Note: Descriptions are shown in the official language in which they were submitted.
2050~55
1-METHYLCARBAPENEM DERIVATIVES AND
PROCESS FOR PREPARATION THEREOF
This invention relates to 1-methylcarbapenem derivatives
- having excellent antimicrobial activities and a process for
the preparation thereof.
It is known that thienamycin, one of the carbapenem
derivatives, has excellent antimicrobial activities against a
wide range of pathogenic microorganisms including Gram
positive bacteria and Gram negative bacteria. Because of it's
high activity against cephem-resistant bacteria, great
attention has been given to it. However, thienamycin is
easily deactivated by dehydropeptidase I present in the human
body and shows no activity via oral administration. From this
viewpoint, many researchers have extensively studied new
carbapenem derivatives which have excellent antimicrobial
activities against various microorganisms via oral
administration and are stable to dehydropeptidase I. For
example, there are disclosed in Japanese Patent First
Publication (Kokai) No. 49783/1990 6-(l-hydroxyethyl)-1-
methylcarbapen-2-em-3-carboxylic acids which are substituted
by a 2-oxo-pyrrolidin-4-ylthio group at the 2-position
thereof. However, there has never been known any compound
having a 2-thioxo-pyrrolidin-4-yl group at the 2-position of a
1-methylcarbapenem nucleus.
The present inventors have intensively studied new 1-
methylcarbapenem derivatives and have found that the 1-
methylcarbapenem derivatives bearing a 2-thioxopyrrolidin-4-yl
group at the 2-position of the carbapenem nucleus have
superior antimicrobial activities to the known carbapenem
derivatives and have superior stability against dehydro-
peptidase I with high absorbability by oral administration.
An object of the invention is to provide novel 1-methyl-
carbapenem derivatives having excellent antimicrobial
activities, excellent stability against dehydropeptidase I and
high absorbability by oral administration. Another object of
the invention is to provide a process for preparing the novel
~A ~
2 20502~5
1-methylcarbapenem derivatives. These and other objects and
advantages of the invention will be apparent to those skilled
-- in the art from the following description.
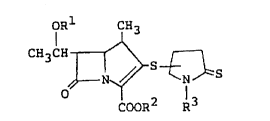
The l-methylcarbapenem derivatives of the invention
have the following formula:
IRl CH3
C~3C~ S~S ~1 ~
COOR R
wherein R1is a hydrogen atom or a hydroxy-protecting group,
RZis a hydrogen atom or an ester residue, R3is a hydrogen atom
or a lower alkyl group.
Among the compounds [I] of the invention, the compounds
of the formula [I] wherein R1is a hydrogen atom and R2is a
hydrogen atom or an ester residue which is hydrolyzed by
metabolism in the human body, or salts thereof have excellent
antimicrobial activities and are useful as medicaments. The
compounds of the formula [I] wherein R1is a protecting group
and/or RZis an ester residue which may be a carboxyl-
protecting group are useful as intermediates in the
preparation of the above compounds having excellent
antimicrobial activities.
The hydroxy-protecting group for R1in the compounds [I]
includes, for example, a lower alkoxycarbonyl group, a
halogeno-lower alkoxycarbonyl group, a substituted or
unsubstituted phenyl-lower alkyl group (e.g. a benzyl
optionally having a substituent selected from a nitro and a
lower alkoxy group), a tri(lower alkyl)silyl group, a
substituted or unsubstituted phenyl-lower alkoxycarbonyl group
(e.g. a benzyloxycarbonyl optionally having a substituent
selected from a nitro and a lower alkoxy group), and the like.
The ester residue for R2includes an ester residue which
can be hydrolyzed by metabolism in the human body and an ester
residue which may be a carboxyl-protecting group. The
,`! A
3 20aO2~5
hydrolyzable ester residue includes, for example, groups of
the formulae: -A-oCoR4, -A-OCOOR40r -A-O-R4wherein A is a
- lower alkylene group, R4is a lower alkyl group, a cycloalkyl
group, a lower alkenoyl group, a lower alkoxy-lower alkyl
5- group, or a lower alkanoyloxy-lower alkyl group. Suitable
examples of these ester residues are a lower alkanoyloxy-lower
alkyl group, a cycloalkylcarbonyloxy-lower alkyl group, a
lower alkenoyloxy-lower alkyl group, a lower alkoxy-lower
alkanoyloxy-lower alkyl group, a lower alkanoyloxy-lower
alkoxy-lower alkyl group, a lower alkoxy-lower alkyl group, a
lower alkoxy-lower alkoxy-lower alkyl group, a lower
alkoxycarbonyloxy-lower alkyl group, a lower alkoxy-lower
alkoxycarbonyloxy-lower alkyl group, and the like.
The ester residue which may be a carboxyl-protecting
group includes, for example, a lower alkyl group, a lower
alkenyl group, a halogeno-lower alkyl group, a nitrobenzyl
group, and a lower alkoxybenzhydryl group.
Throughout the specification and claims, the lower alkyl,
lower alkylene and lower alkoxy groups preferably have 1
to 6 carbon atoms, more preferably l to 4 carbon atoms, and
the lower alkanoyl and lower alkenyl groups preferably have 2
to 8 carbon atoms, more preferably 2 to 6 carbon atoms, and
the lower alkenoyl and cycloalkyl groups preferably have 3 to
8 carbon atoms, more preferably 3 to 6 carbon atoms.
The compounds [I] of this invention can be used as
medicaments either in the free form or in the form of a
pharmaceutically acceptable salt thereof. The pharma-
ceutically acceptable salts include, for example, non-toxic
metal salts, e.g. sodium salt, potassium salt, calcium salt,
magnesium salt, aluminum salt, and the like; salts with
nontoxic amines, for example, trialkylamines (e.g.
triethylamine, etc.), pyridine, ethanolamine, triethanolamine,
dicyclo-hexylamine, and the like; addition salts with basic
amino acids, e.g. lysine, arginine, and the like. The salt of
the compounds [I] may also be a salt with a resin, e.g. a
polystyrene resin having an amino or quaternary amino group.
The compounds [I] or pharmaceutically acceptable salts
r
r~` ~
20502~5
thereof can be administered by oral or parenteral routes
(e.g. intravenous, intramuscular or subcutaneous routes). The
- daily dose of these compounds is in the range of about 0.002
to 0.04 g/kg of body weight, preferably about 0.005 to
0.01 g/kg of body weight. They can be used in the form of a
pharmaceutical preparation suitable for oral or parenteral
administration in admixture with a pharmaceutically acceptable
carrier or diluent, for example, solid preparations (e.g.
tablets, granules, capsules, etc.), and liquid preparations
(e.g. solutions, suspensions, emulsions, etc.).
The compounds [I] include various isomers owing to the
asymmetric carbon and this invention also includes these
isomers and mixtures thereof. In the case of use as a
medicament, however, the compounds [I] preferably have R, S
and S configurations at the 1-, 5- and 6-positions of the
carbapenem nucleus, respectively, and R configuration at the
l-position of the 6-substituent and further S or R
configuration at the substitution position in the pyrrolidine
ring.
The compounds [I] of this invention can be prepared by
reacting a reactive derivative at the 2-position of a ketone
compound of the formula [II]:
ORl CH3
~H3C~,~ ~O 1 I I ]
~2
COOR
wherein R1and RZare as defined above, with a mercaptan
compound of the formula [III]:
~S~S 1 III ]
1 3
wherein R3 is as defined above, or a salt thereof.
b,
~A
2050255
The reactive derivative at the 2-position of the ketone
compound [II] includes any conventional compound, for example,
a compound of the formula [II-a]:
~l CH3
CH3C~
¦ /~ OX [II-a~
0~ ~
COOR
wherein R1and R2are as defined above, and X is a
diphenylphosphoryl group, a di(lower alkyl)-substituted
phenylphosphoryl group, a di(lower alkyl)phosphoryl group, a
lower alkanesulfonyl group, a phenylsulfonyl group, or a lower
alkyl-substituted phenylsulfonyl group, which can be prepared
by reacting the ketone compound [II] with the corresponding
phosphoryl halide or sulfonic acid compound in the presence or
absence of a base (e.g. a tri(lower alkyl)amine, a 4-di(lower
alkyl)aminopyridine, etc.).
The salt of the mercaptan compound [III] includes, for
example, an alkali metal salt, a tri(lower alkyl)ammonium
salt, and the like.
The reaction of the reactive derivative of the ketone
compound [II] and the mercaptan compound [III] or a salt
thereof is carried out in the presence or absence of a base.
The base includes any conventional base, preferably a
tri(lower alkyl)amine, a 4-di(lower alkyl)aminopyridine,
and the like. The reaction is usually carried out in an
appropriate solvent or without solvent under cooling
(e.g. -5C to 0C). The solvent includes any conventional
inert solvent, for example, anhydrous acetonitrile,
tetrahydrofuran, methylene chloride, and the like.
The compounds [I] thus prepared wherein R1is a hydroxy-
protecting group and/or R2is an ester residue which may be a
carboxyl-protecting group may be subjected to removal of the
protecting group and/or the ester residue to give the
~' ~
~; ~
6 20~02~5
compounds of the formula [I-a]:
fRl CH3
C~3C~ ~ S ~1 ~ S [I-a]
COOH R3
wherein R~and R3are as defined above, or a salt thereof.
Removal of the protecting group or ester residue can be
carried out in a usual manner.
The compounds [I-a] thus prepared may be esterified by a
conventional method to give compounds of the formula [I-b]:
ORl CH3
O ~ ~ ~ ~ [I-b~
COOR 1 R3
wherein R1and R3are as defined above, and R21is an ester
residue.
Among the starting compounds, the ketone compound [II]
can be prepared in the same manner as described in
HETEROCYCLES, Vol. 21, page 29, 1984, and the mercaptan
compound [III] can be prepared by reacting an N-substituted or
unsubstituted hydroxy-2-pyrrolidone with thioacetic acid in
the presence of triphenylphosphine and diethyl azodicarboxy-
late, followed by treating with Lawesson's reagent [= 2,4-
bis(4-methoxyphenyl)-1,3-dithia-2,4-diphosphotane-
2,4-disulfide] and further followed by deacetylation.
The compounds of this invention are illustrated by the
following Examples and Reference Examples, but should not be
construed to be limited thereto.
Example 1
(1) (lR, 5R, 6S)-6-[(lR)-1-Hydroxyethyl]-1-methyl-2-oxo-
carbapenam-3-carboxylic acid 4-nitrobenzyl ester (7.6 g) was
20502S5
dissolved in anhydrous acetonitrile (53 ml), and diisopropyl-
ethylamine (2.9 g) and diphenylphosphoryl chloride (6.1 g)
- were dropwise added thereto in that order under nitrogen gas
below 0C. After stirring the mixture at the same temperature
for 30 minutes, a solution of (4S)-4-mercaptopyrrolidine-2-
thione (2.5 g) and diisopropylethylamine (2.9 g) in anhydrous
acetonitrile (53 ml) was added dropwise to the reaction
mixture [which contains (lR, 5R, 6S)-6-[(lR)-l-hydroxyethyl]-
l-methyl-2-diphenylphosphoryl-oxycarbapen-2-em-3-carboxylic
acid 4-nitrobenzyl ester] below -15C. The mixture was
stirred at the same temperature for 1.5 hours, and water
(53 ml) was added to the reaction mixture. The mixture was
concentrated under reduced pressure to remove acetonitrile.
The residue was extracted with ethyl acetate, and the extract
was washed with brine, dried and evaporated under reduced
pressure. The residue was purified by silica gel column
chromatography (eluant, chloroform : ethanol = 20 : 1) to give
(lR, 5S, 6S)-2-[(4S)-pyrrolidine-2-thion-4-ylthio]-6-[(lR)-l-
hydroxyethyl]-1-methylcarbapen-2-em-3-carboxylic acid 4-
nitrobenzyl ester (4.7 g) as an amorphous powder.
NMR (CDCl3) ~ppm: 1.29 (3H, d), 1.36 (3H, d), 2.90
(lH, dd), 3.23-3.34 (2H, m), 3.61-3.71 (2H, m), 4.00-4.32
(5H, m), 5.23, 5.49 (2H, d), 7.65, 8.23 (4H, d), 7.83
(lH, br.s)
(2) A mixture of the product obtained above (0.5 g),
potassium hydrogen carbonate (0.105 g), tetrahydrofuran
(10 ml), ethanol (10 ml) and 10 % palladium-carbon (1 g) was
hydrogenated at room temperature under atmospheric pressure
for one hour. After removing the catalyst by filtration, the
organic solvent was evaporated under reduced pressure. The
aqueous layer was washed with ethyl acetate and evaporated to
dryness under reduced pressure. The residue was purified with
a column packed with a nonionic adsorbing resin (CHP-20P*
manufactured by Mitsubishi Kasei Corporation) (eluant, water)
to give (lR, 5S, 6S)-2-[(4S)-pyrrolidine-2-thion-4-ylthio]-6-
*Trade mark
2050255
[(lR)-l-hydroxyethyl]-l-methylcarbapen-2-em-3-carboxylic acid
potassium salt (0.11 g) as an amorphous powder.
- NMR (D2O) ~ppm: 1.20 (3H, d), 1.2B (3H, d), 2.84 (lH, dd),
3.24-3.47 (3H, m), 3.62-3.72 (lH, m), 4.08-4.27 (4H, m)
Example 2
(1) In the same manner as described in Example 1-(1)
except that (4R)-4-mercaptopyrrolidine-2-thione (1.1 g) was
used instead of (4S)-4-mercaptopyrrolidine-2-thione, there was
obtained (lR, 5S, 6S)-2-[(4R)-pyrrolidine-2-thion-4-ylthio]-6-
[(lR)-l-hydroxyethyl]-l-methylcarbapen-2-em-3-carboxylic acid
4-nitrobenzyl ester (1.8 g) as colorless needles.
M.p. 168 - 170C
NMR (DMSO) ~ppm: 1.15 (6H, d), 2.74 (lH, dd), 3.30-3.51
(4H, m), 3.97-4.31 (4H, m), 5.08 (lH, d), 5.29, 5.47 (2H, d),
7.70, 8.24 (4H, d), 10.39 (lH, br.s)
(2) The product obtained above (0.5 g) was treated in
the same manner as described in Example 1-(2) to give (lR, 5S,
6S)-2-[(4R)-pyrrolidine-2-thion-4-ylthio]-6-[(lR)-l-hydroxy-
ethyl]-l-methylcarbapen-2-em-3-carboxylic acid potassium salt
(0.12 g) as an amorphous powder.
NMR (D2O) ~ppm: 1.20 (3H, d), 1.29 (3H, d), 2.94 (lH, dd),
3.28-3.67 (4H, m), 4.06-4.27 (4H, m)
Example 3
To a mixture of (lR, 5S, 6S)-2-[(4R)-pyrrolidine-2-thion-
4-ylthio]-6-[(lR)-l-hydroxyethyl]-l-methylcarbapen-2-em-3-
carboxylic acid potassium salt (0.34 g), N,N-dimethylformamide
(3 ml) and potassium carbonate (0.12 g) was added dropwise
isobutyryloxymethyl iodide (0.27 g) under ice cooling. After
stirring the mixture at the same temperature for 30 minutes,
ethyl acetate (10 ml) was added to the reaction mixture. The
mixture was washed with water, and the organic layer was dried
and evaporated under reduced pressure to remove the solvent.
The residue was purified by silica gel flash column
chromatography (eluant, chloroform : ethanol = 20 : 1),
followed by crystallization from isopropyl ether and ethyl
acetate to give (lR, 5S, 6S)-2-[(4R)-pyrrolidine-2-thion-4-
ylthio]-6-[(lR)-l-hydroxyethyl]-l-methylcarbapen-2-em-3-
205025S
g
carboxylic acid isobutyryloxymethyl ester (0.12 g) ascolorless needles.
- M.p. 158 - 159C
ExamPles 4 to 8
In the same manner as described in Example 3 (lR, 5S,
6S)-2-[(4R)-pyrrolidine-2-thion-4-ylthio]-6-[(lR)-l-
hydroxyethyl]-l-methylcarbapen-2-em-3-carboxylic acid
potassium salt was treated with various alkanoyloxymethyl
iodide to prepare various ester compounds as shown in the
following Table 1.
Table 1
OH H H CH3
Ex. ~ ~ ~
No. ~ ~ S ~ S
COOR H
R Physical properties, etc.
4 -CH2OCOCH2CH2CH3 m.p. 115 - 117C
-cH2ococH2cH2cH2cH3 m.p. 75 - 77 C
6 -CH2OCOC(CH3)3 m.p. 140 - 142C
7 -CH2OCOCH2CH3 amorphous powder
NMR (CDC13) ~ppm: 1.16 (3~, t),
1.28 (3H, d), 1.35 (3H, d), 1.68
(lH, s), 2.41 (2H, q), 2.97 (lH,
dd), 3.23-3.44 (3H, m), 3.60-3.64
(lH, m), 4.01-4.13 (2H, m), 4.21-
4.30 (2H, m), 5.85, 5.94 (2H, d),
7.79 (lH, s)
8 -CH2OCOCH2cH(cH3)2 m.p. 122 - 125C
- lO 20~0255
Examples 9 to 28
In the same manner as described in Example 3, (lR, 5S,
6S)-2-[(4S)-pyrrolidine-2-thion-4-ylthio]-6-[(lR)-l-
hydroxyethyl]-l-methylcarbapen-2-em-3-carboxylic acid
-5 potassium salt was treated with various substituted methyl
iodided or 1-substituted ethyl iodide to prepare various ester
compounds as shown in the following Table 2.
. r
~ A
11 20502~5
Table 2
- OH H H CH
- Ex.
No. O/
COOCH2OR H
R Physical properties, etc.
9 -COCH2CH3 amorphous powder
NMR (CDCl~) ~ppm: 1.16 (3H, t), 1.28
(3H, d), I.34 (3H, d), 2.14 (lH, s),
2.41 (2H, q), 2.90 (lH, dd), 3.23-3.43
(3H, m), 3.59-3.68 (lH, m), 4.00-4.11
(2H, d), 4.22-4.30 (2H, m), 5.84, 5.94
(2H, d), 7.90 (lH, s)
-COCH(CH3)2 m.p. 143 - 144C (decomp.)
11 -COC(CH3)3 m.p. 152 - 153C (decomp.)
12 -COCH(CH3)CH2CH3 amorphous powder
NMR (CDCl~) ~ppm: 0.90 (3H, t), 1.14
(3H, t), I.27 (3H, d), 1.32 (3H, d),
1.40-1.85 (2H, m), 2.35-2.55 (lH, m),
2.80-3.00 (lH, m), 3.20-3.35 (2H, m),
3.20-3.40 (lH, m), 3.50-3.75 (lH, m),
4.00-4.20 (2H, m), 4.20-4.35 (2H, m),
5.80-5.95 (2H, m)
13 -CO ~ NMR (CDC13) ~ppm: 0.85-1.05 (2H, m),
1.05-1.15 (2H, m), 1.27 (3H, d), 1.34
(3H, d), 1.60-1.80 (lH, m), 2.80-3.00
(lH, m), 3.20-3.35 (2H, m), 3.20-3.40
(lH, m), 3.60-3.75 (lH, m), 3.95-4.20
(2H, m), 4.20-4.35 (2H, m), 5.83, 5.92
(2H, ABq)
14 -COCH2CH=CH2 amorphous powder
NMR (CDC13) ~ppm: 1.20-1.40 (6H, m),
2.80-3.10 (lH, m), 3.15-3.25 (2H, m),
3.25-3.35 (2H, m), 3.35-3.50 (lH, m),
3.60-3.70 (lH, m), 3.80-4.20 (2H, m),
4.20-4.35 (2H, m), 5.10-5.30 (2H, m),
5.75-6.05 (3H, m)
- to be continued -
~, r~ -
~t~
~
12 20502~s
Table 2 ( continued)
OH H H CH3
Ex.
No.CH3CH ~ S ~
~ N ~ N S
O l l
COOCH2OR H
R Physical properties, etc.
-COCH2OCH3
NMR (CDCl~) ~ppm: 1.25 (3H, d), 1.33
(3H, d), ~.85-3.00 (lH, m), 3.20-3.40
(2H, m), 3.30-3.50 (lH, m), 3.60-3.70
(lH, m), 3.95-4.20 (2H, m), 4.20-4.35
(2H, m), 4.10 (3H, s), 5.80-6.10 (2H, m)
16 -COCH2CH(CH3)2 amorphous powder
N~R (CDCl~) ~ppm: 0.96 (6H, d), 1.27
(3H, d), ~. 34 (3H, d), 2.00-2.20 (lH,
m), 2.20-2.30 (2H, m), 2.80-3.00 (lH,
m), 3.20-3.35 (2H, m), 3.35-3.55 (lH,
m), 3.55-3.75 (lH, m), 3.95-4.20 (2H,
m), 4.20-4.35 (2H, m), 5.85, 5.93 (2H,
ABq)
17 -COCH3 amorphous powder
NMR (CDCl~) ~ppm: 1.27 (3H, d), 1.33
(3H, d), 2.13 (3H, s), 2.80-3.00 (lH,
m), 3.20-3.35 (2H, m), 3.30-3.45 (lH, m)
18 -COCH2CH2CH2CH3 amorphous powder
NMR (CDC13) ~ppm: 0.91 ( 3H, t, J=7.2Hz),
1.28 (3H, d, J=7.3Hz)j 1.33 (3H, d, J=
6.2Hz), 1.54-1.70 (4H, m), 2.39 (2H, t,
J=7.7Hz), 2.83-2.96 (lH, m), 3.25-3.42
(3H, m), 3.59-3.71 (lH, m), 4.03-4.10
(2H, m), 4.24-4.30 (2H, m), 5.83, 5.93
(2H, ABq, J=5.6Hz), 8.10 (lH, br)
- to be continued -
~, ~
~ A
13 20~02~5
Ta~le 2 (continued)
OH H H CH3
Ex - ~ r - ~
No. o~~N~ ~s
COOCH2OR H
R Physical properties, etc.
19 -COCH2CH2cH3 amorphous powder
NMR (CDC13) ~ppm: 0.96 (3H, t, J=
7.4Hz), 1.28 (3H, d, J=7.3Hz), 1.33
(3H, d, J=6.2Hz), 1.54-1.76 (2H, m),
2.37 (2H, t, J=7.5~z), 2.8g (lH, dd,
J=4.9, 18.4Hz), 3.25-3.52 (3H, m),
3.64-3.67 (lH, m), 4.02-4.10 (2H, m),
4.23-4.29 ~2~, m), 5.83, 5.g3 ~2H,
ABq, J=5.6Hz), 7.99 (lH, br)
20 -COCH2OCH2CH3 amorphous powder
NMR (CDC13) ~ppm: 1.21-1.35 (9H, m),
2.87 (lH, dd, J=5.0, 18.0Hz), 3.26-
3.41 (3H, m), 3.57-3.71 (3H, m), 4.02-
4.30 (6H, m), 5.86, 6.02 (2H, ABq,
J=5.6Hz), 8.11 (lH, br)
1 -CH2CH2OCOCH2CH3 amorphous powder
NMR (CDC13) ~ppm: 1.05-1.15 (3H, m),
1.27 (3H, d), 1.33 (3H, d), 2.35 (2H,
q), 2.80-3.00 (lH, m), 3.15-3.30 (2H,
m), 3.25-3.40 (lH, m), 3.55-3.70 (lH,
m), 3.80-4.15 (4H, m), 4.15-4.35 (4H,
m), 5.30, 5.61 (2H, ABq)
22 -CH3 m.p. 166 - 168C
23 -CH2cH2OcH3 amorphous powder
NMR (CDC13) ~ppm: 1.27 (3H, d), 1.33
(3H, d), 2.80-3.00 (lH, m), 3.15-3.30
(2H, m), 3.40 (3H, s), 3.50-3.70 (4H,
m), 3.80-3.95 (2H, m), 4.00-4.15 (2H,
m), 4.15-4.35 (2H, m), 5.31, 5.61 (2H,
ABq)
- to be continued -
14
20502~5
Table 2 (continued)
-
OH H H CH
Ex. ~ -
No. CH3C ~ S
COOCHOCOR H
R Physical properties, etc.
24 -CH3 amorphous powder
NMR (CDC13) ~ppm: 1.24-1.34 (6H, m),
1.54-1.58 (3H, m), 2.08, 2.11 (3H, s),
2.83-2.96 (lH, m), 3.28-3.41 (3H, m),
3.62-3.68 (lH, m), 4.06 (2H, m), 4.25
(2H, m), 6.92-7.08 (lH, m), 8.15 (lH,
br)
25 -CH(CH3)2 amorphous powder
NMR (CDC13) ~ppm: 1.14-1.36 (12H, m),
1.54-1.58 (3H, m), 2.49-2.66 (lH, m),
2.83-2.93 (lH, m), 3.25-3.42 (3H, m),
3.59-3.67 (lH, m), 4.03-4.10 (2H, m),
4.22-4.28 (2H, m), 6.91-7.01 (lH, m)
8.20 (lH, br)
26 -OCH(CH3)~2 amorphous powder
~MR (CDC13) ~ppm: 1.25-1.35 (12H, m)
1.60 (3H, t, J=4.9Hz), 2.88 (lH, dd,
J=4.6, 18.2Hz), 3.25-3.42 (3H, m),
1.65 (lH, m), 4.06-4.10 (2H, m), 4.24-
4.28 (2H, m), 4.83-4.98 (lH, m), 6.83-
6.88 tlH, m), 8.19 (lH, br)
27 OCH2CH3 amorphous powder
NMR (CDC13) ~ppm 1.22-1.37 (9H, m),
1.60 (3H, t, J=4.4Hz), 2.87 (lH, dd,
J=4.7, 18.2Hz), 3.25-3.42 (3H, m),
3.62-3.66 (lH, m), 4.06-4.30 (6H, m),
6.84-6.88 (lH, m), 8.13 (lH, br)
28 OcH2cH2ocH2cH3 amorphous powder
N~-~R (CDC13) ~ppm: 1.17-1.35 (9H, m),
1.58-1.62 (3H, m), 2.86 (lH, dd, J=
4.6, 18.3Hz), 3.26-3.42 (3H, m), 3.49-
3.71 l5H, m), 4.07-4.11 (2H, m), 4.24-
4.34 (4H, m~, 6.61-6.91 (lH, m), 8.13
(lH, br)
~;. A
- lS 20~0235
Example 29
(1) (lR, 5R, 6S)-6-[(lR)-l-Hydroxyethyl]-l-methyl-2-oxo-
carbapenam-3-carboxylic acid 4-nitrobenzyl ester (25.2 g) was
dissolved in anhydrous acetonitrile (173 ml), and diisopropyl-
5- ethylamine (13 ml) and diphenylphosphoryl chloride (15.5 ml)
were added dropwise thereto in that order under nitrogen gas
below 0C. After stirring the mixture at the same temperature
for 30 minutes, a solution of (4R)-N-methyl-4-mercaptopyrroli-
dine-2-thione (13 g) and diisopropylethylamine (11.5 g) in
anhydrous acetonitrile (173 ml) was added dropwise to the
reaction mixture below -5C. After stirring the mixture at
0C for 1.5 hours, the reaction mixture was poured into a
phosphate buffer (pH 7.0, one liter). The precipitated
crystals were separated by filtration and dissolved in a
mixture of chloroform (2 liters) and tetrahydrofuran
(1 liter), and the solution was washed with water, dried and
concentrated to dryness under reduced pressure. The residue
was crystallized from diethyl ether to give (lR, 5S, 6S)-2-
[(4R)-N-methyl-pyrrolidine-2-thion-4-ylthio]-6-[(lR)-l-
hydroxyethyl]-1-methylcarbapen-2-em-3-carboxylic acid
4-nitrobenzyl ester (23.5 g) as colorless needles.
M.p. 157 - 158C
(2) A mixture of the product obtained above (6 g),
tetrahydrofuran (250 ml), ethanol (250 ml), sodium hydrogen
carbonate (1.03 g), water (250 ml) and 10 % palladium-carbon
(water content 52%, 22.8 g) was hydrogenated at room
temperature under atmospheric pressure for 1.5 hours. After
removing the catalyst by filtration, the organic solvent was
evaporated under reduced pressure. The aqueous layer was
washed with ethyl acetate, treated with activated charcoal and
filtered. The filtrate was concentrated under reduced
pressure, and the residue was crystallized from ethanol to
give (lR, 5S, 6S)-2-[(4R)-N-methylpyrrolidine-2-thion-4-
ylthio]-6-t(lR)-1-hydroxyethyl]-1-methylcarbapen-2-em-3-
carboxylic acid sodium salt (3.2 g) as colorless needles.
M.p. 170 - 180C (decomp.)
.~ I
16 20502~5
Example 30
(lR, 5S, 6S)-2-[(4R)-N-methylpyrrolidine-2-thion-4-
- ylthio]-6-[(lR)-l-hydroxyethyl]-1-methylcarbapen-2-em-3-
carboxylic acid sodium salt (400 mg) was suspended in N,N-
dimethylformamide (5 ml), and potassium carbonate (146 mg) wasadded thereto. Acetoxymethyl iodide (275 mg) at 5 - 7C was
added to the mixture, and the mixture was stirred at the same
temperature for one hour. The reaction mixture was poured
into a phosphate buffer (pH 7.0) and extràcted with ethyl
acetate. The organic layer was washed with water, dried and
concentrated to dryness under reduced pressure. The residue
was purified by silica gel column chromatography (eluant,
chloroform - chloroform : ethanol = 98: 2), followed by
crystallization from tetrahydrofuran and diethyl ether to give
(lR, 5S, 6S)-2-[(4R)-N-methylpyrrolidine-2-thion-4-ylthio]-6-
[(lR)-1-hydroxyethyl]-1-methylcarbapen-2-em-3-carboxylic acid
acetoxymethyl ester (250 mg) as colorless needles.
M.p. 124 - 126C
Examples 31 to 37
In the same manner as described in Example 30, (lR, 5S,
6S)-2-[(4R)-N-methylpyrrolidine-2-thion-4-ylthio]-6-[(lR)-1-
hydroxyethyl]-l-methylcarbapen-2-em-3-carboxylic acid sodium
salt was treated with various substituted methyl iodides to
prepare various ester compounds as shown in the following
Table 3.
17 20502~.5
Table 3
OH H H CH
Ex. ~ ~ - r 3
No. O ~ I
COOR CH3
R Physical properties, etc.
31 -CH2OCOCH2CH3 m.p.93 - 97C
32 -cH2ococH2cH2cH3 m.p.148 - 149C
33 -CH2OCOCH(CH3)2 m.p.138 - 141C
34 -CH2OCO ~ m.p.133 - 135C
-CH2OCO(CH2)3cH3 m.p.122 - 123C
36 -CH2OCOCH2cH(cH3)2 m.p. 130 - 132C
37 -CH2OCOC(CH3)3 m.p.159 - 160C
Example 38
(1) In the same manner as described in Example 29-(1),
(lR, 5S, 6S)-6-[(lR)-1-hydroxyethyl]-1-methyl-2-oxo-
carbapenam-3-carboxylic acid 4-nitrobenzyl ester was treated
with (4S)-N-methyl-4-mercaptopyrrolidine-2-thione to give
(lR, 5S, 6S)-2-[(4S)-N-methyl-4-mercapto-pyrrolidine-2-
thion-4-ylthio]-6-[(lR)-1-hydroxyethyl]-1-methylcarbapen-
2-em-3-carboxylic acid 4-nitrobenzyl ester as an amorphous
powder.
10NMR (CDCl3) ~ppm: 1.29 (3H, d), 1.36 (3H, d), 3.00-3.15
(lH, m), 3.25 (3H, s), 3.15-3.35 (lH, m), 3.40-3.60 (lH, m),
3.65-3.80 (lH, m), 3.95-4.00 (lH, m), 4.05-4.20 (lH, m), 4.20-
4.35 (2H, m), 5.22, 5.50 (2H, ABq), 7.64, 8.23 (4H, A2B2q)
(2) The product obtained above was treated in the same
15manner as described in Example 29-(2) to give (lR, 55, 6S)-
2-[(4S)-N-methylpyrrolidine-2-thion-4-ylthio]-6-[(lR)-l-
hydroxyethyl]-1-methylcarbapen-2-em-3-carboxylic acid sodium
,~
18 2~5~2~.~
salt as colorless needles.
M.p. 185 - 190C (decomp.)
Examples 39 to 46
In the same manner as described in Example 30, (lR, 5S,
6S)-2-[(4S)-N-methylpyrrolidine-2-thion-4-ylthio]-6-[(lR)-1-
hydroxyethyl]-1-methylcarbapen-2-em-3-carboxylic acid sodium
salt was treated with various substituted methyl iodides to
prepare various ester compounds as shown in the following
Table 4.
Table 4
OH H H CH3
Ex. C ~ ~
No. 1 ~ S ~ l ~S
COOR CH3
R Physical properties, etc.
39 -CH20COCH3 m.p. 165 - 167C
-CH2OCOCH2CH3 m.p. 138 - 139C
41 -cH2ococH2cH2cH3 m.p. 127 - 128C
42 -CH20cOcH~cH3)2 m.p. 163 - 164C
43 -CH20CO ~ m.p. 130 - 133C
44 -CH2OCO(CH2)3CH3 m.p. 119 - 121C
-cH2ococH2cH(cH3)2 m.p. 133 - 136C
46 -CH2OCOC(~CH3)3 m.p. 167 - 169C
ExamPle 47
(1) (lR, 5R, 6S)-6-[(lR)-l-hydroxyethyl]-l-methyl-2-oxo-
carbapenam-3-carboxylic acid 4-nitrobenzyl ester (8.3 g) was
dissolved in anhydrous acetonitrile (50 ml), and
diisopropylethylamine (4 ml) and diphenylphosphoryl chloride
(4.8 ml) was added dropwise thereto in that order under
~~ '
~, ~
20502~5
nitrogen gas below 0C. After stirring the mixture at the
same temperature for 30 minutes, a solution of (3R)-3-
mercaptopyrrolidine-2-thione (4 g) and diisopropylamine (4 ml)
in anhydrous acetonitrile (50 ml) was added dropwise to the
reaction mixture below -15C. The mixture was stirred at the
same temperature for 1.5 hour, and water (50 ml) was added to
the reaction mixture. The mixture was concentrated under
reduced pressure to remove acetonitrile. The residue was
extracted with ethyl acetate, and the extract was washed with
water, dried and evaporated to dryness under reduced pressure.
The residue was purified by silica gel column chromatography
(eluant, chloroform : methanol = 99 : 1) to give (lR, 5S, 6S)-
2-[(3R)-pyrrolidine-2-thion-3-ylthio]-6-[(lR)-l-hydroxy-
ethyl]-l-methylcarbapen-2-em-3-carboxylic acid 4-nitrobenzyl
ester (4.3 g) as an amorphous powder.
NMR (CDCl3) ~ppm: 1.20-1.40 (6H, m), 1.68 (lH, br.s),
2.20-2.40 (lH, m), 2.60-2.80 (lH, m), 3.30-3.50 (lH, m), 3.30-
3.40 (lH, m), 3.60-3.90 (2H, m), 4.20-4.40 (2H, m), 4.40-4.S0
(lH, m), 5.22 (lH, d), 5.50 (lH, d), 7.62 (2H, d), 8.12
(lH, br.s), 8.22 (2H, d)
(2) A mixture of the product obtained above (2 g), water
(78 ml), tetrahydrofuran (39 ml), ethanol (39 ml), potassium
hydrogen carbonate (0.42 g) and 10 % palladium-carbon (4 g)
was hydrogenated at room temperature under atmospheric
pressure for 2 hours. After removing the catalyst by
filtration, the organic solvent was evaporated under reduced
pressure. The aqueous layer was washed with ethyl acetate and
concentrated to dryness under reduced pressure. The residue
was purified with a column packed with a nonionic adsorbing
resin (CHP-20P manufactured by Mitsubishi Kasei Corporation)
(eluant, water) to give (lR, 5S, 6S)-2-[(3R)-pyrrolidine-2-
thion-3-ylthio]-6[(lR)-l-hydroxyethyl]-1-methylcarbapen-2-em-
3-carboxylic acid potassium salt (0.9 g) as an amorphous
powder.
NMR (D20) ~ppm: 1.20-1.40 (6H, m), 2.20-2.40 (lH, m),
2.60-2.80 (lH, m), 3.30-3.50 (2H, m), 3.70-3.90 (2H, m), 4.20-
4.40 (2H, m), 4.50-4.60 (lH, m)
~ r~
20502~
Example 48
(lR, 5S, 6S)-2-[(3R)-Pyrrolidine-2-thion-3-ylthio]-6-
[(lR)-l-hydroxyethyl]-l-methylcarbapen-2-em-3-carboxylic acid
potassium salt (0.5 g) was suspended in N,N-dimethylformamide
(5 ml). Isobutyryloxymethyl iodide (338 mg) was added to the
suspension under ice cooling, and the mixture was stirred at
the same temperature for 30 minutes. To the reaction mixture
was added ethyl acetate, and the mixture was washed with
water, dried and concentrated to dryness under reduced
pressure. The residue was purified by silica gel flash column
chromatography (eluant, chloroform : methanol = 20 : 1) to
give (lR, 5S, 6S)-2-[(3R)-pyrrolidine-2-thion-3-ylthio]-6-
[(lR)-l-hydroxyethyl]-1-methylcarbapen-2-em-3-carboxylic acid
isobutyryloxymethyl ester (0.14 g) as an amorphous powder.
NMR (CDCl3) ~ppm: 1.20-1.40 (12H, m), 2.20-2.40 (2H, m),
2.60-2.90 (2H, m), 3.30-3.40 (2H, m), 3.60-3.90 (2H, m), 4.20-
4.30 (2H, m)j 4.40-4.50 (lH, dd), 5.87 (lH, d), 5.98 (lH, d),
8.15 (lH, br.s)
Example 49
In the same manner as described in Example 47, (lR, 5R,
6S)-6-[(lR)-l-hydroxyethyl]-l-methyl-2-oxocarbapenam-3-
carboxylic acid 4-nitrobenzyl ester was treated with (3S)-3-
mercaptopyrrolidine-2-thione to give (lR, 5S, 6S)-2-[(3S)-
pyrrolidine-2-thion-3-ylthio]-6-[(lR)-l-hydroxyethyl]-l-
methylcarbapen-2-em-3-carboxylic acid potassium salt.
NMR (D20) ~ppm: 1.20-1.40 (6H, m), 2.20-2.40 (lH, m),
2.70-2.90 (lH, m), 3.40-3.50 (lH, m), 3.60-3.90 (3H, m),
4.20-4.40 (3H, m)
Example 50
In the same manner as described in Example 48, (lR, 5S,
6S)-2-[(3S)-pyrrolidine-2-thion-3-ylthio]-6-[(lR)-l-
hydroxyethyl]-l-methylcarbapen-2-em-3-carboxylic acid
potassium salt was treated with isobutyryloxymethyl iodide to
give (lR, 5S, 6S)-2-[(3S)-pyrrolidine-2-thion-3-ylthio]-6-
[(lR)-l-hydroxyethyl]-l-methylcarbapen-2-em-3-carboxylic acid
isobutyryloxymethyl ester.
~ r-
~. A
- 21 20~02S5
NMR (CDCl3) ~ppm: 1.10-1.40 (12H, m), 2.10-2.40 (2H, m),
2.50-2.80 (2H, m), 3.20-3.30 (lH, m), 3.60-3.90 (3H, m), 4.20-
4.40 (3N, m), 5.84 (lH, d), 5.94 (lH, d), 7.82 (lH, br.s)
Reference Exam~le 1
(1) (4R)-4-Hydroxy-2-pyrrolidone (4.5 g) was suspended
in tetrahydrofuran (300 ml), and triphenylphosphine (23.4 g)
was added thereto. After stirring the mixture for 10 minutes,
diethyl azodicarboxylate (14 ml) was added dropwise to the
mixture below -10C, and the mixture was stirred at the same
temperature for 10 minutes. Thioacetic acid (6.3 ml) was
added dropwise to the reaction mixture below -10C, and the
mixture was stirred at the same temperature for 2 hours. The
solvent was evaporated under reduced pressure. The residue
was crystallized from diisopropyl ether, and the precipitate
was removed by filtration. The filtrate was concentrated
under reduced pressure, and the residue was purified by
silica gel column chromatography (eluant, chloroform :
ethanol = 98 : 2) to give (4S)4-acetylthio-2-pyrrolidone
(3.8 g) as an oil.
NMR (CDCl3) ~ppm: 2.29 (lH, dd), 2.35 (3H, s), 2.81
(lH, dd), 3.31 (lH, dd), 3.88 (lH, dd), 4.10-4.23 (lH, m),
7.02-7.17 (lH, b)
(2) A mixture of the product obtained above (4.8 g),
toluene (100 ml) and Lawesson's reagent [= 2,4-bis(4-
25 methoxyphenyl)-1, 3-dithia-2,4-diphosphetane-2,4-disulfide]
(6.1 g) was refluxed for 15 minutes, and thereafter the
solvent was evaporated to dryness under reduced pressure.
The residue was purified by silica gel column chromatography
(eluant, chloroform : ethyl acetate = 95 : 5) to give
30 (4S)-4-acetylthiopyrrolidine-2-thione (3.6 g) as colorless
needles.
M.p. 91 - 92C
ta]23 -57.5 (c = 1, methanol)
(3) A mixture of the product obtained above (3.6 g) and
16 % ammonia-methanol solution (36 ml) was stirred under ice
cooling for 30 minutes. After evaporation of the solvent, the
residue was coevaporated with toluene (36 ml) to give (4S)-4-
205025~
- 22
mercaptopyrrolidine-2-thione (2.7 g). The crude product thus
obtained was used in the subsequent step without further
purification.
Reference Example 2
(1) (4S)-4-Hydroxy-2-pyrrolidone (1.7 g) was treated in
the same manner as described in Reference Example 1-(1) to
give (4R)-4-acetylthio-2-pyrrolidone (2 g) as an oil.
NMR (CDCl3) ~ppm: 2.29 (lH, dd), 2.35 (3H, s), 2.80
(lH, dd), 3.31 (lH, dd), 3.89 (lH, dd), 4.10-4.23 (lH, m)
(2) The product obtained above (1.9 g) was treated in
the same manner as described in Reference Example 1-(2) to
give (4R)-4-acetylthiopyrrolidine-2-thione (1.8 g) as
colorless needles.
M.p. 91 - 93C
[~]D5 +57 7 (c = 1, methanol)
(3) The product obtained above (1.7 g) was treated in
the same manner as described in Reference Example 1-(3) to
give (4R)-4-mercaptopyrrolidine-2-thione (1.3 g). The crude
product thus obtained was used in the subsequent step without
further purification.
Reference Example 3
(1) A mixture of (4S)-N-benzyloxycarbonyl-4-hydroxy-2-
pyrrolidone (116 g), t-butyldimethylsilyl chloride (81.4 g),
imidazole (67.4 g) and dimethylformamide (350 ml) was stirred
at room temperature for 17 hours, and the solvent was
evaporated in vacuo. Ethyl acetate was added to the residue,
and the mixture was washed with water, dried and concentrated
under reduced pressure. The residue was purified by silica
gel column chromatography (eluant, n-hexane : ethyl
acetate = 4 : 1 to 2 : 1) to give (4S)-N-benzyloxycarbonyl-4-
t-butyldimethylsilyloxy-2-pyrrolidone (160 g) as colorless
needles.
M.p. 59 - 60C
(2) The product obtained above (160 g) was dissolved in
methanol (1 liter) and palladium black (5 g) was added
thereto. The mixture was subjected to catalytic hydrogenation
(hydrogen pressure 3.5 kg/cm2, at room temperature, for one
~- r 7
20502~5
- 23
hour). After filtering off the catalyst, the filtrate was
concentrated under reduced pressure, and the residue was
purified by silica gel column chromatography (eluant,
n-hexane : ethyl acetate = 2 : 1 to 1 : 2) to give (4S)-4-t-
5. butyldimethylsilyloxy-2-pyrrolidone (51 g) as colorless
needles.
M.p. 90 - 93C
(3) The product obtained above (51 g) was added to a
suspension of sodium hydride (62%, 10.1 g) in dimethyl-
formamide (510 ml) at -40C, and after 10 minutes, methyl
iodide (37 g) was added thereto. The mixture was heated with
stirring to 40C over a period of 1.5 hours, and acetic acid
was added thereto. The reaction mixture was poured into ice
water (1.5 liter) and extracted with ethyl acetate. The
organic layer was washed with water, dried and concentrated
under reduced pressure. The residue was purified by silica
gel column chromatography to give (4S)-N-methyl-4-t-butyl-
dimethylsilyloxy-2-pyrrolidone (41 g) as a colorless oil.
NMR (CDCl3) ~ppm: 0.00 (6H, s), 0.81 (9H, s), 2.20-2.32
(lH, m), 2.45-2.60 (lH, m), 2.78 (3H, s), 3.12-3.20 (lH, m),
3.45-3.50 (lH, m), 4.30-4.42 (lH, m)
(4) The product obtained above (41 g) was dissolved in
methanol (250 ml), and 6N hydrochloric acid (40 ml) was added
thereto under ice cooling. The mixture was stirred at room
temperatue for lO minutes. After completion of the reaction,
the reaction mixture was neutralized with sodium hydrogen
carbonate (30 g), and evaporated under reduced pressure. The
residue was washed with ethyl acetate and evaporated to
dryness under reduced pressure. Acetone was added to the
residue and undissolved substances were removed by filtration.
The filtrate was concentrated under reduced pressure, and the
residue was purified by silica gel column chromatography
(eluant, chloroform : ethanol = 10 : 1) to give (4S)-N-methyl-
4-hydroxy-2-pyrrolidone (21 g) as a colorless oil.
NMR (CDCl3) ~ppm: 2.30-2.40 (lH, m), 2.60-2.75 (lH, m),
2.65 (3H, s), 3.25-3.35 (lH, m), 3.60-3.70 (lH, m), 4.40-4.50
(lH, m), 4.64 (lH, d)
~; ~
20502~5
24
(5) The product obtained above (21.3 g) was treated in
the same manner as described in Reference Example 1-(1) and
-(2) to give (4R)-N-methyl-4-acetylthiopyrrolidine-2-thione
(26 g) as a yellow oil.
5. NMR (CDCl3) ~ppm: 2.35 (3H, s), 2.90-3.05 (lH, m), 3.26
(3H, s), 3.40-3.55 (lH, m), 3.60-3.70 (lH, m), 4.05-4.15
(lH, m), 4.15-4.30 (lH, m)
(6) A mixture of the product obtained above (16.9 g) and
16 % ammonia-methanol solution (100 ml) was stirred at room
temperature for 20 minutes. After evaporation of the solvent
under reduced pressure, acetonitrile (50 ml) was added to the
residue, and the mixture was concentrated to give (4R)-N-
methyl-4-mercaptopyrrolidine-2-thione (13 g). The crude
product thus obtained was used in the subsequent step without
further purification.
Reference Example 4
(1) (3R)-3-Acetylthiopyrrolidin-2-one (9 g) was treated
in the same manner as described in Reference Example 1-(2) to
give (3R)-3-acetylthiopyrrolidine-2-thione (6.4 g).
M.p. 98 - 100C
[~]D3 -123.1 (c = 0.72, methanol)
(2) A mixture of the product obtained above (4 g) and
16 % ammonia-methanol solution (100 ml) was allowed to stand
at room temperature for 40 minutes. After evaporation of the
solvent under reduced pressure, the residue was washed with
diisopropyl ether to give (3R)-4-mercaptopyrrolidine-2-thione
(4 g). The crude product thus obtained was used in the
subsequent step without further purification.
Reference ExamPle 5
(3S)-3-Acetylthiopyrrolidin-2-one was treated in the same
manner as described in Reference Example 1-(2) to give (3S)-3-
acetylthiopyrrolidine-2-thione.
M.p. 97 - 100C
[a]D3 -126.6 (c = 0.56, methanol)
Reference ExamPle 6
A mixture of isopropoxycarbonyloxyethyl chloride
20502~5
(=2-chloroethyl isopropylcarbonate) (1.67 g), carbon
tetrachloride (10 ml), sodium iodide (2.10 g) and anhydrous
zinc chloride (0.08 g) was stirred at room temperature for 2
hours, and thereafter, ice water was added to the mixture. The
organic layer was separated, and the aqueous layer was
extracted twice with carbon tetrachloride. The combined
organic layers were washed, dried and evaporated under reduced
pressure to give isopropoxycarbonyloxyethyl iodide (= 2-
iodoethyl isopropyl-carbonate) (1.48 g) as an oil.
NMR (CDCl3) ~ppm: 1.31 (6H, d, J=6Hz), 2.20 (3H, d,
J=6Hz), 5.00 (lH, m), 6.82 (lH, q, J=6Hz)
The compounds [I] of this invention which have a 2-
thioxopyrrolidin-4-ylthio group at the 2-position of the 1-
methylcarbapenem nucleus and pharmaceutically acceptable salts
thereof have excellent antimicrobial activities against a wide
range of microorganisms including Gram positive and Gram
negative bacteria, e.g. microorganisms of the genera
Escherichia, Salmonella, Shiqella, Klebsiella, Proteus,
Morganella, Providencia, Citrobacter, Bacteroides,
StrePtococcus, Staphylococcus, Enterobacter, Serratia,
Pseudomonas, and the like, and further have high antimicrobial
activities against clinically isolated pathogenic strains, and
hence, are useful for the treatment of infectious diseases by
these microorganisms.
For example, the compounds of this invention have
superior antimicrobial activity of 2 to 4 times higher against
Staphylococcus aureus, Staphylococcus epidermidis, Escherichia
coli, Proteus vulgaris and Pseudomonas aeruginosa, in
comparison with the compounds having a 2-oxopyrrolidin-4-
ylthio group at the 2-position as disclosed in Japanese Patent
First Publication (Kokai) No. 49783/1990.
Besides, the compounds [I] and their pharmaceutically
acceptable salts of this invention have advantageously higher
stability against dehydropeptidase I owing to the introduction
of a 2-thioxopyrrolidin-4-ylthio group at the 2-position of
the l-methylcarbapenem nucleus. For example, the compounds
[I] of this invention are at least twice as stable against
20~02~5
26
dehydropeptidase I in comparison with the compounds disclosed
in the above Japanese Patent First Publication No. 49783/1990.
Further, the compounds tI] and their pharmaceutically
acceptable salts of this invention are characterized in that
they are effective for treating inflammatory diseases of the
biliary tract because of their high distribution into bile.
Furthermore, the compounds [I] and their pharmaceutically
acceptable salts of this invention show high therapeutic
effects because of their high absorbability on oral
administration. For example, when orally administered to mice
infected with Staphylococcus aureus, the compounds [I] of this
invention show 2 to 8 times superior therapeutic effects in
comparison with the compounds disclosed in Japanese Patent
First Publication (Kokai) No. 49783/1990.
Moreover, the compounds [I] and their pharmaceutically
acceptable salts of the invention also have lower toxicity and
hence higher safety. For example, when one of the compounds
of this invention, (lR, 5S, 6S)-2-[(4R)-pyrrolidine-2-thion-4-
ylthio]-6-[(lR)-1-hydroxyethyl]-l-methylcarbapen-2-em-3-
carboxylic acid isobutyryloxymethyl ester was orally
administered to mice in a dose of 4,000 mg/kg, no deaths were
observed even after 7 days.
Thus, the compounds [I] and their pharmaceutically
acceptable salts of this invention are useful as antimicrobial
drugs, for example, for the prophylaxis and treatment of
various infectious diseases induced by various microorganisms
as a chemotherapeutic drug for mammals including human beings
and also as an additive for animal feeds.