Note: Descriptions are shown in the official language in which they were submitted.
Side-Chain Homologous Vitamin D Derivatives, Process for their
Production, Pharmaceutical Preparations Containing these
Derivatives and their Use as Pharmaceutical Agents
This invention relates to side-chain homologous vitamin D
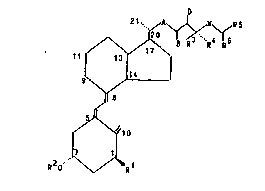
derivatives of formula I
,~ Y~
9`~ Il),
~.9
~o
2 ~ 1
in which . -
Rl means a hydrogen atom, a hydroxy or an acyloxy group with
1 to 9 carbon atoms,
R2 means a hydrogen atom or an acyl group with 1 to 9 carbon
atoms,
R3 or R4 means a hydroxy or acyloxy group with 1 to 9 carbon
atoms, and the respective other substituent is a hydrogen atom or
R3 and R4 together mean an oxygen atom,
R5 and R6, independently of one another, each mean a linear
or branched alkyl radical with up to 5 carbon atoms, a
trifluoromethyl group or together a saturated, unsaturated or
aromatic carbocyclic 3-, 4-, 5- or 6-member ring formed with the
tertiary carbon atom or with the inclusion of 1 or 2 N, O or S
atoms a heterocyclic 3, 4, 5 or 6-member ring,
B and D either mean a hydrogen atom each or together a
second bond (E-configured double bond) and
either --
A means a direct bond between carbon atoms 20 and 22 and
X means an alkylene oxy radical -(CH2)nO- with n = 1 to 3
or
A means a methylene bridge (-CH2-) between carbon atoms 20
and 22 and
X means an alkylene radical -(CH2)n- or an alkylene oxy
adical -(CH2)nO- with n = 1 to 3, or if A stands for a direct
~5
bond and B and D together stand for a second bond ~ 6
means one of radicals -CH2-0-CH2- ~ , -(CH2)2- or -(CH2)2-
as well as a process for their production, pharmaceutical
preparations that contain these compounds and their use for the
production of pharmaceutical agents.
The acyloxy or acyl groups possible for radical~ Rl, R2 and
within radicals R3 or R4 are derived in particular from saturated
carboxylic acids or also from benzoic acid. Other suitable acyl
radicals in Rl, R2, R3, R4 comprise those which are cyclic,
acyclic, carboxyclic or heterocyclic -- all optionally also
unsaturated. The preferred radicals are derived from Cl- to Cg-,
preferably C2- to C5-, alkanecarboxyclic acids, such as, for
example, acetyl, propionyl, butyryl.
If R5 and R6 form, together with the tertiary carbon atom, a
saturated carboxylic ring, then the cyclopropyl or cyclohexyl
ring is especially referred to. As alkyl groups for R5 and R6,
those with 1 to 5 carbon atoms, which can be straight-chain or
branched, are especially suitable. By way of example, there can
be mentioned the methyl, ethyl, propyl and t-butyl group.
Preferred according to this invention are side-chain
homologous vitamin D derivatives of general formula I, in which
Rl, R3 or R4 stands for a hydroxy group or
R5 and R6 stand for a methyl group or, together with the
tertiary carbon atom, for a cyclopropyl ring,
R2 stands for a hydrogen atom
and n is 1 or 2.
Between carbons atoms 22 and 23 (when A means a direct bond) :
or between carbon atoms 23 and 24 (when A means a methylene
group), there is preferably a double bond. Especially preferred
are the compounds
24-(l(R)-Hydroxy-4-methylpentyl)-9,10-secochola-5Z,
7E,10(19),23E-tetraene-l(S),3(R)-diol,
24-(1(S)-hydroxy-4-methylpentyl)-9,10-secochola-5Z,
7E,10(19),23E-tetraene-l(S),3(R)-diol,
24-(l(R)-hydroxy-3-methylbutyl)-9,10-secochola-
SZ,7E,10(19),23E-tetraene-l(S),3(R3-diol,
24-(l(S)-hydroxy-3-methylbutyl)-9,10-secochola-
SZ,7E,10(19),23E-tetraene-l(S),3(R)-diol,
24-(l(R)-hydroxy-3-methylbutyl)-9,10-secochola-5Z,7E,10(19)-
tri.ene-l(S),3(R)-diol,
24-(l(S)-hydroxy-3-methylbutyl)-9,10-secochola-5Z,7E,10(19)-
triene-l(S),3(R)-diol,
24-(l(R)-hydroxy-3-isopropoxypropyl)-9,10-secochola-
SZ,7E,10(19),23E-tetraene-l(S),3(R)-diol,
24-(l(S)-hydroxy-3-isopropoxypropyl)-9,10-secochola-
5Z,7E,10(19),23E-tetraene-l(S),3(R)-diol,
24-isopropoxymethyl-9,10-secochola-5Z,7E,10(19),22E-
tetraene-l(S),3(R),24(R)-triol,
24-isopropoxymethyl-9,10-secochola-SZ,7E,10(19),22E-
tetraene-l(S),3(R),24(S)-triol,
24-(2-isopropoxyethyl)-9,10-secochola-5Z,7E,10(19),22E-
tetraene-l(S),3(R),24(R)-triol,
24-(2-isopropoxyethyl)-9,10-secochola-5Z,7E,10(19),22E-
tetraene-l(S),3(R),24(S)-triol,
26,27-cyclo-24a,24b-dihomo-9,10-secocholesta-
5Z,7E,10(19),23E-tetraene-l(S),3tR),24a(R~-triol,
26,27-cyclo-24a,24b-dihomo-9,10-secocholesta-
5Z,7E,10(19),23E-tetraene-l(S),3(R),24a(S)-triol.
Natural vitamins D2 and ~3 (cf. general formula V) are
biologically inactive in and of themselves and are converted only
after hydroxylation in 2-position in the liver or in l-position
in the kidneys into their biologically active metabolites. The
effect of vitamins D2 and ~3 consists in stabilizing the plasma
Ca~ level and the plasma phosphate level; they counteract a
decrease in the plasma Ca~+ level.
r~'
,~ H
p ~v
H~)~ ~R~
ergocalciferol: Ra = Rb = H,~c = CH3, vitamin-D2
double bond C-22/23
cholecalciferol: Ra z Rb = Rc = H vitamin D3
25-hydroxycholecalciferol: Ra = Rc = H, Rb = OH
lalpha-hydroxycholecalciferol: Ra = OH, Rb = Rc = H
lalpha,25-dihydroxycholecalciferol:Ra = Rb = OH, Rc = H calcitriol
Besides their pronounced effect on the calcium and phosphate
metabolism, vitamins D2 and D3 and their synthetic derivatives
also have proliferation-inhibiting and cell-differentiating
effects (H.F. De Luca, The Metabolism and Function of vitamin D
in Biochemistry of Steroid Hormones, publisher H.L.J. Makin, 2nd
Edition, Blackwell Scientific Publications 1984, pages 71-116).
But overdose phenomena can occur when using vitamin D
(hypercalcemia).
lalpha-Cholecalciferols hydroxylated in 24-position already
follow from DE-A~-25 26 981; they have a lower toxicity than the
corresponding nonhydroxylated lalpha-cholecalciferol. The
hydroxylated compounds show a selective activation of the
intestinal calcium absorption and a weaker bone absorption effect
than lalpha-cholecalciferol. The 24-hydroxy vitamin D analogs
described in international patent application W0 87/00834 can be
used for treating disorders caused by abnormal cell proliferation
and/or cell differentiation in humans and animals.
For various 1,25-dihydroxy-homo vitamin D derivatives, a
dissociation with respect to the properties of the bone
absorption effect and HL 60 cell differentiation has already been
briefly mentioned by De Luca. The in vitro bone absorption
effect here is a direct measurement for the in vivo calcium
mobilization.
It has now been found that the side-chain homologous vitamin
D derivatives of general formula I according to the invention
surprisingly exhibit a more favorable spectrum of action compared
with the vitamin D derivative calcitriol (lalpha,25-
dihydroxycholecalciferol). While the effects on the calcium and
phosphate metabolism are clearly weakened (decrease of the side
effects from overdosing or necessary higher dosage), the
proliferation-inhibiting and cell-differentiating effects are
approximately maintained (dissociation).
The vitamin D activity of the compounds according to the
invention is determined by the calcitriol receptor test. It is
performed by using a specific receptor protein from the intestine
of rachitic chickens.
A binding protein containing a receptor is incubated with
3H-calcitriol (0.5 ng/ml) in a reaction volume of 0.575 ml in the
absence and in the presence of the test substances for one hour
in a test tube. To separate the free calcitriol from the
calcitriol bound to the receptor, a charcoal-dextran absorption
is performed. For this purpose, 200 microliters of a charcoal-
dextran 6uspension is fed to each test tube and incubated for 30
minutes at 22C. Then, the samples are centrifuged at 1,500 x g
for 10 minutes at 4C. The supernatant fluid is decanted and
measured in a beta-counter after about 1 hour of equilibration in --
atom light. --
The competition curves obtained with various concentrationsof the te~t ~ubstance and of the reference substance (unlabeled
calcitriol) with a constant concentration of the standard
substance (3H-calcitriol) are placed in relation to one another
and a competition factor (KF) is determined.
It is defined as the quotient of the concentrations of the
respective test substance and of the reference substance that are
necessary for 50% competition:
concentration of the test substance at 50% competition
KF =
concentration of the reference substance at 50% competition
According to this,
24-(1-hydroxy-3-methylbutyl)-9,10-secochola-
5Z,7E,10(19),23E-tetraene-l(S),3(R)-diol (compound A) has a KF
value of 2 .0 and
24-(1-hydroxy-3-methylbutyl)-9,10-secochola-
SZ,7E,10(19),23E-tetraene-l(S),3(S)-diol (compound ~) has a KF-
value of 3.6.
To determine the antiproliferative power of the compounds
according to the invention, the test described below is performed
instead with compounds A and B as test substances: -
Keratinocytes of newborn mice are prepared and cultivated in
a modification of the method of Yuspa, S. and Harris, C.C.,
"Altered differentiation of mouse epidermal cells treated with
retinyl acetate in vitro," Exp. Cell Res. 86: 95-105, 1974.
Neonatal NMRI mice of both sexes are killed by decapitation,
the ~kin is removed, washed in an antibiotic-antimycotic solution
and, with the dermal side facing down, incubated overnight at 4C
in dispase II solution (1.2 U/ml in tissue culture medium Ml99
+25 mmol/l HEPES + 15% fetal calf serum (~CS) + 50 U/ml of
penicillin/streptomycin (P/S) (preparation medium, PM) The
epidermis is removed and a single-cell suspension is produced by
trypsinization. After centrifuging, the cell sediment is
resuspended, the number of living, small, round cells is
~etermined after trypan blue staining and the cells are sown in a
density of 4x105 cells/cm2 in Primaria 24-hole plates in tissue
culture medium (M199 + 15% FCS + 50 U/ml of P/S). After 24 hours
of incubation at 37C, the cells are washed with phosphate-
buffered saline solution (PBS) and incubated for another 24 hours
in serum-free tissue culture medium (Ml99 ~ 50 U/ml of P/S + 0.5%
ethanol) with and without test substances at 32.5C. Then, 0.4
microcuries/50 microliters of 3H-methylthymidine (40 Ci/mmol) is
added. After 4 hours, the medium is suctioned off and the
reaction is ended by adding 500 microliters of ice-cold 10%
L~
trichloroacetic acid (TCA). The cells are washed with TCA and
PBS, lysed by incubation in a proteinase K-solution (10 mmol/l of
tris-HCl, 10 mmol/l of EDTA, 10 mmol/l of NaCl, 0.2% triton-X
100, pH 8.0, 50 micrograms/ml of protein kinase K) and the lysate
is clarified by centrifuging. In the supernatant fluid, the
radioactivity is determined by scintillation photometry and,
after specific staining of the DNA with diamidinophenylindole
(DAPI), the DNA concentration is determined by fluorescence
photometry. -
Accordingly depending on the dose, calcitriol and compoundsA and B inhibit the 3H-thymidine incorporation in DNA with the
following IC50 values:
calcitriol 2 x 10-9mol/l
compound A 1 x 10 8mol/l
compound B 3.2 x 10 9mol/l
The effects of calcitriol and of the compounds according to
the invention that stimulate the differentiation
26,27-cyclo-24a,24b-dihomo-9,10-secocholesta-
5Z,7E,10(19~,23E-tetraene-l(S),3(R),24a(R)-triol (compound C) and :
26,27-cyclo-24a,24b-dihomo-s,lo-secocholesta-
5Z,7E,10(19),23E-tetraene-l(S),3(R),24a(S)-triol (compound D)
practically do not differ.
It is known from the literature (Mangelsdorf, D.J. et al, J.
Cell. Biol. 98: 391-398 (1984)) that the in vitro treatment of
human leukemia cells (promyelocytic cell line HL 60) with
calcitriol induces the differentiation of the cells into
macrophages~
To quantify the differentiation-stimulating effect of
calcitriol analogs, the test indicated below was performed:
NL 60 cells are cultivated in tissue culture medium (RPMl -
10% fetal calf serum) at 37C in an atmosphere of 5% C02 in air.
To test the substance, the cells are separated by
centrifuging and 2.8 x 105 cells/ml are taken up in phenol red-
free tissue culture medium. The test substances are-dissolved in
ethanol and diluted with tissue culture medium without phenol red
to the desired concentration. The dilution stages are mixed with
the cell suspension in a ratio of 1:10 and 100 microliters each
of this cell suspension mixed with substance is pipetted into an
indentation of a 96-hole plate. As a control, a cell suspension
is mixed analogously with the solvent.
After incubation for 96 hours at 37C in s% Co2 in air, 100
microliters of an NBT-TPA solution (nitro blue tetrazolium (NBT),
final concentration in the batch 1 mg/ml, tetradecanoyl~-phorbol
myristate-13-acetate (TPA), final concentration in the
batch 2 x 10-7 mol/l~ is pipetted into each indentation of the
96-hole plate into the cell suspension.
By incubation for 2 hours at 37C and 5% C02 in air, the NBT
is reduced to insoluble formazan because of the intracellular
oxygen radical release, stimulated by TPA, in the cells
differentiated into macrophages.
To end the reaction, the indentations of the 96-hole plate
are drained and the adhering cells are fixed by adding methanol
and dried after fixation.
To dissolve the intracellular formazan crystals formed, 100
microliters of potassium hydroxide (2 val/l) and lO0 microliters
of dimethyl sulfoxide are pipetted into each indentation and are
exposed to ultrasonic waves for l minute. The concentration of
formazan is measured by spectrophotometry at 650 nm.
The concentration of formazan formed is regarded as a
measurement for the differentiation induction of the HL 60 cells
into macrophages. The relative effectiveness of the test
substance results from the quotient of ED50 test substance/EDso
calcitriol.
According to this, calcitriol, compound C and compound D
have the ED50 values 1.8 x 10 9 mol/l, 2.2 x lo 9 mol/l or 2.5 x
10-9mol/l.
Because of the reduced risk of hypercalcemia, the substances
according to the invention are especially suited for the
production of pharmaceutical agents for treating diseases which
are characterized by a hyperproliferation, e.g.,
hyperproliferative diseases of the skin (psoriasis) and malignant
tumors (leukemia, colon cancer, breast cancer). In an especially
preferred embodiment of the invention, calcitriol receptors are
detected in the target organ before the treatment.
This invention thus also relates to pharmaceutical
preparations that contain at least one compound according to
1~
general formula I together with a pharmaceutically compatible
vehicle. The compounds can be formulated as solutions in
pharmaceutically compatible solvents or as emulsions, suspensions
or dispersions in suitable pharmaceutical solvents or vehicles or
as pills, tablets or capsules that contain solid vehicles in the
way known in the art. For a topical use, the compounds are
advantageously formulated as creams or ointments or in a similar
pharmaceutical agent form suitable for topical use. Each such
formulation can also contain other pharmaceutically compatible
and nontoxic auxiliary agents such as, e.g., stabilizers,
antioxidants, binders, dyes, emulsifiers or flavoring substances.
The compounds are advantageously administered by injection or
intravenous infusion of suitable sterile solutions or as oral
dosage through the alimentary tract or topically in the form of
creams, ointments, lotions or suitable transdermal plasters, as
described in EP-A-0387 077.
The daily dose is
0.1 microgram/patient/day - 1,000 micrograms (1
mg)/patient/day,
preferably
1.0 microgram/patient/day - 500 micrograms/patient/day.
The compounds according to the invention are generally
administered analogously to the administration of the known agent
"calcipotriol" for the treatment of psoriasis.
Further, the invention relates to the use of the compounds
according to formula I for the production of pharmaceutical
ag~nts.
The production of side-chain homologous vitamin D
derivatives of formula I is performed according to the invention
in that a compound of general formula IV
~X' Y ~
~ ~ I
~IV).
\~oa2
in which
Rl means a hydrogen atom or a protected hydroxy group and
R2 means a hydroxy protecting group and
A, X and R5 and R6 have the meaning given in formula I,
optionally after selective hydrogenation of the double bond in
the side chain, is converted into a compound of general formula
IVa
~Iv~
1~1 ~oR2
in which Rl, R2, A, X and R5 and R6 have the meaning given in
formula IV and
optionally, after reduction of the carbonyl function and
optionally after separation of the mixture of the epimeric
hydroxy compounds of general formulas IIIA and IIIB formed by the
reduction
XyR
OH R
H = ~ - ~
b~ <~OH: O-C~l
R ` I\~OR~
1~
in which
R~, R2, A, X and R5 and R6 have the meaning given in formula
IV and B and D have the meaning given in formula I,
by irradiation with ultraviolet light with reversal of the
stereoisomerism at the 5,6 double bond, is converted into a
compound of general formula II
~ ~ R5
y ~lI),
oR2~' ~ R
in which
Rl, R2, A, B, D, X and R5 and R6 have the meaning given in
formula IIIa/IIIb,
and then the latter, by the cleaving of existing hydroxy
protecting groups and optionally by partial or complete
esterification of the hydroxy groups r is converted into a
compound of general formula I.
The reduction of the side-chain carbonyl function in the
compound of general formula IV is performed for example with
cerium(III) chloride/sodium borohydride in a polar solvent. Both
the R and the S hydroxy isomer of general formula IIIa or IIIB
result during the reduction. Both isomers can be separated
chromatographically.
Optionally, before reduction o~ the carbonyl function, the
double bond in the side chain can be selectively hydrogenated.
As hydrogenation agent, lithium-tri-tert-butoxy-aluminum hydride
in a polar solvent is suitable, among others.
The following conversion of a compound of general formula
IIIa/IIIb into a compound of general formula II is performed,
e.g., by irradiation with ultraviolet light in the presence of a
so-called "triplet sensitizer." In the framework of this
invention, anthracene is used for this purpose. By cleaving the
pi-bond of the 5,6 double bond, rotating the A ring by 180
around the 5,6 single bond and reestablishing the 5,6 double
bond, the stereoisomerism at the 5,6 double bond is reversed.
Then, available hydroxy protecting groups are cleaved,
preferably by using tetra-n-butyl-ammonium fluoride and
optionally the free hydroxy groups are esterified according to
current processes partially or completely with the corresponding
carboxylic acid halide (halide = chloride, bromide) or carboxylic
acid anhydride.
1~
Production of the initial materials
1. l(S),3(R)-bis-(tert-Butyldimethylsilyloxy)-20(S)-formyl-9,10-
secopregna-5E,7E,10(19)-triene 1:
The production of ~ is performed according to M.J.
Calverley, Tetrahydron 43, 4609 (1987); see also international
patent application W0 87/00834. The production of the initial
compound in which Rl is a hydrogen atom is also described there.
2. l(S),3(R)-bis-(tert-Butyldimethylsilyloxy)-20(R)-methyl-9,10- -
secopregna-5E,7E,10(19)-triene-21-carbaldehyde 2 --
Aldehyde 2 is produced according to a new process.
a. A solution of 15.57 g of diethyl phosphonoethoxy ethyl
acetate (produced according to W. Grell and H. Machleidt, Liebigs
Ann. Chem. 699, 53 (1965) in 200 ml of THF is instilled at 25C
in a suspension of 1.8 g of sodium hydride (80~ in oil) in 70 ml
of abs. THF. After adding, it is stirred another 90 minutes at
60C, cooled again to 25C and a solution of 6.2 g of 1 in 70 ml
of THF is added drop by drop. It is stirred for 2 hours under
reflux, the cooled reaction solution is then poured into water
and extracted with ethyl acetate. After drying (Na2SO4) and
concentration by evaporation, the crude product obtained is
chromatographed on silica gel with hexane/ethyl acetate. The
main fraction yields 5.2 g of l(S),3(R)-bis-(tert-
butyldimethylsilyloxy)-23-(ethoxy-9,10-secochola-5E,7E,10(19)-
tetraene-24-acid ethyl ester as an oily mixture of the C-22
double bond isomers.
b. 5.2 g of the product obtained under a. is dissolved in
120 ml of toluene and at 0C slowly mixed with 20 ml of a 20%
solution of diisobutylaluminum hydride in toluene. After 30
minutes at 0C, the reaction solution is poured carefully into
NH4Cl solution and extracted with ethyl acetate. After the usual
working up, 4.88 g of l(S),3(R)-bis-(tert-~utyldimethylsilyloxy)-
23-ethoxy-9,10-secochola-5E,7E,10(19),22-tetraen-24-ol is
obtained as a colorless, oily isomer mixture that is used in the
next step without further purification.
c. The compound produced under b. (4.88 g) is stirred in a
mixture of 55 ml of dichloromethane and 55 ml of a 70% aqueous
acetic acid for 4 hours at room temperature. Then it is
neutralized by adding NH3 solution and extracted with
dichloromethane. The crude product is chromatographed on silica
gel with hexane/ethyl acetate. In this way, 2.02 g of l(S),3(R)-
bis-(tert-butyldimethylsilyloxy)-24-hydroxy-9,lo-secochola-
5E,7E,10,(19)-trien-23-one 5 is obtained as colorless oil.
IH-NMR (CDC13): = 0.01 ppm (s, 12H, Si-CH3), 0.52 (s, 3H,
H-18), 0.81 and 0.84 (s; 9H, Si-t-butyl each~, 0.90 (d, J = 7Hz,
3H, H-21), 3.09 (t, J=5Hz, lH, OH), 4.10 (dd, lH, H-24), 4.16 (m,
lH, H-3), 4.21 (dd, lH, H-24), 4.39 (m, lH, H-1), 4.88, 4.93 (s;
lH, H-l9 each), 5.77, 6.39 (d, J=llHz; lH, H-6, H-7 each).
d. The product obtained under c. (2.02 g) is dissolved in 25
ml of methanol and 25 ml of THF and mixed at 0C with 300 mg of
sodium borohydride. It is stirred for 1.5 hours at 0C, the
reaction mixture is then poured into NH4Cl solution and extracted
2~
with ethyl acetate. 1.75 g of l(S), 3(R)-bis-(tert-
butyldimethylsilyloxy~-9,10-secochola-SE,7E,10,(19)-triene-23,24
diol 6 is obtained as a colorless, oily mixture of the 23-epimers
that is used as such in the next reaction.
e. 1.75 g of the product obtained under d. is dissolved in
40 ml of toluene and 1.23 g of lead tetraacetate is added in
portions with ice water cooling. It is stirred for 30 ~inutes,
1.0 g of Pb(OAC)4 is again added and it is stirred for another 15
minutes at +5 to +10C.
For the working upr it is mixed with NaHCO3 solution, the
resulting suspension is filtered over cellite and the filtrate is
extracted with ethyl acetate. The crude product is
chromatographed on silica gel with hexane/ethyl acetate. After
crystallization of the main fraction from ethanol, 560 mg of
l(S~,3(R)-bis-(tert-butyldimethylsilyloxy)-20(R)-methyl-9,10-
secopregna-5E,7E,10,(19)-triene-21-carbaldehyde with a melting
point of 101-104C is obtained.
The reaction of aldehyde 1 or 2 with a phosphorane of
formula
0 ~5
~3 ~ x ~ ~t
leads to the compounds of general formula IV (Wittig reaction).
Production of the phosphorus ylides used:
1. Isobutylcarbonylmethylenetriphenyl phosphorane
a. Bromomethylisobutyl ketone
50 ml of isobutylmethyl ketone in 240 ml of methanol is
mixed at 0C with 20 ml of bromine and, after being added, is
stirred for another 1.5 hours at +10C. After this, 360 ml of
water is added and it is stirred for another 16 hours at room
temperature.
For the working up, the reaction mixture is mixed with
saturated common salt solution, the organic phase that
precipitates is separated and the aqueous phase is extracted with
ether. The combined organic phases are washed with 10% Na2C03
solution and dried on Na2S04. After filtration, the solvent is
removed in the water jet vacuum and the residue is distilled.
The main fraction contains 53.7 g of bromomethylisobutyl ketone
of bpl5 200f 67-69C.
b. Isobutylcarbonylmethyltriphenylphosphonium bromide
Bromomethylisobutyl ketone (S3.6 g) and triphenylphosphine
(78.5 g) are intimately mixed in a 500 ml round-bottom flask and,
after the initially strong heat tonality subsides, are left for
12 hours under nitrogen at room temperature. After that, the
solid reaction mass is taken up in 330 ml of methylene chloride
and refluxed for 30 minutes. After adding 500 ml of ether, it is
allowed to cool to room temperature and the product is isolated
by filtration. After drying, 111.7 g of the phosphonium salt
with a melting point of 244-245C is obtained.
c. Isobutylcarbonylmethylenetriphenyl phosphorane
~'
111.6 g of the phosphonium bromide obtained under b. is
mixed successively with 1500 ml of methylene chloride and 1500 ml
of 2N NaOH and stirred for 30 minutes at room temperature. The
organic phase is separated, washed with water and dried on
Na2S04. The solid residue obtained after concentration by
evaporation is recrystallized from tert-butyl methyl ether and
yields 72.2 g of the ylide with a melting point of 120-121C.
2. Isoamylcarbonylmethylenetriphenyl phosphorane
The formation of the title compound is performed analogously
to the process described under 1. by bromation of isoamylmethyl
ketone, reaction of the bromide with triphenylphosphine to
phosphonium salt and formation of the ylide with 2N NaOH.
After distillative purification, 54.68 g of l-bromo-S-
methyl-hexan-2-one of bpl5-20of 80-86C is obtained from 50.0 ml
of isoamylmethyl ketone and 18.2 ml of bromine.
91.6 g of the phosphonium salt with a melting point of 230-
233C is obtained from 54.58 g of the bromide and 74.14 g of
triphenylphosphine.
After treatment with NaOH and recrystallization of the crude
product from methylene chloride/ester, 69.8 g of the title
compound with a melting point of 64-67C is obtained from 91.6 g
of phosphonium salt.
3. Isopropoxymethylcarbonylmethylenetriphenyl phosphorane
2.43 g of sodium is dissolved in 150 ml of isopropanol.
After adding 20.0 g of chloromethylcarbonylmethylenetriphenyl
phosphorane ketone (R. F. Hudson et al., J. Org. Chem. 28 2446,
1963) dissolved in 200 ml of isopropanol, is refluxed for 8
hours.
The cooled reaction mixture is poured into a common salt
solution and extracted with ethyl acetate. The oily residue
obtained after concentration by evaporation is chromatographed on
silica gel with ethyl acetate. 9.53 g of the title compound with -
a melting point of 134C is obtained.
4. (2-Isopropoxyethyl)-carbonylmethylenetriphenyl phosphorane
a. l-Bromo-4-isopropoxy-butan-2-one
A solution of 68.2 g of 4-isopropoxy-2-butanone (F.B. Hasan
et al., J. Biolog. Chem. 256, 7781, 1981) in 315 ml of methanol
is mixed by instillation at oC with 26.9 ml of bromine and then
stirred for 1.5 hours at +10C. Then 470 ml of water is
instilled in the reaction solution and it is stirred for 16 hours
at room temperature. For working up, it is poured into saturated
common salt solution and extracted with ether. Distillation of
the crude product yields 78.07 g of the bromine derivative of
bpl5-20of 95C.
b. 4-Isopropoxy-2-oxo-butyl-triphenylphosphonium bromide
According to the process described under 1, 133.35 g of
phosphonium salt with a melting point of 183C is obtained from
78.0 g of the bromide obtained under a. and 97.85 g of
triphenylphosphine.
c. (2~Isopropoxyethyl)-carbonylmethylenetriphenyl
phosphorane
The phosphonium bromide (1~3.2 g) obtained under b. is
treated as described under 1. with 2N NaOH in methylene chloride.
After recrystallization of the crude product from ethyl acetate,
64.38 g of the title compound with a melting point of 97C is
obtained.
5. (l-Ethylpropoxymethyl)-carbonylmethylenetriphenyl phosphorane
A solution of 3.04 g of sodium in 100 ml of 3-pentanol is
reacted with 25.0 g of chloromethylcarbonylmethylenetriphenyl
phosphorane analogously to the production of --
isopropoxymethylcarbonylmethylenetriphenyl phosphorane. The
title compound is obtained as crystallized oil with a melting
point of 66-70C.
6. Cyclopropylmethoxymethylcarbonylmethylenetriphenyl
phosphorane
A solution of 5.58 g of sodium in 25.0 g of
cyclopropylmethanol and 200 ml of toluene is reacted with 30.0 g
of chloromethylcarbonylmethylenetriphenyl phosphorane analogously
to the production of isopropoxymethylcarbonylmethylenetriphenyl
phosphorane. The title compound is obtained as solid with a
melting point of 121C.
7. (3-Butinyl)-carbonylmethylenetriphenyl phosphorane
20.0 g of methylcarbonylmethylenetriphenyl phosphorane is
dissolved in 628 ml of tetrahydrofuran and mixed by instillation
at -78C with 41.3 ml of butyllithium (1.6 molar solution in
hexane)O Then, 5.0 ml of propargyl bromide is instilled. The
reaction mixture is added to an ice/common salt solution after
G5
heating to room temperature, and the mixture is extracted with
ethyl acetate. ~fter drying the organic phase with sodium
sulfate, 23.4 g of solid is obtained. Column chromatographic
purification (silica gel/ethyl acetate) yields 15.4 g of the
title compound with a melting point of 135-136C.
8. (3-Butenyl)-carbonylmethylenetriphenyl phosphorane
By reaction of 15.0 g of methylcarbonylmethylenetriphenyl
phosphorane in 471 ml of tetrahydrofuran with 31.0 ml of
butyllithium and 4.28 ml of allyl bromide analogously-to 7, the
title compound is obtained as crystallized oil with a melting
point of 92-93C.
By varying the keto component used for the production of the
Wittig reagent, other phosphoranes, which can be reacted with
aldehyde 1 or 2 analogously to other compounds of general formula
IV as described below, can be obtained in a similar way.
EXAMPLE ~
A solution of 1.6 g of l(S),3(R)-bis-(tert-
butyldimethylsilyloxy)-20(R)-methyl-9,10-secopregna-
SE,7E,10,(19)-triene-21-carbaldehyde in 50 ml of toluene is
stirred for 16 hours at 80C under argon after adding 3.02 g of
isoamylcarbonylmethylenetriphenyl phosphorane. Then, the solvent
is removed under reduced pressure and the residue is
chromatographed on silica gel with hexane/ethyl acetate. The
main fraction yields 1.15 g of tl(S~,3(R)-bis-(tert- -
butyldimethylsilyloxy~-9~lo-secochola-5E~7E~lo~(l9)~23(E)
tetraen-24-yl]-4-methyl-pentan-1-one as colorless oil.
IH-NMR (CDC13): ~ = 0.01 ppm (s,12H,Si-CH3), 0.56 (s,3H,H-
18), 0.87 (s,18H,Si-t.-butyl); 0.88 (d,J=7Hz,6H,C-(CH3)2), 0.95
(d,J=7Hz,3H,H-21); 4.25 (m,lH,H-3); 4.55 (m,lH,H-1); 4.94 and
5.00 (s; lH, H-19 each); 5.82 and 6.46 (d,J=llHz; lH,
H-6, H-7 each); 6.10 (d,J=16Hz,lH,H-24); 6.80 (m,lH,H-23).
EXAMPLE 2
572 mg of cerium(III)-chloride-heptahydrate is dissolved in 10 ml
of methanol and the compound (1.10 g) produced according to
example 1 dissolved in 5 ml of methanol is added. After adding
61 mg of sodium borohydride, it is stirred for 30 minutes at 0C.
For the working up, it is poured into water, extracted with
dichloromethane, dried (Na2S04) and concentrated by evaporation.
The mixture of diastereomeric alcohols thus obtained is separated
by chromatography on silica gel with hexane/ethyl acetate. In
the elution sequence, 290 mg of l(S),3(R)-bis-(tert-
butyldimethylsilyloxy)-24-(1-hydroxy-4-methylpentyl)-9~10-seco-
5E,7E,10(19),23(E)-cholate-traene (epimer A) and 120 mg of epimer
B are obtained. The epimers show identical NMR spectra.
IH-NMR (CDC13): S = 0.01 ppm (s,12H,Si-CH3), 0.49 (s,3H,H-
18), 0.86 (s,18H,Si-t.-butyl); 0.86 (d,J-7Hz,6H,C-(CH3)2); 0.88
(d,J=7Hz,3H,H-21); 4.16 (m,lH,H-3); 4.48 (m,lH,H-l); 4.88 and
4.93 (s; lH, H-l9 each); 5.40 (dd,J=15.5 and 7Hz,lH,H-24~; 5.55
(m,lH,H-23~; 5.77 and 6.40 (d,J=llHz; lH, H-6, H-7 each).
EXAMPLE 3
A solution of 290 mg of the product (epimer A) obtained
under example 2 in 80 ml of toluene is irradiated in a pyrex
immersion reactor by a high pressure mercury vapor lamp (Philips
HPK 125) after adding 44 mg of anthracene and 0.01 ml of
triethylamine. The irradiation time is 3.5 minutes, the thorough
mixing of the solution is guaranteed by introducing a nitrogen
stream. After concentration by evaporation and chromatography on
silica gel with hexane/ethyl acetate, 241 mg of l(S),3(R)-
bis(tert-butyldimethylsilyloxy)-24-(1-hydroxy-4-methylpentyl)-
9,10-secochola-5Z,7E,10(19),23(E)-tetraene is obtained as
colorless oil.
[ ~X ]D20+ 49.6 (CHC13, c=0.425).
Analogous treatment of 120 mg of the polar isomer (epimer B)
obtained according to example 2 yields 113 mg as colorless oil.
[~ ]D20+ 41.4 (CHC13, c=0.285)
EXAMPLE 4
A solution of 225 mg of the product obtained according to
example 3 from epimer A in 5 ml of THF is stirred for 60 minutes
at 60C after adding 1.31 ml of a lM solution of
tetrabutylammonium fluoride in THF. After cooling, it is poured
into a saturated common salt solution and extracted with ethyl
acetate. The crude product is chromatographed on silica gel with
hexane/ethyl acetate and yields 85 mg of 24~ hydroxy-4-
methylpentyl)-9,10-secochola-5Z,7E,10(19),23E-tetraene-l(S),3(R)-
diol as white foam.
IH-NMR (CDC13): ~ = 0.57 ppm (s,3H,H-18), 0.84
(d,J=7Hz,3H,H-21); 0.92 (d,J=7Hz,6H,C-(CH3)2); 4.03 (m,lH,H-25);
4.23 (m,lH,H-3); 4.43 (m,lH,H-l); 5.00 and 5.33 (s; lH, H-l9
each); 5.45 (dd,J=15.5 and 7Hz,lH,H-24); 5.60 (m,lH,H-23); 6.02
and 6.38 (d,J=lHz; llH, H-6, H-7 each).
Analogous treatment of the product (95 mg) obtained
according to example 3 from epimer B yields 35 mg of the epimeric
triol as colorless oil. The NMR spectra of the epimers are
identical.
EXAMPLE 5
Analogously to the process described under example 1, 2.05 g
of l(S~,3(R)-bis-(tert-butyldimethylsilyloxy)-20-(R)-methyl-9,10-
secopregna-5E,7E,10(19)-triene-21-carbaldehyde in 53 ml of
toluene is reacted with 3.4 g of
isobutylcarbonylmethylenetriphenyl phosphorane. After
~9
chromatographic purification, [l(S),3(R)-bis-(tert-
butyldimethylsilyloxy)-9,10-secochola-5E,7E,10(19),23(E)-tetraen-
24-yl3-3-methyl-butan-1-one with a melting point of 79-81C is
obtained (from ethanol).
[C~]D20 + 52.6 (CHC13, c=0.500)
EXAMPLE 6
By reduction of 1.75 g of the product obtained under example
5 under the conditions of example 2, l(S~,3(R)-bis~tert-
butyldimethylsilyloxy)-24-(1(R,S)-hydroxy-3-methylbutyl)-9,10-
secochola-5E,7E,10(19),23E-tetraene is obtained as an oily
mixture of the epimers. By chromatography on silica gel with
hexane/ethyl acetate, 780 mg of epimer A and 600 mg of epimer B
are obtained in the elution sequence as colorless oils, which
cannot be differentiated by NMR spectroscopy.
EXAMPLE 7
By triplet-sensitized photoisomerization analogously to
example 3 and subsequent silylether cleavage analogously to
example 4, 240 mg of 24-(1-hydroxy-3-methylbutyl)-9,10-secochola-
5Z,7E,10(19),23(E)-tetraene-l(S),3(R)-diol (compound A) with a
decomposition interval of 119-125C, is obtained from 700 mg of
epimer A produced according to example 6 [ ~ ~D20+ 38.8
(methanol, c=0.505).
Analogous treatment of 330 mg of epimer B yields 129 mg of
24-(1-hydroxy-3-methylbutyl)-9,10-secochola-5Z,7E,10(19j,23(E)-
^ ~)
tetraene-l(S),3~S)-diol (compound B) with a decomposition
interval of 139-145C, [C~ )D20+ 54.8 (methanol) c=0.5G5).
EXAMPLE 8
A solution of 170 mg of the product obtained according to
example 5 in 5 ml of THF is stirred for 90 minutes at room
temperature after adding 200 mg of lithium-tri-tert-butoxy-
aluminum hydride. For working up, it is mixed with 0.8 ml of
saturated NH4Cl solution, filtered and the filtrate is
concentrated by evaporation. Chromatography of the crude product
on A12O3 (Merck, neutral, step III) yields 108 mg of 1-
[l(S),3(R)-bis-(tert-butyldimethylsilyloxy)-9,10-secochola-
5~,7E,10(19)-trien-24-yl]-3-methyl-butan-1-one as colorless oil.
IH-NMR (CDC13): ~ = 0.53 ppm (s,3H,H-18); 4.22 tm,lH,H-3);
4.54 (m,lH,H-1); 4.93 and 4.98 (m; lH, H-19 each); 5.8Z and 6.46
(d,J=llHz; lH, H-6, H-7 each).
EXAMPLE 9
From 100 mg of the product of example 8, photochemical
double bond isomerization analogously to example 3 and silylether
cleavage analogously to example 4 yield 50 mg of 1-[l(S),3(R)-
dihydroxy~9,10-secochola-5Z,7E,10(19)-trien-24-yl]-3-methyl-
butan-1-one.
W (methanol): = 212 nm ( ~ =14 300), 265 (15 860).
EXAMPLE 10
The reaction of 1.6 g of l(S),3~R)-bis-(tert-
butyldimethylsilyloxy)-20(R)-methyl-9,10-secopregna-5E,7E,10(19)-
triene-21-carbaldehyde with (2-isopropoxyethyl)-
carbonylmethylenetriphenyl phosphorane analogously to example 1
yields 1.15 g of 1-[l(S),3(R)-bis-(tert-butyldimethylsilyloxy)-
9,10-secochola-5E,7E,10(19),23(E)-tetraen-24-yl]-3-isopropoxy-
propan-l-one as colorless oil~ -
IH-NMR (CDC13): ~ = 0.01 ppm (s,12H,Si-CH3), 0.55 (s,3H,H-
18), 0.86 and 0.90 (s; 9H, Si-t.-butyl each); 0.96 (d,
J=7Hz,3H,H-21); 1.15 (d,J=7Hz,6H,C(CH3)2); 3.60(m,1H,CH-0); 3.73
(t,J=7Hz,2H,CH2-0); 4.23 (m,lH,H-3); 4.55 (m,lH,H-l); 4.95 and
5.00 (m; lH, H-l9 each); 5.83 and 6.46 (d,J=ll Hz; lH, H-6, H-7
each); 6.11 (d,J=15.5Hz,lH,H-24); 6.87 (m,lH,H-23).
EXAMPLE 11
By reduction analogously to example 2, photoisomerization
analogously to example 3 and silylether cleavage analogously to
example 4, 143 mg of 24-1l(R,S) hydroxy-3-isopropoxypropyl)-9,10-
secochola-5Z,7E,10(19),23-tetraene-l(S),3(R)-diol is obtained
from 1.05 g of the product produced according to example 10 as a
1:1 mixture of the diastereomers that are separated by high-
pressure liquid chromatography. The isomers exhibit identical
NMR spectra.
IH-NMR (CDC13): ~ = 0.57 ppm (s,3H,H-18),0.94
(d,J-7Hz,3H,H-213; 1.15 (d,J=7Hz,6H,C(CH3)2), 4.17 (m,lH,H-3);
3~
4.21 (m,lH,H-25); 4.38 (m,lH,H-l); 4.98 and 5.29 (m; lH, H-l9
each); 5.45 (dd,J=15.5 and 7Hz,lH,H-24); 5.63 (m,lH,H-23); 6.02
and 6.38 (d,J=llHz; lH, H-6, H-7 each).
EXAMPLE 12
Starting fron aldehyde 1 and
isopropoxymethylcarbonylmethylenetriphenyl phosphorane, isomer B
(5Z,7E,22E-l(S),3(R),24(S)-9,10-seco-24a,24b-dihomo-24b- -
oxacholesta-5,7,10(19),22-tetraene-1,3,24-triol) with a melting
point of 131-132C is obtained analogously to the sequence of
examples 1-4.
EXAMPLE 13
Starting fron aldehyde 1 and (2-isopropoxyethyl)-
carbonylmethylenetriphenyl phosphorane, isomer B (5Z,7E,22E-
l(S3,3(R),24(S)-9,10-seco-24a,24b,24c-trihomo-24c-oxacholesta-
5,7,10(19),22-tetraene-1,3,24-triol) with a melting point of 125-
126C is obtained analogously to the sequence of examples 1-4.
EXAMPLE 14
Analogously to example 1, 0.85 g of l(S),3(R)-bis-(tert-
butyldimethylsilyloxy)-20(R)-methyl-9,10-secopregna-5E,7E,10(19)-
triene-21-carbaldehyde is reacted with 4.5 g of
cyclopropylmethylcarbonyltriphenyl phosphorane. After
chromatographic purification on silica gel with hexane/ethyl
acetate, 500 mg of l(S),3(R3-bis-(tert-butyldimethylsilyloxy)-
26,27-cyclo-24a,24b-dihomo-s,lO-secocholesta-5E,7E,10(19),23E-
tetraen-24a-one is obtained as colorless foam.
IH-NMR (CDCl3): ~ = 0.01 ppm (s,12H,Si-CH3); 0.09 and 0.50
(m; 2H, H-26 and H-27 each); 0.50 (s,3H,H-18); 0.83 and 0.85 (8;
9H, Si-t.-butyl each); 0.91 (d,J=7.3Hz,3H,H-21); 0.96 (m,lH,H-
25); 2.47 (d,J=6Hz,2H,H-24b); 4.16 (m,lH,H-3); 4.47(m,1H,H-l);
4.89 and 4.93(~; lH, H-l9 each); 5.77 and 6.40 (d,J=llHz; lH, H-6
and H-7 each); 6.08~d,J=15.SHz,H-24);
6.75(ddd,J=15.5,9,6.5Hz,lH,H-23).
EXAMPLE 15
Reduction of the product obtained under example 14
analogously to example 2 yields 200 mg of l(S),3(R)-bis-(tert-
butyldimethylsilyloxy)-26,27-cyclo-24a,24b-dihomo-9,10-
secocholesta-5E,7E,10(19),23E-tetraen-24a(R,S)-ol as an oily
mixture of the epimers that cannot be differentiated by NMR
spectroscopy.
IH-NMR (CDCl3): ~ = 0.01 ppm (s,12H,Si-CH3); 0.09 and 0.40
(m; 2H, H-26 and H-27 each); 0.50 (s,3H,H-18); 0.68 (m,lH,H-25);
0.81 and 0.86(s; 9H, Si-t.-butyl each); 0.88 (d,J=7Hz,3H,H-21);
1.40 (t,J=7Hz,H-24b); 4.13 (m,lH,H-24a); 4.17 (m,lH,H-3); 4.49
(m,lH,H-l); 4.88 and 4.93 (s; lH, H-l9 each); 5.45 (dd,J=15.5,
6.5Hz,lH,H-24); 5.59 (ddd,J=15.5, 7, 6.5Hz,lH,H-23); 5.77 and
6.40 (d,J=llHz; lH, H-6 and H-7 each).
34
EXAMPLE 16
Analogously to example 3, by triplet-sensitized
photoisomerization and cleavage of the protecting groups
analogously to example 4, 86 mg of 26,27-cyclo-24a,24b-dihomo-
9,10-secocholesta-5Z,7E,10(19~,23E-tetraene-l(S),3(R),24a(R,S)-
triol is obtained from 190 mg of the compound described under
example 15 as a 1:1 mixture of the diastereomers that are
separated by high-pressure liquid chromatography. The NMR
spectra of both diastereomers are identical.
IH-NMR (CDC13): J = o. og and 0.49 (m; 2H, H-26 and H-27
each); 0.53 (s,3H,H-18); 0.70(m,1H,H-253; 0.93 (d,J=7Hz,3H,H-21);
4.18 (m,lH,H-24a); 4~22 (m,lH,H-3); 4.43 (m,lH,H-l); S.00 and
5.32 (s; lH, H-19 each); 5.50 (dd,J=15.5, 6.5Hz,H-24); 5.64
(ddd,J=15.5, 7, 6.5Hz,lH,H-23); 6.02 and 6.38 (d,J=llHz; lH, H-6
and H-7 each).
EXAMPLE 17
Starting from aldehyde 1 and (l-ethylpropoxymethyl)-
carbonylmethylenetriphenyl phosphorane, isomer B (5Z,7E,22E-
l(S),3(~),24(S)-26,27-dimethyl-24a,24b-dihomo-24b-oxa-9,10-
secocholesta-5,7,10(19),22-tetraene-1,3,24-triol) with a melting
point of 103-105C is obtained analogously to the sequence of
examples 1-4.
EXAMPLE 18
Starting from aldehyde 1 and
cyclopropylmethoxymethylcarbonylmethylenetriphenyl phosphorane,
isomer B (5Z,7E,22E-l(S),3(R),24(S)-26,27-cyclo-24a,24b,24c-
trihomo-24b-oxa-9~lo-secocholesta-5~7~lo(l9)~22-tetraene-l~3~24
triol) is obtained analogously to the sequence of examples 1-4.
IH-NMR (DMS0-~): S=0.16 ppm (m,2H); 0.43 (m,2H); 0.53
(S,3H); 1.00 (d,J-6Hz,3H); 3.21 (m,4H); 4.00 (m,2H); 4.19 (m,lH);
4.51 (d,J=5Hz,lH); 4.70 (d,J=5~z,lH); 4.75 (m,lH); 4.82
(d,J=5Hz,lH); 5.21 (m,lH); 5.39 (m,2H); 5.98 (d,J=llhz,lH); 6.18
(d,J=llhz,lH).
EXAMPLE 19
Starting from aldehyde 1 and (3-butinyl)-
carbonylmethylenetriphenyl phosphorane, isomer B (5Z,7E,22E-
l(S),3(R),24(S)-24-(3-butinyl)-9,10-secochola-5,7,10(19),22-
tetraene-1,3,24-triol) with a melting point of 115-118C is
obtained analogously to the sequence of examples 1-4.
EXAMPLE 20
Starting from aldehyde 1 and (3-butenyl)-
carbonylmethylenetriphenyl phosphorane, isomer B (5Z,7E,22E-
l(S),3(R),24(S)-24-(3-butenyl)-9,10-secochola-5,7,10(19),22-
tetraene-1,3,24-triol) with a melting point of 146-147C is
obtained analogously to the sequence of examples 1-4.