polyphenylene
oxide; polyphenylene sulfide; polyether imide; or a
fluorine-containing resin.
The thermosetting resin according to the
present invention can be molded by known molding
methods such as compression molding, transfer molding,
extrusion molding or injection molding, and then
effectively put to practical use.
The present invention will be described in
further detail by reference to the following examples.
However, these examples should not be construed as in
any way limiting the invention.
-19- 2028313
Example lA
In a reactor equipped with a stirrer, a
reflux condenser, a thermometer, a Dean/Stark water
separator and a nitrogen inlet pipe, there were placed
1410 g of N,N-dimethylformamide from which water had
been removed, 207.6 g (0.6 mole) of 1,4-bis(4-hydroxy-
~,~-dimethylbenzyl)benzene, 193.7 g (1.23 mole) of
4-chloronitrobenzene and 248.8 g (1.8 moles) of
potassium carbonate, and 50 g of toluene was then added
thereto.
The resulting mixture was heated, and
stirring was continued for 5 hours, while a temperature
of from 140 to 150-C was maintained, in order to carry
out the reaction. Water produced by the reaction was
successively removed from the system by azeotropy with
toluene.
After completion of the reaction, the
resulting inorganic salts were removed by hot
filtration. The filtrate was then heated again. While
a temperature of from 90 to 95 C was maintained, 210 g
of water was added thereto over 2 hours in order to
precipitate 1,4-bist4-(4-nitrophenoxy)-~
dimethylbenzyl]benzene. This solution containing the
precipitate was slowly cooled, and the resulting light
yellow crystals were collected by filtration and then
wa~hed with a mixed solvent of N,N-dimethylformamide
and methanol. Afterward, the resulting sludge was
washed with 1 liter of methanol, filtered and then
dried, thereby obtAining 335 g of light yellow powder.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.3%.
The analytical results were as follows:
-20- 2028313
Melting point: 186.5 to 188.5~C
Values of elemental analysis:
C H N
Calcd. (%) 73.475.44 4.76
Found (%) 73.285.56 4.96
IR (KBr tablet method)
1330, 1500 cm~l (a nitro group)
1240 cm~l (an ether bond)
In a hermetic reduction reactor equipped with
a stirrer and a thermometer there were placed 294 g
(0.5 mole) of 1,4-bis[4-(4-nitrophenoxy)-
a,~-dimethylbenzyl~benzene obtained from the preceding
step, 1175 g of dimethylformamide and 17.5 g of a 5%
Pd/C catalyst. A hydrogen gas was then introduced
thereinto, while the solution was stirred vigorously.
Reaction was continlle~ at a reaction
temperature of from 30 to 40 C for 4 hours. At this
point, 67.2 liters of hydrogen had been absorbed by the
solution. Since further absorption was not observed,
the reaction was brought to an end.
After completion of the reaction, filtration
was effected at room temperature to remove the Pd/C
catalyst from the solution.
The resulting reaction solution was heated to
80 to 90 C. While this temperature was maintained, 500
g of water was added dropwise thereto over 2.5 hours in
order to precipitate 1,4-bist4-(4-aminophenoxy)-a,~-
dimethylbenzyl]benzene. This reaction solution was
slowly cooled, and the resulting white crystals were
collected by filtration and then washed with a mixed
solution of N,N-dimethylformamide and methanol and
afterward with methanol, and dried, thereby obtaining
242.8 g of 1,4 bis[4-(4-aminophenoxy)-~,~-dimethyl-
-21- 2028313
benzyl]benzene.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.2%
(total yield 88.3%). The analytical results were as
5 follows:
Melting point: 189 to 190.5C
Values of elemental analysis:
C H N
Calcd. (%) 81.82 6.82 5.30
Found (%) 81.90 6.75 5.21
IR (KBr tablet method)
1620, 3320-3430 cm~l tan amino group)
1230 cm~l (-an ether bond)
EXAMPLE lB
In a reactor equipped with a stirrer, a
reflux condenser, a thermometer, a Dean/Stark water
separator and a nitrogen inlet pipe, there were placed
600 g of N,N-dimethylformamide from which water had
been removed, 207.6 g (0.6 mole) of 1,3-bis(4-hydroxy-
a,a-dimethylbenzyl)benzene, 193.7 g (1.23 mole) of
4-chloronitrobenzene and 248.8 g (1.8 moles) of
potassium carbonate, and 50 g of toluene was then added
thereto.
The resulting mixture was heated, and
stirring was then continued for 5 hours, while a
temperature of from 140 to 150-C was maintained, in
order to carry out the reaction. Water produced by the
reaction was successively removed from the system by
azeotropy with toluene.
After completion of the reaction, the
resulting inorganic salts were removed by hot
filtration. The filtrate was then heated again. While
2~28313
-22-
a temperature of from 90 to 95-C was maintained, 260 g
of water was added thereto over 2 hours in order to
precipitate 1,3-bis[4-(4-nitrophenoxy)-a,a-
dimethylbenzyl]benzene. This solution containing the
precipitate was slowly cooled, and the resulting light
yellow crystals were collected by filtration and then
washed with a mixed solvent of N,N-dimethylformamide
and methanol. Afterward, the resulting sludge was
washed with 1 liter of methanol, filtered and then
dried, thereby obtaining 328 g of light yellow powder.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.1%.
The analytical results were as follows:
Melting point: 154.5 to 156.0-C
Values of elemental analysis:
C H N
Calcd. (%) 73.47 5.44 4.76
Found (%) 73.34 5.20 4.79
IR (KBr tablet method)
1330, 1490 cm~l (a nitro group)
1230 cm~l (an ether bond)
In a reactor equipped with a stirrer, a
thermometer, a reflux condenser and a dropping funnel,
there were placed 294 g (0.5 mole) of 1,3-bist4-
(4-nitrophenoxy)-a,a-dimethylbenzyl]benzene obtained
from the prece~ng step. This material was then mixed
with 1500 g of methyl cellosolve, 29.4 g of active
carbon and 2.9 g of ferric chloride-hexahydrate.
Afterward, stirring was continued for 3 hours, while a
temperature of from 100 to 105-C was maintained.
While the temperature was maintained, 150.2 g
Qf 80% hydrazine monohydrate was added dropwise over 3
hours. Furthermore, the solution was aged at this
-23- 2 028 313
temperature for 1 hour. Solids were then removed by
hot filtration. Afterward, the resulting filtrate was
concentrated and then recrystallized with isopropyl
alcohol. White crystals of 1,3-bis~4-(4-amino-
phenoxy)-a,a-dimethylbenzyl]benzene were obtained which
was the desired compound.
The white crystals were then filtered, washed
with isopropyl alcohol, and dried to obtain 228.5 g of
1,3-bis[4-(4-aminophenoxy)-a,a-dimethylbenzyl)benzene.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.0%
(total yield 81.4%).
The analytical results were as follows:
Melting point: 103 to 105.5-C
Values of elemental analysis:
C H N
Calcd. (%) 81.82 6.825.30
Found (%) 81.86 6.455.22
IR (KBr tablet method)
1620, 3340-3440 cm~1 (an amino group)
1240 cm~1 (an ether bond)
Example lC
In a reactor equipped with a stirrer, a
reflux condenser, a thermometer, a Dean/Stark water
separator and a nitrogen inlet pipe, there were placed
400 g of N,N-dimethylformamide from which water had
been removed, 161.0 g (0.4 mole) of 1,4-bis(4-hydroxy-
3,5-dimethyl-a,a-dimethylbenzyl)benzene, 138.7 g (0.88
mole) of 4-chloronitrobenzene and 66.3 g (0.48 mole) of
potassium carbonate, and 50 g of toluene was then added
thereto.
The resulting mixture was heated, and
stirring was then continued for 5 hours, while a
-24- 2028313
temperature of from 140 to 150C was maintained, in
order to carry out the reaction. Water produced by the
reaction was successively removed from the system by
azeotropy with toluene.
After completion of the reaction, the
resulting inorganic salts were removed by hot
filtration. The filtrate was cooled in to precipitate
1,4-bis~4-(4-nitrophenoxy)-3,5-dimethyl-~
dimethylbenzyl]benzene.
The precipitated light yellow crystals were
collected by filtration, washed with 500 ml of
methanol, and then dried. 245 g of light yellow powder
was obtained.
Using high-speed liquid chromatography, the
purity of the product was determined as 99.5%. The
analytical results were as follows:
Melting point: 224 to 226-C
Values of elemental analysis:
C H N
Calcd. (%) 74.51 6.25 4.34
Found (%) 73.94 5.92 4.54
IR (KBr tablet method)
1345, 1515 cm~1 (a nitro group)
1250 cm~l (an ether bond)
In a hermetic reduction reactor equipped with
a stirrer and a thermometer there were placed 129.0 g
(0.2 mole) of 1,4-bist4-(4-nitroph~nsYy)-3,5-
dimethyl-~ dimethylbenzyl]benzene obtained from the
prece~ng step, 322 g of N,N-dimethylformamide and 2.6
g of a 5% Pd/C catalyst. A hydrogen gas was then
introduced thereinto, while the solution was stirred
vigorously.
Reaction was continued at a reaction
temperature of from 30 to 40 C for 4 hours. At this
- 2028313
-25-
point, 26.9 liters of hydrogen had been absorbed by the
solution. Since further absorption was not observed
the reaction was brought to an end.
After completion of the reaction, filtration
s was effected at room temperature to remove the Pd/C
catalyst from the solution.
The resulting reaction solution was heated to
80 to 90 C. While this temperature was maintained, 80
g of water was added dropwise thereby over 2 hours in
order to precipitate 1,4-bist4-(4-aminophenoxy)-3,5-
dimethyl-~,~-dimethylbenzyl]benzene. The reaction
solution was slowly cooled, and the resulting light
yellow crystals were collected by filtration, washed
with 200 ml of methanol, and then dried, thereby
obtaining 104.7 g of 1,4-bis{4-(4-aminophenoxy)-3,5-
dimethyl-~,~-dimethylbenzyl]benzene.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.2 %
(total yield 85.0%). The analytical results were as
20 follows:
Melting point: 228 to 230-C
Values of elemental analysis:
C H N
Calcd. (%) 82.15 7.58 4.79
Found (%) 82.09 7.13 4.84
IR (KBr tablet method)
1630, 3320-3430 cm~l (an amino group)
1240 cm~l (an ether bond)
ExamDle lD
In a reactor equipped with a stirrer, a
reflux condenser, a thermometer, a Dean/stark water
separator and a nitrogen inlet pipe, there were placed
-' 202g313
-26-
400 g of N,N-dimethylformamide from which water had
been removed, 138.4 g (0.4 mole) of 1,4-bis(4-hydroxy-
a,a-dimethylbenzyl)benzene, 147.8 g (0.88 mole) of
m-dinitrobenzene and 66.3 g (0.48 mole) of potassium
carbonate, and 50 g of toluene was then added thereto.
The resultinq mixture was heated, and
stirring was then continued for 5 hours, while a
temperature of from 140 to 150-C was maintained, in
order to carry out the reaction. Water produced by the
reaction was successively removed from the system by
azeotropy with toluene.
After completion of the reaction, the
resulting inorganic salts were removed by hot
filtration. Then, the filtrate was heated again.
While a temperature of from 90 to 95C was maintained,
112 g of water was added thereto over 2 hours in order
to precipitate 1,4-bist4-(3-nitrophenoxy)-a,a-
dimethylbenzyl]benzene. This solution was slowly
cooled, and the resulting faintly brown crystals were
collected by filtration, washed with 500 ml of
methanol, and then dried. 220 g of faintly brown
powder was obtained.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.3%.
The analytical results were as follows:
Nelting point: 123 to 125-C
Values of elemental analysis:
C H N
Calcd. (%) 73.45 5.48 4.76
Found (%) 73.53 5.39 4.74
IR (XBr tablet method)
1355, 1525 cm~l (a nitro group)
1240 cm~1 (an ether bond)
~ 2028313
-27-
In a hermetic reduction reactor equipped with
a stirrer and a thermometer, there were placed 118 g
(0.2 mole) of 1,4-bis[4-(3-nitrophenoxy)-~
dimethylbenzyl]benzene obtained from the preceding
step, 295 g of N,N-dimethyl-formamide and 2.4 g of a 5%
Pd/C catalyst. A hydrogen gas was then introduced
thereinto, while the solution was stirred vigorously.
Reaction was continued at a reaction
temperature of from 30 to 40-C for 4 hours. At this
point, 26.9 liters of hydrogen had been absorbed by the
solution. Since further absorption was not observed,
the reaction was brought to an end.
After completion of the reaction, filtration
was effected at room temperature to remove the Pd/C
catalyst from the solution.
The resulting reaction solution was heated to
80 to 90 C. While this temperature was maintained, 126
g of water was added dropwise thereto over 2 hours in
order to precipitate 1,4-bis[4-(3-aminophenoxy)-
~
dimethylbenzyl]benzene. The reaction solution was
slowly cooled, and the resulting light yellow crystals
were collected by filtration, washed with 200 ml of
methanol, and then dried, thereby obtaining 90.3 g of
1,4-bis[4-(3-aminophenoxy)-~,~-dimethylbenzyl]benzene.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.2%
(total yield 80.0%).
The analytical results were as follows:
Melting point: 138 to 140-C
Values of elemental analysis:
C H N
Calcd. (%) 81.79 6.86 5.30
Found (%) 81.43 6.79 5.27
IR (KBr tablet method)
-28- 2028313
1630, 3320-3430 cm~l (an amino group)
1240 cm~l (an ether bond)
Exam~le lE
In a reactor equipped with a stirrer, a
reflux condenser, a thermometer, a Dean/Stark water
separator and a nitrogen inlet pipe, there were placed
400 g of N,N-dimethylformamide from which water had
been removed, 138.6 g (0.4 mole) of 1,3-bis(4-hydroxy
-~,~-dimethylbenzyl)benzene, 147.8 g (0.88 mole) of
m-dinitrobenzene and 66.3 g (0.48 mole) of potassium
carbonate, and 50 g of toluene was then added thereto.
The resulting mixture was heated, and
stirring was then continued for 5 hours, while a
temperature of from 140 to 150-C was maintained, in
order to carry out the reaction. Water produced by the
reaction was successively removed from the system by
azeotropy with toluene.
After completion of the reaction, the
resulting inorganic salts were removed by filtration.
Then, the filtrate was heated again. While a
temperature of from 90 to 95 C was maintained, 112 g of
water was added thereto over 2 hours in order to
precipitate 1,3-bis[4-(3-nitrophenoxy)-~,~-dimethyl-
benzyl]benzene. This solution was slowly cooled, andthe resulting faintly brown crystals were collected by
filtration, washed with 200 ml of methanol, and then
dried. 215 g of faintly brown powder was obtained.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.1%.
The analytical results were as follows:
Melting point: 84 to 86-C
Values of elemental analysis:
9 ~ 2028313
C H N
Calcd. (%) 73.45 5.48 4.76
Found (%) 73.70 5.40 4.46
IR (KBr tablet method)
1330, 1510 cm~l (a nitro group)
1245 cm~l (an ether bond)
In a hermetic reduction reactor equipped with
a stirrer and a thermometer, there were placed 118 g
(0.2 mole) of 1,3-bis-[4-(3-nitrophenoxy)-~
dimethylbenzyl]benzene obtained from the preceding
step, 295 g of N,N-dimethylformamide and 2.4 g of a 5%
Pd/C catalyst. A hydrogen gas was then introduced
thereinto, while the solution was stirred vigorously.
Reaction was continued at a reaction
temperature of from 30 to 40 C for 5 hours. At this
point, 26.9 liters of hydrogen had been absorbed by the
solution. Since further absorption was not observed
any more, the reaction was brought to an end.
After completion of the reaction, filtration
was effected at room temperature to remove the Pd/C
catalyst from the solution.
The resulting reaction solution was heated to
80 to 90-C. While this temperature was maintained, 126
g of water was added dropwise thereto over 2 hours in
order to precipitate 1,3-bist4-(3-aminophenoxy)-
~dimethylbenzyl]benzene. The reaction solution was
slowly cooled, and the resulting light yellow crystals
were collected by filtration, recrystallized from
methanol, and then dried, thereby obtaining 86.6 g of
1,3-bis~4-(3-aminophenoxy)-~,~-dimethylbenzyl)benzene.
Using high-speed liquid chromatography, the
purity of the product was determined as being 99.0%
(total yield 75.0%).
The analytical results were as follows:
Melting point: 94 to 96-C
2028313
-30-
Values of elemental analysis:
C H N
Calcd. (%) 81.82 6.825.30
Found (%) 81.35 6.96 5.44
IR (KBr tablet method)
1625, 3340-3440 cm~l (an amino group)
1235 cm~l (an ether bond)
Example 2A
In a reactor equipped with a stirrer, a
thermometer, a reflux condenser having a water
separator and a dropping funnel there were placed 26.5
g (0.27 mole) of maleic anhydride, 1.6 g of
p-toluenesulfonic acid and 150 ml of toluene, and the
temperature of the solution was increased to maintain
lS the reflux condition of toluene. A solution prepared
by dissolving 52.8 g (0.1 mole) of 1,3-
bis[4-(4-aminophenoxy)-a,~- dimethylbenzyl]benzene
formed in Example lB in 100 ml of toluene was added
dropwise to the above-mentioned solution through a
dropping funnel over 7 hours. After completion of the
addition, the solution was aged for 2 hours. Water
produced by the reaction was collected by a water
separator provided on the reflux condenser from the
beg; nn; ~g of dropping to the end of aging. After
completion of the reaction, the solution was cooled to
70-C, and 100 ml of warm water was added thereto.
Stirring followed at this temperature for 30 minutes.
After stAn~ing, the lower layer (the water layer) of
two separate layers was drawn out, and 100 ml of warm
water was again added thereto. Washing and liquid
separation followed in like manner.
The resulting upper layer of a toluene
solution was finally concentrated in vacum at a
2028313
r?
-31-
temperature of not more than 130C to obtain 68 g of
reddish brown transparent resinous crude 1,3-
bis~4-(4-maleimidophenoxy)-~,~-dimethylbenzyl]
benzene which was the desired compound. Its yield was
quantitative, and its purity according to HLC was 91%.
Afterward, the crude crystals were purified
by recrystallization to obtain pure light yellow
powder. The analytical results were as follows:
Melting point: 172 to 175C
Values of elemental analysis (C44H36N206):
C H N
Calcd. (%) 76.70 5.27 4.07
Found (%) 76.20 5.43 4.22
A mass spectrum is shown in Fig. 1, and an IR
spectrum is shown in Fig; 2.
Example 2B
In the same reactor used in Example 2A, there
were placed 26.5 g (0.27 mole) of maleic anhydride, 0.7
g of phosphoric acid and 150 ml of mixed xylene. A
solution was then added dropwise thereto under reflux
of xylene, the solution having been prepared by
thermally dissolving 52.8 g (0.1 mole) of 1,4-bis~4-
(4-aminophenoxy),a,~-dimethylbenzyl)benzene obtained in
Example lA in a mixture of 100 ml of mixed xylene and
50 ml of N,N-dimethylformamide. A dropping time of 5
hours and an aging time of 2 hours were used in order
to carry out the reaction. The reaction solution was
concentrated to about 1/2 the original amount and then
cooled. When a temperature of 80-C had been reached;
50 g of water was added thereto. Afterward, the
solution was slowly cooled with stirring, so that
crystals were precipitated. The crystals were then
cooled to room temperature, filtered, and washed with
- 2028313
-32-
cold methanol. 66.5 g of desired crude 1,4-bis[4-(4--
maleimidophenoxy)-~,~-dimethylbenzyl]benzene was
obtained. Its yield was 96.7%, and its purity
according to HLC was 97.5%.
The product was then purified by
recrystallization from toluene in order to obtain pure
light yellow powder. The analytical results were as
follows:
Melting point: 214 to 216-C
Values of elemental analysis (C44H36N206):
C H N
Calcd. (%) 76.70 5.27 4.07
Found (%) 76.02 5.51 4.18
The results of a mass spectrum were the same
as for the compound of Example 2A (M/Z; M+ 688, 673,
408).
Experiment 1
With regard to the bismaleimides obtained in
Examples 2A and 2B, their solubility in some solvents
were investigated. The results are set forth in Table
1.
33 2028313
Table 1
Solubility of Bismaleimide in Solvent*
(g/loo ml)
Compound Compound N,N'(Methylene-
of of di-P-phenylene)-
Solvent Example 2A Example 2B bismaleimide
Toluene >50 8 <1
Tetra- >50 12 6
hydro-
furan
Methylene <50 15- 8
Chloride
* Measurement was made at 25-C.
Example 2C
In a reactor equipped with a stirrer, a
thermometer, a reflux condenser having a water
separator and a dropping funnel there were placed 26.5
g (0.27 mole) of maleic anhydride, 1.6 g of p-toluene-
sulfonic acid and 150 ml of toluene. The temperature
of the solution was increased to maintain the reflux
condition of toluene. A solution prepared by
dissolving 58.8 g (0.1 mole) of 1,4-bis~4-(4-
aminophenoxy)-3,5-dimethyl-~,~-dimethylbenzyl~benzene
formed in Example lC in 100 ml of toluene was added
dropwise to the above-mentioned solution through a
dropping funnel over 7 hours. After completion of the
~ 2028313
-34-
addition, the solution was aged for 2 hours. Water
produced by the reaction was collected by a water
separator provided on the reflux condenser from the
beginning of dropping to the end of aging. After
completion of the reaction, the solution was cooled to
70-C, and 100 ml of warm water was added thereto.
Stirring followed at this temperature for 30 minutes.
After standing, the lower layer (the water layer) of
two separate layers was drawn out, and 100 ml of warm
water was again added thereto. Washing and liquid
separation followed in like manner.
The resulting upper layer of a toluene
solution was cooled in order to precipitate light
yellow crude 1,4-bis[4-(4-maleimidophenoxy)
-3,5-dimethyl~ -dimethylbenzyl]benzene, which was
collected by filtration and then dried. 65.2 g of
product was obtained.
Its yield was 87.5%, and its purity according
to HLC was 96.4%.
Afterward, the crude crystals were purified
by recrystallization to obtain a pure light yellow
powder. The analytical results were as follows:
Melting point: 232.5 to 235-C
Values of elemental analysis (C48H44N206):
C H N
Calcd. (%) 77.38 5.95 3.76
Found (%) 77.05 6.08 3.66
An IR spectrum of the product is shown in
Fig. 3.
Example 2D
In a reactor equipped with a stirrer, a
thermometer, a reflux condenser having a water
separator and a dropping funnel there were placed 26.5
g (0.27 mole) of maleic anhydride, 1.6 g of
2028313
p-toluenesulfonic acid and 150 ml of toluene. The
temperature of the solution was increased to maintain
the reflux condition of toluene. A solution prepared
by dissolving 52.8 g (0.1 mole) of 1,3
bis[4-(3-aminophenoxy)-~,~-dimethyl-benzyl]benzene
formed in Example lE in 100 ml of toluene was added
dropwise to the above-mentioned solution through a
dropping funnel over 7 hours. After completion of the
addition, the solution was aged for 2 hours. Water
produced by the reaction was collected by a water
separator provided on the reflux condenser from the
beginning of dropping to the end of aging. After
completion of the reaction, the solution was cooled to
70-C, and 100 ml of warm water was added thereto.
Stirring followed at this temperature for 30 minutes.
After standing, the lower layer (the water layer) of
two separate layers was drawn out, and 100 ml of warm
water was again added thereto. Washing and liquid
separation followed in like manner.
The resulting upper layer of a toluene
solution was finally concentrated in vacuo at a
temperature of not more than 130C to obtain 68.2 g of
reddish brown transparent resinous crude
1,3-bist4-(3-maleimidophenoxy)-a,~-dimethyl-
benzyl]benzene. This was the desired compound.
Its yield was quantitative, and its purity
according to MLC was 96.3%.
Afterward, the crude crystals were purified
by column chromatography to obtain a pure light yellow
30 resinous product.
Elemental analysis (C44H36N206)
C H N
Calcd. (%) 76.70 5.27 4.07
Found (%) 76.41 5.44 4.01
-36- ~028313
Mass spectrum
M/Z; M+ 688, 673, 408
An IR spectrum of the product is shown in
Fig. 4.
Example 2E
In the same reactor used in Example 2D, there
were placed 26.5 g (0.27 mole) of maleic anhydride, 0.7
g of phosphoric acid and 150 ml of mixed xylene. A
solution was then added dropwise thereto, the solution
having been prepared by thermally dissolving 52.8 g
(0.1 mole) of 1,4-bist4-(3-aminophenoxy)-~,~-dimethyl-
benzyl]benzene obtained in Example lD in a mixture of
100 ml of mixed xylene and 50 ml of N,N-dimethyl-
formamide. A dropping time of 5 hours and an aging
time of 2 hours were used in order to carry out the
reaction. The reaction solution was concentrated to
about 1/2 the original amount and then cooled. When a
temperature of 80C had been reached, 50 g of water was
added thereto. Afterward, the solution was slowly
cooled, so that crystals precipitated. The
precipitated crystals were then cooled to room
temperatures filtered; and washed with cold methanol.
67.1 g of desired crude 1,4-bist4-(3-maleimi-
dophenoxy)-~,~-dimethylbenzyl]benzene was obtained.
Its yield was 94.8%, and its purity according
to HLC was 92.0%.
The product was then purified by
recrystallization from toluene in order to obtain pure
light yellow powder. The analytical results were as
follows:
Melting point: 192 to 193C
Elemental analysis (C44H36N206)
_37_ 2028313
C H N
Calcd. (%) 76.70 5.27 4.07
Found (%) 76.66 5.33 4.18
The results of a mass spectrum were the same
as from the compound from Example 2D (M/Z; M+ 688, 673,
408).
Examples 3A to 3C and Comparative Exam~les 3A
and 3B
In a reactor equipped with a stirrer, a
reflux condenser and a nitrogen inlet pipe, there were
placed 1,3-bis[4-(4-maleimidophenoxy)-~,~-dimethyl-
benzyl]benzene obtained in Example 2A and
1,3-bis[4-(4-aminophenoxy)~,a-dimethylbenzyl]benzene
obtained in Example lB in the molar ratio set forth in
Table 2. They were then heated and melted at 180-C for
20 minutes. Then, they were defoamed at 150C under
reduced pressure (from 10 to 15 mmHg) for 30 minutes.
Afterward, the mixture was cooled to room temperature,
thereby obtaining a brown transparent glassy solidified
resin forming composition. A mold which had been
heated to 180-C was filled with the resin
forming composition, while the resin composition was
heated and melted. The composition was maintained at
200 C under 50 kg/cm2 for 30 minutes. After this
compression molding, the molded material was taken out
o~ the mold and postcured in an oven at 250-C for 4
hours. Cured test pieces (length 127 mm, width 12.7
mm and thickness 6.4 mm) were obtained.
For these test pieces, h~n~ing strength,
flexural modulus, the starting temperature of thermal
decomposition and hygroscopicity were measured, and the
results are set forth in Table 2.
~- 2028313
-38-
COMPARATIVE EXAMPLE 3C
N,N',4,4'-Diphenylmethanebismaleimide and
4,4'diaminodiphenylmethane were used to prepare a
composition (molar ratio 2/1) shown in Table 2.
S Afterward, the same operation explained with respect to
Examples 3A to 3C was followed, and the results set
forth in Table 2 were obtained.
COMPARATIVE EXAMPLE 3D
Kelimide 601 (made by Nippon Polyimide Co.,
Ltd.) was used as a resin forming composition.
afterward, the same operation explained with respect to
Examples 3A to 3C was followed, and the results set
forth in Table 2 were obtained.
EXAMPLES 3D TO 3G
In a stainless steel reactor equipped with a
stirrer, a reflux condenser and a nitrogen inlet pipe,
there were placed each of the bismaleimide compounds
prepared in Examples 2A, 2D, 2C and 2E and each of the
diamine compounds prepared in Examples lA, lC and lB in
the molar ratios shown in Table 3. N-methyl-2-pyrroli-
done was poured into the solution in such an amount
that the concentration of a resin was 55% by weight,
followed by heating at 150C for 50 minutes. The
resulting varnishy solution was discharged into water
to form a precipitate. The precipitate was then
collected by filtration, washed with water, and then
dried with hot air at 80-C for 15 hours. The product
wa~ further dried at 110C for 20 minutes, and
additionally at 130-C for 30 minutes. It was then
ground in a mortar and then p~Cse~ through a sieve of
60 mesh, to obtain a polyaminobismaleimide type
thermosetting resin forming composition. Afterward,
the same operation described with respect to Examples
2028313
-39-
3A to 3C was followed, and the results set forth in
Table 3 were obtained.
In these examples, physical properties of
thermosetting resins and the like were measured in the
following manner.
Ren~ing strength and flexural modulus were
measured in accordance with ASTM-D-790.
Starting temperature of thermal
decompositions was determined as the temperature at
which weight begins to decrease and was measured at a
temperature rise rate of 10C/minute in air in
accordance with the TGA method.
Izod impact strength was measured in
accordance with ASTM-D 256.
Hygroscopicity was measured in accordance
with ASTM-D570-63.
2028313
--40--
C
o ~ ~
c~ E
~ n
o -- _
~ o --
._ ~ o ~
~ ~ ~ _
~ o ._ ~ ~,
~ E ~ O o o _
-- E 111 n
o ~ .. o
a u
_ -- ~ ~ ~ ._
o ~
~ ._Cl~ ~ ._
E ._ ._
E 111~ ~1
._ . , _
o ~~ ~
._
E
~ _
O -- X
o
~ O
n _ ~ c
C ~.
o ~
o
._ - n
o. ~ _ . o
0 _
o ~ ~
o _ ~ E E
~ ~ o o o o ~. ._
E O ~ J v-- ~ _
n v ~ ~ o._ o
._ ~ _._._ _ _ _ ~ ~o
._ . _ 0 U
o
0 ~
_ ._ ~, ~ ._
Y
o
< ~ U
m o
. . . .
X X X X
~1. ~~. . . . .
E E E~ ~ O. C-
~0 ~11 ~ E E E E
X X XO
Tcble 2 ~Continued)
l~od impDct Temp. at
~olcrstrength Bending Fl-xur-l 0.5% ~eight Hygro-
retio(no notch) 6trength modulus decre-5e scopicity
l/IV~kg.cm/cm) (k9/~m2) ~ks/mm2) (-C) (X)
Excmple 3A 0.3 37 15.0 318 360 0.26
Exsmple 3B 0.5 41 16.1 ~ 316 357 0.29
Ex~mple 3C 1.0 38 15.4 317 362 0.30
Comp. Ex. 3A 1.5 37 15.8 305 315 0.27
Comp. Ex. 3B 0 2 5.2 460 412 0.90
Co~p. Ex. 3C 0.5 11 9.2 372 359 0.89
Comp. Ex. 3D 9 8.1 342 366 0.90
t`g
o
C~
~- 2028313
--42--
_ U ^ ~ o ~ o
~' C ~ _ ~ C ~ C
X U X ~ X ~ X ~D
o ... o C o .... o ~
C C C V C C C C
o U o o o ~ o o
c n c E C D c ~
Q. _ o. ~ C _
o -- o ~ o -- o --
~C ~ C C >. C
OE C E ~ E C E C
~ o~ o 3 o
C E . n., c. n , ~
o~ --~ -- o ~t -- ~t --
o ~ _ ~
--~ c C c
_ O ~ o
C-- ~ -- ~ ~ -- o -- o
o E _ ~o E ~ E
E~-- _~_ ._ _._ ._ ._ ._
_~o n ~~ ~ ~n 1~ n v
._ , .. . .~ . . . .
o
o _ ~_ ~ n~
E
o
o
^ x x X x
c ~ o o o o
~_ _ c ~ c ~ c . c o
U C ~D C ~ -- O C
c ~ --o -- ~ -- O
E ~ c~ ~ c Q ~
c o co c o ~ o C
o ~ o ~ o
o ~_ n ~_ n ~- E ~- n
O ~ E _E _ E ~-- E _
E ~-- -- ~_ _ ._ ~ ._ _
O o >. o ~ u o
~ c~ c ~o ~ ~ c
c o E uE oE ~J O E o
~ . n. ~ ., c . n
C ~ ~
U
O _ --~
oo Eu~ E _u~ E
~c .__ ._ ._._ ._ _ ._ ._
rll D ~ n ~~ ~ ~ n ~
O ~ ~
_ ~ n-- ~
o o o o
E E E E
a
x x X x
Table 3 ~Continued)
Izod i-p-ct Te-p t
~ol-ratrength Oending Fl-xur-lO SX ~ ht Hygro-
r-tio~no notch) strengthuodulua decrease scopicit
I~IV~kg c-/c-~ ~ks/--2) ~ks/uu2) ~-C) ~X)
Ex--ple 3D 0 537 15 5 325 362 0 27
Ex--ple 3E 0 535 15 7 320 359 0 24
Ex-~ple 3F 0 5 35 15 0 340 360 0 29
Ex--pl- 3C 0 536 14 8 332 356 0 26