Note: Descriptions are shown in the official language in which they were submitted.
205~126
CePhem compounds, Their Production and Use
FIELD OF T~IE INVENTION
This invention relates to a novel cephem compound
or a salt thereof for oral administration, which is
useful as a chemotherapeutic agent.
BACKGROUND OF THE INVEN~ION
While there are a number of cephem-type
preparations having excellent antibacterial activity,
most of them are injections and preparations for oral
administration are relatively less in number. On the
other hand, the antibacterial activity and spectrum of
cephem-type preparations for oral administration are
lower than those of cephem-type preparations for
lS injection. For solving this problem, extensive studies
have been carried out ~or leading the carboxyl group at
the 4-position of an injectable cephem compound or a
cephem compound which is hardly absorbed by oral route
to such an ester as bein~ de-esterified in vivo (e.~.
cefuroxime axetil, cefotiam hexetil, cefpodoxime
proxetil, cefteram pivoxil, etc.), which is so-called
"ester-type prodrug". However, no satisfactory
compound has yet been obtained, from the viewpoints of
antibacterial activity and antibacterial spectrum.
While it has been known that 7~-[2-(2-
aminothiazol-4-yl)-(Z)-2-hydroxyiminoacetamido]-3-[(2-
methyl-1,3,4-thiadiazol-5-yl)thiomethyl]-3-cephem-4-
carboxylic acid (hereinafter called "said cephem
carboxylic acid") represented by the formula,
H2N\~
N~5/CONH~S~ N--N
N o N~CH2SI~s~LCHs
OH CQ~I
0
2054126
has an excellent antibacterial activity and a broad
antibacterial spectrum, the said cephem carboxylic acid
was not a compound which can be applied as it is for
the use of oral administration. And, while the
pivaloyloxymethyl ester of the said cephem carboxylic
acid and l-(ethoxycarbonyloxy)ethyl ester of the said
cephem carboxylic acid have been synthesized as ester-
type prodrugs [Japanese Published Unexamined patent
application No. 9698/1985], these esters are not
satisfactory in respect of absorbability from
intestinal tube. Circumstances being such as above,
appearance of cephem agents for oral use, which have
excellent antibacterial activity and broad
antimicrobial spectrum and are improved in oral
absorbability, has been desired.
SUMMARY OF THE INVENTION
The present inventors have continued diligent
study on cephem compounds for oral administration, and,
as the result, they found that the ester compound
obtained by a method characterized by selecting the
said cephem carboxylic acid from an exceedingly large
number of cephem compounds and by introducing into the
carboxyl group at the 4-position of thus-selected
cephem carboxylic acid the group represented by the
formula:
C~lOCO-R2
Il 11
R O
wherein Rl stands for a Cl 3 alkyl group, RZ stands for a
30 C3 s alkyl group branched at a-position or a C3 5
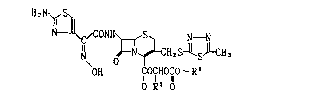
cycloalkyl group, i.e. the cephem compound represented
by the formula,
- 3 - 20~4126
1~, Ny~3
N `~C~coNH\r-~s` N--N
N oD N~LCH7S~S~ 3 ~ I ]
OH IlOGHOCO- R2
O R' O
wherein the symbols are of the same meaning as defined
above, or salts thereof have, unexpectedly, various
excellent properties as cephem compounds for oral
administration, as exemplified by
(i) good absorbability from intestinal tube,
(ii) high blood concentration of the said cephem
carboxylic acid and long half-life thereof,5 (iii) good urinary recovery rate of the said cephem
carboxylic acid,
(iv) good protective activity by oral administration
even against intraperitoneal infections by
Staphylococcus aureus or Escherichia coli, and
(v) less side-effects and high safety.
And, as the characteristic features of the group
constituting the ester bringing about these excellent
properties, the following points are mentioned, i.e.
(1) R2 is bonded via CHOC and O,
R O
(2) R2 stands for a C3s alkyl group,
(3) the C35 alkyl group is branched, and
(4) the branched alkyl group shown by R2 (including C35
cycloalkyl) is branched at a-position, and, based on
the foregoing, the present invention has been
accomplished.
DETAILED DESCRIPTION OF THE INVENTION
In the above formulae, examples of Cl3 alkyl
groups shown by Rl include methyl, ethyl and propyl,
preferably methyl.
2054126
Examples of the C3 5 alkyl group branched at its a-
position shown by 1~2 include isopropyl, l-methylpropyl
and l-ethylpropyl, preferably isopropyl.
Examples of the C3 5 cycloalkyl group shown by R2
include cyclopropyl, cyclobutyl and cyclopentyl,
preferably cyclopentyl.
And, the compound [I] can be used as salts
thereof. In this case, salts with acids, among others,
can be used. Examples of such acids include mineral
acids such as hydrochloric acid, sulfuric acid or
phosphoric acid and organic acids such as maleic acid,
acetic acid, citric acid, succinic acid, tartaric acid,
malic acid, malonic acid, fumaric acid, benzoic acid,
mandelic acid, ascorbic acid or methanesulfonic acid,
which are known, in the fields of penicillin or
cephalosporin, as acids forming pharmaceutically
acceptable salts.
In the compound [I] or salts thereof, due to the
existence of an asymmetric carbon at the ester moiety
of the carboxyl group at the 4-position of the cephem
skeleton, there are two kinds of optically active
compounds (R-isomer, S-isomer) based thereon.
Therefore, while the compound [I~ or a salt thereof may
usually be in the state of a diastereomer mixture
having a suitable ratio, the R-isomer or the S-isomer
isolated by a conventional method can be used.
The compound [I] or salts thereof are readily
absorbed from intestinal tube, and the ester moiety of
the carboxyl group at the 4-position is promptly
hydrolyzed by the enzyme in the body so that the
compound [I] or salts thereof are led to a non-ester
compound of the compound [I], i.e. the said cephem
carboxylic acid.
The compound [I] or salts thereof are valuable
antibiotics showing excellent antibacterial activities
against both gram-positive and gram-negative bacteria
5 205~126
including ~hose isolated clinically, which are used as
medicines for ~lumans and domestic animals and can be
used safely as antibacterial agents for the therapy and
prophylaxis of infections caused by various bacteria.
The compound [I] or salts thereof can be used, as
therapeutic agents of bacterial infections, for the
therapy and prophylaxis of, for example, respiratory
infections, urinary tract infections, suppurative
diseases, bile tract infections, intestinal infections,
gyneco-obsterical infections, otorhinological
infections and surgical infections.
The compound [I] or salts thereof of the present
invention can be orally administered, and they can be
prepared into, for example, capsules, powders,
triturations, granules and tablets by conventional
methods after suitably mixing with ~E se known
pharmaceutically acceptable excipients te.g. starch,
lactose, calcium carbonate or calcium phosphate),
binders (e.g. starch, gum arabic, carboxymethyl
cellulose, hydroxypropylcellulose or crystalline
cellulose), lubricants (e.g. magnesium stearate or
talc), disintegrators (e.g. carboxymethylcellulose
calcium or talc), or the like.
And, a compound [I] or a salt thereof can be
prepared into granules by a conventional method after
incorporating a solid organic acid (e.g. citric acid,
malic acid, tartaric acid, succinic acid, ascorbic acid
and mandelic acid) in an amount of about 1 to 5 times
as much as the compound [I] or its salt. These
granules can be prepared into capsules, tablets, etc.
by conventional methods.
The oral dosage of the compound [I] or a salt
thereof per day for infectious diseases varies with the
conditions, body weight, etc. of the patient, and it
ranges from about 0.5 mg to 100 mg, preferably 1 mg to
20 mg, of the active component (the compound [I] or a
20~4126
-- 6
salt thereof) per kg of body weight of an adult human,
divided into 1 or 3, preferably 1 or 2 times.
The compound [I~ or salts thereof can be produced
by the methods se~ forth below.
Method 1
A compound [l] or a salt thereof can be produced
by allowing a compound represented by the formula:
H2N~ ~
O N ~/CONH ~ S N-N
N 0~) N ~ H 2 S~S~H 3 ~ ~ 3
OH ~OH
or a salt thereof to react with a compound represented
by the formula:
X-CHOCO-R
Il 1l [III]
R O
wherein X stands for a leaving group, Rl and R2 are of
the same meaning as defined above.
In the above formula [III], as the leaving group
shown by X, use is made of, for example, halogen such
as chlorine, bromine or iodine, C6l0 aryl sulfonyloxy
group such as phenylsulfonyloxy group or p-
toluenesulfonyloxy group, or C~6 alkylsulfonyloxy group
such as methylsulfonyloxy group. ~mong these, iodine
is especially preferable as X.
And, since the compound [III] has an asymmetric
carbon atom, it is subjected to optical resolution by a
E~ se conventional means, then used for the reaction
as the R-isomer or S-isomer or a mixture thereof.
On the other hand, the starting compound [II] may
be subjected to the reaction as a salt with a base, for
example, an alkali metal such as sodium or potassium,
an alkaline earth metal such as calcium or magnesium,
an organic amine such as triethylamine, trimethylamine,
2054126
pyridine, colidine or lutidine.
Usually this reaction is conducted desirably in a
inert solvent to the reaction. As the inert solvent,
any one can be used so long as it does not hamper the
reaction, and examples of suitable solvents include
amides such as N,N-dimethylformamide (hereinafter
abbreviated as 'DMY') or N,N-dimethylacetamide
(hereinafter abbreviated as "DMA"), halogenated
hydrocarbons such as dichloromethane or chloroform,
ethers such as dioxane or tetrahydrofuran (hereinafter
abbreviated as 'THF ), nitriles such as acetonitrile,
sulfoxides such as dimethylsulfoxide (hereinafter
abbreviated as "DMSO"), sulfolane, liquefied sulfur
dioxide.
The compound [III] is used preferably in an amount
of 1 to 5 mols. relative to 1 mol. of the compound [II]
or a salt thereof. While the reaction temperature
varies with various conditions, for example, the
starting compounds and kinds of solvents, it ranges
usually from -30C to to 60C, preferably -20C to
30C. While the reaction time varies with various
conditions, for example, the starting compounds, kinds
of solvents and reaction temperatures, it ranges
usually from one minute to 24 hours, preferably 5
minutes to 5 hours.
The starting compound [II] which is used in this
reaction can be synthesized by the method disclosed in
GB-A-1587941 or a method analogous thereto. And, the
starting compound [III] can be easily produced by a
conventional method (e.g. EP-A-128029, Japanese
Published Unexamined Patent Application No.
239490/1985) or a method analo~ous thereto.
Method 2
A compound [I] or a salt thereof can be produced
by allowing a cephem compound [IV] represented by the
formula:
2Q5~26
-- 8
~N~ ~ K 2 S~S~H 3 [N]
co~aoco- R~
0 R' O
wherein the symbols are of the same meaning as defined
above, or a salt thereof to react with a compound of
the formula:
R3]~
l ll
N ~ /CO~H
N\ [V]
OR'
wherein R3 stands for hydrogen atom or a protecting
group of the amino group and R4 stands for hydrogen
atom or a protecting group of hydroxyl group, or a salt
thereof or a reactive derivative at its carboxyl group,
and, further, when necessary, by removing the
protecting group or groups.
In the formula [V], as the amino-protecting group
shown by R3, use is suitably made of, for example,
those employable in the fields of ~-lactam and peptide,
and, among them, chloroacetyl, tert-butoxycarbonyl,
benzyloxycarbonyl, p-methoxybenzyloxycarbonyl, p-
nitrobenzyloxycarbonyl, 2-trimethylsilyl-
ethoxycarbonyl, 2,2,2-trichloroethoxycarbonyl or trityl
is preferable. Among them, chloroacetyl, tert-
butoxycarbonyl or trityl is especially preferable.
As the hydroxy-protecting group shown by R4, use
is made of, for example, chloroacetyl, benzyl, p-
nitrobenzyl, methylthiomethyl, trimethylsilyl, tert-
butyldimethylsilyl, 2-tetrahydropyranyl, 4-methoxy-4-
tetrahydropyranyl, or trityl. Among them, 2-
tetrahydropyranyl, 4-methoxy-4-tetrahydropyranyl and
2~54126
g
trityl are especially preferable.
As salts of the compound [IV], use is made of, for
example, salts with acids similar to those used in the
case of the above-mentioned salts of the compound tI].
As salts of the compound [V], salts similar to those
described in the above-mentioned [II].
As reactive derivatives at the carboxyl group of
the compound [V], use is made of, for example, acid
halides, acid anhydrides, active amides, active esters,
active thioesters, etc., which are more specifically
described as below.
1) Acid halides:
For example, acid chlorides, acid bromides etc.
are employed.
2) Acid anhydrides:
For example, mono-lower alkylcarbonic acid mixed
anhydrid are used.
3) Active amides:
For example, amides formed with pyrazole,
imidazole, 4-substituted imidazole, dimethylpyrazole,
benzotriazole, etc. are used.
4) Active ester:
For example, esters such as methoxymethyl esters,
benzotriazole esters, 4-nitrophenyl esters, 2,4-
dinitrophenyl esters, trichlorophenyl esters,
pentachlorophenyl esters, etc., as well as esters
formed with 1-hydroxy-lH-2-pyridone, N-
hydroxysuccinimide, N-hydroxyphthalimide or the like.
5) Active thioesters:
For example, thioesters formed with, for example,
heterocyclicthiols such as 2-pyridylthiol, 2-
benzothiazolylthiol, etc. are used.
In this reaction, the compound ~V] or a salt
thereof or a reactive derivative at its carboxyl group
is used in an amount of one mol or more, preferably in
a range of from about one mol. to 4 mols. relative to 1
205~126
-- 10 --
mol. of the compound [IV] or a salt thereof. This
reaction may be conducted usually in a solvent which is
exemplified by water, ketones such as acetone, etc.,
ethers such as THF, dioxane, etc., nitriles such as
acetonitrile, etc., halogenated hydrocarbons such as
dichloromethane, chloroform, 1,2-dichloroethane, etc.,
esters such as ethyl acetate, etc., amides such as DMF,
DMA, etc. These solvents may be used singly or in
combination of two or more of them in a suitable
mixture ratio. When the compound [V] is used as the
free acid or as a salt thereof, the reaction may
preferably carried out in the presence of a condensing
agent which is exemplified by N,N-
dicyclohexylcarbodiimide, N-cyclohexyl-N-morpholino-
ethylcarbodiimide, N-cyclohexyl-N-(4-diethylaminocyclo-
hexyl)carbodiimide, N-ethyl-N-(3-dimethylaminopropyl)-
carbodiimide, etc. The reaction can also be carried
out in the presence of a base which includes, for
example, alkali metal carbonates such as sodium
carbonate, potassium carbonate, sodium
hydrogencarbonate, potassium hydrogencarbonate, etc.,
tertiary amines such as triethylamine, tributylamine,
N-methylmorpholine, N-methylpiperidine, etc., and
aromatic amines such as pyridine, picoline, lutidine,
colidine, N,N-dimethylaniline, etc. These bases serve
to accelerate the reaction, to neutralize the acid
formed during the reaction or to make the starting
materials to be readily soluble. The amount of such a
base may usually be about 0.01 to 5 mols., preferably
about 0.1 to 2 mols. relative to 1 mol. of the compound
[V] or a salt thereof.
While the reaction temperature varies with
reaction conditions including the starting materials
and kinds of the solvents then employed, it ranges from
-50C to 100C, preferably -20C to 50C. While the
reaction time varies with reaction conditions including
20~126
11
the starting materials, kinds of the solvents, reaction
temperatures, etc., it ranges from one minute to 24
hours, preferably five minutes to 10 hours.
The starting compound [IV] to be used for this
reaction can be synthesized in accordance with, for
example, the method described in Japanese Published
Unexamined Patent Application No. 239490/1985. And,
the starting compound [V] can be synthesized by, for
example, the method described in GB-A-1580621.
Method 3
~ compound [I] or a salt thereof can be produced
by allowing a starting compound represented by the
formula:
YC~ 2 CO\c/c~N~{ ~ S N N
N ~N~LCH2SJ~SlLCH~ ~ VI ]
OH COC~OCO--R2
o Rl o
wherein Y stands for halogen atom, and the other
symbols are of the same meaning as defined above, to
react with thiourea in an inert solvent.
As the halogen atom of Y, use may be made of, for
example, chlorine, bromine or iodine, preferably, for
example, chlorine or bromine.
Cyclization of the compound [VI] with thiourea can
be carried out in accordance with a Per se known method
(e.g. the method disclosed in, for example, GB-A-
1587941.
While the reaction temperature varies with various
conditions including types of the starting compounds
and kinds of the solvents then employed, it ranges from
-20C to 50C, preferably 0C to 30C. While the
reaction time varies with various conditions including
types of the starting compounds, kinds of the solvents
and reaction temperatures then employed, it ranges from
- 12 - 20~412 6
5 minutes to 24 hours, preferably lO minutes to 5
hours. The reaction is preferably conducted in an
inert solvent. As the inert solvent, while any one may
be used so long as it does not hamper the reaction,
such amides, ketones and acetonitriles as described in
the above Method 2 are preferable.
The starting compound [VI] can be produced by
allowing diketene to react with halogen (especially
chlorine and bromine are preferable) in accordance with
a ~er se known method (e.g. the method disclosed in
British Patent No. 15~7941), by allowing the resultant
product to react with a compound [IV] or a salt thereof
to give a compound represented by the formula:
YcH,c~ll. CON~
cc~oco-R~ t~]
0 R'0
wherein the symbols are of the same meaning as defined
above, then by subjecting the compound [VII] to
nitrosation.
When protecting groups are contained in the
reaction products of the above-mentioned Methods 1 to
3, the compound [I] or a salt thereof can be obtained
by, when necessary, removing the protective group or
groups by a conventional means.
The object compound [I] or its salt thus obtained
can be isolated and purified conventional method, for
example, solvent-extraction, pH-change, phase transfer,
salting out, crystallization, recrystallization,
chromatography, etc.
In the above-mentioned Methods 1 to 3, the
compound [I] (syn[Z]-isomer) or a salt thereof is
obtained sometimes as a mixture with its anti[E]-
205~126
- 13 -
isomer. For isolating the desired syn-isomer (i.e. the
compound [I] or a salt thereof) from the mixture, a per
se conventional method or a method analogous thereto
can be applied, as exemplified by separation utilizing
the difference in solubility, crystallizability, etc.
or isolation by means of chromatography, etc.
The compound [I] or salts thereof obtained by the
present invention have excellent properties as cephem
compounds for oral administration. As concrete
examples of those properties, therapeutic effects
against intraperitoneal infections in mice by oral
administration, and the urinary recovery rate of the
said cephem carboxylic acid and effects of maintaining
the concentration of the said cephem carboxylic acid
in blood after oral administration of the compound [I]
in mice are described as follows in comparison with
control compounds.
(1) Therapeutic Effects against Intraperitoneal
Infections in Mice Test animals: Slc : ICR, 4-week old
male mice (body weight 20 to 22 g) were employed.
Induction of experimental infections (infected mice):
Staphylococcus aureus 308A-1 or Escherichia coli
O-lll was cultured in Brain Heart Infusion broth
(manufactured by Difco Co.) at 37C for 20 hours. Each
of the organisms was suspended in a 8% mucin solution
(manufactured by Difco in) at a concentration of 2 x
108 Colony Forming Units (hereinafter abbreviated as
"CFU")/ml and 2 x 10 CFU/ml, respectively, and 0.5 ml
of each suspension was administered intraperitoneally.
Administration of Test compounds: ~ givell ~ ount of
each test compound was dissolved inla small ~41un~of
DMSO, and each solution was diluted in distilled water
to prepared two fold dilutions of each compound in the
range of 2.5 mg/ml - 0.156 mg/ml in the case of
infection with Staphylococcus aureus and in the range
of 0.156 mg/ml - 0.01 mg/ml in the case of infection
2034126
- 14 -
with Escherichia coli. 0.2 ml volume of each dilution
was once adminis~ered orally using a tube into the
stomach of five mice each in each group immediately
after the infection.
Determination of the effect: Thus-treated mice were
observed for five days, and the survival rates in the
respective amounts of administration were counted, then
ED50(mg/kg) was calculated by the method of Reed and
Muench. The ED50 value of each compound was that in
terms of the said cephem carboxylic acid.
The results are shown in the following table.
N ~ C' 1~ N-N
N ~ Cll2S ~ ~ Cll3
OH C-OR
Or~anis~: 5taphylococcus aureus 3U8/l- 1
Test Comi ound ~, D 60 (~ng/kg)
lix . 1 - CH0C00CH ( Cll l ) 2 7 0 2
__
~x . 2 -C130COOCII (CH~ ) z 5 . ;) 7
The Compound C~a
(HCl sa I t )
o~this ~K.3 ~ o~oo -O ~ 8'I
Invent ion ~x . 5 -CHOCOO ~ 1 4 . O
(IICl salt)
~x 6 -CHOCOOCIl ( C2 l~s ) 2 1 2 5
(IICI sal t)
_ . .
20~4126
~ _ ~ 2 , . A . ~ _ _ A r A . _ . . 2 _ ` A ~ C _ S ~i ~
_ ._ _ _ _. .~ ._ _
r.. t Col~po~nd R 15 D ~ k~).
.... _ _ . _ . _
CB~ -H
lSB794 1 (N~ J~J t ) ~ .~ 5 . O
~he kno~ ~t . 16 .
_ . ,. ._ .. . _
. JP~ ~ COC~C~
Co~Dou~d 9~9811985 H . 2 2 . 3
rR Ex.3 .
. . .. _ ..
JP_~,_ -CHOCOOC, Hs
9698Jlga5 C~, ~ 2 5 . O
P~ ~x.~ .
. . . . ~ . _ ,
1 S - GHOCOO~ ~ 2 5 . O
. _c~toc~cn~ . _ .
~rul ~ . 3 2 5 O
~ICl ~alt)
20a~lo~ous -c~OCOO-O , . __
H 1 7.7
oo~oun~ (HCI s~ I t ) .
~5 . -CI~OCOOCN~ ~ 2 5 . O
(HCI ~a I t) .
.~ . _ . __ _ _
~JP-A-: J-P~neJe p~lishe~ unex~lol~d Pate~t ~PD Icttlo~ nO.
PR ~X.: Ptop~r~tlo~ ple )
.
::~3 '~ 3~'`. 'A~ S ^3~ 9~ : 2AQ35~4ll2,26-~'^
- 16 -
nr~-cl~n ~ ~.ch r 1 ch 1~ Col i O--1 1 1
'~-~Co~9 ~ol ~ _ /10,=
~EI.1 -fHOC~OC~(CN,~, 0^1 4
5~h- Cl~oou~ltt --~ i c 1
. ~1. 2 -CHOCOOC~(C~
~1' th~J ~ ~ ~H O 1 3 0
0l~ve~t lo~
C~-A- ~
. 1 5879~ 1 ~Na Ja I t ~ 1 . 2 5
T~ Xnoun ~Y.16 .
JP_~._ ~ COC(~ ' . _
96~8J 198S H O .. 2 7 9
Co~gol~nd P~ Ex . 3
. I J~ -C~IO~OOC~ H5
9h98J19J5 CH~ 0.3 4 9
_ PR ~s . 4
~_ _ ~ . .._ .
. . H~ 0 ~7 8 ~)
2 5110~ol tHCl s~ l t )
. ~__
~n~ o~6 )~ .~ ~ 5 5
. ~HCl ~nl t~
~~Donn~ ~--
X ~)~48
_tXCl ~lt)
~JP~ ; J-p~ C ~9111151~e ;=~ llc~tloD No.
PR 8~ rr-p~r~tlo~ plt )
205~12~
~ h$~ te~t~t ~how~ th~t the ~omp~un-d ~ I ) c~r ~lt~s
~ ve ~xcellent therapeutic ef~ects, comPared
with th4 kno~,m compound~ and tho nov~l ~nalogouE3
compound~.
S ( 2 ) Vxln~y neco~e~r rc~te ln miG~
~ Cl~ W~:II~K ~1~ ~ mlc~3 t
g body weight) were employed.
Dosage : lO~g of each test compound wa~ di~solved in
0.5 ml of DMS0. The respective solution~ were diluted
10 timo~ wlth ~o~(v/v) of a~ueous solutlon of
polyethyleneglycol 200. A ~olume of 0.1 ml~10 g ~ody
weight of e~ch of th~ re~pective dllution~ wa~
ad~inistered once to noxmal mice (five in one qr~u~
ox~lly into stomach by u~ing ~ tube.
Recovery of urino~ Aft~r admini~tration of the drug,
each ~e~t animal wa~ placed separately in a cage, a~d
urine wa~ collec~ed d~ring 24 hour~.
~ot~rm~ n~ ~ I o~ ruy concentratlon: The concentr~tion
of the sAid cephem carboxylic acid in urine wa~
determ~ned by the a~ar well method U8~ ng, ~Y te~t
bacteri~ r Escherichia coli NIHJ and Proteus rett~eri
ATCC 9250.
The resul~s are set forth as follow~.
~ C,CONU ~ ~ N
N ~J~c~l2s~$~L
o~r
o
2~59126
~ 18 --
. , ~ ~
toAt C~l-po~nd R Crln-r~ ex~re~on~l)
_ ~
E~ 1 -CHOCOOCN(~H~)~ I li . 2
._ _ _ , _ ..
..~h~ Col~pou~a Ex. 2-CHOCOOC~
Cl~ 2 ~; 7
O~ th l ~ (h~ 1 t )
_
~. 3 -FNOCOO--O2 5 . 5
I~v~ l oll GH
. _~
. Cl~. 5 -CHOCOO--O
CH, 2 1 .1
~CI ~a l t )
~ A- _ _ _
l~i ~5~79~ ~ lt) ;~
~h~ ItDO~ _ __ -_ ~ _
JP-A- -I ~COC~CH~ ),
. g~9~1g~5 4 1 3 .8
2 0 co~rou~ I~ . 3
. JP-A- -fHCCo~CtH5
~, . 1 1 . 6
~R tJ~ . ~
, . _ ~ . ....
-~NOCOO~
2S CH~ 7 . B 1 .
I~o~l ~ .. __ _
-C~OCOCH,
oroY- 11 ~ ~ O
~NCi ~ )
3 0 C~noou~t ~
. ~ ~ ~3
~ __ ~C~ s~lt~
(Jr-A- ~ J~s-ne~ ~ubli~h~
3S ~ ~X.~ Ps~p~r~tlo~
T~ls 'cest re~sult ~hows that the co2npound ~I3 has a
hlq)l url n~ry reco~rery rate, comp~red w~th the kno~n
coml?oun~l~ an~ tho no~rol 4n~10gous compound~.
2054126
(3) ~f~cct~of-ma~ ning-th~¦Blood concentration
~bloo~ of the said cephem carboxylic acid ~
Test animals: Sic : ICR, 4-week old male mice (20 - 22
g body weiqht) were employed.
Dosage : ~ givo~ r3~ of each test compound was
dissolved in ~ om~ll volumq of DMSO. The respective
solutions were diluted~ ith ~istillc~ w~tcr ~nd~ ~
aqueous solution of polyethylene glycol 200. 0.1 ml/10
g body weight of each of the respective dilution
solutions was administered once to normal mice (five in
one group) orally into stomach by using a tube.
Recovery of test materials: Blood was collected in a
given period after administration of drugs.
Determination of concentration of drug: The blood
concentration of the said cephem carboxylic acid was
determined by the agar well method using, as test
bacteria, Escherichia coli NIHJ and Proteus rettgeri
ATCC 9250.
The results arP 5et fnrth as fr,l ID~S.
N~C,CONll ~ N--h
ll U~ LCU2S~SJ C]~
`Ol~ OK
Concentration In blood ~,ugjml)
Tesl Con~oulld ` ~... Hour 0 75 0 5-- 2 4 AIIC
30The Compound -CHOCOOCH( CH3), _
of this l 35.5 36.9 25.510.2 2.99 58.4
Inven~lun CH9 _
JP-A- -CHoCO/CH9 i.
rhe l~nol~n . 9698~ 1 988 5 1 2 2 . t 19 4 15 . 4 b . 4 2 . 41 3; . 2
Compound J~-A- -CHOCOOCzHF
9698/~985 l ~5. 28.8 19 0 6.11 2.56 ~0.3
3 5 PR Ex . 4 _ _
(Jp-A-: dapanese published unexalnlDed Patent Application 110.
PR EX.: Pr~p~rntion Exn~le )
- 2~ - 20S4126
This test result shows that administration of the
compound [I] or salts thereof serve to maintain high
blood concentration of the said cephem carboxylic acid
for a long period of time, compared with the known
compounds.
Examples
By the following working examples, the present
invention will be described in further detail.
However, these are nothing more than mere examples and
not to intend to limit the scope of this invention in
any manner.
In the following working examples, NMR spectrum
was measured by means of Gemini 200 (200MHz)-
spectrometer, and all the ~ values were shown by ppm.
Symbols in the examples have the following meanings.
s : singlet
d : doublet
t : triplet
ABq : AB type quartet
dd : double doublet
m : multiplet
br. : broad
J : coupling constant
sh : shoulder
Example 1
l-(Isopropoxycarbonyloxy)ethyl 7~-[2-(2-
aminothiazol-4-yl)-(Z)-2-hydroxyimino-acetamido]-3-[(2-
methyl-1,3,4-thiazol-5-yl)thiomethyl]-3-cephem-4-
carboxylate
In DMA (3.5 ml) was dissolved sodium 7~-[2-(2-
aminothiazol-4-yl)-(Z)-2-hydroxyiminoacetamido]-3-[(2-
methyl-1,3,4-thiazol-5-yl)thiomethyl]-3-cephem-4-
carboxylate (0.2 g). To the solution was added, with
stirring under ice-cooling, 1-
(Isopropoxycarbonyloxy)ethyliodide (0.3 g). The
mixture was stirred for 30 minutes at the same
- 2-2 ~ ~ 05ql ~6
temperature, to which was added ice-water, followed by
extraction with ethyl acetate. The organic layer was
washed with brine, dried over anhydrous sodium sulfate,
then the solvent was distilled off under reduced
pressure. The residue was dissolved in acetone (3 ml),
to which was added isopropyl ether (30 ml) with
stirring at room temperature (20C). Precipitates were
collected by filtration and washed with isopropyl
ether, followed by drying to afford 135 mg of the
above-titled compound as a mixture of two diastereomers
(about 1:1) as a pale yellow powder.
IR(KBr)cml: 3325, 3200, 2980, 1780, 1755, 1670, 1610,
1530
IH-NMR(DMSO-d6) ~: 1.20-1.25(6H,m), 1.50 and 1.53(3H,
each d,J=5Hz), 2.68 and 2.69(3H, each s),
3.71(2H,ABq,J=18Hz), 4.34 and 4.36(2H, each
ABq,J=14Hz), 4.71-4.86(1H,m), 5.16 and 5.19(1H, each d,
J=5Hz), 5.79-5.88(1H,m), 6.66(1H,s), 6.77-6.92(1H,m),
7.25(2H,brs), 9.48 and 9.49(1H, each d, J=8Hz)
Example 2
l-(Isopropoxycarbonyloxy)ethyl 7~-[2-(2-
aminothiazol-4-yl)-(z)-2-hydroxyimino-acetamido]-3-[(2
methyl-1,3,4-thiadiazol-5-yl)thiomethyl]-3-cephem-4-
carboxylate hydrochloride
In ethyl acetate (15 ml) was dissolved the
compound (100 mg) obtained in Example 1, to which was
added, with stirring under ice-cooling, 0.4N
hydrochloric acid solution in ethyl acetate (0.5 ml).
Powdery precipitates were collected by filtration,
washed with ethyl acetate, and dried under reduced
pressure to afford 78 mg of the above-titled compound
as a mixture of two diastereomers (about 1:1) as a pale
yellow powder.
IR(KBr)cm : 2980, 1780, 1750, 1670, 1620, 1520
IH-NMR(DMSO-d6) ~: 1.22-1.26(6H,m), 1.51 and 1.53(3H,
each d,J=5Hz), 2.69(3H,s), 3.59-3.89(2H,m), 4.34 and
- 23 - 2 0 ~ 4 ~ 2 fi
4.40t2H, each ABq,J=13Hz), 4.72-4.87~1H,m), 5.21 and
5.23tlH, each d,J=5Hz), 5.79-5.89tlH,m), 6.81 and
6.88(1H, each q,J=5Hz), 6.85(1H,s), 9.72 and 9.73 tlH,
each d, J=8Hz)
Example 3
l-(Cyclopentyloxycarbonyloxy)ethyl 7~-[2-(2-
aminothiazol-4-yl)-tz)-2-hydroxyimino-
acetamido]-3-[(2-methyl-1,3,4-thiadiazol-5-
yl)thiomethyl]-3-cephem-4-carboxylate
In the same procedure as in Example 1, using 1-
(Cyclopentyloxycarbonyloxy)ethyliodide in place of 1-
(Isopropoxycarbonyloxy)ethyliodide the above-titled
compound was obtained as a mixture of two diastereomers
(about 1:1).
IRtKBr)cml: 3325, 3190, 2970, 2875, 1780, 1755, 1675,
1610, 1530
lH-NMRtDMSO-d6) ~: 1.4-1.9tllH,m), 2.69t3H,s), 3.55-
4.60(4H,m), 4.96-5.07tlH,m), 5.17 and 5.19(lH, each
d,J=5Hz), 5.80-5.90tlH,m), 6.66(1H,s), 6.80 and
6.88(1H, each q, J=5Hz), 7.21(2H,brs), 9.48 and
9.49tlH, each d,J=8Hz)
Example 4
1-tCyclopentyloxycarbonyloxy)ethyl 7~-[2-(2-~
aminothiazol-4-yl)-tZ)-2-hydroxyiminoacetamido]-3-[t2-
2S methyl-1,3,4-thiadiazol-5-yl)thiomethyl]-3-cephem-4-
carboxylate hydrochloride
Using the compound obtained in Example 3, a
reaction was allowed to proceed in the same procedure
as that in Example 2 to afford the above-titled
compound as a mixture of two diastereomers tabout 1:1).
lRtkBr)cm : 2970, 1785, 1750, 1675, 1620, 1520
lH-NMRtDMSO-d6)~: 1.45-l.90(llH,m), 2.69(3H,s), 3.74
and 3.75(2H, each ABq, J=18Hz), 4.33 and 4.39(2H, each
ABq, J=13Hz), 4.96-5.06(lH,m), 5.78-5.88(lH,m), 6.77-
6.91(1H,m), 6.84(1H,s), 9.05t2H,brs), 9.73tlH,d,J=8Hz)
Example 5
- 2~ - 205A 126
l-(Cyclobutyloxycarbonyloxy)ethyl 7~-[2-(2-
aminothiazol-4-yl)-(Z)-2-hydroxyiminoacetamido]-3-[(2-
methyl-1,3,4-thiadiazol-5-yl)thiomethyl]-3-cephem-4-
carboxylate hydrochloride
Using l-(Cyclobutyloxycarbonyloxy)ethyliodide in
place of l-(Isopropoxycarbonyloxy)ethyliodide in
Example 1, a reaction was allowed to proceed in the
same procedure as that in Example 1 and Example 2 to
afford the above titled compound as a mixture of two
diastereomers (about 1:1).
lR(KBr)cml: 2990, 1780, 1760, 1675, 1625, 1525
lH-NMR(DMSO-d6)~: 1.5-2.4(6H,m), 1.51 and 1.53(3H, each
d, J=5Hz), 2.69(3H,s), 3.74 and 3.75(2H, each ABq,
J=18Hz), 4.33 and 4.40(2H, each ABq, J=14Hz), 4.75-
4.96(1H,m), 5.20 and 5.22(1H, each d, J=5Hz), 5.78-
5.88(1H,m), 6.75-6.91(1H,m), 6.85(1H,s), 9.72 and
9.73(1H, each d, J=8Hz)
Example 6
l-(1-ethylpropoxycarbonyloxy)ethyl 7~-[2-(2-
aminothiazol-4-yl)-(Z)-2-hydroxyiminoacetamido]-3-t(2-
methyl-1,3,4-thiadiazol-5-yl)thiomethyl]-3-cephem-4-
carboxylate hydrochloride
Using l-(l-ethylpropoxycarbonyloxy)ethyliodide in
place of 1-(Isopropoxycarbonyloxy)ethyliodide in
Example 1, a reaction was allowed to proceed in the
same procedure as that in Example 1 and Example 2 to
afford the above titled compound as a mixture of two
diastereomers (about l:l).lR(KBr)cm : 2975, 1780,
1750, 1675, 1620, 1525
lH-NMR(DMSO-d6)~: 0.78-0.89(6H,m), 1.45-1.65(7H,m),
2.69(3H,s), 3.73 and 3.75(2H, each ABq, J=18Hz), 4.14-
4.60(3H,m), 5.21 and 5.23(1H,each d,J=5Hz), 5.78-
5.88(1H,m), 6.80 and 6.88(1H, each q, J=6Hz), 6.84 and
6.85(1H,each s), 9.72 and 9.74(1H,each d, J=8Hz)
Reference Example 1
- ~. ? i a^~ . 2 0 5 4 12 6
2g ~
Uslng l-(cyclohexyloxy~arbonyloxy)ethyl~odide
ln~tead of l~ opropoxycaronylo~y)~t~yliodide, a
reAc~ion ~as ~llowed to proceed in a m~nner ~milar ~o
that o~ ~ample 1 to a~or~
(cycloh~xyloxycaxbonyloxy)ethyl 7~-~2-(2-aminothi~zol-
4-yl)-(Z)-2-hydroxylminoacetam1do]-3-~(2-methyl]-1,3,4-
thl~diazol-5-yl)thiome~hyl~ ophem-4-c~rb~xylate ~ a
mix~ur~ of two diaste~eomers (about 1s1).
lR(~sr)cm-': 3325, 2~930, 2860, 1780, 175S, 1675, 1620,
15Z~
l~-~R(DM~o-d5)~ 1.10-1.94(13~,m), 2.69(3H,~), 4.13-
4.6S(3H,m), 5.17 ~nd 5.20(1~ ch d,J-5~), S.79-
5.88(1H;m), 6.70 and 6.71 (lH,each ~), 6.81 ~nd
6.88(lH,each q,J-6Hz), 7.2-7,7(2H,br), 9.54 and
1~ 9.55(1N,ea~h d,J~8H2)