Note: Descriptions are shown in the official language in which they were submitted.
20~69~
SUBSTITUTED-ACYCLIC TERPENE COMPOUND
FIELD OF THE ART
The present invention relates to novel substitut-
ed-acyclic terpene compounds. More particularly, the
present invention is directed to substituted-acyclic terpene
compounds useful as intermediates for producing sarcophytol
A which have an anti-carcinogenic promotor activity and
anti-tumor activity.
BACKGROUND OF THE INVENTION
The sarcophytol A was reported to exhibit anti-
carcinogenic promotor activity [Cancer Surveys, 2, 540
(1983); Taisha, Vol. 25, Special Edition, Gan '88,3 (1988)]
and anti-tumor activity [Japanese Patent Publication
20213/1988], whereby it has been regarded as a useful
anti-tumor agent. As can be seen from the following
structure, sarcophytol A is a cembrane type
diterpene-alcohol containing one conjugated double bond and
other two double bonds in the 14-membered ring.
OH
Sarcophytol A
The present inventors had been studied with the
aim of developing a synthetic method of sarcophytol A and
proposed a synthetic route shown by the following synthetic
20~6~5
-- 2 --
route 1 [JP Patent Appln. 181710/1989; filing date: July 14,
19~1 ] .
Reaction Route 1
type reaction ~ t-BuOH
CO2R7
(A) (B)
halogenation reduction
Ho ~\ srulfonYl~ xlJ~! >
CO2R7 esterification CO2R7
(C) (D)
trimethyl-
silyl-
oxidation I I I I nitrile
x~,~1 > x~
OH CHO
(E) (F)
X' ~ ~ CN
(G) oR8 (H)
Iysis ~ re ~ r
(J) sarcophytol A
_ 3 _ 2045~9~
wherein R7 is Cl - C4 lower alkyl group or phenyl group; X1
is a halogen atom or a leaving group such as OS02R9 and the
like; R8 is a hydrogen atom, or trimethylsilyl group or
l-ethoxyethyl group; and R9 is lower alkyl group such as
methyl group or ethyl group, substituted alkyl group such as
trifluoromethyl group, phenyl group or substituted phenyl
group such as toluyl group, mesityl group or the like.
Although the previously proposed method according
to the above synthetic route 1 gives the aimed sarcophytol
A, it has some problems as follows:
1) it requires as the starting material a valuable
compound (A), namely "E,E'-farnesal" of a structure essen-
tial for the production of sarcophytol A;
2) the oxidation of the terminal methyl group of
compound (B) with selenium dioxide is poor in both the
selectivity and yield.
3) the process to prepare the Compound (F) by
reducing compound (D) to Compound (E), and oxidizing the
latter is complicated and inefficient.
Thus, the process shown by the synthetic route 1,
especially that concerned to the production of the interme-
diate (F) from the starting compound (A) is not optimal for
the industrial production of sarcophytol A and more effi-
cient method for preparing the compound (F) has been demand-
ed.
Under these circumstances, the present inventors
have continuously investigated intimately with the aim of
2~45695
-- 4
developing more efficient and simple method for producing
the intermediate ~F), thereby providiny a process applicable
to the industrial production of sarcophytol A, and have now
found that certain novel substituted-acyclic terpene com-
pounds are useful for the establishment of the purpose of
the invention.
DISCLOSURE OF THE INVENTION
The present invention provides acyclic terpene
compounds of the general formula (I):
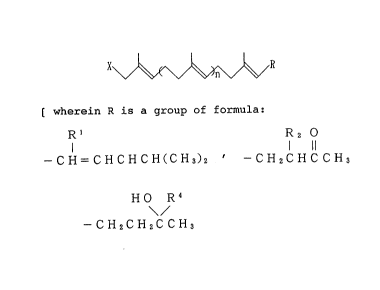
X ~ R
[ wherein R is a group of formula:
Rl R2 0
1 11
--CH=CHCHCH(CH3)2 1 --CH2CHCCH3
\ /
or --CH2CH2C CH3
(wherein Rl is cyano group or formyl group; R2 is a hydrogen
atom or -C02R3; R3 is Cl - C4 alkyl group; R is -C_CH or
-CH=CH2 ); X is a hydrogen atom, hydroxyl group, a halogen
atom, or a group of formula: -oR5 or -OS02R6
(wherein R5 is a hydrogen atom, l-alkoxyalkyl group,
tetrahydrofuryl group, tetrahydropyranyl group or acyl
group,
_ 5 204~ 6~ ~
silyl group substituted with Cl - C5 alkyl group or phenyl
group; R6 is Cl - C4 alkyl group optionally substituted with
halogen atom, or phenyl group optionally substituted with C
- C4 alkyl group); and n is an integer of O to 2 with the
proviso that when R is a group of formula:
R2 0 H O R4
I 11 \/
--CH2CHCCH3 or --CH2CHaCCH3
X must be -oR5 and n must be 0; when R1 is formyl group, X
is not a halogen atom or -OS02R6; when R5 is a hydrogen
atom, R6 is not a hydrogen atom; and when R5 i5
1-ethoxyethyl group, R3 is not a methyl group].
The terms used for the definition of the compound
(I) are explained below.
In the definition of R3, examples of "C1 to C4
lower alkyl group" include a straight or branched alkyl
groups containing 1 to 4 carbon atoms, for example, methyl
group, ethyl group, n-propyl group, isopropyl group, butyl
group, isobutyl group, sec-butyl group, tert-butyl group and
the like.
In the definition of R5, the term "Cl to C5 lower
alkyl group~' refers to the above "Cl to C4 lower alkyl
group" and pentyl group, isopentyl group, neopentyl group
and 1,2-dimethylbutyl group. Examples of "l-alkoxyalkyl
group" include methoxymethyl group, 1-ethoxyethyl group and
the like. Examples of "silyl group substituted with C1 - C5
alkyl group or phenyl group" include trimethylsilyl group,
2~69~
-- 6 --
t-butyldimethylsilyl group, t-butyldiphenylsilyl group and
the like. Examples of llacyl group" include acetyl group,
propionyl group, benzoyl group and the like.
In the definition of R6, the term ~'halogen atom~
include fluorine, chlorine, bromine and the like. Examples
of "Cl - C4 alkyl group optionally substituted with halogen
atom" include methyl group, ethyl group, propyl group,
trifluoromethyl group, trichloromethyl group and the like.
Examples of ~phenyl group optionally substituted with C1 -
C4 alkyl group" include phenyl group, p-tolyl group and the
like.
PREFERRED EMBODIMENT OF THE INVENTION
Typical compounds represented by the general
formula (I) are shown below. However, these are given only
for the illustrative purpose and never restrict the scope of
the invention.
(1) Compound (I) wherein R is
R '
--C H = C H C H C H ( C H 3) 2
204~63~
1) R'=CN, n=O
X~
CN
Compound No. X
--H
2 --0 H
3 - C ~
4 -0 S 02CH3
--0 S 0 2~ C H 3
6 --0 S i ( C H 3 ) 3
-o o
7 \0
8 - 0 C H C H 3 ( 0 ) C 2 H 5
2~4~69~
2) R'=CN, n=l
X~ ~
CN
Compound No X
9 - H
1 0 --OH
1 1 -C~
1 2 --Br
1 3 --O S 02CH3
1 4 -OS02~CH3
1 5 - O C H C H 3 ( O E t)
1 6 --OCH20CH3
7 -~
20~569~
g
3 ) R ' - C N, n = 2
X ~
CN
Compound No X
-
1 8 --H
1 9 --OH
2 0 - C e
2 1 --O S 02CH3
2 2 --O C O C H 3
4) Rl= CHO, n= O
X~
CHO
Compound No. X
2 3 -H
2 4 -OH
2 5 -OSi(CH3)2 C4Hs'
2 6 --O C H C H 3 ( O C 2 H s )
-0,~
- lO 204~69~
5) R'= C H O, n= 1
X ~
CH0
Compound No. X
2 8 - H
2 9 - O H
3 0 - O C H C H 3(0 C 2H s)
3 1 -OCH20CH3
-o o
3 2 ~
3 3 --OCOCH3
3 4 -OCO~
6) R'=CHO. n= 2
X / /~
CH0
Compound No. X
. . . _
3 5 --H
3 6 --OH
3 7 --O C H C H 3 ( O C 2 H 5 )
3 8 - O C O C H 3
11- 20456~
(2) Compound (I) wherein R is R2 o
1 11
C H2C H C C H3
R s o\~
R2
Compound
R2 Rs
NO.
--C 02C H3 --C H20 C H3
2 --C 02C H3 ~]
3 - C 02C H3 --Si(C H3)2C4Hs'
4 --CO2C2Hs --Si(CH3)2C4Hg'
--CO2CH3 --H
6 --C 02C H3 --C O C H3
7 -CO2CH3 --CO~
O C2Hs
8 H
--CHCH3
9 H --C H20 C H3
O~
H ~
11 H -Si(CH3)2C4Hs'
12 H --C O C H3
14 H --CO~)
- 12 - 2~5~5
( 3 ) Compound ( I ) wherein R is \/
-CH2CH2CCH3
HO R4
Rs~ ~
Compound R 4 R 5
No.
O CaHs
1~ --CH=CH2 CHCH3
16 --C H = C H 2 ~
17 --C H = C H2 --C H2O C H 3
18 -CH= CH2 --Si(CH3)2C4H4t
19 --CH= CH2 --H
--C H = C H2 --C O C H 3
21 --CH=CH2 --CO~
O C2Hs
2~ --C - C H C H C H3
23 - C--C H ~
204~69~
_ 13 -
24 --C--C H --C H 2 C H 3
--C--C H --S i ( C H 3 ) 2 C 4 H ~ '
26 --C--- C H --H
27 --C----C H --C O C H 3
28 --C--CH --CO~
Although all the compounds of the formula (I)
including those illustrated in the above are useful as
intermediates for the production of sarcophytol A, there are
certain preferable compounds, that is, for example, those
wherein n is 0 or 1. Especially preferred compounds can be
found among the illustrated ones as follows:
(1): compound Nos. 1, 2, 3, 9, 10, 11, 12, 13, 14,
15, 17, 23, 24, 28, 29, 30, 32 and 33; and
(27: compounds Nos. 2, 6, 8, 10, 12, 15, 16, 19,
20, 22 and 23.
Preparaion of the compound (I) of the present
invention is described below according to the type of the
directed compound.
(1) Compound of general formula (I) wherein R is a
group of formula:
R I
--C H = C H C H C H ( C H 3 ) a
1) Compounds wherein Rl is CN and X is H
2045695
- 14 -
Among the compounds of this t~pe, those wherein n
is 0, 1 or 2 can be prepared from corresponding startin~
materials, that is, those wherein n is O are from geranial,
those wherein n is 1 are from farnesal, and those wherein n
is 2 are from geranyl geranial, by reacting the each
starting material with 1 to 10 mol equivalent of Wittig-
Horner reagent in the presence of less than 1 mol equivalent
of a base (for the Wittig-Horner reagent) in an appropriate
solvent.
The Wittig-Horner reagent which can be used is,
for example, 2-tdimethylphosphono)isovaleronitrile~ 2-
(diethylphosphono)isovaleronitrile, or the like. Generally 1
to 10 mol equivalent of such a reagent is used for the
starting material.
Examples of appropriate solvents include ether
solvents such as tetrahydrofuran (THF), diethyl ether and
the like, hydrocarbon solvents such as benzene, toluene,
n-hexane and the like and aprotic polar solvents such as
dimethylformamide (DMF) and the like. Preferred solvents are
hydrocarbon solvents such as toluene, n-hexane and the like.
Examples of bases include metal hydrides such as
sodium hydride, potassium hydride and the like, organic
metals such as n-butyllithium, lithium diisopropylamide,
lithium-bis-(trimethylsilyl)amide, potassium
bis-(trimethylsilyl)amide and the like, metal alkoxides such
as sodium methoxide, potassium t-butoxide and the like.
Generally, less than 1 mol equivalent of such a base is used
2~456~
- 15 -
for the Wittig-Hornar reagent. In this reaction, it is
possible to control the steric isomerism at the double bond
of the product by selecting the solvent and the base.
The reaction is usually carried out at temperature
from -100 to 100C, preferably from -80 to 50C, more
preferably -70 to 0C.
Each starting compound, when reacted with anion
which generates during the reaction between the compound and
a selected base in the presence of a selected Wittig-Horner
reagent at temperature within the cited range in a selected
solvent, gives the corresponding product. Under these
conditions, the reaction usually completes in ~he period
from 30 minutes to 12 hours.
2) Compound (I) wherein Rl is CHO and X is H
Compounds of this type can be prepared, for
example, by reacting a compound prepared in above 1) with l
to 10 mol equivalent of a metal hydride such as
diisobutylaluminium hydride at temperature from -100 to 150
C in a hydrocarbon solvent such as toluene, n-hexane,
heptane, benzene or the like, which is followed by hydroly-
sis .
3) Compounds (I) wherein X is OH
Compounds of this type can be prepared, forexample, by reacting a compound prepared in above l) or 2)
with an equivalent amount to 50 mol equivalent of t-
butylhydroperoxide in the presence of 0.01 to 0.1 mol
equivalent of selenium dioxide at temperature from -20 to 50
204~59~
- 16 -
c over a period of 1 to 100 hours in a solvent such as
methylenechloride or the like.
4) Compounds (I) wherein R1 is CN and X is a
halogen atom
Compounds of this type can be prepared, for
example, from an alcohol wherein R1 is CN, obtained in above
3), by halogenating said allylic alcohol without allyl
rearrangement. Such a reaction can be carried out by
reacting the alcohol with 1.0 to 10 mol equivalent of carbon
tetrahalide in the presence of 1.0 to 10 mol equivalent of
triphenylphosphine at temperature from room temperature to
100C over a period of 1 to 8 hours in an inert solvent such
as ace onitrile or the like. In case of chlorination~ carbon
tetrachloride can be used as a solvent. Alternatively, it
can be carried out by reacting 1.0 to 10 mol equivalent of
methanesulfonyl chloride together with a metal halide and
y-collidine at temperature from -40C to room temperature
over a period of 1 to 10 hours.
5) Compounds wherein R1 is CN and X is OSO2R6 (R6
is as defined above)
Compounds of this type can be prepared, for
example, by reacting an alcohol wherein R1 is CN obtained in
above 3) with 1.0 to 10 mol equivalent of sulfonyl chloride
such as methanesulfonyl chloride, p-toluenesulfonyl chloride
or sulfonyl anhydride such as trifluoromethanesulfonic
anhydride in the presence of 1.0 to 10 mol equivalent of
amine such as triethylamine, pyridine or the like at
2 ~ 5
- 17 -
temperature from -40C to room temperature over a period of
1 to 10 hours in an ether solvent such as ethyl ether,
tetrahydrofuran or the like or a halogen solvent such as
methylenechloride, chloroform or the like, or pyridine in
case it is used as a base.
6) Compounds wherein X is oR5 (R5 is as defined
above)
a) Compounds wherein R5 is substituted silyl group
Compounds of this type can be prepared by reacting
a compound obtained in above 3) with 0.5 to 10 mol equiva-
lent of a substituted silyl chloride such as
trimethylchlorosilane, t-butyldimethylchlorosilane or the
like in the presence of 0.5 to 10 mol equivalent of a base
such as triethylamine, pyridine, imidazole or the like at
temperature from -50 to 50 C in an ether solvent such as
ethyl ether, THF or the like, an aprotic polar solvent such
as dimethylformamide or the like, a halogen solvent such as
dichlorometahne, chloroform or the like.
b) Compounds wherein R5 is l-alkoxyalkyl group
Compounds of this type can be prepared by reacting
a compound obtained in above 3) with 0.5 to 10 mol equiva-
lent of l-haloalkyl ether such as chloromethylmethyl ether
or chloromethyl-(2-methoxyethyl) ether or the like together
with 0.5 to 10 mol equivalent of a base such as sodium
hydride, potassium hydride, diisopropylamine, triethylamine
or the like at temperature from -50 to 50 in a solvent such
as THF, DMF or the like or without solvent; or with 1 to 10
- 18 - 204~S9~
mol equivalent of l-alkenylalkyl ether such as vinylethyl
ether, dihydropyrane or the like in the presenae of a
catalytic amount to equivalent amount of mineral acid such
as hydrochloric acid, sulfuric acid or the like, an organic
acid such as p-toluenesulfonic acid, camphorsulfonic acid or
the like or a salt such as pyridinium salt of p-
toluenesulfonic acid or the like at temperature from -20 to
100 C in an ether solvent such as diethyl ether, THF or the
like, an ester solvent such as ethyl acetate or the like, or
a halogen solvent such as dichloromethane, chloroform or the
like.
c3 Compounds wherein R5 is acyl group
Compounds of this type can be prepared by reacting
a compound obtained in above 3) with 1 to 10 mol equivalent
of acyl halide such as acetyl chloride, benzoyl chloride or
the like or acid anhydride such as acetic anhydride,
trichloroacetic anhydride or the like in the presence of 1
to 10 mol equivalent of a base such as triethylamine,
pyridine or the like at temperature from -20 to 100 C in a
halogen solvent such as dichloromethane, chloroform or the
like or an ether solvent such as ethyl ether, THF or the
like or a hydrocarbon solvent such as benzene, toluene,
n-hexane or the like, or without solvent where a base serves
as a solvent.
(2) Compound of general formula (I) wherein R is a
group of formula: R 2 o
1 11
--CH2CH C CH3
204~95
- 19 -
1) Compounds (I) wherein R5 is l-alkoxyalkyl group
, tetrahydrofuranyl group or tetrahydropyranyl group, silyl
group substituted with Cl - C5 alkyl group or phenyl group
and R2 is C02R3 (wherein R3 is as defined above)
Compounds of this type can be prepared by substi-
tuting the hydroxyl group at the 8 position of
8-hydroxygeranyl acetate with -oR5 ~R5 is as defined above)
according to either of the following methods.
a) 8-Hydroxygeranyl acetate is reacted with 0.1 to
10 mol equivalent of 1-haloalkyl ether such as
chloromethylmethyl ether, chloromethyl-2-methoxyethyl ether
or the like in the presence of 0.5 to 10 mol equivalent of a
base, for example, a metal hydride such as sodium hydride,
potassium hydride or the like, amines such as
diisopropylamine, triethylamine or the like, or pyridine or
the like at temperature from -20 to +100 over a period of 5
minutes to 24 hours in a halogen solvent such as methylene
chloride, chloroform or the like, an ether solvent such as
diethyl ether, tetrahydrofuran or the like, or ethyl acetate
or dimethylformamide or the like, or without solvent.
b) 8-Hydroxygeranyl acetate is reacted with 0.1 to
10 mol equivalent of vinyl ether such as ethylvinyl ether,
dihydropyrane or the like in the presence of a catalytic
amount to equivalent amount of mineral acid such as hydro-
chloric acid, sulfuric acid or the like, an organic acid
such as p-toluenesulfonic acid, camphorsulfonic acid or the
like, or a salt of a strong acid such as pyridinium salt of
2~4~9~
- 20 -
p-toluenesulfonic acid or the like at temperature from -20
to +100 C in a halogen solvent such as dichloromethane,
chloroform or the like, an ether solvent such as diethyl
ether, tetrahydrofuran or the like, or ethyl acetate or
dimethylformamide or the like, or without solvent.
c) 8-Hydroxygeranyl acetate is reacted with 0.1 to
10 mol equivalent of trialkylsilyl halide such as trimethyl-
silyl chloride, t-butyldimethylsilyl chloride or the like in
the presence of 0.1 to 10 mol equivalent of a base such as
nitrogen-containing compound such as triethylamine,
dimethylaminopyridine, imidazole or the like, or metal
hydride such as sodium hydride, potassium hydride or the
like at temperature from -20 to +100 C over a period of 5
minutes to 24 hours in a halogen solvent such as methylene
chloride, chloroform or the like, a hydrocarbon solvent such
as hexane, benzene or the like, an ether solvent such as
diethyl ether, tetrahydrofuran or the like, or ethyl ace-
tate, dimethylformamide, dimethyl sulfoxide or the like.
The resultant compound, when reacted, for example,
with an alkali metal salt of acetoacetic ester, gives the
desired compound. Thus, the compound is reacted with an
alkali metal salt of an acetoacetic ester at temperature
from -70 to +100C over a period of 30 minutes to 48 hours
in an aprotic polar solvent such as diethyl ether,
tetrahydrofuran, dimethylformamide, dimethyl sulfoxide or
the like to give the directed compound, where the alkali
metal salt of an acetoacetic ester can be prepared by
2 ~ 9 ~
- 21 -
reacting an acetoacetic ester such as ethyl acetoacetate,
ethyl acetoacetate or the like with a metal hydride such as
sodium hydride or the like, or a strong base such as n-
butyllithium, lithium diisopropylamine or the like in the
presence of a palladium (o) chelate such as
tetrakis(triphenylphosphine) palladium and the like as a
catalyst at temperature from -70 to +100C in an aprotic
polar solvent such as diethyl ether, tetrahydrofuran,
dimethylformamide, dimethyl sulfoxide or the like.
2) Compounds (I) wherein R5 is H and R2 is Co2R3
(R3 is as defined above)
Compounds of this type can be prepared by reacting
a compound obtained in above 1) with 0.1 to 10 mol equiva-
lent of a mineral acid such as hydrochloric acid, sulfuric
acid or the like, an organic strong acid such as p-
toluenesulfonic acid or the like or a salt of a strong acid
such as pyridinium salt of p-toluenesulfonic acid or the
like in an alcohol solvent such as methanol, ethanol or a
the like or water, or a mixed solvent thereof. Alterna-
tively, it can be prepared by reacting the compound obtained
in 1) with 0.1 to 10 mol equivalent of tetraalkylammonium
fluoride such as tetrabutylammonium fluoride or hydrogen
fluoride in a protonic polar solvent such as methanol,
ethanol, water or the like, an ether solvent a~ diethyl
ether, tetrahydrofuran or the like, or a mixed solvent
thereof.
2~569~
- 2~ -
3) Compounds (I) wherein R5 is acyl group and R
is Co2R3 (R3 is as defined above)
Compounds of this type can be prepared by reacting
a compound obtained in above 2) with 0.1 to 10 mol equiva-
lent of acyl halide such as acetyl chloride, benzoyl chlo-
ride or the like or acid anhydride such as acetic anhydride
or the like in the presence of 0.1 to 10 mol equivalent of a
base such as triethylamine, pyridine or the like at tempera-
ture from -20 to +100 C in a halogen solvent such as
dichloromethane or the like, an ether solvent such as
diethyl ether or the like, a hydrocarbon solvent such as
benzene, n-hexane or the like, or without solvent where a
base serves as a solvent.
4) Compounds (I) wherein R5 is a hydrogen atom,
1-alkoxyalkyl group, tetrahydrofuranyl group,
tetrahydropyranyl group, silyl group substituted with Cl
C5 alkyl group or phenyl group or acyl group and R2 is a
hydrogen atom
Compounds of this type can be prepared through the
decarboxylation or decarboalkoxylation of a compound
obtained in above 1), 2) or 3). The decarboxylation can be
carried out by reacting said compound with 0.1 to 10 mol
equivalent of a metal hydroxide such as sodium hydroxide,
potassium hydroxide or the like, metal alkoxide such as
sodium methoxide or the like at temperature from 0 to 100C
over a period of 10 minutes to 24 hours for the hydrolysis
2~6~
- 23 -
or ester-exchanging reaction, and heating at temperature
from 100 to 250C over a period of 30 minutes to 10 hours.
The decarboalkoxylation which is carried out by reacting a
compound with 0.1 to 10 mol equivalent of a metal halide
such as sodium chloride, sodium iodide or the like at
temperature from 50 to 250C in an aprotic polar solvent
such as dimethylformamider dimethylsulfoxide or the like.
(3) Compounds (I) wherein ~ is a group of formula:
\ /
- C H2C H2C C H3
1) Compounds (I) wherein R5 is a hydrogen atom,
1-alkoxyalkyl, tetrahydrofuranyl or tetrahydropyranyl group,
or silyl group substituted with C1 - C5 al y g p
phenyl group and R is -CH=CH2
Compounds of this type can be preparcd by sub~ect-
ing a compound obtained in (2), 4) to an addition reaction,
which is conducted by reacting said compound with 0.1 to 10
mol equivalent of a vinyl anion such as vinyl lithium, vinyl
magnesium bromide or the like at temperature from -50 to 100
C over a period of 30 minutes to 48 hours in an ether
solvent such as diethyl etherr tetrahydrofuran or the like
or a hydrocarbon solvent such as n-hexane , benzene or the
like.
2) Compounds (I) wherein R5 is a hydrogen atom,
1-alkoxyalkyl group, tetrahydrofuranyl group,
tetrahydropyranyl group or silyl group substituted with Cl -
C5 alkyl group or phenyl and R4 is -C-CH
2~4~S9~
- 24 -
Compounds of this type can be prepared by subject-
ing a compound obtained in above (2), 4) to an addition
reaction which is conducted by reacting said compound with
0.1 to 10 mol equivalent of a metal acetylide such as
lithium acetylide, ethynyl magnesium bromide or the like at
temperature from -50 to +100 C over a period of 30 minutes
to 48 hours in an ether solvent such as diethyl ether,
tetrahydrofuran or the like or a hydrocarbon solvent such as
n-hexane, benzene or the like.
3) Compounds (I) wherein R5 is acyl group and R4
is a group of -CH=CH2 or -C-CH
Compounds of this type can be prepared by reacting
a compound obtained in above ~3),1) or 2), in which R5 is a
hydrogen atom and R4 is -CH=CH2 or -C-CH, with 0.1 to 10 mol
equivalent of acyl halide such as acetyl chloride, benzoyl
chloride or the like or acid anhydride such as acetic
anhydride or the like in the presence of 0.1 to 10 mol
equivalent of a base such as triethylamine, pyridine or the
like at temperature from -20 to +100 C in a halogen solvent
such as dichloromethane or the like, an ether solvent such
as diethyl ether or the like, a hydrocarbon solvent such as
benzene, n-hexane or the like, or without solvent where a
base serves as a solvent.
The above are examples of preferred procedures for
the production of the compounds of formula (I) of the
invention. As one of skill will appreciate, the present
invention is not restricted to the compounds (I) produced
- 25 _ 20~5~
by the above methods, but include any compounds of formula
(I) prepared by other methods known to the art.
As mentioned above, the present invention makes it
possible to obtain Compound (F), the key intermediate in the
synthetic route 1 for the production of sarcophytol A, from
a monoterpenoid which is cheap and easy to obtain by an
improved and efficient process with avoiding the oxidation
of alcohol to aldehyde of the previously provided method,
reducing the total steps, and in high yield.
Thus, the present invention provides an industri-
ally advantageous synthetic route for preparing sarcophytol
A.
Typical procedures for the production of the
intermediate (F) in the synthetic route 1 from various
compounds ~I) of the invention as the starting material, and
that for the production of the final product, sarcophytol A,
will hereinafter be described.
(1) When the starting material is a compound (I)
wherein X is a halogen atom, n is 1 and R is a group of
formula: C N
I
_ r ~ - r ~ r T7 ~ TT ~ ~ TT ~
v ~ n ~, n ~ ~, n 3 J 2
Compound (F) in the above synthetic route 1 can be
prepared by reacting a compound (I) as defined above with
0.1 to 10 mol equivalent of diisobutylaluminium hydride in a
solvent such as toluene, benzene, n-hexane or the like at
temperature from -100 to 100 C and hydrolyzing the product.
2~4~6~
- 26 -
(2) When the starting material is Compound (I') which
is shown by the formula (I) wherein R5 is l-alkoxyalkyl
group and R is a group of formula:
HO C--CH
--CH2CH2C CH3
or Compound (I") which is shown by the formula (I) wherein
R2 is l-alkoxyalkyl group and R is a group of formula:
HO C--CH
\/
--C H2C H2C C H3
~ 27 _ 2~4~
Svnthetic Route 2 HO CH=CH2
R50 J ~ ~ oxidation
HO C-CH
R50 ~ ~ \/ R50 ~ ~ ~ CHO >
\~\ . ~ , \~/
eplmerlzatlon
(I') (A')
Rs~ > Rs~ >
CN CHO
(B') (C')
HO~ > X ~ >
(E~ ) CHO (F) CHO
a process of JA181710/1989 ~ ~ ~
> ~, ~OH
sarcophytol A
(wherein R5 and X are as defined above).
a) When the starting material is Compound tI')
Compound (A') can be prepared by subjecting the
Compound (I') to rearrangement reaction in the presence of
0.01 to 1 mol equivalent of silylvanadate such as
tris(triphenylsilyl)vanadate, poly{(diphenylsilyl)vanadate~
2~4~9~
- 28 -
or the like at temperature from 100 to 300C over a period
of 30 minutes to 24 hours in a hydrocarbon solvent ~uch as
undecane, xylene or the like, an ether solvent such as
bis[2-(2-methoxyethoxy~-ethyl] ether or a mineral oil.
b) When the starting material is Compound (I")
An unsaturated aldehyde (A') can be prepared by
reacting the Compound (I") with chromic oxides such as
pyridinium chlorochromate or the like at temperature from 0
to 100C over a period of 30 minutes to 24 hours in a
halogen solvent such as methylene chlorider chloroform or
the like, a hydrocarbon solvent such as n-hexane, banzene or
the like, or dimethylformamide or the like.
c) Compound (B') can be prepared by reacting
Compound (A') with 0.1 to 10 mol equivalent of Wittig-Horner
reagent such as 2-(dimthylphosphono)isovaleronitrile,
2-(diethylphosphono)isovaleronitrile or the like in an ether
solvent such as tetrahydrofuran, diethyl ether or the like,
a hydrocarbon solvent such as toluene r n-hexane or the like
or an aprotic polar solvent such as dimethylformamide,
dimethyl sulfoxide or the like at temperature from -100 to
+100C, in the presence of less than 1 mol equivalent (for
the Wittig-Horner reagent) of a base, for example, metal
hydride such as sodium hydride, potassium hydride or the
like, organic metal (e.g. n-butyllithium, lithium
diisopropylamide) or metal alkoxide such as sodium
methoxide, potassium t-butoxide or the like while allowing
to react Compound (A') with a generated anion.
2~4~g9~
- 29 -
Aldehyde (C') is prepared by reacting Compound
(B') with 0.1 to 10 mol equivalent of a metal hydride
complex such as lithium aluminum hydride or the like at
temperature from -70 to +100C in an ether solvent such as
diethyl ether, tetrahydrofuran or the like or reacting with
0.1 to lO mol equivalent of a metal hydride such as
diisobutylaluminium hydride or the like at temperature from
-70 to +100C over a period of 5 minutes to 5 hours in a
hydrocarbon solvent such as n-hexane, benzene or the like.
~ ldehyde (C') is converted into alcohol (E') when
treated with 0.1 to 10 mol equivalent of a mineral acid such
as hydrochloric acid, sulfuric acid or the like, an organic
strong acid such as p-toluenesulfonic acid or a salt of a
strong acid such as a pyridinium salt of p-toluenesulfonic
acid or the like in a solvent such as aqueous methanol,
aqueous ethanol, aqueous tetrahydrofuran, or a mixed solvent
thereof.
Compound (F) can be prepared from Compound (E') by
halogenating the allylic alcoholic without allyl rearrange-
ment. For example, Compound (E') is reacted with 0.1 to 10
mol equivalent of carbon tetrahalide in the presence of 1.0
to 10 mol equivalent of triphenylphosphine in a solvent such
as acetonitrile, dichloromethane or the like, in case of
chlorination, with carbon tetrachloride without solvent, at
temperature from -10 to +100C over a period of 10 minutes
to 12 hours. Alternatively, Compound (E') is reacted with
0.1 to 10 mol equivalent of sulfonyl halide such as
20~G95
- 30 -
methanesulfonyl chloride, p-toluenesulfonyl chloride or the
like together with a metal halide such as lithium chloride
in the presence of an amine such as pyridine, y-collidine,
lutidine or the like at temperature from -40 to +30C over a
period of 1 to 12 hours.
The final product, sarcophytol A, can be prepared
be treating Compound (F) according to the procedure shown by
the synthetic route 1 as shown below.
Process of the svnthetic route 1
a) Preparation of Compound (G)
Thus, Compound (G) whexein R8 is trimethylsilyl
group is prepared, for example, by treating Compound (F~
with 1.0 to 10 mol equivalent of trimethylsilylnitrile in
the presence of a catalytic amount of metal cyanide 18-
crown-6-ether complex, an ammonium salt such as
tetraalkylammonium cyanide or the like at temperature from
-20 to 50C over a period of 30 minutes to 5 hours in a
solvent such as methylene chloride, chloroform, ethyl
acetate or the like, or without solvent. The resultant
product can be converted into cyanohydrin wherein R8 is
hydrogen by treating with 0.1 - 3N aqueous mineral acid such
as hydrochloric acid, sulfuric acid or the like at 0C to
room temperature over a period of 5 minutes to 5 hours or by
treating with a catalytic amount to 10 mol equivalent of
tetraalkylammonium salt such as tetrabutylammonium fluoride
or the like at temperature from -20C to room temperature in
a solvent such as tetrahydrofuran, dioxane or the like.
- 31 - 2a4~69~
Compound (G) wherein R8 is 1-ethoxyethyl group can
be prepared by reacting the above cyanohydrin with 1.0 to 10
mol equivalent of ethyl vinyl ether in the presence of a
catalytic amount of mineral acid such as hydrochloric acid,
sulfuric acid or the like, an organic strong acid such as
p-toluenesulfonic acid or the like, or a salt of strong acid
such as p-toluenesulfonic acid or the like pyridinium salt
at temperature from -20C to room temperature over a period
of 30 minutes to 5 hours in a solvent such as ethyl ether,
ethyl acetate or the like.
b) Preparation of Compound (H~
Compound (H) wherein R8 is trimethylsilyl or
l-ethoxyethyl group can be prepared by reacting Compound (G)
wherein R8 is trimethylsilyl group or l-ethoxyethyl group
with 1.0 to 10 mol equivalent of a base such as lithium
diisopropylamide, lithium bis-(trimethylsilyl) amide, sodium
hydride or the like at temperature from -70 to 100C over a
period of 5 minutes to 10 hours in an ether solvent such as
ethyl ether, tetrahydrofuran or the like, an aromatic
hydrocarbon solvent such as benzene, toluene or the like or
a saturated hydrocarbon solvent such as n-hexane, n-heptane
or the like.
Compound (H) wherein R8 is a hydrogen atom is
prepared by treating the compound obtained above with 0.1 -
3N aqueous mineral acid such as hydrochloric acid, sulfuric
acid or the like at temperature from 0C to room temperature
over a period of 5 minutes to 5 hours in a solvent such as
2~69~
- 32 -
tetrahydrofuran, methanol or the like or by treating with a
catalytic amount to 10 mol equivalent of tetraalkylammonium
salt such as tetrabutylammonium fluoride at temperature from
-20C to room temperature in a solvent such as tetrahydro-
furan, dioxane or the like.
c) Preparation of a ketone, Compound (J)
The ketone (J) can be prepared by treating a
solution of Compound (H) wherein R8 is a hydrogen atom in an
organic solvent such as ethyl ether, ethyl acetate or the
like with aqueous sodium bicarbonate at temperature from 0C
to room temperature over a period of 5 minutes to 5 hours,
or by treating Compound H wherein R8 is trimethylsilyl group
with a catalytic amount to 10 mol equivalent of an
alkylammonium fluoride such as tetrabutylammonium fluoride
in a solvent such as aqueous tetrahydrofuran, dioxane or the
like.
d) Preparation of sarcophytol A
Sarcophytol A can be prepared by reacting the
ketone (J) thus obtained with 1.0 to 10 mol equivalent of a
metal hydride such as diisobutylaluminum hydride or the like
or a metal complex such as lithium aluminum hydride or the
like at temperature from -70 to 50C over a period of 5
minutes to 5 hours in an ether solvent such as ethyl ether,
tetrahydrofuran or the like, an aromatic hydrocarbon solvent
such as benzene, toluene or the like or a saturated hydro-
carbon solvent such as n-hexane, n-heptane or the like.
204~9~
- 33 -
Further, sarcophytol A in native form shown below
is prepared by subjecting ketone Compound (J) to asymmetric
reduction with an asymmetrically-modified metal hydride or
metal hydride complex.
~ ~ OH
~1
Sarcophytol A in native form
Examples of asymmetrically-modifying reagents used
for preparing asymmetrically-modified metal hydride or metal
hydride complex, which are used in the asymmetric reduction,
include asymmetric amino alcohols prepared by converting
carboxy group of optically-active amino acid such as L- or
D-proline, valine or the like into substituted alcohol group
or substituted amino group [Bull. Soc.Chim.Belq. 97: 691
(1988); J. Chem. Soc. Perkin I 1673: (1983)]; asymmetric
diamines [Bull. Chem. Soc. Japan 51: 1869 (1978);
Tetrahedron 37: 4111 (1981)], asymmetric alkaloids such as
L- or D-methylephedrine and the like [Chem.Pharm.Bull. 31:
837 (1983)]; and (S)- or (R)-l,l'-bis-2-naphtol and the
like.
Examples of metal hydrides or metal hydride
complexes include diisobutylaluminium hydride, lithium
aluminium hydride, sodium borohydride and the like. An
asymmetric reducing reagent can be prepared by reacting a
2~4~6~
- 34 -
metal hydride or metal hydride complex with 0.1 to S mol
equivalent, preferably 0.5 to 1.5 mol equivalent of the
above-mentioned asymmetrically-modifying reagent, optionally
in the presence of an additive such as alkyl-substituted
aniline, substituted aminopyridine, stannous chloride or the
like at temperature from -50 to 50C, preferably from -20C
to room temperature over a period of 10 minutes to 5 hours
in an appropriate solvent to obtain a coordinated complex of
said asymmetrically-modifying reagent and metal hydride or
metal hydride complex. Examples of appropriate solvents
include ether solvents such as diethyl ether,
tetrahydrofuran and the like and hydrocarbon solvents such
as benzene, toluene, n-hexane and the like. A halogen
solvent such as dichloromethane and chloroform is also
available in case metal hydride is used. Illustrative
combinations are listed in the Table 1 below.
204~69~
- 35 -
Table 1
_
metal hydride axymmetric
or metal modifying additive
hydride compl~ x reagent
O H
L iA lH 4 Ph ~ HsC 2 N
NMe2
~
H A l(i- B u)2 A ~ ~
(D I B A L) ~ N ~ S nCl2
. ~
~ B H 9 \;
20~69~
- 36 -
Although the amount of the asymmetric reducing
reagent to be reacted with the macrocyclic ketone ~hown by
the structure (J) is not critical, it is preferable to u~e 1
to 2 mol equivalent of asymmetric reducing reagent for the
ketone considering the recovery of un-reacted starting
materials and yield of the product. The reaction is usually
conducted at temperature from -150 to 100C, preferably from
-100C to room temperature over a period of 10 minutes to 5
hours in the same solvent as that used for ~he preparation -
of the asymmetric reducing reagent. No regularity can be
found between the absolute configuration of the product
~arcophytol A ( its native form is expressed by IR and
non-native form IS as shown below) and that of the asymmet-
ric reducing reagent, which i8 attributable to the original
compound in L- or D-form. The absolute configuration of the
product varies depending on the combination of the asymmet-
ric reducing reagent and metal hydride or metal hydride
complex.
The by-product of the present method, sarcophytol
A in non-native form of formula:
~ ~~
-OH
J (I ~)
, when subjected to the conventional epimerization reaction
2 ~ ~ 5 S ~ ~
- 37 -
for hydroxyl group, easily gives the optically-active
sarcophytol A (Is) in native form after the inversion.
(3) When the starting material is a compound of
formula (I) wherein n is 1 and R i8 a group of formula:
R I
- C H = C H C H C H ( C H 3) 2
wherein R1 is as defined above.
- 38 - 2(~ i6~5
Synthetic Route 3
CHO (K)
epoxidation
(L)
Ae(O-C3H7)3 )~
OH (M) OH
sulfonylesterification
(N) OR~
etherification ~ )
J
(O)
[2.3]Wittig rearrangement ~
OH
sarcophytol A
20~69~
- 39 -
R : substituted sulfonyl group such as methanesulfonyl group
or p-toluenesulfonyl group.
Compound ~K) in the above synthetic route 3 can be
prepared by, for example, reacting a compound of formula (I)
wherein n is 1, X is a hydrogen atom and R is a group:
C H 0
- C H = C H C H C H (C H3)2
with either of 1.0 to 10 mol equivalent of metal hydride
such as dibutylaluminium hydride or the like, or a metal
complex such as lithium aluminium hydride or the like at
temperature from -70 to 50 C over a period of 5 minutes to
5 hours in a ether solvent such as ethyl ether,
tetrahydrofuran or the like, an aromatic hydrocarbon solvent
such as benzene, toluene or the like or a saturated hydro-
carbon solvent such as n-hexane, n-heptane or the like, or
with 0.5 to 10 mol equivalent of metal hydride complex such
as sodium borohydride or the like at temperature -70 to
100C in a solvent such as methanol, ethanol or the like.
The resulting Compound (K) is converted into
Compound (L) through the epoxidation which is conducted by
halogenating the Compound K with 0.1 to 1 mol equivalent of
a halogenating agent such as N-bromosuccinimide, N-
chlorosuccinimide or the like at temperature from -20 to
100C over a period of 30 minutes to 5 hours in an aqueous
solvent of a water-miscible solvent such tetrahydrofuran,
dimethoxyethane or the like, followed by treating with an
aqueous solution of a base such as sodium hydroxide,
2~Gg~
- 40 -
potassium hydroxide, sodium carbonate or the like, or after
the halogenation, isolating halohydrin, and treating it with
a base such as sodium carbonate, sodium methoxide or the
like in a solvent such as methanol or tetrahydrofuran or the
like; or by treating with 0.1 to 1 mol equivalent of organic
peracid such as m-chloroperben~oic acid, peracetic acid or
the like at temperature from -50 to 50C over a period of 30
minutes to 10 hours.
Compound (M) can be prepared by reacting Compound
(L) with 0.1 to 10 mol equivalent of metal alkoxide such as
aluminium triisopropoxide or the like at temperature from 50
to 200 C in a solvent such as toluene, xylene or the like;
or with 0.1 to 10 mol equivalent of a metal amide such as
lithium diisopropylamide, lithium diethylamide or the like
at temperature from -70 to 100 C in a solvent such as
diethyl ether, tetrahydrofuran or the like.
Compound (N) can be prepared by the
sulfonylesterification of the diallyl alcohol (M). For
example, Compound (M) is reacted with 0.1 to 1.5 mol equiva-
lent of a substituted sulfonyl chloride such as
methanesulfonyl chloride, p-toluenesulfonyl chloride or the
like in the presence of 0.1 to 10 mol equivalent of a base
such as triethylamine, pyridine or the like at temperature
from -70 to 100 C in a halogen solvent such as
dichloromethane, chloroform or the like or a ether solvent
such as diethyl ether, tetrahydrofuran or the like.
20~69~
- 41 -
Compound (0) can be prepared, for example, by
treating Compound (N) with 0.1 to 10 mol equivalent of metal
hydride such as sodium hydride, potassium hydride or the
like or organic metal such as n-butyllithium, ethyl magnesi-
um chloride or the like at temperature from -50 to 150 C in
an ether solvent such as diethyl ether, tetrahydrofuran or
the like, a hydrocarbon solvent such as benzene, toluene,
n-hexane or the like or an aprotonic polar solvent such as
dimethylformamide, dimethyl sulfoxide or the like.
The resultan~ Compound (o) can be converted into
sarcophytol A by reacting said Compound (o) with 0.1 to 10
mol equivalent of organic metal such as n-butyl lithium,
sec-butyl lithium, lithium diisopropyl amide or the like at
temperature from -100 to 100 C in an ether solvent such as
diethyl ether, tetrahydrofuran or the like, a hydrocarbon
solvent such as benzenel toluene, n-hexane or the like, or
further adding hexamethylphosphoric triamide or the like to
the solvent.
(4) When the starting material is a compound of
formula I wherein n is 0 and R is a group of formula:
1 1
- C H = C H C H C H (C H 3)2
wherein Rl is as defined above.
204~fi9~
- 42 -
Synthetic route 4
CN CN
(A")
epoxidation ~ Claisen Rearrangement
> /~/\
OH CN
(B")
OHC ~ Wittig Reaction
CN
(C")
~ H
CO2CH3 CN
(D")
HO ~ PPh3, CCe4
CHO
(E")
CB ~ a route of JA 181710/1989
CHO
(F)
OH
Sarcophytol A
20~6~
- 43 -
Compound (A") in the above synthetic route 4 can
be prepared from a compound (I) wherein R1 is -CN, n is 0
and X is a hydrogen atom as mentioned above through
epoxidation as follows. Thus, Compound (A") i9 prepared by
halogenating a compound (I) with 0.1 to 1 mol equivalent of
a halogenating agent such as N-bromosuccinimide, N-
chlorosuccinimide or the like at temperature from -50 to
50C over a period of 30 minutes to 5 hours in an aqueous
solvent of a water-miscible solvent such tetrahydrofuran,
dimethoxyethane or the like, followed by treating with an
aqueous solution of a base such as sodium hydroxide, potas-
sium hydroxide, sodium carbonate or the like, or after the
halogenation, separating halohydrine, and reducing it with a
base such as sodium carbonate, sodium methoxide or the like
in a solvent such as methanol or tetrahydrofuran or the
like; or by treating the compound with 0.1 to 1 mol equiva-
lent of organic peracid such as m-chloroperbenzoic acid,
peracetic acid or the like at temperature from -50 to 50C
over a period of 30 minutes to 10 hours.
Compound (B") is prepared by treating the above
epoxy compound (A") with 0.1 to 10 mol equivalent of metal
alkoxide such as aluminium triisopropoxide or the like at
temperature from 50 to 200 C in a solvent such as toluene,
xylene or the like; or with 0.1 to 10 mol equivalent of a
metal amide such as lithium diisopropylamide, lithium
diethylamide or the like at temperature from -70 to 100 C
204569~
- 44 -
in a solvent such as diethyl ether, tetrahydrofuran or the
like.
~ he aldehyde compound (C") can be prepared by, for
example, through the Claisen rearrangement, which is con-
ducted by reacting Compound (B") with 1.0 to 100 mol equiva-
lent of alkyl vinyl ether such as ethyl vinyl ether or the
like in the presence of 0.1 to 5 mol equivalent of a mercury
salt such as mercury acetate or the like at temperature from
0 to 100 C to give the vinyl ether of Compound (s") or
leading said Compound (B") to 3-alkoxyacrylic acid according
to a known method [J. Orq. Chem., 48: 5406 (1983)], ~ollowed
by heating at temperature from 100 to 250 C in the presence
of a catalytic amount of hydroquinone in each case.
Compound (D") is prepared by reacting the aldehyde
(C") with 0.5 to 5 mol equivalent of Wittig reagent such as
carbomethoxyethylidene triphenylphosphorane or the like or
an anion made from Wittig-Horner reagent such as ethyl 2
-(diethylphosphono)propionate, ethyl 2-(dimethylphos-
phono)propionate or the like at temperature from -50 to
100C in a solvent such as diethyl ether, THF, DMF,
dichloromethane or the like.
Compound (D"), when treated with 0.5 to 10 mol
equivalent of metal hydride complex such as lithium
aluminium hydride or the like at temperature from -70 to 100
C in an ether solvent such as diethyl ether, THF or the
like or with 0.5 to 10 mol equivalent of metal hydride such
as dibutylaluminium hydride or the like at temperature from
204~G9~
- 45 -
-70 to 100 C in a hydrocarbon solvent such as benzene,
toluene, n-hexane, n-heptane or the like, gives Compound
(E~), which is a compound o~ formula I wherein n is 1, X is
hydroxyl
group and ~ is a group of formula:
C H O
- C H = C H C H C H(C H 3)2
, which is the same as Compound (E~) in the synthetic route
2.
Compound (F) can be prepared from Compound (E") by
halogenating the allylic alcoholic without allyl rearrange-
ment as previously described in the synthetic route 2.
Compound (E"), when treated in the same manner as mentioned
above, gives sarcophytol A.
As can be seen from the above, sarcophytol A can
be prepared effectively from the compound (I) of the inven-
tion through various processes using or without using the
intermediate F, which demonstrates that the compound (I) is
highly useful and important for the attainment of the
purpose of the invention.
Following Examples are provided for purposes of
illustration only and are not to be construed as limiting
the scope of the instant invention in any way.
PreParation 1
HO~ THPOJ~
OCOCH 3 OCOCH 3
2~69~
- 46 -
A mixture of 8-acetox~-2,6-dimethyl-2,6-octadien-
1-ol (1.81 g, 8.52 mmol) and dihydropyran (1.17 ml, 12.8
mmol) in dichloromethane (6 ml) was stirred, and
p-toluenesulfonic acid (40 mg) was added thereto, and the
mixture was stirred at room temperature for 30 minutes.
After addition of saturated aqueous sodium bicarbonate (30
ml), the product was extracted with hexane/ether (5:1.2) (30
ml). The extract was dried over Na2S04 and evaporated in
vacuo to remove the solvent to give a residue, which was
purified with silica gel column chromatography to give
l-acetoxy-8-(2-tetrahydropyranyl)oxy-3,7-dimethyl-2,6-octad-
iene (2.42 g, 96%~.
Pre~aration 2
HO ~ > H3COH2CO ~
OCOCH3 OCOCH3
A mixture of 8-acetoxy-2,6-dimethyl-2,6-
octadien-l-ol (110 mg, 0.52 mmol) and triethylamine (0.25
ml, 1.83 mmol) and chloromethyl methyl ether (0.069 ml, 0.92
mmol) in acetonitrile (2 ml) was refluxed with stirring for
4 hours. After addition of water (3 ml) to the reaction
mixture, the product was extracted several times with ether
(5 ml). The extract was dried over Na2S04 and evaporated in
vacuo to remove the solvent to give a residue, which was
then sub~ected to silica gel column chromatography to give
the aimed l-acetoxy-8-(2-methoxymethyl)oxy-3,7-dimethyl-2,6-
octadiene (109 mg, 82~).
20~6~5
- ~7 -
Preparation 3
CH3
HO~OCOCH3 C4H9 '-SiO~V OCOCH3
8-Acetoxy-2,6-dimethyl-2,6 octadien-l-ol in
dimethylformamide (4 ml) was stirred on an ice bath. To the
solution were added imidazole (338 mg, 4.96 mmol) and
chlorodimethyl t-butylsilane (410 mg, 2.73 mmol), and the
mixture was stirred at room temperature for one hour. After
addition of water ~30 ml) to the reaction mixture, the
product was extracted with hexane (20 mlx2). The extract
was dried over MgSO4 and evaporated in vacuo to remove the
solvent to give a residue, which was subjected to silica gel
column chromatography to obtain l-acetoxy-8-(dimethyl
t-butylsilyl)oxy-3,7-dimethyl-2,6-octadiene (606 mg, 75%).
Exam~le 1
0~ ~ 0~
OCOCH3 ~/
CO2CH3
To a solution of 1-acetoxy-8-(2-
tetrahydropyranyl)oxy-3,7-dimethyl-2,6-octadiene (1.08 g,
3.64 mmol) in tetrahydrofuran (6 ml) were added under
nitrogen atmosphere triphenylphosphine (105 mg, 0.4 mmol)
and tetrakis(triphenylphosphine)paradium (168 mg, 0.15
mmol), and the mixture was stirred at room temperature for
15 minutes. To the mixture were added a sodium salt of
2 0 ~ ~ 6 9 ~
- 48 -
methyl acetoacetate in tetrahydrofuran (25 ml) which has
been prepared from sodium hydride (305 mg, 12.7 mmol) and
methyl acetoacetate (1.57 ml, 14.6 mmol), and the mixture
was refluxed for 5 hours. After addition of ~ater (10 ml)
and ether (30 ml), the reaction mixture was stirred well,
and the organic layer was separated. The aqueous layer was
extracted with ether (5 ml), and the extract was dried over
Na2SO4 and evaporated in vacuo to remove the solvent to give
a residue, which was then subjected to silica gel column
chromatography to obtain purified methyl
2-acetyl-5,9-dimethyl-10-(2-tetrahydropyranyl)oxy-4,8-decad-
ienate (1.06 g, 83~).
IR(film)cm 1; 2950, 2870, 1750, 1722, 1440, 1360, 1201,
1150, 1022.
NMR(CDC13, 250MHz)~ppm; 1.45-1.92(m, 6H,
C(O)H2-Ca2-CH2-CH2-CHO-), 1.63, 1.65(2s, 6H, 2x-CH3C=CH-),
1.94-2.15(m, 4H, -C=CH-CH2-CH2-C=CH-), 2.22(s, 3H, CH3C=O),
3.46(t, J=7.5Hz, lH, -CHCO2-), 3.53(m, lH, -CH2-CHaHb-O-),
3.73(s, 3H, CO2CH3), 3-83, 4-09(2d~ J=11-8Hz~ 2H~
-OCa2C=CH-), 3.82-3.94(m, lH, -CH2-CHaHb-O-), 4.60(t,
J=3.4Hz, lH, -OCHO-), 5.04(t, J=7.3Hz, lH, -C=CH-CH2-),
5.38(t, J=6.2Hz, lH, -C=CH-CH2-).
ExamPle 2
o
H3COH2CO~ H3COH2CO ~
OCOCH 3 C 2CH 3
20~69~
- 49 -
To a solution of l-acetoxy-8-(methoxymethyl)oxy-
3,7-dimethyl-2,6-octadiene t600 mg, 2.34 mmol) in
tetrahydrofuran (6 ml) were added under nitrogen atmosphere
triphenylphosphine (60 mg, 23 mmol) and
tetrakis(triphenylphosphine)paradium ~108 mg, 0.09 mmol),
and the mixture was stirred at room temperature for 15
minutes. To the mixture were added a sodium salt of methyl
acetoacetate in tetrahydrofuran (25 ml) which has been
prepared from sodium hydride (225 mg, 9.36 mmol) and methyl
acetoacetate (1.26 ml, 11.7 mmol), and the mixture was
refluxed for 2 hours. After addition of water (40 ml) and
ether (50 ml), the reaction mixture was stirred well, and
the organic layer was separated. The aqueous layer was
extracted with ether (50 ml), and the extract was dried over
Na2SO4 and evaporated in vacuo to remove the solvent to give
a residue, which was then subjected to silica gel column
chromatography to obtain purified methyl
2-acetyl-5,9-dimethyl-10-(methoxymethyl~oxy-4,8-decadienate
(670 mg, 92%).
IR(film)cm 1; 2930, 1745, 1720, 1438, 1355, 1208, 1150,
1100, 1040, 920.
lH NMR(CDC13, 250MHz)~ppm; 1.63, 1.65(2s, 6H, CH3C=CH-x2),
1.93-2.17(m, 4H, -C=CH-CH2-CH2-C=CH-), 2.22( 6, 3H, COCH3),
2.56(t, J=7.4Hz, 2H, -C=CH-CH2-CH(CO2CH3)-), 3.38(s, 3H,
CH3OCH2O-), 3.46(t, J=7.SHz, lH, CH(CO2CH3)), 3.73(s, 3H,
CO2CH3), 3.92(~, 2H, -OCH2C=CH-), 4.61(s, 2H, -OCH2O-),
2045695
- sn -
5.04(t, J=7.3Hz, lH, -C=CH-CH2-), 5.38(t, J-6.7Hz, lH,
-C=CH-CH2- ) -
ExamPle 3
CH3 CH3 0
C4H~ '-SiO I I ~ C4H9 '-SiO ~ ~ ~
C~3\~\ococl99 CH3\~\
CO2Cl13
To a solution of 1-acetoxy-8-(dimethoxy
t-butylsilyl) oxy-3,7-dimethyl-2,6-octadiene (600 mg, 1.84
mmol) in tetrahydrofuran (5 ml) were added under nitrogen
atmosphere triphenylphosphine (47 mg, 0.18 mmol) and
tetrakis(triphenylphosphine)paradium (81 mg, 0.07 mmol), and
the mixture was stirred at room temperature for 15 minutes.
To the mixture was added a sodium salt of methyl
acetoacetate in tetrahydrofuran (20 ml) which has been
prepared from sodium hydride (92 mg, 8.0~mmolj;and methyl
acetoacetate (0.99 ml, 9.20 mmol), and the mixture was`
refluxed overnight. After addition o water (10 ml) and
ether (30 ml), the reaction mixture was stirred well, and
the organic layer was separated. ~he aqueous layer was
extracted with ether (5 mlx2), and the extract was dried
over Na2S04 and evaporated in vacuo to remove the solvent to
give a re~idue, which was then subjected to silica gel
column chromatography to obtain purified methyl
2-acetyl-10-(dimethyl t-butylsilyl)oxy-5,9-dimethyl-4,8-
decadienate (620 mg, 88%).
- 51 - 2~69~
IR(film)cm 1; 2970, 2940, 2910, 2860, 1745, 1722, 1435,
1360, 1250, 1065, 837, 775.
H NMR(CDC13, 250MHz)~ppm; 0.06, (s, 6H, (CH3)2Si), O.90(s,
9H, (CH3)3CSi), 1.59, 1.63(2s, 6H, 2xCH3C=CH-), 1.92-2.15(m,
4H, -C=CH-CH2-CH2-C=CH-), 2.22(s, 3H, -CHCH3), 2.55(t,
J=7.4Hz, 2H, -C=CH-CH2-CH(C02CH3)), 3.46(t, J=7.4H~, lH,
-CH(CO2CH3)), 3.73(s, 3H, -CO2CH3), 3.99(s, 2H, SiOCH2),
5.04(t, J=6.7Hz, lH, -C=CH-CH2-), 5.33(t, J=6.8Hz, lH,
-C=CH-CH2-)-
ExamPle 4
O . O
C02CH3
To a solution of methyl 2-acetyl-5,9-dimethyl-10-
(2-tetrahydropyranyl)oxy-4,8-decadienate (370 mg, 1.05 mmol)
in methylsulfoxide (2 ml) were added sodium chloride (180
mg, 3.08 mmol) and water (0.1 ml), and the mixture was
stirred at 150C. After four hours, the reaction mixture
was allowed to cool to room temperature, and water (15 ml)
was added thereto. The product was extracted with ether (20
mlx2). The extract was dried over Na~SO4, and concentrated
to give a residue, which was then purified with silica gel
column chromatography to obtain
6,10-dimethyl-11-(2-tetrahydropyranyl)oxy-5,9-undecadien-2-
one (70~).
204~9~
~ 52 -
IR(film)cm 1; 2950, 2880, 1720, 1442, 1358, 1120, 1078,
1024, 90s, 870, 815.
lH NMR(CDC13, 250MHz)~ppm; 1.45-l.90(m, 6H,
-OCH2-cH2-ca2-ca2-cH(o))r 1.62, 1.65(2s, 6H,
(Ca3)C=CH-CH2-CH2-(CH3)c=cH-), 1.96-2.20(m, 4H,
-C=CH-CH2-CH2-C=CH-), 2.14(s, 3H, COCH3), 2.26(q, J=7.lHz,
2H, -C=CH-CH2-CH2CO-), 2.46(t, J=7.lHz, 2H, -CH2COCH3),
3.45-3.55(m, lH, OCHaHb-CH2-CH2-CH2-CH(0)), 3.84, 4.10(2d,
J=11.5Hz, -OCa2C=CH-), 3.80-3.95(m, lH,
CHaHb~cH2-cH2-cH2-)~ 4.60(t, J=3.6Hz, lH, CH(O)), 5.08(t,
J=7.lHz, -C=Ca-CH2-CH2-C=CH-), 6.9(t, J=6.9Hz, lH,
-C=CH-CH2-CH2-C=ca-)-
Example 5
O O
H3COH2CO ¦ ¦ ¦¦ H3COH2CO ¦
CO2CH3
To a solution of methyl 2-acetyl-5,9-dimethyl-10-
(methoxymethyl)oxy-4,8-decadienate (420 mg, 1.37 mmol) in
methylsulfoxide (4 ml) were added sodium chloride (160 mg,
2.74 mmol) and water (0.1 ml), and the mixture was stirred
at 150C. After five hours, the reaction mixture was
allowed to cool to room temperature, and water (10 ml) was
added thereto. The product was extracted with ether (20
mlx2). The extract was dried over Na2S04, and concentrated
to give a residue, which was then purified with silica gel
column chromatography to obtain 6,10-dimethyl-11-
20~695
- 53 -
(methoxymethyl)oxy-S,9-undecadien-2-one (358 mg, 67%).
IR(film)cm 1; 2940, 1720, 1440, 1358, 1150, 1100, 1050,
920.
~ NMR(CDCl3, 250MHz)~ppm; 1.62, 1.66(2s, 6H, 2xCH3C=CH-),
1.94-2.32(m, 6H, -C=CH-CH2-CH2-C=CH-CH2-), 2.14(s, 3H,
COCH3), 2.46(t, J=7.3Hz, 2H, -CH2COCH3), 3.38(s, 3~, CH30),
3.92(s, 2H, OCH2C=CH-), 4.61(s, 2H, -OCH2C-), 5.08(t,
J=6-1Hz~ lH~ -C=Ca-CH2-), 5.40(t, J=6.7Hz, lH, -C=CHCH2-).
ExamPle 6
CH3 0 CH3 0
C4Hg'-Si0 ~ > C4Hg'-Si0
CH3 ~ CH
C02CH3
To a solution of methyl 2-acetyl-10-(dimethyl t-
butylsilyl)oxy-5,9-dimethyl-4,8-decadienate (96 mg, 0.25
mmol) in hexamethylphosphoric triamide (0.5 ml) were added
sodium iodide (45 mg, 0.30 mmol) and water (0.01 ml), and
the mixture was stirred at 150C. After two hours, the
reaction mixture was allowed to cool to room temperature,
and water (2 ml) was added thereto. The product was ex-
tracted with ether (5 mlx2). The extract was dried over
Na2SO4, and concentrated to give a residue, which was then
purified with silica gel column chromatography to obtain
6,10-dimethyl-11-(dimethyl t-butylsilyl)oxy-undecadien-2-one
(57 mg, 70%).
IR(film)cm 1; 2970, 2950, 2910, 2870, 1725, 1465, 1360,
1255, 1155, 1110, 1070, 837, 775, 662.
2045695
- 54 -
H NMR(CDCl3, 250MHz)~ppm; 0.06(s, 6H, (CH3)2Si), 0.91(5,
9H, (CH3)3CSi), 1.59, 1.62(2s, 6H, 2x-C=CH-CH2-), 1.92
-2.32(m, 6H, -CH=CH-CH2~CH2-C=CH-CH2-), 2-14(s, 3H, COCH3)~
2.46(t, J=8.7Hz, 2H, CH2COCH3), 4.00(s, 2H, SiOCH2-), 5.08,
5.35(2m, 2H, -C=CH-CH2-x2).
ExamPles 7 and 8
The procedures described in Examples 4-6 were
repeated except that methyl 2-acetyl-5,9-dimethyl-10-
acetoxy-4,8-decadienate or methyl 2-acetyl-5,9-
dimethyl-10-(benzoyl)oxy-4,8-decadienate was employed as a
starting material to give 6,10-dimethyl-11-acetoxy-
5,9-undecadien-2-one and 6,10-dimethyl-11-(benzoyl)oxy-
5,9-undecadien-2-one.
,ExamPle 9
0 ~ > H0 ~
To a solution of 6,10-dimethyl-ll-(2-
tetrahydropyranyl) oxy-5,9-undecadien-2-one (362 mg, 1.23
mmol) in a mixture of methanol (5 ml) and water (l ml) was
added p toluensulfonic acid (20 mg), and the mixture was
stirred at room temperature overnight. After addition of
saturated aqueous sodium bicarbonate (20 ml), the product
wa~ extracted with ethyl acetate (20 mlx2). The extract was
dried over MgSO4 and evaporated in vacuo to remove the
solvent to give a residue, which was subjected to silica gel
2~4~69~
- 55 -
column chromatography to provide
2,6-dimethyl-2,6-dodecadien-10-on-1-ol (234 mg, 90%).
IR(film)cm 1; 3430, 2930, 2860, 1715, 1440, 1360, 1160,
1080.
lH NMR(CDCl3+D20, 250MHz)~ppm; 1.62, 1.66(2s, 6H,
2xCH3C=CH-), 1.98-2.34(m, 6H, -C=CH-CH2-CH2-C=CH-CH2-),
2.13(s, 3H, COCH3), 2.47(tl J=7.2H~, 2H, -CH2COCH3), 3.98(s,
2H, -CH2OH), 5.06(t, J=7.lHz, lH, -C=CH-CH2-), 5.33(t,
J=6.9Hz, lH, -C=CH-CH2-)-
Exam~le 10
o
C-CH
A solution of 6,10-dimethyl-11-(2-
tetrahydropyranyl) oxy-5,9-undecadien-2-one (90 mg, 0.31
mmol) in tetrahydrofuran (5 ml) was stirred on an ice bath
under argon atmosphere. To the solution was added lithium
acetylide ethylenediamine complex (180 mg, 1.95 mmol), and
the mixture was warmed to room temperature and stirred for 3
hours. After addition of saturated aqueous ammonium chlo-
ride (2 ml), the reaction mixture was extracted with ether.
The extract was dried over Na2SO4 and evaporated in vacuo to
remove the solvent to give a crude product, which was
purified with silica gel column chromatography to obtain
3,7,11-trimethyl-12-(2-tetrahydropyranyl)oxy-6,10-
dodecadien-1-in-3-ol (75 mg, 75%).
2a~5~9~
- 56 -
IR(film)cm 1; 3440, 3320, 2950, 2880, 2200, 1440, 1450,
13~2, 1360, 1260, 1200, 1180, 1115, 1075, 1020, 905, 865,
810.
H NMR(CDC13, 250MHz)~ppm; 1.50(s, 3H, CH3C(OH)), 1.45
-1 90(m, 8H, OCH2-CH2-CH2-CH2-cH(O)~ CH2C( )~
2xCH3C=CH-), 2.00-2.40(m, 7H, -C=CH-CH2-CH2-C=CH-CH2-, OH),
2.46(s, lH, -C~_C-H), 3.45-3.57(m, lH, -OCHaHb-CH2-), 3.84,
4.10(2d, J=11.5Hz, 2H, OCH2C=CH-), 3.80-3.94(m, lH,
-OCHaHb-CH2-), 4.60(t, J=3.4Hz, lH, -OCHO-), 5.19~t,
J=6-7Hz~ lH~ -C=CH-CH2-), 5-41(t, J=6.7Hz, lH, -C=CH-CH2_).
Example 11
H3COH2CO ~ > H3COH2CO ~ C-CH
OH
A solution of 6,10-dimethyl-11-
(methoxymethyl)oxy-5,9-undecadien-2-one (67 mg, 0.26 mmol)
in tetrahydrofuran (2 ml) was stirred on an ice bath under
argon atmosphere. To the solution was added lithium
acetylide ethylenediamine complex (30 mg, 0.33 mmol), and
the mixture was warmed to room temperature and stirred for 3
hours. After addition of saturated aqueous ammonium
chloride (2 ml), the reaction mixture was extracted with
ether. The extract was dried over Na2S04 and evaporated in
vacuo to remove the solvent to give a crude product, which
was purified with silica gel column chromatography to obtain
3,7,11-trimethyl-12-(methoxymethyl)oxy-6,10-dodecadien-l-in-
2~6~
- 57 -
3-ol (61 mg, 83~).
IR(film)cm l; 3450, 3300, 2940, 1445, 1370, 1148, 1045,
918.
H NMR(CDCl3, 250MHz)~ppm; 1.05(s, 3H, CH3C(O)), 1.55
-1.84(m, 9H, 2xCH3C=CH-, OH), 2.00-2.40(m, 6H,
-C=CH-CH2-CH2-C=CH-CH2-), 2.46(s, lH, -C--C-H), 3.78(s, 3H,
CH30), 3.92(s, 2H, -OCH2C=CH-), 4.61(s, 2H, OCH2O), 5.19(t,
~=6.2Hz, lH, -C=CH-CH2-), 5.41(t, J=6.2Hz, -C=CH-CH2-).
Example 12
CH3 O CH3
C4H9'-SiO ~ C4Hg~-SiO ~ ~
CH3 ~ >CH3\V~ V\~ \C-CH
OH
A solution of 6,10-dimethyl~ (dimethyl t-
butylsilyl)oxy-5,9-undecadien-1-one (71 mg, 0.22 mmol) in
tetrahydrofuran (2 ml) was stirred on an ice bath under
argon atmosphere. To the solution was added lithium
acetylide ethylenediamine complex (90 mg, 0.98 mmol), and
the mixture was warmed to room temperature and stirred for 4
hours. After addition of saturated aqueous ammonium chlo-
ride (2 ml), the reaction mixture was extracted with ether.
The extract was dried over Na2SO4 and evaporated in vacuo to
remove the solvent to give a crude product, which was
purified with silica gel column chromatography to obtain
3,7,11-trimethyl-12-(dimethyl t-butylsilyl)oxy-6,10-
dodecadien-1-in-3-ol (51 mg, 67~).
- 58 - 20~
IR(film)cm 1; 3450, 3320, 2960, 2940, 2910, 2860, 1460,
1360, 1250, 1110, 1065, 835, 775.
H NMR(C~C13, 250MHz)~ppm; 0.06(s, 6H, (CH3)2Si), 0.91(s,
9H~ (CH3)3Si), 1.50(s, 3H, -CH2-C(OH)(CH3)-C-H), 1.59,
1.66(2s, 6H, 2xCH3C=CH-), 1.68-1.76(m, 2H, -CH2C(OH)-),
1.94-2.36(m, 7H, -C=CH-CH2-CH2-C=CH-CH2-, OH), 2.46(s, lH,
-C_CH), 4.00(s, 2H, OCH2-C=CH-), 5-19(t~ J=7.1Hz, lH~
-C=CH-CH2-), 5.36(t, J=6.9Hz, lH, -C=CH-CH2-).
Example 13 and 14
The procedure described in Example 11 was repeated
except that 6,10-dimethyl-11-acetoxy-5,9-undecadien-2-on or
6,10-dimethyl-11-(benzoyl)oxy-5,9-undecadien-2-one was
employed as a starting amterial to give
3,7,11-trimethyl-12-acetoxy-6,10-dodecadien-1-in-3-ol and
3,7,11-trimethyl-12-(benzoyl)ox~-6,10-dodecadien-1-in-3-ol.
Example 15
The procedures described in Examples 10 and 12
were repeated except that
2,6-dimethyl-2,6-dodecadien-10-on-1-ol was employed as a
starting material to give
2,6,10-trimethyl-2,6-dodecadien-11-in-1,10-diol.
ExamPle 16
o
~yO~ > ~,J~
A solution of 6,10-dimethyl-11-(2-
2 0 ~
- 59 -
tetrahydropyran~l) oxy-5,9-undecadien-2-one (80 mg, 0.27
mmol) in tetrahydro~uran (3 ml) was stirred on an ice bath
under argon atmosphere. To the solut.ion was added
vinylmagnesium bromide in tetrahydrofuran (O.3 ml, 0.3 mmol,
1.0 M), and the mixture was warmed to room temperature and
stirred for 10 hours. After addition of saturated aqueous
ammonium chloride (2 ml), the reaction mixture was extracted
with ether. The extract was dried over Na2SO4 and
evaporated in vacuo to remove the solvent to give a crude
product, which was purified with silica gel column
chromatography to obtain 3,7,11-trimethyl-12-(2-
tetrahydropyranyl)oxy-1,6,10-undecatrien-3-ol (61 mg, 70%).
IR(film)cm ; 3460, 2950, 2880, 1200, 1118, 1075, 1022,
905, 865, 810.
lH NMR(CDC13, 250MHz)~ppm; 1.28(s, 3H, C(OH)CH3), 1.65
-1.92(m, 8H, -OCH2-CH2-CH2-cH2-cH(O)-~ CH2-C(OH))~ 1-95
-2.40(m, 7H, C=CH-CH2-CH2-C=CH-CH2-, OH), 3.45-3.55(m, lH,
-OCHaHb-CH2-), 3.84, 4.10(2d, J=11.6Hæ, 2H, -OCH2C=CH2-),
3.80-3.95(m, lH, -OCHaHb-CH2-), 4.60(t, J=3.4Hz, lH,
OCH(O)), 5.06(dd, J=1.3, 10.7Hz, lH, -CH=CHaHb), 5.14(m, lH,
-C=CH-CH2-), 5.22(dd, J=1.3, 17.4Hz, lH, -CH=CHaHb), 5.41(t,
J=6.3Hz, lH, -C=CH-CH2-), 5.92(dd, J=10.7, 17.4Hz, lH,
-CH=CH2)-
ExamPle 17
H3COH2CO ~ ~ H3COH2CO
OH
2Q4~9~
- 60
A solution of 6,10-dimeth~l-11-
(methoxymethyl)oxy-5,9-undecadien-2-one (60 mg, 0.24 mmol)
in tetrahydrofuran (2 ml) was stirred on an ice bath under
argon atmosphere. To the solution was added vinylmagnesium
bromide in tetrahydrofuran (1.0 ml, 1.0 mmol, 1.0 M), and
the mixture was warmed to room temperature and stirred for 4
hours. After addition of saturated aqueous ammonium
chloride (2 ml), the reaction mixture was extracted with
ether. The extract was dried over ~a2SO4 and evaporated in
vacuo to remove the solvent to give a crude product, which
was purified with silica gel column chromatography to obtain
3,7,11-trimethyl-12-(methoxymethyl)oxy-1,6,10-dodecatrien-
3-ol (54 mg, 80%).
IR(film)cm 1; 3480, 2940, 1450, 1370, 1210, 1150, 1100,
1045, 920, 845, 685.
lH NMR(CDC13, 250MHz)~ppm; 1.28(s, 3H, CH3C(OH)), 1.60,
1.66(2s, 6H, 2xCH3-CH=C-), 1.52-1.72(m, 2H, -CH2-C(OH)),
1.95-2.20(m, 7H, -C=CH-CH2-Ca2-C=CH-CH2-, OH), 3.38(s, 3H,
CH30), 3.92(s, 2H, -OCH2C=CH-), 4.61(s, 2H, -OCH2O-),
5.06(dd, J=1.3, 10.7Hz, lH, -CH=CHaHb), 5.14(t, J=7.2Hz, lH,
-C=CH-CH2-), 5.20(dd, J=1.3, 17.4Hz, lH, -CH=CHaHb), 5.41(t,
J=6.9Hz, lH, -C=CH-CH2-), 5.92(dd, J=10.7, 17.4Hz, lH,
-CH=CH2-)~
Example 18
C4H9'-SIiO~A C Hg~-~O~
OH
2~4~
- 61 -
A solution of 6,10-dimethyl-11-(dimethyl-t-
butylsilyl) oxy-5,9-undecadien-2-one (48 mg, 0.15 mmol) in
tetrahydrofuran (2 ml) was stirred on an ice bath under
argon atmosph~re. To the solution was added vinylmagnesium
bromide in tetrahydrofuran (10 ml, 1.0 mmol, l.O M), and the
mixture was warmed to room temperature and stirrsd for 4
hours. After addition of saturated aqueous ammonium chlo-
ride (2 ml), the reaction mixture was extracted with ether.
The extract was dried over Na2SO4 and evaporated in vacuo to
remove the solvent to give a crude product, which was
purified with silica gel column chromatography to obtain
3,7,11-trimethyl-12-(dimethyl-t-butyls~lyl)oxy-1,6,10-
dodecatren-1-3-ol t32 mg, 60%).
IR(film)cm 1; 3420, 2970, 2940, 2860, 1462, 1360, 1250,
1150, 1070, 920, 835, 775, 662.
lH NMR(CDCl3, 250MHz)~ppm; 0.06(s, 6H, (CH3)2Si), 0.91(s,
9H, ~CH3)3CSi), 1.2~(s, 3H, -C(OH)(CH3)-CH=CH2), 1.60(s, 6H,
2xCH3C=CH-), 1.46-1.73(m, 3H, -CH2C(OH)(CH3))-, 4.00(s, 2H,
SiOCH2-), 5.06(dd, J=1.3, 10.7Hz, lH, -CH=CHaHb), 5.14(m,
lH, -C=CH-CH2-), 5.22(dd, J=1.3, 17.4Hz, lH, -CH=CHaHb),
5.36(m, lH, -C=CH-CH2-), 5.92(dd, J=10.7, 17.4Hz, lH,
-CH=CH2-)-
Example 19
The procedure described in Example 16 was repeated
except that 6,10-dimethyl-11-acetoxy-5,9-undecadien-2-one or
6,10-dimethyl-11-(benzoyl)oxy-5,9-undecadien-2-one was
employed as a starting material to give 3,7,11-trimethyl-
204~S~
- 62 -
12-acetoxy-1,6,10-undecatrien-3-one and
3,7,11-trimethyl-12-~benzoyl)oxy-1,6,10-undecatrien-3-one.
Example 20
The procedures described in Examples 17 and 18
were repeated except that 2,6-dimethyl-2,6-dodecadien 10-
on-l-ol was employed as a starting material to give
2,6,10-trimethyl-2,6,11-dodecatrien-1,10-diol.
Exam~le 21
~ CH0 >
To a solution of 2-
(diethylphosphono)isovaleronitrile (6.54 g, 30 mmol) in
toluene (55 ml) was added a 0.5 M solution of potassium
bis(trimethylsilyl)amide in toluene (56 ml) with stirring on
a cooling bath at -70~C. After 30 minutes, geranial t3.80
g, 25 mmol) was added thereto with continuous stirring at
the same temperature, and then the reaction mixture was
warmed up to room temperature. After addition of water to
the mixture, the organic layer was extracted. The organic
extract was washed with saturated aqueous sodium bicarbonate
and saturated aqueous sodium chloride, dried over MgSO4, and
filtered. The filtrate was concentrated to give a residue,
which was then subjected to silica gel column chromatography
(solvent: n-hexane/ethyl acetate=100:1) to give
204~6~
- 63 -
2-(1-methylethyl)-5,9-dimethyl-2,4,8-decatrienenitrile (4.87
g, 90%, 2Z:2E=22.4:1).
Spectral data of 2Z compound
IR(film)cm 1; 2980, 2940, 2890, 2220, 1640, 1450, 1390,
1375, 1295, 1225, 1105, 1030.
lH NMR(CDC13, 250MHz)~ppm; 1.17(d, J=6.8Hz, 6H, CH(CH3)2),
1.61, 1.69(each bs, each 3H, -C=CCH3), 1.83(d, J=1.2Hz, 3H,
-C=CCH3)~ 2--12-2(m, 4H, -CH2~H2-), 2.53(hep, J=6.8Hz, lH,
CH(CH3)2), 5.08(m, lH, -C=CHCH2-), 6.28, 6.82(each d,
J=11.5Hz, each lH, =CH-CH=).
Exam~le 22
/~ ~ ~
CN CH0
To a solution of 2-(1-methylethyl)-5,9-dimethyl-
2,4,8-decatrienenitril (2Z compound, 217 mg, 1 mmol) in
n-hexane (4 ml) was added a 1 M solution of
diisobutylaluminium hydride in toluene (2 ml) with stirring
under argon atmosphere at -70C. After two-hour-stirring at
the same temperature, water (0.8 ml) was added to the
mixture followed by removal of the cooling bath and vigorous
stirring. The resultant while precipitates were filtered
and washed with n-hexane. The filtrate was combined with a
10% aqueous solution of oxalic acid and stirred for 3 hours.
The organic layer was extracted and separated, washed with
water, dried over MgSO4, and concentrated.
204~69~
- 64 -
The above manipulation was conducted under argon atmosphere.
The resultant residue was subjected to silica gel column
chromatography (~olvent: n-hexane/ethyl acetate=50:1) to
obtain the aimed 2-tl-methylethyl)-S,9-dimethyl-2,4,8-
decatrienal (198 mg, 90%).
IR(film)cm 1; 2980, 2940, 2880, 1670, 1630, 1455, 1375,
1295, 1235, 1135, 1105, 1075.
NMR(CDC13, 250MHz)~ppm; 1.07(d, J=6.8Hz, 6H, -CH(CH3)2),
1.62, 1.69(each bs, each 3H, -C=CCH3), 1.89(d, J=l.OHz, 3H,
-C=CCa3), 2.-12.3(m, 4H, -CH2CH2-), 2.91(hep, J=6.8Hz, lH,
-Ca(CH3)2), S.lO(m, lH, =CaCH2-), 6.83, 7.14(each d,
J=12.0Hz, each lH, =Ca-CH=), 10.29(s, lH, -CHO~.
Example 23
CHO
CN
~ o a solution of 2-
(diethylphosphono)isovaleronitrile (8.72 g, 40 mmol) in
toluene (75 ml) was gradually added a 0.5 M solution of
potassium bis(trimethylsilyl)amide in toluene (75 ml) with
stirring at -70C under argon atmosphere. The cooling bath
was removed, and the reaction mixture was stirred at room
temperature for 30 minutes. The reaction mixture was cooled
to -70C again, and farnesal (5.88 g, 26.7 mmol) was added
thereto with stirring, and the mixture was allowed to warm
to room temperature. After addition of water, the organic
204~69~
- 65 -
layer was separated, washed with saturated aqueous sodium
bicarbonate and then saturated aqueous sodium chloride, and
dried over MgSO4. The organic layer was separated from
MgSO4 by filtration and concentrated to give a residue,
which was purified with silica gel column chromatography
(solvent: n-hexane/ethyl acetate=100:1) to obtain the aimed
2 -(1-methylethyl)-5,9,13-trimethyl-2,4,8,12-
tetradecatetraenenitrile (7.23 g, 96~; 2Z:2E=25.6:1).
SPectral data of 2Z compound
IR(film)cm 1; 2980, 2940, 2210, 1640, 1450, 1390, 1290,
1225, 1110, 1030.
NMR(CDC13, 250MHz)~ppm; 1.14(d, J=6.8Hz, 6H, CH~CH3)2),
1.58(bs, 3Hx2, -C=CCH3), 1.65(bs, 3H, -C=CCH3), 1.81(d,
J=1.2Hz, 3H, -C=CCH3), 1.9-2.2(m, 8H, -CH2CH2-x2), 2-50(hep~
J=6.8Hz, lH, -CH(CH3)2), 5-06(m, 1~, =CHCH2-), 6-26~ 6-80(
each d, J=11.5Hz, each lH, =CH-Ca=~.
ExamPle 24
CN CH~
To a solution of 2-(1-methylethyl)-5,9,13-
trimethyl-2,4,8,12-tetradecatetraenenitrile (856 mg, 3.0
mmol) in n-hexane (30 ml) was added a 0.5 M solution of
diisobutylaluminium hydride in toluene (6 ml) with stirring
at -70C under argon atmosphere. After one hour, water (3
ml) was added, the cooling bath was removed, and the
204~95
- 66 -
reaction mixture was stirred well. Resultant white
precipitates were filtered and washed. The filtrate was
concentrated to give a residue, which was dissolved in
n-hexane (10 ml). The n-hexane so~ution was combined with a
10% aqueou~ solution of oxalic acid (5 ml) and stirred for 3
hours. The organic layer was extracted and separated,
washed with water, dried over MgSO4, and concentrated. The
resultant residue was subjected to silica gel column
chromatography ~solvent: n-hexane/ethyl acetate=10:1) to
obtain 2 -(1-methylethyl)-5,9,13-
trimethyl-2,4,8,12-tetradecatetraenal (865 mg, 84%).
IR(film)cm 1; 2980, 2940, 2210, 1640, 1450, 1390, 1290,
1225, 1110, 1030.
NMR(CDC13, 250MHz)~ppm; 1.07(d, J=6.8Hz, 6H, -CH(CH3)2),
1.59, 1.61, 1.67(each bs, 3Hx3, -C=CCH3), 1.89(d, J=l.OHz,
3H, -C=CCH3), 2.0-2.2(m, 8H, -CH2CH2-x2), 2.91(hep, J=6.8Hz,
lH, -CH(CH3)2), 5.10(m, lH, -C=CCH3), 6.81, 7.16(each d,
J=12.0Hz, each lH, =CH-CH=), 10.29(s, lH, -CHO).
ExamPle 25
> HO~
CN CN
To a suspension of selenium dioxide (58 mg) and
2-hydroxybenzoic acid (365 mg) in methylene chloride (10.5
ml) wa~ gradually added an aqueous solution of 80~ t-butyl
204~6~
- 67 -
hydroperoxide (11.6 ml) with stirring on a water bath.
After 30 minutes, 2-(1-methylethyl)-5,9,13-trimethyl-
2,4,8,12-tetradecatetraenenitrile (7.56 g, 26.8 mmol)was
added to the mixture, which was then allowed to stand at
room temperature for 30 hours. Most of the solvent was
removed by evaporation in vacuo, and the residue was
dissolved in ether. The organic layer was washed with
saturated aqueous sodium bicarbonate, dried over MgSO4, and
concentrated. The resultant residue was subjected to silica
gel column chromatography to give the aimed
14-hydroxy-2-(1-methylethyl)-5,9,13-trimethyl-2,4,8,12-tetr-
adecatetraenenitrile (2.53 g, 31%). The starting material
was also recovered (3.10 g, 40~) in the column
chromatography. The yield of the aimed product based on the
consumed starting material was 52%.
IR(film)cm 1; 3450, 2975, 2930, 2880, 2210, 1635, 1445,
1385, 1220, 1020.
NMR(CDC13, 250MHz)~ppm; 1.17(d, J=6.7Hz, 6H, CH(CH3)2),
1.62, 1.67(each ~s, each 3H, -C=CCH3), 1.84(d, J=1.2Hz, 3H,
-C=CCH3), 2.0-2.2(m, 8H, -CH2CH2-x2), 2.S3(hep, J=6.7Hz, lH,
-CH(CH3)2), 3.99(bs, 2H, -CH2OH), 5.11(m, lH, -CHCH2-),
5.39(bt, J=5.5Hz, lH, -CHCH2-), 6.28, 6.83(each d, J=11.5H~,
each lH, =CH-CH=).
Exam~le 26
H0 ~ ce~
CN CN
2~6~
- 68 -
To a solution of 14-hydroxy-2-(1-methylethyl)-
5,9,13-trimethyl-2,4,8,12-tetradecatetraenenitrile (904 mg,
3.0 mmol) in carbon tetrachloride (2 ml) was added
triphenylphosphine ~1.02 g, 3.9 mmol), and the mixture was
heated under reflux f or one hour. Most of carbon tetra-
chloride was removed by evaporation in vacuo, and n-hexane
was added to the residue. The resultant mixture was f il-
tered and washed and the f iltrate was concentrated to give a
residue, which was then subjected to silica gel column
chromatography (solvent: n-hexane/ethyl acetate=10:1) to
obtain the aimed 14~chloro compound (890 mg, 93~).
IR(film)cm ; 2980, 2940, 2880, 2215, 1635, 1445, 1390,
1265, 1025.
NMR(CDC13, 250MHz~ppm; 1.14(d, J=6.8Hz, 6H, CH(CH3)2),
1.59, 1.64(each bs, each 3H, -C=CCH3), 1.81(d, J=l.OHz, 3H,
-C=CCa3), 1.9-2.2(m, 8H, -CH2CH2-x2), 2.50(hep, J=6.8Hz, lH,
-CH(CH3)2), 3.96(bs, 2H, -CH20H), 5.08(m, lH, -CHCH2-),
5.36(bt, J=5.5Hz, lH, =CHCH2-), 6.25, 6.80(each d, J=11.5Hz,
each lH, =CH-CH=).
The following Reference Examples illustrate a
method of preparation of sarcophytol A by the use of the
compounds obtained in the foregoing Examples.
Reference ExamPle 1
H3COH2CO ~ H3COH2CO ~ CHO
OH
204~69~
- 69 -
A solution of 3,7,11-trimethyl-12-
(methoxymethyl)oxy-6,10-dodecadien-1-in-3-ol (175 mg, 0.62
mmol)/ tris (triphenylsilyl)vanadate (49 mg, 0.062 mmol),
and benzoic acid (7.6 mg, 0.062 mmol) in xylene (2 ml) was
stirred on an oil bath at 140C. After two-hour-stirring,
the solution was allowed to cool to room temperature and
concentrated. The resultant residue was subjected to silica
gel column chromatography to obtain 12-(methoxymethyl)oxy-
3,7,11-trimethyl-2,6,10-dodecatrienal (88 mg, 50%).
IR(film)cm 1; 2950, 1675, 1450, 1385, 1198, 1157, 1120,
1105, 1050, 925, 850.
lH NMR(CDC13, 250MHz)~ppm; 1.57, 1.62, 2.13(each s, each
3H, CH3C=CH-), 1.93-2.30(m, 8H, 2x-C=CH-CH2-CH2-~, 3.33(s,
3H, CH30), 3.88(s, 2H, -OCH2C=CX-), 4.57(s, 2H, -OCH20-),
5.06, 5.36(each m, each lH, -C=CH-CH2-), 5.84(d, J=8.2Hz,
lH, -C=CH-CHO), 9.96(d, J=8.2Hz, lH, -C=CH-CHO).
Reference ExamPle 2
H3COH2CO ~ H3COH2CO ~ CHO
OH
A solution of 3,7,11-trimethyl-12-
(methoxymeth~l)oxy-1,6,10-dodecatrien-3-ol (460 mg, 1.6
mmol) in dishloromethane (30 ml) was added pyridinium
chlorochromate (690 mg, 3.2 mmol), and the mixture was
stirred at room temperature for 8 hours. After addition of
a mixture of hexane, ethyl acetate, and ether (3:1:1) (lOO
2~4~9~
- 70 -
ml), the mixture was stirred and insoluble materials were
filtered. ~he filtrate was concentrated ln vacuo, and the
residue was purified with silica gel column chromatography
to give 12-(methoxymethyl~oxy-3,7,11-trimethyl-2,6,10-
dodecatrienal (233 mg, 52%). The physico-chemical
properties of the product was the same as those described in
Reference Example 1.
Reference Exam~le 3
H3COH2CO ¦ I i H3COH2CO
CHO >
CN
To a solution of 2-
(diethylphosphono)isovaleronitrile (316 mg, 1.44 mmol) in
toluene (1 ml) was dropwise added a 1.0 M solution of
lithium bis(trimethylsilyl)amide in hexane (1.3 ml, 1.3
mmol) with stirring at -70C. After 30 minutes,
12-(methoxymethyl)oxy-3,7,11-trimethyl-2,6,10-dodecatrienal
(130 mg, 0.46 mmol) in toluene (2 ml) was added at the same
temperature, and the mixture was allowed to warm to room
temperature over about 3 hours. An aqueous ammonium chlo-
ride (6 ml) was added and the mixture was extracted with
hexane. The extract was dried over Na2SO4 and evaporated in
vacuo to remove the solvent to give a residue, which was
then subjected to silica gel column chromatography (solvent:
n-hexane/ethyl acetate=20:1) to obtain the aimed 2
204~6~
- 71 -
-(1-methylethyl)-14-(methoxymethyl)oxy-
5,9,13-trim0thyl-2,4,8,12-tetraenenitrile (135 mg, 85%).
IR(film)cm ; 2980, 2945, 2900, 2310, 1640, 1150, 1050.
lN NMR(CDC13, 250MHz)~ppm; 1.17(d, J=6.8Hz, 6H, (CH3)2CH-),
1.61, 1.67, 1.84(each s, each 3H, CH3C=CH-), 1.96-2.21(m,
8H~ -C=CH-CH2-CH2-C=CH-CH2-CH2-)~ 2.53(hep, J=6.8Hz, lH,
-CH(CH3)2), 3.38(s, 3H, CH30-), 3.92(s, 2H, -OCH2O-),
4.62~s, 2H, -Q-CH2-C=CH-), 5.10(brs, lH, -C=CH-), 5.42(brt,
J=6.4H~, lH, -C=CH-), 6.28, 6.82(each d, J=11.5Hz, each lH,
-C-CH-CH=C(CN)-).
~eference Example 4
H3COHaC0 ~ ~ H0 ~
CN CN
To the product obtained in Reference Example 3
(135 mg, 0.39 mmol) in methanol (5 ml) was added a trace
amount of conc. HCl, and the mixture was heated with
stirring at 60C for 6 hours. After addition of saturated
aqueous sodium bicarbonate (20 ml), the reaction mixture was
extracted with ether (20 mlx2). The extract was dried over
MgSO4 and evaporated in vacuo to remove the solvent. The
resultant residue was purified with silica gel column
chromatography (solvent: n-hexane/ether=5:1) to obtain the
14-hydroxy compound (96 mg, 82%).
IR(film)cm 1; 3460, 2980, 2930, 2210, 1635, 1450, 1020.
2~4569~
- 72 -
H NMR(CDCl3, 250MHz)~ppm; 1.14(d, J=6.8Hz, 6H, (CH3)2CH-),
l.S8(m, 4H, CH3C=CH-, -OH), 1.65(s, 3H, Ca3C=CH-), 1.81(d,
J=l.lHz, 3H, CH3C=CH-), 1.92-2.25(m, 8H,
-2 CH2 (CH3)C=CH-Ca2-Ca2-), 2.51(hep, J=6.8Hz lH
(CH3)2CH-), 3.97(d, J=5.9Hz, 2H, CH2OH), 5.08(brs, lH,
-C=CH-), 5.36(m, lH, -C=CH-), 6.26, 6.80(each d, J=ll.SHz,
each lH, -C=CH-CH=C(CN)).
Reference Example 5
H0 ~ > H0 ~
CN , CH0
To the hydroxy compound obtained in Reference
Example 4 in toluene (S ml) was dropwise added a 1 M
solution of diisobutylaluminium hydride in toluene (2.0 ml,
2.0 mmol) at -70C under argon atmosphere. After
two-hour-stirring at -70C, a 1 M aqueous solution of oxalic
acid (4.0 ml) was added, and the mixture was allowed to warm
gradually to room temperature with stirring under argon
atmosphere. The organic layer was washed with water and
saturated aqueous sodium bicarbonate, dried over Na2SO4, and
evaporated in vacuo to remove the solvent. The resultant
residue was purified with silica gel column chromatography
(solvent: n-hexane/eth~l acetate=7:1) to obtain the aimed
formyl compound.
IR(film)cm 1; 3430, 2960, 2920, 2870, 1670, 1630, 1450,
1390, 1295, 1230, 1130, 1070, 1010.
73 204~695
lH NMR(CDC13, 250MHz)~ppm; 1.04(d, 6H, J=6.8Hz, -CH(CH3)2),
1.59(d, J=0.6Hz, 3H, CH3-C=), 1.63(brs, 3H, CH3-C=), 1.86(d,
J=1.2Hz, 3H, CH3-C=), 1.7-2.2(m, 8H, -CH2CH2-),
2.88(hep.J=6.8Hz, lH, -CH(CH3)2), 3.95(brs, 2H~ -CH2OH)~
5.09(m, lH, -CH2CH=), 5.38~brt, J-6.8Hz, lH, -CH2CH=),
6.80(d, J=12.0Hz, lH, =CH-CH=), 7.11(d, J=12.0Hz, lH,
=CH-CH=), 10.25(s, lH, -CHO).
7 2~
-- 4
~eference ExamPle 6
~0~ > C~
C~0 CH0
~ solution of dry lithium chloride (64 mg, 1.5
mmol), 2, 6-lutidine (0.23 ml, 2.0 mmol) and hydroxy formyl
compound (305 mg, 1.0 mmol) in dimethylformamide (1.0 ml)
was chilled on an ice water bath and mixed with methane-
sulfonyl chloride (160 mg, 1.4 mmol) with stirring in argon
atmosphere. About 8 hours later, the starting material was
confirmed to disappear, and the reaction mixture was dis-
solved in water and ether. The organic layer was washed
with water, dried over magnesium sulfate and concentrated.
The residue was chromatographed on a column of silica gel
eluting with n-hexane: ethyl acetate (15:1) as an eluent to
give the objective chloroformyl com~ound (281 mg, 87%).
IR (film)cm- ; 2970, 2930, 2880, 1670, 1630, 1445, 1390,
1295, 1265, 1135.
MMR (CDC13, 250MHz)~ppm; 1.04 (d, J = 7.0Hz, 6H, -
CH(CH3)2), 1.59, 1.70 (each bs, each 3H, -C = CCH3), 1.87
(d, J = 1.3Hz, 3H, - C = CCH 3), 1.9 - 2.2 (m, 8H, -
CH2CH2-), 2.89 (hep, J = 7.0 Hz, lH, - CH(CH3)2), 3.98 (bs,
2H2, - CH2Cl), 5.09 (m, lH, - C = CHC H2-), 5.47 (bt, J =
6.5Hz, lH, - C = CHCH2-), 6.82 (d, J = 12.0Hz, lH, - C = CH
2Q~6~
- 75 -
- CH = C(CHO) -), 7.11 (d, J = 12.0Hz, -C = CH - CH -
C~CHO)-), 10.27 ts, lH, - CHO).
Reference Exam~l~ 7
ce~ ~ > c~
CN CH0
To a solution of the nitrile (14-chloro-2-(1-
methylethyl)-5,9,13-trimethyl-2,4,8,12-tetradecatetraene-
nitril)(890 mg, 2.78 mmol) in n-hexane (30 ml) was dropwise
added gradually a lM solution (4.2 ml) of diisobutylaluminum
hydride in toluene at -70C under argon atmosphere. One
hour later, 2 ml of water was added to the mixture, and the
bath was removed. The reaction mixture was vigorously
stirred, and the resultant solid was filtered and washed
with n-hexane. ~he resultant filtrate was stirred still
with 10% oxalic acid. The organic layer was washed, dried,
filtered and concentrated. The residue was chromatographed
on a column of silica gel eluting with n-hexane: ethyl
acetate (20 : 1) to give the objective formyl compound (781
mg, 87%).
IR(film)cm 1; 2970, 2930, 2880, 1670, 1630, 1445, 1390,
1295, 1265, 1135.
NMR(CDC13, 250MHz)~ppm; 1.04(d, J=7.0Hz, 6H, -CH(CH3)2),
1.~9, 1.70(each bs, each 3H, -C=CCH3), 1.87(d, J=1.3Hz, 3H,
-C=CCH3), 1.9-2.2(m, 8H, -CH2CH2-), 2.89(hep, J=7.OHz, lH,
-CH(CH3)2), 3.98(bs, 2H, -CH2Cl), 5.09(m, lH, -C=CHCH2-),
2a4~59~
- 76 -
5.47(bt, J=6.5Hz, lH, -CH=CHCH2-), 6.82(d, J-12.0Hz, lH,
-C=CH-CH-C(CHO)-), 7.11(d, J-12.OHz, -C=CH-CH=C(CHO)-),
10.27( 6, lH, -CHO~.
Reference Example 8
C~ ~ ce i
CHO CN
OSi(CH3)3
The above formyl compound, 14-chloro-2~
methylethyl)-5,9,13-trimethyl-2,4,8,12-tetradecatetraenal
(640 mg, 2.0 mmol) was dissolved in trimethylsilylnitril
(0.35 ml, 2.6 mmol). To the solution on an ice-water bath
was added with stirring under argon atmosphere a trace
amount of potassium cyanide/18-crown 6-ether complex. Two
hours later, disappearance of the starting compound was
confirmed. Excessive trimethylsilylnitrile was evaporated
off to obtain crude 15-chloro-3-(1-methylethyl)-6,10,14-
trimethyl-2-(trimethylsiloxy)-3,5,9,13-
pentadecatetraenenitrile (647 mg, quantitative).
IR(film)cm 1; 2960, 2930, 2880, 2320, 1445, 1255, 1080,
875, 845.
NMR(CDC13, 250MHz)~ppm; 1.11, 1.15(each d, J=6.9Hz, each
3H, -CH(CH3)2), 1.60, 1.71, 1.77(each s, each 3H, -C=CCH3),
1-9-2-2(m~ 8H, -CH2CH2-), 2-64(hep, J=6.9Hz, lH, -CH(CH3)2,
3.99( 8 , lH, -CH2Cl), 5.11(m, lH, -C=CHCH2-), 5.33(s, lH,
-CHCN), 5.48(bt, J=6.5Hz, lH, -C=CHCH2-), 6.04, 6.25(each d,
J=11.3Hz, each lH, -C=CH-CH=C-).
204~fi9~
Reference Example 9
'C~
,/~CN
OSi (CH9) 3
~ ~ k
\~<CN + ~ ~<Cl~
OSi (CH3) 3 ~ / OH
A solution of the crude cyanohydrine trimethyl-
5ilyl ether (647 mg, 2.00 mmol if it is 100% pure), which
was obtained in Reference Example 8 in tetrahydrofuran (25
ml) was dropwise added with stirring at 50-55C under argon
atmosphere over 30 minutes to a solution of lM lithium
bis(trimethylsilyl)amide in tetrahydrofuran, which had been
diluted with 25 ml of tetrahydrofuran. After completion of
the dropwise addition, the tetrahydrofuran was evaporated
off in vacuo, and the residue was dissolved in ethyl ether
(30 ml), and the solution was washed with cooled lN HCl,
water, and then saturated aqueous sodium chloride. The
organic layer was dried over MgSO4 and then concentrated to
give a residue, which was then subjected to silica gel
column chromatography (solvent: n-hexane/ethyl
acetate=50:1-5:1) to obtain the aimed cyclized
2-(1-methylethyl)-5,9,13-trimethyl-1-
trimethylsiloxy-2,4,8,12-cyclotetradecatetraen-1-
2Q4~9~
- 78 -
carbonitrile (496 mg, 64%) and desilylated analogue (56 mg,
9%) .
NMR s~ectrum of l-trimethYlsiloxY comPound
NMR(CDC13, 250MHz)~ppm; 0.23(s, 9H, -Si(CH3)3), 1.09,
1.15(each d, J=6.7Hz, each 3H, -CH(C ~ )2)' 1.50, 1.62(each
bs, each 3H, -C=CCH3), 1.70(d, J=1.3Hz, 3H, -C=CCH3),
2-0-2-2(m, 8H, -CH2CH2-), 2-51(hep, J=6.7Hz, lH, -CH(CH3)2),
2.55, 2.65(each d, J=14.2Hz, each lH, -CHa Hb CN-), 4.94(bt,
J=6.lHz, lH, -C=CHCH2-), 5.15(bt, J=5.6Hz, lH, -C=CHCH2-),
6.17, 6.44(each d, J=11.8Hz, each lH, -C=CH-CH=C-).
NMR sPeCtrum of l-hvdroxy comPound
NMR(CDC13, 25OMHz)~ppm; 1.15, l.l9(each d, J=6.7Hz, each
3H, CH(CH3)2), 1.55, 1.63, 1.69(each s, each 3H, CH3-C=C-),
1.94-2.35(m, 8H, CH2-C=C-), 2.51(hep, J=6.7Hz, lH,
CH(CH3)2), 2.66, 2.73(each d, J=14.1Hz, 2H, CHa Hb CCN),
2.89(brs, lH, OH), 4.93, 5.24(each brt, J=5.3Hz, each lH,
-C=CH-CH2-), 6.22, 6.42(each d, J=ll.lHz, each lH,
-C=CH-CH=C-).
Reference ExamPle 10
~ /~
Si(CH3)3 ~ ~
The above cyanohydrine trimethylsilyl ether
compound, 2-(1-methylethyl)-5,9,13-trimethyl-1-
trimethylsiloxy-2,4,8,12-cyclotetradecatetraen-1-
2 0 ~
- 79 -
carbonitrile (657 mg, 1.7 mmol) was dissolved in 10% aqueous
tetrahydrofuran (10 ml). To the solution on an ice-water
bath was added a solution of lM tetra n-butylammonium
fluoride in tetrahydrofuran (0.02 ml)~ and the mixture was
stirred and then allowed to stand at room temperature for 2
days. Most of the tetrahydrofuran was removed in vacuo and
the residue was dissolved in ethyl ether. The ether layer
was dried over MgS04, filtered, concentrated, and subjected
to silica gel column chromatography (solvent: n-hexane/ethyl
acetate=30:1) to obtain the ketone compound, 2
-(1-methylethyl)-5,9,13-trimethyl-2,4,8,12-
cyclotetradecatetraen-1-one (411 mg, 85~).
Reference Example ll
> ~ OH
To the above ketone compound, 2-(1-methylethyl)-
S,9,13-trimethyl-2,4,8,12-cyclotetradecatetraen-l-one (137
mg, 0.48 mmol) in dry toluene (2.5 ml) was dropwise added
with stirring on a cooling hath at -70C a solution of lM
diisobutyl aluminium hydride in toluene (0.6 ml). One hour
later, disappearance of the starting material was confirmed.
After addition of water (0.25 ml) and removal of the cooling
bath, the reaction mixture was stirred, followed by drying
over MgS04, filtration, and concentration to give a residue,
2~ 6~
- 80 -
which was subjected to silica gel column chromatography
(solvent: n-hexane/ethyl acetate=12:1) to obtain the aimed
sarcophytol A (125 mg, 88%)~
Reference Exam~le 12
Lithium aluminium hydride (80.0 mg, 2.11 mmol) was
added to diethyl ether (S ml) under argon atmosphere, and
the mixture was stirred. To the suspension was dropwise
added at room temperature over 5 minutes a solution of
(lR,2S)-(-)-N-methylephedrine (308 mg, 2.12 mmol) in diethyl
ether (5 ml). After one hour reflux of the reaction mixture
with stirring, N-ethylaniline (0.53 ml, 4.23 mmol) was
dropwise added thereto over 5 minutes, and the mixture was
refluxed with stirring additional one hour. The mixture was
then cooled to -72C, and a solution of the ketone compound
(136 mg, 0.475 mmol) obtained in Reference Example 10 in
diethyl ether (3 ml) was gradually added thereto, and the
mixture was stirred for 6 hours at -72C. After addition of
lN HCl (9 ml), the organic layer was separated, washed with
3N HCl (5 mlx2~, and dried over Na2SO4. Removal of the
solvent in vacuo gave a residue, which was then subjected to
silica gel column chromatography to give optically active
~arcophytol A (81 mg, 60%) and nonreacted ketone compound
(51 mg, 37%).
Optical purity of the optically active sarcophytol
A was determined to be 87% by means of high pressure liquid
chromatography u~ing a separation column for optical
isomers, specifically CHIRALCELL OD (commercially available
204~
- 81 -
from Daisel Kagaku Kogyo), said analysis being referred to
as "HPLC analysis using CHIRALCELL OD~ hereinafter.
Reference Example 13
A solution of lithium aluminum hydride in diethyl
ether (2.26 ml, 1.40 mmol, 0.62M) was stirred under argon
atmosphere. To the solution was dropwise added
(S)-2-(anilinomethyl)pyrolidine (296 mg, 1.68 mmol) in
diethyl ether (3 ml) at room temperature over 10 minutes.
The reaction mixture was stirred at room temperature addi-
tional one hour and then cooled to 72C. To the mixture
was gradually added the ketone compound (162 mg, 0.56
mmol)in diethyl ether (5 ml), which had been prepared in
Reference Example 10. After one hour stirring at -72C,
saturated aqueous sodium bicarbonate (1 ml) was added, and
the mixture was stirred at room temperature for 10 minutes.
After addition of lN HCl (15 ml) and diethyl ether (20 ml),
the organic layer was separated. The aqueous layer was
extracted with diethyl ether (20 ml), and the extract was
washed with saturated aqueous sodium chloride (20 ml), dried
over Na2SO4, and evaporated in vacuo to remove the solvent.
The resultant residue was subjected to silica gel column
chromatography to obtain optically active sarcophytol A (126
mg, 78%).
Optical purity of the thus obtained sarcophytol A
was 92% when measured by HPLC analysis using CHIRALCELL OD.
[~] 4D: +209.9 (c=0.372, CHCl3)
Reference Example 14
2~4~35
- 82 -
A solution of lithium aluminium hydride in diethyl
ether (2.94 ml, 2.0 mmol, 0.68M) was stirred under argon
atmosphere, and to the solution was gradually added
(S)-2-(2,6-xylidinomethyl)pyrrolidine (490 mg, 2.4 mmol) at
room temperature, and the mixture was stirred at room
temperature for 2 hours. The reaction mixture was cooled to
-74C, and to the mixture was dropwise added over 10 minutes
a solution of the ketone compound (69 mg, 0.24 mmol) in
diethyl ether (3 ml), which had been prepared in Reference
Example 10. After one hour stirring at -74C, saturated
aqueous sodium sulfonate (1 ml) was added, and the resultant
mixture was stirred at room temperature for a while. After
addition of diethyl ether (10 ml) and diluted HCl l20 ml),
the organic layer was separated, and the aqueous layer was
extracted with diethyl ether (20 ml). The extract was
washed with saturated aqueous sodium chloride (20 ml), dried
over Na2SO4, and evaporated in vacuo to remove the solvent
to give a residue, which was subjected to silica gel column
chromatography to obtain optically active sarcophytol A (61
mg, 88%).
Optical purity of the optically active sarcophytol
A was 93% according to HPLC analysis using CHIRALCELL OD.
~] D: +204.4 (c=0.27, CHCl3)
Reference ExamPle 15
A suspension of tin (II) chloride (382 mg, 2.01
mmol) and (R)-1-methyl-2-(piperidinomethyl)pyrrodine (366
mg, 2.01 mmol) in dichloromethane (6 ml) was cooled to -72C
20~95
~ 83 -
under argon atmosphere. To the suspension was added
diisobutylaluminum hydride in toluene (1.0 mmol), and the
mixture was stirred for ten minutes. To the mixture was
gradually added at -72C a solution of the ketone compound
~100 mg, 0.349 mmol) in dichloromethane (3 ml). The
reaction mixture was stirred for 4 hours, and the stirring
was continued at room temperature for 30 minutes after
addition of saturated aqueous sodium chloride (3 ml).
Resultant precipitates were filtered by the use of sellite,
and the filtrate was dried over Na2SO4 and evaporated in
vacuo to remove the solvent. The resultant residue was
purified with silica gel column chromatography to give
optically active sarcophytol A (79.2 mg, 79%).
Optical purity of the sarcophytol A thus obtained
was 42% according to HPLC analysis using CHIRALCELL OD.
[a]25D: +101.9 (c=0.54, CHCl~)
Industrial UtilitY
As stated above, the compounds (I) of the present
invention are very useful as intermediates for preparing
sarcophytol A which possesses an anti-carcinogenic promotor
activity and anti-tumor activity. Thus, the present
invention pro~ides a method suitable for industrial
production of sarcophytol A.