Note: Descriptions are shown in the official language in which they were submitted.
X-8265A
TIT~
6-SUBSTITUTED-HEXAHYDROBENZ~CD]INDOLES
Field of the_Invention
This invention relates to the fields of
synthetic organic chemistry and pharmaceutical
chemistry and involves hexahydrobenz[cd]indoles which
are useful in treating conditions requiring regulation
L0 of the serotonin function in the body.
BackgrQ~nd of th~ Inv~.ntion
Over the last several years it has become
apparent that the neurotransmitter serotonin (5-
hydroxytryptamine -- 5-HT) is associated directly or
indirectly with a number of physiological phenomena,
including appetite, memory, thermoregulation, sleep,
sexual behavior, anxiety, depression, blood pressure
lowering and hallucinogenic behavior [Glennon, R. A.,
.~. Med~ ~h~m~, lQ, 1 (1987)].
It has been recognized that there are
multiple types of 5-HT receptors. These receptors have
been classified as 5-HT1, 5-HT2, and 5-HT3 receptors,
with the former being further divided into the sub-
classes 5-HT1A, 5-HT1B, 5-HT1C, and 5-HT1D. The
:: .
~-8265A 2 ~0~9~7~
binding affinity of a compound for one or more 5-HT
receptors can provide a desirable physiological effect
or minimize an undesirahle effect. Therefore it is
desirable to provide compounds which can bind to 5-HT
receptors to act as serotonin agonists or antagonists.
Flaugh in U.S. Patent No. 4,576,959 (issued
1986) disclosed a family of 6-substituted-4-
dialkylamino-l,3,~,5-tetrahydrobenz[cd]indoles which
are described as central serotonin agonists. Leander
in U.S. Patent 4,745,126 (1988) disclosed a method for
treating anxiety in humans employing a 4-substituted-
1,3,4,5-tetrahydrobenz[cd]indole-6-carboxamide
derivative.
Certain indolines have been reported, as in
U.S. Patent No. ~,110,339 of Bach e~ al. (197~), Flaugh
et al., J. Med. Che~.. ~1, pp 1746-1753 (1988), Flaugh
in U.S. Patent No. 4,576,959 and ~uropean Patent Ap-
plication 153083 (published 1985). These were used as
intermediates in the preparation of the corresponding
indoles.
It has now been found that certain 6-sub-
stituted- and particularly 6-acyl-substituted-4-amino-
hexahydrobenz[cd]indoles (indolines) particularly
certain stereoisomers of such indolines are useful in
treating conditions requiring modification of serotonin
function in the body.
Summarv of the I~vention
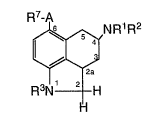
This invention relates to a compound of the
Formula I
' ,: :, `
~',
X-8265A 3
7~
~N p~1 R2
F~3N~
~1 I
wherein:
R1 is hydrogen, C1-C~ alkyl, C3-C4 alkenyl,
cyclopropylmethyl, phenyl-s~stituted C1-C4 alkyl,
- CoR4 r - ( CH2) nS(Cl-C4 alkyl), or -(CH2) nCoNR5R6;
R2 is hydrogen, C1-C4 al]cyl, C3-C4 alkenyl,
or cyclopropylmethyl;
R3 iS hydrogen, Cl-C4 alkyl or an amino-
blocking group;
n is 1-4;
R4 is hydrogen, C1-C4 alkyl, Cl C4 haloalkyl,
C1-C4 alkoxy or phenyl;
R5 and R6 are independently hydrogen, a C1-C4
alkyl, or a C~-C8 cycloalkyl;
R7 iS Cl-C8 alkyl, substituted C1-C8 alkyl, aryl,
substituted aryl, aryl (Cl-C4 alkyl), substituted aryl (Cl-C4
alkyl), C3-C7 cycloalkyl-substituted methyl, or C3-C7
cycloalkyl with the proviso that when A is C-C then R7 is Cl-
C7 alkyl, substituted Cl-C7 alkyl, aryl, aryl (Cl-C3 alkyl),
substituted aryl, substituted aryl (Cl-C3 alkyl), or C3-C7
cycloalkyl;
A is C=O, CHOH or C-C; and
pharmaceutically acceptable salts thereof.
In a further embodiment, the instant invention
comprises a compound of Formula I wherein
(a) R1, and ~2 are independently ~ydrogen or a
Cl-C4 alkyl;
(b) R3 is hydrogen;
(C) R7 iS Cl-C8 alkyl, substituted C1-C~ alkyl,
phenyl, phenyl (C1- C4 alkyl);
(d) n is 2-4; and
.
X-8265A 4 20~17~
(e) A is C=O; and pharmaceutically acceptable
salts thereof.
The invention also provides a pharmaceutical
formulation comprising a compound of Formula I and a
pharmaceutically acceptable excipient therefor.
A further embodiment of the invention is a
method for effecting a biological response at a 5-HT
receptor by adminlstering an effective amount of a
compound of Formula I. Further embodiments involve the
treatment of disease states with re~uire regulation of
serotonin function in the body.
Detailed Descriptio~ of tk~ In~en~iQn
As used herein, the term "alkyl" represents a
straight or branched alkyl chain having the indicated
number of carbon atoms. For example, "Cl-C~ alkylll
groups are methyl, ethyl, n-ProPyl, isopropyl, ~-butyl,
sec.-butyl, isobutyl and ~gr~-butyl. "Cl-C8 alkylll -
groups include those listed for Cl-C4 alkyl as well as
~-pentyl, 2-methylbutyl, 3-methylbutyl, g-hexyl, 4-
methylpentyl, ~-heptyl, 3-ethylpentyl, 2 methylhexyl,
2,3-dimethylpentyl, ~-octyl, 3-propylpentyl, 6-methyl-
heptyl, and the like.
The term " C3 - c4 alkenyl" refers to
olefinically unsaturated alkyl groups such as -CH2CH=CH2
-CH(CH3)CH=CH2, -CH2CH2CH=CH2 and the like.
The term ~aryl~ means an aromatic carbocyclic
structure ha~ing one or two rings with a total of six to ten
carbon atoms in the rings. Examples of such ring structures
are phenyl, naphthyl, indan~l, and the like.
The term l'cycloalkyl" means an aliphatic
carbocyclic structure ha~ing the indicated number of
carbon atoms in the ring. For example, the term " C3 - C7
cycloalkyl" means cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl, and cycloheptyl.
The term "aryl (C1 C4 alkyl)" means an
, ~ ,
~ ;,
~9~7~
X-8265A 5
aromatic carbocyclic structure joined to a Cl-C4 alkyl
group. Examples of such groups are benzyl,
phenylethyl, a-methylbenzyl, 3-phenylpropyl, a-
naphthylmethyl, ~-naphthylmethyl, 4-phenylbutyl, and
the like. Similarly the term ~aryl (C1-C3 alkyl)"
means an aromatic carbocyclic st m cture joined to a C
-C3 alkyl.
The C1-C8 alkyl, the aryl, the aryl (C1-C4
alkyl) , and aryl (C1-C3 alkyl) groups can be
substituted by one or two moieties. Typical aryl
and/or alkyl substituents are C1-C3 alkoxy, halo,
hydroxy, C1-C3 thioalkyl, and the like. Moreover, the
aryl, aryl (C1-C4 alkyl) and aryl (C1-C3 alkyl) groups
can also be substituted by a Cl-C3 alkyl or a
trifluoromethyl group.
In the foregoing, the term "C1-C3 alkyl"
means any of methyl, ethyl, ~-propyl, and isopropyl;
the term "CL-C3 alkoxy" means any of methoxy, ethoxy,
n-propoxy, and isopropoxy; the term ~halo" means any oE
fluoro, chloro, bromo, and iodo; and the term "C1-C3
thioalkyl~ means any of methylthio, ethylthio, n-
propylthio, and isopropylthio.
Examples of substituted Cl-C8 alkyl are
methoxymethyl, trifluoromethyl, 6-chlorohexyl, 2-
bromopropyl, 2-ethoxy-4-iodobutyl, 3-hydroxypentyl,
methylthiomethyl, and the like.
Examples of substituted aryl are ~-bromo-
phenyl, ~-iodophenyl, ~-tolyl, n-hYdroxYphenYl, ~-(4-
hydroxy)naphthyl, ~-(methylthio)phenyl, m- trifluoro-
methylphenyl, 2-chloro-4-methoxyphenyl, a-(5-chloro)-
naphthyl, and the like.
Examples of the substituted aryl (Cl-C4
alkyl) are ~-chlorobenzyl, Q-methoxybenzyl, m-
(methylthio)-a-methyl-benzyl/ 3-(4l-trifluoromethylphenyl)-
propyl, o-iodobenzyl, ~-met~ylbenzyl, and the like.
The term "amino-blocking group" is used as it
X-8265A 6
is fre~uently used in synthetic organic chemistry, to
refer to a group which will prevent an amino group from
participating in a reaction carried out on some other
functional group of the molecule, but which can be
removed from the amine when it is desired to do so.
Such groups are discussed by T. W. Greene in chapter 7
of Pro~ective Groups in Orqanic Synthesi~, John Wiley
and Sons, New York, 1981, and by ~. W. Barton in
chapter 2 of PrQtective Grou~s in Orgam ç Chemistry. J.
F. W. McOmie, ed., Plenum Press, New York, 1973, which
are incorporated herein by reference in their entirety.
Examples of such groups include those of the formula
-COOR where R includes such groups as methyl, ethyl,
propyl, isopropyl, 2,2,2-trichloroethyl, l-methyl-l-
phenylethyl, isobutyl, ~-butyl, ~-amyl, vinyl, allyl,
phenyl, benzyl, ~-nitrobenzyl, o-nitrobenzyl, and 2,4-
dichlorobenzyl, benzyl and substi~uted benzyl such as
3,4-dimethoxybenzyl, o-nitrobenzyl, and triphenyl-
methyl trityl; acyl groups and substituted acyl such as
formyl, acetyl, chloroacetyl, dichloroacetyl,
trichloroacetyl, trifluoroacetyl, benzoyl, and p-
methoxybenzoyl; and other groups such as
methanesulfonyl, ~-toluenesulfonyl, ~-
bromobenzenesul~onyl, ~-nitrophenylethyl, and p-
toluenesulfonylaminocarbonyl. Preferred amino-blocking
groups are benzyl (-CH2C6H~), trityl, acyl [C(O)R] or SiR3
where R is Cl-C4 alkyl, halomethyl, 2-halo-substituted
alkoxy, or phenyl.
The compounds of the instant in~ention have
at least 2 chiral centers and therefore at least four
stereoisomers can exist for each. Chiral centers exist
at position 2a and 4 as in Formula I. If a substituent
group contains a chiral cen~er, then additional stereo-
isomers can of course exist. Racemic mixtures as well
as the substantially pure stereoisomers of Formula I
are contemplated as within the scope of the present
X-8265A 7
invention. The term ~substantially pure" refers to at
least about 90 mole percent, more preferably at leas~
about 95 mole percent, most preferably at least about
98 mole percent of the desired stereoisomer being
present compard to the other stereoisomers present.
Particularly preferred stereoisomers of Formula I are
those in which the configuration of the chiral center
at position 2a is S and at position 4 is R, i.e., 2aS,
4R.
The terms "~" and "S" are used herein as
commonly used in organic chemistry to denote specific
configuration of chiral center. The term ~'R" refers to
"rightl~ and refers that configuration of a chiral
center with a clockwise relationship of group
priorities (highest to second lowest) when viewed along
the bond toward the lowest priority group. The term
"S" or l'left'l refers to that configuration of a chiral
center with a counterclockwise relationship of group
priorities (highest to second lowest) when viewed along
the bond toward the lowest priority group. The
priority of groups is based upon their atomic number
(heaviest isotope first). A partial list of priorities
and a discussion of stereo chemistry is contained in
the book: The Vocabulary Qf Qrganic Chemi~trv Orchin,
et al. John Wiley and Sons Inc., publishers, page 126,
w~lich is incorporated herein by reference.
While all of the compounds of the in~ention
are useful for the purposes taught herein, certain of
the present compounds are preferred for such uses.
Preferably R1 and R2 are both C1-C~ alkyl, and
especially ~-propyl. R3 is preferably hydro~en, R7 is
preferably C1- C4 alkyl, substituted Cl- C4 alkyl, or C3
-C7 cycloalk~l. Although compounds in which A is CHOH
or CaC have activity, their primary utility is as
intermediates in the preparation of compounds in which
A is C=O. Other preferred aspects of the present
- X-8265A 8 ~ 37~
invention are noted hereinafter.
As pointed out above, this invention includes
the pharmaceutically-acceptable salts of the compounds
of Formula I. Since the compounds of this invention
are amines, they are basic in nature and accordingly
react with any number of inorganic and organic acids to
~orm pharmaceutically acceptable salts such as hydro-
chloric acid, nitric acid, phosphoric acid, sulfuric
acid, hydrobromic acid, hydroiodic acid, phosphorous
acid and others, as well as salts derived from non-
toxic organic acids such as aliphatic mono and
dicarboxylic acids, amino acids, phenyl-substituted
alkanoic acids, hydroxyalkanoic and hydroxyalkandioic
acid, aroma~ic acids, aliphatic and aromatic sulfonic
acids. Such pharmaceutically-acceptable salts thus
include sulfate, pyrosulfate, bisulfate, sulfite,
bisulfite, nitrate, phosphate, monohydrogenphosphate,
dihydrogenphosphate, metaphosphate, pyrophosphate,
chloride, bromide, iodide, acetate, propionate, capry-
late, acrylate, formate, tartrate isobutyrate, caprate,
heptanoate, propiolate, oxalate, malonate, succinate,
suberate, sebacate, fumarate, maleate, mandelate,
butyne-1,4-dioate, hexyne-1,6-dioate, hippurate,
benzoate, chlorobenzoate, met~ylbenzoate, phthalate,
terephthalate, benzenesulfonate, toluenesulfonate,
chlorobenzenesulfonate, xylenesulfonate, phenylacetate,
phenylpropionate, phenylbutyrate, citrate, lactate, b-
hydroxybutyrate, glycolate, malate, naphthalene-1-
sulfonate, naphthalene-2-sulfonate and mesylate.
The following list illustrates representative
compounds of the present invention:
4-(di-~-propylamino)-6-acetyl-1,2,2a,3,4,5-
hexahydrobenz[cd~indole;
4-(di-n-propylamino)-6-(2,2-dimethylpropanoyl!-
1,2,2a,3,4,5-hexahydrobenz[cd]indole;
4~(diethylamino)-6-propanoyl-1,2,2a,3,4,5-
~ X-8265A 9 2~ 7~
hexahydrobenz[cd]indole;
4-(di-n-propylamino)-6-benzoyl-1,2,2a,3,4,5-
hexahydrobenz[cd]indole;
(2aS,4R)-4-(~-propylamino)-6-(2-methylpropanoyl)-
1,2,2a,3,4,5-hQxahydrobenz[cd]indole;
~.-methyl-4-(di-~-propylamino)-6-benzoyl-
1,2,2a,3,4,5-hexahydrobenz[cd]indole;
l-methyl-4-(n-propylamino)-6-(3-methylbutanoyl)-
1,2,2a,3,4,5-hexahydrobenz~cd]indole;
(2aS,4R)-4-(di-~-propylamino)-6-(2,2-dimethyl-
propanoyl)-1,2,2a,3,4,5-hexahydrobenz[cd]indole;
(2aS,4R)-4-(di-n-propylamino)-6-benzoyl-
1,2,2a,3,4,5-hexahydrobenz[cd]indole; and
4-(N-~-propyl-N-cyclopropylmethyl)amino-6-
propanoyl-1,2,2a,3,4,5-hexahydrobenz[cd]indole;
(2aS,4S)-4-(di-n-propylamino)-6-acetyl-
1,2,2a,3,4,5-hexahydrobenz[cd]indole;
(2aS,4R)-4-(di-~-propylamino)-6-(2-phenylethanoyl)-
1,2,2a,3,4,5-hexahydrobenz[cd]indole; and
Scheme 1 depicts a process for preparing
compounds of the present in~ention wherein Rl, R2 and
R7 are as defined abo~e and Z is an amino-blocking
group as defined hereinabove.
According to one route of this pro~ess, a 4-
amino-6-bromohexahydrobenz[cd]indole 1 is combined with
an e~uimolar to slight excess amount of potassium
hydride in diethyl ether. The reagents are generally
combined at a cold tempera~ure, typically in the range
of about -20C to about 10C, pre~erably at about 0C.
The resulting mixture is cooled to a temperature in the
range of about -100C to about -60C, preferably at
about -78C, and cornbined with a lithiatin~ reagent,
preferably in at least a two molar excess amount.
Suitable lithiating reagents include ~-butyllithi~,
the preferred t-butyllithium, and other similar organo-
lithium compounds is preferred. The reaction is
- X-8265A 10 ~7~ -
preferably conducted at a temperature in the range of
about -100C to about -20C~ more preferably at about
-60~ to about -40C.
The 4-amino-6-lithiohexahydrobenz~cd]indole
thus prepared is then contacted with an appropriate
electrophile such as L-C(o)R7 wherein ~7 iS defined
above and L is a good leaving group such as chlorine
bromine, methoxy, phenoxy and the like. Typically, a
solution of the compound ~ at a temperature in the
range of about -100C to about -60C, preferably at
about -80C, is added to a solution of this reagent in
a mutual solvent. If an excess amount of the
electrophile is employed in the reaction, the 1-amino
group will also be acylated (ie Z is the acyl group
R7C(o) in compound ~) and a subse~uent hydrolysis
reaction is required to provide the free indoline, I.
A 1:1 ratio of electrophile to lithiated indoline
(compound 2) can be used to minimize acylation of the
l-nitrogen. The reaction is preferably conducted at a
temperature in the range of about -40C to about 10C.
The desired compound iæ purified by quenching the
reaction mixture with, for example, ice water when a
1:1 ratio is used. With a higher ratio in which
significant l-acylation is obtained, the product is
hydrolyzed using an acid such as sulfuric acid or a
base such as sodium hydroxide. The mixture is then
washed with a water-immiscible organic solvent. The
organic phase is extracted with acid; the aqueous
phases are combined and made basic; and the desired
compound is extracted with a water immiscible organic
solvent. The organic solvent is then concentrated,
typically under vacuum, and the desire~ compound I is
further purified, if necessary, by standard procedures.
In an alternative route, the l-nitrogen can be
"blocked" or l'protectedll before initiating the
metallation reaction. A blocking group (depicted as
:
, ~
:
X-8265A 11 ~7~i
"Z") such as SiR3, C(O)R, or CH2(C6H~) where R is C3-C4
alkyl or phenyl (C6H5) is preferably used to provide
compound 1~ Compound la is then reacted with a
lithiating agent as described above to provide compound
~a. Compound ~a can then be acylated by contacting
with a suitable electrophile as described hereinabove.
The resultin~ compound 3a is then deprotected by
hydrolysis when Z is SiR3. When Z is benzyl, compound
~ can be subjected to hydrogenolysis over a catalyst
such as palladium to remove the benzyl group. The
desired compound is isolated by standard conditions and
purified by crystallization from common solvents or
column chromatography over solid supports such as
silica gel or alumina.
An alternative synthesis of the compounds I
is depicted in Scheme 2 and involves treatment of the
6-lithio derivatives 2 and ~a (depicted in Scheme 1)
with an aldehyde~ R7CHo, to form an alcohol 4 or ~.
Oxidation of the alcohol can be accomplished with
oxidants known to those skilled in the art for such
purposes such as pyridinium chlorochromate, dimethyl-
sulfoxide and oxalyl chloride, an aqueous solution of
chromic acid and sulfuric acid, and the like. Depro-
tection of the 1-amino group provides the free amine
2S compounds I.
- X-8265A 12 ~ 0~917~i
~cheme 1
Br Br
,NR1R2 ~ NR1R2
I!~J Protect~ I~J
HN~ 1 ,N~ ~
KH
¦ LiC4Hg
1 ~
~,NR1R2 ~NR1Rr
11 Z
R7CX
1 ~ 1 ~ :
R7~o ~7~
NR1R2 ~ NR1R2
~t ~
-~N~ x ,N~
~;' ' -
2~ L7~
X-8265A 13
Scheme 2
Li Li
~g~NR1R2 ~,NR1~2
KN ~ ZN ~
lR7CHo lR CHO
\/ R7\/oH
p~2 ~,N ~1 R2
'r _~Deprotect ~
HN I ~ ZN I ~a
I [o]
[o]
R7~o R7~o
NP~1R2 ~ R2
~ Deprot~ct ~J
HN I ZN
The alcohol intermediates 4 and ~a can alter-
natively be prepared as depicted in Scheme 3 by
addition of an organometallic reagent (R7M) such as an
alkyl lithi~m R7Li or a Grignard reagent R7MgX to
aldehyde 5 and ~a, respectively.
,
x-826sA 14 Z~4917~
S~heme 3
~NRIR2 CHO N~ R2
~ M 1R7-M
\~ R7~oH
~NRlR2 ,~ R1R2
~lJ ~ ~U
T T
HN~ Deprotect l l
Various routes can be used to prepare
aldehydes 5 and ~a. The methods disclosed herein are
not intended to be exhaustive and other procedures may
be apparent to those skilled in the art. One route
involves treating 6-lithioderivatives ~ and ~a with
dimethylformamide followed by aqueous work up. Anoth~r
method depicted in Scheme 4 involves the preparation of
the 6-nitrile deriva~ive ~ followed by partial reduction and
hydrolysis.
,
..
- . :
X-8265A 15 ~ 7
~cheme 4
NR1~2 N~1R2
E~r~H cuc I N~H
B~ z 6
(~,N H2
Hl\l NRlR2 NR1~2
h H 3~1 H2SO4 )( h H
H~ -- -D H ~
E~z 7 H s
The l-benzoyl-6-bromo-derivative 1 is con-
tacted with a mixture of cuprous cyanide and cuprous
iodide in dimethylformamide at about 140C. The re-
sulting 6-nitrile ~ is llydrogenated over Pd/C in the
presence of semicarbazide to provide the 6-
semicarbazone, compound 7. This is hydro:lyzed using
sulfuric acid to provide aldehyde ~.
In an alternative method of preparation,
depicted in Scheme 5, the 6-nitrile derivative ~ is
contacted with a reducing agent [H] such as diisobutyl-
aluminum hydride. The resulting aldehyde ~ can be
contacted with an organometallic reagent such as a
Grignard reagent, R7MgBr, to provide alcohol ~a which
is oxidized as described hereinabove to the l-blocked-
6-acyl derivative ~.
: :
,
. ; ~
:
X-8265A 16
~h~
C~NRlye ~NR1RZ
Z-N ~ Z-N ~
¦R7MgBr I 7
R MgBr
R~NR~RZ R¢~NRlRZ
ZN ;~ ZN 4a
Another method of preparation of compounds of
Formula I involves the Friedel-Crafts acylation of the
6-~ indoline ~ as depicted in Scheme 6. The indoline
8, wherein R1~ R2 and Z are as defined hereinabove, is
contacted with an acylating agent such
- ,. . .
:
,. . : . ' ~:' '
X-8265A 17 ~9~7~
~çhçme 6
o
R7-C//
N~1 R2
Lewis Acld
8 ZN
/ydrolysis
R7- ~/
~NRl R2
HN
aæ a carboxylic acid anhydride [(R7Co) 2~ or a
carboxylic acid halide, particularly the acid chloride
R7C(O)Cl, in the presence of a Lewis ACid. Preferred
Lewis Acids include aluminum chloride, aluminum
bromide, BF3, SnCl4, HF, TiCl~, and the like. The
reaction is preferably conducted in a solvent commonly
used for such acylation reactions, such as
nitrobenzene, and the like. The reaction is normally
conducted at a temperature in the range of 20C to
reflux. Preferably the 1-amino group is protected with
a blocking group depicted as Z in Scheme 6. A
preferred blocking group is the benzoyl group. The
blocking group can be removed from compound ~a by
hydrolysis, preferably using a base such as sodi~
hydroxide, to provide compound I.
Alternatively, certain com~ounds of Formula I
can be prepared using the 6-iodo derivative ~ as
,
X-826sA 18
depicted in Schemes 7 and 8 wherein R1, R2 and Z are as
defined hereinabove. In Scheme 7 a method is shown in
which a 6-alkyne derivative is prepared. This method
provides 6-acyl compounds in which there is a methylene
group adjacent to the carbonyl group. In this method
the 1-amino group is protected with a group
(represented by Z) such as a benzoyl group. This
compound ~ is contacted with a palladium catalyst
Pd(PPh3) 4 [where Ph is phenyl] and the tin alkyne
compound R7a-C3C-Sn(CH3)3. R7a is a Cl-C7 alkyl,
substituted Cl-C7 alkyl, aryl, aryl (Cl-C3 alkyl),
substituted aryl, substituted aryl (C1-C3 alkyl), or C3-C7
cycloalkyl group. This reaction is normally
conducted in a solvent such as toluene at an elevated
temperature, eg. about 100C. Typically an excess of
the tin alkyne is used along about 0.25 equivalents of
the palladium compound based on compound ~. The 6-
alkyne lQ is then contacted with HgS0~ in water or with
a~ueous acid to provide the ketone 11. The 1-blocking
group can be removed by hydrolysis with base as
described above to provide compound I.
7~
x-826sA 19
Scheme 7
R7a
111
P7-C-C-Sn(Ca3)3 ~ ,NR1RZ
;Z-N ~ Z-N L~
~120
HgSO4
R7a C~NRl R2 R7a-CH2 C~ NR~ RZ
HN I ~-N 11
In Scheme 8 a preparative method is depicted in
which a vinyl ether is reacted with the 6-iodo derivative 9.
R1, R2 and Z are as defined hereinabove wi~h Z preferably a
benzoyl group, except as provided below. This method
provides a 6-tl-alkoxyalkenyl)derivative ~1 which can then be
hydrolyzed and deprotected to provide the desired con~oound of
Formula I. Alternatively, ~he derivative 81 can be
deprotected, with for example butyl lithium, and then the
vinyl group hydrolyzed. In this method the 1-amino group is
protected with an amino protecting group, preferably a
benzoyl group. This compound 9 is then contacted with a
palladium catalyst and the desired vinyl e~her. The vinyl
' - ~
.
X 8265A 20 ~:049~
ethers useful in this method include those in which Rc is a
Cl-C4 alkyl and Q is hydrogen or an alkyl tin, alkyl or
alkoxy boron, zinc halide, or magnesium halide, for example
tributyltin. When Q is zinc halide or magnesium halide, it
is preferred that Z be a group such as trityl. Ra and Rb can
independently be hydrogen, Cl-C6 alkyl, substituted Cl-C6
alkyl, aryl, aryl (Cl-C2) alkyl, substituted aryl,
substituted aryl (Cl-C2) alkyl, or C3-C7 cycloalkyl group.
The palladium catalyst used can be palladium powder (black)
or Pd(PPh3)4 [where Ph is phenyl]. The Pd(PPh3)4 is commonly
used with toluene at reflux. The Pd-black can be used with
triphenylphosphine in toluene at reflux or in a mixture of
acetonitrile and triethyl amine at about 100C. Similar
reactions are reported in ~11. Chem, ~o~. ~n. (1987), Q,
767-768, incorporated herein by reference
.
~(~4~
X-826sA 21
Scheme 8
Ra
NR1R2 ~ C\ R ~ ORC
Pd - Black/PPh
ZN - I or
9 Pd (PPh3) 4 7N
Ra F~a l
R C~c~,O RC~c~O
~NR1 R2
HN ~N
:~a
In another preparation method depicted in
Scheme 9, the 6-iodo derivative ~ can be used to
prepare certain 6-acyl compounds directly. This i9
accomplished by contacting the 6-iodo compound with
trialkyltin-alkyl complex and car~on monoxide in the
presence of a palladium catalyst Pd(PPh3)4 [where Ph is
phenyl] as described in the literature for arylhalides.
[A. Schoenberg and R. F. Heck, ~. Orq Ch~m., ~, p.
3327 (1974); and A, Schoenberg, I. Bartoletti, and R.
F. Heck, ~. Or~. Cham., ~, p. 3318 (1974)~. The
blockin~ group Z which is preferably benzoyl for this
method can ~e removed as described hereinabove to
provide cqm~pund I.
J
X-8265A 22 ~9~ 7
~h~m~
o
NR1 2 R7 C
co~ ~,NRl R2
Z-N R3~nRl Z-N
O ,~/
R7- ~
~ NR1R2
HN
The processes depicted in Schemes l-9 can
result in a mixture of products which require
purification by standard methods of purfication, for
example, crystallization or chromatographic techniques
as appropriate.
Scheme lO illustrates a preparation of the s~arting
material for reaction Scheme l.
, ~ . . .
., ~ `
,
X-826sA 23
Scheme 10
~ $~f~
R8
~NH2 ~ ~ NHR~ ~N
3,N ~7 R3,N ~ F,3,N lE
~,NH2
,N
R3
Epoxides of formula ~3 are known in the art
or can be prepared from compounds such as ketone 12,
which is known to the art, using common rea~ents and
techniques. For example, Flaugh, ~ al.,
~. Med. Chem~ , ~1, 1746 (1988); Nichols Q~
Org. Prep. a~d ~roG ~ ~ Int., 9, 277 (1977); and Leanna
e~ al., T~t. ~ett~, 30, No. 30, 3935 (1989), teach
methods of preparation of various embodiments of com-
pounds of formula 1~. Those ski1led in the art of
organic chemis~ry will recognize tha~ there are four
L5 stereoisomers of formula 1~:
x-826sA 24 ~ ~ ~ 9 ~ 7
~ D ~ ~ O
B lla B L~b B l~c B l~d
Formulae L~a and 1~ are herein referred to
collectively as the exo-isomers; similarly, formulae
1~ and 13d are the endo-isomers. Leanna ~_al~,
~ara, teach the preparation of epoxides of formula 13
which are substantially exo or substantially endo, as -
desired. A preferred starting material is the compound
of formula 13 wherein R3 is benzoyl; the most preferred
starting material is the mixture of substantially the
exo-isomers thereof.
Amino alcohols of formula 1~ are formed by reacting
an epoxide of formula 13 with an amine of formula R8~2, where
R8 can be hydrogen, a Cl-C4 alkyl, or a Cl-C~ alkyl
substituted with one to three groups selected from halogen,
nitro or phenyl. Such amines are readily available. Opening
of the epoxide ring proceeds substantially regiospecifically
with the amino group at the 5-position and the hydroxyl
group at the 4-position. The reaction is also
stereospecific in the sense that stereoisomers of
formulae 14a-d are predictably formed from, re-
spectively, stereoisomers of formulae l~a-d.
~HR8 NHR~ NHR8 NHR8
OH $~
B L4a B ~Lb B ~c ~3 ~Ld
25A stereoselective synthesis of the amino
alcohol of formula 1~, and hence of all the subsequent
7~j
~-8255A 25
intermediates and products of Scheme 10, can be effected
by using a substantially pure enantiomer of an amine of
the formula R8NH2, wherein R8 contains at least one
chiral center. A particularly preferred amine is (+)
or (-) l-phenylethylamine. The diastereomers of the
resulting amino alcohol can then be separated by a
number of means known in the art, for example by
chromatography or crystallization. Suitable solvents
for recrystallization include those such as diethyl
ether, butanol, and mixtures of hexane and ethyl
acetate. An alternative method of achieving a
stereospecific synthesis comprises conversion of all
the diastereomers of formula 1~ to corresponding
diastereomers of formula 1~, followed by the separation
of said diastereomers of formula 1~; that alternative
method is discussed below. If a stereoselective syn-
thesis is not desired, then separation of the stereo-
isomers of the amino alcohol of formula 1~ is not
required and the amine R8NH2 need not be optically
active.
A particularly efficient stereoselective
process for a highly preferred compound of formula 14,
l-benzoyl-4-hydroxy-5-(1-phenylethyl)amino-1,2,-
2a,3,4,5-hexahydrobenz[cd]indole, comprises the
reaction of a mixture of substantially the exo-isomers
of the corresponding epoxide of formula 1~, or a
mixture of substantially the endo-isomers of the
corresponding epo~ide of formula 1~, with a
substantially pure enantiomer of l-phenethylamine in
the solvent butanol and the subseguent selective
crystallization of one of the two isomers of the amino
alcohol. The temperature of the reaction is preferably
from about 50 to about 150C, more preferably in the
range of about 80 to about L00C.
After the reaction is complete, as determined for
example by thin layer chromatography or liquid
. ~
X-8265A 26 ~9~7~
chromatography, the desired amino alcohol is crystallized at
about -20 to about ~0C; the preferred temperature for the
crystallization is about 0 to about 15C. Therefore
this process has the valuable attribute that the
reaction and the separation of stereoisomers occur
efficiently in a slngle step. By the proper selection
of the epoxide isomers, exo or endo, and the enantiomer
of l-phenylethylamine, R or S, one can determine which
of the stereoisomers of the compound of formula 14
precipitates from the reaction mixture. For example, a
preferred stereoisomer of l-benzoyl-4-hydroxy-5-(1-
phenylethyl)amino-1,2,2a,3,4,5-hexahydrobenz[cd]indole,
the (2a-S,4-R,5-R)-isomer can be selectively prepared
by reacting the exo-epoxides with S-l-phenylethylamine.
A number of methods of forming aziridines such as
those of formula 1~ from amino alcohols such as those of
formula 14 are known to the art. Two examples are the
use of diethyl azodicarboxylate and triphenylphosphine
(O. Mitsunobu, 9~n~h9EiQ, January, 1981, page 1), and
the use of bromine and triphenylphosphine (J. P.
Freemer and P. J. Mondron, a~n¢h~gi~, December, 1974,
page 894).
A particularly efficient alternative to the above
mathods involving treating a compound of formula 1~ with a
tertiary amine in an inert solvent followed by the addition
of methanesulfonyl chloride. The stereoisomers l~a-d of
the a2iridine 1~ arise respectively from the
stereoisomers of formula l~a-d with retention of
configuration at any chiral center in the substituents
R3 or R8 as well as at position 2a:
X-8265A 27 ~ 76
R8 N,R ,R8 ~N'
~H r ~ H r H
B ~,~a B ~b B ~,~c B L~d
Suitable tertiary amines include those of the
formula (R9)3N, where the R9 group~ are independently C1-C~
alkyl. Suitable solvents are chlorinaked hydro-
carbons such as methylene chloride, chloroform, carbon
tetrachloride, and dichloroethane; aromatic
hydrocarbons such as benzene, toluene, and the xylenes;
and ethers such as tetrahydroEuran, diethyl ether, and
methyl ~-butyl ether. The reaction can be conducted at
a temperature from about -35 to about 45C. In ~he
preferred embodiment, the amino alcohol is treated with
triethylamine in methylene chloride a~ about -20 to
about 0C, then the reaction mixture is warmed to about
15 to about 35C for ~he completion of the reaction.
If desired, tne product, an aziridine of formula 1~,
can be crystallized from an appropriate solvent such as
acetonitrile or isopropanol after an aqueous workup.
In the event that R8 contains at least one chiral
center in substantially a single stereoconfiguration,
the individual stereoisomers of the aziridine of
formula 1~ can be separated by methods such as
chromatograp~y and crystallization, thereby providing a
stereospecific synthesis of the aziridine of formula l~
and subsequent products.
The aziridine ring can b~ opened to form an
intermediate secondary amine of formNla 16. A number of
methods of opening aziridines are commonly known. It is,
however, crucial that the method used for opening the
aziridine to form a secondary amine of formula 16 be
,. ~ ..
. :
X-8~65~ 28 Z ~
substantially regiospecific, i.e., the aziridine must be
opened to form substantially the ~-amino compound rather than
the 5-amino compound. One such method is catalytic
hydrogenolysis as taught by Y. Sugi and S. Mitsui,
Bull. Chem.~oc. Ja~ , pp. 1489-1496 (1970).
Catalysts which are suitable are the usual
hydrogenation and hydrogenolysis catalysts, such as the
noble metal catalysts; the preferred catalyst is
palladium. Suitable solvents include hydrocarbons such
as hexanes and heptanes; aromatic hydrocarbons such as
benzene, toluene, xylenes, ethylbenzene, and t-
butylbenzene; alcohols such as methanol, ethanol, and
isopropanol; and mi~tures of solvents such as acetic
acid mixed with said alcohols. Preferred solvents for
preparing the compound of formula 1~, wherein R3 is
benzoyl, and R8 is l-phenylethyl, include glacial
acetic acid or a mixture of methanol and phosphoric
acid~ The source of hydrogen can be an atmosphere of
elemental hydrogen supplied at a pressure of about 1
atmosphere or higher, or the source of hydrogen can be
compounds which are suitable to serve as hydrogen
donors in a catalytic transfer hydrogenolysis reaction,
such as formic acid, cyclohexene or hydrazine. The
preferred hydrogen source is an atmosphere of hydrogen
gas supplied at about 1 to about 10 atmospheres pres-
sure. The t~mperature of the reaction may be from
about -20 to about 80C; the preferred temperature for
the hydrogenolysis of the aziridine wherein R3 is
~enzoyl and R8 is l-phenylethyl is about -20 to about
0C.
The con~ersion of compounds of formula 1~ to
compounds of formula 1~ proceeds without disturbing the
stereochemical configuration of the chiral centers at
the 2a- or 4- positions of the formula 1~ or of the
chiral centers that may be present in any of the
substituents.
7~i
~-8265A 29
If desired, the compound of formula 16 can be
isolated by the usual methods such as crystallization. The
secondary amine at position 4 of formula 16 can be converted
to a primary amine of formula 17 b~ a number of methods known
to the art of organic chemistry, or alternatively the
secondary amine itself can be isolated. However, a preferred
method is to convert the secondary amine of formula 16 to the
primary amine of formula 11 without isolating the
secondary amine, but rather by simply continuing
without interruption the hydrogenolysis reaction that
produced the compound of formula 1~. Therefore, the
preferred solvent and catalyst are the same as those
for the preparation of the secondary amine of formula
1~. It may be desirable to conduct the hydrogenolysis
of the secondary amine of formula 1~ at a different
temperature or a different pressure or different
temperature and pressure than the hydrogenolysis of the
aziridine of formula l.S. For the hydrogenolysis of the
preferred compound of formula 1~ wherein R3 is benzoyl
and R8 is 1-phenylethyl, the preferred temperature and
pressure are about 50~ to about 60C and about 1 to
about 20 atmospheres. Under th~se conditions, the
hydrogenolysis of compounds of formula 1~ to compounds
of formula 17 proceeds without disturbing the
stereochemical configuration of the chiral center at
the 4-position.
The isolation of the compound of formula 17 can be
accomplished by the usual methods such as crystallization.
If desired, the compound of formula 17 can be further
purified, for example by recrystallization.
The compound of formula 1~ can be halogenated to
provide, for example, the 6-bromo or 6-iodo derivative 1~.
Iodination of compound 17 can be accomplished by using iodine
and orthoperiodic acid in the presence of an acid such as
sulfuric acid or trifluoroactic acid, in a solvent such as
, ~
,
3~
X-~265A 30
acetic acid. Another method of iodination involves the use of
N-iodosuccinimide in the presence of trifluoroacetic
acid. The 6-bromo derivative can be prepared using
bromine in acetic acid or using N-bromosuccinimide.
Of course, as those skilled in the art will
reco~nize, variations of any of the Schemes discussed
herein may be desirable or necessary for certain em-
bodiments of the invention. Such variations are con-
templated as within the scope of the present invention.
Compounds of Formula I can be prepared from the
appropriate compound of formula 1~, whether it exists
as a mixture of stereoisomers or as a substantially
pure diastereomer using common reagents and methods
well known in the art. A preferred intermediate to the
compounds of the instant invention is the 6-bromo-
derivative of 1~ although the 6-iodo derivative is
preferred if the carbonylation reaction of Scheme 8 is
used. PreEerably R~ is an amino-blocking group such as
benzoyl. Amino blocking groups can be added, if
desired, to the 4-amino substituent using such methods
as those disclosed by Gree~e, su~ra, and Barton, su~ra.
Alkyl groups can be added, if desired, to the 4-amino
substituent using such common methods as reac~ion of
the 4-amine with the appropriate halide as discussed by
Morrison and Boyd, Chapter 22, O~aanic Chemistry, Third
Edition, Allyn and Bacon, Boston, 1973. If desired,
the benzoyl group can be removed from the 1-position
using known methods and optionally replaced with other
amino-protecting groups. The amino-protecting groups
and alkyl groups can be added either before or after
the bromination, as desired.
The 4-amino-6-bromohexahydrobenz[cd]indole
startin~ materials used to prepare the compo~nds of the
invention can be readily prepared by other processes
such as disclosed in United States Patent No. 4,576,959
and EPO ~pplication 153083 of Flaugh, each of which is
.
: ` :
.
,
7~
X-82~s~ 31
incorporated herein by reference in its entirety.
The following examples further illustrate the
preparation of the compounds of this invention. The
examples are provided for purposes of illustration only
and are not to be construed as limiting the scope of
the instant invention in any way.
The terms and abbreviations used in the
instant examples have their normal meaning unless
otherwise designed, for example, "C" refers to degrees
celsius; ~N~ refers to normal or normality; "mmol~
referes to millimole; "g" referes to gram; "ml" means
milliliter; "M" refers to molar; "min" refers to
minutes; ~hr~ refers to hours; "EtOAc refers to ethyl
acetate; "RT" refers to room temperature; "sat'd" means
saturated; ""ppt" means precipitate; "Et2O" re~ers to ethyl
ether; "THF" refers to tetrahydrofuran; "MsCl" refers to
mesyl chloride; "NMR" refers to nuclear magnetic resonance;
"IR~ refers to infrared spectroscopy; "U.V." refers to
ultraviolet spectroscopy; and "m.s." refers to mass
spectrometry.
Example 1
Preparation of mixture of (2aS,4R)-,
(2aR,4S)-l-Ben~oyl-~-cyano-4-(di-n-propylamino)-
1,2,2a,3,~,5-hexahydrobenz[cd]indole.
To a solution of dimethyl formamide (lOOmL)
containing a mixture of (2aS,4R)- and (2aR,4S)-1-benzoyl-6-
bromo-4-(di-n-propyl-amino)hexahydroben2[cd]indole under a N2
atmosphere were added 3.4g (37.5 mmol) of CuCN and 7.lg
(37.5 mmol) of CuI. The reaction mixture was then
stirred at 140C for 6 hours. The reaction mixture was
poured onto ice, diluted with water, CH2C12 was added
and the mixture stirred for 30 minutes. The mixture
w~s filtered through a diatomaceous earth (tradename
"Celite") pad and the filtrate was extracted twice with
CH2Cl2. The organic ~olution was dried o~er MgSO4 and
.. : ~ . ~
.
!. ~,
. ' .
X-8265A 3~ ~33L~7
then evaporated to provide 4g of solid. Chromatography
of this crude product over silica gel with 1:19 MeOH/CH2Cl2 as
eluent gave 3g (62%) of product.
~Im~
Preparation of mixture oE (2aS,4R)-,
(2aR,4S)-6-cyano-4-(di-n-propylamino)-1,2,2a,3,4,5-
hexahydrobenz[cd]indole.
To a stirred solution of 4.8g (0.0124 mol) of
l-benzoyl-6-cyano-~-(di-n-propylamino)-1,2,2a,3,4,5-
hexahydrobenz[cd]indole prepared as in Example 1 in
200 mL of ~HF cooled to -78C under N2 atmosphere, was
added 16 mL (0.025 mol) of 1.6~ solution of n-butyl-
lithium in hexane. The reaction mixture was stirred at
-78C for 30 minutes and then allowed to warm to -20C.
To the reaction mixture was added 100mL in 1~ HCl. The
mixture was extracted once with ethyl ether. The
acidic solution was made alkaline with the addition of
cold 5N NaOH. The basic mixture was extracted twice
with CH2C12. The coIribined organic solution was washed
with saturated aqueous NaCl solution. qhe CH2C12
solution was dried over ~qgS04 and evaporated to give 4g
of an oil. Chromatography of this oil over silica gel
with ethyl acetate as eluent gave 3g (85~) of product
as an oil, which upon standing solidified.
E~sam~
Preparation of mixture of (2aS,4R)-,
(2aR,4S)-6-acetyl-4-(di-n-propylarnino)-1,2,2a,3,4,5-
hexahydrobenz[cd]indole.
A solution of 0.5g (1.8 ~nol) of 6-cyano-4-
(di-n-propylamino)-1,2,2a,3,4,s-hexahydrobenz[cd]indole
prepared as in Exam;ple 2 in 75mL of benzene was treated
with 5mL of 2.0M methylmagnesium bromide in diethyl
ether. The reaction mixture was refl~n~ea for 2 days.
The reaction mixture was cooled and excess Grignard
X-8265A 33
reagent was decomposed with addition of saturated
aqueous NH4Cl solution. The benzene layer was
separated and washed once with saturated aqueous NaCl
solu~ion. m e organic solution was evaporated to an
oil. The oil was dissolved in 25mL of 5N HCl and the
solution was stirred at room temperature for 30
minutes. The acidic solution was made alkaline with
the addition of excess concentrated aq~leous NH40H
solution. The basic mixture was extracted twice with
CH2Cl2. The combined organic solution was washed once
with saturated aqueous NaCl solution and dried over
MgSO4. The CEI2C12 solution was evaporated to yield
0.5g of an oil. Chromatography of this oil over silica
gel with ethyl acetate as eluent gave 0.4g (75%) of
product as an oil, which upon standing solidified, m.p.
76-77C.
AnalySiS for (ClgH28N2O)
Theory: C, 75.96; H, 9.39; N, 9.32
Found: C, 75.66; H, 9.33; N, 9.38
NMR: (300 M~z, CDC13) d 0.89 (t, 6H, CCH3), 1.46
(mult, 5H, 3a-H & CH2Me), 2.16 (br d, lH, 3~-H), 2.49
(mult, 4H, CH2Et), 2.50 (s, 3H, COCH3), 2.87 (dd, lH,
5a-H), 3.15 (mult, lH, 2~-h), 3.19 (mult, 2H, 2a-H &
2~-H), 3.~2 (dd, lH, 5~-H), 3.73 (mult, lH, 4-H), 4.04
(br s, lH, l-H), 6.43 (d, lH, 8-H), 7.63 (d, lH), 7-H).
M.S.: m/e = 300 (fd).
Exa~
Preparation of (2aR,4R)-6-acetyl-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
A. A mixturs of l-benzoyl-4,5-(endo)epox~-1,2,2a,3,4,5-
hexahydrobenz[cd]-indole (2lg, 0.076mol) and (+)-R-l-
phene~h~lamine (18g, 0.15mol) in 400ml of n-butanol was
refluxed under N2 for 16h. The reaction was concentrated in
,
X-8265A 34
vacuo ~o provide 30g of an oil as an equal mixture of two
dias~ereomeric amino alcohols.
The mixture of amino alcohols was dissolved in
300ml of CH2Cl2 and Et3N (30g, 0.225mol) was added at once
under N2. The reaction mixture was cooled to -10C then
MsCl(12.9g, 0.011) was slowly added dropwise. The rate of
addition was such as to maintain a reaction temperature
between -10 and 5C. Upon complete addition of MsCl, the
reaction mixture was stirred for an additional 30min a~ -5C
and then 30 min at ambient temperature. To the reaction
mixture was added 200ml of water and the mixture was stirred.
The CH2Cl2 solution was separated and washed successively
sat'd NaHCO3 sol and brine sol. The organic sol was dried
(MgSO4) and concentrated to dryness to provide a mixture of
two diastereomeric aziridines. The mixture was separated by
preparative HPLC (silica gel; hexanes/EtOAc gradient). The
first diastereomer of the aziridines to be eluted was
designated isomer l; 6.6g, mp 162-163C from i-PrOH. The
second diastereomer to be eluted was designated as isomer ~;
7.4g, mp 144-145C from isopropyl alcohol.
B. (2aR,4R)-4-amino-1-benzoyl-1,2,2a,3,4,5-
hexahydrobenzl[cd]indole
A solution of aziridine isomer 1 (9.4g,0.025mol) in
90ml of glacial acetic acid was hydrogenated at 60psi and at
60C over 5% Pd/C f~r 16h. The reaction mixture was filtered
and the filtrate was evaporated to a residual oil. The
residue was dissol~red in lN HCI and the acidic mixture was
extracted once wi~h EtOAC. The acidic solution was made
alkaline with addition of concentrated N~40H. The basic
mixture was extracted with CH2C12. The CH2C12 solu~ion was
washed with brine solution and dried (MgSO4). The organic
solution was evaporated to dryness to provide 2aR,4R-4-amino-
1-benzoyl-1,2,2a,3,4,5-hexhydrobenz[cd] indole; 5.2g as an
oil.
- X-8265A 35 ~ 7~3
C. (2aR, 4R)-4-amino-1-benzoyl-6-bromo-
1,2,2a,3,~,s-hexahydrobenz[cd]indole
A solution o~ (2aR, 4R)-4-amino-1-benzoyl-
1,2,2a,3,g,5-hexahydro-benz[cd]indole(5.2g,0.019mol) and
sodium acetate (6.2g,0.076) in 40mL glacial acetic acid
(HOAc) and 10mL of MeOH was cooled to 10C. to the reaction
mixture was added dropwise a solution of bromine(3g,
0.019mol) in 10mL oE glacial HOAc. The reaction temperature
was maintained at 10C during addition of the bromine. The
reaction was then stirred at ambient temperature for lh. The
solvents were evaporated and the residue was dissolved in
water. The acidic solution was made alkaline with cold 50%
aqueous NaOH. The basic mixture was extracted twice with
CH2C12. The organic solution was washed with brine solution,
dried (MgSO4) and concentrated in vacuo to provide 6.8g
(2aR,4R)-6-bromo compound as an oil.
D. (2aR, 4R)-l-benzoyl-6-bromo-4(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
A reaction mixture of (2aR, 4R)-4-amino-1-benzoyl-
6-bromo-1,2,2a,3,4,5-hexahydrobenz[cd]indole(6.8g, 0.019mol),
K2CO3(8.2~g, 0.06mol) and n-propyliodide(10.2g, 0.06mol) in
200mL of CH3CN was stirred at reflux temperature ~or 16h.
The reaction mixture was filtered and solvent was evaporatQd.
The residue was dissolved in EtOAc and the solution was
extracted with dilute HCl. The acidic solution was made
alkaline with concentrated NH40H. Th~ basic mixture was
extracted with EtOAc. The organic solution was washed with
brlne solution and dried (MgSO4). ~he EtOAc was evaporated
to provide a residual oil. Chromatography (silica gel-EtOAc)
gave product, 2.4g.
, ~ .
':
.
, : ~
X-8265A 36
E. (2aR,4R)-l-Benzoyl-6-cyano-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
To a solution of (2aR,4R)-1-senzoyl-6-bromo-4-(di-
n-propylamino)-1,2,2a,3,4,5-hexa-hydrobenz[cd]indole
(2.4g;5mmol) in 100mL of dimethyl formamide (DMF) was added
CuCN (1.34g,15mmol) and CuI (2.85g, 15mmol). The reaction
mixture was stirred at reflux under a N2 atmosphere for 16hr.
qhe reaction mixture was poured into 500mL of water. The ppt
was collected and washed several times with water. The ppt
was suspended in dil NH40H and slurried with EtOAc. The
whole mixture was filtered thru a celite pad. The EtOAC sol
was separated and washed with brine sol. The EtOAc sol was
dried(MgSO4) and conc to dryness to provide 1.7g of nitrile
as an oil.
F. (2aR,4R)-6-Cyano-4-(di-n-propylamino)-
1,2,2a,3,4,5-hexahydrobenz~cd]indole
To a stirred solution of 1.7g (4.4~nol) of
(2aR,4R)-6-cyano-4-(di-n-propylamino)-1,2,2a,3,4,5-
hexahydrobenz[c,d]indole in ~5mL of THF cooled to -78C under
a N2 atmosphere was added 5.SmL (8.8~nol) of 1.6M solution of
n-BuLi in hexane. The reaction mixture was stirred at -78C
for 30 min. and then allowed to warm to -20C. To the
reaction mixture was added 20~L of lN HCl. The mixture was
extracted once with Et2O. The acidic solution was made
alkaline with the addition of cold 5N NaOH. The baslc
mi~ture was extracted twice with CH2C12. The coInbined
organic solution was washed with sat'd NaCl solution. The
CH2C12 solution was dried over MgSO4 and evaporated to give
1.3g of an oil. Chromatography of this oil over silica gel
with EtOAc as eluent gave lg (~096) of product as an oil.
G. (2aR,4R)-1-Trityl-6-cyano-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydro~enz[cd]indole
-- X-8265A 37 ~ 7~
To a sol of (2aR,4R~ trityl-6-cyano-4-(di-n-
propylamino)-1,2,2a,3,4,s-hexahydrobenz[cd~indole (lg,
3.5mmol) and Et3N (354mg, 3.5mmol) in 50mL of methylene
chloride was added a sol of triphenylmethyl chloride (trityl
chloride)(0.98~,3.5mmol) in 10mL of methylene chloride
dropwise at RT. The reaction mixture was stirred for 16hr at
RT. The reactlon mixture was extracted with water and cold
lN HCl. The organic sol was washed with sat'd NaHCO3 sol and
with sat~d brine sol. The organic sol was dried (MgSO4) and
conc to dryness in vacuo to give a residue. The residue was
slurried with warm hexanes, cooled and filtered to remove
insolubles. The filtrate was conc to an oil. The oil was
chromatographed (silica gel, 20% EtOAc in hexanes) to provide
1.5g of (2aR,4R)-l-trityl-6-cyano-4-(di-n-propylamino)-
1,2,2a,3,4,5-hexahydrobenz-[cd]indole.
H. (2aR,4R)-6-acetyl-4-(di-n-propylamlno)-
1,2,2a,3,4,5-hexahydrobenz[c,d]indole
A solution of 1.6g(3mmol) (2aR,4R)-l-trityl-6-
cyano-4-(di-n-propylamino)-1,2,2a,3,4,5-
hexahydrobenz~cd]indole in 100ml of THF was treated with 20mL
of 2.0M methylmagnesium bromide in diethyl ether. The
reaction mixture was refluxed for 16hr. The reaction mixture
was cooled and excess Grignard reagent was decomposed with
addition of sat'd NH4Cl solution. The reaction mixture was
extracted with EtOAc. The organic solution was evaporated to
an oil. The oil was dissolved in 25mL of 5N HCl and the
solution was stirred at room tempera~ure for 30 min. The
acidic solution was made al~aline with the addition of excess
conc N~40H solution. The basic mixture was extracted twice
with EtOAc. The combined organic solution was washed once
with satld NaCl solution and dried over MgS04. The EtOAc
solution was evapora~ed to yield 0.9g of an oil.
Chromatography of this oil over silica gel with EtOAc as
X-8265A 38
eluent gave 600mg of product. Recryst from hexanes to yield
228mg (-) ketone.
mp 85-~6;[a] D= -4.94(CH30H)
Example 5
Preparation of (2aS,~S)-6-acetyl-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
A. Aziridine isomer ~ from Example 4A (8.5g,
0.022mol) was hydrogenated to provide (2aS,4S)-4-amino-1-
benzoyl-1,2,2a,3,4,5-hexahydrobenz~cd]indole(4.5g) as an oil.
B. (2aS,4S)-4-amino-1-benzoyl-6-bromo-
1,2,2a,3,4,5-hexahydro~enz[cd]indole
Usin~ the procedure of Example 4C, (2aS,~S)-4-
amino-1-benzoyl-1,2,2a,3,4,5-hexahydrobenz[cd]indole
(4.5g,0.016mol) was halogenated to yield 5.4g (2aS,4S)-6-
bromo compound as an oil.
C. (2aS,4S)-1-benzoyl-6-bromo-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
Using the procedure of E~ample 4D, the reaction of
(2aS,4S)-4-amino-1-benzoyl-6-bromo-1,2,2a,3,4,5-
hexahydrobenz[cd]indole(5.4g, 0.015mol) with n-propyliodide
(10.2g, 0.06mol) in the presence of K2CO3(8.28gr 0.06mol) in
200ml of CH3CN gave, after chromatography, 3.lg of product.
D. (2aS,4S)-1-Benzoyl-~-cyano-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd~indole
Using the procedure of Example 4E, (2aR,4R)-1-
benzoyl-6-bromo-4-(di-n-propylamino)-1,2,2a,3,4,5-
hexahydrobenz[cd]indole (3.1g, 7~mol) with CuCN (l.g,21mmol)
and CuI (4g, 21mmol) in lOOml DMF gave 2.sg of nitrile as an
oil.
X-8265A 39
E. (2aS,~S)-6 Cyano-4-(di-n-propylamino)-
1,2,2a,3,4,s-hexahydrobenz~cd]indole
The procedure of Example ~F was followed using 2.5g
(6.5mmol) of (2aS,4S)-l-benzoyl-6-cyano-4-(di-n-propylamino)-
1,2,2a,3,4,5-hexahydrobenz[cd]-indole and 8.lml (13mmol) n-
butyl lithium to provide 1.6g of an oil. Chromatography of
the oil over silica gel with EtOAc as eluent gave lg (54%) of
product as an oil.
F. (2aS,4S)-l-trityl-6-cyano-4-(di-n-propylamino)
-1,2,2a,3,4,5-hexahydrobenz[cd]indole
The procedure of Example 4G was followed using the
product from Example 4E (lg, 3.5mmol) to provide 1.6g of
product.
G. Formation of (2aS,4S)-6-acetyl-4-(di-n-propyl-
amino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
The procedure of Example 4X was followed using
product from Example 4F (1.6g, 2.9mmol) to provide l.Og of an
oil. Chromatography of the oil over silica gel with EtOAc as
eluent gave 700mg of product. Recrystallization from hexanes
yielded 24Omg of the (~) ketone.
mp 85-86C
[a] D = ~ 5.15(CH30H)
~x~m~Q
Preparation of (+)-(2aS,4R)-6-acetyl-4-(di-n-
propylamino) 1,2,2a,3,4,5-hexahydrobenz [cd]indole.
The above described procedure was used to prepare
(2aS,4R)-l-benzoyl-6-bromo-4-(di-n-propylamino)-1,2,2a,3,4,5-
hexahydrobenz [cd]indole. The procedures of Example 4 ~ere
used to form (+) (2aS,~R)-1-trityl-6-cyano-4-(di-n-
propylamine)-1,2,2a,3,4,5-hexah~drob~nz[cd]indole a solution
of which (2.4g,4.6mmol) in lOOml of THF was treated with 25m~
X-8265A 40 ~r~
of 2.0M methylmagnesium bromide in diethyl ether. The
reaction mixture was refluxed for 16hr. The reaction mixture
was cooled and excess Grignard reagent was decomposed with
addition of saturated NH4Cl solution. The reaction mixture
was extracted with ethyl acetate. The organic solution was
evaporated to an oil. The oil was dissolved in 25mL of 5N
HCl and the solution was stirred at room temperature for
30min. The acidlc solution was made alkaline with the
addition of excess concentrated NH40H solution. The basic
mixture was extracted twice with ethyl acetate. The combined
organic solution was washed once with saturated NaCl solution
and dried over MgS04. The ethyl acetate solution was
evaporated to yield 1.4g of an oil. Chromatography of this
oil over silicia gel with ethyl acetate as eluent gave 1.2g
(87%) of product. Recrystallization from hexane yielded 840
mg of the product (+)ketone.
mp = 121-122C
[a] D = + 66.60(CH30H)
~am~le 7
Preparation of (-)(2aR,4S)-6-acetyl-4-(di-n-
propylamino)-1,2,2a,3,4,s-hexahydroben~[cd]indole
The above described procedure was used to prepare
(2aR,4S)-l-benzoyl-6-bromo-4-(di-n-propylamino)-1-2,2a,3,4,5-
hexahydrobenz[cd]indole. I~te procedures of Example 4 were
used to prepare (2aR,4S)-l-trityl-6-cyano-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole a solution
of which (3.~g,6.smmol) in 100ml of THF was treated with 40mL
of 2.0M methylmagnesium bromide in diethyl ether. ~te
reaction mixture was refluxed for 16hr. The reaction mixture
was cooled and excess Grignard reagent was decomposed with
addition of satld NH4Cl solution. The basic mixture was
extracted twice with EtOAc. ~he combined organic solution
was washed once with sat~d NaCl solution and dried over
~Q~ 7~
X-8265A 41
Mg~O4. The EtOAc solution was evaporated to yield l.9g of an
oil. Chromatography of this oil over silicia gel with EtOAc
as eluent gave 1.8g of product which was recrystallized from
hexane to yield 1.4g of product.
mp 120-121C
[a]D = -64.48(CH30H)
Examwle ~
Preparation of (~)-(2aS,4R)-6-(2-methylpropanoyl)-
4-(di-n-propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
(2aS,4~ trityl-6-cyano-4-(di-n-propylamino)-
1,2,2a,3,4,5-hexahydrobenz[cd]indole was prepared as in
Example 6. A solution of this hexahydrobenz[cd]indole
(9.5g,0.018mol) in 200mL of THF was treated with 30mL of 2.OM
isopropylmagnesium chloride in diethyl ether. The reaction
mixture was refluxed for 16hr. The reaction mixture was
cooled and decomposed with addition of 50mL of 5N HCl then
warmed for 30min on a steam bath. The acidic mixture was
extracted twice with EtOAc. The combined organic solution
was washed once with sat~d NaCl solution and dried over
MgS04. The EtOAC solution was evaporated to yield l.9g of an
oil. Chromatography of this oil over silicia gel with ~OAc
as eluent gave 0.9g of product. Recrystallization from
hexanes to yield 360mg of product.
mp 87-89C
[a]D = ~52.72(CH30H)
~lm~he ~
Preparation of (-)-6-(2-methylpropanoyl)-~-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
X-826~A 42
The procedure of Example 8 was followed with (-)-1-
trityl-6-cyano-4-(di-n-propylamino)-1,2,2a,3,4,5-
hexahydrobenz[cd]indole (7g,13mmol), isopropylmagnesium
chloride (50 mL, 2 molar in ethyl ether), THF (150mL) to give
3.8 g of crude product. Chromatography with silica gel using
EtOAc as eluent gave 0.8 g of material whch was
recrystallized from hexanes to give 400 mg of product.
mp = 8~-89C
[~]D - -51.0 (CH3OH)
Exam~le 1~
Preparation of (-)-(2aR,4S)-6-(propanoyl)-4-(di-n-
propylamino)-1,2,2a,3,~,5-hexahydrobenz[cd]indole
A solution of (-)-(2aR, 4S)-l-trityl-6-cyano-4-(di-
n-propylamino)-1,2,2a,3,4,5-hexahydrobenz[c,d]indole (1.5g,
2.7mmol) in 200ml of THF was treated with 25 mL of 2.OM
ethylmagnesium bromide in diethyl ether. The reaction
mixture was refluxed for 16 hr. The reaction mixture was
cooled and decomposed with addition of 50mL of 5N HCl then
warmed for 30 min on a steam bath. The acidic mixture was
extracted with EtOAc. The acidic solution was made alkaline
with the addition of excess conc NH40H solution. The basic
mixture was extracted ~wice with EtOAc. The combined organic
solution was washed once with satld NaCl solution and dried
over MgS04. The EtOAc solution was evaporated to yield 0.6g
of an oil. Chromatography of this oil over silica gel with
EtOAc as eluent gave 0.4g of product. Recrystallization from
hexanes gave 300mg (-) ketone.
mp 90-91C
[~]D = -63.68(CH30E)
Exam};)1 e l L
Preparation of (+)-(~aS,4R)-6-(pentanoyl)-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
X-8265A 43 ~ ~ 4 ~ ~ 7
A solution of (+)-(2aS,4R)-l-trityl-6-cyano-4-(di-
n-propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole (1.0g,
2mmol) in 4Oml of THF was added dropwise to a solu~ion of n-
butylmagnesium iodide (25mmol) in 25mL diethyl ether. Thereaction mixture was refluxed for 16hr. The reaction mixture
was cooled and decomposed with addition of 50mL of SN HCl
then warmed for 30min on a steam bath. The acidic mixture
was extracted with EtOAc. The acidic solution was made
alkaline with the addition of excess conc NH40H solution.
The basic mixture was extracted twice with EtOAc. The
combined organic solution was washed once with sat'd NaCl
solution and dried over MgS04. The EtOAc solution was
evaporated to yield 0.4g of an oil. Chromatography of this
oil over silica gel with EtOAc as eluen~ gave 70mg of
product. Recrystallization from hexane gave 25mg ketone.
mp 104-105C
[a] D = + 35.7(CH30H)
Exam~le 12
Preparation of (~)-(2aS,4R)-6-(benzoyl)-4-(di-n-
propylamine)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
A solution of (+)-(2aS,4R)-l~trityl-6-cyano-4-(di-
n-propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole (1.5g,
2.7mmol) in 30m~ of THF was treated with 10 mL of 3.0M
phenylmagnesium bromide in diethyl ether. The reaction
mixture was refluxed for 16hr. The reaction mixture was
cooled and decol~posed with addition of 50mL of 5N HCl then
warmed for 30min on a steam bath. The acidic mixture was
extracted with EtOAc. The acidic solution was made alkaline
with the addition of excess conc NH40~ solution. The basic
mixture was extracted twice with ~tOAc. The comhined organic
solution was washed once with sat'd NaCl solutlon and dried
over MgSO4. The EtOAc solution was evaporated to yield 0.6g
of an oil. Chromatography of this oil over silica gel with
. . , . ~ ~ .
.. , ': ' '- ' ~ :
X-8265~ 44
EtOAc as eluent gave 0.3g of product. Recrystallization from
h0xanes gave 360mg (+) ketone.
mp 161-162C
[a]D = + 93.66(CH30H)
Exam~le 13
Preparation of (+)-(2aS,4R)-6-(2-phenyle~hanoyl)-4-
(di-n-propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
A solution of (~)-(2aS, 4R)-l-trityl-6-cyano-4-(di-
n-propylamino)-1,2,2a,3,4,5-he~ahydrobenz[c,d]indole
(1.0g,2mmol) in 40mL of THF was added dropwise to a solution
of benzylmagnesium chloride (25mmol) in 25mL diethyl ether.
The reaction mixture was refluxed for 16hr. The reaction
mixture was cooled and decomposed with addition of 50mL of 5N
HCl then warmed for 30min on a steam bath. The acidic
mixture was extracted with EtOAc. The acidic solution was
made alkaline with the addition of excess conc NH40H
solution. The basic mixture was extracted twice with EtOAc.
The combined organic solution was washed once with sat~d NaCl
solution and dried over MgSO~. The EtOAc solution was
evaporated to yield 0.6g of an oil. Chromatography o~ this
oil over silica gel with EtOAc as eluent gave 0.4g of
product. Recrystallization ~rom hexanes gave 225mg (+)
ketone.
mp 104-105C
[~] D = ~ 47.62(CH30H)
Preparation of (2aS,4R)-6-ethynyl-4-(di-n-
propylamino)-1,2,2a,3,4,5-hexahydrobenz[cd]indole
l-benzoyl-(2aS,4R)-6-iodo-4-(di-n-propylamino)-
1,2,2a,3,4,5-hexahydrobenz[cd]indole (100 mg, 0.205mmol) and
trimethyltin acetylene trimethylsilane (272 mg, 1.0 m~ol, 3
eq) was disso]ved in anhydrous toluene (5 mL), to which was
X-8265A 45
then added tetrakis-triphenylphosphine palladium (20 mg,
0.017 mmol, 0.05 eq). The resulting light yellow solution
was brought to raflux under N2 atmosphere. Af~er 4 hr, the
reaction mixture was cooled to room temperature, filtered and
concentrated to dryness. The residue was chromatographed
over silica gel with hexanes:ethyl acetate (1:1) to afford
the desired product (79mg,84%). This material was dissolved
in a 1~ solution (5mL) of tetrabutylammonium fluoride in THF,
and stirred at room temperature overnight (12 h). The
solution was diluted with EtOAc (10 mL) and rinsed
successively with H20 (3xlOmL), brine (10 mL) and dried over
Na2SO4. The residue was chromatographed over silica gel with
hexanes:ethyl acetate (1:1) to afford a mixture of the 1-
benzoyl (61%) and the N-deprotected indoline (33%).
The present compounds of Formula I have been
found to have selective affinity for the 5HT receptors
in the brain with much less affinity for other
receptors. Because of their ability to selectively
bind to 5~T receptors, the compounds of Formula I are
useful in treating disease states which require
alteration of 5-HT1A receptor function but without the
side effects which may be associated with less
selective compounds. It has been further found that certain
of the instant compounds have su~stantial affinity for both
the 5-HT1A and 5-HT1D receptors and are useful in treating
disease states which can benefit from an alteration of the
receptors. The alteration of the 5~T1A and 5HTlD receptors
may involve mimicking (an agonist) or inhibiting (an
antagonist) the function of serotonin. The disease states
include anxiety, d~pression, excess gastric acid secretion,
hyper~ension, nausea, sexual dysfunction, cognition,
senile dementia, consumptive disorders such as appetite
disorders, alcoholism and smoking. The foregoing
conditions are treated with a pharmaceutically
effective amount of a compound of Formula I.
The term ~'pharmaceutically effective amount",
'
~ ` ~
.
X-8265A 46 ~ ~ ~ 9 7
as used herein, represents an amount of a compound of
the invention which is capable of diminishing the
adverse symptoms of the particular disease. The par-
ticular dose of compound administered according to this
invention of course be determined by the particular
circumstances surrounding the case, including the
compo~md administered, the route oE administration, the
particular condition being treated, and similar con-
siderations. The compounds can be administered by a
variety of routes including the oral, rectal, trans-
dermal, subcutaneous, intravenous, intramuscular or
intranasal routes. A typical single dose for
prophylactic treatment, however, will contain from
about 0.01 mg/kg to about 50 mg/kg of the active
compound of this invention when administered orally.
Preferred oral doses will be about 0.01 to about
3.0 mg/kg, ideally about 0.01 to about 0.1 mg/kg. When
a present compound is given orally it may be necessary
to administer the compound more than once each day, for
example about every eight hours. For IV administration
by bolus, the dose will be from about 10 ~g/kg to about
300 ~g/kg, preferably about 20 ~g/kg to about 50 ~g/kg.
The following experiments were conducted to
demonstrate the ability of the compounds of the present
invention to interact with the serotonin lA and/or lD
receptors. The affinities of the compounds at the cen~ral 5-
HT1A receptors were determined using a modification of the
binding assay described by Taylor, ~ . Pharma5s1
EXD. The~. 236:118-125, 1986). Membranes for the binding
assay were prepared from male Sprague-Dawley rats (150-250
g). The animals were killed by decapitation, and the brains
were rapidly chilled and dissected to obtain the hippocampi.
The hippocampi were either prepared that day or stored frozen
(-70C) until the day of prepara~ion. Membranes were
prepared by homogenizing the tissue in 40 volumes of ice-cold
Tris-HCl buffer (50 mM, pH 7.4 at 22C) using a Techmar
, ~ . . ~ . .
X-8265A 47 ~ 7~i
Tissumizer (setting 65 for 15 sec), and the homogenate was
centrifuged at 39800xg for 10 min. The resulting pellet was
then resuspended in the same buffer, and the centrifugation
and resuspension process was repeated three additional times
to wash the membranes. Between the second and third washes
the resuspended membranes were incubated for 10 min at 37C
to facilitate the removal of endogenous ligands. The final
pellet was resuspended in 6'7mM I'ris-HCl, pH 7.4, to a
concentration of ~ mg of tissue original wet weight/200 ~l.
This homogenate was stored frozen (-70C) until the day of
the binding assay. Each tube for the binding assay had a
final volume of 800 ~l and contained the following: Tris-HCl
(50 mM), pargyline (10 ~M), CaCl2(3 m~), [3H]8-o~-DPAT (1.0
nM), appropriate dilutions of the compound being evaluated,
and membrane resuspension eguivalent to 2 mg of original
tissue wet weight, for a final pH of 7.4. The assay tubes
were incubated for 10 min at 37C, and the contents were then
rapidly filtered through GF/s filters (pretreated with 0.5%
polyethylenimine), followed by four one-mL washes with ice
cold buffer. The radioactivity trap~ad by the filters was
quantitated by liquid scintillation spectromet~y, and
specific [3H]8-OH-DPAT binding to the 5-HT1A sites was defined
as the difference between [3H]~-oH-DPAT bound in the presence
and absence of 10 ~M 5-HT.
The affinity of the particular compound at the 5-HTlA
receptor is expressed as IC50 value, i.e., the concentration
required to inhibit 50% of the binding. The IC50 values were
determined from 12-point competition curves using nonlinear
regression (SYSTAT, SYST~T, INC., Evanston, IL). The results
from this determination are provided in Table I.
The affinities of the compounds at the central 5-~T1D
binding sites were determined using a modification of the
binding assay described b~ ~euring and Peroutka (~L ~xQSCi.
7:894-903, 1987). Bovine brains were obtained from Pel-
Freeze Biologicals, and ~he caudate nuclei were dissected out
and frozen at -70C until the time that the membranes were
.
.
X- 8265A 48
prepared for the binding assays. At that time the tissues
were homogenized in ~0 volumes of ice-cold Tris-HCl buffer
(50mM, pH 7.4 at 22C) with a Techmar Tissumizer (setting 65
for 15 sec), and the homogenate was centrifuged at 39,800g
for 10 min. The resulting pellet was then resuspended in the
same buffer, and the centrifugation and resuspension process
was repeated three additional times to wash the membranes.
Between the second and third washes the resuspended membranes
were incubated for 10 min at 37C to facilitate the removal
of endogenous 5-HT. The final pellet was resuspended in Tris
buffer to a concentration of 25 mg of original tissue wet
weight/ml for use in the binding assay. Each tube ~or the
binding assay had a final volume of 800 ~l and contained the
following: Tris-HCl (50mM), pargyline (10 ~M), ascorbate
(5.7 mM), CaCl2 (3 mM), 8-OH-DPTA (100 nM to mask 5-HTlA
receptors), mesulergine (100 nM to mask 5-HT1C receptors),
[3E~] 5-HT (1.7-1.9 nM), appropriate dilutions of the drugs of
interest, and membrane suspension equivalent to 5 mg of
original tissue wet weight, for a final pH of 7.4. The assay
20 tubes were incubated for 10 min at 37C, and the contents
were then rapidly filtered through GF/B filters (pretreated
with 0.5% pulyethylenlmine), followed by four one-mL washes
with ice-cold buffer. The radioactivity trapped by the
filters was quantitated by liquid scintillation spectrometry,
25 and specific [3H] 5 -HT binding to the 5-HT1D sites was defined
as the difference between [3H]5-HT bound in the presence and
absence of 10 ~M 5-HT.
The affinities of compounds at the 5 -HTlD receptor
are expressed as IC50 values, i.e., the concentration required
to inhibit 50% of the 'oinding. These values were determined
from 12-point competition curves using nonlinear regression
(SYSTAT, SYSTAT, Inc., Evanston, IL)~ The results from this
determi.nation are provided in Table I.
X-8265A 49
TABLE I
Exam~le NQ. ~ A(1)
3 0.63 7.47
4 0.80 236.38
0.31 129.24
6 0.3 6.25
7 6.61 8500.0
8 0.25 1.24
9 54.88 3125.00
9.47 9000-00
12 0.34 1.78
13 0.98 2.7
(1) IC50 in nanomoles per liter
The compound of Example 1~ was evaluated for its
ability to interact with serotonin lA receptor using the
following procedure which is generally set forth in Wong 8
~1~, J. Neural ~r~nsm., I1, 207-218 (1988). Male Sprague-
Dawley rats (110-150 g) from Harlan Industries (Cumberland,
IN) were fed a Purina Chow ad libitum for at least 3 days
before being used in the studies. Rats were killed by
decapitation. The brains were rapidly removed, and the
cerebral cortices were dissected out at 4C.
Brain ~issues were homogenized in 0.32M sucrose.
After centrifugation a~ 1000 x g for 10 min and then at 17000
x g for 20 min, a crude synaptosomal fraction was sedimented.
The pellet was suspended in 100 vol of 50 mM Tris-HCl, pH
7.4, incubated at 37C for 10 min, and centrifuged at 50000 x
g for 10 min. The process was repeated and ~he final pellet
was suspended in ice-chilled 50 mM Tris-HCl, pH 7.4. By the
radioligand binding method, sites specifically labeled by
tritiated 8-hydroxy-2-dipropylamino-1,~,3,4-
X-8265A 50 ~ O ~ 9 ~ 7 ~
tetrahydronaphthalene (3H-8-OH-DPAT) have been identified as
5-HT1A receptors.
Binding of (3H-8-OH-DPAT) was performed according
to the previously described method [Wong et al., J. ~eural
TLans{n. ~:251-269 (1~85)1. Briefly, synaptosomal membranes
isolated from cerebral cortex were incubated at 37C for 10
min. in 2 mL of 50 mM Tris-HCl, pH 7.4; 10 ~M pargyline; 0.6
~M ascorbic acid; 0.4 nM 3H-8-oH-DPAT; and from 1 to 1000 mM
of test compound. Binding was terminated by filtering
samples under reduced pressure through glass fiber (GFB)
filters. The filters were washed twice wi~h 5 mL of ice cold
buffer and placed in scintillation vials with 10 mL of PCS
(Amersham/Searle) scintillation fluid. Radioactivity was
measured with a liquid scintillation spectrometer. Unlabeled
8-OH-DPAT at 10 ~ was also included in separate samples to
establish non-specific binding. Specific binding of 3H-8-oH-
DPAT iS defined as the difference of radioactivity bound in
the absence and in the presence of 10 ~M unlabeled 8-OH-DPAT.
The result is provided in Table II. The value is
the IC50, i.e. the concentration in nanomoles of the compound
necessary to inhibit the binding of 3H-8-oH-DPAT by 50~.
Table II
E~am~le IC~Q
14 0.5
Experiments were conducted to demonstrate the
serotonin against properties of ~he instant compounds.
Certain compounds were evaluated to determine their ability
to affect the 5-hydroxyindoles serotonin, 5-hydroxyindole
acetic acid (5HIAA) and serum corticosterone, in vivo, using
the following procedures.
Compounds in aqueous solution were injected
subcutaneously into male albino rats. Rats were decapitated
one hour later. Trunk blood was collected and allowed to
.~ .
- ~
X-8265A 51 ~ 7~,
clot; after centrifugation, serum was stored frozen prior to
analysis. Whole brain was removed and frozen on dry ice,
then stored frozen prior to analysis. Serum corticosterone
concentration was measured spectrofluorometrically (J.H.
Solem and T. Brinch-Johnsen, ~An evaluation of a method for
determination of free corticosteroids in minute quantities of
mouse plasma," ~ d. J. ~lin~_La~. Invest. (Suppl. 80),
1.14 (1965).) 5-Hydroxyindoleacetic acid (5HIAA)
concentration in whole brain was measured by liquid
chromatography with electrochemical detection. (Ray W.
Fuller and Kenneth W. Perry, ~Effects of buspirone and its
metabolite, ~-(2-pyrimidinyl)piperazine, on brain monoamines
and their metabolites in rats", ~ Pharmacol. Exp. Thex.
248, 50-56 (1989).) The results are provided in Table III.
Table III
Brain 5-hydroxyindoles
(n moles/g)
Serium
F~m~le No. Corti~ost2rone
(dose m~/R~] erotonin 5~Laa (~g/lnnml)
Control 2076 iO.122.13 + 0.10 3.8 + 0.2
Example 6
(0.003) 2.46 ~ 0.141.81 ~ 0.145.8 ~ 1.0
(0.03) 2.99 ~ 0.061.58 i 0.08*10.6 i 2.0*
(0.3) 3.08 i 0.04*1041 i 0.03*42.2 i 1.1*
Example 7
(0.003) 2.75 ~ 0.052.~6 ~ 0.133.8 i 0.5
(0.03) 2.57 ~ 0.101.87 ~ 0.076.5 ~ 2.6
(0.3~ 2.85 ~ 0.081.77 + 0.178.4 i 4.0
Control 1.66 * 0.04L.68 ~ 0.123.4 i 0.2
Example 8
(0.003) 1.88 + 0.05*1.56 ~ 0.103.6 ~ 0.7
(0.03) 2.26 ~ 0.06~1.34 ~ 0.06*27.1 i 6.4*
X-8265A 52 ~49~
(0.3) 2.26 ~ 0.16* L.30 ~ 0~07* 42.0 i 0.4*
Example 9
(0.003) 1.83 ~ 0.08 1.68 + 0.10 4.1 ~ 0.5
(0.03) 1.90 ~ 0.10 1.91 i 0.06 6.0 -~ 1.6
(0.3) 1.69 + 0.06 1.74 ~ 0.04 6.7 ~ 2.0
*Significant difference from control group (P<0.05)
The compounds of the present invention are
preferably formulated prior to administration. There-
fore, yet another embodiment of the present invention
is a pharmaceutical formulation comprising a compound
of the invention and a pharmaceutically acceptable
excipient therefor.
The present pharmaceutical formulations are
prepared by known procedures using well known and
readily available ingredients. In making the composi-
tions of the present invention, the active ingredient
will usually be mixed with an excipient, diluted by an
excipient or enclosed within such a carrier which can
be in the form of a capsule, sachet, paper or other
container. When the excipient serves as a diluent, it
can be a solid, semi-solid or li~uid material which
acts as a vehicle, carrier or medium for the active
ingredient. Thus, the compositions can be in the ~orm
of tablets, pills, powders, lozenges, sachets, cachets,
elixirs, suspensions, emulsions, solutions, syrups,
aerosols (as a solid or in a liquid medium), ointments
containing for example up to 10% by weight of the
active compound, soft and hard gelatin capsules,
suppositories, sterile injectable solutions and sterile
packaged powders.
Some examples of suitable excipients include
lactose, dextrose, sucrose, sorbitol, mannitol,
starches, gum acacia, calcium phosphate, alginates,
tragacanth, gelatin, calcium silicate, microcrystalline
.
,
~ :.,' , '' ':
X-8265A 53 ~L~ 7
cellulose, polyvinylpyrrolidone, cellulose, water,
syrup, and methyl cellulose. The formulations can
additionally include lubricating agents such as talc,
magnesium stearate and mineral oil, wetting agents,
emulsifying and suspending agents, preserving agents
such as ~ethyl- and propylhydroxybenzoates, sweetening
agents or flavoring agents. The compositions of the
invention may be formulated so as to provide quick,
sustained or delayed release of the active ingredient
after administration to the patient by employing
procedures well known in the art.
The compositions are preferably formulated in
a unit dosage form, each dosage containing from about
0.5 to about 50 mg, more usually about 1 to about
10 mg, of the active ingredient. The term "unit dosage
formll refers to physically discrete units suitable as
unitary dosages for human subjects and other mammals,
each unit containing a predetermined quantity of active
material calculated to produce the desired therapeutic
effect, in association with a suitable pharmaceutical
carrier.
The following formulation examples are illus-
trative only and are not intended to limit the scope of
the invention in any way.
For~ul~ion 1
Hard gelatin capsules are prepared using the
following ingredients:
Q~ y (mg/ca~ule)
6-acetyl-4-(di-~-propylamino)-
1,2,2a,3,4,5-hexahydrobenz-
[cd]indole 25
Starch, dried 42
Magnesium stearate _1~
Total 460 ~g
The above ingredients are mixed and filled
X-8265A 54 ~9~7~
into hard gelatin capsules in 460 mg quanti~ies.
Formulation 2
A tablet formula is prepared using the in-
gredients below:
Ouantitv (mq/ta~let)
4-(di-n-propylamino)-6-(2,2-
dimethylpropanoyl)-1,2,2a,3,4,5-
hexahydrobenz[cd]indole 25
Cellulose, microcrystalline 625
Colloidal Silicon Dioxide10
Stearic acid 5
m e components are blended and compressed to
form tab]ets each weighing 665 mg.
Formulatio~_~
A dry powder inhaler formulation is prepared
containing the following components:
Weight
4-(diethylamino)-6-propanoyl-
1,2,2a,3,4,5-hexahydrobenz[cd]-
indole 5
Lactose 95
The active compound is mixed with the lactose
and the mixture added to a dry powder inhaling ap-
plicance.
Formul~ion 4
Tablets each containing 60 mg of active ingredien~
are made up as follows:
4-(n-propylamino)-6-~2-methyl-
propanoyl)-l,2,2a,3,4,5-hQxahydro-
benz[cd]indole tartrate salt 60 mg
Starch 45 ~g
Microcrys~alline cellulose35 mg
: . ~ .
X-8265A 55 ~.9~7~
Polyvinylpyrrolidone (as 10~
solution in water) 4 mg
Sodium carboxymethyl starch 4.5 mg
Magnesium stearate 0.5 mg
Talc 1 mq
Total 150 mg
The active ingredient, starch and cellulose
are passed through a No. 20 mesh U.S. sieve and mixed
thoroughly. The solution of polyvinylp~rrolidone is
mixed with the resultant powders which are then passed
through a No. 4 mesh U.S. sieve. The granules so
produced are dried at 50-60C and passed through a No.
16 mesh U.S. sieve. The sodium carboxymethyl starch,
magnesium stearate and talc, previously passed through
a No. 30 mesh U.S. sieve, are then added to the
granuleswhich, after mixing, are compressed on a tablet
machine to yield tablets each weighing 150 mg.
Formulation ~
Capsules each con~aining 20 mg of medicament
are made as follows:
(2aS,4R)-4-~di-n-propylamino)-
6-(2,2-dimethylpropanoyl)-
1,2,2a,3,4,5-hexahydrobenz-
~cd)indole 20 mg
Starch 163 mg
Magnesium stearate
Total 190 mg
The active ingredient, cellulose, starch and
magnesium ste~rate are blended, passed through a No.
20 mesh U.S. sieve, and filled into h~rd gelatin
capsules in 190 mg quantities.
' :~ ~ ' -
~ X-8265A 56 ~049~7&i
Fonmulation_6
Suppositories each containing 225 mg of
active ingredient are made as follows:
54-(di-n-propylamino)-6-benzoyl-
1,2,2a,3,4,S-hexahydrobenz[cd]-
indole 225mg
Saturated fatty acid
glycerides to 2,000 mg
The active ingredient is passed through a No.
60 mesh U.S. sieva and suspended in the saturated fatty
acid glycerides previously melted using the minimum
heat necessary. The mixture is then poured into a
suppository mold of nomi.nal 2 g capacity and allowed to
cool.
Formula~ion 7
Suspensions each containing 50 mg of
medicament per 5 mL dose are made as follows:
1-methyl-4-(n-propylamino)-6-
(3-methylbutanoyl)-1,2,2a,3,4,5-
hexahydrobenz~cd]indole 50 mg
Xanthan Gum 4 mg
Sodium carboxymethyl cellulose (11%)
Microcrystalline Cellulose (89%) 50 mg
Sucrose 1.75 g
Sodium Benzoate 10 mg
Flavor q.v.
Color q.v~
Purified water to 5 mL
The medicament, sucrose and xanthan gum are
blended, passed through a No. 10 mesh U.S. sieve, and
then mixed with a previously made solu~ion of the
microcrystalline cellulose and sodium carboxymethyl-
cellulose in water. The sodium benzoate, flavor andcolor are diluted with some of the water and added with
:
X-8265A 57 ~49~7~
stirring. Sufficient water is then added ~o produce
the required volume.
Formula~ion 8
Capsules each containing 150 mg of medicament
are made as follows:
4-(di-n-propylamino)-6-acetyl-
1,2,2a,3,4,5-hexahydrobenz[cd]-
indole50 mg
lo Starch507 mg
Magnesium stearate 3 mq
Total 560 mg
The active ingredient, cellulose, starch and
magnesium stearate are blended, passed through a No. 20
mesh U.S. sieve, and filled into hard gelatin capsules
in 560 mg quant~ties.
: : .