Note: Descriptions are shown in the official language in which they were submitted.
WO ~/11~7 PCT/FRg0/002~
. ?
', ` ~ `, . i
Inhibiteurs de la mono-amine oxydase, leur procédé de
pr¶tion et leur utilisation en thérapeutique
DOMAINE DE L'INVENTION
La présente invention a trait à de nouveau.~ inhibiteurs
de la mono-amine oxydase. Elle concerne ëgalement leur
utilisation en thérapeutique et leur procédé de
préparation.
Les produits selon l'invention agissent sur le système
ner-eux central en tant qu'inhibiteurs de la mono-amine
o~dase A (en abrégé I~AO-~) et/ou B (en abrégé IMAO-B)
et sont utiles notamment dans le traitement des
patholo~ies liées au~ processus dégénératifs et à
certains états dépressifs, seuls ou en association a~ec
des précurseurs de mono-amines biogènes.
A~T ANTERIEUR
On connait du document de bre~et DE-A-253~339 le
composé ~-[(3-chlorophényl~méthoxyl-a-méthyl-benzène
éthanamine et le composé ~-[(4-chlorophényl)méthoxy]-a-
méthyl-benzèneéthanamine en tant qu'intermédiaires de
synthèse dans la préparation de l-(aralkoxyphényl)-2-
~ou-3)-(bisarylalkylamino)alkanes utiles en tant
qu'agents antihypertenseurs. Dans ce document, il n'est
pas décrit d'application thérapeutique pour lesdits
produits intermédiaires.
OBJET DE L'INVENTION
On propose à present des inhibiteurs de mono-amine
o~ydase différents des composés antihypertenseurs
décrits dans DE-A-263~339 (i) par leur s-~ructure et
~ par leur utilité thérapeutique en tant
:: :
. . . . ;`': . ~', . '.:
'; : :: .,: ,~ : ' :
WO90/11~7 pCT/F~/00209
~ 2
qu inhibiteurs de la mono-amine o~dase .~ etiou ~ qui
est un domaine therapeutique totalement different de
l'h~pertension.
Seion un premier aspect de i'inven~ion on ~ise, en tant ~
que produits industriels nou~eau~, les composés de -~ -
formule Io ci-après.
Selon un second aspect, on ~ise le procédé de
préparation et l'utilisation en thérapeutique des
composés de formule I ci-après.
Les nouveau~ inhibiteurs de la mono-amine o~ydase selon
l'invention sont caractérises en ce qu'ils sont choisis ~ :
parmi l'ensemble constitué par : :
.,
(i) les composés de formule :
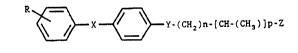
~X~3Y~CH2~ [~ ]p~z ( } )
dans laquelle :
__ - R représente un atome d'halogène, un groupe
alko~y en Cl-C~, un groupe cyano, un groupe nitro
ou un groupe trifluorométhyle,
- ~ représente un groupe -~CH2 )20- OU un
groupe -CH20-,
_.ij - Z représente
(i) un groupe NRlR2 (où Rl et Rz, identiques ou
différents, representent chacun l'atome
d'hydrogène, un groupe alk~le en Cl-C~, un
groupe -~CH2)~-CH20H ou v est egal à 1, 2 ou 3, Rl
et R2, considérés ensemble, pouvant former avec
l'atome d'azote auquel ils sont liés un groupe
N-héterocyclique de 5 à 7 sommets susceptible de
' . ,' ' ' ~' .' ' ~ . ' ,, ; . ' ' '' , ' ' ~ '. ' ' '' " " ', ', ' ': ' '
. ' . : ' .,, '. . , '. . ;."' ' , , .: , ' ' .
WO90/11~7 PCT/~/00209
contenir un second heteroatome choisi parmi ~, O
et S) et ~ represente
- une liaison simple, auguel cas n = 0, 1, ~
ou 3 et p = 0 ou l avec la condition
supplémentaire que, lorsque ~ représen~e le
groupe CH20, n = l, p = l et R représente le
groupe ~-Cl ou le groupe 3-Cl, alors l'un au :
moins des groupes R1 ou Rz est différent de
l'atome d'hydrogène ;
_~ - l'atome d'o~y~ène, auquel cas n = l et
p = 1 i
- le groupe CONH ou le groupe NHC0, auquel
cas n = 2 et p = 0 ; et,
les sels d'addition formés avec le groupe NRlR2;
-
(ii) un groupe OR~ (ou R3 représente l'atomed'hydrogène, un groupe alkyle en Cl-C~ ou un
groupe -COall;- où alk représente un ~roupe all~yle
en C1-C~1 et Y représente
_. - une liaison simple, auquel cas n = 1, 2 ou
3 et p = 0 ou l ;
- le ~roupe CO~H ou le groupe NhCO, auquel
cas n = 2 et p = 0.
_ _
DESC~IPTION DETAILLEE DE L'INVENTION
Par halogene, on entend les atomes de fluor, de chlore
et de brome et de préférence les atomes de chlore et de
fluor.
Par groupe alko~y en C1-C~, on entend un groupe de
formule CtH2t~10 ou t peut prendre les valeurs entières
1 à ~ et où la partie Ct H2t~1 est linéaire ou ramifiee.
_~ Le ~roupe alkox~ préfére est le groupe méthoxy.
~ : '~ ',' . ,
W090/11~7 PCTn~/0020g
~ yl ~ . .
~ar aroupe all;~le en ~1-C~, on entend un groupe de
formule CtHze-1, lineaire ou ramifié, où t peut prendre
ies ~aieurs entières 1 a ~. Le groupe nl1i~-le prefére
est le oroupe méth~le.
Lorsque R1 et Rz forment a~ec l'atome d'azote auquel
ils sont liés un groupe hétérocyclique, on préfère le
groupe morpholinyle et le groupe pipéridinyle. Parmi
les autres groupes NRlR2 hétérocycliques qui
j con-iennent selon l'invention on peut notamment citer
les ~roupes p~rrolidin~le, thiomorpholinyle,
pipérazinyle, ~-(2-h~droxyéthyl~pipérazin~le,
chlorophényl)pipérazin~le et hexaméth~lèneimino.
_- Les produits selon la formule ~Io) où Z représente un
groupe ~R1R2 sont salifiables par les acides minéraux
ou organiques. Parmi les acides- mineraux, on préfère
l'acide chlorhydrique. Parmi les acides organiques, on
préfère l'acide fumarique, l'acide maléique, l'acide
_~ methanesulfonique, l'acide oxalique et l'acide
citrique.
Parmi les composés de formule Io selon l'invention on
préfère les familles telles que :
_5
l) ~ représente le groupe CHzO, Y représente une
liaison simple, n = 0, l, 2 ou 3, p = l, Z
représente un groupe NRlR2 et R, Rl et R2.ont les
significations indiquées ci-dessus,
_ _
2) ~ représente le groupe CHzO, }- représente
l'atome d'oxygène, n = l, p = l, Z représente un
~roupe ~RlRz et R, Rl et Rz ont les significations
indiquées ci-dessus,
,, . ~, . : : . . . . . .
.
'. : :
~ ~: . . . .
WO ~11~7 PCT/F~/002
3) ~ représente le groupe (CH2 )20, ~ représente
une liaison simple, n = 2, p = 1, Z represente un
groupe .~IR~Rz et R. Rl e~ R2 ont les significat1ons
indiquées ci-aessus,
. . .
I) ~ représente le oroupe CH20, ~i représente le
groupe CONH, n = 2, p = 0, Z représente un groupe
NRlR2 et R, Rl et R2 ont les significations
indiquées ci-dessus,
_ ~
~) ~ représente le groupe CH20, Y représente le
groupe CONH, n = 2, p = 0, Z représente un oroupe
OR3 et R et R3 ont les significations indiquées
ci-dessus,
l_
6) ~ représente le groupe CH2O, Y représente le
groupe NHCO, n = 2, p = 0, Z represente un groupe
NRlR2 et R, Rl et R2 ont les significations
indiquées ci-dessus,
_ .,
7) ;~ représente le groupe CH20, Y représente le
groupe NHCO, n = 2, p = O, Z represente un groupe
OR3 et R et R3 ont les significations indiquées
ci-dessus,
_ --
8) ~ représente le groupe CH2 0, Y représente une
liaison simple, n = 1, 2, 3 , p = O, Z représente
un groupe OR3 et R et R3 ont les significations
indiquées ci-dessus,
_ ~,
9) ~ représente le groupe CH20, Y représente une
liaison simple, n = 2, p = 1, Z représente un
groupe OR3 et R et R3 ont les significations
indiquées ci-dessus,
_i
WOgO/11~7 PCT~0/0~2
ca i, ~ ' ` i ~, ` ! ' ~
10~ ~ represente le groupe CH2~, ~ représente une
liaison simple, n = 3, p = 0, Z represente un
groupe ~RlR2 et ~, Rl et R2 ont les significations
indiquées ci-dessus.
~elon l'in~ention on preconise une composi~ion
thérapeutique caractérisée en ce qu'elle renferme, en
association a~ec un e~cipient ph~siologiquement
acceptable, au moins un compose choisi parmi l'ensemble
constitué par :
les composés de formule : -
~ ~ ~ ~ Y-(CH2)n-~CH-(C~3)]p-Z (I)
;
dans laquelle : ~
- R représente un atome d'halogene, un groupe :~ ;
: al~o~y en C1-C~, un groupe cyano, un rgroupe nitro
ou un groupe trifluorométh~-le,
- .~. représente un groupe -(CH2)zO- ou un
groupe -CH20-
- Z représente
~_ (i) un groupe N~1~2 ~ou R1 et Rz, identiques ou
différents, représentent chacun l'atome
d'hydrogène, un groupe alLyle en C1-C~, un groupe
-(CH2~-CH20H où v est é~al à l, 2 ou 3, ~1 et ~2,
considérés ensemble, pouvant former avec l'atome
7C~ d'azote auquel ils sont liés un groupe N-
hétérocJ~clique te ~ à 7 sommets susceptible de
coontenir un second hétéroatome choisi parmi ~, 0
et S~ et Y représente
- une liaison simple, auquel cas n = 0, 1, 2
__ ou 3 et p = 0 ou 1 ;
- l' atome d'o~ygène, auquel cas n = 1 et
p = l;
WO ~/11~7 PCT/F~/00209
7 ! '
- le groupe CONH ou le groupe NHCO, auquel
cas n = 2 et p = 0 ; et,
les sels d'addition ~ormes avec le ~roupe NR1R. ;
(ii) un groupe OR~ (oi~ R3 represente l'atome
_ d'h~dro~ène, un groupe alkyie en Cl-C~ ou un
groupe -COall;- ou all; represente un ~roupe alk~le
en C1-C~ et Y represente
- une liaison simple, auquel cas n = l, 2 ou
3 et p = 0 ou l ;
- le groupe CONH ou le groupe ~HCO, auquel
cas n = 2 et p = 0.
.
Les composés de formuie I peu~ent être préparés selon
des methodes connues en soi par application de
mécanismes réactionnels classiques. Selon l'in~ention
on préconise de preparer les composés de formule I de
la manière sui~ante :
Variante A, lorsque ~ représente le groupe CH20, Y
représente une liaison sim~le, n = O, p = 1, Z
représente un groupe NR1R2 et R, R1 et R2 ont les
significations indi~uées ci-dessus,
a) on soumet le phénol de formule :
O (I1)
j
à une substitution nucléophile par un chlorure de
benz~le de formule :
R ~
_S ~ CH2Cl (III)
', ' .':'. ~ '` ,'' : :. ,,:, ~, . ' '' . : '
WO ~/11~7 PCT/F~/00209
~;~,``, ' ` 3
où R a la si~nification indiquée ci-dessus,
par e.~emple selon une reaction de WILLI~,~1SO,~, pour
obtenir un ether de formule :
_
~ C~2 ~ CI (IV)
e où R a la signification indiquée ci-dessus,
. .
b) on traite la cétone de formule I~' par le formamide ~.
selon une reaction de LE~CEART pour obtenir un composé
de formule :
~ 3C~ (V~ ~
OU R a la signification indiquée ci-dessus,
c~ on procède à une déform~lation du composé de formule
V, selon les méthodes connues de l'homme de l'art, par
e~emple par action de l'acide chlorhydrique à une
temperature comprise entre la température ambiante (15-
25 C) et la température d'ébullition du milieu
réactionnel, pour obtenir un composé de formule I où ~
représente le groupe CH20, Y représente une liaison
simple, p = l, n = 0, Rl = R2 = H et R a la
signification indiquée ci-dessus, -
d) si nécessaire, on procède à une mono- ou une di-
alk~lation selon les méthodes connues de l'homme de
l'art, par e~emple par action d'un compose mono ou di-
halo~ené, ou par protection du groupement amino,
notamment au moyen de di-tert-butyl-dicarbonate de
formule O~CO2C~CH3)3]z, puis alkylation suivie (i)
. .
. : ,. .. : : . .:: :,,
WO90/11~7 ~ ! . PCT/F~/002~
d~une réduction ou (ii) d'une deprotection sui~ie d'une
seconde al1iylalion, pour obtenir un compose de formule
I où ~ represente le ~roupe CH2O, Y represente une
liaison simple, p = l, n = 0 et R, R1 et R2 ont les
significations indiquées ci-dessus,
e) si nécessaire, on salifie l'amine ainsi obtenue.
On peut aussi accéder aux composés de formule I où R
représente un halogène, un groupe alko~y en C1-C~ ou un
groupe nitro et R2 a la si~nification indiquee ci-
dessus, ~ représente lé groupe CHzO, Y représente une
liaison simple, p = l, n = 0 et R1 = CH3 par
al~ylation, par exemple au moyen d'un iodure d'alkyle,
du composé de formule V puis réduction du composé ainsi
obtenu, par exemple au moyen d'aluminohydrure ae
lithium ou de trihydrure de bore. .
On peut également accéder au~ composés de formule I où
~ représente le groupe CHzO, ~ représente une liaison
simple, p = l, n = 0, R1 = H, Rz = CH3 et R a la
signification indiquée ci-dessus par réduction du
composé de for~ule V, par exemple au moyen
d'aluminohydrure de lithium ou de trihydrure de bore.
A S
On peut encore accéder au~ composés de formule I où ~
représente le groupe CHzO, Y représente une liaison
simple, p = l, n = 0, a1 = H, R2 = CH2-alk' ~où alk'
représente un atome d'hydrogène ou un groupe alkyle
_j linéaire ou ramifié en Cl-C3) et R a la signification
indiquée ci-dessus par acylation du composé de formule
I où ~ représente le groupe CH2O, Y représente une
liaison simple, p = l, n = 0 et Rl = R2 = H selon les
méthodes connues de l'homme de l'art puis réduction du
_5 composé obtenu, par exemple au moyen d'aluminohydrure
de lithium ou de trihydrure de bore.
WO ~/11~7 PCT/FRgO/00209
r., ~
S~
`.'~r~ ante ~, ior~que ~: -eFresen-~ le ~rcu~e CH~0. ~:
~ .t~ .e ' 3'~C~. simp'e ! ~ P
r^~r^^-cr.~e u. ,-oupe !:.~ . reFresente un h~log_ne _u
u-. ,r-uFe li!;o~ er. C1-''; et P.~ ont les
~' 'Yn ' i_31 :~r.s _nd ~uees 1-~ -dessus .
~! -r. soumet le -:séncl de rormule : ~ -
HO ~ C (VI) : .
~ une sub~t tution n~lcl~ophile pa- un ^hlorure de
Den~ e de formule :
~ C~2 C~
.. ou R a 13 si3nifl_~tion lndi~uce ci-dcssus,
.-~r ~;c~lc selon une réacti3n de ~7ILLI.~SO~I, pou-
c~~^-.i- un éther de formule :
~CN2~3c/~ (V10
-i~ ~ 13 si~nif_^atior. indi~uee ci-dcssus,
WO ~/11997 PCT/FR~/002~
~ 1 ;' -- ' ! ~; i
b1 on traite l'aldé11~de de formule ~II par le
nitroéthane, par e~emple selon une reaction de
~NOE~'ENAGEL, pour obtenir un oompose de formule :
~CNz~ClCC~lo (~11~1)
où R a la signification indiquée ci-dessus,
c) on réduit le nitro-alcène VIII ainsi obtenu,
notamment en présence d'aluminohydrure de lithium
pour obtenir un composé de formule I où ~
représente le oroupe CHzO, Y représente une
~- liaison simple, p = 1, n = l, R1 = Rz = H et R a
la signification indiquée ci-dessus,
d) si nécessaire, on procède à une mono- ou une
di-alX~lation, notamment par action d'un composé
mono ou di-halogéné, ou par protection du
~roupement amino, notamment au moyen de di-tert-
but~l-dicarbonate, puis alk~lation suivie (i~
d'une réduction ou (ii) d'une déprotection sui~ie
d'une seconde alk~lation, pour obtenir un composé
'~ de formule I où ~ représente le groupe C~2O, Y
représente une liaison simple, p = l, n = l et R,
R~ et Rz ont les significations indiquées ci-
dessus,
~' e) si nécessaire, on salifie l'amine ainsi
obtenue,
::
Y~rian~ , lorsque X représente le groupe CHzO ou le
groupe (CHz)zO, Y représente une liaison simple, n = 2
Z~ ou 3, p = l, Z represente un groupe NR1Rz et R, R~ et
Rz ont les significations indiquées ci-dessus,
, ,,,. ., ,.. . , ,: , , , . : . ~ ::
WO90/1199~ PCT/~/00209
,., . ~
a) on soumet le phénol de formule :
HO ~ (CH2)" C CH3 (IX)
.,,
où n = ~ ou 3,
à une substitution nucléophile par un compose de
i~j formule : .
~>~ . , .
~ (CH2)u-L ~ -:
"~
ou u et R ont les significations indiquees ci-
dessus et où L représente un groupe labile, comme : -
par exemple l'atome de chlore ou le groupe mésyle,
_o par exemple selon une réaction de WILLIAMSON, pour
obtenir un éther de formule : -
CH2 ~ (CH2)n- C~ 3 (X) ~
où n = 2 ou 3 et R a la signification indiquée ci- -
dessus,
_~ b) on soumet la cétone X ainsi obtenue à une -~.
amination réductrice, par e~emple celon la méthode f
de BORCH, pour obtenir un composé de formule I où
X représente le groupe CH2O, Y represente une
liaison simple, p = 1, n = 2 ou 3 et R, R~ et R2
'S ont les significations indiquées ci-dessus,
:' , , :
WO ~/11~7 ~ , PCT/~/00209
cl si necessaire, on salifie l'amlne ainsi
obtenue.
- ,Variante n, lorsque X represente le groupe CH20, ~- :
reprësente l'atome d'o~-gène, n = l, p = l, Z
représente un groupe NR1R2 et R, R~ et Rz ont les
significations indiquees ci-dessus, .-
~O a) on soumet l'hydroquinone de formule : ~
~o ~ OH (XI) :~ :
à une substitution nuciéopAile par un chlorure de : : .
benzyle de formule : ~ `
:G ~ C~2Cl (III)
où R a la signification indiquée ci-dessus, par
exemple selon une réaction de WILLIAMSON, pour
_J obtenir un éther de formule :
_-,
où R a la signification indiquée ci-dessus, ~
WO90/11~7 PCT/F~/00209
" ~, ,
b) on soumet le phenol ~II ainsi obtenu à une
substitution nucléophile par la chloroacétone de
formule :
_ /C~3 (XIII)
C1--CH2 ~~
O ' ~ '
pour obtenir un éther de formule : ~
~, :
~0 - Cl2 - I~/ (XIV)
_ où R a la signification indiquée ci-dessus,
C~ on soumet la oétone ~IV ainsi obtenue à une
amination réductrice, par exemple selon la méthode
de BORCH, pour obtenir un composé de formule I ou
_ X représente le groupe CH2O, ~- représente l'atome
d'oxygène, n = 1, p = l, Z représente un groupe
NRlR2 et R, R~ et R2 ont les significations
intiquées ci-dessus,
,j d) si nécessaire, on salifie l'amine ainsi
obtenue.
, lorsque X représente le groupe CH2O, Y
représente une liaison simple, n =-l, 2, 3 ou ~, p = l,
_~ Z représente un groupe NR1R2 et R, R1 et R2 ont les
significations indiquées ci-dessus,
.. . . : . : ............................... :
: ., . .: . . : , - ~ ~ :
.. .. ..
W O 90/11997 ~, ` ' `f; PC~r/FR90/00209
~5
:
a) on soumet la cetone de formule :
H3C - O ~ ~CH3
où n est égal à 1, 2, 3 ou ~
à une amination réductrice, par exemple selon la :~.
méthode de ~ORCH, pour obtenir un composé de
formule :
H3C - O ~ CH
~2
où n, Rl et Rz ont les sianifications indiquées
ci-dessus,
,,; . .
b) le composé de formule ~VI est soumis à une
déméthylation selon une méthode connue en soi,
notamment en présence d'un acide pour obtenir un :
phénol de formule : ~ :
~o ~ (C~2)r--C1r-~ (XVII~
_O où n, Rl et R2 ont les significations indiquées
ci-dessus, :
WO ~/11~ pCT/~/002
c~ si necessaire on procède à une protection du
groupement amino, notamment au moyen de
diterbut~ldicar~onate pour obtenir un phenoi de
formule :
HO ~ (~z~n- CH- N\ (XVIII~
1~ où n a la signification indiquée ci-dessus et où
R'l et R'2 ont les significations indiquées ci-
dessus ou peuven~ représenter un groupe protecteur
comme par e~emple-le t-but~lox~carbonate,
_ d) le phénol de formule ~III est soumis à une
substitution nucléophile par un chlorure de , -.
benz~le de for~ule :
_ ~ CH2Cl (III)
où R a la signification indiquée ci-dessus,
_~ par e~emple selon une réaction de WILLIA~ISON pour
obtenir un éther de formule :
~ CH2 ~ ( CH2 )n~ 11/ (XIX) '
_; ~2
où R, n, Rl et Rz ont les significations indiquées
ci-dessus,
_~ e) si necessaire on procède à une deprotection
pour obtenir un composé de formule I où
represente le groupe CHzO, Y représente une
: . ' ~' ' ' . "' ' ':
'. ,
! ' '. ' :
.' . ~ ' ' ~, :
' . ` . ' ' . ' . . ' ' ! , ~ ' '
WOgO/119g7 ~ n, . pCT/FRV0/0020
,~7
liaison simple, p = 1, n = 1, ^, 3 ou 1, Z
représente un groupe ~R1R2 et R, R1 et Rz ont les
significalions in~iquee~ ci-dessus,
_ f) si nécessaire, on salifie l'amine ainsi
obtenue.
Var-ante F, lorsque X represente le groupe CH2O, Y
représente le groupe CONH, n = 2, p = 0, Z représente
un groupe NR1R2 ou un groupe OH et R, ~1 et R2 ont les
significations indiquées ci-dessus,
a) on soumet le phénol de formule :
L_ :
HO ~ C - O - CH3 (XX)
~ '
_s à une substitution nucléophile par un chlorure de
benzyle de formule :
~ C8~C1 (III)
où R a la signification indiquée ci-dessus,
par exemple selon une réaction de WILLIA~ISON, pour
_j obtenir un éther de formule :
. . ,
~ C82 ~ I -O-CH, (XXI~
où R a la signification indiquée ci-dessus,
.... .. ... . . - . .. . -. - . .
WOg0/11~7 PCT/FR90/00209
b) on soumet l'ester ainsi obtenu à uné
substitution nucléophile par une base de ~ormule :
H2N--(CH2)2 Z (XXII)
où Z' représente un groupe OH ou un groupe NRl R2
ou R1 et R2 ont les significations indiquées ci-
, dessus ou représentent un groupe protecteur, comme
par e~emple le groupe t-butylox~carbonate,
- c) si nécessaire on déprotège le composé ainsi
obtenu pour obtenir un composé de formule I où X
_ représente le groupe CH2 0, Y représente le groupe
CONH, n = 2, p = 0, Z represente un groupe NR1R2
ou un groupe OH et R, R1 et Rz ont les
significations indiquées ci-dessus,
-! d) si nécessaire, on salifie l'amine obtenue quand
Z' représente un groupe NR~ R2 .
Varinnte G, lorsque X représente le groupe CH20, Y
_~ représente le groupe NHCO, n = 2, p = 0, Z représente
un groupe NR1 R2 ou un groupe OR ' 3 où R'3 représente
l'atome d'hydrogène ou un groupe alkyle en C1-C~ et R,
R1 et R2 ont les significations indiquées ci-dessus,
~C a) on soumet une amine de formule :
NG2 (XXIII)
, _
où R a la signification indiquée ci-dessus,
.
' : . . "'
, ,
.:
W090tll~7
~9 ',
à une ac~-lation 2ar un composé de formule :
A - C-(CH2)2-~'' (XXIV)
où .~ représente un atome d'halosène, un ;roupe OH
ou une liaison simple, Z" represente un atome
d'oxygène lié par A au groupe CO lorsque
_` représente une liaison simple ou Z" represente un
groupe NRlRz où R1 et R2 ont les significations
indiquées ci-dessus ou représentent un groupe
protecteur, notamment le groupe t-but~-lo~;-
carbonate, ou Z" represente un groupe OR'3 où R'3
a la signification indiquée ci-dessus,
b) si nécessaire on déprotège le composé ainsi
obtenu pour obtenir un composé de formule I où X
représente le groupe CH20, Y représente le groupe
NHCO, n = ~, p = 0, Z représente un groupe NRIR2
ou un groupe OR'3 et R, Rl, R2 et R'3 ont les
sisnifications indiquées ci-dessus,
. .
c) si nécessaire on salifie l'amine ainsi obtenue
~3 quand Z représente un groupe NRlR2. ~:
Variante H, lorsque X représente le groupe CH20, Y
représente une liaison simple, n est égal à 1, 2, 3 ou
~' 4, p = 0, Z représente un groupe OR3 et R, R3 ont les
significations indiquées ci-dessus,
a) on soumet le phenol de formule : :
' ' ~ t CH2 )n--OH ( XXV ) : ,,
..... .. . ...... ,. . . ~ ., , ,, ~ ....... . .
WO ~/11~7 PCT/FR90/00209
2 0
où n a la signification indiquée ci-aessus,
à une substitulion nucléophile par un chlorure de
benzyle de formule :
~ C~Cl (Ili)
où R a la signification indiquée ci-dessus,
par e~emple selon une réaction de WILLIAMSON,
_ pour obtenir un composé de formule I où R3
représente l'atome d'hydrogène,
b~ si nécessaire, on soumet le composé ainsi
obtenu : -
_ _
~i) à une alk~lation selon les méthodes connues de
l'homme de l'art, notamment par action d'un iodure
d'al~yle, pour obtenir un composé de formule I où
R3 représente un groupe alkyle en C1-C4, ou
,_
(ii~ à une estérification selon les méthodes
connues de 1'ho~me de 1'art, notamment par action
d'un anhydride ou d'un chlorure d'acide, pour
obtenir un composé de formule I où ~3 représente
un groupe -COalk- où alk représente un groupe
al~yle en C1-C4.
WO ~/11~7 s~ PCT/~/00209
21 ~ ~ .
V.~r.ia~e__I, lorsque .~ represente le aroupe CH2~
représente une liaison slmple. n est é~al a ~, p = l, Z
représente le groupe 0~3 et R1, ~3 ont les
significations indiquees ci-dessus,
a) on soumet le phenol de formule :
HO ~ CH2 CHz ICI (XXVI)
i-J O
à une substitution nuciéophile par un chlorure de
benzyle de formule :
R
C~2Cl ~III) :
. :~ ' ' -,,
:::-. .
où R à la signification indiquée ci-dessus, :
par e~emple selon une réaction de WILLIAMSON,
. ~ .
b~ on soumet la cétone ainsi obtenue à une
réduction selon une méthode connue en soi, :
~5 notamment par action du tétrahydroborure de : :
sodium,
c) si nécessaire on soumet le composé ainsi
obtenu :
~, .
(i) à une alkylation selon les méthodes connues de
l'homme de l'art, par e~emple par action d'un
iodure d'alkyle, pour obtenir un composé de
formule I où R3 représente un groupe alkyle en
_,_ Cl--C~, ou
WO90/11~7 PCT/FF90/00209
~' 22
(iii à une esterification seion les méthodes
connues de l'homme de l'ar~, notamment par action
d'un ann~dride ou d'un chlorure d'acide, pour
obtenir un compose de r`ormuie I où R3 represente
un ~roupe -COall;- où al~ represenle un ~roupe
alk~-le en C1-C~.
L'~.' L'invention sera mieu.~ comprise à la lecture qui va
suivre d'exemples de préparation d'un certain nombre de
composés de formule I et `de résultats des essals
- biochimiques entrepris avec un certain nombre de
composés selon l'in-ention.
l_
Ces e~emples et résultats sont donnés pour illustrer
l'in-~ention sans toutefois la limiter dans sa portée.
En particulier, l'homme de l'art pourra, à l'aide de
ses connaissances et sans sortir du cadre de
l'invention, apporter des modifications qui pourront
néanmoins s'écarter sensiblement de la description qui
en est donnée.
WO 90/11997 PCI'/1;~90/00209
r~ .
pP~EPAR~TTON I
Obtention de méthyl (4-(3-chlorobenzyloxy)phényl)
méthanone
A une suspension de ~,9 g (246 mmoles) d'hydrure de
sodium dans 200 ml de diméthylformamite anhydre à 0 C,
est ajoutée en 1,5 heure sous atmosphère d'azote, une
solution de 30 g (220 mmoles) d'acétyl-4-phénol dans
10 300 ml de diméthylformamide anhydre. Le mélange est -
agité 1 heure à 0 C, puis on ajoute une solution de
40,6 g (252 mmoles) de chlorure de métachlorobenzyle
dans 50 ml de diméthylformamide anhydre. Le mélange est
agité une nuit à température ambiante puis est versé
sur 1,5 litre d'eau glacée. On obtient 56,38 g
(rendement : 98 X) du produit attendu ~ous forme d'un
solide fondant à 78 C. -
20 PRlzpl~RATIol~l TT
Obtention de N-tl-(4-(3-chlorobcnz~losy)phén~l)éthyl]-
for~a~ide
Un mélange de 13 g (50 mmoles) du produit obtenu à la
préparation I, de 15,75 g t350 mmoles) de formamide et
6,9 g (150 mmoles) d'acide formique est agité à 190-C
pendant 7 heures. Le milieu réactionnel refroidi à
température ambiante (15-25-C) est dilué par 200 ml
d'eau. Le produit est extrait par 2 fois 200 ml
d'acétate d'éthyle. Les phases organique~ ~ont lavées 3
fois avec une solution squeu~e saturee en NaCl, séchées
sur ~ulfate de magnésium et évaporées sou~ vide. On
obtient 14,5 g de produit brut. Par recri~tallisation
de l'éther~ on obtient 9,05 g (rendement : 62 X) du
produit attendu fondant à 79 C.
. .: : .
' :.' ' , . ' ~ ; ' ' ' :
. , :. . . : . .
WO ~/11~7 PCT/F~/00209
. . . ~ ~, , .
~ ~ .4
Obtention de 4-~(3-chlorophenyl)métho~y~-a-~éthyl-
benzèneméthanamine (esemple 4)
Une suspension de 2,31 g (8 mmoles) du produit obtenu à
la préparation II, dans 100 ml d'une solution d'acide
chlorhydrique 3 M est portée à 100-C pendant 45
minutes. Le ~ilieu réactionnel refroidi à température
ambiante est extrait par 3 fois 200 ml d'acétate
d'éthyle. Les phases organiques- sont ~échées sur
sulfate de magnésium et évaporées souq vide. Le solide
obtenu est trituré avec de l'éther, filtré puiq partagé
entre une solution saturée de bicarbonate de sodium et
de l'éther. La phase agueu~e est extraite 3 fois
l'éther. Les phases organigues sont lavées à l'eau,
~échées sur sulfate de magnésium et évaporées sous
vide. On obtient 1,59 g (rendement : 76 X) d'une huile
qui cristallise à -18-C. Le chlorhydrate de cette huile
(exemple 4') fond à 141-C.
EBE~ ,
Obtention de N--éthrl-4-l(3-chloroph~nyl)~étho~y~-~-
méthyl-benzène~th~namine (e~enple 1)
: . ,
A une suspension de 0,39 -g (10,4 Immole_)
d'aluminohydrure de lithium dans 30 ~1 d'éther anhydre
est ajoutée en 0,5 heure sous azote une ~olution de
l,S g (5,2 ~moles) du produit obtenu à la préparation
II dans 75 ml d'éther anhydre. Le mlélangc est porté à
reflux de l'éther pendant 1 heure. Le- milieu
réactionnel ost refroidi à tempé~rature ambisnte puiq
est hydrolysé par addition très lente de 0,38-ml d'eau
puis 0,38 ml d'une ~olution aqueuse de soude ~ 15 %,
, . :: . : . . . ~, .
,: , ~ ' , : ., ': ' : .
: . . ' : .: ' ` ` . . `: ~ ''. :',. . . :
WO ~/11~7 PCT/F~/00209
_5 ~
puis 1,2 ml d'eau. Le mélange est agité l houre puis du
sulfate de magnésium est ajouté et le mélange est à
nouveau agité l heure. L'alumine et le sulfate de
magnésium formés sont filtrés et lsvés à l'éther a~ant
; d'être écartés. Les phases organigues rassemblées sont
évaporées sous vide et on obtient l,l g (rendement :
77 %) du produit attendu sous forme d'une huile dont on
peut former le maléate (exemple 1') fondant à l05 C
puis 126-C (double point de fusion).
PRFP~l~TTON V'
Obtention de N~ (4-(3-chlorobenzylosy)phényl)
éthyl]-acétamide
Un mélange de 0,80 g (3,06 mmoles) du produit obtenu à
la préparation III, de 0,63 g (6 mmoles) d'anhydride
acétique, de 0,71 g (9 mmoles) de pyridine et de io ml
de chlorure de méthylène anhydre est agité pendant
5 heures à température ambiante. Le mélange réactionnel
est dilué par 200 ml d'acétate d'éthyle et lavé 2 fois
par une solution d'acide chlorhydrique l M, puis à la
saumure. La phase organique est ~échée sur sul~ate de
magnésium et évaporée. On obtient 0,80 g (rendement :
86 X) du produit attendu fondant à 103-104-C.
P~XP~R~TON VT
Obtention de N-éthrl-4-1(3-chlorophén~l)~ithosyl-~-
méthyl-benzèneméthana ine (esemple 5)
A une suspension de 0,38 g (10 mmoles) d'aluminohydrure
de lithium dans 20 ml de tétrahydrofurane anhydre, une
solution de 0,74 g (2,44 moles) du produit obtenu à la
préparation V dans 20 ml te tétrahydro~urane anhydre
. , - . - . . -, . . . . .
, . , . ~ . .. . ~ - . ,
- . . , ~
., ~ .
',
W~90/11997 PCT/F~/00209
,.~ .~, ,
~ '6
est ajoutée en l0 minutes. Le melange resultant est
porté à reflux durant 3,5 heures puis refroidi à
température s~biante et hydrolysé avec précaution par
sddition successive de 0,38 ml d'eau, 0,38 ml d'une
solution aqueuse de soude à l5 X (p/v) et l,2 ml d'eau.
Le mélange est agité l heure puis est dilué par de
l'éther. Le solide formé est filtré puis lavé à
l'éther. Les phases organiques sont évaporées sous
vide. On obtient 0,68 g (rendement : 96 %) d'une huile
incolore dont on peut former le chlorhydrate (exemple
5') gui fond à 203 C.
pU~ VT T
~5
Obtention de N-méthyl-N-~l-(4-(3-chlorobenzylox~
phén~l)éthyl1-form~ide
A une suspension refroidie à 0 C de 0,36 g (15,2
mmoles) d'hydrure de sodium dans 20 ml de
diméthylformamide anhydre est ajoutée sous atmosphère
d'azote une solution de 4 g (l3,8 mmoles) du produit
obtenu à la préparation II dans 50 ml de
diméthylformamide anhydre. Le mélange est agité 4 -
heures à température ambiante, refroidi à 0-C, puis une
solution de 4,26 g (30 mmoles) d'iodure de méthyle dans
lO ml de diméthylformamide anhydre est additionnée. Le
mélange est agité 12 heures à température ambiante,
puis est versé sur de l'eau glacée. Le composé attendu
est extrait à l'éther. Les phases organiques sont
lavées avec une solution aqueuse saturée en NaCl,
séchées sur sulfate de magnésium puis évaporées ~ous
vide. On obtient 4,l g (rendement : 97 %) du produit
attendu fondant ~ 70-72 C.
WO ~/11~7 PCT/FRgO/00209
r 'A .'.' I'~ '~' A~
~R~p~BATIQ~ VT T T
Obtention de N,N-diméthyl-4-[(3-chlorophénYl)métboxy~-
a-~éthyl-benzèneméthana~ine (e~emple 7)
Selon le mode opératoire décrit à la préparation IV, au
départ de 4,19 g (13,8 mmoles) du produit obtenu à la
préparation VII et de 1,05 g (27,6 mmoles)
d'aluminohydrure de lithium, on obtient 3,60 g
(rendement : 90 %) du produit attendu sous forme d'une
huile dont le citrate (exemple 7') fond à 114-C.
~ . :
Obtention de l-chloro-3-t[4-(2-nitro-1-propenyl)
phéno~yl-~éthyl]-benzène
Un mélange de 2,47 g ~0,01 mole) de 4-(3-
chlorobenzyloxy)-benzaldéhyde, de 0,45 g d'acétate
d'ammonium et de 10 ml de nitroéthane est chauffé
pendant 1 heure et 30 minute~ à 100-C. Le milieu
réactionnel obtenu est partiellement évaporé sous vide
et 20 ml d'éther sont ajoutés. Après 3 heures à 20-C : .-.
les cristaux obtenus sont filtrés puis di~sous dans
100 ml de chlorure de méthylène. La phaQe organique ~:
obtenue e~t lavée à l'eau puis séchée sur sulfate de
magnésium et évaporée. On obtient 1,51 g (rendement :
50 %) du produit attendu fondant à 118-C. .:
~ .-
.
.~ ., . : ,
WO ~/11~7 PCT/F~/002
~ 8
PREpARATToN
Obtention de 4-~(3-chlorophényl)métho~yl-a-méthyl-
benzèneéthanamine (esemple 2~
Une ~olution de 1,~0 g (0,493 mmole) du produit obtenu
à la préparation IX dsns 20 ml de tétrahydrofuranne est
ajoutée lentement sous argon à une suspension de 0,85 g
~22 mmoles) d'aluminohydrure de lithium. Après 20
minutes d'agitation à 20-C, le milieu est hydrolysé par
addition de 4 ml d'eau puis 2 ml de NaOH lN. Après une
heure d'agitation, le précipïté est filtré pui~ lavé à
l'éther. Les filtrats obtenu~ sont séchés sur carbonate
de sodium puis les ~olvants sont évaporé~ sous pression
réduite. On obtient 1,20 g (rendement : 88 %) d'une
huile dont le maléate ~exemple 2') fond à 148-C.
: ~ '
Obtention de N-(l,1-diméth~léthos~carbonrl)-4-[(3-
chlorophenyl)métho~]-~-méth~l-bens~neéthAnA ine
A une solution refroidie à O-C de 5,69 g (20,6 mmoles)
du produit obtenu à la préparation X dans 100 ml de
dioxanne et 15 ml d'eau sont ajoutéeQ ~ucces~ivement
3,2 g (31,7 mmoles) de triéthylamine puis une solution ~ `~
de 5,4 g (24,7 mmoles) de di-tert-butyl-dicarbonate
dans 10 ml de dioxanne. Après 1,5 heure d'agitation à
O-C, le mélange réactionnel est concentré sous pression
réduite pour donner un ré~idu qui e~t partagé entre de
l'eau et de l'acétate d'éthyle. La phase organigue eQt
lavée avec une solution d'hYdrogénosulfate de ~odium à
5 X dans l'eau, à l'eau saturéo de chlorure de sodium,
séchée ~ur ~ulfate de magnésium et évaporée sou~ vide.
Le ré~idu obtenu est purifié par chromatographie cur
silice (de granulométrie moyenne 70-35 ~m) en éluant
,, . ~ . ,,` : '' , : .
`
O ~/11~7 PCT/F~/002
r~ ' r~,
9 i~
avec un mélange acetate d'éthyle/hexane (15/85) (v/v).
On obtient 5,08 g (rendement : 67 X) du produit attendu
qui fond à 85 C.
Obtention de N-(l,1-di-éthylétho~ycarbonyl)-N-méth~1-4-
t(3-chloroPhénrl)méthoxy]-a-méthyl-benzèneéthanamine
Selon le mode opératoire décrit à la préparation III,
au départ de 4,72 g (0,0125 mole) du produit obtenu à
la préparation XI, de 0,53 g (0,0175 mole) d'hydrure de
sodium et de 0,94 cm3 d'iodure de méthyle on obtient
4,3 g (rendement : 88 %) du produit attendu sous forme
d'une huile dont la pureté est jugée suffisante pour
poursuivre les synthèses.
II~-- - IL L~
Obtention de N--éthyl-4-1(3-chlorophén~l)méthoxy]-a-
méthyl-ben~èneétbananine ~e~enple 10)
A une solution refroidie à O-C de 3,95 g (10,1 mmoles)
du produit obtenu à la préparation XII dans 20 ml de
chlorure de méthylène est additionnée sous atmosphère
d'azote, une solution de 10 ml d'acide
trifluoroacétigue dans 10 ml de chlorure de méthylène.
Le mélange est con~ervé 0,5 heure à O-C puis concentré
sous vide. Le résidu est partagé entre une solution
d'hydroxyde de sodium 1 M dans l'eau et de l'acétate
d'éthyle. Après extraction à l'acétate d'éthyle, lavage
avec un peu d'eau saturée de chlorure do sodium puis
évaporation du solvant sous pression réduite, on
obtient 2,7B g (rendement : 94 X) du produit attendu
... : i : :
,
WO ~/11~7 PCT/~ 02~
. . ~ .
~ 30
sous forme d'huile dont le fumarate (exemple 10') fond
à 174-C.
PR~P~TTO~
Obtention de N,N-diméthyl-4-[(3-chlorophényl)méthoxy~-
a-méthyl-benzèneéthanamine (e~emple 14)
:
A une suspension de 1,01 g (26,5 mmoles)
d'aluminohydrure de lithium dans 100 ml d'éther anhydre
est additionnée à 0 C sous atmosphère d'azote une
solution de 3,47 g (8,9 mmoles) du composé obtenu à la
préparation XII dans 50 ml d'éther anhydre. Le mélange
est porté au reflux de l'éther durant 3 heure~ puis,
après refroidissement, est hydrolysé avec précaution
par addition de 2,8 ml d'eau, puis 2,8 ml d'une
solution d'hydroxyde de sodium à 15 % (p~v) dans l'eau
puis 10,8 ml d'eau. Après 1 heure d'agitation, le
solide formé est filtré, lsvé plusieurs fois à l'ether.
Les phases organiques sont évaporées sous pression
réduite. On obtient 2,12 g (rendement : 78 %) du
produit attendu sous forme d'une huile dont le fumarate
(exemple 14') fond à 159-C.
PRl2P~R~TTON lrv
.
Obtention de 4-(4-(3-chlorobenzylo~y)phényl)-2-butanone
A une solution refroidie à -20 C, de 4-(4-
hydroxyphényl)-2-butanone dans 170 ml de
diméthylformamide anhydre on ajoute 2,52 g (105 mmoles)
d'hydrure de sodium par petites fractions ~ous
atmosphère d'azote en 1 heure. Le mélange est agité 2
heures à -10-C puis 0,25 heure a température ambiante.
Le mélange est refroidi à nouveau à -20 C et est
~0 ~/11~7 PCTiF~/00209
31
additionné d'une ~olution de 17,71 g (110 mmoles) de
chlorure de 3-chlorobenzyle dans 15 ml de
diméthylformamide anhydre. Le mélange réactionnel est
gardé 24 heures à 5 C puis est versé sur de l'eau
glacée. Le solide formé e~t filtré puis recri~tallisé
de l'hexane. On obtient 27 g (rendement : 93 %) du
produit attendu fondant à 86-C.
10 PRRPAR~TTOli XVT
Obtention de 4-(4-(3-mëthosybenzyloxy)phényl)-2-
~ butanone
Selon le mode opératoire décrit dans la préparation XV,au départ de 15,4 g (0,094 mole) de 4-(4-
hydroxyphényl)-2-butanone, de 16,25 g ~0,104 mole) de -
chlorure de 3-méthoxybenzyle et de 2,96 g (0,123 mole)
d'hydrure de sodium on obtient 24,2 g (rendement
91 %) du produit attendu fondant à 59-C.
~.II .
Obtention de 4-(4-(2-chlorobenzylos~)phényl)-2-butanone
Selon le mode operatoire décrit dan~ la préparation XV,
au départ de 16,2 g (0,0987 mole) de 4-(4-
hydroxyphényl)-2-butanone, de 17,5 g (O,108 mole) de
chlorure de 2-chlorobenzyle et de 3,11 g (0,129 mole)
- d'hydrure de sodium on obtient, aprè~ recristalli~ation
d'un mélange toluène/hexane, 21,1 g (rendement : 74 %)
du produit attendu fondant à 66-C.
.
' ~: ; ;` ,: '
WO ~/11~7 -
~,. . . .
~ ~2
Obtention de 4-(4-(4-chlorobenzyloxy)phényl)-2-butanone
Selon le mode opératoire décrit dan~ la préparation XV,
au départ de 15,77 g (0,096 mole) de 4-(4-
hydroxyphényl)-2-butanone, de 17,01 g (0,106 mole) de
chlorure de 4-chlorobenzyle et de 3,02 g (0,126 mole~
d'hydrure de sodium, on obtient, aprèis
10 recristallisation d'un mélange toluène~hexane, 22,3 g - ~
(rendement : 80 X) du produit attendu fondant à 70'C. .
~.
- .: .
:~
Obtention de 4-(4-(3-cyaDobenz~lo~y)phényl)-2-butsnone : :
. . .
Selon le mode opératoire décrit dans la proparation XV, : -::~
au départ de 6 g (0,0365 mole) de 4-(4-hydroxyphényl)- . :
2-butanone, de 6,09 g (0,040 mole) de chlorure de 3-
cyanobenzyle et de 1,15 g (0,0382 mole) d'hydrure de
i30dium on obtient, aprè~ recrii3tallisation d'un mélange
toluène/hexane, 8,60 g (rendement : 84 X) du produit
attendu fondant à 85-C.
'
Obtention de 4-(4-(3-nitrobenzJloxy)phényll-2-but one
Selon le mode opératoire décrit dan~ la préparation XV, . -
au départ de 5 g (0,03 mole) de 4-(4-hydroxyphényl)-2-
butanone, de 5,66 g (0,033 mole) de chloruro de 3-
nitrobenzyle et de 0,95 g (0,0315 mole) d'hydrure de
~odium, on obtient 8,13 g du produit attendu
(rendement : 90,5 X) fondant à 84-C.
. . ~ . .
WO ~/11~7 PCTA~/002
Obtention de 4-(4-(3-fluorobenzyloxy)phényl)-2-butanone
Selon le moàe operatoire décrit dans la préparation ~
au départ de ~ ~ (0,03 mole) de ~ -h~dro~yphényl)-2-
butanone, de ~,77 g (0,033 molel de chlorure de 3-
fluorobenzyle et de 0,96 g (0,031~ mole) d'h~drure de
i sodium, on obtient ~,6 g du produit attendu
(rendement : 68,5 Xl fondant à 79 C.
., ~ .
Obtention de 4-(4-(4-fluorobenzyloxy)phényl)-2-butanone
Selon le mode opératoire décrit dans la préparation .~,
au départ de 6 g (0,0365 mole) de ~ -h~dro~yphényl~-
_! 2-butanone, de ~,81 g (0,040 mole) de chlorure de 4-
fluorobenzyle et de 1,1~ g (0,0383 mole) d'hydrure de
sodium, on obtient 7,28 g du produit attendu
(rendement : 73 %) fondant à 46;C.
n~
Obtention de N-13-(4-(3-chlorobenzyloxy)phényl)-1-
méthylpropyll-morpholine (exemple 13)
~C~
Un mélange de ~ g (17,3 mmoles) d~ produit obtenu à la
preparation XV, de 10,7 g (86,6 mmoles) de chlorhydrate
de morpholine et de 1,63 g (25,9 mmoles) de
cyanoborohydrure de sodium dans 1~0 ml de methanol
_~ anhydre est agité ~8 heures à température ambiante. Le
méthanol est évaporé et le résidu est partagé entre une
solution d'hydro~de de sodium 1 ~ et de l'acétate
.-, ~, . .
:. : - ~ . ,
., , ~
.
WOg0/11~7 PCT/FR90/00209
, ..~ ~....
~,~' 34
d'éthyle Ls pha~e aqueuse est extraite ~ l'acétate
d'éthyle Les phaqes organiques qont lavées à l'eau,
séchées sur sulfate de magnésium et évaporées On
obtient 6,1 g (rendement 98 %) du produit attendu
_ous forme d'huile dont le fumarate (exemple 13') fond
à 132-133 C
' . -' .
Obtention de N-méthyl-4-t(3-méthoxyphényl)méthoxy]-a-
méthyl-benzènepropanamine (exemple 8)
-
Selon le mode opératoire técrit dans la préparation
XXIII, au départ de 5 g (0,0175 mole) de 4-(4-(3-
méthoxybenzyloxy)phényl~-2-butanone obtenu à la
préparation XVI, de 5,94 g ~O,088 mole) de chlorhydrate
de méthyla~ine et de 1,66 g (0,0264 mole) de
cyanoborohydrure de ~odium on obtient 4,74 g
~rendement 89 X) du produit attendu sous forme d'une
huile dont le fumarate (exemple 8') fond à 101-103 C
PRl; PAR~l'IO~
Obtention de N-méthrl-4-[(2-chlorophényl)méthoxy]-
~méthyl-benzènepropana ine (esemple 11)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 5 g (0,0173 mole) de 4-(4-(2-
chlorobenzyloxy)phényl)-2-butanone obtenu à la
préparation XVII, de 6,84 g ~0,086 mole) de
chlorhydrate de méthylamine et de 1,63 g ~0,026 mole)
de cyanoborohydrure de sodium on obtient 4,87 g
(rendement 93 %) du produit attendu sous forme d'une
huile dont le fumarate (exemple 11') fond à 134-137 C
.WO ~/11~7 ~ PCT/~/002
~5
Obtention de N-~éthyl-4-[(4-chlorophényl)méthoxy~-a-
méthyl-benzènepropana~ine (e~emple 12)
Selon le mode opératoire décrit dan~ la préparation
XXIII, au départ de 5 g (0,0173 mole) de 4-(4-(4-
chlorobenzyloxy)phényl)-2-butanone obtenu à la
préparation XVIII, de 5,84 g (O,086 mole) de
chlorhydrate de methYlamine et de 1,63 g (0,026 mole)
de cyanoborohydrure de ~odium on obtient 4,2 g
(rendement : 80 X) du produit attendu sousi forme t'un
~olide dont le fumarate (exemple 12') fond à 170-171-C.
~}:~
Obtention de N-prop~1-4-[t3-chlorophényl)n~tho~y]-a-
méthyl-benzènepropan~mine (esemple 19)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 2 g (0,0069 mole) de 4-~4-(3-
chlorobenzyloxy)phényl)-2-butanone obtenu à la
préparation XV, de 3,3 g (O,0345 mole) de chlorhydrate
de n-propylamine et de 0,65 g (0,010 mole) de
cyanoborohydrure de ~odium, on obtient 2,28 g
(rendement : 99 %) du produit sttendu dont le fumarate
(exemple 19') fond ~ 97 C pUi9i 115-C (double point de
fusiion).
. ~ ; .
,'' ~ ' ~ :; '
'
WO ~/11~7 PCT/F~/~2
~ 36
PR~PAR~.IQN~Y~YI.II
Obtention de N-l3-(4-(3-chlorobenzyloxy~phényl)-1-
méthylpropyll-pipéridine (exemple 18~
Selon le mode opératoire décrit dans la preparation
.~III, au départ de 2 g (0,0069 mole) de 4-(l-(3-
chlorobenzyloxy~phényl~-2-butanone obtenu à la
préparation ~V, de ~,21 g (0,03~ mole) de chlorhydrate
!_ de pipéridine et de 0,6~ g (0,0103 mole) de
cyanoborohydrure de sodium, on obtient ~,9~ g
(rendement : 35 %~ du produit attendu dont le fumarate
(exemple 18 ) fond à 130;C.
~RF.PARATION ~IX
Obtention de N-(2-hydroxyéthyl)-4-~(3-chlorophényl~
méthoxy]-a-méthyl-benzènepropana~ine (exe~ple 26)
,
Selon le mode opératoire décrit dans la préparation
~III, au départ de 2 g (0,0069 mole) de ~-(4-(3-
chlorobenzyloxy)phényl~-2-butanone obtenu à la
préparation XV, de 3,38 g ~0,03~6 mole) de chlorhydrate
_~ de 2-hydroxyéthylamine et de 0,66 g (0,0103 mole) de
cyanoborohydrure de sodium, on obtient 2t33 g
(rendement ~ 100 %~ du produit attendu dont le fumarate
te~emple 26 ) fond à 11~-118 C.
.G
P~EPARATTON X~X
Obtention de N-éthyl-4-t(3-chlorophényl~méthoxy~-a-
méthyl-benzènepropanamine ~exemple 9)
, c
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 2 g t0,0069 mole) de 4-(4-(3-
, , , ,, , ~ .
:
WO ~tll~7 ~ i`' PCT~ ~2~
37
chlorobenzyloxy)phényl~-2-butanone obtenu à la
préparation ~V, de 2,85 g (0,035 mole) de chlorhydrate
d'éthylamine et de 0,65 g (0,0103 mole) de
cyanoborohydrure de ~odium, on obtient 1,7 g
(rendement 70 X) du produit attendu dont l'oxalate
(exemple 9') fond de 101 C a 107 C
Obtentio~ de 4-[(3-chlorophényl)méthosy]-a--éthyl-
benzènepropanamine (esemple 3)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 8 g (0,017 mole) de 4-(4-t3-
chlorobenzyloxy)phényl)-2-butanone obtenu à la
préparation XV, de 32 g (0,59 mole) de chlorure
d'a~monium et de 10 g (0,159 mole) de cyanoborohydrure
de sodium, on obtient 7,5 g du produit attendu 80US
forme d'huile dont le chlorhydrate (exemple 3') fond à
165 C
Obtention de N,N-diméthyl-4-[~3-chloroph~ényl)méthoxyl-
a-néth~l-bcDzènepropanaaine (esemple 6)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 8 g (0,027 mole) de 4-(4-(3-
chlorobenzyloxy)phényl)-2-butanone obtenu à la
préparation XV, de 48 g (O,58 mole) de chlorhydrate de
diméthylamire et de 10 g ~0,159 mole) de
cyanoborohydrure de sodium, on obtient 3 g (rondemont
35 %) du produit attendu dont le citrate (exemple 6')
fond à 84-86 C
. ~ .. - . . . . .
- ~ , .
,. ~
'.
... . . . . .
', , ; '
WO 90/11997 PCr/~PgO/00209
~1
Obtention de N-~éthyl-4-[(3-chlorophényl)méthosy]-a-
~éthyl-benzènepropana~ine lesemPle 15)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 8 g (0,027 mole) de 4-(4-~3-
chlorobenzyloxy3phényl)-2-butanone obtenu à la
préparation XV, de 9,~ g ~0,140 mole) de chlorhydrate
de méthylamine et de 2,3 g (0,036 mole) de
cyanoborohydrure de sodium, on obtient 6,5 g
~ (rendement : 79 %) du produit attendu dont le
chlorhydrate (exemple 15') fond à 148-149-C.
Obtcntion de 4-t(3-iluorophényl)métho~y]-a-méth~l-
benzènepropsna ine (esenple 25)
Selon le mode opératoire técrit dan~ la préparationXXIII, au départ de 1,8 g (0,0066 mole) de 4-(4-(3-
fluorobenzyloxY)phényl)-2-butanone obtenu .à la
préparation XXI, de 1,77 g (0,033 mole) de chlorure
d'ammonium et de 0,62 g (0,0098 molo) de
cyanoborohydrure de sodium, on obtient 1~63 g
(rendement : 90,35 %) du produit attendu dont le
fumsrate (exemple 25') fond à 185-187-C.
. . -, : . , ,, , :
WO ~/11~7 PCT/F~/00209
Obtention de N-~ethyl-4-~(3-fluorophényl)~éthoxy~-a-
methyl-benzènepropanamine (esemple 22)
Selon le mode operatoire décrit dans la préparation
XXIII, au départ de 2,4 g (0,0088 mole) de 4-(4-(3-
fluorobenzyloxy)phényl)-2-butanone obtenu à la
préparation XXI, de 3 g (O,044 ~ole) de chlorhydrate de
méthylamine et de 0,9 g (0,0143 mole) de
cyanoborohydrure de sodium, on obtient 2,39 g
~rendement 94,~ %) du produit attendu dont le
fumarate (exemple 22') fond à 129 C
Obtention de N-méthyl-4-[(4-fluorophénJl)méthosy]-a-
éth~l-benzènepropan~ ine (esemple 23)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 2 g (0,00934 mole) de 4-(4-~4-
fluorobenzyloxy)phénrl)-2-butanone obtenu à la
préparation XXII, de 2,48 g (O,0367 mole) de
chlorhydrate de méthylamine et de 0,69 g (0,0100 mole)
de cyanoborohydrure de ~odium, on obtient 2,09 g
(rendement 99,6 %) du produit attendu dont le
fumarate (exemple 23') fond à 165 C
~=:~
Obtention de 4-~(4-fluorophénJl)motho~-a-méthJl-
benzèneprop ~a~ine (esemple 24)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 2 g (0,00734 mole) de 4-(4-(4-
'~ ' "; '' ' ~ '
:`
.
:: , :' :
WO ~/11~97 PCT/FR~/~0209
fluorobenzyloxy)phényl)-2-butanone obtenu à la
préparation ~YXII, de 1,96 g (O,036 mole) de chlorure
d'smmonium et de 0,69 g ~0,0109 mole) de
cyanoborohydrure de ~odium, on obtient 2,09 g
(rendement ~ 100 ~) du produit attendu dont le fumarate
(exemple 24') fond à 175-179'C.
PRRPAR~l'TON lmvTTT :~
Obtention de 4-[(3-cyanophényl)méthoxy]-a-méthyl-
benzènepropanamine (exemple 1i~
Selon le mode opératoire décrit dsns la préparation
XXIII, au départ de 2 g (0,0072 mole) de 4-(4-(3-
cyanobenzyloxy)phényl)-2-butanone obtenu à la
préparation XIX, de 1,93 g (0,036 mole) de chlorure
d'ammonium et de 0,68 g (0,0108 molel de
cyanoborohydrure de sodium, on obtient 1,8 8
(rendement : 89 X) du produit attendu dont le fumarate
(exemple 17') fond à 167-C.
p~p~TION SX~TY
Obtention de N-méthyl-4-t(3-cyanophényl)méthosy]-a-
m~thyl-benzènepropana~ine (esemple 16)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 2 g (0,072 mole) de 4-(4-(3-
cyanobenzyloxy)phényl)-2-butanone obtenu à la
préparation XIX, de 2,42 g (0,036 mole) de chlorhydrate
de mé~thylamine et de 0,68 g (0,0108 molei de
cyanoborohydrure de sodium, on obtient 2,12 g
(rendement : 100 %) du produit attendu dont l'oxalate
(exemple 16') fond à 132-136'C.
! ' : '` ' ` ' , ~ 1 .' ' ' ,' i . '' 1 . : '' `
WO90/11~7 PCT/~/00209
e =e~
Obtention de 4-{(3-nitrophényl)méthoxy3-a-méthyl-
benzènepropanamine (exemple 21)
Selon le mode operatoire décrit dans la prépsration
XXIII, au départ de 2,06 g (O,0069 mole) de 4-(4-(3-
nitrobenzyloxy)phényl)-2-butanone obtenu à la
préparation XX, de 1,84 g (0,034 mole) de chlorure
d'ammonium et de o,~ g (0,0103 mole) de
cyanoborohydrure de ~odium, on obtient 2,05 g
(rendement : 99 X) du produit attendu dont le fumarate
(exemple 21') fond à 176-179-C.
Obtention de N-méthrl-4-[(3-nitrophényl)nethox~]-a-
méthyl-benzènepropanaoine (exemple 20)
Selon le mode opératoire décrit dans la prépsration
XXIII, au départ de 2,02 g (0,00675 mole) de 4-(4-(3-
nitrobenzyloxy)phényl)-2-butanone obtenu à la
préparation XX, de 2,26 g (0,0334 mole) de chlorhydrate
de méthylamine et de 0,63 g (0,0100 mole) de
cyanoborohydrure de sodium, on obtient 2,04 g
(rendement : 96 X) du produit attendu dont le fumarate
(exemple 20') fond à 112-C.
,, ~, . . .
, ~..... '."'. ~',, " '' . ~ ' .
WO90/11~7 PCT/F~100209
~ ` 42
Obtention de N-méthyl-(4-méthosy)-~-méthyl-benzène
butana~ine
On ajoute à une solution de 5,7 g (0,03 mole) de 4-
méthoxy-benzène-2-pentanone dans 300 cm3 de méthanol
anhydre, 10,13 g (0,15 -moleJ de chlorhydrate de
méthylamine et 2,83 g ~0,045 mole) de cyanoborohydrure
de _otium. Après 12 heureq d'agitation à température
ambiante, le méthanol est évaporé sous pression
réduite. Le résidu obtenu est dissous dans un mélange
acétate d'éthyle/soude lN. La pha_e organique est lavée
plusieurs fois au moyen d'une solution de soude lN,
puis à l'eau et évaporée sous pression réduite après
_échage _ur qulfate de magnésium. On obtient 5,9 g
(rendement : 95 %) du produit attendu sous forme
d'huile (n24 C = 1,5169).
0~ T~
Obtention de bromhydrate de N-m~thyl-(4-hydrosy)-a-
méthyl-benzènebutanaxine
5,1 g (0,0246 mole) du N-méth~ 4-méthoxy)-Q-méthyl-
benzènebutanamine obtenu à la préparation XXX$II sont
di~sous dans 70 ml d'une solution d'acide bromhydrique.
La qolution obtenue est portée à 130-140-C pendant 4
heures. L'acide bromhydrique est ensuite évaporé sous
pression réduite et l'on obtient une huile brune qui
e~t rincée au moyen de toluène. On obtient, après
cristalli~ation, 6,5 g (rendement : 96,4 %) du produit
attendu sous forme de cristaux brun~ fondant à 95-98-C.
. .. . . ; . . .
.
WO ~/11g97 , .~. ~ PCT/FR~/00209
~3
pR~pAR~rToN mn~Tv
Obtention de N-(l,l-diméthyléthoxycarbonYl)-N-méthrl-
(4-hydro~y)-a-~éthyl-benzènebutana-ine
A une solution de 6,5 g (0,024 mole) de bromhydrate de
N-méthyl-~4-hydroxy)-a-méthyl-benzènebutanamine obtenu
à la préparation XXXXIII dansi un mélange H20/dioxanne à
O-C sont ajoutés succe_sivement 5,34 g ~0,053 mole) de
triéthylamine en qolution dans du dioxanne, puis après
lS minutes sous agitation, 6,28 g (0,0287 mole) de di-
tert-butyl-dicarbonate en solution dans du dioxanne.
~ Après 12 heures d'agitation à température ambiante, le
milieu réactionnel esit concentré sous preqsion réduite
pour donner un résidu qui est dissous danis un mélange
H20/acétate d'éthyle. La phase organique iqolée est
lavée à l'eau, au moyen d'une solution aqueuse siaturée
de NaCl puis au moyen d'une ~olution aqueuse
d'hydrogénosulfate de potassium, qéchée sur sulfate de
magné~ium, filtrée puis évaporée sous pression réduite.
On obtient 7,17 g (rendement ~ lOO %) du produit
attendu qui fond à 86-87-C aprèq lavage à l'hexane.
~ 31~
Obtention de N-(l,l-diméth~létbiosycsrbonyl)-N-méth~l-4-
[(3-chlorophén~l)méthosy]-a-m~th~l-benzènebut iDe
A une suspension de O,95 g (O,0396 mole) de NsH dans
200 cm3 de diméth~lformamide à - 25-C sous atmosphère
d'argon sont ajoutées siucce~i~ivement une solution de
7,l g (0,024Z mole) de N-(l,}-diméthyléthoxycarbonyl)-
N-(méthyl)-(4-hydroxy)-a-méthyl-benzènebutanamino
obtenu à la préparation XXXXIV dan~ 70 cm3 de
diméthylformamide puis après 2 heures d'agitation à
- lO-C, lO minutes à température ambiante et aprè~
WO ~/11~7 PCT/F~/002
' ? ~ 4 4
a~oir abaisse 1B température à environ - 25-C, une
solution de 4,28 g (0,026 mole) de chlorure de
3-chlorobenzyle dans 10 cm3 de diméthylformamide. Après
2 heures d'asitation à -25'C, puis 12 heures
'agitation à température ambiante le milieu
reactionnel est hydrolysé par un mélange H20/glace puis
le produit attendu est extrait 8U moyen d'acétate
d'éthyle. La phase organique obtenue est lavée à l'eau,
puis au moyen d'une solution saturée de NaCl, séchée
sur sulfate de magnésium, filtrée et évaporée sous
pression réduite. On obtient 10 g du produit attendu
(rendement : 64,3 %) sous forme t'huile
(n2~-C = 1,5285).
~: .
~} 1~ =11 ' :
:
Obtention de N-(méthrl)-4-[(3-chlorophénrl)uéthosy]-a-
méthyl-benzènebutana ine (e~euple 28)
A une solution de 6,5 g ~0,015~ mole) de N-(1,1-
diméthyléthoxycarbonyl)-N-méthyl-4-l(3-chlorophényl)
méthoxy]-a-méthyl-benzènebutanamine dans 30 cm3 de
chlorure de méthylane à 0-C est ajoutée une solution de
cm3 d'acide trifluoroacétique dans 15 cm3 de
chlorure de méthylène. Après 1 heure 30us agitation,
l'acide trifluoroacétigue est é~aporé sous pression
réduite. On obtient une huile gui est solubilisée dans
de l'acétate d'éthyle. La phase organique obtenue est
lavée au moyen d'une solution de bicarbonate de
potassium, puis à l'eau, puis séchée sur sulfate de
magnésium et évaporée sous pression réduite. On obtient
4,42 g ~rendement : 89,7 %) du produit attendu dont le
fumarate (exemple 28') fond à 98 C.
:' ., : ' ' "
WO ~/11997 ~. ~PCT/FR~/00209
PREpARATIo~l ~T T
Obtention de l'acide 4-~(3-chloroPhényl)méth
benzènepropanoïque
A une solution de 15 g (0,090 mole) d'acide 4-hrdroxy-
benzènepropanoïque dans 400 cm3 d'éthanol à 95 % on
ajoute successivement 38 g (0,275 mole) de carbonate de
potassium, 1,4 g d'iodure de sodium puis 25,3 g (0,157
mole) de chlorure de 3-chlorobenzyle. Après 12 heure~
sous agitation à reflux, l'éthanol est évaporé sous
pression réduite. On obtient un solide pâteux blanc qui
est trituré dans une solution d'acide chlorhydrigue lN
glacée. Le produit attendu est extrait au moyen
d'acétate d'éthyle. La phase organique obtenue est
lavée au moyen d'une solution ~aturée de chlorure de
sodium, puis ~échée sur sulfate de magné~ium et
évaporée sous vide. On obtient, après recristallisation
d'un mélange toluène/hexane, 20,5 g (rendement : 76 X)
du produit attendu fondant à 94-97-C.
Obtention de N-(éthyl)-4-t(3-chlorophényl)~éthosy]-
bonzènepropanauide
A une solution de 10 g ~0,0344 mole) d'acide 4-t(3-
chlorophényl~méthoxy]-benzènepropanoïque obtenu à la
préparation XXXVII dans 200 cm3 de chlorure de
méthylène, on ajoute successivemeQt 4,88 g (0,045 mole)
de chloroformate d'éthyle pui~ à 0 C, 4,86 g
(0,048 mole) de triéthylamine. ~e mélange obtenu est
ensuite maintenu aous agitation pendant 1 heure à 0 C
et est ajouté goutte à goutte qur une solution ~ 0 C de
68,9 g (1,53 mole) d'éthylamine dans 100 cm3 de ~ -
chlorure de méthylène. Après 12 heures sous agitation à
. . . .. . ~ . , . . ,... .,: ,, . , . : . :
WO ~/11~7 PCT/F~/00209
?:
température ambiante le mélan~e obtenu est hydrolysé au
moyen d'une solution d'acide chlorhydrique 3N glacée.
La phase aqueuse obtenue est extraite au moyen de
chlorure de méthylène. Les pha~es organigues obtenues
sont rassemblées, lavées successivement au moyen d'une
solution d'acide chlorhydrique lN, d'une solution de
soude lN puis d'une solution saturée de chlorure de
sodium, séchées par sulfate de magnésium, filtrée~ et ~-
évsporées sous pression réduite. On obtient, après
recristallisation d'un mélange toluène/hexane, 6 g
(rendement : 46 %~ du produit attendu fondant à ll9 C.
~ .
Obtention de N-(éthyl)-4-~(3-chlorophényl)métho~y]-
benzènepropana~ine (esemple 27)
A une suspension de 1,1 g (0,029 mole) d'aluminohydrure
de lithium dans 150 cm3 de tétrahydrofuranne on ajoute
goutte à goutte une solution de 4,43 g (0,0140 mole) de
N-éthyl-4-1(3-chlorophényl)méthoxy~-benzènePropanamide
obtenu à la préparation XXXXVIII dans 150 cm3 de
tétrahydrofuranne. Après 1 heure sous agitation à
température ambiante et 2,5 heures à reflux, le milieu
réactionnel est refroidi à température ambiante. On
ajoute 1,5 cm3 d'eau, 1,5 cm3 d'une solution de soude
lN puis 4,5 cm3 d'eau. Le mélange obtenu e~t soumiQ à
une ~igoureuse agitation pendant 1 heure puis filtré
sur Célite~. La phase organigue obtenue est sechée sur
sulfate de magnésium, filtrée et évaporée sous pression
réduite. On obtient 3,97 g (rendement : 94 X) du
produit attendu dont le maléate (exemple 29') fond à
118 C.
., ~ , ~ . . , ' I !
''
,, ~ ' ,' ' .' ' "' :,
': ' ' ' . ~ " , ' ~ ` '
WO ~/11~7 PCT/~/00209
;'~ `' :' . ' i '
47 r,i -~
Obtention de 4-[(3-chlorophényl)~éthoxy~-phénol
5 Un melange de 10 g 0,091 mole) d'hydroquinone, de
}4,65 g (0,090 mole~ de chlorure de 3-chlorobenzyle et
de 25,15 g (0,18 mole~ de carbonate de potassium dans
400 cm3 d'éthanol à 95 %, en présence de 0,27 g
d'iodure de sodium est porté a reflux 1,5 h SOU8 srgon.
Le précipité formé est filtré, leq eaux-mères sont
évaporées et le ré~idu obtenu est di~sous dans de
l'scétate d'éthyle. La phase organique obtenue est
~ lavée à l'eau, séchée sur ~ulfate de magnésium, filtrée
pui~ évaporée sous vide. On obtient 16,1 g de cristaux
lS qui sont purifiés par "flash chromatography" en éluant
avec un mélange hexane/acétate d'éthyle (1/3) (v/v). On
obtient 5,8 g ~rendement : 28 %) du produit attendu qui
fond à 117-C.
P~2F.PAR~ To~ T.T
Obtention de 4-t(3-chloroPhén~l)métho~phén
propanone
2S
Un mélange de 6,5 g (0,0277 mole) de 4-~(3-
chlorophényl)méthoxyl-phénol obtenu à la préparation L,
de 5,36 g ~0,039 mole) de carbonate de potassiun, de
0,8 g d'iodure de sodium et de 5,S3 g (0,059 mole) de
chloroacétone dans l'acétone e~t porté à reflux sous
azote pendant 2 jours. Le précipité formé e~t filtré,
le~ eaux-mères sont évaporées puis le réqidu obtenu e~t
dissous dans l'éther. La phaae organique obtenue est
lavée à la soude lN, puis à l'eau, séchée sur sulfate
de magnésium et évaporée sous pression réduite. On
obtient 7,8 g ~rendement : 96 %) du produit attendu
sous forme de cristaux jaunes qui fondent à 73-74-C.
. ~, . . . . . . . . .
WO ~/11~7 PCT/F~/n0
48
P~EpRATToN T.IT
Obtention de N-méthyl-1-[4-{(3-chlorophényl)méthoxy]
phéno~y3-2-propanamine (exemple 29)
Un mélange de 2,6 g (0,0089 mole) de 4-[(3-
chlorophényl)méthoxy]phénoxy-propanone obtenu à la
préparation LI, de 0,81 g (0,013 mole) de
cyanoborohydrure de sodium et de 2,9 g (0,043 mole) de
chlorhydrate de méthYlamine dans 150 cm3 de méthanol
anhydre est agité à température ambiante ou~ azote
pendant 5 heures. Le méthanol est ensuite évaporé sous
lS pression réduite puis le résidu est dissou~ dans de
l'acétate d'éthyle et de la soude lN. La phase aqueuse
est extraite plusieurs fois à l'acétate d'éthyle puis
les phases organiques sont réunies, lavées à la soude,
à l'eau puis séchées sur sulfate de magnésium et
évaporées sous pression réduite. On obtient 2,80 g
~rendement ~ 100 %) du produit attendu dont le fumarate
(exemple 2g') fond à 122-C.
e~ ~ 3~9~L~
Obtention de méthane~ulfo~ate de 3-chlorobenzèneéthyle
A une ~olution de 8,57 g (0,055 mole) de 3-
chlorobenzèneéthanol dans 100 cm3 de chlorure deméthylène refroidi à 0-C, on ajoute successivement
8,35 g (0,0826 mole) de triéthylamine en-solution dans
le chlorure de méthylène puis, après 15 minutes
d'agitation, 10,65 g (0,093 mole~ de chlorure de
l'acide méthsnesulfonique. Après 15 minutes sou~
agitation à 0-C, le milieu iéactionnel est hydrolysé
par une solution d'hydrogénosulfate de potassium dans
^~ ~ ,.- .. . , . . ~, , .
: . : ,: . - : . . . : . . . ~ .
. . . : - , : : , , .: :
.. : .
..
..
WO ~/11~7 PCT/F~/00209
4g ~ ``
l'eau glacée. Le produit ~ttendu est extrait au
chlorure de méthylène. Les phases organiques obtenues
sont lavée~ à l'eau, puis au moyen d'une ~olution
saturée de chlorure de sodium, séchées sur sulfate de
magnésium, filtrées et évaporées sous pre~sion réduite.
On obtient 13,85 g (rendement # 100 %) du produit
attendu sous forme d'huile dont la pureté est jugée
suffisante pour poursuivre les synthèse~.
P~ ATION ~IV
Obtention de 4-[4-~2-(3-chlorophényl)éthYloxY1phénYl]-
2-butanone
A une suspension de 0,5~ g (0,023 mole) d'hydrure de
sodium dans 20 cm3 de diméthylformamide refroidie à
-25 C, on ajoute successivement 2,2 g (0,0134 mole) de
4-(4-hydroxyphényl)-2-butanone, puis après 3 heures
sous agitation à -l0-C et après avoir abaissé la
température du milieu à -25-C, 3,~ g (0,0149 mole) de
méthanesulfonate de 3-chlorobenzèneéthyle préparé selon
la préparation LIII. Le milieu est maintenu sous
agitation pendant 2 heures à -l0-C puis 12 heures à
température ambiante puis hydroly~é par un mélange
eau/glace. Le produit attendu est extrait au moyen ~ -
d'acétate d'éthyle. La phase organique obtenue est
lavée à l'eau, au moyen d'une solution saturée de
chlorure de sodium puis séchée sur sulfate de magnésium
et évaporée sous pression réduite. On obtient le
produit attendu ~ous forme d'huile qui e~t purifiée par
"flash chromatography" en éluant par un mélange
hexane/acétate d'éthyle 85/15 puis 80/20 (v/v).
, ,, ,. ~ . . ................................ .
.
WO ~/11997 PCT/F~/00209
~1~ 0~
~'~ 50
~B~
Obtention de N-éthyl-4-~2-(3-chlorophényl)éthyloxy]-a-
méthyl-benzènepropanamine (exemple 30)
Selon le mode opératoire décrit dans la préparation
XXIII, au départ de 0,33 g (0,00108 mole) de 4-~4-[2-
(3-chlorophényl)éthyloxy]phényl]-2-butanone obtenu à la
préparation LIV, de 0,900 g (0,0ll mole) de
chlorhydrate d'éthylamine et de 0,22 g (0,0035 mole) de
cyanoborohydrure de sodium, on obtient 0,30 g
(rendement : 36 X) du produit attendu sous forme
- d'huile dont l'oxalate ~exemple 30') fond à 90-C.
I;~P ~ T T ON I ~VI
Obtention de l'ester méthylique de l'acide 4-1(3-
chlorophényl)~éthoxy]-benzoïque
A une suspension de 3,6 g (0,12 ~ole) d'hydrure de
~odium dan~ 60 ml de diméthylformamide refroidie à 0 C
on ajoute 15,2 g (0,0l mole) d'ester méthyligue de
l'acide 4-hydroxybenzoïgue en solution dans 50 ml de
diméthylformamide, l00 ml de diméthylformamide puis,
après l,5 heure sous agitation à 20-C, 17,7 g
(0,ll mole) te chlorure de 3-chlorobenzyle dissous dans
ml de diméthylformamide. Après 20 heures ous
agitation à température ambiante, le milieu réactionnel
est hydrolysé par un mélange glace/eau (l litre
environ) et le précipité formé est filtré, lavé à l'eau
puis à l'hexane et séché ~ou~ vide. On obtient, après
recristallisation du méthanol, 19,2 g du produit
attendu (rendement : 69 %) fondant à 70 C.
... . ~ . : , . . . :
.. ,, - ~ ~ ., j . . ~, ... .
- ~ . .: ~ ~ . . ,
;:
., ... ~, . . .
WO ~/11~7 PCT/F~/002~
~1 f; .,
PREpARATTo~ T~VT T
Obtention de N-(2-aminoéthyl)-4-[(3-chlorophényl)
métho~y3-benzamide (e~e~ple 31)
Une suspension de 2,76 g (0,0l mole) d'ester méthylique
de l'acide 4-~(3-chlorophényl)méthoxyl-benzoïque obtenu
à la préparation LVI dans 5 ml de 2-amino-éthylamine
sous azote est portée à 130-C pendant 5 heures puis le
milieu réactionnel obtenu est versé dans l'eau. ~e
précipité formé est filtré et séché puis partagé entre
une solution d'acide chlorhydrique lN, du méthanol et
- de l'acétate d'éthyle. La phase organique isolée est
extraite de nombreuses fois par une solution d'acide
~5 chlorhydrique lNt puis les pha~es aqueuses acides sont
rendues basiques par addition de soude puis extraites
au moyen d'acétate d'éthyle. Le~ pha~es organiques
rassemblées sont séchées et évaporées sous pression
réduite. On obtient l,8 g du produit attendu qui est
purifié sur colonne d'alumine en éluant avec un mélange
chloroforme/méthanol 9/1 (v/v). On obtient un ~olide
fondant à 94-96-C (base) dont le chlorhydrate fond à
204-205 C (rendement : 20 %).
, ,:
PREpA~TToN l .VT T ~
Obtention de N-(2-hydro~yéthyl)-4-L(3-chlorophén~l)
m~tho~y3-~enza ide (esemple 32)
Une suspension de 2,76 g (0,0l mole) d1ester méthylique
de l'acide 4-t(3-chlorophényl)méthoxy~-benzoïque obtenu
à la préparation LVI dans l0 ml d'aminoéthanol est
chauffée pendant 2 heures à 130-C. Le uelange
réactionnel refroidi est hydrolysé par un melange acide
chlorhydrique lN/glace. On obtient un précipité qui est
filtré, lavé à l'eau puis di~sous dans 200 ml d'acétate
' ' ~ : , . ' , , : , ' ,
.: . . . : '' ` ' . : ~ ' '
WO ~tll~7 PCT/FR90/00209
C?~ : ~`" ` 52
)
d'éthyle. La phase organique obtenue est séchée sur
sulfate de magnésium. On obtient 2,63 g de produit
blanc cristallisé (rendement : 86 X) qui est
recristallise deux fois du methanol. On obtient 1,40 3
5 du produit attendu, pur, fondant à 129-C.
pQEpARl~TTo~ ~.Tl~
Obtention de N-~4-l(3-chlorophényl)métho~ylphényl]-3-
tN-(tert-butYlQx~csrbonyl)aminol-propanamide
A un mélange de 1,52 g (0,008 mole) d'acide N-~tert- -
butyloxycarbonyl)-~-aminopropionique et de 1,04 g
(0,0096 mole) de chloroformate d'éthyle refroidi à 0-C,
on ajoute 1,05 g (0,0104 mole) de triéthylamine puis
après 30 minutes sous agitation, 1,8 g (0,008 mole) de
4-1(3-chlorophényl)méthoxy~-aniline. Après 2 heures
sous agitation à température ambiante, le milieu
réactionnel est hydrolysé sur bicarbonate de potassium,
extrait au moyen de chloroforme, séché et évaporé sous
pression réduite. On obtient 2,7 g du produit attendu
(rendement : 84 X) fondant à 148-C.
PRFp~ TToN ~Y,
Obtention de N-[4-[(3-chlorophényl)m~thorr]phényl]-3-
~ino-propanamide (exemple 33)
~ -
Un mélange de 2,6 g (0,00643 mole) de N-14-[(3-
chlorophényl)méthoxy~phényl]-3-lN-(tert-butyloxy-
carbonyl)amino~-propanamide obtenu à la-préparation LIX
et de 5 cm3 d'acide trifluoroacétique dans 20 cm3 de
chlorure de méthylène est agité 3 heures à température
ambiante puis évaporé sous pression réduite. On obtient
une huile qui est dissoute dans 30 ml d'eau, puis on
',, : , :' , ' :. ' '
. . , : , ~
... :' : ~ '~' '
.,:' ~ :
WO ~/11997 PCT/FR~/00209
~3
ajoute du bicarbonate de ~odium solide jusqu'à pH
basique. Le solide beige obtenu est filtré et rincé à
l'éther. O~ obtient 1,7 g ~rendement : 90 %) du produit
attendu dont le fumarate (exemple 33') fond à 180-
182'C.
. .
,
Obtention de N-[4-[(3-chlorophényl)méthoxy~phényl]-3-
hydro~ypropanamide (e~emple 34)
A une solution de 3 g (0,013 mole) de 4-~3-
chlorophényl)méthoxy~-benzènamine dan~ 30 cm3 de
toluène on ajoute 4,6 g (0,064 mole) de ~-propiolactone
et l'on porte à reflux sous agitation pendant 3 heures.
Après complet refroidissement du milieu réactionnel, on
ajoute 150 cm3 d'acétate d'éthyle. La phase organique
obtenue est lavée au moyen d'une solution aqueu~e de
bicarbonate de potassium, séchée et évaporée sous
pression réduite. On obtient 3,1 g de cristaux que l'on
recristallise successivcment du toluène, puis de
l'éthanol. On obtient 1,4 g (rendement : 35 X) du
produit attendu fondant a 144-C.
~ TTON T .ST T
Obtention de 4-[(3-chlorophényl)méthosyl-benzène
propanol (exemple 3i5)
Un mélange de 2 g (0,013 mole) de 4-hydroxy-
benzènepropanol, de 2,11 g (0,013 mole) de chlorure de
3-chlorobenz~le, de 3,59 g (0,0259 mole~ de carbonate
de potassium et de 0,19 g (0,0013 mole) d'iodure de
sodium dans 80 cm3 d'éthanol à 95 %, est porté ~ reflux
pendant 2 jours. Le précipité formé est filtré. Les
.. . . .
WO ~/11~7 PCT/F~/OD2
~ ~^ 54
eaux-mères sont évaporees sous pres~ion réduite et le
residu obtenu est dissous dan~ de l'acétate d'éthyle.
La phase organique obtenue est lavée à l'aide de soude
lN, puis d'une solution aqueuse saturée en chlorure de
sodium et enfin à l'eau, puis séchée sur sulfate de
magnésium et évaporée sous pression réduite. Après
recristallisation du cyclohexane, on obtient 3,2 g
~rendement : 89 %) du produit attendu fondant à 54'C.
PRl;.P~ TTON T.YTTT
Obtention de 4-{(3-chlorophényl)méthoxyl-benzène
méthanol (esemple 36)
Selon le mode opératoire décrit dans la préparation
LXII, au départ de 2,2 g (0,016 mole~ de 4-
hydroxybenzèneméthanol, de 2,6 g (0,016 mole) de
chlorure de 3-chlorobenzyle, de 3,7 g (0,027 mole) de
carbonate de potassium et de 0,20 g (0,0135 mole)
d'iodure de potassium, on obtient, après
recristallisation du mélange toluène/hexane 1/1 (v/v),
3,38 g (rendement : 85 %) du produit attendu fondant à
76-C.
2S
Obtention de 1-(3-méthoxypropyl)-4-~(3-chlorophényl~
~étho~y]-benzène (e~emple 37)
0,5 g (1,8.10-3 mole) de 4-[(3-chlorophényl)méthoxy]-
benzèneméthanol obtenu à la prépsration LXIII et 3 cm3
de diméthylformamide sont introduits dans un mélange de
0,06 g (Z.10-3 mole) d'hydrure de sodium et de 2 ~m3 de
diméthylformamide. Le milieu résctionnel obten~ est
agité 15 minutes à température ambiante. On obtient une
, . ~ , .
:. .. . .
; ,
:, ~ ' ' ' '
WO ~/11~7 PCT/FR~/00209
suspension jaunatre tans laquelle on introduit 0,28 g
(2.10-3 mole) d'iodure de méthyle. Après 12 heures
d'agitation, le milieu est hydrolysé par de l'eau. La
phase organique obtenue est lavée au moyen d'une
solution de bicarbonate de sodium, puis à l'eau, séchée
sur sulfate de magnésium et évaporée sous pression
réduite. On obtient, après purification par
chromatographie sur colonne de silice en éluant avec de
l'éther, 0,4 g (rendement : 80 %) du produit attendu
sous forme d'huile.
- p~Rp~RATTO~ T.X~
Obtention de 4-[(3-chlorophényl)métho~y]-benzène
éthanol (e~emple 38)
Selon le mode opératoire décrit dans la préparation
LXII, au départ de 4,9 g (O,035 mole) de 4-hydroxy-
?0 benzèneéthanol, de 6,19 g (0,0385 mole) de chlorure de3-chlorobenzyle, de 10 g (0,070 mole) de carbonate de
sodium et de 0,6 g d'iodure de sodium, on obtient,
après recristallisation du cyclohexane, 5,7 g
(rendement : 50 X) du produit attendu fondant à 78 C.
Obtention de 4-t(3-nitroPhénYl)méthos~l-b~n~èncpropanol
(e~emple 39)
Selon le mode opératoire décrit dans la préparation
LXII, au départ de 3,5 g (O,023 mole) de 4-hydroxy-
benzènepropanol, de 4,35 g (0,0253 mole) de chlorure de
3-nitrobenzyle, de 6,5 g (0,045 mole) de carbonate de
potassium et de 0,35 g d'iodure de ~odium, on obtient,
après recristallisation d'un mélange chloroforme~
WO ~11~7 PCT/FR~/002
pentane, 3,7 g (rendement D6 X ) du protuit attendu
fondsnt à 42 C
I-~-Y~C~
Obtention de 4-l(3-cyanophényl)méthoxy]-benzènepropanol
(e~emple 40)
Selon le mode opératoire décrit dans la préparation
LXII, au départ de 2,8 g t0,0184 mole) de 4-hydroxy-
benzènepropanol, de 3,6 g (0,~183 mole) de carbonate de
potassium et de 0, 3 g d'iodure de odium, on obtient,
après recristallisation de l'éther isopropyligue, 1,8 g
(rendement 36 %) du produit attendu fondant à 64 C
~ .
Obtention de 4-l(3-chlorophényl)méthoxy]-benzènebutanol
(exemple 41)
Selon le mode opératoire décrit dans la préparation
LXII, au départ de 2,3 g (0,014 mole) de 4-hydroxy-
benzènebutanol, de 2,7 g (0,0168 mole) de chlorure de3-chlorobenzyle, de 3,5 g (0,025 mole) de carbonste de
potassium et de 0,6 g d'iodure de ~odium, on obtient,
après recristallisation du cyclohexane, 2 g
(rendement 50 X) du produit attendu fondant à 63 C
.: . , ::
. . ' :.
:
WO ~/11997 PCT/~/00209
57 ~,.
Obtention de 4-~E3-(trifluorométhYl)phényl]méthoxy~-
benzènepropsnol ~e~emp1e 42)
Selon le mode opératoire décrit dans la préparation
LXII, au départ de 1,05 g (0,0069 mole) de 4-hydroxy-
benzènepropanol, de 1,68 g (0,0086 mole) de chlorure de
3-(trifluorométhyl)benzyle, de 2 g (0,0145 mole~ de
carbonate de potassium et de 0,11 g d'iodure de sodium,
on obtient, après recristallisation de l'hexane, 1 g
(rendement : 46 %) du produit attendu fondsnt à 48 C.
7 5 PRl;~Pl~RATTO~ T.Y'f
Obtention de l'acétate de 4-[(3-nitrophényl)méthox~3-
benzènepropanol (exemple 43)
A une solution de 1 g (0,0035 mole) de 4-E(3-
nitrophényl)méthoxy~-benzènepropanol obtenu à la
préparation LXVI dans 15 cm3 de pyridine, on ajoute
2 cm3 ~0,021 mole) d'anhydride acétique. ~e milieu
réactionnel obtenu est agité 12 heures à température
ambiante, puis hydrolysé par une solution d'acide
chlorhydrique lN à 0-C. Le produit attendu est extrait
à l'acétate d'éthyle. La pha~e organique est lavée au
moyen d'une solution aqueu~e saturée en chlorure de
sodium, puis séchée sur sulfate de magnésium et
évaporée ~ous pression réduite. On obtient, après
recristallisation de l'éthanol à 95 X (v/v), 0,8 g
(rendement : 70 %) du produit attendu fondant à 65-C.
. . ' ,~. ~. : . ' ' .
WO ~/11~7 PCT/FR~/00209
~ 8
Obtention de l'acétate de 4-[(3-chlorophénrl)méthoxy]-
benzènepropanol (exemple 44)
-
Selon le mode opératoire décrit dans la préparation
LXX, au départ de 2 g (0,0072 mole) de 4-[(3-
chlorophényl)méthoxyl-benzènepropanol obtenu à la
préparation LXII et de 4,4 g (0,043 mole) d'anhydride
acétique, on obtient, après recristallisation de
l'hexane, 2 g (rendement : 87 %) du produit attendu
fondant à 36 C.
15 p~p,~R~TTO~11 l.lnrTT
.
Obtention de diméthrl-2,2-propanoate de 4-l(3-
chlorophényl)métho~y]-benzènepropanol (esemple 45)
1,3 g (0,0108 mole) de chlorure de diméthyl-2,2-
propanoyle sont ajoutés à 0-C à une solution de 2 g de
4-t(3-chlorophényl)méthoxy]-benzènepropanol obtenu à la
préparation LXII dan~ 15 cm3 de pyridine. Après 12
heures d'agitation à température ambiante le milieu
réactionnel est hydrolysé par une solution aqueuse
d'acide chlorhydrique 3N. Le produit attendu est
extrait à l'acétate d'éthyle. La phase organique
obtenue est lavée au moyen d'une solution d'acide
chlorhydrique lN, d'une solution aqueuse ~aturée en
bicarbonate de sodium puis d'une solution aqueuse
saturée en chlorure de ~odium, séchée sur sulfate de
magnésium, pui8 évaporée sous pression réduite. Après
purification par chromatographie sur colonne de silice
en éluant avec un mélange éther/hexane 9/7 (v/v), on
obtient 1,6 g (rendement : 61 %) du produit attendu
~ous ~orme d'huile.
: , ' . , : ,. ..
,
?
, . . .
.
WO ~/11~7 PCT/~/002
59
~R~ AT 10~
Obtenti~n de dimethyl-2,2-propanoate de 4-[(3-
nitrophényl)méthoxy3-benzènepropanol ~exemple 46)
Selon le mode operatoire décrit dans la prepsration
L~II, au départ de 1,28 g (~,~6.10-3 mole) de ~-[(3-
nitrophényl)méthoxy3-benzènepropanol obtenu à la
!~j préparation L~VI et de 0,8 g (6,68.10-3 mole) de
chlorure de diméths~l-2,2-propanoyle, on obtient, après
purification par chromatographie sur coionne de silice
- en éluant avec un méiange éther/he~ane 3/~ ~v/v), 1,3 g
(rendement : 80 %) du produit attendu sous forme
_ d'huile.
''' .
- '
e~Q~XXIY : .
Obtention de l-(4-méthoxypropyl)-4-tl3-cyanophényl)
méthoxy]-benzène (exemple 47) :
Selon le mode opératoire décrit dans la prëparation
L~I~, au départ de l ~ (0,0037 mole) de ~-~(3-
cyanophényl~méthoxy3-benzènepropanol obtenu à la
preparation LXVI, de 0,12 o (0,006 mole) d'hydrure de
sodium et de 0,766 g ~0,006~ mole~ d'iodure de méthyle,
on obtient, après purification par chromatographie sur
colonne de silice en éluant au moyen d'éther, 0,86 g
(rendement : 81 %) du produit attendu sous forme
d'huile.
- -~ ., - . . ~ . . . ..... . . . . . . . .
.: . . . . .
WO ~/11~7 PCT/F~ 0209
c~ ` 60
.~ . . .
.
Obtention de 4-{4-{(3-chlorophényl)méthoxylphényl]-2-
butanone
3,84 g (0,16 mole) d'hydrure de sodium qont ajoutés à
-25 C dans un mélange de 20 g (0,1218 mole) de 4-
hydroxyphényl-2-butanone dans 100 cm3 de diméthyl
formamide. Après 2 heures d'agitation à -10-C, 21,57 g
(0,134 mole) de chlorure de 3-chlorobenzyle sont
ajoutés au milieu réactionnel à -25 C. Après 12 heures
sous agitation, le milieu réactionnel est hydrolysé sur
un mélange glace/eau. Le précipité formé est
recristallisé de l'hexane. On obtient 27,68 g
(rendement : 78,7 %) du produit attendu fondant à 87 C.
PRF~l?Al~ATIoN T.~VT
Obtention de 4-t(3-chloroPhényl)méthoxyl-a-méth
benzènepropanol (esemple 48)
A un mélange de 6 g (0,02 mole) de 4-l4-[(3-
chlorophényl)méthoxy~phényl~-2-butanone obtenus à la
préparation LXXV dans 150 cm3 de méthanol, on ajoute
0,86 (0,023 mole) de tétrahydroborure de sodium. Après
30 minutes d'agitation à température ambiante le milieu
réactionnel est hydroly~é sur un mélange de 5- B de
chlorure d'ammonium solide, 1 C~3 d'eau et 100 cm3
d'éther. Le précipité obtenu est filtré. Les solvants
sont évaporés sous pression réduite et le résidu obtenu
est dissous dans l'éther. La phase éthérée est séchée
sur sulfate de magnésium, filtrée puis évaporée sous
pression réduite. Aprè~ recristallisation d'un mélange
éther isopropylique/hexane, on obtient ~,~ g
(rendement : 83,3 X) du produit attendu 90US forme de
cristaux blancs fondant à 62 C.
- . .
~. . ., A
' ' ' ., " ,~' :' '',;, '; :
,. .: ' :, . ':,: .
; .. '- ,.:' , : . . .
.. . .
:~ :
WO90/~1997 PCT/FR~/~0209
",, _, r,
ol
On a regroupé dans les tableaux I à III ci-après un
certain nombre de produits selon l'invention.
On a résumé dans le tableau IV ci-après lec résultats
des essais biochimiques entrepris avec le~ composés
selon l'invention.
~E~UIre de l~;nh;b;tion deY activités des MAO-A et M~O-R
Les activités des MAO-A et MAO-B sont déterminées dan~
le cerveau de rat Wistar mâle selon la méthode décrite
par DOSTERT Ph. et al (J. Pharm. Pharmacol. 1983, 35 :
161-165).
-
La sérotonine ~5-HT = 480 yM) et la ~-phényléthylamine
(B-PEA = 12 yM~ sont utilisées respectivement com~e
substrat vis-à-vis de la MAO-A et de la MAO-B.
L'effet inhibiteur de tous les produits testés est
déterminé après différents temps de préincubation à
37-C entre le produit et 1'enzyme, notamment 60
minutes.
':
La puissance inhibitrice est donnée par l'IC 50
~concentration en mole~l de produit qui inhibe de 50 %
l'activité témoin).
La sélectivité des produits pour l'effet IMAO~B) par
rapport à l'effet IMAO(A) est donnée par le rapport
IC 50 (A)
.
IC 50 (B)
WO90/11~7 PCT/FR90/00209
b 2
Les produits selon l'invention sont utiles en
thérapeutique. Ils peuvent etre utilisés seuls ou en
association avec deq précurseurs de mono-amines
biogènes, tels que Le tryptophane, la l-dopa ou la
phénylalanine, ainsi qu'en association avec des
excipients physiologiquement acceptables, notamment
dans certains états dépressifs et dans- les pathologies
liées aux processus dégénératifs dus à l'environnement
ou à l'âge, comme par exemple la maladie de Parkinson
et les démences séniles et les prédémences séniles.
'- .
. . . : .. .. . . . .
WO g0/11997 PCT/FK9,0~0020
TABLEAU I
:. -
R~ 2~3tCH2)n--[CH~ 3)]P--N/
~22
. .
lS Exemple R n R1 R~
N- .
_ .' ~
' , " '
l 3-Cl 0 1 H CH3
26 2 3-Cl 1 l H H
3 3-Cl 2 l H H :
4 3-Cl 0 1 H H
3-Cl 0 1 H CH2CH3 ~:
6 3-Cl 2 l CH3 CH3
7 3-Cl 0 1 CH3 CH3
8 3-OCH3 2 1 H CH3 :
9 3-Cl 2 1 H CH2CH3
3-Cl 1 l H CH3
ll 2-Cl 2 l H CH3 ..
l2 4-Cl 2 1 H CH3
l3~ 3-Cl 2 l CH2CH2-O-CH2-CH2
. l .,
WO 90/11997 PCT/FR90/00209
s~ 64
TA~LEAU I (fin)
Exemple R n P R1 R2
N-
.
l4 3-Cl l 1 CH3 CH3
3-Cl 2 l H CH3
l6 3-CN 2 l H CH3
l7 . 3-CN 2 l H H
l8~' 3-Cl 2 l CX2-CHz-CH2-CH2-CH2
19 3-Cl 2 l H CH2CH2CH3
3-NO2 2 1 H CH3
2l 3-NOz 2 1 H H
22 3-F 2 1 H CH3
23 4-F 2 1 H CH3
24 4-F Z 1 H H
3-F 2 1 H H
26 3-Cl 2 1 H CH2CH2OH
27 3-Cl 3 0 H CH2CH3
28 3-Cl 3 _ _ CN3
* NR1R2 représente le reste morpholino
** NR1R2 représente le reste pipéridino
WO ~/11~7 PCT/F~/00209
~5 ~ ?~ 3
TABLEAU II
Sels correspondants aux bases du tableau I
, : -.
. . .
Exemple Acide F-C . :
N- . :-
:. ''
'':',', ' '.,
1' maléique 105 puis 126 (a) ~
2' maiéique 148 :-:
3'chlorhydrique 165 .
4'chlorhydrique 141 ~ .
5'chlorhydrique 203 . .
20 6'citrique 84-86
7'citrigue 114
8'fumarique 101-103 . .
9'oxaligue 101-107
10'fumarique 174
2511'fumarique 134-137
12'fumarique 170-171
13'fumarique 132-133
14'fumarique 159
15'chlorhydrique 148-149
3016'oxalique 132-136
.. ,. . . . .. . . , . . .. - , . . . . -. ,.. .. . . ... . .. ~ . , .
WO90/11997 PCT/FR90/00209
~6
`' ` '`
TA~LEAU II tfin)
~Exemple Acide F-C
N^
17' fumsrique 16~
18' fumarique 130
19' fumarique 97 puis 115 (a)
20' fumarique 112
21' fumarique 176-179
22' fumarique 129
23' fumarique 166
24' fumarique 175-179
25' fumarique 185-187
26' fumarique . 115-118
27' maléique 118
28' fumarique 98
(a) double point de fusion
~: '
WO 90/11997 PCI`/F1~90/00209
67 ~,; ,
TABLEAU I I I
R ~
~C~2~3(CX2)n--[CH-(CH3) ]p--OR3
Zxe~lple ~ R ~ n ~ p ~F- C
C l H 3 0 54 - .
36 C l H 1 O 76
37 Cl CH3 3 0 huile ~ 1) I .
38 Cl H 2 O 78
39 NOz H 3 0 42
CN H 3 O 64
41 Cl H 4 O 63
42 CF3 H 3 O 48 ¦ -
43 NOz COCH3 3 0 65
"O 44 Cl COCH3 3 O 36
Cl COC~CH3 )3 3 O huile ~2)
46 NOz COC~CH3 ~3 3 0 huile ~ 3)
47 CN CH3 3 O huile ~ 4)
S 4 ~ C 1 H Z 1 62
NQ$es : ~1) n23 C = 1,5542(3) nD3-C = 1,5418
(2) nZ3 c = 1,5341 (4) nZ~ c = 1,559
.
.. .. .. . . . .
; . , , ~, .~ ,. , , ~ .
WO ~119g7 PCT/FR90/002~
.. .
TABLEAU IV
IC 50 (A)
5 Exemple IC 50 (A) IC 50 (B)
IC 60 (B)
1'7.10-5 1,7.1o-6 41
2'2,5.10-5 6,5.10-7 38
3'5.10-5 2,7.10'7 185
4'4,2.10-5 3,5.10-7 120
5'8,8.10-5 1,3.1o-6 68
6'9,5.10-5 1,2.10-6 79
7'4,5.10-5 4.10-6 11
8'6,7.10-5 5,2.10-7 129
9'1,3.10-~ 1.10-7 1 300
10 '4.10- 5 7.10- 7 57
11' 1.10-~ 4,2.10-7 238
12' 3.10-5 8.10-7 37
13' 3.10-~ 2,5.10-7 1 200
14' 4.10-5 3,2.10-7 125
15'6,5.10-5 3.10-7 217
16'3,5.10-5 4,5.10-6 7,8
17'1,2.10-5 2,7.10-6 4,4
18 '2,8.10-4 6.10- 7 467
19'5,7.10-5 1,2.10-6 48
20'1,1.10-5 4,3.10-7 26
21'6,5.10-6 6,3.10-7 10
2~'7,5.10-5 1,6.10-7 469
23'2,1.10-5 1,4.10-7 150
24 '1,2.10- 5 9,5.10- 8 ( * )126
25 '3.10- 5 2,3.10- 7 130 ;
' '.
: .. :. .: .: . . ~ :
,~,~"~.,,, : : r~ r~v/uu~u~
og
T~8LEAU IV (fin)
IC 50 (A)
Exemple IC 60 (A) IC 60 (~)
IC 50 (B)
. ..
26'8,5.10-5 1,9.10-7 447
~0 27'8,1 1o-6 2,5.10-7 ~32
28'1,8.10-4 3,1.10-7 581
29'5.10-5 1,1.10-7 455
30'3.10-5 5,4.10- 8 656
31'1,3.10-4 3,6.10-7 361
32'2,2.10-5 2,1.10- 6 10
33'1,7.10-4 8,8.10-7 193
349,2.10-5 3,4.10-7 271
368.10-5 10-9 (*) 8.10-4
368,5.10-5 7,3.10-7 116
37> 3.10-4 4,1.10-7(*) > 732
38 10-4 5,8.10-7(*) 172 ~-
393,8.10-6 9,6.10-3(*) 396
404,3.10-6 l,2.10-7 36
411,2.10-4 5,8.10-8(*) 2 069
427,2.10-5 3.10-9 à2,4.10-4 à 2,4.10-6
3.10-1l(*) -
436,2.10- 6 2.10- 8 ~310 ~ 1 550
4.10-9(*)
44> 3.10-5 3,2.10-~(*)> 93g
45> 3.10-5 8,5.10-7 > 35
461,3.10-5 1,5.10-7 87
475,2.10-5 1,5.l0-6 35
4~1,9.10-4 1,4.10-7 1 357
~5
: (*) facteur de pente très different de 1