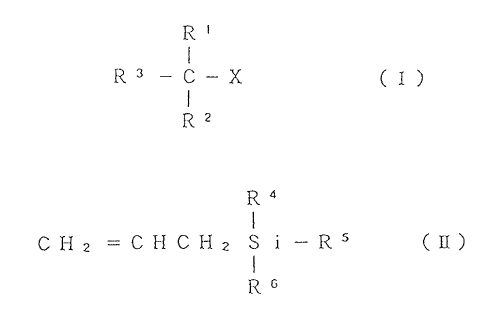Note: Descriptions are shown in the official language in which they were submitted.
DESCRIPTION
PROCESS FOR PREPARATION OF A POLYMER
HAVING REACTIVE TERMINAL GROUP
Field of the Invention
The present invention relates to an isobutylene-type
allyl-terminated polymer.
Background Art
Functional group-terminated polymers such as polymers
having hydroxyl groups or the like introduced to both ends
o.f the molecule are useful as materials for polyurethane,
adhesives, modifiers, coating compositions, sealing agents,
etc.
Isobutylene-type unsaturated group-terminated polymers
are known as such functional group-terminated polymers.
For example, a process for preparing an isobutylene-
type functional group-terminated polymer is reported
(specification of U.S. Patent No.4524188) in which a CP-
terminated polymer is prepared by the Inifer method
(specification of U.S. Patent No.4276394) comprising
cationically polymerizing isobutylene in the presence of
1,4-bis(a-chloroisopropyl)-benzene (hereinafter referred to
as "p-DCC") serving as an initiator and concurrently as a
chain transfer agent, and BCP3 serving as a catalyst, and the
obtained polymer is subjected to reaction for removal of HCP
from the polymer.
-2-
However, the reported method involves numerous reaction
steps, and therefore is not convenient.
A simplified method for introducing unsaturated groups
to an isobutylene-type CP-terminated polymer is known in
which the isobutylene-type polymer with CP groups at both
ends obtained by the Inifer method is reacted with
allyltrimethylsilane in the presence of a Lewis acid to
thereby convert into a polymer having allyl groups at both
ends (Japanese Unexamined Patent Publication
No.105005/1988).
Our research revealed, however, that the process
disclosed in Japanese Unexamined Patent Publication
No.105005/1988 has problems. For example, unsaturated
groups are introduced to ends of the molecule in a low
ratio, and it is necessary to use an expensive material.
Disclosure of the Invention
An object of the present invention is to provide a
process as simple as the process described in Japanese
Unexamined Patent Publication No.105005/1988 for preparing
an isobutylene-type unsaturated group-terminated polymer.
Another object of the invention is to provide a process
for preparing an isobutylene-type unsaturated group-
terminated polymer having the unsaturated groups introduced
in a high ratio.
A further object of the invention is to provide an
~~''~~iz.~, i ice
-3-
inexpensive process for preparing an isobutylene-type
unsaturated group-terminated polymer.
Other objects and features of the present invention
will become more apparent from the following description.
In view of the foregoing present situation, we
canducted extensive research and found that all of the
above-mentioned objects can be accomplished by carrying out
a specific reaction process using a specific Lewis acid.
According to the present invention, there is provided
a process for preparing an isobutylene-type allyl-terminated
polymer, the process comprising mixing:
(A) a cationically polymerizable isobutyl.ene-containing
monomer;
(B) an organic compound serving as an initiator and
concurrently as a chain transfer agent, the organic compound
being represented by the formula (I)
R~
I
R 3 -c- x Cz)
R2
wherein X is a halogen atom, a RCOO- group (wherein R is a
monovalent organic group, the same hereinafter) or a RO-
group, R3 is a polyvalent aromatic ring group or a
substituted or unsubstituted polyvalent aliphatic
hydrocarbon group, and R~ and R'- are the same or different
~~~1~:~~~
_4_
and each represent a hydrogen atom or a substituted or
unsubstituted monovalent hydrocarbon group, provided that
when R3 is a polyvalent aliphatic hydrocarbon group, R' and
RZ can not be concurrently a hydrogen atom;
(C) at least one Lewis acid selected from the group
consisting of (CZHS)ZAPCP, (CZHS)APCP2, SnCP4 and TiCP4; and
(D) a compound serving as an end capping agent, the compound
being represented by the formula (II)
Ra
C H z - C H C H 2 S i - R'
R6 _
wherein R°, RS and R6 are the same or different, and each
represent a monovalent organic group or a monovalent organic
group having 1 to 3 carbon atoms replaced by silicon atoms,
whereby the cationically polymerizable monomer is
polymerized.
The foregoing process of the invention has features of
being convenient, low in costs because of using an
inexpensive Lewis acid, capable of introducing unsaturated
groups in a high ratio to the ends, and also an excellent
feature of giving an isobutylene-type polymer which has a
narrow molecular weight distribution. With a narrow
molecular weight distribution, the polymer has the
advantages that the polymer is low in viscosity and
i~~)~I~~K:~
-5-
therefore easy to handle, for example easily kneadable in
mixing the components and that when crosslinked, the polymer
provides a cured product which is outstanding in mechanical
properties and the like.
It is possible in the invention to reduce the amount of
the component (C), hence economical. Since the
polymerization rate can be controlled by adjusting the
amount of the component (C) which participates in the
polymerization reaction, the rate of heat evolution in the
polymerization reaction can be regulated. The reduction of
heat evolution rate can control the polymerization rate
without use of a large-scale cooler in the large-scale
manufacture of isobutylene-type allyl-terminated polymer,
thereby making the process economical.
The process of the invention has an additional
advantage that even when the polymerization reaction is
conducted at a relatively high temperature of -40 to 10°C,
an isobutylene-type polymer with terminal functional groups
formed in a high ratio can be obtained.
The term "cationically polymerizable isobutylene-
containing monomer" used herein includes not only a monomer
consisting of isobutylene alone, but a monomer comprising a
combination of isobutylene and up to 50% by weight (percent
by weight being simply indicated hereinafter by °'o") of a
cationically polymerizable monomer which is copolymerizable
~iSa.~~~~
-6-
with isobutylene.
Examples of the cationically polymerizable monomer
which is copolymerizable with isobutylene are olefins having
3 to 12 carbon atoms, conjugated dienes, vinyl ethers,
aromatic vinyl compounds, vinyl silanes, etc. among which
olefins having 3 to 12 carbon atoms, conjugated dienes and
the like are preferred.
Specific examples of the cationically polymerizable
monomer which is copolymerizable with isobutylene are
propylene, 1-butene, 2-butene, 2-methyl-1-butene, 3-methyl
2-butene, pentene, 4-methyl-1-pentene, hexene,
vinylcyclohexane, butadiene, isoprene, cyci.opentadiene,
methyl vinyl ether, ethyl vinyl ether, isobutyl vinyl ether,
styrene, a-methylstyrene, dimethylstyrene,
monochlorostyrene, dichlorostyrene, (3-pinene, indene,
vinyltrichlorosilane, vinylmethyldichlorosilane,
vinyldimethylchlorosilane, vinyldimethylmethoxysilane,
vinyltrimethylsilane, divinyldichlorosilane,
divinyldimethoxysilane, divinyldimethylsilane, 1,3-divinyl-
1,1,3,3-tetramethyldisiloxane, trivinylmethylsilane,
tetravinylsilane, y-methacryloyloxypropyltrimethoxysilane,
y-methacryloyloxypropylmethyldimethoxysilane, etc. Among
these monomers, for example, propylene, 1-butene, 2-butene,
styrene, butadiene, isoprene, cyclopentadiene, etc. are
suitable. These canonically polymerizable monomers which
i~o~o~~~~~
are copolymerizable with isobutylene can be used singly or
at least two of them are usable in mixture.
Examples of useful organic compounds of the formula (I)
which are used as an initiator and also as a chain transfer
agent in the invention include:
compounds of the formula (III)
AYn (III)
wherein A is a group having 1 to 4 aromatic rings, Y is a
group attached to an aromatic ring represented by the
~ formula (IV)
R
C _. ~ (fir)
R~
(wherein R' and R8 are the same or_ different and each
represent a hydrogen atom or a monovalent hydrocarbon group
having 1 to 20 carbon atoms and X is a halogen atom such as
F, CP, Br or I, a RCOO- group or a RO- group) and n is an
integer of 1 to 6;
compounds of the formula (V)
BZm (V)
wherein B is a hydrocarbon group having 4 to 40 carbon
atoms, Z is a halogen atom, a RCOO- group or a RO- group,
all attached to the tertiary carbon atom and m is an integer
of 1 to 4;
-g_
oligomers having a-halostyrene units; etc.
to which the compounds of the formula (I) are not limited.
These compounds can be used singly or at least two of them
are usable in mixture.
The group A having 1 to 4 aromatic rings in the
compound of the formula (IIT) may be either a condensation
product or a non-condensation product. Examples of such
aromatic ring-containing groups are a monovalent to
hexavalent phenyl group, biphenyl group, naphthalene group,
anthracene group, phenanthrene group, pyrene group, group of
Ph-(CHZ)2-Ph (wherein Ph is a phenyl group and P is an
integer of 1 to 10), etc. These aromatic ring-containing
groups may be substituted with a straight- and/or branched-
chain aliphatic hydrocarbon group of 1 to 20 carbon atoms,
or a group having a functional group such as hydroxyl group,
ether group, vinyl group or the like.
The group Z in the compound of the formula (V)
represents a ha7.ongen atom such as F, CP, Br or I, a RCOO-
group or a RO- group, all attached to the tertiary
hydrocarbon group. The group B in the formula (V) is a
hydrocarbon group having 4 to 40 carbon atoms among which an
aliphatic hydrocarbon group is preferred. When 'the
hydrocarbon has less than 4 carbon atoms, the halogen atom,
RCOO-group or RO-group is not attached to the tertiary
carbon atom with the result that polymerization fails to
~I~~a~l~~~
-9-
smoothly proceed. Thus the compound of the formula (v)
having such hydrocarbon is not proper for use.
Oligomers having a-halostyrene units which are usable
as an initiator and as a chain transfer agent include, for
example, oligomers of a-chlorostyrene and oligomers produced
by copolymerization of a-chlorostyrene with a monomer
copolymerizable therewith.
When a compound of the formula (I) having halogen
atoms, RCOO- groups or RO- groups as attached which are each
at least two in number, or a compound of the formula (I)
having halogen atom, RCOO- group or RO- group as attached
and another reactive functional group is used, as an
initiator and concurrently as a chain transfer. agent in the
process of the invention, a polymer having functional groups
at both ends, the so-called telechelic polymer, can be
obtained whicY~ has terminal functional groups introduced in
a high ratio. Thus such compound can be effectively used.
Examples of the foregoing compound useful both as an
initiator and as a chain transfer agent are compounds of the
following formulas
C I-I s C I-i 3 C I-I a C ( C I-I s ) a
?~,'-C~-C-X X-C-C
I
C I-I s C I-I s ~ C I-I s C X ( C I-I .>
~;~:3~~~3
-lo-
CHs CX (CHs ) z
I
X -- C -~'~
I
CHs CX (CHs ) z
CHs CH: CI-is CHa
X_C C_X X_C~--~~-~.~/C_X
( I I I
CHs CHs . CHs CHs
CHs CHs
I I
X - C -~~ C I-i z C I-I z ~ ~ C - X
I . I
CI-Is CHa
CHs CI-Is
I I
X - C -~~ C I-I z C I-I z C I-I z ' ~ C -
I I
C I-I a C I-I ~ ,
CI-Ij CHJ CHj
X-C-CI-iz CI-iz -C-X CI-I~ -C-x
I I I
C I-I ~ C I-1 3 , C I-( ~ ,
Cn-C~ I-I(~) (»_-Cs I-I~~)
I I
X-C-CI-Iz CI-(z CI-Iz CI-Iz --C-X
I ~ I
~ n - C Q I-I ~ .I ) ( n -- C s I-I ~ .; )
~~~e~a~~~
-11-
CHJ CHs CIIJ
X-C-CHz CHz -C-CI-Iz CI-Iz -C-
I I I
CHs '~ CI-is
- oligomers of a-chlorostyrene and like halogen-containing
organic compounds and RCOO- group-containing organic
compounds which are not limitative. Among these compounds,
preferred are aromatic compounds having the structure which
~ is unlikely to produce an indane-type skeleton as a by-
product, such as those of the following formulas
CHa CC ~ (CHa )
C L - C -~~
CI'~3 CC~ (CI-Is ) 2
C I-I .; C ( C I-I :, )
/
Ce-C
CII;, CCL~ (CI-(s ) z
cH3 C B ~ (CI-I3 ) 2
i /
B r - c -~_
C ~I s C B r ( C 1-I 3 )
~~.~i~.~~~
-12-
CHI C (CHs ) s
B r-C-~~
I
CHa C B r (CPIs ) 2
CH3 C (CHs ) 2 OCI-I;,
I
CHa 0-C /
CHa C (CHs ) z OCH.;
0
0 CHs C (CI-Is ) 2 OCC~I:,
II I _
CI-I~ C0-C
CI-is C (CI-I3 ) z 0 C CHI
0
halogen-containing organic compounds having -C(CH;),Ce or
C(CH3),Br which are likely to produce stable carbon cations,
such as the compounds of the following formulas
C ( C ( .>
I J I
c --- --.~; -- c
~ ~ - n
- -
C ( C I ,
f- ~ L
~~~~i~8
-13-
CHs CI-Is CH3
I I I
C.~-C-CH2 CH2 -C-CH2 CI-I2 -C-C~
I I
CHs C.~ CHs
and compounds of the following formulas
0 CHs CHs CI-Is 0
II I I I II
CIIs CO-C-CI-I2 -C-CH2 -C-0-CCHs
CHa CHs CHa ,
0 CH3 CI-Is CI-i3 0
II I I I il
CI-i3 C0-C-CHZ -C-CI-I2 -C-0-CCHs
C I-I 3 0 C I-I a
I
C=0
I
C I-I ;, ,
C I-I ~ C I-1 s C I I
Clvs 0-C-CI-I? -C-CIU~ -C--OCI-(3
C 11 .; C I-I .; C 1
i~~a~a~..~ad~
-14-
These compounds are used as the component which can act
as an initiator and concurrently as a chain transfer agent.
In the invention, the aforesaid compounds can be used singly
or at least two of them are used in mixture. The molecular
weight of the resulting polymer can be controlled by
adjusting the amount of such compound to be used. In the
present invention, the foregoing compound is used in an
amount of about 0.01 to about 25%, preferably about 0.1 to
about 15%, based on the cationically polymerizable
isobutylene-containing monomer.
The Lewis acid is the component to be used as a
catalyst in the invention, and is at least one class
selected from the group consisting of (CzHs)ZAPCP, (CZH;)APCP2,
SnCP4 and TiCP4. Preferred are TiCe4 and SnCP4. The amount
of the Lewis acid used is 0.0001 to 0.5 equivalent based on
the mole number of X in the organic compound of the formula
(I) which is used as an initiator and concurrently as a
chain transfer agent.
Tlsed as the end capping agent in the present invention
is a compound of the formula (II)
1Z "
I
C I-I 2 - C I-I C I-~ 2 S i - iZ '' C ll )
~Z s
~~~a~~~~
-15-
wherein R4, RS and R6 are as defined above. Examples of such
compounds are those of the following formulas
CHa
S 1 ( C H 3 ) s ~ ~ S 1 --
I
CHs
\
S i --~ S i ' \ ) n
CI-IJ
S 1 ( C H 2 C ~I 3 ) 3
~-- S i ( C I-I 2 C I I 2 C I I a ) s .
S i ( C I-I 2 C I-I 2 C H 2 C ~I s ) s .
-- S i --(-~~~ ) a ,
~_ S i ( C I-I z C i-I 3 ) ~ .
~S i (Cll? CII2 C1-I;, )
i~~a~~~~~
-16-
S i (CHI CH2 CHz CHs )
S i '
CHs CHs
~-- S i ~ S i -
I
CH:, CI~3
C~IJ C~Ij
S 1 - C I-I 2 - S 1 -
I
CH.> CHs
Among these compounds, preferred are those of the
following formulas
C 1'I
J
i~~.jWl~~
-17-
S z S i -{~) a
CHs
A preferred mole ratio of X in the organic compound of the
formula (I) as the component (B) to the_compound of the
formula (II) as the component (D) in the present invention
((X)/(II), in terms of mole numbers of X and compound of the
formula (II)] is in the range of 2.0 . 1 to 0.2 . 1.
Solvents useful in the invention include, for example,
hydrocarbon solvents such as aliphatic hydrocarbon and
halogenated hydrocarbon among which halogenated hydrocarbon
is desirable, and chlorine-containing chlorinated
hydrocarbon is more preferable. Specific examples of such
aliphatic hydrocarbons are pentane, hexane, etc. Examples
of useful halogenated hydrocarbons are chloromethane,
chloroethane, methylene chloride, 1,1-dichloroethane,
chloroform, 1,2-dichloroethane, etc. These hydrocarbons can
be used singly or at least two of them are usable in
mixture. The hydrocarbon can be used conjointly with a
small amount of another solvent. Examples of such solvents
are acetates such as ethyl acetate, nitro-containing organic
compounds such as nitroethane, sulfoxides such as
~:~ 3~~"~
-18-
dimethylsulfoxide, sulfones, amide-containing organic
compounds such as dimethylformamide and dimethylacetoamide,
nitrite-containing organic compounds such as acetonitrile,
etc.
A low polymerization temperature (i.e. 20 to -100°C) is
preferred in the present invention. Yet a relatively high
polymerization temperature (i.e. 10 to -40°C) is more
preferred in the manufacture of polymers.
The polymerization time is in the range of about 0.5 to
about 120 minutes, preferably about 1 to about 60 minutes.
The monomer concentration in the polymerization is
preferably about 0.1 to about 8 moles/ , more preferably
about 0.5 to about 5 moles/e.
The polymerization reaction may be conducted batchwise
(or semi-batchwise), or in a continuous manner such that the
components (A), (B), (C) and (D) are continuously charged
into the reactor.
According to the invention, an isobutylene-type polymer
having unsaturated groups introduced in a high ratio to ends
can be produced at low costs by a simplified process. The
process of the invention gives an isobutylene-type polymer
having a narrow molecular weight distribution.
Examples
The present invention will be described below in more
detail with reference to the following examples.
~~~~K~i~ef~
-19-
Example 1
A three-way cock was attached to a 200 ml-vol.
pressure-resistant glass reactor for polymerization and the
reactor was evacuated with use of a vacuum line and dried by
heating at 100°C for 1 hour. The reactor was cooled to room
temperature and the internal pressure was restored to normal
pressure with nitrogen using the three-way cock.
Thereafter, while feeding nitrogen to the reactor
through the three-way cock, ~0 ml of 1,1-dichloroethane
dried over calcium hydride was introduced as a main solvent
into the reactor with a syringe. A 5 mmoles quantity of
allyltrimethylsilane obtained by distillation and
purification was added, followed by addition of 10 ml of a
solution of 2 mmoles of TCC (Compound A) in 1,1
dichloroethane.
A 7 g quantity of isobutylene dehydrated by passage
through a barium-oxide containing column was placed into a
pressure-resistant glass tube for collection of liquefied
gas equipped with a needle valve. The tube was connected to
the three-way cock with a pressure-resistant rubber tube.
Thereafter, the reactor body was immersed in a dry ice-
acetone bath maintained at - 70°C to cool the contents of
the reactor for 1 hour. After cooling, the internal
pressure of the reactor was reduced with a vacuum line and
the needle valve was opened to transfer the isobutylene from
~i~~~a~ICo~
-20-
the liquefied gas-collecting pressure-resistant glass tube
to the reactor. The internal pressure of the reactor was
restored to normal pressure by introducing nitrogen through
the three-way cock into the reactor. The reactor was
immersed in a dry ice-acetone bath at - 10°C for 1 hour to
raise the temperature in the reactor to - 10°C.
Next, 1.9 g (10 mmoles) of TiCl4 was fed to the reactor
with a syringe through three-way cock to initiate the
reaction for polymerization. After a lapse of 60 minutes,
methanol cooled to not higher than 0°C was added. to complete
the reaction.
Thereafter, the reaction mixture was collected in an
eggplant type flask and the unreacted isobutylene, 1,1-
dichloroethane, allyltrimethylsilane and methanol were
distilled off. The remaining polymer was dissolved in 100
ml of n-hexane and the obtained solution was washed with
water repeatedly until the solution was made neutral. The
n-hexane solution was concentrated to 20 ml and the
concentrate was added to 300 ml of acetone. The resulting
mixture was stirred to precipitate the polymer from the
mixture.
The polymer thus obtained was dissolved in 100 ml of n-
hexane again, the resulting solution was dried over
anhydrous magnesium sulfate and the solids content was
separated by filtration. The n-hexane was distilled off
~:~;3;~N~3
-21-
under reduced pressure, giving an isobutylene-type polymer.
The yield of the obtained polymer was calculated from
the amount thereof. Mn and Mw/Mn were determined by gel
permeation chromatography and the structure of the terminal
in the polymer was identified by 1H-NMR (300 MHz) analysis.
Table 2 shows the results.
Examples 2 to 16
A polymer was prepared and evaluated in the same manner
as in Example 1 with the exception of using varied kinds of
the compounds serving as an initiator and concurrently as a
chain transfer agent, catalyst, and additional solvent in
the amounts and employing the polymerization temperatures,
as shown below in Table 1. Table 2 shows the results.
Comparison Examples 1 to 7
Polymers were produced in the same manner as in Example
1 except that allyltrimethylsilane was not used and that the
kind and amount of compounds serving as an initiator and
concurrently as a chain transfer agent and catalyst and
polymerization temperature were changed as shown below in
Table 1. Table 2 shows the results.
Comparison Example 8
A polymer was prepared and evaluated in the same manner
as in Example 8 with the exception of using BC13 (l0 mmoles)
as a Lewis acid. Table 2 shows the results.
In Table 1, the compounds A to E used as an initiator
~:~~~~,8
-22-
and concurrently as a chain transfer agent are the compounds
represented by the following formulas.
Compound A
CHs C C ~ (CH3 )
C .~ - C ~ ~ (T C C)
CH3 CCp (CHa )
Compound B
CHs CH,;
Compound C
Compound D
C ~ - C -~~~- C - C ~
C I-i s C I-I 3
CI-IJ C CHJ ) s
C .~ C
CI-Ia C C ~ (CI-Is ) z
C I~ a C I-I
cI-I:~ co--c-ff~-c-occl-I
I. I I
o c I-I ~ c I-I ; o
Image
;~Q'~;.2~3~,
-24-
o 0 0 o 0 0 o o o
U r, ~ .-,.-, 0 ~
, , , , , , , , ,
o
~o
.
Y
v~ , , , , o
, , , ~ , , ,
0
0 7
is
G
0
N
b O O
,b
a ~ , , ~ , , , , , x , , ,
x
U U
0 0 0 0 0 0 0 0 0 0 0 0
~n v m ~nv~ v7v7 v~v mnv7 v~
0
>
7
0
N ~ N N N N N N N ,~N N
U U U U U U U U U U U U
x x x x x x x x x x x x
U U U U U U U U U U U U
x x x x x x x x x x x x
U U U U U U U U U U U U
d U U d > d V U
U U U U a U U U a a U
H H E~ cnW W H H H W W E-
U
0
w
b N N SV N N N N N N N N N
'. C
~
O
o: o
~
.
p
m
cV
N
a a a a a a m m r~ ~ w U
b b b b b b b b b b b
c. C G C C A G b C G C G
O O O ~ ~ ~ O O O
C1 L1f3 O O O
t3C1
. , , . ,
~" x '
"
U F. ~ .' C P.
C..' ~
C1 O O O O O O O O O O O O
N
U U U U U U U U U U U U
a~
'
N M V'V'1vDt~ COOv ~ N
.
w
ir:~a~~~~~
_25_
N
U o 0 0 0 0 0 0 0 0 0 0 0
f .-n.-.I,-,r..~......~.-.1f .-..1f f
a n I
1 I 1 1 1 1 1 1 1 I
v
N ~ I p In1 I n I I n n n a
O O
O
c~
C
O
b
b ~~
//rr
d C 1 x x I I I 1 1 I I I 1
h~1
U U
U p O O O O O O O O O O O
~n v mnIm n In v~v~ ~nv~ In nn
C C
U
>
>
O
rn
C
y ca .~ ~ .~.~ .~.~ ~'..~.-:.~
U U U U U U U U U U U U
x x x x x x x x x x x x
U U U U U U U U U U U U
x x x x x x x x x x x x
U U U U U U U U U U U U
rr
a~
cs
o ~ .... ..~ a ~ v Cj U ,.,
T
U U U U U U U U U ~ d (j
H H E-Wn H H w H H ~' .r 0.1
U W W
0
w
N N N N N N N N N N N N
4
d
C
of
v,
U U ~1W d d d as cncn m as
b b b v b b v b b v ~ b
b p .O C C C C C C C C C C C C
y 7 C C C C ~ ~ ~ C C O C
O O O O O O O O O O O O
x C, d. C2,G1 C1G. L1.L7,n.L1,CL Cl,
ti E ~ ~ ~ ~ ~ ~ ~ ~ C ~ C
C
C a) O O O O O O O O O O O O
U U U U U U U U U U U U
a~
t. M d In~ "'N cnV In~D f o0
' O
~ _ _ _
O
a: a~ ~ a~a~ f W W W W W W W
X j
a a ~ ~ a a
0 0 0 0 0 0 0 0
W o W W W W U U U U U U U U
I
U
-26-
s~ ~
it o 0 0 0 0 0 0
0 0
* !~ b~
H
O
-X
b~ *
N '-i N wlf-iw-I
p . . . . p
rt$ ~ p O O O O O O
O U b~
.,.d
U
N
x
U
Il
o x
x
m o, o co cro, coo,
U
x I x N N M N N N '-irl
O W U -U -U
O rtf I
r-1
N
O S-1
U 4-~
O
-ri
O to O tn O O COO
'
M rl r-iN M V N N
' " Wi v-Ir-I.-i.-1rlri
'
~
F
r-1
47
~.
W
O
~'.
'~
.
N
I
a)
rt5
1 0 0 0 0 0 0 0 0
j'
.
, O O O O O O O O
,
,
rl 01O ~OM COe-I~OCO
I~.'
N M N M N M M M
3
~
rtf
O
!~
''i o o u10 o m u10
~
~ rn ~ o~ a~~ coof
N
Q,
r1N M d' lI1~O f~CO
',
~a
z
x
w
~~~~~I~~'e~
-27-
O
rtj p O O O O
O ~ O O
H
O
~i
H ~ ~ O O O O O O O
O
O
O
p
U
w
x
U
II
O
U
0 0~ ~ rn o 0 0~o~
'
~
'O ~ x I n7~-I~ .-IN N r-ie-1
x
4a U-U-U
O rtf I
H
Q '~
O S-a
O
la O
U
U ,y a
N
Qy ~
O
H
H
~
~
~
' 1f1O O t!1O ~ l(W
f7
'
.-IIn M N N r-iM d
.,.~
' H H H ~-IH rd '-iri
~
r1
O
~,~
3
-a
1
O
b
~
O'~ 0 0 0 0 0 0 0 0
~
U
'
-~ ~ro0 0 0 0~~r m
~~
.R
fa
M N M d' M M M M
3
z
~d
o
o m un o o mn
r o~ o~o, a~~ coa~
O r-IN M ~cl'111~D
~
~s
z
x
w
~~~~
-28-
CO 10N r-irl
(~ O O O O
'[~ ~ O O O O O
* r~ LT
H
O
Sa *
*
~
., UJ lD 01l~ M d' N
~ ~?
.,
H ~ O x
N O N O O O O U
x
O ~' U - U
x
U U
I
N
x
>~
o x
x ~' x
I I I 1 . I.1 U '
I a x
U U-U
U
W I
-U-
U ~ O rd I
~ N
O f.~
f-~ O
C:
C7 W
O 5.~ O
U ~ w
)~
N ~ r-1
~ ~
.
O O ll1O II1tf1O O
co co ~ ~r ~rcoM ,-1
_ al O
.-I
.,.
~
C'7N N M N N N N
H ~
a
1
I
~
O
3
-I ~u O
O
O
~ f~
-V U rd
5. 0 o 0 o 0 0 o 0
~
t7~
,--1
O O O O O O O O ri N
~2 ~ N coco M d wo u~ O C~
S-~
U
~
I~
U co ~ d t m vom M
3
i
z
o
r
d
R,
>~
~a -~t
m o m o 0 o in o
O
o, o~ ~ o~ o~a~o~ o~ ~
O
z
*
* *
00 1
~ I
r-i
.
r-IN M d' t11~0!~ CO
Paz o
oW z
U
~~a~~~~~
-29-
The results shown in Table 2 reveal the followings.
When a specif is Lewis acid is used in the process of the
present invention, side reactions are inhibited by the
presence of allylsilane in the polymerization reaction
system in advance despite the use of a compound serving as
an initiator and concurrently as a chain transfer agent, and
even polymerization at a relatively high temperature gives
an oligomer in a high yield which oligomer has allyl
terminal groups introduced in a high ratio and a narrow
molecular weight distribution (Examples 1 to 18 and
Comparison Examples 1 to 9).
In Comparison Examples l, 3 and 9 (described
hereinafter), the polymerization was terminated by
deprotonation, preferentially producing olefin terminal
groups (isopropenyl and internal olef.in), but little or no
C1 terminal group. Similarly, the groups represented by the
formulas
CH3 CH3
vv~.~ C-H, ~,~w C-CzHs
CH3 CH3
and the like were preferentially produced due to protonation
and alkylation when the polymerization was terminated in
Comparison Examples 6 and 7, but substantially no Cl
terminal group was found (A similar reaction was carried out
using a low-molecular model compound to confirm this
~:~3~~~3
-3 0-
behavior according to GAS-MASS analysis).
The aforementioned behavior in Comparison Examples 1 to
9 and the results obtained in Examples 1 to 18 indicate the
following.
In the polymerization in the reaction system having
allyltrimethylsilane present therein as in the process of
the invention, a reaction between the cation-containing
component and the allysialne (termination reaction) occurs
in competition with a reaction between the cation-containing
component and the monomer (propagation reaction). In brief,
allyl groups are introduced by direct attack against
rations, and it can be said that C1 groups are not replaced
with allyl groups after the formation of tertiary C1 groups.
This suggests that an isobutylene-type polymer
containing allyl groups is obtained by a mechanism clearly
different from the one as described in the foregoing
Japanese Unexamined Patent Publication No. 105005/1988 (the
mechanism including the formation of tertiary Cl groups).
Example 17
A three-way cock was attached to a 200 ml-vol.
pressure-resistant glass reactor and the reactor was dried
by heating at 100°C for 1 hour while being evacuated by
means of a vacuum line. The reactor was cooled to room
temperature and the internal. pressure was restor_ ed to normal
pressure with nitrogen.
~~~~~8
-31-
Thereafter, while feeding nitrogen to the autoclave
through the three-way cock, the autoclave was charged with
a solution of 0.308 g (1 mmol) of TCC (Compound A) in 30 ml
of methylene chloride dried over calcium hydride and serving
as a solvent far polymerization and with 0.51 ml (3.2
mmoles) of allyltrimethylsilane using a syringe.
A 5 g quantity of isobutylene dehydrated by passage
through a barium oxide-containing column was placed into a
pressure-resistant glass tube for collection of liquefied
gas equipped with a needle valve. The tube was connected to
the three-way cock with a pressure-resistant rubber tube.
The reactor body was immersed in a dry ice-acetone bath
maintained at - 30°C and the contents of the reactor were
cooled with stirring for 1 hour. After cooling, the
internal pressure of the reactor was reduced with a vacuum
line and the needle valve was opened to transfer the
isobutylene from the liquefied gas-collecting pressure-
resistant glass tube to the reactor. The internal pressure
was restored to normal pressure by introducing nitrogen
through the three-way cock into the reactor.
A solution of 0.055 ml (0.5 mmol) of titanium
tetrachloride in 5 ml of methylene chloride was added with
a syringe to initiate a polymerization reaction.
After a lapse of 60 minutes, the solution obtained by
the polymerization was added to 100 ml of an aqueous
'~!~;~3~~~
-32-
solution of saturated sodium hydrogencarbonate. The
resulting mixture was shaked and the organic layer was
washed with 100 ml of water twice. The organic layer was
concentrated to 10 ml and added to 300 ml of acetone,
followed by stirring to precipitate the polymer from the
mixture.
The polymer thus obtained was dissolved in 80 ml of n-
hexane and the solution was dried over anhydrous magnesium
sulfate to separate the solids content by filtration. The
n-hexane was distilled off under reduced pressure, giving an
isobutylene-type polymer.
The yield of the obtained polymer was calculated from
the amount thereof. Mn and Mw/Mn were determined by gel
permeation chromatography and the structure of the terminals
was identified by 1H-NMR (300 MHz) analysis. Table 3 below
shows the results.
A polymer having allyl groups introduced in a high
ratio was obtained in a high yield.
The allyl-terminated polyisobutylene obtained in this
example is represented by-the following formula (VI)
C I-I 3 C ~-r 3
I I
_- _- C __ ~' ~ o
w I C ~~I
C I-I :, C I-f 3
CR" (CI-I3 ) 2
~~~'~a~~~
-33-
wherein R9, R'° and R" are each a polyisobutylene chain having
allyl groups at its ends, and the isobutylene chains
represented by R9, R'° and R" may be the same or different in
length.
Comparison Example 9
A polymer was prepared in the same manner as in Example
17 with the exception of not using allyltrimethylsilane, and
the structure of the obtained polymer was analyzed. Table
3 shows the results. Use of titanium tetrachloride in a
catalytic amount as in this example resulted in the
preparation of the desired product in a satisfactory yield.
However, the obtained polymer was brand in molecular weight
distribution and various functional terminal groups were
formed.
Comparison Example 10
A polymer was produced and evaluated by the same
procedure as in Comparison Example B except that 0.51 ml
(3.2 mmoles) of allyltrimethylsilane was added after
completion of the polymerization reaction and that the
reaction mixture was stirred at room temperature for 6 hours
in a nitrogen atmosphere. The results are shown in Table 3.
The obtained polymer had allyl groups introduced in a low
ratio to ends and was broad in molecular weight
distribution.
Example 18
~a~~~~~
-34-
A polymer was prepared in the same manner as in Example
17 with the exception of using allyltrimethylsilane in an
amount changed to 0.71 ml (4.5 mmoles) , and the structure of
the obtained polymer was analyzed. Table 3 shows the
results. A larger amount of allyltrimethylsilane used
resulted in the production of a polymer having a lower
number average molecular weight. However, the polymer is
substantially the same as the product obtained in Example 17
in the number of moles. This means that al.lyltrimethyl.-
silane acted as a short-stop.
i~~~~~~~~
-35-
>~
w
N
O o 0
I ~-I
N
>~
Iw
>~
w
tU
r~lr-i~-It~ cD
O
I O O O O
_
N
Iw
z
0
0
r-irl CO N
lL"., O
U o 0 0
M f~
fs.
H
I
N N O
iw
M ('
ri~ a~
W
U o 0 0 0
>~o O o 0
I Wo ~rm
d' N d' d'
O ~DO O
~ O V'O O
o\~
-1 ri .-I
I~ c0 O
-a ,-io~ .
s~ sa
k k O O
k k
W tx7U U
W W
;~~~5~;.~i~~
-36-
Note -- 1) Yield of monomers
-- 2) Allyl terminal group
CHs
~~~~~ P I B ~~~~~ C H 2 - C - C H 2 C H = C H 2
CH3
Cl terminal group
CHa
~P I B~wCH2 -C-C 2
CHa
1-Olefin
~CI~?
~P z B~CH2 --C~
j
2-Olefln
~CH2
.",M, p I B ~M~, C I-I 2 - C ~
C I-I ;,
Example 19
A 5 2-vol. flask for use as a polymerization reactor
was provided with an agitating blade, a three-way cock and
a vacuum line. While being evacuated with use of the vacuum
line, the polymerization reactor was dried by heating at
i~~a~~~~~
-37-
100°C for 1 hour. The reactor was cooled to room
temperature and the internal pressure was restored to normal
pressure with nitrogen passed through the three-way cock.
While feeding nitrogen to an autoclave through the
three-way cock, the autoclave was charged, using a syringe,
with 1700 ml of 1,1-dichloroethane which was a solvent
dried over calcium hydride. A 35.5 ml (224 mmoles) quanity
of allyltrimethylsilane which was distilled and purified was
added thereto, followed by addition of a solution of 21.3 g
(69 mmoles) of TCC (Compound A) in 1,1-dichloroethane.
A 330 g quantity of isobutylene dehydrated by passage
through a barium oxide-containing column was placed into a
pressure-resistant glass tube for collection of liquefied
gas equipped with a needle valve. The tube was connected to
the three-way cock with a pressure-resistant rubber tube.
The reactor body was immersed in a dry ice-acetone bath
maintained at - 70°C and the contents of the reactor were
cooled with stirring for 1 hour. After cooling, the
internal pressure of the reactor was reduced by means of the
vacuum line and the needle valve was opened to transfer the
isobutylene from the liquefied gas-collecting pressure-
resistant glass tube to the reactor. The internal pressure
of the reactor was restored to normal pressure by
introducing nitrogen through the three-way cock into the
reactor and the reactor was immersed in a dry ice-acetone
~~a~~a~~~
-38-
bath at - 30°C for 1 hour to raise the temperature in the
reactor to - 30°C.
Thereafter, 50 ml of a solution of 3.9 ml (35 mmoles)
of titanium tetrachloride in 1,1-dichloroethane was added
dropwise at a constant rate over a period of 30 minutes so
as to maintain the polymerization temperature at - 30 to
25°C. The resulting solution was stirred at - 30°C for 1
hour, 2 $ of a saturated aqueous solution of sodium
hydrogencarbonate was added and the resulting mixture was
vigorously agitated.
The organic layer was collected in an eggplant type
flask and the unreacted isobutylene, 1,1-dichloroethane,
allyltrimethylsilane were distilled off. The remaining
polymer was dissolved in 1500 ml of n-hexane and the
solution thus obtained was washed with water repeatedly
until the solution was made neutral. The n-hexane solution
was concentrated to 600 ml and the concentrated solution was
added to 3 $ of acetone, followed by stirring to precipitate
the polymer from the solution.
The polymer thus obtained was dissolved in 1000 ml of
n-hexane again and dried over anhydrous magnesium sulfate,
and the solids content was separated by filtration. The n-
hexane was distilled off under reduced pressure, giving an
isobutylene-type polymer.
The yield of the obtained polymer was calculated from
-39-
the amount thereof. Mn and Mw/Mn were determined by gel
permeation chromatography and the structure of the terminals
in the polymer was identified by 1H-NMR (300 MHz) analysis.
Table 4 shows the results.
Example 20
In this example, a polymer was prepared and the
structure thereof was analyzed in the same manner as in
Example 19 except that 55 ml (500 mmoles) of titanium
tetrachloride was added at one time and that the initial
polymerization temperature was adjusted to - 70°C in order
to avoid a hazard occurring owing to the rise of temperature
during the polymerization reaction. Table 4 shows the
results. In Example 20, the rise of the reaction
temperature amounted to 72°C and the obtained polymer had
allyl groups introduced in a slightly lower ratio.
Table 4
Initial Rise of NMR
polymerization tempera- Fn (allyl)
temperature (C) ture (C)
Example 19 - 30 5 3.0
~
Example 20 - 70 72 2.2
The results obtained in Example 20 show that the
polymerization reaction conducted in this example entailed
a great degree of heat evolution. However, the rise of
temperature during the polymerization can be suppressed to
-40-
not more than 5°C by the procedure of Example 19.
Example 21
First, a monomer solution (a) and a solution of
titanium tetrachloride (b) were prepared.
The monomer solution (a) comprised 80 g (1430 mmoles,
2.86 M) of isobutylene, 5.24 g (17 mmoles, 34 mM) of
tricumyl chloride, 9.4 ml (60 mmoles, 120 mM) of
allyltrimethylsilane and 400 ml of methylene chloride dried
over calcium hydride. The solution of titanium
tetrachloride (b) comprised 1.1 ml (10 mmoles, 50 mM) of
titanium tetrachloride and 200 ml of methylene chloride
dried over calcium hydride.
The monomer solution (a) and the solution of titanium
tetrachloride (b) were introduced into a glass tube for
polymerization as shown in Fig. 1 using a constant delivery
pump. In this case, the monomer solution (a) and the
solution of titanium tetrachloride (b) were fed to the glass
tube so that the two solutions were mixed together only in
the tube. The monomer solution (a) was introduced at a rate
of about l0 ml/min, and the solution of titanium
tetrachloride (b) at a rate of about 4 ml/min. The glass
tube for polymerization was a spiral one having an inside
diameter of 4 mm and a length of 10 m, and was immersed in
an acetone bath maintained at - 35°C.
The polymerization reaction was completed during the
~~3~~~~3
-41-
passage of the mixed solutions (a) and (b) through 'the tube.
The obtained solution containing an isobutylene-type polymer
was transferred from the tube to an aqueous solution of
sodium hydrogencarbonate and the resulting mixture was
vigorously stirred.
Thereafter, the polymer eventually obtained was
purified and the structure thereof was analyzed in the same
manner as in Example 19. Table 5 below shows the results.
Table 5
Yield GPC NMR
)
Mn Mw/Mn Fn Fn (Cl + 1-
(allyl) olef in +
2-Olefin)
Ex. 21 100 4700 1.4 3.0 ~ 0.1
The results shown in Table 5 reveal that the process of
the present invention, when conducted by a continuous
polymerization method, can provide polymers in a high yield
which are uniform in molecular weight and which had allyl
groups introduced in a high ratio to ends.
Brief Description of the Drawing
Fig. 1 is a view schematically showing an apparatus for
continuous polymerization used in Example 21.