Note: Descriptions are shown in the official language in which they were submitted.
WO9~n8121 2 ~ ~ ~ 5 ~19 PCT/GB90/00063
Process for th~ production of fatty alcohols
- !
This invention relates to a process for the
production of fatty alcohols.
Fatty alcohols, or higher alcohols as they are
sometimes designated, are monohydric aliphatic alcohols
containing six or more carbon atoms which are derived either
from natural sources or are synthesised from petroleum
feedstocks. They are often classified by their market
usage. As the primary end use of primary alcohols
containing between about 6 and about 11 carbon atoms is the
production of plasticiser esters, such alcohols are often
termed plasticiser alcohols. For higher alcohols
containing, for example, from about 11 up to about 20 carbon
atom~, the major use is for the production of synthetic
detergents: hence such alcohols are often termed detergent
alcohols. The distinction between plasticiser alcohols and
detergent alcohols is somewhat arbitrary and there is some
production of phthalate esters from a C13 "oxo" alcohol and
also some production of, for example, nonionic surfactants
from C8 to Clo alcohols.
Although there are some natural products which
contain esters which can be hydrogenated to produce alcohols
in the plasticiser range, these are more usually produced
synthetically from petroleum feedstocks by, for example, the
so-called "oxo" process, a process which is also termed
oxonation or hydroformylation. Detergent range alcohols, on
the other hand, are typically produced by hydrogenation of
lower molecular alkyl esters of fatty acids. Such esters
can be produced by transesterification of natural
triglycerides or by esterification of the fatty acids
obtained by hydrolysis of such triglycerides. Examples of
triglycerides which can be used as raw materials include
natural oils, such as coconut oil, rape seed oil, and palm
oils, and animal fats, such as lard, tallow, and fish oil.
As such natural raw materials usually contain mixtures of
... .
. .
.
' .
j
~-'090/~121 PCT/GB90/0006~ _
204~3 - 2 -
triglycerides, the alcohol products obtained upon
hydrogenation are also mixtures of n-alkanols of differing
molecular weight. Such mixtures of alkanols are acceptable
for production of detergents without prior separation of the
alkanols one Irom another.
Whatever the commercial end use of the fatty
alcohol or fatty alcohol mixture the user generally insists
that the alcohol product must have as low an acid value as
possible and also as low a saponification value as possible.
The acid value ~AV) is a measure of the free acid content of
the alcohol product and is defined as the num~er of mg o~
XOH required to neutralise the free fatty acid in l g of
alcohol. The saponification value (SV) gives, together with
the acid value, a measure of the free ester content of the
alcohol product and is defined as the number of mg of KOH
required to saponify the esters and acids in l g of alcohol.
The ester value (EV) is the number obtained by subtracting
the acid value from the saponification value (EV = SV -
AV). In all cases the lower the value is (AV, SV, or EV),
the better is considered to be the quality of the alcohol
product. Another measure of purity of saturated alcohols is
the iodine value (IV), i.e. the number of g of I2 absorbed
by l00 g of the alcohol. ~he iodine value indicates the
ethylenic double bond content of the alcohol product.
Again, it is generally considered desirable to have as low
an iodine value as possible for a saturated alcohol. ;~
Examples of commercial fatty alcohol products are
the products sold under the following trade names:
- . - , - , . , .,, , ~ ~ ,
" :, - ., . . , .. ,, , , . , . ~ , .
' ; . ... , :: ~ , . ~ ; ,
, ' , , ~ ~ :
;- . . : . .
. c.
,. ' . ` ' - .
WO 90/OXI~1 PCT/GB90/00063
- 3 - 2~ 4~5 ~3
.U~
U~ o , ~ ~ ,
~ ~,
o
g
,
ct~
d~_~ ~ I~ I~ ~ :)
o U
dP'
3 ~ .
C
o
._,
o
oE ~r
U ~
. C~
O
Q_I ~ ~ ~ _
C~
E u~ uq
O ~ _~
~
O O
~5 JJ
~J ~ 3 ~
~ C C C C O C h ~ O
,~ O O O O _I O ~
)-I U OU U _~ U J~ ~ O
o o o o la o ,a a u
~) U U ~ U C C ,1
I~
U --I
C~
S~ ~ l O
~ C
X~r ~CD In cr~ 1~ ~) D~ Q) ~ '
t/~ I C I I I I r C ~
,, o c)o o ~: o a~ ~D U 1-
E~ o ~ a ~ c u
~, o
E ~ R
Z
~1 1 1
~ O ~
:> C O O O O O O O O
._~ _1 10 C C C C C C C C
J~ O U tO G) ~
C ~ O U C~ C) U ` '
u ~ ~ ~a ~ ~ ~ ~ ~ ~a In
u a~
LO ~ ~ ~ X X ~ ~ J- V V ~
o~ a~ u Q~ ~ U U U ~ U O
O ~ c O O O O O Z
SUBSTITUTE SHEET
.
.
.
,
WO90/08l21 PCT/GB90/00063 -
-- 4
'~45~
The esters usually used as raw materials for the
production of detergent range alcohols are the methyl
esters. A problem arises in refining of the product alcohol
mixtures because the boiling point of one or more of the
methyl esters present in the ester mixture which is
hydrogenated will usually be close to that of one of the
product alcohols. Hence it becomes difficult, if not
impossible, to separate by distillation any unconverted
methyl esters from the product alcohol mixture.
As an illustration of the difficulty of separating
fatty alcohols from methyl fatty acid esters, particularly
from mixtures containing a major amount of a mixture of
fatty alcohols and a minor amount of a mixture of methyl
fatty acid esters, reference may be made to the following
list of boiling points:
_
Substance Boilinq Point Pressure mm Hq tbar)
l-dodecanol 150C 20 tO.027)
methyl laurate 149C 20 (0.~27)
l-tetradecanol 167C 15 tO.020)
methyl myristate 170C 15 (0.020)
l-hexadeca~ol 189.5C 15 (0.020)
methyl palmitate 192C 15 (0.020)
l-octadecanol 210C 15 (0.020)
methyl stearate 213C 15 (0.020)
A mixture containing all of these components, such
as might be produced by hydrogenation of a mixture of methyl
esters of C12- to C18-fatty acids produced by hydrolysis of ~ -
a natural triglyceride, is difficult tif not impossible) to
separate satisfactorily by distillation without recourse to
use of multiple distillation columns. To avoid the expense
of multiple distillation columns, one of two approaches is
normally adopted. The first approach involves use of ~ -
somewhat vigorous hydrogenation conditions, including use of
high pressures and temperatures so as to ensure that as
.. - ~,, .:
. . . ' '.
'
~ .
~Vo~O/nNl~l PCT/CB90/00063
2~5~9
small a proportion of unconverted methyl esters remains in
the hydrogenation product. Although this largely obviates
the problem of separating the methyl esters from the product
alcohols, the use of vigorous hydrogenation conditions has
drawbacks, particularly in that such conditions also tend to
increase the yield of alkane and ether byproducts which
represent a significant loss of potentially valuable
alcohols. In addition catalyst consumption is rather high
and the use of high pressure equipment increases the capital
and running costs of the plant.
The second approach to the problems associated with
the presence of unconverted esters in the alcohol
hydrogenation product is to use less vigorous hydrogenation
conditions, which reduces the loss of alcohol product by
formation of alkane and ether byproducts, with subsequent
removal of the unconverted ester by hydrolysis ~ith hot
aqueous alkali, such as hot sodium hydroxide solution. In
this case the remaining ester is converted to a fatty acid
salt which is lost in the aqueous phase. In addition this
procedure involves consumption of sodium hydroxide or other
alkali. Finally, as the sodium or other alkali metal salts
of the fatty acids act as soaps, problems may arise in
separating the aqueous phase from the alcohol product due to
formation of emulsions.
In the esterification of fatty acids perhaps the
most widely used catalysts are sulphuric acid and organic
sulphonic acids, such as P-toluenesulphonic acid. Although
these catalysts are efficient, they are homogeneous
catalysts and a neutralisation step is necessary before
ester purification can ~e attempted. Typically washing with
an alkali, such as sodium hydroxide solution, is used in
such a neutralisation step. As esterification is an
equilibrium process, a disadvantage of this procedure is
that the washing step also results in removal of any
unreacted fatty acid in the wash liquor. Normally it is
.. . .
,, . . ~ . .
WO90/nX1'l PCT/GB90/0006~ _
2 ~ 4 ~ 6 -
uneconomic to attempt to recover the unreacted acid from its
salt in the wash li~uor so that this may represent a
significant loss of process efficiency. In addition some
ester may be lost in this washing step. The losses of ester
in the aqueous alkali phase will depend on the solubility of
the ester in such solutions. Furthermore the disposal of
the wash liquor may present environmental problems which may
be aggravated by the presence of the organic carboxylic acid
salt in the wash liquor. In addition, particularly when
long chain fatty acids are involved, problems may arise in
the washing step due to formation of emulsions that are
stabilised by the al~ali metal fatty acid salts, which are
surface active, and that are often difficult to separate
into their component aqueous and organic phases. The
stability of such emulsions is known to vary in an erratic
way, thus making the design of the organic phase~a~ueous
phase separation equipment difficult. Therefore it is
difficult to practise an esterification process with a
homogeneous catalyst on a continuous basis. As a result
batch processing is usually adopted, a factor which may
affect product quality from batch to batch. An additional
disadvantage of the use of such homogeneous catalysts as
sulphuric acid and ~-toluenesulphonic acid is the risk of
contamination of the ester with sulphur-containing
components. Such sulphur-containing components can
interfere seriously with subse~uent hydrogenation.
For further background information about the
production of fatty alcohols reference may be had to the
following reviews: -
1. "Fatty alcohols", by J.A. Monick, J. Am. Oil
Chemists' Soc., November 1979, Vol. 56, pages 853A to 860A;
2. "Natural fats and oils route to fatty alcohols"~ by
Henning Buchold, Chemical Engineering, February 21, 1985,
pages 42 and 43:
3. "Manufacture of Fatty Alcohols Based on Natural
- . - ' . - - -
: . ' ': ' ,: ,
- :. .: . . . .. - .
- ' ' .
.
W090/0812l PCr/GB90/00063
'~0~4'~
Fats and Oils", by Udo R. Kreutzer, JAOCS, Vol. 61, No. 2
(February 1984), pages 343 to 348;
4. "Production of Fatty Alcohols from Fatty Acids", by
Theodor Voeste and Henning Buchold, JAOCS, Vol. 61, No. 2
(February 1984), pages 350 to 352;
5. "Alcohols, higher aliphatic", Kirk-Othmer
Encyclopedia of Chemical Technology, Third Edition (1978),
Vol. 1, (published by J. Wiley & Sons. Inc., New Yor~),
pages 716 to 739; and
6. "Technical Processes for the Manufacture of Fatty
Alcohols" by H.-D. Kompp and H.P. Kubersky, in "Fatty
Alcohols - Raw Materials, Methods, Uses" published in 1982
by Henkel KGaA, D}sseldorf, at pages 49 to 74.
It would be desirable to provide a method enabling
substantially ester free fatty alcohols to be produced by
hydrogenation of a methyl fatty acid ester feedstock under
relatively mild conditions with less formation of byproduct
alkane than occurs in conventional methods of manufacture of
fatty alcohols by this route, even though the selected
hydrogenation conditions result in the presence of
significant amounts of unconverted methyl fatty acid ester
in the crude hydrogenation product. The provision of such
an improved method would result in improved yield of fatty
alcohols, since the process losses due to alkane byproduct
formation would be reduced.
The present invention accordingly seeks to provide
an improved process for production of fatty alcohols by
hydrogenation of lower alkyl esters, especially methyl
esters, of fatty acids derived from natural triglycerides,
under conditions which minimise formation of byproduct
al~anes and ethers followed by refining of the resulting
ester containing hydrogenation product.
According to the present invention there is
provided a process for the production of fatty alcohols in
which a fatty acid or fatty acid mixture is esterified in an
WO90/0~121 PCT/G~90/00~63
2~5 ~9 8 -
esterification step with a lower alkanol to form the
corresponding lower alkyl fatty acid ester or esters, in
which the resulting lower alkyl fatty acid ester or esters
is or are subjected to hydrogenation in the presence of a
heterogeneous ester hydrogenation catalyst to yield an ester
hydrogenation product comprising a fatty alcohol or
alcohols, and in which the ester hydrogenation product is
subjected to product refining for recovery of fatty alcohol
or alcohols therefrom, characterised in that the
esterification step includes continuously supplying the
Eatty acid or fatty acid mixture in liquid phase to an
esterification zone maintained under esterification
conditions and containing a charge of a solid esterification
catalyst containing sulphonic acid groups and/or carboxylic
acid groups in countercurrent to a vaporous stream
containing vapour of the fatty alkanol, that the
esterification zone is supplied with a feed stream of lower
alkanol vapour having a water content of less than about 5
mole %, that a vaporous exit stream containing lower alkanol
vapour and water of esterification is recovered from the
esterification zone, that a lower alkyl fatty acid ester
stream is recovered from the esterification zone that
contains at least about 99 mole % of lower alkyl fatty acid ~
ester, that lower alkyl fatty acid ester or ester mixture ~ .
recovered from the esterification step is vaporised in a
stream of hydrogen and passed in vapour form through a
hydrogenation zone containing a charge of a solid ester
hydrogenation catalyst under vapour phase hydro~enation
conditions such that the vaporous mixture in contact with
the catalyst is always above its dew point, that the
resulting hydrogenation product is collected and contains at
least about 0.5 mole % of unreacted lower alkyl fatty acid
ester in addition to product fatty alcohol or alcohols, that
the hydrogenation product is subjected to
transesterification in a first transesterification zone
.
,
..
WO90/08121 P~T/GB90/00063
20q55~19
g
maintained under transesterification conditions, thereby to
convert unreacted lower alkyl fatty acid ester in the
hydrogenation product by reaction with product fatty alcohol
or alcohols into a wax ester or wax esters derived from the
or a product alcohol and a fatty acid, that unreacted lower
alkanol is evaporated from the resulting mixture, and that
the now substantially lower alkanol free mixture is further
distilled to yield (i) an overhead fraction that contains
the product alcohol or alcohols and is substantially free
from lower alkyl fatty acid ester and ~ii) a distillation
residue comprising fatty alcohol or alcohols and wax ester
or esters.
In a particularly preferred process the
distillation residue (ii) is subjected to ~ ~-
transesterification in the presence of added lower alkanol
in a second transesterification zone maintained under
transesterification conditions, thereby to reconvert wax
ester or esters to lower alkyl fatty acid ester or esters
and to fatty alcohol or alcohols, followed by evaporation of
lower alkanol to yield a liquid residue that is
substantially îree from lower alkanol and then by
distillation of fatty alcohol or alcohols and lower alkyl
fatty acid ester or esters present in this liquid residue to
produce (a) an overhead product containing a mixture of
lower alkyl fatty acid ester or esters and fatty alcohol or
alcohols and (b) a relatively involatile residue. This
relatively involatile residue contains wax esters and-
possibly also a transesterification catalyst if one is used
in the second transesterification zone.
In this specification all figures in "mole %" are
calculated on a lower alkanol free basis, except where the
context indicates otherwise. Also the term "fatty alcohol"
means a linear alkanol containing from about 6 to about 26
carbon atoms. Preferred fatty alcohols contain from about
10 to about 20 carbon atoms. Typical fatty alcohols include
:' ' ., :.'
. .
,
! ~ .
WO90/08121 PCT/GB90/00063 -
- 10 -
'~ 0 ~
l-decanol, l-dodecanol, l-tetradecanol, l-hexadecanol, l-
octadecanol, l-octadecenol and the like, and mixtures
thereof. The term "lower alkyl" means Cl- to C4-alkyl,
including methyl, ethyl, n-propyl, lso-propyl, n-butyl,
so-butyl and sec-butyl. The preferred lower alkyl radic l
is methyl. Similarly the term "lower alkanol" embraces C
to C4 alkanols, including methanol~ ethanol, n-propanol,
iso-propanol, n-butanol, iso-butanol, and sec-butanol.
Methanol is the preferred lower alkanol. By the term "fatty
acids" we mean linear saturated, unsaturated or
polyunsaturated aliphatic acids, such as linear alkyl,
alkenyl, or hydroxyalkenyl carboxylic acids containing from
about 6 to about 26 carbon atoms, preferably about lO to -~
about 20 carbon ato~.s. Examples of such fatty acids are
decanoic acid (capric acid), dodecanoic acid (lauric acid),
tetradecanoic acid (myristic acid), pentadecanoic acid,
hexadecanoic acid (palmitic acid), heptadecanoic acid
(margaric acid), octadecanoic acid (stearic acid or
isostearic acid), octadecenoic acids (oleic acid, linoleic
acid or linolenic acid), hydroxyoctadecenoic acid
(ricinoleic acid), eicosanoic acid (arachidic acid) and
docosanoic acid (behenic acid). Mixtures of fatty acids are
of especial importance as raw materials Erom which the lower
alXyl fatty acid esters used as starting material in the
hydrogenation step are prepared. Such mixtures of acids can
be obtained by hydrolysis of naturally occurring
triglycerides such as coconut oil, rape seed oil, palm oils,
tallow, lard and fish oils. If desired, such mixtures of
acids can be subjected to distillation to remove lower
boiling acids having a lower boiling point than a chosen
temperature and thus produce a "topped" mixture of acids ! or
to remove higher boiling acids having a boiling point higher
than a second chosen temperature and thus produce a "tailed"
mixture of acids, or to remove both lower and hiqher boiling
acids and thus produce a "topped and tailed" mixture of
:~ : - - : .
. .
,
WOgo~o#l~I PCT/G~90/nO063
2~55~
-- 11 --
acids.
In a preferred process according to the invention
esterification of the fatty acid or fatty acid mixture with
the lower alkanol (e.g. methanol) is effected by a procedure
in which the fatty acid or fatty acid mixture and lower
alkanol are passed in countercurrent flow through a column
reactor provided with a plurality of esterification trays
mounted one above another, each adapted to hold a
predetermined liquid volume and a charge of solid
esterification catalyst thereon, liquid downcomer means
associated with each esterification tray adapted to allow
liquid phase to pass down the column reactor from that
esterification tray but to retain solid esterification
catalyst thereon, and vapour upcomer means associated with
each esterification tray adapted to allow vapour to enter
that esterification tray from below and to agitate the
mixture of liquid and solid esterification catalyst on that
tray, in which the fatty acid or fatty acid mixture is
supplied in liquid phase to the uppermost one of said
plurality of esterification trays whilst the lower alkanol
is supplied in vapour form beneath the lowermost one of said
plurality or esterification trays, in which vapour
comprising lower alkanol and water of esterification is
recovered from an upper part of the column reactor, and in ~ .
which a lower alkyl fatty acid ester or ester mixture is
recovered from a lower part of the column reactor. ~:
In such a procedure the water content of the lower
alkanol vapour supplied to the column reactor should be less
than about 5 mole % and the number of esterification trays
and the reaction conditions should be selected so that the
stream of lower alkyl fatty acid ester or esters has a low
acid content of less than about 1 mole %, calculated on a
lower alkanol free basis, and an ester content, also
expressed on an alkanol free basis, of at least about 99
mole %.
wo90/n81'1 PCT/GB90/00063 _
20~S~9 - 12 -
The process of the invention utilises the vaporous
stream of the lower alkanol to carry away water of
esterification produced in the esterification reactor but
without carrying with it significant quantities of the fatty
acid or acids or of the lower alkyl fatty acid ester or
esters.
The esterification conditions used in the column
reactor will normally include use of elevated temperatures
up to about 160C, for example a temperature in the range of
from about 80C to about 140C, preferably in the ran~e of
rrom about 100C to about 125C. Such operating
temperatures will be determined by such factors as the -
thermal stability of the esterification catalyst, the
kinetics of the esterification reaction and the vapour
temperature of the lower alkanol fed to the base of the
column reactor at the relevant inlet pressure. ~ypical
operating pressures at the vapour inlet of the colu~n
reactor range from about 0.1 bar to about 25 bar. A liquid
hourly space velocity through the column reactor in the
range of from about 0.1 hr 1 to about 10 hr 1, typically
from about 0.2 hr~l to about 2 hr 1, may be used.
The fatty acid or fatty acid mixture is supplied in ~-
liquid form to an upper part of the column reactor or in
admixture with lower alkanol, in solution in recycled ester
product, or in solution in an inert solvent or diluent
therefor. It is possible to pre-react the lower alkanol and
the fatty acid or fatty acid mixture prior to introduction
to the column reactor. The resulting reaction mixture
contains a mixture of lower alkyl fatty acid ester or ester
mixture, water, and lower alkanol. Usually it is convenient
o pre-react the lower alkanol and the fatty or fatty acid
mixture to equilibrium in the presence of an acidic ion
exchange resin containing -S03H and/or -COOH groups prior to
introduction of the resulting equilibrium mixture to the
column reactor.
,
WO90/081~1 PCT/GB90/00063
- 13 ~ 20~;~S~
In such an esterification process a vaporous
mixture exits the column reactor as an overhead product.
Provision may be made for scrubbing such vaporous mixture
with lower alkanol in liquid form in order to wash traces of
fatty acid ester and of fatty acid back into the column
reactor. This overhead product from the column reactor can
be condensed and treated in known manner to separate its
constituents, the recovered water of esterification being
rejected and the lower alkanol being recycled for re-use in
as dry a form as is practicable within the relevant economic
constraints. The lower ~he water content of the lower
alkanol vapour that is supplied to the lowermost one of said
esterification trays, the further towards 100% conversion to
ester the esterification equilibrium reaction can be driven
and the lower the residual acidity of the ester containing
product recovered from the bottom of the column reactor will
be. However, a balance may often have to be struck between
the cost of providing, for example, a substantially dry
lower alkanol for vaporisation into the column reactor, on
the one hand, and the cost of providing and operating any
additional downstream processing facilities that may be
required to upgrade the ester product to the required
quality if a less dry alkanol is used. This will vary from
lower alkanol to lower alkanol and will depend upon the
interaction between water and lower alkanol (e.g. azeotrope
formation) aDd its effect upon alkanol/water separation. In
any case, the water content of the lower alkanol vapour
supplied to the reactor is less than about 5 mole ~, and
even more preferably is less than about l mole %.
The column reactor has a plurality of
esterification trays. Although two or three trays may
suffice in some cases, it will typically be necessary to
provide at least about 5 up to about 20 or more
esterification trays in the column reactor. Typically each
esterification tray is designed to provide a residence time
,
:. : .
- : ,
~'090/0~l~1 PCT/GB90/0006~ -
20~ 9 - 14 -
for liquid on each tray of from about l minute up to about
120 minutes, preferably from about 5 minutes to about 60
minutes.
The solid esterification catalyst may be a granular
ion exchange resin containing -SO3H and/or -COOH groups.
Macroreticular resins of this type are preferred. Examples
of suitable resins are those sold under the trade marks
"Amberlyst", "Dowex", "Dow" and "Purolite", such as
Amberlyst l~, Amberlyst 66, Dow C351 and Purolite Cl50.
Different solid esterification catalysts may be
used on different trays of the column reactor. Moreover
different concentrations of solid esterification catalyst
can be used on different trays.
The charge of solid particulate or granular
esterification catalyst on each tray is typically sufficient
to provide a catalyst:liquid ratio on that tray
corresponding to a resin concentration of at least about
0.2% w/v for example, a resin concentration in the range of
from about 2% w/v to about 20% w/v, preferably 5% w/v to 10%
w/v, calculated as dry resin. Sufficient catalyst should be
used to enable equilibrium or near equilibrium conditions to
be established on the tray within the selected residence
time at the relevant operating conditions. On the other
hand not so much catalyst should be used on each tray that
it becomes difficult to maintain the catalyst in suspension
in the liquid on the tray by the agitation produced by the
upflowing vapour entering the tray from below. For a
typical resin catalyst a resin concentration in the range of
from about 2% v/v to about 20% v/v, preferably 5% v/v to 10%
v/v may be used.
The particle size of the catalyst should be large
enough to facilitate retention of the catalyst on each tray
by means of a screen or similar device. However, as larger
catalyst particle sizes are more difficult to maintain in
suspension and have lower geometrical surface area per gram,
WO90/0X121 PCT/GB90/00063
lS- 2~5~19
it is expedient to use not too large a catalyst particle
size. A suitable catalyst particle size is in the range of
from about 0.1 mm to about 5 mm.
One or more wash trays may be provided above the
esterification trays in order to prevent loss of product,
solvent and/or reagents from the column reactor.
In the column reactor the vapour upcomer means
associated with each esterification tray may comprise a
sparger positioned so that, in operation, it will lie below
the surface of the mixture of liquid and solid
esterification catalyst on that tray and so that vapour
bubbles emerging therefrom will agitate said mixture of
liquid and solid particulate catalyst. The sparger may be a
ring sparger. At least one baffle means may be mounted in
the vicinity of the sparger to enhance the mixing action
thereof. For small scale operation a sparger on the axis of
the column reactor under a cylindrical baffle can be used.
In one embodiment the sparger is a ring sparger and
inner and outer annular baffle means are positioned in the
vicinity of the sparger and define an upflow zone in the
region of upflowing vapour bubbles and adjacent downflow
zones within and outside the upflow zone.
It is important to avoid stagnant zones where solid
esterification catalyst can settle out because this can lead
to excessive formation of by-products or to occurrence of
hot spots. Although mechanical stirrers can be provided on
each tray to maintain the catalyst particles suspended in
liquid, this adds somewhat to the complexity of the reactor.
It is possible, however, by suitable design of the sparger
and tray to ensure that the upflowing vapour provides
sufficient agitation in passage through the liquid on the
tray to maintain the catalyst particles in suspension. To
achieve this end it is convenient that at least a part of
the floor of one or more (and preferably all) of the
esterification trays slopes towards a zone where there is ;
.::
.. . , ' ~ .
- . . : :. . ~ .
'''' :' ~ '
WO90/OXl2l PCT/GB90/00063 _
20~ 9 - 16 -
turbulence caused by the upflowing vapour such as is to be
found under the sparger. The angle of slope is preferably
selected so as to be equal to or greater than the angle or
repose of the solid particulate esterification catalyst
under the liquid in the esterification tray. The adoption
of such a slope will tend to ensure that all of the catalyst
is in dynamic contact with the liguid during operation and
that no stagnant zones of catalyst are formed. Such
stagnant zones are undesirable because they can enable
undesirable side reactions or even thermal runaways to occur
in certain instances.
In a preferred apparatus the vapour upcomer means
of one or more (and preferably all) of the esterification
trays is or are provided with a liquid suckback preventer
means.
A screen means may be provided on at least one
esterification tray to hinder loss of solid esterification
catalyst from that esterification tray via its associated
downcomer means. In this way downward flow of the solid
catalyst from one esterification tray to the next lower one
can be substantially prevented.
Means may be provided for withdrawing or adding
resin to one or more of the trays during operation of the
column reactors. For example, a conduit having a down
turned open end can extend into the interior of a respective
tray with its open lower end positioned at a low point
within the tray. By this means a slurry of catalyst and
li~uid can be withdrawn in controlled manner from the tray
intermittently or continuously as desired or further
catalyst can be introduced in slurry form to the trays, as
desired. Catalyst withdrawn from a given tray can be re-
introduced into the column reactor, either into the same
tray or to a lower or higher one, possibly after being given
a regeneration treatment.
In the hydrogenation step of the process of the
WO 9t)/0XI'I PCT/GB90/00063
- 17 - 2~45~
invention lower alkyl fatty acid esters are hydrogenated
under vapour phase hydrogenation conditions in which the
composition of the gas stream is selected so that at all
times the material in contact ~ith the hydrogenation
catalyst is above the dew point, preferably at least about
5C above the dew point. Suitable hydrogenation catalysts
include known ester hydrogenation catalysts such as reduced
copper oxide-zinc oxide ~see GB-~-2116552 and WO-A-
82/03854), and copper chromite, and promoted copper chromite
catalysts. The preferred catalysts are reduced copper
oxide-zinc oxide catalysts of the type disclosed in GB-B-
2116552 and WO-A-82/03854. Such catalysts include reduced
mixtures of copper oxide and zinc oxide derived from
mixtures comprising, before reduction, (a) from about 10 to
about 70 percent by weight CuO and about 90 to about 30
percent by weight ZnO, ~b) from about 65 to about 85 percent
by weight CuO and about 15 to about 35 percent by weight
ZnO, and (c) from about 40 to about 50 percent by weight
each of CuO and ZnO and 0 to 20 percent by weight of
alumina. The preferred copper chromite catalysts are those
containing from about 25 to about 45 percent by weight of
copper and from about 20 to about 35 percent by weight of
chromium, calculated as metal. Typical vapour phase
hydrogenation conditions include use of temperatures of up
to about 260C, such as temperatures in the range of from
about 140C to about 240C, and pressures in the range of
from about 5 bar to about 100 bar. Typically the H2:ester
mole ratio in the vaporous feed to the hydrogenation zone is
at least about 200:1 up to about 2000:1 or more.
The hydrogenation mixture obtained by hydrogenating
a lower alkyl fatty acid ester or mixture of esters
contains, in addition to a fatty alcohol or fatty alcohol
mixture, also lower alkanol, such as methanol. The methanol
is separated in any known manner, as by distillation in one
or more stages, from the fatty alcohol or alcohols to yield
.
~`~
. , :-" ',
': ` :
?
WO90~08121 PCT/GB90/0006~ -
- 18 -
~5~
a fatty alcohol fraction suitable for use in the process of
the invention. Such a fatty alcohol fraction typically
contains, besides possibly a minor molar amount of methanol
or other lower alkanol (usually less than about S mole ~), a
major molar amount of a fatty alcohol or alcohols (usually
about 90 mole ~ or more) and a minor molar amount of
unreacted lower alkyl fatty acid ester or esters (usually
from about 0.5 mole % up to about 5 mole ~).
In the hydrogenation step of the process of the
invention vapour phase conditions are used. In order to
maintain all components in the vapour phase two important
factors are (a) the H2:ester molar ratio of the vaporous
mixture to the hydrogenation zone and (b) the temperature
thereof. In general, the high the molecular weight of the
lower alkyl fatty acid ester is, the less volatile it is and
the higher its boiling point. Hence, for example, when
using methyl laurate as a feedstock to the hydrogenation
zone, a lower H2:ester molar ratio and a lower inlet
temperature to the hydrogenation zone can be used than when
a higher boiling ester, such as methyl stearate, is to be
hydrogenated. In practice a plant operator may wish to have `
the freedom to operate the process using fatty acids derived
from different sources at different times. For example, he
may wish to operate at different times using fatty acids
f rom any of the common sources, such as tallow, lard, fi Sh
oil, coconut oil, rape seed oil or palm oil. A plant
capable of handling such a range of acid feedstocks must be
capable of hydrogenating the highest boiling methyl or othe~
lower alkyl ester of a fatty acid that is likely to be used.
Hence it must have an ester vaporisation section that can
operate over a range of H2:ester molar ratios and that can
deliver to the hydrogenation zone a vaporous inlet mixture
at the appropriate temperature, i.e. a higher inlet
temperature and a higher H2:ester molar ratio for methyl
stearate, for example, than for methyl laurate.
' '' ' '
woso/o8l~l PCT/GB90/00063
2~5~3
- 19 -
The hydrogenation zone may comprise a single
reactor operated under adiabatic conditions and containing a
sinqle bed of an ester hydrogenation catalyst, such as
copper chromite or a reduced CuO-ZnO catalyst. In this
case, however, the bed of catalyst must be sized so as to
enable hydrogenation to be completed so far as possible by a
single passage of the vaporous mixture therethrough at the
design feed rate when opèrating at the lowest design
temperature. In addition provision has to be made in
designing the plant for any catalyst deactivation that may
occur with ageing of the catalyst. If this approach is
adopted then, with a catalyst charge that is sized for
operation at a temperature suitable for a relatively low
boiling ester, such as methyl laurate, it will be understood
that, at the higher operating temperatures and higher
H2:ester molar ratios needed to maintain a high boiling
ester, such as methyl stearate, in the vapour phase,
hydrogenation occurs faster so that it is mainly the front
end of the catalyst bed that is playing a part in the
hydrogenation reactor, whilst the back end of the catalyst
oed plays essentially no part. A disadvantage of this
design approach is that, when operating with a high boiling
ester, such as methyl stearate, the hot reaction mixture
remains in contact with the catalyst for a significant time
at the back end of the catalyst bed, although the
hydrogenation reaction has effectively gone to completion,
with the result that the conversion to by-products is
correspondingly higher.
To overcome this problem it is proposed, in a
preferred process according to the invention, to conduct
hydrogenation using a hydrogenation zone having a plurality
of beds, or sections of catalyst bed, of hydrogenation
catalyst arranged in series which can be brought into use as
required. In one arrangement the hydrogenation reactor has
a main inlet and a main outlet, a plurality of beds of
-'' ' ~ ,
~<)yn~oxl2l PCT/GB90/00063
.
2 0 ~ 20 -
hydrogenation catalyst in the path of gas flowing between
the main inlet and the main outlet, and one or more
secondary flow connections each located between a respective
pair of catalyst beds. the vaporous mixture containing
hydrogen and lower alkyl fatty acid ester can be fed to ~he
hydrogenation reactor by means of the main outlet whilst the
reaction product is withdrawn either via the main outlet, so
that all of the catalyst beds are used, or via one of the
secondary flow connections, so that one or some only of the
catalyst beds is or are used, depending upon the volatility
of the ester, and hence upon the H2:ester molar ratio and
the inlet temperature of the vaporous mixture.
Alternatively the reaction mixture can be withdrawn from the
main outlet whilst the vaporous mixture is fed to one of the
secondary flow connections. any catalyst beds which are not
in active use are maintained under an appropriate pressure
of hydrogen. In this way the plant operator can readily
select the appropriate number of beds of catalyst to suit
the nature of the fatty acid feedstock currently being used.
In the first transesterification zone the fatty
alcohol fraction is subjected to transesterification. Such
transesterification can be carried out in the absence of
added catalyst by heating to elevated temperature, for
example to a temperature o~ about 250C or higher.
Normally, however, it will be preferred to effect
transesterification in the first transesterification zone in
the presence of a transesterification catalyst. Any known
transesterification catalyst may be used. Examples include
alkyl titanates, alkali metal alkoxides, and metallic tin
and stannous hydroxide. Although acids, such as sulphuric
acid and sulphonic acids, have been proposed as liquid
phasetransesterification catalysts in the prior art, the use
of such catalysts is best avoided since there is a risk of
the fatty alcohol product becoming contaminated with
sulphurous impurities. Other transesterification catalyst
. .
WO90/08121 PCT/~B90/00063
2~5~9
- 21 - t '
systems which have been proposed, but are not preferred,
include bases, compounds OL alkali and alkaline earth
metals, water, and metals such as zinc, cadmium, lead and
their compounds. It is also conte~plated that acidic resins
containing -SO3H and/or -COOH groups or basic resins
containing substituted ammonium groups can be used as
transesterification catalysts.
A particularly preferred class of
transesterification catalyst is the alkyl titanates. Any
alkyl titanate may be added as catalyst but, as the alkyl
titanate will itself participate in ester interchange, the
al.~oxide radicals originally present in the alkyl titanate
will tend to undergo exchange with alkoxide radicals derived
from the fatty alcohol or alcohols during the operation of
the process of the invention.
Another particularly preferred class of
transesterification catalyst is the alkali metal alkoxides,
such as sodium methoxide or sodium ethoxide. Again exchange
of alkoxide radicals in the catalyst with alkoxide radicals
derived from the fatty alcohol or alcohols will tend to
occur with time in the first transesterification zone.
Alternatively there may be used an alkali metal alkoxide
derived from the fatty alcohol product itself, or from one
or more of them if a mixture of fatty alcohols is to be
produced.
The transesterification conditions used in the
first transesterification zone will to a large extent depend
upon the use or otherwise of a transesterification catalyst
and upon the activity o~ the transesterification catalyst
Although the use of elevated pressures is not ruled out, it
will normally be preferred to operate at a substantially
atmospheric pressure or below, for example a pressure in the
range of from about 0.1 bar to about 1.2 bar. In this way
the vaporisation of methanol or other lower alkanol is
facilitated during the course of the transesterification
. . :: ~.-,,.,,~., ,
,
.
WO90~0X121 PCT/GB90/0006~ -
2~ 4S5 ~ 22 -
reaction. Removal of the lower alkanol during
transesterification drives the transesterification reaction
towards completion.
When using an alkyl titanate a temperature of up to
about 240C, such as a temperature in the range of from
about 120C to about 200C, is typically used in the first
transesterification zone, for example a temperature of from
about 170C to about la0C. Alkali metal alkoxides enable
use of lower operating temperatures, e.g. in the range of
from about 40C to about 100C, but normally require
introduction of extra processing steps as will be further
explained below.
Similar transesterification conditions can be used
in the second transesterification zone. A
transesterification catalyst will normally be used, such as
one of those listed above for use in the first
transesterification zone. However, it is also contemplated
to operate without such a catalyst. It will usually be
preferred to employ in the second transesterification zone a
superatmospheric pressure, for example a pressure of from
about l.5 bar to about 50 bar, in order to maintain the
lower alkanol (e.g. methanol) in the liquid phase in the
second transesterification zone.
An advantage of the use of an alkyl titanate as
transesterification catalyst in the first and second
transesterification zones is that the subsequent
distillation and/or evaporation steps can be conducted
without prior removal of the catalyst. However, when using
an alkali metal alkoxide as transesterification catalyst, it
is preferable to neutralise the catalyst prior to
distillation and/or evaporation. Conveniently this
neutralisation step can be effected by passing the catalyst
containing material through a bed of an ion exchange resin
containing -SO3H and/or -COOH groups, thus removing the
alkali metal from the liquid mixture:
- : -:
: .
Wo9n/oxl2l PCT/GB90/00063
- 23 - 20~5~
R-SO3H + NaOR' = RSO3Na + HOR',
where R represe~ts the resin and -OR' represents an alkoxide
radical.
A further advantage of the use of alkyl titanates
is that the catalyst remaining in the distillation residue
(ii) or in the relatively involatile residue (b) can be used
to form at least a part of the transesterification catalyst
used in the first transesterification zone. The balance of
any amount of catalyst required can then be supplied by make
up al~yl titanate. Control of "heavies" in the process can
be achieved by purging a part of the relatively involatile
residue of (b); the remainder of this relatively involatile
residue can be recycled for use in the first
transesterification zone.
When using an alkali metal alkoxide as
transesterification catalyst, on the other hand, there will
usually be no residual catalyst in the relatively involatile
residue (ii) as neutralisation will usually be practised
prior to any distillation step. Similarly, if a resin
catalyst is ~lsed as a transesterification catalyst, there -
will be no catalyst dissolved in the relatively involatile
residue (ii). Hence recycle of the relatively involatile
residue has no benefit in these cases and the relatively
involatile residue (ii) can be purged from the plant and
used as fuel.
If an alkyl titanate transesterification catalyst
is used, then the evaporation and distillation steps
downstream from the second transesterification zone can be
carried out without prior removal of the catalyst. In this
case it is best to operate with as short residence times as
possible in these steps so as to minimise the risk of
substantial reversion of the transesterification reaction
with consequent re-formation of wax esters in these steps.
~ence it is preferred to effect this evaporation step by
flash distillation so as to minimise the residence time in
-' : ' '~; :: .
U 090~081'1 PCT/~B90/00063 --
~4~ 24 -
this step and to effect this distillation step! for similar
reasons, in a falling film or wiped film evaporator.
Distillation of the substantially lower alkanol
free mixture to yield overhead fraction (i) and distillation
residue (ii) and of the liquid residue to produce overhead
product ta) and relatively involatile residue (b) are
normally effected at or near atmospheric pressure or below,
for example at a pressure in the range of from about 0O005
bar to about 1.2 bar.
In order that the invention may be clearly
understood and readily carried into effect some preferred
forms of alcohol production plant designed to operate
according to the teachings of the present invention will now
be described, by way of example only, with reference to the
accompanying drawings, in which:- -
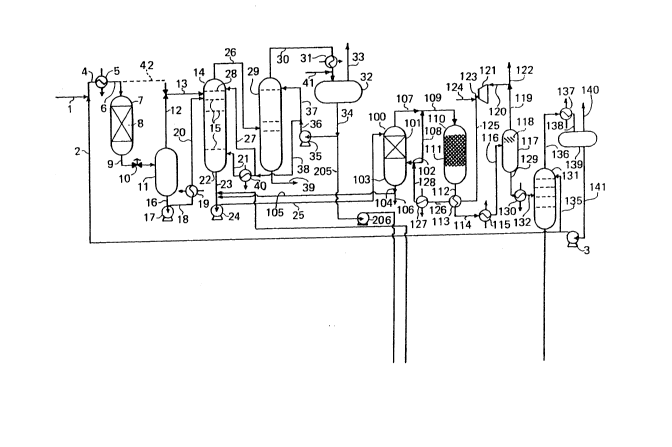
Figure 1 is a flow diagram of a plant f~r the
production of fatty alcohols constructed in accordance with
the teachings of the invention:
Figures 2 to 4 show details of parts of two
designs of esterification reactor that can be used in the
plant of Figure l;
Figure 5 is a flow diagram of another plant;
Figure 6 shows part of a modified form of plant
designed to be capable of operating using different fatty -
acid feedstocks;
Figures 7 and 8 each show part of respective
further forms of fatty alcohol production plant;
Figure 9 is a vertical section through a modified
form of hydrogenation reactor for use in a fatty alcohol
production plant; and
Figure 10 is a horizontal section through the
reactor of Figure 9.
It will be understood by those skilled in the art
that the drawings are diagrammatic and that further items of
equipment such as reflux drums, pumps, vacuum pumps,
U~Ogo/OX1~l PCT/GB90/00063
204~ 9
- 25 -
temperature sensors, pressure sensors, pressure relief
valves, control valves, flow controllers, level controllers,
holding tanks, storage tanks, and the like may be required
in a commercial plant. The provision of such ancillary
items of equipment forms no part of the present invention
and is in accordance with conventional chemical engineering
practice.
Referring to Figure 1 of the drawings, a fatty acid
mixture is supplied to the plant in line 1 and is admixed
with recycled methanol in line 2, which is pumped by pump 3,
to form a mixed feed to the plant in line 4. The fatty acid
mixture is, for example, a mixture of fatty acids obtained
by hydrolysis of a naturally occurring triglyceride, e.g. -
coconut oil, followed by "topping and tailing". Such a
fatty acid mixture contains approximately 65 mole %
dodecanoic acid, 25 mole % tetradecanoic acid, and 10 mole %
hexadecanoic acid. The mixed feed in line 4 flows on to a
heat exchanger 5, in which its temperature is raised to
110C. The heated acid/methanol mixture flows on in line 6
into primary esterification reactor 7, which contains a
charge 8 of an ion exchange resin containing sulphonic acid
and/or carboxylic acid groups, such as Amberlyst 13. (The
word "Amberlystn is a trade mark). The pressure in reactor
7 is 5 bar.
In reactor 7 part of the acid mixture is esterified
by reaction with methanol to yield a corresponding mixture
of methyl fatty acid esters. There exits from reactor 7 in
line 9 a mixture of methyl esters, unreacted fatty acid,
water produced by esterification and unreacted methanol.
This mixture passes through a pressure let down valve 10
into a vapour/liquid separator 11. A vapour phase
comprising methanol and water is fed at 1.3 bar by way of
lines 12 and 13 to an upper part of an esterification
reactor 14. Reactor 14 is provided with a number of
esterification trays 15; two possible forms of
: .
~ . . .
~ 90/OX121 PCT/GB90/00063 -
2a ~55 ~9 ` 26 -
esterification tray 15 are illustrated in Figures 2 and 3
and will be described in greater detail below. In reactor
14 of Figure 1 there are six esterification trays 15 tnot
all of which are shown); however, a greater or lesser number
of such trays may be provided, depending upon the nature of
the fatty acid or fatty acid mixture and the reaction
conditions selected.
The liquid phase from vapour/liquid separator 11 is
fed by way of line 16, pump 17 and line 18 to heat exchanger
19, in which it is heated by steam to a temperature of up to
150C, (e.g. 120C), and then by means of line 20 to reactor
14 at a point below the entry point of line 13.
In reactor 14 the downflowing unreacted fatty acids
in the mixture in line 20 pass downwardly from each
esterification tray 15 to the next lower tray 15 against an
upflowing current of vapour comprising methanol and water of
esterification, i.e. water produced as a result of the
esterification reaction. Dry methanol vapour is supplied to
reactor 14 in line 21. Each esterification tray 15 holds a
charge of an acidic ion exchange resin, such as a resin
containing sulphonic acid groups. Amberlyst 13 is a
suitable resin. (Amberlyst is a trade mark). In passage
down column 14 any unreac'ed ~ree acid encounters
progressively drier methanol vapour on each tray lS. By
designing each tray 15 to provide an appropriate liquid hold
up, it is possible to regulate the residence time on each
tray 1~. By selecting a suitable number of trays 15 it is
further possible to design reactor 14 so that essentially no
free fatty acid remains in the liquid passing downwards from
the bottom tray 15 into the sump 22 of reactor 14. Methyl
ester product (i.e. a mixture of methanol and methyl esters
derived from the mixed fatty acids supplied in line 4) is
removed from sump 22 in line 23 and pumped onward by pump 24
via line 25 to the hydrogenation stage of the plant.
A mixture of methanol vapour and the water released in the
,' ' , . ,,, .; ' ' ~ ~ .~
- ' ' - ' '' . ,
-
WO90/081'1 PCT/CB90/00063
- 27 - 20~
esterification reaction is recovered overhead from reactor
14 in line 26. Liquid methanol is supplied in line 27 to an
upper part of reactor 14 above the point of connection of
line 13 to provide liquid methanol on wash tray 28.
The vapour in line 26 is fed to a methanol/water
separation column 29 which is operated at l.3 bar and at a
head temperal:ure of 70C. Dry methanol vapour is recovered
overhead in line 30 and is condensed in condenser 31. The
resulting condensate is collected in drum 32 which is vented
as indicated at 33. Dimethyl ether produced as byproduct
isvented in line 33. Methanol which would otherwise be lost
along with the dimethyl ether can be recovered by providing
a chilled condenser (not shown) in line 31. Part of the
condensed methanol is recycled to column 29 from drum 32 as
a reflux stream in line 34 by means of pump 35 and lines 36
and 37. Part of the condensed methanol is pumped in line.
205 by high pressure pump 206 to line 180 for use in the
second transesterificatlon reactor. The remainder is pumped
back for re-use in line 38.
~ he sump product from column 29 consists
essentially of water. This is withdrawn in line 39. Part
is recycled to column 29 by way of a steam heated reboiler
(not shown); the remainder is passed on for effluent :~
treatment.
Some of the dry methanol in line 38 is passed
through vaporiser 40 to provide the stream of dry methanol
vapour in line 21. The rest flows on to provide the reflux
stream in line 27. Make-up methanol for the plant is
supplied throuoh line 41 to reflux drum 32.
In a modification of the plant of Figure l reactor
7 and vapour/liquid separator ll are omitted and the mixture
of fatty acids and optionally some methanol is fed by way of
line 42 to line 13. In this case the balance of the recycle
methanol in line 2 is passed through vaporiser 40 and the
number of esterification trays 15 in reactor 14 is increased
,- .. ~ .
.. . . ' ~
WO 90/OXI~1 PCI`/G890/00063 -
-- 28 --
2 ~ r~
by (say) 4 or 5.
Figure 2 illustrates one form of construction of a
tray 15 of reactor 14 of the plants of Figure 1. A
horizontal diaphragm or partition 50 extends within wall 51
of reactor 14 and closes off the cross section of reactor 14
completely except for a downcomer 52 for liquid and a vapour
upcomer 53. Partition 50 has an axial frusto-conical part
54 surrounding vapour upcomer 53 and an annular sloping
portion 55 adjacent wall 51. Tray 15 can thus retain a
volume of liquid whose surface is indicated at 56 and whose
volume is determined by the height of the overflow level of
downcomer 52 above the partition 50. Each tray 15 also
supports a charge of an acidic ion exchange resin containing
-SO3H groups, such as Amberlyst 13, whose particles are
indicated diagrammatically at 57. Such ion exchange
particles are kept in suspension in the liquid on tray 15 as
a result of agitation caused by the upcoming vapour as will
be described below. To prevent escape of ion exchange
particles 57 with the liquid overflowing down downcomer 52
the top of downcomer 52 is provided with a screen 58. The
slope of frusto-conical part 54 and of sloping portion 55 is
equal to or greater than the angle o~ repose of the
Amberlyst 13 or other solid particulate esterification
catalyst under the liquid on esterification tray 15.
Vapour upcomer 53 conducts upcoming vapour to a
circular sparger 59, which surrounds frusto-conical part 54,
by way of spider tubes 60. Suckback of liquid down upcomer
53 is prevented by means of an anti-suckback valve 61.
Annular draught shrouds or baffles 62 and 63 are
positioned within the body of liguid on tray 15, one inside
and one outside circular sparger 59 to promote agitation of
the liquid/resin suspension by the upcoming vapour. The
vertical extent of shrouds 62 and 63 is not critical but
should generally be between one third and three ~uarters of
the vertical height between diaphragm 50 and liguid surface
~o so/nxl21 PCr/GB90/00063
2 ~ 3
- 29 -
56. It is prefe~red that shrouds 62 and 63 should be placed
in a symmetrical or near symmetxical vertical position. In
the annular zone between shrouds 62 and 63 the liquid flow
is generally upward whilst inside shroud 62 and outside
shroud 63 the general direction of liquid flow is downward.
Preferably the area of the annular zone between shrouds 62
and 63 approximately equals the sum of the areas inside
shroud 62 and outside shroud 63.
Reference numeral 64 indicates a downcomer from the
next tray above the one illustrated in Figure 2. The liquid
level in downcomer 64 is indicated at 65, the height H of
this liquid level above liquid level 56 on tray 15 being
fixed by the liquid level on the tray which feeds downcomer
64 (i.e. the txay above the illustrated tray 15) plus the
pressure drop through the sparger 59 on that tray (i.e. the
one above the illustrated tray 15) and the frictional
pressure drop.
In operation of reactor 14 a liquid containing a
fatty acid or mixture of fatty acids is typically passed
downwards in countercurrent to an upflowing vaporous stream
of lower alkanol e.g. methanol. Each tray 15 acts as an ,~
esterification zone containing a respective charge oE
esterification catalyst which catalyses the esterification
reaction and the release of water of esterification. Under
the countercurrent conditions prevailing in the reactor 14
such water of esterification is vaporised and carried
upwards through reactor 14 with the upflowing lower alkanol
vapour. The liquid passes downwards from one tray 15 to the
next downward tray 15 and the free acid concentration in the
liquid on each tray 15 is lower than the corresponding acid
concentration in the liquid on the next higher tray 15. In
addition the liquid encounters drier and drier lower alkanol
vapour on each tray 15 as it passes down through reactor 14
In this way the equilibrium of the esterification reaction
is pushed further towards ester formation, the reverse
:
- ~ ' '
. .
wos~o~ PCT/GB90/00063
- 30 -
2 ~ 9
hydrolysis reaction being effectively supp~essed because the
water concentration in the liquid on the trays 15 decreas~s
from tray to tray in the downward direction.
By selecting a suitable number of trays 15 in
column 14 and designing each tray 15 to provide a sufficient
liquid hold up to provide the requisite residence time on
each tray it is possible to design reactor 14 so that the
product in line 25 contains less than about 1 mole % of
fatty acid, together with fatty acid esters and lower
alkanol as its principal components. By providing an
adequate upflow rate for lower alkanol vapour the agitation
caused by the vapour bubbles 66 emerging from circular
sparger 59, coupled with the liquid circulation induced by
the presence of draught shrouds 62 and 63, can suffice to
maintain the acidic ion exchange resin particles
sufficiently in suspension for esterification to proceed
successfully. The surfaces of sections 54 and 55 slope
towards the zone under the sparger 59 and ensure that there
are no stagnant zones where significant quantities of resin
can settle out of suspension. (It will be appreciated th~t, -
although Figure 2 only shows resin particles 57 in
suspension in the zone between draught shrouds 62 and 63,
they would in practice be present in suspension in the
li~uid phase outside this zone). If necessary, the volume
of the upflowing vapour can be boosted by inert gas or by
other vaporisable inert material, conveniently an inert
material that is a byproduct of the process. For example, ~,
it is often found that an ether is found amongst the
byproducts, as acidic catalysts can promote formation of an
ether from the alcohol used. Thus dimethyl ether is a
potential byproduct if methanol is used as the alcohol,
whilst diethyl ether can be formed in reactor 14 if ethanol
is the alcohol used: either material can be used, if
necessary, to boost vapour upflow to provide additional
agitation on trays 15 or to provide additional vapour to
~o 90/n8l2l PCr/~B90/0006~
- 31 - 20~ 19
carry away water of esterification.
In Figure 3 there is illustrated an alternative
design of esterification tray 15 suitable for use in a
relatively small scale reactor 14. In this case a frusto-
conical partition or diaphragm 70 extends within wall 71 of
reactor 14 and closes off the cross section of reactor 14
completely except for a downcomer 72 for liquid and a vapour
upcomer 73. The slope of frusto-conical diaphragm 70 is
equal to or greater than the angle of repose of the solid
particulate catalyst under the liquid present on tray 15.
The vapour upcomer 73 includes an axial sparger 74 provided
with a bubble cap 75 and is fitted with an anti-suckback
valve 76. Optionally bubble cap 75 can be surrounded by a
mesh screen (not shown) to prevent ingress of catalyst
particles interfering with the operation of valve 76. A
cylindrical baffle 77 surrounds sparger 74 symmetrically and
is positioned beneath the liquid level 78, the height of
which is determined by the height of the upper end of
downcomer 72. A screen 79 is fitted to the top of downcomer
72 to retain solid esterification catalyst, e.g. ~mberlyst
13, on tray 15. Reference numeral 80 indicates the
downcomer from the next higher esterification tray 15 (not
illustrated). In a similar manner to that described in f
relation to Figure 3 the bubbles 81 of vapour agitate the
liquid on tray 15 and maintain particles 82 of catalyst in
suspension. Baffle 77 defines an upflow zone within baffle
77 and a downflow zone outside baffle 77. Preferably the
areas of the two zones are substantially equal. This design
ensures that, so far as is possible, no stagnant zones where
catalyst particles can sediment are formed.
If desired the feed line 20 or 13 in the plant of
Figure 1 can be arranged to discharge onto a tray, similar
to tray 15 of Figure 2 or Figure 3, which does not hold a
charge of ion exchange resin. One or more alkanol wash
trays may be provided above the connection of feed line 20
.
: , . - - . :
; . .
' . ,., . ~ ; . . .:
.
,
~Osn/oxl~l PCT/GB90/OOG63 -
'~0 ~ 32 -
or 13 so that the vapours are scrubbed with a minor amount
of liquid alkanol before exiting reactor 14 in line 26 so as
to limit the amount of acid or ester to traces therein.
Reverting to Figure 1, the methyl ester product in
line 25 is fed to the top of a vaporiser 100 and is
distributed over the packing 101 in vaporiser 100. In
vaporiser 100 the descending organic material is vaporised
into an ascending stream of hot hydrogen supplied in line
102. Any liquid that collects in sump 103 of vaporiser 100
is recycled in lines 104 and 105 to the suction side of pump
24. Provision is made for purging a small stream in line
106 in order to limit build-up of "heavies" in the organic
material supplied to vaporiser 100. Typically vaporiser 100-
is operated at a temperature of about 205C and at a
pressure of about 41 bar.
A vaporous organic material/hydrogen stream is
recovered overhead from vaporiser 100 in line 107 and is
admixed with further hot hydrogen from bypass line 108 so as
to dilute this vaporous stream somewhat and to maintain the
resulting diluted mixture in line 109 at a temperature above
its dew point, preferably at least about 5C above its dew
point. From line 109 the vaporous mixture enters
hydrogenation reactor 110 which contains a charge 111 of a
reduced copper oxide-zinc oxide catalyst of the type
disclosed in GB-B-2116552. A typical catalyst contains,
before reduction, about 35 weight % of copper oxide and
about 65 weight ~ of zinc oxide.
In passage through the charge 111 of hydrogenation
catalyst a high proportion of the methyl esters is converted
to fatty alcohols and methanol. Accordingly there exits
from hydrogenation reactor 110 in line 112, at a temperature
of 214C and at a pressure of 40.7 bar, a vaporous reaction
mixture containing fatty alcohols, methanol, a minor amount
of unreacted methyl esters and minor amounts of byproducts.
This mixture is passed through a gas~gas heat exchanger 113
,,' ., ' ~ ' ' " '~ ' '`'
.. . ~
.
wo9o/oxl~l PCT/GB90/00063
20~55~9
- 33 -
and on through line 114 to a cooler 115. The cooled
reaction mixture flows on in line 116 to a gas/liquid
separator 117 which contains a demister pad 118 or other
vapour/liquid separation device.
A hydrogen-rich stream exits gas/liquid separator
117 in line 119 and is passed by line 120 to the suction
side of gas circulator 121. A purge gas stream is taken in
line 122 in order to limit the build-up of inert gases in
the circulating hydrogen. Make-up hydrogen gas is admixed
with the compressed gas in line 123, such make-up hydrogen
gas being supplied from line 124. The combined stream of
make-up and recirculated gas is fed in line 125 to the other
side of gas/gas heat exchanger 113 and then via line 126 to
gas super-heater 127 to provide in line 128 the source of
hot hydrogen for lines 102 and 108.
The make-up hydrogen stream in line 124 can be
produced in conventional manner from synthesis gas followed
by a water gas shift reaction, CO2 removal and, if desired,
further purification by pressure swing absorption. It may
contain one or more inert gases, such as nitrogen, methane
and argon.
A liquid condensate is removed from gas/liquid -
separator 117 in line 129, is partially vaporised in heat ~ ~
exchanger 130 and fed to methanol recovery column 131 in ~ ;
line 132 via a pressure let down valve (not shown).
Methanol vapour from lines 133 and 134 of the product
alcohols recovery and refining stage of the plant (which is
to be described further below) is admixed with the material
in line 132 prior to entry to methanol recovery column 131.
A methanol reflux stream is supplied to column 131 in line
13~.
Methanol vapour from column 131 is taken via line
136, condenser 137 and line 138 to methanol condensate drum
139 in which it accumulates. Vent line 140 allows any
volatiles to escape to the flare stack of the plant via a
: - ' '. ' ., : - ,
.~ . , - .
wo9o/nxl~1 PCT/GB90/00063
æo~5~ 34
vent condenser (not shown). Methanol is withdrawn from drum
139 by pump 3 to provide the recycle streams in lines 2 and
135.
A substantially methanol free stream of fatty
alcohols, containing a minor amount o~ unreacted methyl
fatty acid esters, is pumped via line 142 from the bottom of
column 131 by pump 143 to form the fatty alcohol fraction
stream in line 144 supplied to the product alcohol recovery
and refining stage of the plant.
The crude fatty alcohol stream in line 144 contains
a minor amount of unconverted methyl esters, besides minor
amounts of by-product alkanes, unknowns and "heavies". The
crude fatty alcohol stream passes through heat exchanger 145
in which its temperature is adjusted to about 160C to about~ ~
200C, preferably about 170C to about 190C, e.g. 190C. ~ `
The hot stream in line 146 is admixed with a mixture of
fresh and recycled ester interchange catalyst
(transesterification catalyst), e.g. an alkyl titanate,
supplied in line 147 and passes on in line 148 into a first
ester interchange reactor 149 which provides a first
transesterification zone. Reactor 149 is designed so as to
provide a residence time therein in the range of from about
10 minutes up to about 120 minutes, preferably from about 1
minutes to about 60 minutes. The length of the residence
time depends upon the temperature of the stream in line 148
and in reactor 149 as well as the effective concentration of
the alkyl titanate supplied in line 147. In reactor 149 the
methyl esters of the fatty acids present in the feed stream
in line 148 are converted to wax esters, i.e. fatty alcohol
esters of the acid moieties of the methyl esters, by
transesterification of the methyl esters with product fatty
alcohols. Most of the methanol formed by
transesterification is recovered as a vapour in line 133
from the vapour space in vessel 149 and is recycled for
admixture with the material in line 132 as described above.
.. . . . .
- . ' :
::
W090/08l~l PCr/GB90/00063
- 35 -
20~5~49
The product from the first ester interchange
reactor 149 contains, besides a major molar amount of
product alcohols, also minor molar amounts of alkane by-
products, wax esters and "heavies", as well as traces of
methanol. It is passed via line 150 into a product column
151 which is provided with three beds of structured packing
152, 153 and 154. Light ends, consisting mainly of alkane
by-products, as well as traces of methanol, are recovered
overhead in line 155 and are condensed by means of condenser
156. The resulting condensate in line 157 accumulates in
reflux drum 158 which is vènted to a vacuum pump (not shown)
operating at 0.005 bar by line 159. Some alkanes are
returned to product column 151 via line 160, pump 161 and
line 162 to provide a reflux stream, whilst the net
production of alkanes passes via line 163 to storage.
Product alcohols are withdrawn as vapour from
product column 151 in line 164 and are condensed by means of
condenser 165. The condensate passes on in line 166 to
product drum 167 which is vented to a vacuum unit tnot
shown) by line 168. Liquid product alcohols are passed via
line 169, pump 170 and line 171 to product storage.
~ ottoms product is withdrawn from product column
151 in line 172 and passed via line 173 to a falling film
reboiler 174 which is operated at a temperature in the range
of from about 210C to about 24SC, e.g. 240C. The heated
material from reboiler 174 is recycled to product column 151
in line 175. Part of the bottoms product is withdrawn in
line 17~ and is pumped by pump 177 via line 178 to heat
exchanger 179. Excess methanol from line 180 is admixed
with the hot bottoms product from heat exchanger 179. The
quantity of methanol admixed via line 180 is typically at
least about 5 times the stoichiometric quantity equivalent
to the wax esters present in the bottom product up to about
100 times this stoichiometric quantity, ~or example about 80
times the stoichiometric quantity. In this way the
.. - :- : . . ,: -,
., ~ ....
WO 90/OX I ' I PCI /G B90/00063
-- 36 --
2~ ~5 ~9
equilibrium between wax esters and methanol, on the one
hand, and methyl fatty acid esters, fatty alcohol and excess
methanol, on the other hand, is shifted away from wax ester
formation towards methyl fatty acid ester formation.
In heat exchanger 179 the temperature of the
bottoms product, which still contains alkyl titanate
transesterification catalyst, is adjusted. The mixture of
hot bottoms product and excess methanol passes on to a
second ester interchange reactor 181 which provides a second
transesterification zone and is designed to provide a
residence time of from about 30 minutes to about 240
minutes, preferably from about 60 minutes to about 180
minutes, e.g. about 120 minutes. The temperature in reactor
181 lies in the range of from about 160C to about 195C,
e.g. about 180C. The size, and hence the residence time,
selected for reactor 181 should be sufficient to allow the
ester interchange to proceed to equilibrium at the
temperature selected. The pressure in reactor 181 is
typically about 42 bar. From second ester interchange
reactor 181 the resulting transesterification product
mixture is fed via line 182 through a pressure let down
valve 183 to reduce its pressure to about 1.3 bar. It then
continues in line 184 to a heated flash vessel 185.
Methanol vapour is recovered overhead in line 134 and is
admixed, as described above, with the material in line 132.
The residual liquid phase exits flash column 185 in
line 186 and is pumped by pump 187 through line 188 via a
pressure let down valve (not shown) to falling film
evaporator 189 which is operated at a maximum temperature of
about 240C and at a pressure of about 0.005 bar. A mixture
of vapour and liquid exits falling film evaporator 189 in
line 190 and passes into separation drum 191. The vapour
is recovered in line 192 and condensed by condenser 193.
The resulting condensate is passed in line 194 to drum 195
which is connected to a vacuum system (not shown) by line
~VO 4n/08171 PCI/GB90/00063
-- 37 --
196. The liquid condensate, which comprises a ~Q~et~
product fatty alcohols, methyl esters, some methanol and
traces of by-product5, is recovered in line 197 and pumped
by pump 198 to form a recycle stream in line 199.
The liquid from drum 191 is passed by line 200 and
pump 201 either for waste disposal via line 202 or for
recycle via line 203 to line 147.
Fresh alkyl titanate transesterification catalyst ~ :
can be added as required via line 204.
Line 205 and pump 206 provide the methanol for line
180.
The approximate flow rates of various of the
streams expressed in molar units are summarised in Table 1
below:
-
.
WO90/08121 3 PCl/GB90/00063 _
20'1~r~'~ ~ ~ o I I I I I I ~ I I O o
~,. .... _ ~
O
~ ~ ~ o ~ O a~ o I O I ~ ~ :, ,,
_ ~
N I` O O O CS~ O _I _i
_ _ _ _ - _. . _ _
Oa~ ~ r I I I I O u~
_ _ _ _ _ _
~ I I o _/ In o I I I u~
o~ I -I O ~r I I I I ~ I I o
_l ~ I I _l I I I I I I I I ~" o
~ _ _ _ ._. ,
~ ~ I I ~l o I I I I I o 1 o ~ .: "'
_ . ~ .~
o ~ .
I I o I I I I I ~ ~
~ ~o o O ' ~
Cl~ I I ~ ~ '
_
~ I I ~ I I I i I I I ~" U~ ~
_ _ _ _ . C
_1 I I I I o I I I I I ~ o U~ _
~ Y
. . : , : . .. ':' . : ,
: : - :
.: ,
WO 90/OX12 1 - 3 g - 2 ~ ~ 5 5 4 9 PCr/GB90/000~3
~ . .
_~ IIa~IIIII1'I:..1.o
_ ,_ .. - - ---- I .
~o I I E~ o I O I ~ ~~ O ' '
_ - .......... _ _ . _ _ _
~o I I o I I ,,~ o ~ ~ ~
_ ,_ _ _ . - --:
~ ~ I O I ~0 o ~
_ _ _ _ _
C~ ~ ~ :
~O I O I , I ~ ~r ~. ;
~ - ~ -
_~ ~ I o ~ o o
.. _ . . ~ : ~'
_~ ~IIIoII~C,o ~:
UO~ ~ o o, o. U~ ~ o
_ ._. _ . .
o o o ~ U~ I o U~
o ~ o ~ o
, ~
~ ~ '` R, , o o, , , ~ ~
O ~ v ~ -
.c ~ ~
-
:~ :
~vo90/n8121 PCT/GB90/00063
2~ 9 - 40 -
The plant or Figure 1 can be modified to operate
using an alkali metal alkoxide as the transesterification
catalyst in place of an alkyl titanate. In this case the
temperature of the crude fatty alcohol stream in line 144 is
adjusted to 45C in heat exchanger 145. A solution
containing 10% w/v of sodium methox de in dry methanol is
added in line 147 so as to provide a concentration of 0.05%
w/v of sodium methoxide in the material flowing in line 148.
In this modified form of plant first ester interchange
reactor 149 is designed to provide, typically, a residence
time therein of about 30 minutes. The material exiting
reactor 149 in line 150 is then passed through a bed of ion
exchange resin in vessel (not shown) provided in line 150 to
neutralise the catalyst. The ion exchange resin of this bed
can contain sulphonic acid and/or carboxylic acid groups.
The c~talyst free stream passes on in line 150 to product
column 151.
Removal of the sodium methoxide catalyst prior to
distillation in product column 151 is desirable so as to
obviate the formation of condensation by-products and dark
coloured organic tars, which would be promoted by the
presence of sodium methoxiae in the mixture at the elevated
temperatures prevailing in the product column 151.
Recovery of product alcohol in product column 151
is effected in the same way as for the plant of Figure 1.
The bottom product in lines 172, 176 and 178 is then cooled
to about 50C in heat exchanger 179. A similar
stoichiometric excess of methanol is added from line 180 to
the liquid stream from heat exchanger 179 in the form of a
solution of sodium methoxide in methanol so as to provide a
concentration of about 0.05% w/v sodium methoxide in the
mixed stream before entry to second ester interchange
reactor 181 which is designed for a residence time of about
120 minutes. The interchanged product stream in line 182 is
then passed through a second bed of ion exchange resin (not
- ,'.' '` . ~ ' ~
- . .
W O 90/08121 2 ~ S ,~ ~ Pc~r/GB90/00063
shown) in line 182. This resin contains, for example,
sulphonic acid groups and/or carboxylic acid groups. The
resin removes sodium ions from the liquid phase and
neutralises the sodium methoxide transesterification
catalyst. The neutralised liquid phase passes on in line
182 and 184 to flash column 185.
As the material in line 184 contains no
transesterification catalyst there is no need to recycle
"heavies" via line 203 (as in the plant of Figure 1).
Moreover, as there is no catalyst remaining in the material
in line 184, the risk of reversion of methyl esters to wax
esters and loss of methanol vapour in columns 185 and 189 by
ester interchange with fatty alcohols product is obviated.
In a still further modification of this plant
columns 185 and 189 are replaced by a batch still (not
shown). In this case the neutralised material in line 184
is collected until there is sufficient to justi$y operating
the batch still.
Figure 4 illustrates a further design-of
esterification tray 15 suitable for use in a laboratory
scale reactor 14 or in a commercial scale reactor 14. This
comprises a generally frusto-conical partition or diaphragm
250 which extends within wall 251 of reactor 14. The slope
of the upper surface of diaphragm 250 is greater than the
angle of repose of the solid particulate catalyst. A vapour
upcomer 252 is fitted with a cap 253 with a dependent skirt
of mesh 254. Downcomer 255 is fitted with a mesh cap 256
and with a seal bucket 257. The upper end of downcomer 255
is positioned so as to provide a suitable retention voIume
for liquid on tray 15 whilst mesh skirt 254 and mesh cap 256
retain the charge of resin particles on diaphragm 250.
Methanol vapour flows up upcomer 252 as indicated by arrow
257, through the space between upcomer 252 and cap 253 as
indicated by arrows 258, and through skirt 254 as indicated
by arrows 259, and carries with it water vapour resulting
, , , : ~ , ''' : .
"' ' . ' ,
wo ~)tn8l2l PCT/GB90/00063
2~ 4~5 ~ - 42
from water of esterification formed on a lower tray or
trays.
The plant of Figure 5 is generally similar to that
of Figure 1 and li~e reference numerals have been used in
both Figures to indicate like parts. The feed acid in line
1 is typically an unsaturated fatty acid, such as oleic
acid. -
As the number of theoretical stages in column 14
does not necessarily correspond to the number of trays 15
fitted in column 14, and the number of such theoretical
stages may vary, for a particular column, for different feed
acids supplied in line 4, the acid content of the methyl
ester product in line 23 may vary if the nature of the feed
acid in line 4 is changed.
As already mentioned a by-product of ester
formation in the column is often a dialkyl ether. ~he yield
of such dialkyl ether by-product is found to be dependent
upon the temperature of operation of the reactor 14. Hence
by minimising the temperature of operation of column reactor
14 the yield of by-product ether can be minimised. However,
a corollary of this is that a lower conversion of acid to
ester is obtained at lower operating temperatures. In this
case it is possible to optimise the conversion to ester by
admixing the ester-containing product, which contains
perhaps about 97 mole ~ to about 99 mole % of ester with the
balance being acidic materials, with further alkanol (e.g.
methanol) and passing the resulting mixture containing, for
example, a 2:1 to 4:1, e.g. 3:1, alkanol:ester molar mixture
through a polishing reactor having a fixed bed of a solid
esterification catalyst, such as Amberlyst 13, which can be
operated at a lower temperature than the column reactor. In
this way extremely high overall conversion to ester can be
achieved. Such a modified form of plant is illustrated in
Figure 5.
In the plant of Figure 5 there are six
WO 90/08121 PCl/GB90/00063
- 43 - 2 ~ L~ 4 ~
esterification trays 15 and the methyl ester product in line
25 still contains a minor amount of oleic acid. Typically
the methyl oleate:oleic acid molar ratio is in the region of
~7:3. This mixture is admixed with further methanol
supplied from line 301 to form a mixture having a molar
ratio of methanol:methyl oleate:oleic acid of 3:0~97:0.03.
This mixture is supplied in line 302 at a temperature of
60C and at a liquid hourly space velocity of 1 hr~1 to a
further esterification reactor 303 containing a fixed bed
304 of an acidic ion exchange resin, such as Amberlyst 13.
The resulting mixture flows on in line 305 to a further
distillation column 306. Methanol vapour passes overhead
via line 307 to column 29 via line 26. Liquid methanol to
form reflux streams for columns 29 and 306 and the stream
in line 301 is pumped from condensate drum 32 via line 34 by -
pump 3S through lines 36 and 308. The methanol to form the
reflux streams flows on in line 309 via line 37 to column 29
and via line 310 to column 306. The bottom product from
column 306 in line 311 comprises essentially pure methyl
oleate (of purity at least 99.5 mole ~). Part is recycled
to column 306 by way of line 312 via column reboiler 313 and
line 314, whilst the remainder is passed to vaporiser 100 in
line 315.
Part of a modified form of plant is illustrated in
Figure 6. This is similar to the plant of Figure 5 except
for the hydrogenation section. Like reference numerals
indicate like parts in Figures 5 and 6.
The plant of Figure 6 is designed for use with a
variety of different fatty acid feedstocks. Hence the ester
(or the main constituent of an ester mixture) supplied in
line 315 to vaporiser 100 may at one time be, for example,
methyl laurate, whilst at another time it is methyl
stearate. To ensure that the feed mixture in line 109 to
the hydrogenation reactor 110 is in the vapour phase a
higher ~2:ester molar ratio must be used when the ester used
- ' " "'
,
. .
WO 90/OXI21 PCr/GB90/00063
~ O L~
is, for example, ~ethyl stearate than when it is methyl
laurate, due to the lower volatility of the higher molecular
weight ester. In addition it will usually be expedient to
adjust the heat input to heat exchanger 127 so as to
increase the temperature of the hydrogen supplied in line
128 and hence cause an increase in the temperature of the
feed mixture in line 109. The increased inlet temperature
to hydrogenation reactor 110 contributes to increased rates
of reaction within the catalyst charge 111 with the result
~hat hydrogenation is achieved within a smaller volume of
catalyst at the inlet end of the catalyst charge 111 than is
the case when methyl laurate is the ester or the main
component of the ester mixture. Hence the outlet end of the
catalyst charge 111 is not serving any useful function in
this case. As reactor 110 is operated adiabatically, the
result is that the vaporous reaction mixture is spending an
appreciable time at outlet temperature in contact with the
catalyst. This situation is disadvantageous since it may
lead to increased by-product formation, e.g. to formation of
additional minor amounts of alkanes. To avoid this
situation hydrogenation reactor 110 is provided in the plant
of Figure 6 with three alternative outlet ports 320, 321 and
322, in addition to line 112. These outlet ~orts 320, 321,
and 322 are connected to an outlet manifold 323. A bypass
line 324 allows a supplementary flow of hydrogen containing
gas to be introduced into the bottom of hydrogenation
reactor 110 when one of the outlets, 320, 321 or 322 is in
use. This supplementary flow of hydrogen prevents
condensation of any liquid on the lower part of the catalyst
charge. The catalyst charge 111 itself can be split into
several discreet beds with each supplementary outlet 320,
321 and 322 being connected between a respective pair of
beds. Alternatively the catalyst charge can be a unitary
charge with collector devices, each connected to a
respective one of the outlets 320, 321 and 322, buried in
` 7
.,.. ,, ~ ~ .. .. - . . ....
'' ' ' '
'
'
WO90/l)X1~l PC~/GH90/00063
- 45 - 2~955~
the catalyst charge. Hence i.n this case there are not
separate beds of catalyst in the sense of having separate
discreet charges of catalyst arranged in series but rather
the beds comprise separate sections of a unitary catalyst
charge.
Besides allowing the plant operator to minimise
by-product formation when utilising high boiling fatty acid
feedstocks the provision of supplementary outlets 320, 321
and 322 also permit account to be taken of any loss of
catalyst activity with time due to ageing of the catalyst
charge.
Variants in design of the hydrogenation reactor 110
are illustrated in Figures 7 and 8. Figures 9 and 10 show
details of a form of distributor/collector for one of the
supplementary ports fitted to the reactor 110 shown in
Figures 6 to 8.
In the modified design of Figure 7 it is the point
of inlet of the vaporous mixture to hydrogenation reactor
110 that can be varied, rather than the point of outlet. In
this case reactor 110 has a number of supplementary inlets
325, 326 and 327, connected to an inlet manifold 328, and
flow to reactor 110 is controlled by valves 329, 330, 331
and 332. Also hydrogen bypass line 324 is connected in this
design to line 315, flow through line 324 being controlled
by valve 333. Reference numerals 334 indicate
diagrammatically distributors within catalyst charge 111
each connected to a respective inlet 325, 326 or 327.
Figure 8 illustrates a further modified design of
hydrogenation reactor 110 in which discrete individual
sections of the catalyst charge 111 can be used in sequence
to suit the desired reaction kinetics and equilibria within
an acceptable range of reaction and vaporisation
temperatures. This has a number of supplementary lines 335,
336, 337 and 338 connected to a line 339 which connects
lines 315 and 312. Line 340 connects lines 336 and 338.
.
~0 ~/0~1'1 PCT/GB90/00063 ~
2 0 ~ 46 -
Supplementary hydrogen can be fed to the reactor 110 by
either of lines 341 and 342. Valves 343 to 352 can be used
to control flow through reactor 110~ A typical operating
sequence might be as set out in Table 2.
Table 2
Phase No. Valve No.
343 344 345 346 347 348 349 350 351 352
0 X X X X O X X O X `'
2 X O X O X X X O O X
3 X O X X O X X X X O
4 X o X O X X X X X o
X O O X X X X X X o
6 O X X X X X X X X O
Note: In Table 2 "O" indicates an open valve and "X"
indicates a closed valve.
Figures 9 and 10 show one form of distributor or
collector 334 in part of a hydrogenation reactor 110. This
takes the form of a lozenge ring covered with fine mesh to
prevent entry of catalyst particles. As can be seen from
Figure 9 each distributor/collector is immersed in the
catalyst charge 111.
The invention is further illustrated in the
following Examples.
Example 1
A crude fatty alcohol product containing a minor
amount of unconverted fatty acid methyl est~rs was prepared
by hydrogenating in a laboratory hydrogenation reactor under
vapour phase conditions (i.e. under conditions such that the
reaction mixture in contact with the catalyst was at all
times above its dew point) a mixture of fatty acid methyl
esters obtained from a "topped and tailed" fatty acid
mixture produced by hydrolysis of coconut oil. The catalyst
used was a reduced copper oxide-zinc oxide ester
hydrogenation catalyst. Prior to use the crude alcohol
product mixture was distilled to remove substantially all
-
, - . . ,
~ .
:
:
WO90/08l2l PCT/CB90/00063
_ 47 _ ~ ~5S49
the methanol produced as coproduct in the hydrogenation
step.
Three samples of the substantially methanol free
crude fatty alcohol product were each heated to 200C under
a nitrogen atmosphere for 30 minutes at 0.99 bar with 0.03%
w/w of Tilcom BIP ~trade mark of Tioxide Chemical Division
of British Titan Products p.l.c.). This material is
reported to be a mixed lso-propyl/n-butyl titanate.
Subsequent analysis showed that, in the presence of a large
excess of fatty alcohols and under conditions allowing
methanol to escape from the reaction system, substantially
all of the methyl esters had been transformed into wax
esters. The results are plotted in Table 3 below which
indicates the amounts of the components present in % w/w.
In ~able 3 "C12 Me ester" means methyl dodecanoate, whilst
"C14 Me ester", "C16 Me ester", and "Clg Me ester" represent
respectively the corresponding methyl esters of the C14, C16
and C18 carboxylic acids. There were detected sixteen
unidentified compounds, listed as "Unknowns 1 to 16" in
Table 3, in minor or trace amounts.
, - . . .
..
- :
WO~0/0XI2l PCT/~B90/00063 _
'~ 0~.5 hi~ - 48 -
Table 3
. . _
.
PRODUCT
COMPONENT FEED ¦ Run 1 Run 2 Run 3
_
Methanol 2.19 0.03 0.11 0.02
Cl2 Alkane 0.34 0.27 0.20 0.24
Cl4 Alkane 0.37 0.37 0.34 0.34
Cl6 Alkane 0.31 0.35 0.33 0.33
¦Unknown Compounds
¦l to 6 1.34 1.6B 1.75 1.90
(Cl2 Me Ester +) 1.79* 0.06 0.10 0.04
8 Alkane) 0.04 0.03 0.03
Vnknowns 7+8 0.16 ! 0 3 0-4g 0.33
Unknowns 9+10 0.87 ¦ trace trace trace
C12 Alcohol 57.30 1 56.97 55.86 54.33
C14 Me Ester 0.21 ! trace trace trace
Unknowns ll to 13 0.17 ¦ G.17 0.14 0.23
C14 Alcohol 24.64 1 24.86 24.94 26.52
Cl6 Me Ester 0.09 ¦ trace trace trace
Unknowns 14+15 0.14 0.03 0.03 0.08
C16 Alcohol 9.45 9.42 9.66 9.47
Cl8 Me Ester 0.23 0.02 0.03 0.03
Unknown 16 0.05 trace trace trace
C18 Alcohol 0.35 0.45 0.50 0.46
Wax Esters:
ta) C12 ~ C12 2.78 2.60 3.20
(b) Cl2 ~ C14 1.42 1.72 1.68
(c) Cl2 ~ C16 ~ 0.75 0.80 0.67
) C14 ~ C14 trace trace 0.08
~ l
* Components not resolv ed.
- . . .
.~:
,
wo ~/081~1 PCT/GB90/00063
` - 49 - ~4~
In Table 3 the wax esters are identified variously
as (a) Cl2 - Cl2, (b) Cl2 ~ Cl4, (c) Cl2 ~ Cl6 and (d) C14
Cl4. These materials are thought, by reason of their gas
chromatographic retention ti~es, to represent respectively:
(a) the ester of a Cl2 alkanol with a Cl2 fatty acid;
(b) mixture of estexs of a Cl2 alkanol with a Cl4
fatty acid and of a Cl4 alkanol with a Cl2 fatty
- acid;
(c) mixture of esters of a Cl2 alkanol with a Cl6
fatty acid and of a Cl6 alkanol with a Cl2 fatty
acid; and
(d) the ester of a Cl4 alkanol with a Cl4 fatty acid.
The results plotted in Table 3 were obtained using
a Pye Unicam 4500 Gas Chromatograph fitted with 25 metre
long Nordian NB351 FAME capillary column and with a flame
ionisation detector. The carrier gas was helium at a column
inlet pressure of 2.39 bar. The sample injection volume was
O.4 microlitres. The column was temperature prograrnmed as
as follows: 2 minutes at 80C after sample injection,
followed by heating at 8C per minute to 230C, whereafter
the temperature was maintained at this value. The injection
port temperature was 250C and the detector temperature was
270C. A sample stream split ratio of 40 to 50:1 was used.
It is clear from these results that, under the influence of
the transesterification catalyst, the methyl esters of the
Cl2, Cl4, Cl6 and Cl8 fatty acids are smoothly converted to
wax esters. It should be noted, however, that the gas
chromatographic technique employed, although resolving the
wax esters in total carbon number order, did not enable good
resolution between wax esters containing the same number of
carbon atoms. For example, the resolution achieved between
a Cl2 - Cl6 wax ester and a Cl4 - Cl4 wax ester was
relatively poor.
Example 2
665 grams of crude fatty alcohol product which had
::
:-, : : ;: .. .. . - : , . -
. .
: : : . , :: . : ~
,,, ~ : :: ~. :
: . : : ~ :: . : :
: .' , : ' ' . ~' :
W~90/0812l PCT/GB90/00063
50-
been subjected to transesterification under the co~ditions
outlined in Example 1 were distilled under vacuum in a
simple laboratory distillation unit, the boiler of which was
fitted with a short packed column to prevent droplet
entrainment. The dimensions of the pac~ed column were 2.5
cm ~iameter x 30 cm high, packed with 4 mm Raschig rings.
The results are summarised in Tables 4 and 5 below. The
analysis figures of Table 5 are again expressed as ~ w/w.
The abbreviations used in Table 5 are the same as those used
in Table 3.
Table 4
. _ _ .__
Fraction Fore run Product ¦ Residue
.._ . ..
Pressure (bar) O.G14 0.013 0.013
Temperature up to 141C141 - 170C Not
distilled
..___
Weight (g) 96.1 537.1 ¦ 30.0
.: , ' , ' ':
', : -
WO ~/08121 PCT/GB90/00063
- Sl- 2~55~9
Table 5
ANALYSIS
_ _ , ~
Fraction Fore run Product ~ Residue ¦ I .
_. ._ . _ _ '
Methanol trace _
Vnknown 1 0.84 _ _ ¦ :
C12 Alkane 1.22 0.04 _
Unknown 2 l.S3 _ _
C14 Alkane 1.35 0.21 _
Unknown 3 0.91 _ _
Unknown 4 0.17 _
C16 Alkane 0.47 0.33
Unknown 5 2.36 _ _
Unknown 6 0.46 _
¦C12 Me Ester + .
C18 Alkane 0.03 0.01 1 0.01
Unknown 7-10 0.60 0.40 0.04
C12 Alcohol 75.34 59.66 ' 0.30 ' `
¦C14 Me Ester trace
Unknown 11-12 0.11 0.2 -
C14 Alcohol 10.24 29.51 1.50
C16 Me Ester 0.03 _ : _
Unknown 13 trace trace trace
Unknown 14-15 0.01 _ _
C16 Alcohol 3.16 8.91 6.86
C18 Me Ester 0.09 _ ~ _
C18 Alcohol 0.29 0.06 , 0.88
Wax Esters: .
C12 ~ C12 0.02 _ . 0.37
¦Other wax esters0.28 0.8 i 81.66
¦Other unknowns 0.09 - 8.36
.. _ .. _
.
,
WO90/08121 PCT/GB90~00063
20~5~ 52 -
~ ecause the transesterification catalyst remained
active throughout the distillation and because the lower
alcohols were progressively removed from the system by the
distillation procedure, the wax esters remaining in the
distillation residue were of higher molecular weight than in
the starting material. In other words there was continuous
ester interchange amongst the wax esters during distillation
with a progressive loss of the more volatile fatty alcohol
components to the distillate.
Example 3
The distillation residue of Tables 4 and 5 was
divided into two portions. One portion was heated to 180C
for 2 hours with methanol at a methanol:wax ester mole ratio
of 20:l and the other portion was heated at the same
temperature and for the same time but at a methanol:wax
ester ratio of 40:1. Vpon quench cooling analyses in % w/w
were obtained, using the gas chromatographic technique of
Example 2, as set out in Table 6 below. The abbreviations
in Table 6 are the same as are used in Tables 3 and 5. The
analytical figures are expressed on a methanol free basis.
.
,
W090/0X12l PCT/GB90/00063
2 Q ~ 9
- 53 -
Table 6
... .
?ortion No. ¦ 1 2
-12 Me ester ¦ 32 94 - 34.56
C12 Alcohol 1 0.17 0.13
C14 Me ester 4.48 4.88
C14 Alcohol 2.54 2.43
C16 Me ester 0.56 0.62
C16 Alcohol 41.27 42.75
C18 Me ester 0.33 trace
C18 Alcohol i7.85 8.16
Wax esters
(a) Cl2-cl2 ¦0.02 trace
(b) C12-C14 10.27 0.33
(c) C12 C16 !7 04 5.17
(d) C14-C14 2.52 0.98
It can be seen from these results that, in
comparison with the composition of the residue of Table 5,
treatment with methanol has effected a considerable
cOnversion of the wax esters to C12~ C14' C16 and C18 fatty
alcohols and to the methyl esters of C12, C14, C16 and C18
fatty acids. This conversion ha5, moreover, been effected
without the addition of further alkyl titanate
transesterification catalyst, thus demonstrating that the
transesterification catalytic activity has survived the
vacuum distillation step of Example 2.
Example 4
A laboratory scale column reactor with an internal
diameter of 76.2 mm made of glass QVF components and having
ten trays one above another was used. Each tray had the
form illustrated in Figure 5. The column reactor was lagged
and wound with external electrical heating tapes. Each tray
had its own temperature control system. The top tray
contained no resin and acted as a liquid scrubbing tray to
. .
. - '. - :
.
U~90~081~1 PCT/GB90/00063
20 ~ 54 -
limit losses of the acid feed or of the ester product. The
second tray from the top also contained no resin and was
supplied with the acid feed. The remaining eight trays each
held a charge of Amberlyst 16 ion exchange resin which had
been sieved to remove beads with a particle size less than
355 ym and then washed extensively with methanol and dried
at 105C to constant weight . The mesh size of the
stainless steel mesh of skirt 254 and of cap 256 was 300 ~m.
Dry methanol was vaporised by passage through a coil
immersed in an oil bath at lS0C and the resulting vapour
was fed to the bottom of the column reactor below the
lowermost tray. Each tray held about 240 ml of liquid. The
resin charge on each tray corresponded to 14% by weight
calculated as dry resin based on the liquid charge on each
tray. The overhead vapour from the column reactor, which
consisted of unreacted methanol, water which is produced in
the course of esterification, and a minor amount of by-
product dimethyl ether, was condensed. A constant head
overflow device was used to control the rate of removal of
product esters from the column reactor.
At start up the column reactor was charged with
resin and with methyl laurate. When the methanol flow and
the temperatures of the various trays had stabilised a feed
of 50 mole % methyl laurate, 40 mole % lauric acid, and 10
mole % myristic acid was supplied to the column. This feed
mixture was similar to the mixture in line 20 of Figure 1
when that plant is supplied in line 4 with a mixture of
lauric acid and myristic acid. The level of C14 ester in
the bottoms product from the column reactor was monitored
until an equilibrium level was attained. The liquid on each
tray was analysed. The results are summarised in Table 7
below; the trays are numbered from 1 to 10, tray No. 1 being
the top tray and tray No. 10 being the bottom tray.
-
~'" ' ' ' ~ ' ' '
.~ . ' ' ' '
WO90/0X121 PCT/GB90/00063
- 55 - 20~5~3
Table 7
MeOH Acid mole ratio5:l T 3.6:l ~ 3:1
Residence time (hours)2.6 ¦ 2.2 ¦ 2.0
Tray No.Mole % Conversion
_
98.32 96.53 95.16
6 99.39 98.76 97.05
7 99.62 99.15 97.25
8 99.87 99.45 98.74
9 99.93 99.75 99.48
ND 99.81 99.76
DME make 3.0 ~ 2.7 1.5
In Table 7 and in the following Tables "N.D" means
"not determined", whilst "DME" means "dimethyl ether", the
"DME make" being expressed as a percentage by weight or the
acid feed.
Exam~le 5
The same column reactor as was used in Example 4
was fed with a mixture of natural straight chain fatty acids
of the following composition:
ComDonent % bY wei~ht
C8 acid5.l0
Cl0 acid4.62
C~2 acid40.64
Cl4 acid14.12
Cl6 acid9.57
Cl8 acids 25.0l
Unknowns0.77
H2O 0.l7
The results are summarised in Table 8.
wo9o/n#12l PCT/GB90/00063
- 56 -
20~S~
Table 8
MeOH: 2.7:1 3.8:1 4.2:14.1:1 4.7:1 6.7:1
AactidOmole
Residence 1.9 3.3 3.6 3.5 4.64.7
Time
(hours)
Tray No Mole % Conversion
61.22 66.54 68.89 68.01 69.0~ 79.82
6 ND ND ND ND ND ND
7 86.20 89.74 91.78 90.16 91.78 92~38
8 92.50 94.62 96.14 95.29 96.22 98.07
9 95.28 97.46 98.15 97.68 98.11 99.20
97.53 98.77 99.12 98.90 99.30 99.64
DME
Make 2.0 2.8 2.5 2.7 !
Average _
Temperature
(C) 112 107 104 111 112 113 ,
ExamPle 6
The procedure of Example 5 was repeated using a
51.6:48.4 acid:ester mole ratio feed mixture. Such a `
mixture corresponded to a typical feed mixture in line 20 of
Figure 5. The acids used were a mixture of natural straight
chain fatty acids comprising 65% by weight C12 acid, 25% by
weight C14 acid, and 10% by weight C16 acid. The results ~ -
are shown in Table 9 below. In this Example the amounts of ~ -
resin on a dry basis used on each tray corresponded to 10~
.
.
,-: . . , .. ., ., ~ ,:
. . ~, - . .
. . . :. . : . .
: - ' ' ' '
WO90/081~1 PCT/GB90/00063
2 ~
- 57 -
by weight based upon the liquid retained for each of trays
Nos. 3 to 7 and to 5~ by weight on the same basis for trays
Nos. 8 to 10.
.. .. . . .
-.
.' '
~VO 90/OXI'1 PCI/GB90/00063 .
20455,~3 - 58 -
Table 9
MeOH:Acid mole ratio 3:1 ¦ 2:1
Residence time (hours) 2.5 2.5
.. __
Tray No Mole ~ C~ )nversion
.
3 70.08 67.10
4 83.10 79.21
91.59 88.48
6 96.4 94.46
7 98.55 97.39
8 99.13 98.24
9 99.49 98.87
99.68 99.27 :~
~ :
DME Make 2.1 1.3
.,.
. Average Temperature (C) 110 107
Example 7
Using the column reactor of Example 4 the following
esterification reactions between the specified acid and the
corresponding alcohol component are carried out with : ~
similarly good results with the more volatile reactant in . :
each case being supplied to the bottom of the reactor in
vapour form and the less volatile component being supplied
in liquid form to the second tray of the reactor: '
(a) stearic acid with methanol to methyl stearate;
(b) palmitic acid with ethanol to ethyl palmitate;
(c) arachidic acid with methanol to methyl arachidate.
(d) oleic acid and lso-propanol to yield iso-propyl
oleate;
-
. - . - . .
- -:
,. ~ ' .
.
W090/oXl2l PCT/GB90tO0063
20~5~4~
- 59 -
(e) ricinoLeic acid and methanol to methyl ricinoleate;
and
(f) isostearic acid and methanol to methyl isostearate.
Example 8
An ester mixture obtained by esterification of a
"topped" coconut fatty acid mixture with methanol was
hydrogenated in a laboratory reactor under vapour phase
conditions using a reduced copper oxide/zinc oxide catalyst
obtained by reduction of PG 88/32 catalyst precursor
(obtainable from Davy McKee (London) Limited of Davy House,
68 Hammersmith Road, London, W14 8YW) according to the
supplier's instructions. The ester mixture had the
following analysis in % by weight: methanol 0.16; C10 ester
1.63; C12 ester 54.60; C14 ester 20.24; C16 ester 10.34; C
ester 10.74; C12 acid 0.09; C14 acid 0.05; C16 acid 0.05;
C18 acid 0.42; "unknowns" 1.61; and H20 0.07. The H2:ester
molar ratio was 630:1. The results are summarised in Table : .
10. ~ :"
,
.
'
W090/08121 PCT/GB90/00063
2~ 60 -
Table 10
I _
Run No. ¦ 1 2 3 4 5
. _
Reactor
Temp. (C) 215 215 215 215 215
L~SV (hr 1) 0.22 0.25 0.25 0.30 0.35
(bar) 42.36 42.70 42.70 42.7042.36
conv- C12 99.6 99.2 99.2 98.698.0
Sel. C12 99.2 99.3 99.3 99.499.5
conv- C14 99.8 97.8 97.5 99.699.7 : :
Sel. C14 98.1 98.2 98.2 98.799.1 ~ .
conv- C16 99.7 99.8 99.8 99.799.9
Sel. C16 96.6 96.9 96.9 97.898.5
Conv. C18 99.9 99.3 99.3 99.7 99.8
Sel. C18 91.9 92.6 92.6 94.595.4 ~ ;:
. . .
. .
. . ' - ,. .
.', ''"'
W090/oX121 PCT/GB90/00063
- 61 -
Example 9
Following the teachin~s of Example 8 each of the
esters prepared according to Example 7 above is hydrogenated
according to the general procedure described in Example 8
and then the hydrogenation product is subjected to
transesterification by the procedure of Example 1, followed
by distillation as described in Example 2, and then the
distillation residue is submitted to a second
transesterification step as taught by Example 3. Similarly
good results are obtained.
''' '.
'.' '