Note: Descriptions are shown in the official language in which they were submitted.
.....
FINT B7576EPC
AROMA'PIC CONVERSION PROCESSES
AND CATALXSTS LTSEFL1L THEREIN
FIELD OF THE INVENTION
This invention relates to aromatic conversion processes
including alkylation and transalkylation processes involving aro-
matic compounds including zeolite catalysts useful in such pro-
cesses and more particularly to alkylation-transalkylation pro-
cesses involving alkylation of a benzene feedstock with a C2-C4
alkylating agent and liquid phase transalkylation of resulting
polyalkylbenzenes, treatment of the alkylation product in a
separation zone, and recycle of at least a portion of the trans-
alkylation product to the separation zone.
-2- .... ~Q~~~3~
BACKGROUND OF THE INVENTION
Molecular sieves and their use in aromatic conversion
processes are well known in the chemical processing and refining
industry. Aromatic conversion reactions of considerable commer-
cial importance include the alkylatic>n of aromatic compounds such
as in the production of ethyltoluene, xylene, ethylbenzene, cu-
mene, or higher alkyl aromatics and in disproportionation reac-
tions such as toluene disproportionation, xylene isomerization,
or the transalkylation of polyalkylbenzenes to monoalkylbenzenes.
Such alkylation, transalkylation or other aromatic conversion
processes may be carried out in the liquid phase, in the vapor
phase, or under conditions in which both liquid and vapor phases
exist.
An example of vapor phase alkylation is found in U.S.
Patent No. 4,107,224 to Dwyer. Here, vapor phase ethylation of
benzene over a zeolite catalyst is accomplished in a down flow
reactor, The output from the reactor is passed to a separation
system in which ethylbenzene product is recovered, with the re-
cycle of polyethylbenzenes to the alkylation reactor where they
undergo transalkylation reactions with benzene. The Dwyer cata-
lysts are characterized in terms of those having a constraint
index within the approximate range of 1 to 12 and include, with
the constraint index in parenthesis, ZSM-5 (8.3), ZSM-11 (8.7),
ZSM-12 (2>, ZSM-35 (4.5), ZSM-38 (2), and similar materials.
Various molecular sieves, including, inter alia, zeolite beta
(constraint index 0.6), are disclosed as having constraint indi-
....
ces outside of the range suitable for the Dwyer ethylbenzene pro-
duction process.
U.S. Patent No. 3,551,510 to Pollitzer et al. discloses
an alkylation-transalkylation process in which the output from
the alkylation reaction zone is passed directly to the trans-
alkylation zone. In Pollitzar, alkylation is carried out using
an alkylating agent, characterized as an olefin acting compound,
over a solid phosphoric acid alkylation catalyst. The olefin
acting compound may be selected from materials such as monoole-
fins, diolefins, polyolefins, actylenic hydrocarbons, alkyl hali-
des, alcohols, ethers and esters. The output from the alkylation
reaction zone, which includes polyethylbenzenes, is supplied to a
transalkylation reaction zone along with an aromatic substrate,
e.g., benzene. The transalkylation zone contains an acid ex-
tracted crystalline aluminosilicate catalyst, specifically mor-
denite, and is operated in an upflow mode. Exemplary transalkyl-
ation conditions include a liquid hourly space velocity of 1.0, a
pressure of 500 psig and a temperature of 250°C. The output from
the transalkylation zone is supplied to a separation zone from
which a polyalkylaromatic, e.g., polyethylbenzene, is withdrawn
and recycled to the alkylation zone.
Another alkylation-transalkylation process is disclosed
in U.S. Patent No. 4,008,90 to Ward. Ward, like the patent to
Pollitzer, discloses the use of a solid phosphoric acid catalyst
in the alkylation zone. In Ward, benzene is reacted with propy-
lene to produce curnene. The output from the alkylation reactor
is split so that a portion principally benzene and cumene, is re-
-
cycled to the alkylation reactor. Another portion containing
principally benzene, cumene, propane and di- and tri- isopro-
pylbenzene is supplied to a separation zone. In the separation
zone a di- and tri-isopropylbenzene :rich stream is separated and
supplied along with benzene to a transalkylation zone which con-
tains a solid phosphoric acid catalyst. A cumene rich effluent
from the transalkylation zone is recycled to the separation zone.
U.S. Patent No. 4,169,111 to Wight discloses an alkyla-
tion-transalkylation process for the manufacture of ethylbenzene
employing crystalline aluminosilicates in the alkylation and
transalkylation reactors. The catalysts in the alkylation and
transalkylation reactors may be the same or different and include
low sodium content zeolites, preferably less than 0.5 weight per-
cent Na20, having silica/alumina mole ratios between 2 and 80 and
preferably between 4-12, Exemplary zeolites include molecular
sieves of the X, Y, L, B, ZSM-5 and omega crystal types with
steam stabilized Y zeolite containing about 0.2$ Na20 being pre-
(erred. The alkylation reactor is operated in a downflow mode
and under temperature and pressure conditions in which some
liquid phase is present. The transalkylation reactor, which is
described as generally requiring higher temperatures than the
optimum temperature for alkylation in order to achieve maximum
transalkylation efficiency, is also operai~ed in a downflow mode.
In Wight, the output from the alkylation reactor is cooled and
supplied to a benzene column from which benzene is recycled to
the alkylation rea<aor. The bottoms fraction from the benzene
column is supplied to an ethylbenzene column from which ethylben-
...
-5-
zene is recovered as the process product. The bottoms product
from the ethylbenzene column is supp:l.ied to a third column which
is operated to provide a substantially pure diethylbenzene over-
heads fraction which contains from 10 to 90$, preferably 20 to
60~ of the total diethylbenzene feed to the column. The diethyl-
benzene overheads fraction is recycled to the alkylation reactor
while a side cut containing the remaining diethylbenzene and tri-
ethylbenzene and higher molecular weight compounds is supplied to
the transalkylation reactor along with benzene. The effluent
from the transalkylation reactor is recycled to the benzene
column.
U.S. Patent No. 4,774,377 to Barger et al. discloses an
alkylation/transalkylation process which, like the above-
described Wight process, involves the use of separate alkylation
and transalkylation reaction zones, with recycle of the trans-
alkylated product to an intermediate separation zone. In the
Barger process, the temperature and pressure conditions are ad-
justed so that the alkylation and transalkylation reactions takE
place in essentially the liquid phase. The transalkylation cata-
lyst is an aluminosilicate molecular sieve including X-type, Y-
type, ultrastable-Y, L-type, omega type and mordenite type zeo-
lites with the latter being preferred. The catalyst employed in
the alkylation reaction zone is a solid phosphoric acid contain-
ing material. Aluminosilicate alkylation catalysts may also be
employed and water varying from 0.01 to 6 volume percent is
supplied to the alkylation reaction zone. The output from the
alkylation reaction zone is supplied to first and second separa-
...
-6-
tion zones. Water is recovered in the first separation zone. In
the second reaction zone intermediate aromatic products and tri-
alkylaromatic and heavier products are separated to provide an
input to the transalkylation reaction zone having only dialkyl
aromatic components, or diethylbenzene in the case of an ethyl-
benzene manufacturing procedure or diisopropylbenzene in the
case of cumene production. A benzene substrate is also supplied
to the transalkylation zone for the transalkylation reaction and
the output from the transalkylation zone is recycled to the first
separation zone. The alkylation and transalkylation zones may be
operated in downflow, upflow, or horizontal flow configurations.
As noted previously, various zeolitic molecular sieves
are known for use in alkylation and/or transalkylation proce-
dures. As indicated in the aforementioned U.S. Patent No.
4,107,224 to Dwyer, certain molecular sieves, including zeolite
beta, are characterized as being unsuitable for use in the pro-
duction of relatively low molecular weight alkylbenzenes such as
ethylbenzene. In contrast to the relatively low molecular weight
alkylbenzenes disclosed in the Dwyer et al. patent, U.S. Patent
No. 4,301,316 to Young, discloses the use of molecular sieves to
produce relatively high molecular weight alkylbenzenes which may
be used as precursors for alkyl aryl sulfonate detergents. In
Young, relatively long chain length alkylating agents having one
or more reactive alkyl groups of at least 5 carbon atoms are em-
ployed in the alkylation of benzene in the presence of a
crystalline zeolite alkylation catalyst. The reactants may be in
the vapor phase or the liguid phase and the zeolite catalysts may
-7- .....
be modified or unmodified. Preferred zeolite catalysts include
zeolite beta, ZSM-4, ZSM-20, ZSM-38, and synthetic and naturally
occurring isotypes thereof such as zeolite omega and others. As
described in Young, the zeolites may be subject to various chemi-
cal treatments including alumina extraction and combination with
one or more metal components such as the metals of groups IIB,
III, IV, VI, VIIA, and VIII. The zeolites may also be subjected
to thermal treatments including steaming or calcination in air,
hydrogen or an inert gas. Specifically disclosed is the reaction
of benzene and 1-dodecene over zeolite beta (Si02/A1203 = 175) in
a flow reactor at 250°C and 600 psig.
LJ.S. Patent No. 4,185,040 to Ward et al. discloses an
alkylation process employing a molecular sieve catalyst of low
sodium content which is said to be esgecially useful in the pro-
duction of ethylbenzene from benzene and ethylene and cumene from
benzene and propylene. The Na20 content of the zeolite should be
less than 0.7 weight percent and preferably less than 0.5 weight
percent. Examples of suitable zeolites include molecular sieves
of the X, Y, i~, B, ZSM-5, and omega crystal types, with steam
stabilized hydrogen Y zeolite being preferred. Specifically dis-
closed is a steam stabilized ammonium Y zeolite containing about
0.2$ Na20. The alkylation process may be carried out with either
upward or downward :Flow, the latter being preferred, and prefer-
ably under temperature and pressure conditions so that at least
some liquid phase is present, at least until substantially all of
the olefin alkylating agent is consumed. Ward et al. states that
rapid catalyst deactivation occurs under most alkylating con-
~_. ~~~J~~'~~
_8-
ditions when no liquid phase is present.
Another alkylation procedure is disclosed in European
Patent Application No. 272,830 to Ratcliffe et al. The Ratcliffe
procedure employs molecular sieve alkylation catalysts which have
been treated in a manner to improve selectivity to monoalkyla-
tion, specifically in the propylation of.benzene to produce cu-
mene. Selectivity is said to be increased by at least one
percentage point by first depositing a carbonaceous material on
the catalyst and then subjecting the resultant carbon containing
catalyst particles to combustion. Specific zeolitic crystalline
molecular sieves include those selected from the group of Y
zeolites, fluorided Y zeolites, X zeolites, zeolite beta, zeolite
L, and zeolite omega. The zeolites may be modified to arrive at
products of reduced alumina content and reduced sodium content.
A preferred zeolite is Y zeolite produced by first ammonium ex-
changing to a sodium content of about 0.6-5.0 wt.~, expressed as
Na20, calcining at a temperature of about 315°-900°C in the
pre-
sence of steam, and then ammonium exchanging the steam-calcined
zeolite to obtain a product having less than 1.0 weight percent
and preferably less than about 0.2 weight percent sodium, ex-
pressed as Na20.
Zeolite beta referred to in certain of the references
addressed previously is a crystalline aluminosilicate molecular
sieve zeolite which finds application in a number of industrial
processes including as a catalyst in various hydrocarbon conver-
sion reactions such as hydrocracking, hydroisomerization and de-
waxing. Zeolite beta, like many other molecular sieve zeolites,
...
_g_
is synthesized by the hydrothermal digestion of a reaction mix-
ture comprising silica, alumina, an alkali alkaline earth metal
and an organic templating agent. ThE~ organic agent acts as a
template in the nucleation and growth of the zeolite beta
crystals. Once the crystals are formed, it is conventional prac-
tice to carry out a calcination treatment in order to remove the
organic material from the interstitial channels of the molecular
sieve network.
Crystalline zeolite beta, which is identified by its x-
ray diffraction pattern, and basic procedures for its preparation
are disclosed in U.S. Patent No. 3,308,069 to Wadlinger et al.
The chemical composition of zeolite beta in the as synthesized
form as disclosed in the patent to Wadlinger et al. may be
characterized as follows:
CX m (1.0 1-x) TEAJ A102 ~ ySi02 ' WH20
2
wherein: X is less than 1,
m is at least one cation, usually an alkali metal or
alkaline earth metal, more specifically sodium,
n is the valence of m, y is from about 5 to 100,
W is about 4, and
TEA represents the tetraethylammonium ion.
As described in Wac'tlinger et al., zeolite beta may be formed from
a mixture in water of tetraethylammonium hydroxide and suitable
sources of sodium monoxide (or hydroxide), alumina, and silica.
Typical reaction mixture compositions, in terms of mole ratios,
fall within the following ranges:
_lo_
Si02/A1203 - from about ZO to about 200;
Na20/tetraethylammonium hydroxide (TEAOH) - from about
0-0.1;
TEAOH/Si02 - from about 0.1--1.0;
H20/TEAOH - from about 20 to about 75.
The resulting reaction mixture can be heated at a temperature of
about 75° to about 200°C until crystallization of the molecular
sieve occurs. The crystallized product can be separated from the
reaction mixture by filtration or centrifuging and then washed
with water and dried to remove water from the molecular sieve
network. The product can then be calcined in air or in an inert
atmosphere in order to remove the templating agent as described
above.
The Wadling:r patent discloses that the catalytic mater-
ials can be prepared by calcining the original sodium form of the
zeolite beta and/or replacing the major portion of the sodium
ions with other metallic or ammoniacal ions. Specifically dis-
closed in Wadlinger (Example 2) is a composition containing after
calcination in air at 55°C, 0.7 mole percent Na20. Disclosed in
Example 8 is a product formed by treating a dried product which
was exchanged continuously fox 48 hours with 2~ solution of ammo-
nium chloride. After washing free of excess chlorine ions, the
catalyst was dried and calcined for 3 hours at 1000°F to produce
an acid beta aluminosilicate having 0.07$ Na content.
Various other procedures are known for the synthesis of
zeolite beta. European Patent Application 159,846 by Rubin,
-11-
discloses the synthesis of zeolite beta having a silica/alumina
mole ratio of up to 300 employing a templating agent formed by
the combination of dimethylbenzylamine and benzyl halide. The
hydrothermal digestion procedure in which the crystals are formed
is carried out at a temperature below 175°C in order to avoid the
formation of undesirable side effects. The zeolite beta produced
in accordance with the Rubin application, when employed either as
an absorbent or a catalyst, can be at least partially dehydrated
by heating at a temperature of about 200°-600°C in an air or
nitrogen atmosphere for about 1-48 hours. The inorganic rations
of freshly synthesized zeolite beta can be decomposed by heating
to a temperature up to about 550°C for 1-48 hours. Zeolite beta
prepared in accordance with the Rubin process can have the origi-
nal rations associated therewith replaced by a wide variety of
other rations including hydrogen, ammonium and metal rations and
mixtures thereof.
European Patent Application 165,208 by Bruce et al.,
discloses a procedure for the preparation of zeolite beta simi-
lar to that disclosed in the aforementioned Rubin application ex-
cept that the templating agent is a dibenzyl dimethyl ammonium
halide or hydroxide with the silica/alumina components employed
to provide a silica/alumina mole ratio in the synthesized product
of about 20-250.
U.S. Patent x,642,226 to Calvert et al. discloses a pro-
cess for the preparation of zeolite beta which is similar to
those disclosed in the aforementioned European patent applica-
tions and which employs dibenzyl dimethylammonium hydroxide or
....
-12-
chloride as a templating agent. The reaction mixture in Calvert
is heated at a temperature of about 80° to about 175°C for about
1 to about 120 days. The Calvert patent states that the zeolite
beta can be used in either the organic nitrogen-containing an
alkali metal containing form, the alkali metal form and hydrogen
form or another univalent or multivalent cationic form. Calvert
also discloses that zeolite beta can be used in intimate combina-
tion with a metallic component, e.g., a hydrogenation component
such as tungsten, vanadium, molybdenum, rhenium, nickel, cobalt,
chromium, manganese, or a noble metal such as platinum or palla-
dium. The patent further states that the zeolite beta should be
at least partially dehydrated when employed either as an absor-
bent or as a catalyst or as a hydrocarbon conversion catalyst.
Chlorides, nitrates, and sulfates are disclosed as ion exchange
agents. Calvert et al. discloses zeolite beta of relatively low
sodium content, e.g., 0.14 wt.~ Na and 0.11 wt.~ Na.
Another process for the preparation of zeolite beta is
disclosed in European Patent Application 164,939 by Calvert. The
synthesis procedures disclosed here are similar to those in the
above-mentioned references, except that a tetraethylammonium bro-
mide or hydroxide templating agent is employed to produce a par-
tially crystalline product of extremely high silica/alumina ratio
which is said to be less expensive than fully crystalline zeolite
beta which is dealuminized to provide a corresponding silica/
alumina mole ratio. The digestion period in this procedure is
for a period of about 1-7 days at a temperature of 90°-200°C.
The silica/alumina ratio of the zeolite beta produced here ranges
2,~~~~~~:~~
-13-
from 20-1000 and is preferably greater than 200.
European Application 186,447 by Kennedy et al., disclo-
ses the use of zeolite beta in catalytic cracking processes. The
zeolite beta may be used in the as-synthesized form following
calcination and be of either low or high silica/alumina activi-
ties, It may be synthesized with trivalent framework ions other
than aluminum to form, for example, borosilicates, boroaluminosi-
licates, gallosilicates, or galloaluminosilicates structural iso-
types, which are considered to constitute forms of zeolite beta. .
The zeolite beta may be acid extracted to form the high silica/
alumina products.
As noted previously, various aromatic conversion pro-
cesses may be carried out in either or both of the liquid and
vapor phases. At the relatively high temperatures involved in
vapor phase reactions, it is generally accepted that water pre-
sent in the feed stream is detrimental to the reaction process.
While various reasons are advanced for the adverse impact of
water, the most widely observed detrimental effect is probably
catalyst deactivation due to dealumination. For example, U.S.
Patent I~o. 4,197,214 to Chen et al. discloses a process fox
modifying various crystallined zeolite molecular sieves such as
ZSM-5, ZSM-11, ZSM-:L2, ZSM-35, ZSM-38, faujasite, mordenite, and
erionite by the inclusion of metallic ions such as zinc. Chen et
al. state that high temperature steam functions by way of a
hydrolysis reaction to cause loss of framework aluminum which is
accompanied by the :Loss of the associated protons, leading to a
reduction in catalytic activity. The hydrolysis reaction is said
,..
-14-
to be quite slow at temperatures below about 800°F. However, at
higher temperatures above 900°F, the reaction rate is sufficient-
ly fast to affect long term stability of the zeolite catalyst.
In some cases, water can be tolerated under the high
temperature conditions involved in vapor phase reactions. For
example, the aforementioned patent to Dwyer states that water and
hydrogen sulfide are tolerable if more rapid aging of the cata-
lyst is acceptable, but are moderately detrimental in the pro-
cess. Steam stabilized zeolites are disclosed as useful in aro-
matic conversion processes involving alkylation such as in the
production of ethylbenzene or cumene. Thus, the aforementioned
patent to Ward et al. discloses that steam stabilized hydrogen Y
zeolite is preferred in the alkylation of benzene to produce
ethylbenzene or cumene.
The use of steam stabilized zeolites in the production
of high molecular weight alkyl benzenes is disclosed in the
aforementioned patent to Young in which relatively high molecular
weight alkylating agents are used in either the vapor phase or
the liquid phase. The zeolite catalyst may be subjected to
modifying treatments involving steaming for periods ranging from
about one quarter to about 100 hours in an atmosphere containing
from about 5 to about 100 steam.
In hydrocarbon conversion processes involving olefin
conversion, water m;ay or may not be tolerated in the feed,stream,
depending on the nature of the molecular sieve employed. For ex-
ample, LI.S. Patent No. 4,551,438 to Miller discloses the oligo--
merization of olefins over molecular sieves such as ZSM-5, ZSM-11
s '1
-ls-
and silicalite characterized as intermediate pore size having an
effective pore aperture in the range of about 5 to 6.5 angstroms.
Miller discloses that the feed should contain less than 100 ppm
and preferably less than 10 ppm water, as well as being low in
sulfur and nitrogen. On the other hand, when employing a some-
what larger pore size molecular sieve, specifically steam stabi-
lized zeolite Y in the conversion of C2-C4 olefins to motor
fuels, water is described as an effective cofeed which stabilizes
the catalyst and reduced the deactivation rate. As also describ-
ed in U.S. Patent No. 4,740,648 to Rabo et al., co-fed water is
described as a particularly desirable diluent which tends to aid
in resistance of zeolite Y catalyst to coking and aging.
-16- . ~~~~Ja~'3~x~De.~
SUMMARY OF THE INVENTION
In accordance with the present invention there is pro-
vided an alkylation-transalkylation process involving alkylation
of an aromatic substrate with a C2-C4 alkylating agent coupled
with separation to recover a monoalkylated aromatic product and
liquid phase transalkylation of a polyalkylated product. In one
aspect of the invention, both the alkylation and transalkylation
reactions are carried out in the liquid phase over molecular
sieve aromatic alkylation and transalkylation catalysts. The
output from the alkylation reaction zone is supplied to a separa-
tion zone which is operated to produce a lower boiling fraction
comprising the aromatic substrate, which may be recycled to the
alkylation reaction zone, and a higher boiling fraction compris-
ing a mixture of monoalkylated and polyalkylated aromatics. The
higher boiling fraction is supplied to a second lower boiling
fraction comprising the desired monoalkylated product and a high-
er boiling fraction comprising polyalkylated product.
At least a portion of the polyalkylated fraction includ-
ing substantially all dialkylated and trialkylated aromatics is
supplied, along with the aromatic substrate, to a transalkylation
reaction zone containing a molecular sieve transalkylation cata-
lyst. The transalkylation zone is operated under liquid phase
conditions to cause disproportionation of the polyalkylated frac-
tion to arrive at a disproportionation product having a reduced
polyalkylated aromatic content and an enhanced monoalkylated aro-
matic content. At least a portion of the disproportionation pro-
~~~u.~r.~.~
_1~_
duct is supplied to the first separation zone, In a specific
application of the invention directed to the production of ethyl-
benzene or cumene, the output from the transalkylation zone is
supplied to a third separation zone from which benzene and a
monoalkyl benzene fraction (ethylbenzene or cumene) is recovered
and recycled to the separation zone.
In another embodiment of the invention, a benzene feed-
stock and a C2-C4 alkylating agent are supplied to an alkylation
reaction zone containing a molecular sieve alkylation catalyst
and which is operated to produce an alkylated product comprising
a mixture of monoalkyl and polyalkyl benzenes. In this embodi-
ment of the invention, the alkylation zone may be operated under
liquid phase or vapor phase conditions with the output from the
alkylation zone being subjected to separation steps as described
above. The transalkylation reaction zone is operated at an
average temperature below the average temperature of the alkyla-
tion reaction zone and under conditions to maintain the benzene
in the liquid phase. In a specific application of this embodi-,
ment of the invention to a procedure employing vapor phase ethy-
lation of benzene followed by liquid phase transalkylation, the
average temperature of the transalkylation reaction zone is at
least 100°C less than the average temperature of the alkylation
reaction zone. '
In yet a further aspect of the invention involving the
alkylation of a benzene feedstock with a C2-C4 alkylating agent,
the alkylation catalyst is selected from the group consisting of
zeolite beta, zeolite omega, and zeolite Y and the alkylation re-
-18-
actor is operated under conditions to maintain the benzene feed-
stock in the liquid phase, as described previously. The effluent
from the alkylation reactor is subjected to separation steps
along the lines described above and :subsequent to separation to
recover the desired monoalkylbenzene product, e.g., ethylbenzene
or cumene, at least a portion of the polyalkylbenzene fraction
including substantially all of the dialkylbenzene content and a
predominant portion of the trialkylbenzene content is supplied to
the transalkylation zone containing a transalkylation catalyst
selected from the group consisting of zeolite Y, zeolite beta and
zeolite omega. Preferably, the alkylation catalyst comprises
zeolite beta and the transalkylation catalyst zeolite beta or
zeolite Y.
In a further embodiment of the invention directed speci-
fically to the production of ethylbenzene, in which the alkyla-
tion reaction takes place over a zeolite beta alkylation
catalyst. The output from the alkylation reaction zone is
supplied to a benzene separation zone. A higher boiling traction
comprising an ethylbenzene polyethylbenzene mixture is supplied
from the benzene separation zone to wn ethylbenzene separation
zone. This zone is operated to produce lower boiling product
fraction comprising ethylbenzene and a higher boiling fraction
comprising polyethylbenzene containing no more than 5 wt.~
ethylbenzene. The polyethylbenzene fraction is supplied along
with benzene to a transalkylation reaction zone which preferably
contains a zeolite Y transalkylation catalyst.
In accordance with a further aspect of the invention
_19_
there is provided a modified molecular sieve comprising hydrogen
zeolite beta of high surface area and low content of sodium or
other alkali metal which is of a good activity and aging quality
when used as a catalyst in hydrocarbon conversion reactions.
Also provided in accordance with the invention is a process of
producing such modified zeolite beta and a process for the alky-
lation of aromatic substrates with relatively low molecular
weight alkylating agents under moderate temperature conditions
including liquid phase conditions employing an alkylation cata-
lyst comprising zeolite beta as described herein. A preferred
embodiment of the present invention is in the alkylation of ben-
zene with ethylene to produce ethylbenzene carried out in the
liquid phase and at alkylation condi~ions under which the xylene
make, based upon the amount of ethylbenzene produced, is no more
than 0.03 wt.~.
In another embodiment of the invention, liquid phase
alkylation process is carried out using a plurality of series
connected reaction stages operated at an average temperature of
no more than 300°C with the interstage injection of the C2-C~
alkylating agent in a manner to maintain at least 1 mole percent
and preferably at least 2 mole percent of alkylating agent solu-
bilized in the aromatic substrate.
In one aspect of the present invention, the modified
zeolite beta has a surface area based upon the crystalline struc-
ture of the zeolite beta of at least 600 m2/g and preferably at
least 650 m2/g. A further aspect of the invention provides a
molecular sieve in which the hydrogen zeolite beta is in combina-
~? f i
-20-
tion with a binder and in which the surface area of the zeolite
beta, based upon the molecular sieve and the binder, is at least
450 m2/g.
In yet a further aspect of the invention, there is pro-
vided a hydrogen zeolite beta having a sodium content in the
crystalline structure of the zeolite beta of less than 0.04 wt.~
Na20. Preferably, the sodium content is less than 0.02 wt.~
Na20.
Another embodiment of the present invention involves a
process for the preparation of a hydrogen zeolite beta derived by
modification of an alkali metal containing zeolite beta synthe-
sized by the hydrothermal digestion of a reaction mixture con-
taining an organic templating agent. The synthesized zeolite
beta may be produced by any suitable technique, such as described
in Wadlinger et al. or Calvert et al. or the European patent
applications. In carrying out the process, the synthesized
zeolite beta is treated with an ion exchange medium in order to
protonate at least a portion if the active sites of the zeolite.
by exchanging off alkali metal ions. The ion exchange zeolite is
then calcined at a temperature within the range of 400°-700° for
a period of at least two hours, preferably within the range of
2-10 hours. The calcined zeolite beta is again treated with an
ion exchange medium to protonate an additional portion of the
active sites by exchanging off additional alkali metal ions. The
ion exchanged zeolite from this step is mixed with a binder to
produce a mulled zeolite-binder mixture. This mixture is pelle--
tized by extrusion or any other suitable technique and the re-
a
-21-
sulting pellets are then dried.
In a preferred embodiment of the invention, the ion ex-
change treatment steps are accomplished by repeated submersions
of the zeolite beta in an ion exchange: medium comprising an
aqueous solution of ammonium salt. In a further aspect of the
invention, the intermediate calcination step between the ion ex-
change treatment steps is carried out under time and temperature
conditions sufficient to arrive at a surface area based upon the
crystalline structure of the zeolite beta which has at least
twice the surface area of the zeolite beta in the as synthesized
form.
In a further embodiment of the invention, there is pro-
vided a liquid phase aromatic conversion process employing a
zeolite molecular sieve catalyst having a pore size greater than
6.5 angstroms in which a feedstock containing at least one aroma-
tic compound and having water entrained therein is passed to a
dehydration zone. In the dehydration zone, water is removed to
provide a dehydrated feedstock of a water content no more than
100 ppm, preferably 50 ppm, or less. The dehydrated feedstock is
then supplied to the reaction zone containing the molecular sieve
catalyst which preferably is selected from the group consisting
of zeolite Y and zeolite beta. The reaction zone is operated at
temperature and pressure conditions to maintain the reactor con-
tents in the liquid phase and also sufficient to cause the con-
version reaction to proceed in the presence of the catalyst.
One embodiment of the invention involves the liquid
phase alkylation of an aromatic substrate, specifically the ethy-
?~~~~~
-22-
lation of benzene to produce ethylbenzene with the dehydration of
the feedstock to reduce the water content to no more than 100
ppm, as described above. A further aspect of the invention in-
volves the transalkylation of a feedstock containing a mixture of
a polyalkylbenzene component and a benzene component to produce a
disproportionation product comprising a monoalkylbenzene. At
least a portion of the feed to the transalkylation reaction zone
is dehydrated to provide a feedstock, including both the poly-
alkylbenzene and the benzene components, having a total water
content of no more than 100 ppm.
Another embodiment of the present invention involves a
process for the liquid phase alkylation of an aromatic substrate
with a C2-C4 alkylating agent in a multistage reaction system in
which the aromatic substrate/alkylating agent mole ratio is pro-
gressively decreased when going from one stage to another. In
this aspect of the invention, the reaction stages are operated
under temperature and pressure conditions effective to cause
alkylation of the aromatic substrate with the pressure being '
abave the vapor pressure of the aromatic substrate and below the
vapor pressure of the alkylating agent at the alkylation reaction
conditions. A feedstock comprising the aromatic substrate and
the alkylating agent is supplied to the initial reaction stage in
relative amounts to provide a first mole ratio of aromatic sub-
strate to alkylating agent. Effluent comprising a mixture of the
aromatic substrate and alkylated product is withdrawn from each
reaction zone and supplied to the next succeeding reaction stage,
Alkylating agent is supplied along with the effluent to the next
._.
-23-
succeeding reaction stage to provide an overall progression
across the multistage reaction system of a decreasing mole ratio
of the aromatic substrate to the alkyl.ating agent in an amount to
provide a final mole ratio of aromatic: substrate to alkylating
agent for the final reaction stage which is less than the first
mole ratio supplied to said initial reaction stage. Preferably,
the multistage reaction system in this embodiment of the inven-
tion comprises from 3-8 reaction stages operated under conditions
producing the monoethylation of benzene in which the xylene make
based upon the amount of ethylbenzene produced, is no more than
0.05 wt.~, preferably no more than 0.03 wt.$ as indicated pre-
viously.
~~5~3~~
--24-
BRIEF DESCRIPTION OF THE DRAWINGS
FIGURES la-lc and 2a-2c are cJraphs illustrating the re-
sults of transalkylation experiments carried out using two dif-
ferent zeolite Y catalysts.
FIGURES 3a-3c are graphs illustrating the results of
experimental work carried out in the transalkylation of diethyl-
benzene using another zeolite Y catalyst.
FIGURES 4a-4c are a series of graphs showing experimen-
tal work carried out with a rare earth zeolite.
FIGURES 5a, 5b and 6 are graphs illustrating further
experimental work employing a zeolite Y catalyst.
FIGURE 7 is a simplified schematic flow diagram
illustrating one embodiment of the invention in which a poly-
ethylbenzene fraction is subjected to a residue extraction step
prior to transalkylation.
FIGURE 8 is a schematic illustration of a modification
of the process of FIGURE 7 in which the output from the trans-
alkylation reactor is subjected to a separation step prior to re-
cycle.
FIGURE 9 is a simplified schematic illustration of yet
another embodiment of the invention in which the bottoms fraction
from. an ethylbenzene column is supplied directly to a trans-
alkylation reactor with the output of the transalkylation reactor
being supplied to a downstream separation zone,
FIGURE 10 is a schematic flow diagram showing a modifi-
cation of the embodiment of FIGURE 9.
-25-
FIGURE 11 is a schematic illustration of a specific
embodiment for carrying out the invention employing a plurality
of series connected reaction stages.
FIGURE 12 is a graph showing the results of experimental
work involving dehydration of a feedstream to a transalkylation
reaction carried out over a zeolite Y catalyst.
FIGURE 13 is a graph showing the results of experimental
work involving dehydration of a feedstream to a transalkylation
reaction carried out over another zeolite Y catalyst.
-26- ~~,J~~d~
DETAILED DESCRIPTION
A preferred application of the invention involves liquid
phase alkylation over a zeolite beta alkylation catalyst coupled
with liquid phase transalkylation over a molecular sieve trans-
alkylating catalyst selected from the group consisting of zeolite
Y and zeolite beta. An especially preferred embodiment of the
invention involves the use of zeolite beta as an alkylation cata-
lyst and zeolite Y as a transalkylation catalyst. However, as
will appear below, other molecular sieve catalysts can be employ-
ed in carrying out the present invention. Moreover, while a pre-
ferred application of the invention is in the use of liquid phase
transalkylation in conjunction with liquid phase alkylation, the
invention can be carried out employing vapor phase alkylation, as
disclosed, for example, in the aforementioned patent to Dwyer,
coupled with liquid phase transalkylation and appropriate recycle
of the transalkylated product to a separation zone.
In its more general aspects, the invention involves
transalkylation coupled with aromatic alkylation employing C~-C4
alkylating agents which, broadly stated, can be alkylating agents
of the type disclosed in the aforementioned patent to Pollitzer
et al., such as ole:Eins, alkynes, alkyl halides, alcohols, ethers
and esters. The most widei:y used alkylating agents are ethylene
and propylene applied in the production of ethylbenzene and
cumene, respective7.y. The invention is especially applicable to
the ethylation of benzene under conditions in a manner in which
byproduct xylene yields are reduced and the invention will be
~~ .m
_.
--27-
described specifically by reference to the production of ethyl-
benzene together with the attendant transalkylation of polyethyl-
benzenes.
As noted previously, a conventional process fox the pro-
duction of ethylbenzene involves recycsling polyethylbenzenes,
separated from the ethylbenzene produc;t, to the alkylation reac-
for where they undergo transalkylation to yield ethylbenzene. A
byproduct of this procedure is increased xylene yield in the
effluent from the alkylation reactor. The presence of xylenes
complicates downstream processing and separation steps. A par-
ticular impact of a significant xylene content in the product
stream is that it often mandates operation of the distillation
column from which the ethylbenzene is taken overhead in a manner
to provide a substantial ethylbenzene content, often times 15-20$
or more, in the bottom polyethylbenzene fraction. For example,
ethylbenzene produced in accordance with the present invention
can be employed in the production of styrene by catalytic de-
hydrogenation. The boiling points of ortho xylene and styrene '
are very close, within 1°C of one another. As a practical
matter, the ethylbenzene specifications will call for a very low
xylene content, normally less than 2000 ppm. Tn order to meet
this specification, it is normally necessary to operate the
ethylbenzene column under moderate distillation conditions re-
sulting in a high ethylbenzene content in the bottoms fraction as
described above. Tkie present invention, by carrying out poly-
ethylbenzene transalkylation in a separate reactor under relati-
vely mild liquid phase conditions, minimizes the xylene make in
-28-
the manufacturing process. This enables ethylbenzene recircula-
tion to be reduced by limiting the ethylbenzene content in the
polyethylbenzene fraction to 5 wt.~ or less, and where preferred
catalysts are used to further minimize: xylene make, down to about
2 wt. $. or less ethylbenzene.
A preferred aspect of the present invention involves
supplying the polyethylbenzene fraction, including both diethyl-
benzene and the triethylbenzene and higher molecular weight com-
pounds to the transalkylation reactor as contrasted with
separating out a substantial portion of the diethylbenzene for
recycle to the alkylation zone, as disclosed in the aforemen-
tinned patent to Wight, or separating out trialkylaromatics with
transalkylation only of dialkylbenzene, as disclosed in the
aforementioned patent to Barger. In this respect, depending upon
the configuration of the interface of the transalkylation reactor
and polyethylbenzene or other separation zones, substantially all
of the diethylbenzene and substantially all or most of the tri-
ethylbenzene content will be supplied to the transalkylation
reactor. In either case, the practical effect of this embodiment
of the invention is that recycle to the alkylation reactor is
limited to benzene and lighter components, e.g., ethylene, while
most if not all of the triethylbenzenes together with diethylben-
zenes are retained in the system ultimately for conversion to
benzene and ethylbenzenes. This offers significant advantages
over the prior art processes, not only in terms of reduced xylene
makes as described previously, but also in terms of ultimate pro-
duct yield.
-29-
In experimental work relative to the invention, a number
of catalysts were employed in transalkylation tests carried out
in an upflow, flooded-bed reactor, that is, only a liquid phase
was in contact with the catalyst. The feed employed in this ex-
perimental work was an approximate 1:1 mixture of benzene and the
polyethylbenzene overheads fraction from a commercial operation
employing vapor phase alkylation of benzene to produce ethylben-
zene. A typical feed employed in the experimental work had the
composition as shown below in Table I.
-30-
Table I
Non-Aromatic 0.32
Benzene 50.241
Toluene 0.000
Fthylbenzene 6.117
p+ M-Xylene 0.000
Styrene 0.063
o-Xylene 0.066
Cumene 3.973
n Propylbenzene 7.816
m + p Ethyltoluene 2.053
1,3,5-Trimethylbenzene 0.128
o-Ethyltoluene 0.356
1,2,4-Trimethylbenzene 0.536
1,2,3-Trimethylbenzene 0.401
m-Diethylbenzene 14.808
o + p-Diethylbenzene 7.328
Butylbenzenes 1.653
heavies 4.429
In the experimental work, the average pressure was about
300 psia with a pressure drop across the reactor ranging from
about 5 to 15 psi. The temperature profile across the reactor
was relatively constant with an endotherm from the inlet to the
outlet of less than 10°C and usually less than 5°C. The experi-
mental runs were initiated at relatively low temperatures, usual-
ly less than 100°C and progressively increased as described lat-
-31-
er. The space velocity was maintained relatively constant at a
value of 6 hr.-l (ZHSV) based on the total hydrocarbon feed. Di-
ethylbenzene conversions and selectivity to ethylbenzene were
measured as a function of catalyst age (duration of the run)
along with the production of various other components including
xylenes.
In a first test run, the catalyst used was a commercial-
ly available zeolite Y (identified herein in Catalyst A) in which
the inlet temperature was progressively increased up to about
235°C and stabilized there with an average temperature increase
through the reactor of only 1° or 2°C. The results of this ex-
perimental work axe illustrated in FIGURES la-lc in which percent
diethylbenzene conversion C, percent selectivity to ethylbenzene
S, ortho xylene production O, in ppm, and temperature, T, °C, are
plotted as curves 11, 12, 14, and 16, respectively versus the
catalyst age A, in hours, on the abscissa. As can be seen from
an examination of the data presented in FIGURE 1, the diethylben-
zene conversion stabilized in about the 32-37~ range for a reac-'
for temperature of about 237°C with the catalyst showing very
little deactivation over the duration of the run. The select-
ivity to ethylbenzene was virtually 100. During the run, O-
xylene production stabilized at about 400 to 500 ppm.
Another test run was carried out using an experimental
zeolite Y identified herein as Catalyst B. The results o~ this
run are set forth in FIGURES 2a-2c in which curves 18, 19, 21 and
22 are graphs of diethylbenzene conversion, C, selectivity to
ethylbenzene, S, parts per million O-xylene, O, and temperature,
~~2~
-32-
T, respectively plotted as a function of catalyst Age A. In this
experiment, the catalyst was run for nearly 400 hours with the
temperature, after initialization, increasing slightly with time
to a final value of about 240°C. As can be seen, diethylbenzene
conversion was relatively good, mostly in the 30-40~ range at
relatively moderate temperatures. Selectivity to ethylbenzene
was greater than 90$ and during most of the run was virtually at
100. The 0-xylene content of the product stream stabilized at
about 900 ppm.
Yet another test run was carried out employing another
zeolite Y catalyst identified herein as Catalyst C. The results
herein in terms of diethylbenzene conversion, selectivity and as
a function of time and temperature, are set forth in FIGURES 3a-
3c. In FIGURE 3, curves 24, 25, 27 and 28 are graphs of diethyl-
benzene conversion, selectivity to ethylbenzene, 0-xylene content
(ppm), O, and temperature, T, °C as a function of catalyst age on
the abscissa. As shown in FIGURE 3, diethylbenzene conversion
was, on balance, slightly better than for catalysts A and B, and
fell generally into the 40-50~ range at reactor temperatures
ranging from about 210° to about 236°C. Selectivity to ethylben-
zene was more than 90~ over much of the run at virtually 100.
O-xylene content stabilized at about 800-900 ppm. The catalyst
showed very little deactivation over the life of the run.
A rare earth zeolite Y identified herein as Catalyst D
was employed in yet another test. The results for catalyst D are
set forth in FIGURE~~ 4a-4c with curves 30, 32, 33 and 35 repre-
senting graphs of diethylbenzene conversion, selectivity to
-33- ~"~~t~ ~~~~
athylb~naene, ppr~r ~~xYlane and t~m~aeratur~, raa~eotivaly as a
~una~tioaa ~~ cat~lyat age, cutaly~it t~ ~hov~~a~ ralativelY 9fl~d x~~~
ault~ including diethylbenzetta ~c~s~vgt:~ion in the ~0-50~ ras~ga.
Initial r~1~ctivity way about 1DD0, with ss~lect~.vity fa111ng of~
slightly to abaut 94t tawmgd the nc~d of the t.-un. s~hil~ quad can°~
version and ~alectivity wat~a aoEa.i~~ved, the reaction tampcz'atur~
was substantially taigh~x than tsar the ~rxevlat~a zaralite Y, r~~ing
to abcaut 2°70°t: at the ogr~c~.ugion a~ the roan, xbrrut 210
hours.
~h~ ~eada for the ~acperi~~ntal Worl~ da~iat~c~ in ~xBU~~s
1-4 confvgt~c~ gmnara~lly to ttx~ cs~mpositlon ahawn in ~'abl~ I.
~cwavat, th~ ~a~d fear the ~ira~t teat run tCa.talyat ~1 was ~re~ of
s~rgho. xylana and the feed ~ar the aecouW runt tcatallrnt H 3 oan-~
tainted about 0.02 pe~ra arsd mete xxlent$.
~ddl.tional e~parimental war~C und~z the above~idanti~ied
conditions Was caxri~d oat employing three ~additianal aatalyatat
C~talya~t ~, a canon n~cchange resin avr~i.labl~ ~rb~ ltohra ~ Haas
un~sr the dasigna~xion l~anbexlyat 15, Catalyst ~', a. 9uparacidia
alu~ina available from ~larahawayil,tra~. under rh~ designati~~a 3999
and ~atalyatc ~ a nictcel modi~i~d mordenite a~railable Exam Utsion
Carbi.dc under tote, designation NibCn9044. t~a'talyat ~ sl~a~asd
little c3lathylbanzen~ es~nversinn sand r~o athylbangann psoduatian~
up to thn tune th~ axp~rlit~ant was kar~inat.~d, at about 5t? Maura
dlnd ~ e'.eR1~1~1'~~'.Illr~ 8~ ~.J~~~.i d51~ 1:0 ~X~6.~'~l~l~n~d~.
1~~~=i.~u~t~~~e
Catalyst F praduc~d diethylbsnz~n~ canver~ion~ r~raging txoaa about
to 20~ at temperatures ranging tram about 300"-450°C with, ..
s~l~caivi%y to ethylbenzen~ ~or the moe~t part being ibaa that
s0~. Catalyst. c~ was run tnr 100 hears at te~mperatur~a ranc~ia~g up
to 350~C a~t~d e~hawed almost na dia$hylben~ena conversion.
~Pha ~aoltt~ Y catal,yata ident~.lies3 above as Catalysts ~
-34-
and B were also used in downflow trickle bed reactors where a
substantial gas phase was present. Fresh and regenerated cata-
lysts were used. This experimental work was carried out at
pressures of about 330 psig, nominal space velocities of about 10
hr-1 (LHSV> and average reactor temperatures of about 300°C in
the case of fresh catalyst A, about 300°-400°C in the case of
fresh Catalyst B and about 200°C in the case of the regenerated
catalysts. For fresh catalyst A, initial diethylbenzene conver-
sion was about 24~ but this fell off rapidly after a few hours.
The catalyst was then regenerated and under the less severe tem-
perature conditions of about 200°C, initial diethylbenzene con-
version was high, about 60$ but this, again, reduced to only a
few present after about 24 hours.
When employing fresh catalyst B the initial diethylben-
zene conversion was over 50~, but this fell to about 20~ after
about 5 hours and then decreased further to only a few percent.
The regenerated catalyst B, when run at the lower temperature of
about 200°C, showed an initial diethylbenzene conversion of about
58$ which declined to about 27~ after 29 hours, at which time the
run was terminated.
Yet additional experimental work was carried out employ-
ing zeolite Y identified above as catalyst B in which the feed
was a relatively pure diethylbenzene mixed in approximately equal
parts with benzene. Unlike the feedstock employed in the experi-
mental work of FIGURES 1 through 4, the pure diethylbenzene feed-
stock contained only very small amounts of material susceptible
to cracking or other conversion reactions, e.g., deethylation, to
-35-
produce xylenes and was also free of ~;ylenes. The makeup of the
feedstock in this experimental work is set forth below in Table
II.
Table II
Components Wt,~
Non-aromatics 0.01
Benzene 56.58
Toluene 0.09
Ethylbenzene 0,01
Xylenes 0.0000
n-PR-BZ 0.02
m,p-ethyltoluene 0.03
o-ethyltoluene 0.01
124 trimethylbenzene
sec-BU-BZ 0.47
123 Trimethylbenzene
m,Diethylbenzene 27.62
o,p-diethylbenzene 14.27
n--BU-B Z 0 . 3 5
Heavies 0.54
In this test run,.the inlet and outlet pressures were
held at 310 and 305 psig, respectively. The average temperature
of the reactor was increased approximately linearly with time
from an initial value of about 198° to a final value of about
298°C. The space velocity was generally held within the range of
_.
-36-
about 5.8-6.0 hr'1 (LI-ISV) with the exception of about two-thirds
of the way through the test where it fell to about 5.1 before re-
covering to the higher value.
The results of this test run are set forth in FIGURES 5
and 6. In FIGURE 5a, curve 38 is a graph of temperature, T, ver-
sus catalyst A in hours on the abscissa. In FIGURE 5b curves 40
and 41 are graphs of percent selectivity to ethylbenzene and per-
cent ethylbenzene conversion, respectively. Curve 42 is a graph
of the total xylene make, X, expressed in ppm, based upon the
amount of ethylbenzene produced. FIGURE 6 shows the relationship
between ethylbenzene conversion and temperature. Curve 43 is a
graph of ethylbenzene conversion, C, on the ordinate versus tem-
perature, T, on the abscissa.
As indicated by the data set forth in FIGURE 5, xylene
make remained low throughout the test run. No xylene was pro-
duced until the temperature was increased to about 260°C (which
generally corresponds to the reduction in space velocity to about
5.1 hours-1 as reported previously). Percent conversion remained
good until the temperature was increased above 280°C. As indi-
Gated in FIGURE 6, ethylbenzene conversion appears to remain
above 50$ over a temperature range of about 200°-290°C with the
optimum range appearing to be about 210° to 28U°C.
With further reference to 'the drawings, FIGURES 7
through 10 illustrate schematic flow diagrams illustrating dif-
ferent embodiments o:E the invention. It will be assumed for
purposes of discussion that the invention is applied in the pro-
duction of ethylbenzene by reaction of ethylene with benzene and
~~~~~r~~~a
that the alkylation reaotion in carrtad out i~ ~ E3.oodedwbed
li~u~.d phase al'~tlation r~acte~r employing ~eol9.te b~t~:r ~~o~.ita Y
or ra~o3.it.~ amega ,~a ~th~ alkyla~.i4n catalyst. Hv~a~ver, as noted
previouall' aid as di~cusssd iaa c~rsater de~tnil below, th~ ~llkyla~
Lion step can be oc~nducted as a v~pc~s phase seaotien emplcaying a
catalyst auGh as ~ilicallt~ og ~5~~~.
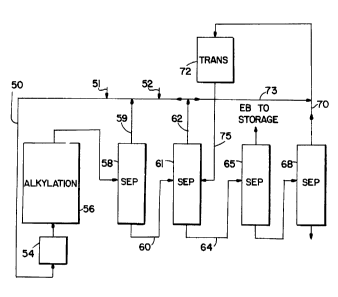
Ftafarxing flrgt tg l~IGIJ~tE '~, a fmedatraam 50 ccantainin~
ethylene and benzene supplied via lanes 51 and 5~o r~~p~cti'~alW
is passed first tc~ a d$hydrator 54r whexe the mater eontant is
reduced to a levsl of ~bnut 1p0 ppm or leas, pr~f~rably about ~0
ppm ~~c legs, and then to an alkylation reaction gone 54. The
alkylation reactor, vthich may comprl,~a a pluxaJ.ity a~f ~arie~ con
nested adiabatic r~act4ra with ir~tarstaga infection of ethylene
an8 mlao interatage ogoling, normally will.,b~ operated at an.
av~rager temperature of abQUt ZZ~°C and under auffi~ciant pressure,
about 6U0 Asia or above, to maia~ta~.n the b~n~eria in the liquid
phase and at least sheaf ~ t~~le~ percent of athylac~s ae~.~ib~.li~~d ,
in the ben~dne, hs an altarnativ~ to aging adiabatic r~aci:orar
one ar m~ra isothermal reao$s~s~ cnn b~ sm~laYad ~rifi~h suitable
oosling .m~arr~ um~d tc~ anaintain a aubatantiaily aonataat
tempara°°
turn (little or t~~ tecnperat~ura dlffereritial~ from the islet to
the r~ut.le~: of the seact.or. "l~ha effluent a~.~eeatm ffom the a1~3~16t~ "
Lion reactor i~ supplied to a pxe~fract~.onatiot~ oolumn 59 wi9loh la
gperat~d tn psovida a .li.ght ~varheacia traatian includine~ banaen~
which la auppli~d via line 59 to the a~.bcY1~tio~~t Yeaot~r input a~ld
a haavi~r lieiuide Zra.mtian containing b~naan~, ~t.hylb~an~~n~
a~parativn tone b~..
Th~ output from the pre~ractionatis~a~ Bona 56 is auppli~d'"
via ilna 60 ~a a bac~rena asparati.on ~~one ~1~ The o~t~rhe~ed, frao-
-38-
tion from column 61 contains the remaining benzene which is re-
cycled via line 62 to the alkylation reactor input. The heavier
bottoms fraction from column 61 is supplied via line 64 to an
ethylbenzene separation zone 65. The overheads fraction from
column 65, of course, comprises ethylbenzene which is supplied to
storage or to any suitable product destination. By way of ex-
ample, the ethylbenzene may be used as a feedstream to a styrene
plant in which styrene is produced by the dehydrogenation of
ethylbenzene. The bottoms fraction containing polyethylbenzenes,
heavier aromatics and preferably only a small amount of ethylben-
zene, no more than 5$ as discussed previously, is supplied to
polyethylbenzene separation zone 68. The bottoms fraction of
column 68 comprises a residue. The overhead fraction from column
68, containing polyethylbenzene, triethylbenzene (usually~in
relatively small quantities) and a minor amount of ethylbenzene
is supplied to a transalkylation reaction zone 72. By minimizing
the amount of ethylbenzene recovered from the bottom of column
65, the ethylbenzene content of the transalkylation feedstream is
kept small in order to drive the transalkylation reaction in the
direction of ethylbenzene production. The polyethylbenzene frac-
tion withdrawn overhead through line 70 is mixed with benzene
supplied via line 73 and then supplied to the transalkylation re-
actor 72. The mole ratio of benzene to polyethylbenzene should
be at least 1:1 and preferably is within the range of 1:1 to 4:1.
The output from the transalkylation reactor containing benzene,
ethylbenzene and diminished amounts of polyethylbenzenes is
supplied via line 75 to the benzene column 61.
~n the proCe~~ depicted in ~gGtlR~ 7, the alkylat:ian re~
actian is carried out in the lfc~uid phase with ~lshyd~'atidn ~f
gees to the elk ylation rsactor...~s ~ot.~d previoaaly, the inven-
tion may bs carried out smp~.oyia~g vapgr ph~as al.kyl~~.ign fol3.ow~d
by liquid phase tranaalkylatis~r~ and in such reactions, dapsndirsg
upon the catalyst ~mployed, sigrai~icant ~qe~antitisa r~~ wats~r may
big included ire ttis feed t~ ~.hs alkwlation Reactor. In this Caaa.
it taay b~ necessary to asparat~ly aacornpliah dehydration of the °
~~sd to the t~ranaal3cyl.ation rbaotor. such dehydrati~n easy take
pZaas at any point upstream at t;.he tranaaikylat~.~n reactor, and
,~ naosaeary, dahydratinn should be aocorapliahsd with r~s~rsct to
the ~rest~ bonssne feed auppli~d via lirse '1~ as wsll~ an with ro-
spsct to the polyethylene comps~nant praduc~d dux~,lng the alkyla~-
tiolt lgaCt.iidn a
~xGUItE 9 diacloa~aa a anodl.fication c~ the pr~aasga dia-
claaed in ~iGo~~ 7 in wriiah tha~ Iran~alkYl.atior~ reactor output is
atab~ectsd to further treatment prior to x~scyci~ to the asparatian
system. ~~a embadi~ant of FYGUI~~ 8 is particu3,ax~.y useful irf
thc~as~ cases in which relatively high oonveraioa~ is achieved in
the tran~~lkylati.on r~acts~r. In flee smbcadi.r~ont o! ~IG~it~ ~, 'the
alkylnti4n rsaator and separation gyatem .ia idsrtti~cal to that of
hYQI~R~ '~ and like oomparis~tts are indioatsd.,by the. $a~a~ xe~srst~as
ahataotars. ~ liowavsx, t3~e output from they tranaaskylation rs~ctor
to 'nup~slisai to a Dsoandary aspara~tion roes ?7 which may take they
form of a diatil,lation o~lumn which is c~peratedt in a inannsr to. .
produce .a bottc~rn' purge stream withdrawn via line ?8 and. a raaycl.s
stream withdrawn via ~.ing 80 and supplied to the bsn~sns ~~lut~n.
elm pur:ge stream containing heavy hydrocarbons is wii~h-
drawn from the ~'yats~, thus providing a ~aartially airaqlg..gaaa
..
-40-
system in which high molecular weight hydrocarbons are not recir-
culated.
FIGURE a illustrates yet another embodiment of the in-
vention in which the polyethylbenzene fraction recovered from the
ethylbenzene column is directly passed to a transalkylation reac-
tor. In FIGURE 9, the same system components as shown in FIGURES
7 and 8 are designated by like reference numerals. As shown in
FIGURE 9, the output from the ethylbenzene column 65 is mixed
with benzene supplied via line 82 and supplied to the trans-
alkylation reactor 84. Here, the entire polyethylbenzene frac-
tion is subjected to transalkylation. The conditions employed in
reactor 84 may be the same as described above with the ratio of
benzene to polyethylbenzene ranging from about 1:1 to 4:1.
It will be recognized that the procedure depicted in
FIGURE 9 is similar to that of FIGURE 8 except that the entire
bottoms fraction from the ethylbenzene column is subjected to the
transalkylation reaction. Limiting the ethylbenzene content of
the input to the transalkylation reactor to no more than 5$, pre-
ferably 2~ or less is especially significant here in establishing
conditions promoting the transalkylation reaction. The output .
from the transalkylation reactor is applied via line 85 to a post
transalkylation separation zone 86 which may take the form of a
distillation column operated to produce an overhead fraction that
is comprised predominantly of benzene and ethylbenzene and a bot-
toms fraction, composed predominantly of Cg and Clp hydrocarbons
such as ethyltoluene, cumene, butylbenzene, etc., which is elimi-
nated from the recycle stream by purge line 88. The overheads
-41-
fraction is recycled through line 89 i.o the benzene column simi-
larly as described above.
The embodiment of FIGURE 10 is similar to that of FIGURE
except that the transalkylation reactor output is split, with
a portion being directly supplied to the benzene column 61 via
line 92 and the remainder to the separation zone 86 which is op-
erated as described above. The configuration of FIGURE 10 provi-
des a means for maintaining a low concentration of Cg and C10
hydrocarbons in the system and reduces the energy costs of opera-
ting column 86. Typically about 60$ or more of the transalkyla-
tion reactor output is recycled directly to the benzene column 61
with the remainder being directed to the separation zone 86.
As noted previously, one of the zeolite catalysts useful
as an aromatic alkylation catalyst is zeolite beta and a specific
embodiment of the present invention involves the use of an aroma-
tic alkylation catalyst comprising a modified zeolite beta, under
relatively mild liquid phase alkylation conditions. The inven-
tion is especially applicable to the ethylation of benzene under
mild liquid phase conditions producing little or no xylene make,
and the invention will be described specifically by reference to
the production of ethylbenzene. Eowever, other alkylation reac-
tions may be utilized in carrying out the invention. For ex-
ample, the invention may be applied to the reaction of propylene
with benzene to produce cumene. Also, while olefinic alkylating
agents normally wil:L be employed, other alkylating agents such as
alkynes, alkyl halides, alcohols, ethers, and esters as disclos-
ed, fox example, in the aforementioned patent to Pollitzer et
.,.
-42-
al., may be used. Other aromatic substrates such as toluene and
xylene may also be subject to alkylat:ion in accordance with the
invention.
The activity of zeolite beta as utilized in the present
invention is in direct contrast to the teachings found in the
aforementioned patent to Dwyer which suggests that zeolite beta,
because of its low constraint index, is unsuitable for use under
the relatively severe conditions involved in the vapor phase
ethylation of benzene, and in the aforementioned patent to Young,
which limits its application to relatively long chain alkylating
agents. Dwyer, which is directed to ethylbenzene production
under relatively severe temperature conditions well above 300°C,
i.e., 650°-900°F, and preferably 700°-850°F,
teaches as noted
above, that zeolite beta cannot be used as a catalyst in the
ethylation of an aromatic substrate even under the high tempera-
ture conditions involved in the Dwyer process.
The present invention proceeds in a manner directly con-
trary to these prior art teachings. In the invention, zeolite
beta is a highly effective catalyst for the alkylation of an aro-
matic substrate with low molecular weight (C2-C4) alkylating
agents. Moreover, it is an effective alkylating agent under mild
liquid phase conditions involving temperatures of 300°C or less,
providing high conversion efficiency and high selectivity to
monoalkylation. As noted previously, these mild reaction con-
ditions permit the production of ethylbenzene with a xylene make
which is negligible and, for all practical purposes, nonexistent.
Crystalline zeolite beta which is identified by its x-
-43-
ray diffraction pattern and basic procedures for its preparation
are disclosed in the aforementioned U.S. Patent No. 3,308,069 to
Wadlinger et al. As described therein, zeolite beta is synthe-
sized by the hydrothermal digestion of a reaction mixture com-
prising silica, alumina, an alkali or alkaline earth metal oxide
or hydroxide and an organic templating agent.
The zeolite beta catalysts employed in 'this embodiment
of the invention preferably are of ultra-low sodium content which
can readily be produced by new procedures as described in detail
below. Low sodium content zeolite betas are in themselves, known
in the art. For example, the aforementioned patent to Wadlinger
discloses zeolite beta formed by treating a dried product result-
ing from the digestion procedure which was exchanged continuously
for 48 hours with a 2$ solution of ammonium chloride. After
washing free of excess chloride ions, the catalyst was dried and
calcined for three hours at 1000°F to produce an acid beta alumi-
nosilicate having 0.07$ Na content. The aforementioned patent to
Culvert et al. also discloses zeolite beta of relatively low
sodium content, e.g., 0.14 wt.~ and 0.11 wt.~ Na.
The preferred zeolite beta of the present invention is
characterized by a sodium content substantially lower than those
disclosed in the patents to Wadlinger et al. and Culvert et al.
It is also characterized in terms of a very high surface area,
specifically at least 600 m2/g based upon the crystalline zeolite
beta. The preferred zeolite beta has a low sodium content of
less than 0.04 wt.~ and preferably less than 0.02 wt.$, expressed
as Na20. The preferred zeolite beta is produced by means of a
~~:'
%~~,~z.~;~r.D
--44-
series of ion exchange and calcination procedures carried out em-
ploying as-synthesized zeolite beta as a starting material. The
synthesized zeolite beta can be produced by the hydrothermal
digestion of a reaction mixture in accordance with any suitable
procedure such as those disclosed in the aforementioned U.S.
patents to Wadlinger et al. and Calvert et al., and the aforemen-
tinned European patent applications.
In producing modified zeolite beta in accordance with
the present invention, the as synthesized zeolite beta is sub-
jected to a plurality of sequential ion exchange and calcination
treatments to arrive at a molecular sieve product of extremely
low sodium content, substantially below the sodium content of the
acid zeolite beta produced in accordance with the patent to
Wadlinger as described above. In addition, the final molecular
sieve product has a substantially higher surface area than that
heretofore attained for zeolite beta. This embodiment of the
invention involves an initial ion exchange treatment of the as-
synthesized zeolite beta followed by calcination, followed by
subsequent ion exchange to arrive at a product of ultra-low
sodium content and high surface area.
While the invention is not to be limited by theory, it
is believed the initial ion exchange treatment removes a substan-
tial portion of the sodium or other alkali metal ions incorporat-
ed during synthesis, such that the subsequent calcination pro-
cedure, carried out under essentially anhydrous conditions, does
not lead to collapse or otherwise undesirable changes in the
crystal structure of the zeolite beta. This calcination pro-
-45-
cedure, together with the initial ion exchange treatment, pre-
ferably results in at least a twofold increase in the zeolite
beta surface area over the surface area of the zeolite in the as
synthesized form.
The intermediate calcination step is effective to decom-
pose the organonitro templating agent within the pore network of
the zeolite beta and opens up the molecular sieve channels so
that they are readily susceptible to subsequent ion exchange
treatment. After the initial calcination step, an ion exchange
treatment is again carried out in order to protonate an addi-
tional portion of the active sites in the zeolite by exchanging
off sodium or other alkali metal ions. This second ion exchange
treatment results in a zeolite beta of extremely low sodium con-
tent, less than 0.04 wt.~ Na20, and high surface area, at least
600 m2/g based upon the zeolite beta crystalline structure. The
preferred ion exchange medium is an ammonium salt. The zeolite
beta resulting from the second ion exchange treatment is mixed
with a binder, pelletized-and dried and then subjected to another
calcination step in order to convert the ammonium exchanged act-
ive sites to acidic iH+> active sites.
As synthesized zeolite beta used as a starting material
for the present invention can be synthesized by the hydrothermal_
digestion of silica, alumiria,, or sodium and other alkyl metal
oxides, and an organic templating agent in accordance with any
suitable procedure :such as those disclosed in the aforementioned
U.S. Patents to Wad7_inger et al. and Calvert et al. and the
aforementioned European patent applications.
-46-
Typical digestion conditions include temperatures rang-
ing from slightly below the boiling point of water to about 170°C
at pressures equal to or greater than the vapor pressure of water
at the temperature involved. The resulting reaction mixture
should be maintained under mild agitai~ion, such as stirring, for
periods ranging from about 1 day to several months to achieve the
desired degree of crystallization to form the zeolite product.
Lower temperatures will normally require longer periods in order
to arrive at the desired crystal formation. For example, at tem-
peratures of about 100°C, crystal growth may occur during periods ,
ranging from about 1 month to 4 months, whereas, at temperatures
near the upper end of the aforementioned range, e.g., about
160°C, the digestion period may be from 1 or 2 days up to about 1
week. At intermediate temperatures of about 120°-140°C, the di-
gestion period may be for several weeks, perhaps 2-4 weeks.
Any suitable templating agent may be used in forming the
zeolite beta molecular sieve crystalline structure and, as indi-
Gated by the references referred to above, appropriate templating
agents include tetraethylammonium hydroxide and halides such as
tetraethylammonium chloride and dibenzyl dimethyl-ammonium
hydroxide or halide such as dibenzyl dimethyl ammonium chloride.
The reaction components may be varied in accordance with tech-
niques well known in the art to provide the zeolite beta product
of varying silica/a7_umina ratios. Typically, the reaction mix-
ture used to synthe:~ize the zeolite beta molecular sieve will
contain formulations within the following retie xanges:
CA 02028935 2000-10-27
-47-
Table III
Si02/A1203: 20-1000
H20/Si02: 5-200
OH-/Si02: 0.1-0.2
M/Si02: 0.01-1.0
R/Si02: 0.1-2.0
In the above table, R is the nitroorgano templating agent, e.g.,
a tetraethylammonium group and M is an alkali metal ion, usually
but not necessarily, sodium. For a further description of
zeolite beta and methods for its synthesis, recourse may be had
to the above patents and patent applications including specifi-
cally, U.S. Patent Nos. 3,308,069 (Wadlinger et al.) and
4,642,226 (Calvert et al.).
As explained above, a critical first step in carrying
out the method of the present invention lies in treating the as
synthesized zeolite beta with an ion exchange medium prior to
high temperature calcination which is designed to remove a predo-
minant portion of the templating agent from the intercrystalline
molecular sieve network. The product at the conclusion of the
hydrothermal digestion procedure leading to crystallization of
the zeolite beta, can be washed and dried at a temperature
usually substantially less than 200°C, e.g., about 150°C, design-
ed to remove water from the product including dehydrating the
product of water retained within the intercrystalline pores.
Higher calcination temperatures, typically on the order of 400°C
;~ ~ :~ ~~ z~ ,~.
"J UzleP~
_48-
or above, which lead to the decomposition of the templating
agent, should be avoided at this stage of the process.
The ion exchange medium may include any suitable agent
effective to protonate active sites in the molecular sieve struc-
ture by exchanging the sodium or other alkaline metal ions incor-
porated during the crystallization procedure. P.mmonium salts, as
described in detail below, are the preferred ion exchange agents
and the invention will be described in detail with reference to
the use of such ion exchange agents. However, it should be
recognized that other ion exchange agents compatible with aci-
difying the active sites in the molecular sieve network can be
used in carrying out the invention. For example, ion exchange
can be accomplished using aqueous solutions of mineral acids,
such as hydrochloric acid, nitric acid, or sulfuric acid or low
molecular weight organic acids such as formic, acetic, or pro-
pionic acid. The use of acids, particularly the strong mineral
acids, may be undesirable in preparing and modifying the zeolite
beta for certain applications in that dealumination of the zeo-
lice may result. in addition, organic salts, such as ammonium
acetate and primary, secondary or tertiary amine salts incor-
porating low molecular caeight alkyl substituents such as methyl
and ethyl groups, may be employed. Examples of such amine salts
include alkyl ammonium chlorides and nitrates such as ethyl ammo-
nium nitrate, methyl ammonium nitrate, trimethyl ammonium nitrate
and like amine salts may be employed. Also included as ion ex-
change agents are quaternary ammonium salts based again on low
molecular weight alkyl groups.
~~~'~t~
-49-
As noted previously, normally inorganic ammonium salts,
such as ammonium nitrate, ammonium sulfate, ammonium carbonate,
or ammonium chloride, will be used as the ion exchange agent.
Ammonium nitrate is particularly preferred since upon heating,
subsequent to the ion exchange step, it decomposes to ammonia and
nitric acid which, in turn, produces water and nitrogen oxides
which evolve from the catalyst product. A salt such as ammonium
sulfate is usually less advantageous because of its substantially
higher decomposition temperature relative to ammonium nitrate.
Also, in some cases, the sulfur may incorporate into the molecu-
lar sieve framework replacing framework oxygen.
Preferably, the initial ion exchange treatment is
carried out in two discrete steps each involving submersing the
zeolite beta in fresh ion exchange solutions. During the first
and preferably during both ion exchange steps, the zeolite beta
remains submersed in the medium until the exchange system
approaches equilibrium between the ammonium and sodium (or other
alkaly metal) ions. By way of example, employing a 2 normal '
ammonium nitrate solution, the zeolite beta may initially be sub-
mersed in the ion exchange solution for a period of about 1-5
hours at a temperature of 50°-90°C. Upon conclusion of the ini-
tial treatment, the zeolite beta is withdrawn from the solution,
caashed with water a;nd then submersed in a fresh solution of 2
normal ammonium nitrate. The time and temperature conditions
here may be the same as for the first submersion.
At the conclusion of the initial ion exchange treat-
men a , the ammonium exchanged zeolite beta is then subjected to a
~~? ~~)~~
-50-
high temperature calcination treatment. The calcination treat-
ment is carried out at a temperature and for a time sufficient to
evolve at least a predominant portion, and preferably substan-
tially all, of the templating agent from the interstitial pore
spaces of the channels of the molecular sieve network. The
calcination temperatures should be at least 400°C. It normally
will be no more than 700°C, although higher temperatures can be
employed. The calcination treatment should be normally carried
out for a period of about 2-10 hours, although at higher tempera-
Lures, shorter periods of down to about 1 hour may be adequate.
The calcination treatment may extend beyond 10 hours,
although there usually will be no reason for the longer calcina-
tion treatment. Preferably, the surface area at the conclusion
of this calcination step is at least twice the surface area of
the zeolite beta in the as synthesized form. As indicated by the
example described below, three-fold or more increase in surface
area can be achieved at the conclusion of the calcination step.
The initial calcination step is followed by a second ion
exchange treatment which results in a further increase in surface
area of the zeolite beta and a further decrease in the sodium, or
other alkali metal ion content. This post-calcination ion ex-
change treatment is, like the initial treatment, preferably
carried out in two :stages by twice submersing the zeolite beta
within fresh ion exchange solutions. The time and temperature
parameters employed in the second set of ion exchange treatments
may be the same as those involved in the first ion exchange
treatment.
-51-
At the conclusion of the second set of ion exchange
treatments, the zeolite beta typically will have a surface area
at least twice that of the original starting material and a very
low sodium content of no more than 0.04 wt.$ calculated as Na20
and usually less than 0.02 wt.~ Na20.
Where the resulting zeolite beta is to be used as a
catalyst, it normally will be mixed with a binder such as alumina
salt, gamma/alumina, or other refractory oxides to produce a
mulled zeolite beta binder mixture. The mixture can then be
palletized by any suitable technique such as extrusion, and the
resulting pellets then dried. At this point, the palletized
binder zeolite product is calcined under conditions sufficient to
decompose the ammonium ions on the active site so the zeolite
beta can arrive at the acid (H+) form. By way of example illus-
trating the present invention, an as synthesized zeolite beta
having a silica/alumina ratio of about 20-50 and containing
tetraethylammonium hydroxide as a retained templating agent, was
used as a starting material. The as synthesized zeolite beta had
an initial surface area of 210 m2/g and a sodium content of about
0.5-1$ Na20. The as synthesized zeolite beta was initially sub-
jected to an ammonium ion exchange treatment by submersing 100
grams of the catalyst in 1 liter of an aqueous solution of ammo-
nium nitrate having a normality of 2. The zeolite beta was sub-
mersed in the ion e~:change medium under mild agitation at 85°C
for a period of two hours. The zeolite beta was then separated
from the ion exchange solution, washed and retreated with a fresh
solution of 2 normal. ammonium nitrate again at 85°C far a period
~~~~~z~
-52-
of two hours. The surface area at the conclusion of the second
ammonium exchange step was 247 m2/g and the sodium content was
less than 0.11. The ammonium exchanged zeolite beta was then
calcined at a temperature of about 560°C for two hours. The sur-
face area at the conclusion of the calcination step was 666 m2/g.
After calcination, the exchanged and calcined zeolite
beta was cooled and then subjected to a second ion exchange
treatment involving a two-stage process, with each stage extend-
ing for two hours using the same ion exchange medium and under
the same conditions as used during the initial treatment. At the
conclusion of the third ammonium exchange procedure (the first
stage of the second treatment>, the surface area of the zeolite
beta cvas further increased to 708 m2/g. The surface area at the
conclusion of the final ion exchange treatment was 815 m2/g. The
sodium content at the conclusion of the final ion exchange step
was reduced to a value where it could not be measured using ato-
mic absorption techniques. Based upon this analysis, the Na20
content was substantially less than 100 ppm.
'The ammonium zeolite beta was mulled with peptized alu-
mina in a proportion of four parts zeolite beta to one part alu-
mina binder. The resulting plastic zeolite binder mixture was
extruded to form pellets having the size of about 1/16" and the
resulting pellets were then calcined at 560°C for two hours. The
surface area of the final product, based upon the zeolite binder
mixture, was 642 m2/g.
Modified zeolite beta produced in accordance with the
present invention can be used in various catalyst applications as
-53-
indicated previously or in other applications, for example, such
as selective absorbent. Where used as a catalyst, it may often
times be desirable to incorporate a metal component into the
zeolite beta binder substrate. Suitable metal components include
those found in groups VIB and VIII of the Periodic Table.
Specific metals include chromium, molybdenum, tungsten, vanadium,
iron, cobalt, nickel, copper, platinum, and palladium.
In experimental work carried out respecting the inven-
tion, the high surface area, ultra low sodium content zeolite
beta described above, was employed as a catalyst in the reaction
of ethylene and benzene to produce ethylbenzene. The experimen-
tal work was carried out in an upflow reactor heated by a sand
bath set at a nominal temperature of 200°C. The reactor contain-
ed 3.6 grams (7.0 ml) of catalyst in the form of 1/16" pellets
based upon the zeolite-binder mixture arid having a surface area
of 642 m2/g as described previously. The benzene was supplied to
the bottom of the reactor at a rate to provide a space velocity
(LHSV) of 10 hr-1. Ethylene was supplied to provide a benzene/
ethylene mole ratio of 5.2 with the actual mole ratio varying
during the experimental work from about 5.0 to 5.3. The results
of the experimental work are set forth in Table IV. The effluent
analysis is set forth in weight percent for benzene and ethylben-
zene and for various other components including toluene, cumene,
meta-diethylbenzene, and ortho and para diethyl benzene in yields
relative to ethylbenzene. The total xylene yield throughout the
run was zero.
The reactor was equipped with four thermocouples spaced
_5a_
evenly from the inlet, TC #1, to the outlet TC #4. From the exo-
therm profile indicated by the thermocouples, it is evident that
most of the alkylation reaction occurred in the lower portion of
the catalyst bed. Thus, the effective space velocity was
substantially higher than the nominal value of 10 hr'1 (LHSV>.
~t~~~~~~~~
N N O ~ N p O N ~ N vDo0h O U7
. t11 t ~.~ M d'N crm O cn
-i T~
u7 N N N N C' ~ t~ cTD O O
O
t~01 O C'.-1O vD
'
N r-i r-ir-4 N
rl O v0N tD.-1O tn a0rlv0cD~ O d
~-1N N O N .-1O tpcn 01V~c~N c0 O rl
c~t17N N N N I~ N I~ t~~t1O tn
r-1I
O ~ w O m ~ O r
rl .-tri N
a
O O h a0N O t0'-I rlN vDOwD O tn
ri cDN O N m O v~OS 01a~m crV~ O (71
O tn N N N N M cnt~ t~O O h
.-i1 Lt7
t'01 O ei'I-IO U7
.-i ri r-1r-I N
O1 O tnr1M r1 riO M rlO lDtn O N
~Drl O rie-IO ritf7 O C~V~COO O Q1
~
COtn N N N N CO c~I'~ N N O C
I .-i
O CDrN O Inr-1O vD
r-I r1 rirl N
CO O inQ~O N I~tD I~tDrlN a0 O O1
t0 O .-iN O C'CD InM lJ~M c~1O ~D
rl~ N N N N t~ m r. O ~D O ~D
h
a001 O ' t11O O tf7
e1ri N
(~ O O c~U7tnr-1V'N Q1O O Q10~ O C
,
O O ~'~N O V'cn t~V' u7~ O tf1
~ c~tn N N N N t0 N ~n c t~ O N
f"I ~ tI1 O O c~ O 00
-.I .-1 r~l
I
u1
I ~ o Inc~P rl a n r-trm ~n o .-I
E.
O~ O rlN O cnt~ ~'err'N m tf)O O~
t0V' N N N N lf1 c''f~o N O1 O t~
M tD
t~o~ O O t~ O cn
.-I r-i .1
In O tnO~h ri N ~ O Wit'tI1d'M O t~
N O O .~N O N M rlN N c~C' O .-1 ,
u~ N N N N O c'~~ tn~D O N
N ~ O O O I~ O c0
N .- and
C' O N cpO~cn .~tn t~C~U'1GOV' O N '
O O r-1N O t~~ ~D.-aN r-tas O O
01t0 N N N N M m vO COtn O h
t~O O O P O O
N
r-1~ O ' t~.-~N O V N 41O~.-1ri O N
C
O O rlf~tO 01In ODm-1tDI~l0 O M
~
r-1tf~'N N N N !'~ ~T.-1 a1v~ O c0
~ oD O ~ ~ cDvD O tn
r-1
'P
(-~ . o
~
'U~ o~dP
r-cnN
~i
~ ~ ~ ~ n ~ ~ z ~ ~ ~ o ~ ~ '
a
U
v N o m N ..v .-a m m o, own m r o w
N ~n r O N N O W O m O~ N m O O O
-- . V c' N N N N O r m tn O N O t'7
h N O~-1 N O N ~ ~ls ~ t.I ~ 4.~ ~.~
m N O \D N r-1 O fn N O m t~ r1 r O m
Nc900NNO~ON crN~NtDNOm
tn tc1 N N N N N N U1 01 r O ~D
~ O O~ONN
N u1 O r a~ m ,--~ '-I r~ O m Uv O O O O
N W O O N N O O In r ~'1 N O m O ov
~''L tf1 N N N N c'~ N N m m O tD
V O O ~1 O N
N r1 .-1 N
.1 01 N r V fn .-! 00 tD ~P N m rl r1 O W
N U1 O O N N O tD U1 tD Wit' N r r O V'
c'f tn N N N N r .-1 N In In O .1
tOD N O a ~ O ~ ..
O .-a tf1 v r m .-I Q1 v0 tn '-1 vD aT cn O r
N V ~ O O N O O O r aT N m tn O cn
~''1 V~ N N N N N N l11 r In O M
r T O O .-~ O N
.-I ri r-~ N
C'
Q1 V' N t~ ep M r-1 m .-i O (''1 O1 N tA O .-4
.y N 01 O N N O O t0 m r N If7 O~ O Uf
tn C~ N N N N Q~ N N r ~D O V~
h N O - .~-1 ~ O N
m tD O tD O c~'1. .-i .-y' O N O N vD O .-1
r-~ O O O N N 'O~ ~ N rl ~"~ cT1 d' r~ O m
c'W n N N N N .-1 C' O O cD O m
m N O ~ O a1 0 01
..~ .-m-t
h O O m O~ tD N Q1 ~O tn V' t~1 M C O r
.-1 O .~-~ O N N O N O r vD tn r tn O O
tn N N N N M V' Ov O~ O O O
M
m .-1 O cn ~'n O r
rl N N d'
~ Q~ If1 O1 N O N V~ N O r T cn N O tn
ri t~ V' O N N O In C V' CT N O1 N O ri
N N N N N N 01 N Q tf1 m O C~
h N O ~ rNi O N
V'
tn t~ t1W O N d .°I h lf1 lf1 O N ~ t~ N ' O N
.-i u'1 O O N N O .-i tf1 N ~O N o~ m O m
N tn N N N N ~ m N tn O~ N O N
O 01 01 O
v o u~ o, o ~ .-wo o m .-mo ~ r o M
.-1 .-1 N O m N O r O v rl N v N o r
N tn N N N N ~ O m t~ N N O T
h ~ O ~ ~ O N
th O1 t11 t~ DD If1 .-1 n ch e! eYf N r~ Q1 O O
.-1 t~ N O r-1 .-1 O .-i imD ~P cn r ~ O a~
r1 t'1 N N N N O ~' W f~ ~! Qv O V'
O cD tf1 O
M ri
.--1 op
r-1 dP
ci ~ ~ ~ aP aP
m N N ~ O, C1.
u~7 c~ ~ ~ ~ ~ a m ~ r z ~ ~ ~ o
!~ ~~ i~ i ~ ~ 'w~
-5'7- .
From an examination of the experimental data presented
in Table IV, and bearing in mind that, for the most part, the
ethylene charge to the reactor was slightly less than 20~ of the
stoichiometrically equivalent amount of benzene, it can be seen
that the zeolite beta catalyst is highly active and shows good
selectivity to ethylbenzene production. The activity of the
catalyst remained substantially undiminished after the duration
of the experimental work.
In carrying out this embodiment of the invention, the
alkylation reaction is carried out at pressures well above the
vapor pressure of the aromatic substrate at the reaction tempera-
ture involved in order to ensure that a liquid phase is retained
in the reactor. In order to provide a complete liquid phase
reaction, a flooded-bed format is used in which the catalyst bed
is completely immersed in liquid. This can readily be
accomplished using an upflow technique such as used in the ex-
perimental work and this usually will be preferred i.n carrying
out the invention. However, downflow flooded bed operation can'
be accomplished by control of the outlet flow rate to ensure that
the catalyst beds are covered by liquid benzene or other aromatic
substrate.
Preferably, a staged reaction format is employed in
order to ensure good solubility of the ethylene (or other alky-
lating agent) in the benzene for other aromatic substrate) so
that the entire reaction takes place in the liquid phase. In
addition, use of multiple stages provides an opportunity for
interstage cooling where adiabatic reactors are used or permits
cal Ls3 s ~'7 ~,
~.~~l~~.a~~~
_5s_
the use of several isothermal reaction stages.
Turning now to FIGURE 11 of the drawings, there is shown
a schematic illustration of a staged reactor system used for the
production of ethylbenzene by the reaction of ethylene with ben-
zene which includes a plurality of adiabatic reactors with inter-
stage cooling and injection of ethylene. More particularly, and
as illustrated in the drawing, ethylene and benzene are supplied
via lines 102 and 104 to the inlet line 105 of a dehydration unit
106 which is operated in accordance with another embodiment as
described hereinafter. The dehydration unit functions to de-
hydrate the input to the alkylation reactors so that it is essen-
tially dry, preferably containing less than 100 ppm, more
preferably, less than 50 ppm, water. By way of example, dehydra-
for 106 may take the form of a packed column packed with a desic-
cant such as silica gel or other suitable hydrophilic medium.
The dehydrator effluent is supplied to a reactor 108,
the first of a plurality of series connected alkylation reactors
operated in an upflow mode. Reactor 108 is operated at an
average temperature of 300°C or less, and preferably, at an
average temperature within the range of 200°-250°C. The pressure
on reactor 108 is sufficient to maintain the benzene in the
liquid phase and preferably is at least 50 psi above the vapor
pressure of the benzene at the reactor temperature. Typically,
the reactor pressure is within the range of about 500-600 Asia.
The remaining downstream reactors normally are operated under
approximately the same conditions as the initial reactor. The
effluent from the initial reactor 108 is withdrawn via line 109
rd 'lJ ~d :.~ c~ r.~
and applied through a h~aat exchanger 112 where it is cooled.
Lthylane is supplied via l3.ns 111 whaxe .lt is mixed with th~
flfgluent from the tirs~ r~ractor, ~lae. Praf~a; ably, the ethylene is
supplied to the r~:actar effluent prier to cr~olinc~ In order to
faci.litat~ distribution c~f the ethylene thr.~ughout the lis~ui,d
bsn~ene. l3aeirably, the cooling axt~ap is carried ~ut en reduced
the temperature of tha feed mir,t.ure supplied to the sac~nd re~G-
tor 11~ t.o a value about the sa~we set the lnls~ t~em~~raturra to the
first reaator 10.9. The average temp~raturs in the seoond xsactor
normmlly wii,l be about the same as that of the First reactor.
fibs preas~ara will, of necssgi:y, be somewhat lc~wsr i.n order to
provide for sufgi~isnt pr8eeure gradient to aGCnmodate tl~w
through the cyst~m. fibs effluent Pram ths~ secaad $enctar 114 is
supplied along with athylenr~ provided via ~llaae 117 to a seacnd
interntage .cooling unit li9 where the shares mixtura~to third
rga~ctar Z20 ie aga~.n aaa~,ed to a te~nnera~ture about ~qusl to the
inlet tempsrature for they first twc~ reactors.
The outgun. frs~~n xeact~or~ 120 is supplied via line 12~ to
a downstream aeparatian and processing unit 12~. rn unit 12d,
eth~lbenzene ie e~parat~~ad and withdrawn ae the produet of the
alkylation plant. Typicalz~,~, $r.hy.lbanzene will b~ used ae the
charge to a dehy~3roganation system syatsm~where It undergoes
catal.ytia d~shYdroget~ation in the pxod~acl:ion of etyren~. Normal
ly, banza»a arid ~thyl~ena will ba aeparat~d in unit 12~ and re-
Cyclad tt~r ue9 in the al3cylst.i~n presses. Heeviaar palystt~yl~~
bsn~ena~ may ba t~canamlky~.ated with ben~ena to produce additional
ethyl.ben$en~~ as described previouei.y.
d is cvnvantianal., n ataichlometria exreasa of b~n~ena
to athylvns Will be supplied in the shares ntoaJc to ttW a~yl~.-
~.60-
tion reactors in drderr tee anisanca seelectivigy ~t~r mona~axkylati~n.
~psrmtion of the seactara tc provide lictuid phtsae alkylation
undar relat3v~ly mild ca~nditiona~'nc~t only minimixea the acylena
ps'oducad in the alkYlatian s;aaction hut a1s~ enables the use of a
somewhat lows b~nxena,/ethYl~na mol~a reties than is usually the
aaao. Usually, the benxan~/~thyi~na mole retie ~'i13, be 5sI ctc
lager and mare pr~farably~ ~sl or less. ~ant~na~athYlane mole
a~atioa as low as about ~ a 1 maY be at~ploy~d . Ratiog graa,t~r than
ssi can bs used. Howcvurr tfaera is usually little incan$ive to
uea e~ttrtmely h3.gh ratios and, ~t~ a practical lita,tte~, the
bans;~ne/ethylsng mole gatio will a~ldom ~~cCaad i.5sl. ~h~
bens:ene/ethylation mole ratios referred to ~tbave era with raapant
to the avsrall system and fcr a mufti-atag~ raaotian ayatern such
ae dapiated in the drasaS.ngr Ch~ banzene/athyl,cne ratio of the
fa$d to sash stage gill be 1~~a than the saverail ratio.
the aanount of ethylenedaolubilixed its the bettzt~sne charges
to each reactor atag~ ~ail1 cepand in part upon the number ~f
reac%~or stagers employed. Prafea't~blyr at least 3 reactor staa~ee,
ae illu$trat.ed, Mill be head. . Additional s:'eaatas~ stages may tie
prov idad, a~'thoug~ th~ total number of atag~a sxsarmally c~il1 not
~xcead 6. t~x~ferablyr the pr~aauxa in each raactior~ stags nrsd
the eraount of ethylene eeyplied that~in ia..auoh as to Provide at
lenat l~mole percent of ettsyler~~ sclubilizea in th~ banaanae
~$ua,lly at ieaa~t 2 mote percent of ethyl~ns vaill ba acalt~bilixad
in the charge i:o ~sxah reactor. ~s~ferablyr unlasis n ggaat 9na~nY
reactor stages era amplolred, ~aaually the amsaunt of ethylene ~alu~
bi.liaud in the liquid banxene ghas$a of each sr~actoz~ e~lll tie at
-61-
least 4 mole percent.
The following Table V gives exemplary conditions and re-
action parameters for a multistage system of the type shown in
the drawing, but employing five reaction stages. As will be dis-
cussed below, Table V also illustrates a preferred mode of opera-
Lion when going from one reaction stage to the next, as well as
advantages accruing in the use of multiple reaction stages in the
liquid phase alkylation process where the reaction pressure,
while above the vapor pressure of the aromatic substrate, is
below the vapor pressure of the alkylating agent at the reaction
conditions.
.
W Ix ( O ~-i N m ~
N O 01 CO r. ~p
UI
-N .~ ~ ~r er ~ ~
NU rt1 O
U w ~, g
N
G C, dP dP dP dP dP
~ O O1 O r1 N C~
LW U ri ri ~ .-a
G
Q~ t/~
N ~ N V~ OD P ~p
~ N r-1 w N a»
N N O
W Cx~ ~ ~ m m N N
~ N N
~ ~
10~.. cDtDC'Nr-1
a ~r
v' a~
v~
I
W
N a
m
a
00000
L1 C.I O OD \O eT N
H In C' W' C' C'
-L1O O O O O
Wit' v~ C~ eH
cr
O N N N N N ' -
U
C1~
E
U
00000
C .a ~ .-a ~ ~
H N N N N N
O
~i
U
N t0
U~
~ U1 ~-i N m C lC1
-63-
In Table V, the idealized reactor conditions for the
ethylation of benzene with ethylene are illustrated in columns
2-5. The benzene feed rate in moles per unit time to each of the
reaction stages is set forth in column 6. Benzene conversion for
each reaction stage is indicated in column 7 and the ethylene
feed rate in moles per unit time to each reaction zone is set
forth in column 8. The last column presents the mole ratio of
benzene to ethylene at the input of each of the successive reac-
tion stages. The data presented in Table V is based upon an
idealized case, assuming that benzene conversion is about 90$ of
theoretical based upon the feed rate of ethylene which, of
course, is the limiting reactant.
At the temperature and pressure conditions depicted in
Table V, the solubility of ethylene in the liquid aromatic com-
pounds involved, including benzene, ethylbenzene and polyethyl-
benzene, is about 10 mole percent. The ethylene feed to the
first reaction stage is controlled in order to provide an amount
of ethylene near the solubility limit. Within the first reaction
zone, 0.36 moles of benzene are converted and the effluent from
the first reaction zone, after cooling as described previously,
is applied to the second reaction zone. The aromatic feed to the
second reaction zone will comprise about 3.64 moles of benzene
and about 0.36 moles of ethylbenzene and,polyethylbenzenes per
unit time. Since the ethylated product can serve to solubilize
the ethylene at the reaction conditions, the ethylene feed rate
can be retained at 0.4 moles per unit time, resulting in a de-
creased benzene conversion rate. The relationships described
-64-
above prevail when going from one reaction stage to the next, re-
sulting in a decreased mole ratio of benzene to ethylene in each
succeeding reaction stage and an increased benzene conversion
rate although in the idealized case presented in Table V, the
interstage injection of ethylene is maintained constant. This
need not necessarily be the case. For example, the ethylene feed
rate can be increased or decreased slightly from one stage to the
next, or alternatively decreased and increased, so long as the
overall progression across the multistage system is one of
decreasing benzene/ethylene ratio with the attendant increase in
benzene conversion. By way of an example of a progressively de-
creasing feed rate and with reference to the system depicted in
Table V, the ethylene feed rate can be progressively decreased by
2-3$ when going from one reaction stage to the next while retain-
ing the characteristic of a decreasing benzene/ethylene ratio as
depicted in the Table. If the ethylene feed rate is increased
when going from one stage to the next, it is preferred to main-
taro the ethylene below the solubility limit at the temperature
and pressure conditions involved in order to avoid multiphase
flow through the catalyst bed.
Multistage ethylation of benzene may also be carried out
in accordance with the present invention employing isothermal
reaction zones. Isothermal reactors can take the form of shell
and tube type heat exchangers with the alkylation catalyst depo-
sited within the tubes and with a heat transfer medium circulated
through the shell surrounding the catalyst filled tubes. The
heat exchange medium will, of course, be supplied through the
-65-
reactors at rates to maintain a relatively constant temperature
across each reaction stage. In this case, interstage cooling
will be unnecessary, although it will be preferred to inject
ethylene at the front of each reaction stage.
As discussed previously, it is desirable in accordance
with certain aspects of the invention to employ a dehydration
step. Aromatic conversion reactions such as alkylation or tran-
salkylation may be carried out in the vapor phase or in the
liquid phase. Intermediate pore sized zeolites such as ZSM-5
(pore size of about 6 angstroms) are effective catalysts for
vapor phase alkylation or transalkylation where movement of aro-
matic molecules in the gas phase through the molecular sieve net-
work takes place by energy vibration. Somewhat larger pore size
molecular sieves appear to be necessary to provide effective
catalysts for processes such as the liquid phase alkylation of
benzene. Thus, benzene, which has a kinetic diameter of about
5.9 angstroms, will enter into the molecular sieve network of an
intermediate pore size molecular sieve such as ZSM-5. However,'
the resulting alkylated product such as ethylbenzene or cumene
will not readily move through the molecular sieve channels by
liquid phase displacement.
The zeolite molecular sieves employed in the present
invention and having a pore size greater than 6.5 angstroms are
effective catalysts under relatively mild conditions for liquid
phase hydrocarbon aromatic hydrocarban conversion reactions such
as the ethylation of benzene or the transalkylation of polyethyl-
benzene. As noted previously, conversion takes place at relati-
~~?~~)~~~
-66-
vely loco temperature conditions of less than 300°C, about 275°C
or less. In fact, effective ethylation or transalkylation reac-
tions can take place in the liquid phase over larger pore size
zeolite molecular sieves employed in the present invention at
temperatures within the range of about 200°-250° C and such reac-
tions can be accomplished without undesirable side reactions as
may be encountered in vapor phase reaction conditions. The
pressure on the reaction zone in which the conversion reaction
takes place is necessarily above the vapor pressure of the aroma-
tic substrate involved. Preferably, the reaction zone pressure
is at least 50 psi above the vapor pressure. Thus, in the ethy-
lation of benzene at 225°C to produce ethylbenzene, the reactor
pressure preferably would be about 350 psig or more. In general,
the reactor pressure may range from about 250-1000 psig.
While, as noted previously, water can be tolerated in
vapor phase reactions, it does under the high temperature con-
ditions encountered in vapor phase reaction, effect the dealumi-
nation of the catalyst with a corresponding decrease in proton-
aced sites and a reduction in acidic catalyst activity. One
would not expect a similar effect to be encountered under the
relatively mild conditions of liquid phase aromatic conversion
reactions and, in fact, it appears that dealumination in the pre-
sence of water does not occur under these conditions, However,
by dehydrating the :Feed stream to the liquid phase reaction zone,
the aging quality o:E the catalyst is substantially increased. In
fact, by decreasing the water content to well below 300 ppm, a
value normally tolerated in vapor phase reactions without sub-
n~ s/~ r~ ~ s~ ~i~
YJ ~..J ".~ LY
-67-
stantial adverse impact upon catalyst aging quality, the aging
quality of the catalyst in the liquid phase condition is
materially enhanced.
As nested previously, the molecular sieves employed in
the present invention have pore sizes greater than 6.5 angstroms
which readily accomodate movement of molecules within the molecu-
lar sieve network by a liquid phase displacement mechanism. The
preferred zeolite molecular sieves, zeolites Y and beta, have a
pore size within the range of 7.0-7.5 angstroms. The catalysts
are not acid extracted to effect dealumination. In additional
experimental work carried out relative to the invention, such
larger pore size zeolite molecular sieves ware employed as cata-
lysts in the liquid phase transalkylation of diethylbenzene. Two
zeolite Y catalysts were used in this experimental work. Zeolite
Y is characterized by a three dimensional channel system and has
an average pore size of about 7.3. Zeolite Y catalysts have
silica/alumina ratios of less than 10, usually about 5-6.
In this experimental work, a mixture of benzene and a .
polyethylbenzene overheads fraction resulting from a vapor phase
alkylation process was passed into a reactor containing a zeolite
Y catalyst. The reactor was operated in a flooded, upflow mode
configuration and under a pressure of about 30 psig to maintain
the aromatic compounds in the liquid phase. The flow rate was
sufficient to provide a space velocity (~HSV) based upon the
total feed of about 3 hr-1. The weight ratio of benzene to
polyethylbenzene overheads was about 4. A typical feed com-
position employed in the experimental work is shown in Table VI.
4'1 !~ ,..
~r/~;'a~~~
-68-
Table VI
Non-Aromatic 0.01
Benzene 78.87
Toluene 0.00
Ethylbenzene 3.40
p-Xylene 0.01
m-Xylene 0.02
Styrene 0.03
o-Xylene 0.04
Cumene 1.67
n Propylbenzene 3.30
m-Ethyltoluene 0.15
p-Ethyltoluene 0.05 .
o-Ethyltoluene 0.04
1,3,5-Trimethylbenzene 0.07
1,2,4-Trimethylbenzene 0.20
sec-Butylbenzene 0.39
1,2,3-Trimethylbenzene 0.32
m-Diethylbenzene 7.03
n-Butylbenzene 0:29
p,o-Diethylbenzene 3.90
Heavies ~ 0.49
The water content of the feed was about 300 ppm. The
temperature was progressively increased during the run as
necessary to maintain the transalkylation reaction at 70~ conver-
sion of diethylbenzene. Over the first 11 days of the experimen-
--69-
tal run, the charge of wet feedstock was first passed into a de-
hydrator filled with a molecular sieve desiccant. The output
from the dehydrator was passed into the reaction zone. The dried
feedstock was estimated to have a water content of about 30 ppm.
Thereafter, and over the remainder of the run, the wet feed was
applied directly to the reactor.
The results of the experimental work employing one of
the zeolite Y catalysts axe set forth in FIGURE 12. In FIGURE
12, curves 126 and 127 are graphs of temperature, T in °C
necessary to maintain 70~ diethylbenzene conversion plotted on
the ordinant versus the age, A, of the catalyst (the duration of
the run) in days plotted on the abscissa. As indicated by curve
126 for the dried feed, the catalyst exhibited an aging quality
of about 1.8°C per day (average daily increase in temperature
necessary to maintain 70~ conversion). Curve 127 of FIGURE 12
indicates the aging quality of the catalyst when the feed stream
was diverted from the dryer so that the wet feed containing about
300 ppm water was directly applied to the alkyaltion reactor. As
indicated by curve 127, the aging characteristic for the catalyst
more than doubled to about 3.9°C/day.
Similar experimental work was carried out using another
zeolite Y catalyst in the liquid-phase transalkylation of poly-
ethylbenzene. The feedstock employed here was the same as the
feedstock used in the experimental work described immediately
above. In this case, the temperature was adjusted as necessary
to maintain the transalkylation reaction at 80$ conversion of di-
ethylbenzene. The space velocity was the same as employed in the
previous zeolite Y experimental work, 3 hr-1 (LHSV?. The trans-
alkylation reaction was carried out at a pressure of 300 psig in
order to maintain the aromatic hydrocarbons in the liquid phase.
In this test, the wet feed, containing about 300 ppm, was ini-
tially applied to the reaction vessel containing the zeolite Y.
At the conclusion of nine days, the feedstream was first directed
to a dehydrator containing silica gel which extracted water from
the feed stream to provide a water content of about 30 ppm. The
run was then continued for an additional 11 days during which de-
hydrated feed was supplied to the reaction zone. The results of
the experimental work carried out for the second zeolite Y are
illustrated in FIGURE 13, in which curves 129 and 130 are graphs
of temperature T, in °C, required for 80$ diethylbenzene conver-
sion of the wet and dry feeds, respectively, plotted against
catalyst age in days. As shown in FIGURE 13, the initial wet
feed caused a very rapid deactivation of the catalyst. However,
at the conclusion of the wet feed injection, the introduction of
dry feed not only materially reduced the catalyst deactivation
rate but actually enhanced the activity of the catalyst.
In addition to transalkylation, this embodiment of the
invention may be employed in the liquid phase alkylation of aro-
matic substrates. As indicated earlier, a particularly important
liquid phase alkylation reach on is the ethylation of benzene
under mild liquid phase conditions which results in little or no
xylene make. Other liquid phase alkylation reactions may be em-
ployed, particularly those involving the use of C2-C,~ alkylating
agents. For example, this embodiment of the invention may be
-71-
employed in the reaction of propylene and benzene to produce
cumene. Usually, alkylating agents will take the form of ole-
fins. However, other al)cylating agents such as alkynes, alkyl
halides, alcohols, ethers and esters .as disclosed, for example,
in the aforementioned patent to Pollitzer, may be employed.
Also, aromatic substrates other than benzene, for example,
toluene or xylene, may also be subject to liquid phase alkylation
in accordance with the invention.
As noted previously, dehydration of an aromatic feed-
stock in accordance with the invention may be carried out in con-
junction with the use of a zeolite molecular sieve other than
zeolite Y having a pore size within the range of 7.0-7.5
angstroms. Specifically, zeolite beta is an effective alkylation
catalyst under the mild temperature conditions involved in liquid
phase alkylation. The preferred zeolite beta alkylation cata-
lysts are, as described earlier, of a very low sodium content,
less than 0.04 weight percent and preferably less than 0,02
weight percent expressed as Na20, and have a high surface area,,
at least 600 m2/g. The zeolite beta has a silica/alumina ratio
of about 20-25.
Zeolite omega, referred to previously in regard to its
use in alkylation and transalkylation is disclosed in U.S. Patent
No. 4,241,036 to Flanigan et al., along with its x-ray diffrac-
tion pattern and basic procedures for its preparation. Zeolite
omega is synthesized by the hydrothermal digestion of a reaction
mixture comprising silica, alumina, an alkali or alkaline earth
metal oxide or hydroxide, specifically sodium hydroxide, and an
a ry r~ ~.
~~~~~,._~~
_72._
alkylammonium component, specifically tetramethylammonium
hydroxide.
The chemical composition of zeolite omega in its pre-
ferred form may be characterized as follows:
[xR2+yM2n0):A1203:5-20Si02:0-8H20
wherein:
x has a value ranging from 0-0.7 and the sum of x and y
ranges from 0.5-1.5;
R represents hydrogen, ammonium, alkylammonium or mix-
tures thereof;
M is a metal compound, usually an alkali metal compound
such as sodium; and
n is the valence of M.
For a further description of zeolite omega and its preparation,
reference is made to the aforementioned U.S. Patent 4,241,036 to
Flanigan et al., the entire disclosure of which is incorporated
herein by reference.
In the alkylation of benzene, both the benzene feedsto~ck
and the ethylene for other alkylating agent) may contain water.
Accordingly, it will be preferred to pass both the benzene and
the ethylene through a dehydration unit. While separate dehydra-
toys may be used for the two feed components, usually the ethy-
lene and benzene will be mixed in the mixed feed stream and
applied to the dehydration unit and from there to the liquid
phase reactor.
In the application of this aspect of the invention to
the transalkylation of polyalkylbenzenes, all or part of the feed
~~~t~~~v
to the transalkylation reactor may be subject to a prior dehydra-
tion step. Normally, the transalkylation of polyalkyl benzenes
will be carried out in conjunction with a prior alkylation step
with the output from the alkylation reactor being subjected to
one or more separation steps resulting in a polyalkylbenzene com-
ponent which is combined with benzene and then passed to the
transalkyaltion reaction zone operated under liquid phase dispro-
portionation conditions as discussed previously.
Where the invention involves a transalkylation process
carried taut in conjunction with a liquid phase alkylation step
proceeded by a dehydration step as described above, the poly-
alkylbenzene component supplied to the transalkylation reactor
should be substantially free of water and it normally will be
necessary to subject only the benzene component to a dehydration
step. However, in other applications of the invention, it may be
necessary to subject the polyalkylbenzene component to a dehydra-
tion step prior to its introduction to the transalkylation reac-
tor. For example, the transalkylation procedure may be carried
out in combination with a vapor phase alkylation procedure which
tolerates water in the feed stream or in which water is addi-
tionally added, for example, as disclosed in the aforementioned
patent to Barger et al. In this case, it may be necessary to
subject both the polyethylbenzene component and the benzene com-
ponent to dehydration prior to passage to the transalkylation
reactor.
Having described specific embodiments of the present
invention, it will be understood that modification thereof may be
suggested to those skilled in the arty and it is intended to
cover all such modifications as fall within the scope of the
appended claimso