Note: Descriptions are shown in the official language in which they were submitted.
20~74~4
The invention relates to a substituted pyridyl-dihydroxy-
heptenoic acid, its salts, a process for its preparation,
and its use in medicaments.
It has been disclosed that lactone derivatives isolated
from fungal cultures are inhibitors of 3-hydroxy-3-
methyl-glutaryl coenzyme A reductase (HMG-CoA reductase)
[mevinolin, EP 22,478; US-4,231,938].
It is additionally known that pyridine-substituted
dihydroxyheptenoic acids are inhibitors of HMG-CoA
reductase tEP 325,130; EP 307,342; EP 306,929].
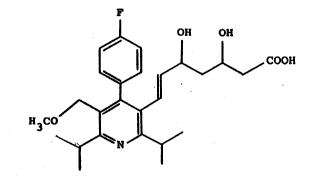
It has now been found that the substituted pyridyl-
dihydroxy-heptenoic acid of the formula
F
1 OH OH
~ ~ COOH
H3CO
~ N ~
and it~ salts, if desired in an isomeric form, have a
superior inhibitory action on HMG-CoA reductase and thus
bring about a surprisingly good lowering of the
cholesterol content in the blood.
Le A 28 042 - 1 -
2057~4~
The substituted pyridyl-dihydroxy-heptenoic acid accord-
ing to the invention can be present in the form of its
salts. In general, salts with organic or inorganic bases
may be mentioned here.
In the context of the present invention, physiologically
acceptable salts are preferred. Physiologically accept-
able salts of the substituted pyridyl-dihydroxy-heptenoic
acid according to the invention can be metal or ammonium
salts. Preferred salts which may be mentioned are sodium,
potassium, magnesium or calcium salts and also ammonium
salts which are derived from ammonia or organic amines,
such as, for example, methylamine, ethylamine, propyl-
amine, isopropylamine, di- or triethylamine, diisopropyl-
amine, di- or triethanolamine, dicyclohexylamine, argin-
ine, lysine or ethylenediamine. Sodium and potassiumsalts are particularly preferred.
The substituted pyridyl-dihydroxy-heptenoic acid accord-
ing to the invention and its salts have two asymmetric
carbon atoms, namely the two carbon atoms to which the
hydroxyl groups are bonded, and can therefore exist in
various stereochemical forms. The invention relates both
to the individual isomers and to their mixtures. Thus,
the substances sccording to the invention can be present,
depen~;ng on the relative position of the hydroxyl
y~OU~~~ in the erythro configuration or in the threo
configuration:
Le A 28 042 - 2 -
20~744~
1 OH OH
~ ~ COOH
H3CO
~ N
Erythro form
F
1 OH OH
~ ~ COOH
H3CO
~ N
Threo form
The erythro configuration is preferred.
Two enantiomers each exist in turn both of the substanceQ
in the threo and in the erythro configuration, namely of
the 3R,5S-isomer or the 3S,5R-isomer (erythro form) and
of the 3R,5R-isomer and the 3S,5S-isomer (threo form). Of
these, the 3R,5S/3S,5R racemates and the 3R,SS enantio-
mers are preferred.
The substances according to the invention can moreover be
Le A 28 042 - 3 -
- 205744~
pre~ent in the E configuration or the Z configuration
owing to the double bond. Those compounds which have the
E eonfiguration are preferred.
The (~)-enantiomers of the substituted pyridyl-dihydroxy-
heptenoic acid in the erythro (E) configuration and its
salts are particularly preferred.
The substituted pyridyl-dihydroxy-heptenoic acid of the
formula (I)
F
1 OH OH
~ ~ COOH
H3CO ~ (I)
~ N ~
and its salt~, if desired in an i~omeric form,
are prepared by
[A] in the case of the racemic products, hydrolysing the
corre~po~;ng racemic ester~ of the formula (II)
1 OH OH
~ I ~ COORl (II)
H3CO
~ N ~
Le A 28 042 - 4 -
20 57 444
ln which
R - represents Cl-C4-alkyl or benzyl,
or
[B] ln the case of the stereoisomerically homogeneous
products sub~ecting a pure diastereomeric amide of the formula
~IV)
~'' ~ NH~3R3
av)
to hydrolysis to glve the enantiomerlcally pure product.
A pure dlasteromeric amlde of the formula (IV) can
be obtained by first converting a racemic ester of the formula
(II) into the corresponding amide and then separating the
mixture of diastereomeric amides into the lndlvldual
stereoisomers. The corresponding amide can be obtained
using the (+)- or (-)-enantlomerlc amlne of the formula (III)
R3 ~,R2
2 ~
23189-7298(S)
_ 2~ 57 44~
in whlch
R2- represents Cl-C4-alkyl which ls optionally substituted by
hydroxyl
and
R3- represents hydrogen, halogen, Cl-C4-alkyl or Cl-C4-
alkoxy. Separatlon of the mixture of diastereomerlc amldes
into the individual stereoisomers can be done by
chromatography or crystallization.
If required, an obtained compound of formula (I) can
be converted lnto a salt thereof or a salt of a compound of
formula tI) can be converted lnto the free acid.
The process ls lntended to be illustrated by way of
example ln the following scheme:
23189-7298(S)
- _ 20S74~
1 OH OH
~ I~/cooc~3
H3CO ~
N ~ Racemate
l)R-(+)-PhenylethYlamine
2)chromatographicseparation
OH OH ~ CH3 OH OH ~ CH3
H3CO ~ ~ ~
oisolllcrA DiastereoisomerB
Hydrolysis
~ ~COO~Na~ ~ OH , COO-N~
H3CO~ H3CO~
(+)-En~domcr (-)-Enantiomer
Le A 28 042 - 7 -
205744~
The hydrolysis of the esters (II) is in general carried
out by treating the esters with bases in inert solvents,
the salts in general being formed initially and then
being converted into the free acid (I) in a second step
by treating with acid.
Suitable solvents for the hydrolysis of the esters are
water or the organic solvents customary for hydrolysis of
esters. These preferably include alcohols ~uch as metha-
nol, ethanol, propanol, isopropanol or butanol, or ethers
such as tetrahydrofuran or dioxane, or dimethylformamide
or dimethyl sulphoxide. Alcohols such as methanol,
ethanol, propanol or isopropanol are particularly
preferably used. It is also possible to employ mixtures
of the solvents mentioned.
Suitable bases for the hydrolysis of the esters are the
customary inorganic bases. These preferably include
alkali metal hydroxides or alkaline earth metal hydrox-
ides such as, for example, sodium hydroxide, potassium
hydroxide or barium hydroxide, or alkali metal carbonates
such as sodium carbonate or potassium carbonate or sodium
hydrogen carbonate, or alkali metal ~lkoxi~s such as
sodium ethQY;~P~ sodium methQYi~e~ potassium methoxide,
potassium ethox;~e or potassium tert.-butoxide. Sodium
hydroxide or potassium hydroxide are particularly prefer-
ably empLoyed.
Hydrolysis of the esters is in general carried out in atemperature range from -10~C to l100~C, preferably from
Le A 28 042 - 8 -
205744~
+20~C to +80~C.
~ydrolysis of the esters is in general carried out at
normal pressure. However, it is also possible to work at
reduced pressure or at elevated pressure (for example
from 0.5 to 5 bar).
When carrying out the hydrolysis, the base is in general
employed in an amount of 1 to 3 mol, preferably of 1 to
5 mol, relative to 1 mol of the ester. Molar amounts of
reactants are particularly preferably used.
When carrying out the hydrolysis, the salts of the acid
according to the invention are formed in the first step
and can be isolated. The acid according to the invention
i~ obtained by treating the salts with customary in-
organic acids. These preferably include mineral acids
~uch as, for example, hydrochloric acid, hydrobromic
acid, sulphuric acid or phosphoric acid. It ha~ proven
advantageous in this case in the preparation of the
carboxylic acid to acidify the basic reaction mixture
from the hydrolysis in a second step without isolation of
the salts. The acid can then be isolated in a customary
manner.
The reaction of the esters (II) with the enantiomerically
pure a~; ne~ (III) to give the diastereomeric amides (IV)
is in general carried out in inert solvents.
Suitable solvents for this purpose are the organic
Le A 28 042 - 9 -
2057444
solvents customary for amidations. These preferably
include ethers such as diethyl ether, dioxane or tetra-
hydrofuran, or chlorinated hydrocarbons such as methylene
chloride or chloroform, or dimethylformamide. However,
the corresponding amine ~III) is particularly preferably
employed in excess, if desired with tetrahydrofuran or
dioxane as solvent.
The reaction is in general carried out in a temperature
range from 0~C to 100~C, preferably from +20~C to +80~C.
The reaction is in general carried out at normal pres-
sure, but it i8 also possible to work at reduced pressure
or elevated pressure.
It has proved advantageous in the reaction either to
employ the amine directly as the solvent in a very large
excess, or else when using a further solvent to work in
an excess of up to 10-fold.
The hydrolysis of the diastereomeric amides (IV) is
carried out by customary methods, for example by treating
the amides with bases or acids in inert solvents.
Suitable inert solvents for this purpose are water and/or
organic solvents. Organic solvents which may be prefer-
ably men~ioned are alcohols such as methanol, ethanol,
propanol or isopropanol, or ethers such as diethyl ether,
dioxane or tetrahydrofuran. Water and water/alcohol
mixtures are particularly preferred.
Le A 28 042 - 10 -
20S7~44
Suitable acids for the hydrolysis of the amides are the
customary inorganic or organic acids. Hydrochloric acid,
hydrobromic acid, sulphuric acid and methanesulphonic
acid or toluenesulphonic acid are preferably used here.
Suitable bases for the hydrolysis of the amides are the
customary inorganic bases such as sodium hydroxide or
potassium hydroxide or sodium methoxide or ethoxide or
potassium methoxide or ethoxide or sodium carbonate or
potassium carbonate.
In the case of the phenethylamides, the hydrolysis of the
amides is preferably carried out in ethanolic hydro-
chloric acid and in the case of the phenylglycinolamides
with sodium hydroxide solution, if desired in the pre-
sence of alcohol.
Hydrolysis of the diastereomeric amides (IV) is in
general carried out in a temperature range from 0~C to
150~C, preferably from +20~C to +100~C.
Hydroly~is of the amides is in general carried out at
normal pressure, but can also be carried out at elevated
or reduced pre~sure.
It i~ moreover al~o possible to prepare the enantiomeri-
cally pure salts of the formula (I) by separating the
corre~po~ g racemates by customary methods of chromato-
graphy.
Le A 28 042 - 11 -
- - - 2057~
The ~ines (III) employed as starting substances are
known or can be prepared by methods known per se. Prefer-
ably, amines of the formula (III) according to the
invention are employed in which R3 represents hydrogen
and R2 represents methyl or hydroxymethyl.
The diastereomeric amides (IV) are new. They are useful
intermediates for the preparation of the enantiomerically
pure substituted pyridyl-dihydroxy-heptenoic acid and its
salts.
The substituted pyridyl-dihydroxy-heptenoic acid
according to the invention, its salts and isomeric forms
have useful pharmacological properties which are superior
compared with the prior art, in particular they are
highly active inhibitors of 3-hydroxy-3-methyl-glutaryl
coenzyme A ( HMG- CoA ) reductase and as a result thereof
inhibitors of cholesterol biosynthesis. They can there-
fore be employed for the treatment of hyperlipoprotein-
aemia or arteriosclerosis. The active compounds according
to the invention additionally bring about a lowering of
the cholesterol content in the blood.
The pharmacological action of the substances according to
the invention were determined in the following tests:
A ) The enzyme activity determination was carried out, in
modified form, according to G.C. Ness et al., Archives
of Biochemistry and Biophysic~ 197, 4g3-499 (1979). Male
Rico rats (body weight 300 to 400 g) were treated for 11
Le A 28 042 - 12 -
- 205~4~
days with altromin powdered feed, to which 40 g of
cholestyramine/kg of feed had been added. After
decapitation, the liver was removed from the animals and
placed on ice. The livers were comminuted and homogenised
3 times in a Potter-Elvejem homogeniser in 3 volumes of
0.1 M sucrose, 0.05 M RCl, 0.04 M K~ phosphate (mixture
of R2HPO4 and RH2PO4 of pH 7.2), O.03 M
ethylenP~i~minetetraacetic acid, 0.002 M dithiothreitol
(SPE) buffer (sucrose-phosphate-ethyle~e~i~minetetra-
acetic acid buffer) pH 7.2. The homogenisate was thencentrifuged for 15 minutes and the sediment was
discarded. The supernatant was sedimented at 100,000 g
for 75 minutes. The pellet is taken up in 1/4 volume of
SPE buffer, homogenised again and then centrifuged again
for 60 minutes. The pellet is taken up with a 5-fold
amount of its volume of SPE buffer, homogenised and
frozen and stored at -78~C (enzyme solution).
For testing, the test compounds (or mevinolin as refer-
ence substance) were dissolved in dimethylformamide with
the addition of 5 vol-% of 1 N NaOH and employed in
various concentrations in the enzyme test using 10 ~1.
The test was started after preincubation of the compounds
with the enzyme at 37~C for 20 minutes. The test batch
was 0.380 ml and contAineA 4 ~mol of glucose-6-phosphate,
1.1 mg of bovine serum albumin, 2.1 ~mol of dithio-
threitol, 0.35 ~mol of NADP (~-nicotinamide a~nine
dinucleotide phosphate), 1 unit of glucose-6-phosphate
dehydkog~nase, 35 ~mol of R~ phosphate pH 7.2, 20 ~1 of
enzyme preparation and 56 nmol of 3-hydroxy-3-methyl-
Le A 28 042 - 13 -
20574~4
- ~ glutaryl coenzyme A (glutaryl-3-1~C) 100,000 dpm.
The mixture was incubated at 37~C for 60 minutes and the
reaction was stopped by addition of 300 ~1 of 0.25 N HCl.
After a post-incubation of 60 minutes at 37~C, the batch
was centrifuged and 600 ~1 of the supernatant was applied
to a 0.7 x 4 cm column packed with 5-chloride anion
exchanger having a part~cle size of 100 to 200 mesh. The
column was washed with 2 ml of dist. water and runnings
plus washing water were treated with 3 ml of a scintilla-
tion fluid and counted in a scintillation counter. IC50
values were determined by intrapolation by plotting the
percentage inhibition against the concentration of the
compound in the test. To determine the relative inhibi-
tory potency, the ICSo value of the reference substance
mevinolin was set at 100 and compared with the simul-
taneously determined IC50 value of the test compound.
B) The subchronic action of the compounds accor~illg to ~e invention on the blood
cholesterol values of dogs was tested in feeding e~ nt~ of several weeks
duration. For this, the subst~r~re to be investigated was given p.o. once daily in a
2 0 capsule to healthy beagle dogs together with the feed over a period of time lasting
several weeks. Cole ly~ e (4 g/100 g of feed) as the gallic acid sequestrant wasadditionally ;9dmix~-~ in the feed during the en~re e~Jelilllental period, i.e. before,
during and after the a~mini~tration period of the substances to be investigated.Venous blood was taken ~om the dogs twice weekly and the serum cholesterol was
2 5 de~e~ ed enzymatically using a co"~ e,.;ial test kit. The serum cholesterol values
duIing ~e ~ ion period were col~pa,~d wi~ ~e serum cholesterol values
before the aAl~ ;cllalion peIiod (con~ols).
The present invention al~o includes pharmaceutical
preparations which contain one or more compounds of the
general formul~ (I) in addition to inert, non-toxic,
pharmaceutically suit~ble auxiliaries and excipients or
which consist of one or more active compounds of the
formula (I), and ~ oces~es for the production of these
preparations.
The active compounds of the formula (I) ffhould be pre~ent
in these preparation~ in a concentration of 0.1 to 99.5%
by weight, preferably from O.S to 95% by weight of the
total mixture.
Le A 28 042 - 14 -
2057~4~
~ 23189-7298
In addition to the active compounds of the formula (I),
the pharmaceutical preparations can also contain other pharma-
ceutical active compounds.
The above-mentioned pharmaceutical preparations can be
prepared in a customary manner by known methods, for example
using the auxiliary(ies) or excipient(s).
The invention also extends to commercial packages
containing a compound of the invention, together with
instructions for its use for treatment of hyperlipoproteinaemia
or arteriosclerosis and for lowering the cholesterol content of
the blood stream.
In general, it has proved advantageous to administer
the active compound(s) of the formula (I) in total amounts from
about 0.1 ~g/kg to about 100 ~g/kg, preferably in total amounts
from about 1 ~g/kg to 50 ~g/kg of body weight every 24 hours,
if appropriate in the form of several individual doses, to
achieve the desired result.
However, it may be advantageous to deviate from the
amounts mentioned, in particular depending on the species and
the body weight of the subject treated, on individual behaviour
towards the medicament, the nature and severity of the disease,
the type of preparation and administration, and the time or
interval at which administration takes place.
Exemplary embodiments
Example 1
Sodium 3R,5S-(+)-erythro-(E)-7-[4-(4-fluorophenyl)-2,6-
diisopropyl-5-methoxymethyl-pyrid-3-yl]-3,5-dihydroxy-hept-6-
enoate
2057~4~
1 OH OH
~ ~ COO~Na+
H3CO
~ N
and
Example 2
Sodium 3S,5R-(-)-erythro-(E)-7-[4-(4-fluorophenyl)-2,6-
diisopropyl-5-methoxymethyl-pyrid-3-yl]-3,5-dihydroxy-
hept-6-enoate
1 OH OH
~ COO~Na+
H3CO
~ N ~
Process variant A - Racemate ~eparation using
R-(+)-phenylethylamine
~0 a) Preparation and ~eparation of the diastereomeric
phenethylAmide~
Le A 28 042 - 16 -
~_ 20~7~
OH OH ~ CH3
H3CO ~V~NH~3
N
Diastereomer Al
F
H3CO ~
Diastereomer B1
4.7 g (10 mmol) of methyl erythro-(E)-4-(4-fluoro-
phenyl)-2,6-diisopropyl-5-methoxymethylpyrid-3-yl]-
3,5-dihydroxy-hept-6-enoate are dissolved in 20 ml
of R-(+)-phenethylamine and heated at 40~C for 72
h. The reaction solution is poured into 150 ml of
water and the solution is adjusted to pH 4 with 1 N
hydrochloric acid. It is then extracted several
times with ether. The combined organic extracts are
washed with saturated sodium chloride solution,
dried over magnesium sulphate and concentrated.
After prepurification on silica gel 63-200 ~ (eluent
Le A 28 042 - 17 -
2Q57~4~
ethyl acetate/petroleum ether 4:6 ~ 6:4), the
residue is separated on a 15 ~ pre-packed column
(eluent ethyl acetate/petroleum ether l:l).
Yield: 2.1 g of diastereomer Al (37.4% of theory),
1.5 g of diastereomer Bl (26.6% of theory).
b) Preparation of the enantiomerically pure sodium
salts (Ex. 1/2)
2.1 g (3.7 mmol) of the diastereomer A1 are dis-
solved in 70 ml of 15% strength ethanol and, after
addition of 13 ml of 1 N hydrochloric acid, heated
under reflux for 48 h. After cooling, the super-
natant solution i8 filtered off and the residue is
stirred several times with ethanol. The combined
ethanol solutions are concentrated and the residue
is taken up in 50 ml of water and 50 ml of dichloro-
methane. The pH of the solution is adjusted to 3.5
using 1 N hydrochloric acid and the solution is then
extracted several times with dichloromethane. The
combined organic solutions are dried over sodium
sulphate and concentrated. The residue is taken up
in 50 ml of tetrahydrofuran/water 1:1 and the pH of
the solution is adjusted to 7.5 using 1 N sodium
hydroxide solution. The tetrahydrofuran is evapora-
ted on a rotary evaporator and the remaining aqueous
solution is lyophilised. The crude lyoph~ ate is
purified on RP 18 (eluent: acetonitrile/water
30:70). After freeze-drying of the product frac-
tions, 850 mg (48% of theory) of the
Le A 28 042 - 18 -
20~7~4~
(+)-enantiomeric sodium salt (Ex. 1) are obt~ine~.
lH-NMR (DMSO-d6): ~ (ppm) = 1.0 (m, lHJ; 1.23 (d,
6H); 1.28 (d, 6H); 1.3 (m, lH); 1.75 (dd, lH); 1.98
(dd, lH); 3.07 (8, 3H); 3.2-3.4 (m, 3H); 3.52 (m,
lH); 4.02 (m, 2H); 5.28 (dd, lH); 6.17 (d, lH); 7.1-
7.3 (m, 4H)-
Specific rotation (EtOH): t~]DO = 24.1 (c=1.0).
800 mg (61.5~ of theory) of the (-)-enantiomeric
sodium salt (Ex. 2) are obt~ine~ as described above
from 1.5 g (2.6 mmol) of the diastereomer Bl.
Specific rotation (EtOH): [~]D = 23.2 (c=1.0).
Process variant B - Racemate separation using S-(+)-
phenylglycinol
a) Preparation of the diastereomeric phenylglycinol-
amides
H3CO ~
Diastereomer A2
Le A 28 042 - 19 -
2057~
H3C0 ~ ~ ~ ~ ~
N
Diastereomer B2
418 g (0.88 mol) of methyl erythro-(E)-7-[4-(4-
fluorophenyl)-2,6-diisopropyl-5-methoxymethyl-pyrid-
3-yl]-3,5-dihydroxy-hept-6-enoate and 360 g
(2.6 mol) of S-(+)-phenylglycinol are dissolved in
1 1 of absol. tetrahydrofuran and the mixture is
heated to 50~C for 96 h. After cooling to room
temperature, 1 1 of water is added, and the solution
is adjusted to pH 4 using 5 N hydrochloric acid and
extracted 3 times using 400 ml of ether each time.
The combined organic phases are washed with 400 ml
of saturated sodium chloride solution, dried over
sodium sulphate and concentrated on a rotary evapo-
rator. The residue (500 g of crude product) is
preseparated (eluent ethyl acetate/petroleum ether
8:2) into two portions on a column (in each case
about 1.8 kg of silica gel). 350 g of prepurified
crude product are thus obtA;ne~, which consists
almost exclusively of the two diastereoi~omeric
amides. The prepurified crude product is separated
into 7 x 50 g portions on a silica gel column (Buchi
Le A 28 042 - 20 -
- - 20~7~
column, length 63 cm, 0 7 cm, silica gel 20 ~,
sample application via a 100 ml sample loop).
Yield: 195 g (38.2% of theory) of the diastereomer
A2. The diastereomer B2 was not isolated pure, but
was recovered as a crude product for possible later
use on washing the columns.
b) Preparation of the enantiomerically pure sodium
salts (Ex. 1/2)
195 g (0.34 mol) of the diastereomerically pure
amide A2 are dissolved in 1 1 of ethanol p.A. and,
after addition of 1.2 1 of 1 N sodium hydroxide
solution, the mixture is heated overnight under
reflux. After cooling to room temperature, the
supernatant solution is decanted off and the oily
residue is stirred 3 times using 50 ml of ethanol
p.A. each time. The solutions are combined and
concentrated. The residue is taken up in 500 ml of
water and 500 ml of methylene chloride and the
- solution is adjusted to pH 3.5 using 1 N hydro-
chloric acid. The organic phase is then separated
off and the aqueous phase is extracted 3 times using
400 ml of methylene chloride each time. The combined
organic phases are dried (Na2S04) and concentrated.
The residue is dissolved in 100 ml of tetrahydro-
furan and the solution is diluted with 500 ml of
water. It is then adjusted to pH 7.5 using 1 N
sodium hydroxide solution, the tetrahydrofuran is
removed on a rotary evaporator and the aqueous
Le A 28 042 - 21 -
20574~4
.
solution which remains is lyophilised.
142 g of crude lyophilisate are obtained which, for
desalting, are further purified and desalted in
27 x 5 g portions and 2 x 3.5 g portions on an RP 18
col D (length 40 cm, 0 3 cm, silica gel RP 18, 30
~, eluent acetonitrile/water 30:70). All product
fractions are combined, the acetonitrile is removed
on a rotary evaporator and the aqueous residue is
lyophilised.
Yield: 102 g (62.5% of theory) of the (+)-enantio-
meric sodium salt (Ex. 1).
Le A 28 042 - 22 -