Note: Descriptions are shown in the official language in which they were submitted.
WO91/09X75 PCT/JP9OtO1726
-- 1 --
~ 5~ 7
DESCRIPTION
MONOCLONAL ANTIBODY, HYBRIDOMAS, THEIR PRODUCTION AND USE
Technical Field
The present invention relates to monoclonal antibodies `~
5 which immuno-neutralize the biological activity of basic `~
fibroblast growth factor ~also briefly referred to as bFGF
in the present specification) proteins and have high binding
sensitivity with bFGF proteins, hybridomas secreting the
same, their production and use thereof.
Backqround Art
bFGF is a basic polypeptide hormone having an affinity
for heparin and a molecular weight of about 17,000 ~D.
Gospodarowicz, Nature 249, 123 (1974)]. It is now known
that bFGF exhibits growth promoting action on almost all
cells derived from mesoblast, and acts as a differentiating
factor to a mesoblast system. For example, bFGF induces the
proliferation of nerve cells, the proliferation of vascular
endothelial cells to cause angiogenesis, and the
proliferation of cancer cells. Coupled with these
functions, therefore, bFGF is involved in diseases such as
tumors. Accordingly, a monoclonal antibody which inhibits
the actlvity of bFGF would be useful as a therapeutic drugs
for such diseases. However, this possibility has not been
recognized till now. Further, as a means for diagnosing
25 these diseases, the determination of blood bFGF `::
concentrations is considered. It is however impossible to
detect bFGF with great sensitivity by previously known
methods.
WO91/09875 pCT/JP90/01726
z~ 2 -
As discussed above bFGF is involved in diseases such as
cancer, so that the inhibition of the growth promoting
- activity of bFGF to the cancer cells presents the
possibility of use as a cancer therapy.
In addition, there is the potential that bFGF itself
may have application as a therapeutic drug for traumas and
burns. In this case, the determination of fundamental
information regarding bFGF is essential for the development
of bFGF as a medicine. Also in the diseases in which bFGF
is invollved, such as cancer, the diagnosis of the disease
becomes possible by tracing the blood bFGF concentrations.
However, since bFGF is adsorbed by heparan sulfate existing
among vascular endothelial cells, bFGF exists in blood only
in trace amounts. Accordingly, there is a need in the art
to develop a highly sensitive quantitative assay for
measuring bFGF in the blood.
SummarY of the Invention
In view of the above-mentioned circumstances, the
present inventors prepared monoclonal antibodies which
2~ highly sensitive determination of bFGF proteins, as well as
immuno-neutralize the activity of the bFGF. The present
invention conducted further researches based thereon, thus
completing the present invention.
The present invention provides:
~l) a monoclonal antibody which has the following
characteristics, it immuno-neutralizes the activity of a
bFGF protein and has high binding sensitivity with the bFGF
: . :
., ., ... . . . ~ .. :: .. : .. . :: . ~ - . :
WO 91to9~7~; PC'r/JI~O/û1726
-- 3
0~ ~
protein:
(a) it has a molecular weight of about 140,000 to
160,000,
(b) it does not cross-react with acidic fibroblast
growth factor,
~ c) it belongs to immunoglobulin class IyGl,
(d) it binds with rhbFGF mutein CS23 (a mutein in which
the cysteine residues at positions 70 and 88 of hbFGF are
substituted for serine residues),
(e) it completely inhibits proliferation of a human
unbilical vein endothelial (HUVE) cell by addition of 50 : :
ng/ml thereof in the presence of 2 ng/ml of bFGF, and
(f) it can detect 20 pg/ml of the bFGF protein by a
sandwich enzyme-linked immunosorbent assay (ELISA) using
monoclonal antibody MoAbl2 (solid phase) and a peroxidase-
labeled antibody;
~ 2) a cloned hybridoma derived from a mammalian spleen
cell and a homogenic or heterogenic lymphoid cell, said
mammal being immunized with a mutein in which at least one ~:
cysteine residue of bFGF is substituted for a serine
; residue;
(3) a method for producing a cloned hybridoma which
comprises fusing a spleen cell from a mammal and a homogenic
or heterogenic lymphoid cell, said mammal being immunized
with a mutein in which at least one cysteine residue of bFGF
is substituted for a serine residue, and selecting the
desired hybridoma, followed by cloning;
WO9~ 7s pCT/JP9~/01726
t Q`~ 4
(4) a method for producing the monoclonal antibody
described in the above item (1), which comprises culturing a ~`
cloned hybridoma comprising a spleen cell
from a mammal and a homogenic or heterogenic lymphold cell
in a liquid culture medium or in a peritoneal cavity of the
mammal to produce the monoclonal antibody, said mammal being
immunized with a mutein in which at least one cysteine
residue of bFGF is substituted for a serine residue, and
recovering the monoclonal antibody;
(5) a method for purifying a bFGF protein, which
comprises contacting the monoclonal antibody described in
the above item (1) with a sample containing bFGF protein;
and
(6) a method for detecting or measuring a bFGF protein,
which comprises using the monoclonal antibody described in
the above item (1).
- Brief Description of Drawinq_
Fig. 1 shows a cDNA se~uence of human aFGF used in
Reference Example l;
Fig. 2 is a schematic representation showing the
construction of plasmid pTB975 obtained in Reference Example
l;
Figs. 3 to S show elution pat~erns obtained in
Reference Example 1;
Fig. 6 is a graph showing the affinity of monoclonal
antibody 3H3 of the present invention obtained in Example 4
for rhbFGF mutein CS23;
`~
:`
., .. , .,, . .. . . ..... .,, . . . .. . ~. . .
WO91/~987~ PCTIJP90/01726
Fig. 7 is a graph s~o~ing the human bFGF-neutralizing
activity of monoclonal antibody 3H3 of the present invention
obtained in Example 6;
Figs. 8~a) and 8(b) are yraphs showing the
proliferation inhibition effect of monoclonal antibody 3~3
of the present invention obtained in Example 6 for HUVE
cells, in which Fig. 8(a) shows the results of cultivation
for 3 days and Fig. 8(b) shows the results of cultivation
for 5 days;
Fig. 9 is a graph showing the relationship between the
concentration and the absorbance of hbFGF, obtained in
Example '~;
Fig. 10(1) and 10~2) are graphs showins the influence
of heparin in a hbFGF determination system, obtained in
Example 10, in which Fig. 10(1) shows the results when MAbl2
is fixed, and Fig. 10(2) shows the results when a 50-50
mixture of MAb52 and MAb98 is fixed;
Fig. 11(1) and 11(2) are graphs showing the reaction in
a hbFGF determination system, obtained in Example 11, in
which Fig. 11(1) shows the results when MAbl2 is solidified,
and Fig. 11(2) shows the results when a 50-50 mixture of
MAbS2 and MAb98 is fixed;
Fig. 12 is a graph showing the anti-tumor effect of an
antibody of the present invention, obtained in Example 13;
and
Fig. 13 is a schematic representation showing the
construction of plasmid p'rB1000 obtained in Reference
Example 2.
~:
WO91/09875 PCT/JP90/01726
~ 6 -
Detailed Descri~tion of the_Invention
The bFGF proteins of the present invention include
bFGFs and muteins in which at least one cysteine residue of
bFGF is substituted for a serine residue and which have bFGF
activity.
As the bFGF proteins, there are preferably used
polypeptides containing the amino acid sequence represented ~:
by formula (I):
Phe-Phe-Leu-Arg-Ile-~is-Pro-Asp-Gly-Arg-Val- `
lOAsp-Gly-Val-Arg-Glu-Lys-Ser-Asp-Pro tI)
The bFGFs of the present invention include bFGFs
derived from mammals. The mammals include humans, monkeys,
pigs, bovines, sheep and horses.
The bFGFs also include bFGFs extracted from various
lS organs existence of which is already clarified, such as
brains and pituitary glands.
The bFGF proteins may be produced by recombinant DNA
techniques.
Examples of amino acid sequences of the bFGFs include
the amino acid sequence represented by formula tII):
Pro-Ala-Leu-Pro-Glu-Asp-Gly-Gly-Ser-
Gly-Ala-Phe-Pro-Pro-Gly-His-Phe-Lys-Asp-
Pro-Lys-Arg-Leu-Tyr-Cys-Lys-Asn-Gly-Gly-
Phe-Phe-Leu-Arg-Ile-His-Pro-Asp-Gly-Arg-
25Val-Asp-Gly-Val-Arg-Glu Lys-Ser-Asp-Pro-
His-Ile-Lys-Leu-Gln-Leu-Gln-Ala-Glu-Glu-
Arg-Gly-Val-Val-Ser-Ile-Lys-Gly-Val-Cys-
WO91/o9875 PCT/JP90/01726
-- 7
~ ;'``;~.7~
Ala-Asn-Arg-Tyr-Leu~Ala-Met-Lys-Glu-Asp-
Gly-Arg-Leu-Leu-Ala-Ser-Lys-Cys-Val-Thr-
Asp-Glu-Cys-Phe-Phe-Phe-Glu-Arg-Leu-Glu-
Ser-Asn-Asn-Tyr-Asn-Thr-Tyr-Arg-Ser-Arg-
Lys-Tyr-X -Ser-Trp-Tyr-Val-Ala-Leu-Lys-
Arg-Thr-Gly-Gln-Tyr-Lys-Leu-Gly-Y Lys-
Thr-Gly-Pro-Gly-~ln-Lys-Ala-Ile-Leu-Phe-
Leu-Pro-Met-Ser-Ala-Lys-Ser (II)
wherein X represents Thr or Ser, and Y represents Ser when X
is Thr, an~ Y represents Pro when X is Ser.
As the bFGF, human bFGF is preferable. The human bFGF
has the amino acid sequence in which X is Thr and Y is Ser
in the above formula (II).
The muteins in which at least one cysteine residue of
the above bFGF is substituted for a serine residue include,
for example, the muteins described in Seno et al., BiophYs.
Res. Commun. 151, 701 (1988) and European Patent Publication
No. 281,822.
In particular, there is preferably used recombinant
human bFGF mutein CS23 (hereinafter also briefly referred to
as rhbFGF mutein CS23) in which each of the cysteine
residues at positions 70 and 88 of the human bFGF is
substituted for a serine residue. With respect to the
number of the positions of the above amino acids, in the
amino acid sequence in which Met is added to the N-terminus
of the peptide in which X is Thr and Y is Ser in the above
formula ~II), the Met is numbered as the first.
~'`', '
:
~091/09M75 PCT/JP90/01726
~ 8 --
In thè bFGFs produced by the above-mentioned genetic
engineering techniques, examples of the human bFGF include
bFGF produced by the methods described, for example, in FEBS
Letters 213, 189 (1987), ~E~y___Res. Commun. 146, 470
(1987) and European Patent Publication No. 237,966.
When mammals are immunized with the bFGF proteins or
protein conjugates, there are used experimental animals such
as sheep, goat, rabbits, guinea pigs, rats and mice, as the
mammals to be immunized. In order to obtain monoclonal
antibodies, however, it is preferred to use rats or mice for
immunization.
When mice are immunized, for example, they can be
immunized by any of the subcutaneous, intraperitoneal,
intravenous, intramuscular and intracutaneous routes.
However, subcutaneous, intraperitoneal and intravenous
injections are preferably used. In particular, subcutaneous
injection is preferable. The immunizing interval and the
immunizing dose are widely variable, and various methods are
available. For example, methods in which immunization is
carried out about 2 to 6 times at intervals of 2 weeks and
spleen cells are removed after about 1 to 5 days, preferably
about 2 to 4 days from the final immunization are frequently
used. As the immunizing dose, it is preferred to use about
0.1 ~g or more, preerably about 10 to 300 ~g of the peptide
per one immunization of a mouse. Further, it is desirable
to carry out the fusion process using the spleen cells after
confirmation of an increase in antibody titer in blood by
WOgltO9875 PCT/JP9~/~t726
jJ?
collecting a portion o~ blood and measuring the antibody
titer before removal of the spleens.
The above spleen cells are then fused wlth lymphoid
cells. For example, the spleen cells removed from the mice ~
5 are fused with lymphoid cell strains such as suitable ~ ;
myeloma cells [for example, P3-X-63-Ag-8UI ~Ichimori et al.,
J. Immun. Method 80, 55 ~1985))~ of the same kind or a
different kind (preferably ~he same kind) having markers
such as hypoxanthine-guanine-phosphoribosyl-transferase
10 deficient (HGPRT ) and thymidine kinase deficient (TK ).
For example, begin a new paragraph the fused cells are
produced in accordance with the method of Kohler and
Milstein [~ature 256, 495 (1975)1. For example, myeloma
_
cells and spleen cells in a ratio of about 1:5 are suspended
15 in a medium prepared by mixing Iskov medium and Ham F-12
medium in a 1:1 ratio (hereinafter referred to as IH
medium), and a fusion accelerator such as Sendai virus or
polyethylene glycol (PEG) is added thereto. It is of course
possible to add other fusion accelerators such as dimethyl
20 sulfoxide (DMSO). The polymerization degree of PEG is ?
usually about 1,000 to 6,000, the fusion time is about 0.5
to 30 minutes, and the concentration of the suspension is
about 10 to 80%. As a preerred condition, the fusion is
carried out efficiently by using PEG 6,000 in a
concentration of about 35 to 55% for about 4 to 10 minutes.
The fused cells can be selectively proliferated using
hypoxanthine-aminopterin-thymidine medium ~HAT medium)
~ ' '
WO 91/09875 PCr/JP90/01726
1 0
,~ f --
[Nature, 256, 4~5 (1975) ].
_ _
The culture supernatant of the proliferated cells is
then screened for the production of the desired antibody.
Screening of the antibody titer can be carried out in the
following manner. First, the presence or absence of the
antibody production by peptide immunization is examined by
radio immunoassays (RIAs) or enzyme immunoassays (EIAs).
For these methods, various modified me~hods are also
available.
As a preferred example of the assays, a method using
the EIA is described below. A rabbit anti-mouse
immunoglobulin antibody is coupled with a carrier such as
cellulose beads according to conventional methods, and then
a culture supernatant or mouse serum to be assayed is added
thereto, followed by reaction at a constant temperature
(about 4 to 40C, the same applied hereinafter) for a
definite time. After the reaction product is thoroughly
washed, an enzyme-labeled peptide (a peptide is coupled with
an enzyme according to conventional methods, followed by
purification) is added thereto, followed by reaction at a
constant temperature for a specified time. After the
reaction product is thoroughly washed, an enzyme substrate
is added thereto, followed by reaction at a constant
temperature for a speciied time. Then, the absorbance or
fluorescence of the color-produced product is measured.
As to the culture supernatant of the proliferated
cells, the screening of the neutralizing activity to the
,. .
. ~:
wogl/0987~ 7~ PCT/JP9OtO1726
bFGF proteins can be carried out in the following manner.
In this case, the neutralizing activity can be examined
using cells whcse proliferation is induced by the bFGF
proteins. As such cells, there can be used vascular
endothelial cells, fibroblasts, nerve cells and the like.
It is however desirable to know whether or not the
additional effect of the bFGF proteins is inhibited by
; measuring the number of cells, using the vascular
endothelial cells begin a new paragraph.
Methods for measuring the number of cells include a
method for measuring directly the number of cells, a method
or determining radioactivity by using tritium thymidine and
a method for measuring colorimetrically using (4,5-dimethyl-
; 2-thiazoly)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (MTT
method). As one example of the preferred assays, there is
; hereinafter described a method for assaying cell
proliferation by the MTT method using human umbilical vein-
derived endothelial cells. After the cells are seeded, the
bFGF protein and the culture supernatant to be assayed are
added thereto, followed by cultivation at a constant
temperature (37C) at a low concentration of oxygen ~about
7% is preferable) for a definite time. Then, the culture
solution are replaced by a medium containing MTT, and
cultivation is further continued, whereby MTT is reduced to
a formazan, a colorimetric material, in intracellular
endogenous mitochondria.
After a specified time has elapsed, SDS is added
WQ91/09875 PCT/JP90/01726
~ 12 -
thereto to dissolve the cellsl and the concentration of the
formazan is made even, the absorbance at 590 nm is measured.
The proliferative property of cells correlates with the
absorbance. Hence, if the solution containing the hybridoma
supernatant is decreased in absorbance compared with a
solution not containing the hybridoma supernatant, the
antibody contained in the supernatant can be said to have
bFGF neutralizing activity.
It is desirable that the cells in wells which
proliferate in a selective medium and which produce
antibodies with immuno-neutralizing activity to the peptide
used for immunization be cloned by a limiting dilution
analysis. The supernatant of the cloned cells is similarly
screened, and the selected cells which show a high antibody
titer are proliferated, whereby monoclonal antibody-
producing hybridoma clones showing the reactivity with the
immunized peptide can be obtained.
The hybridoma cells thus cloned are proliferated in a
liquid medium. Specifically, for example, the hybridoma
ce}ls are cultivated in the liquid medium such as a medium
prepared by adding about 0.1-40% bovine serum to RPMI-1640
[G. E. Moore et al., J. Am. Med. Assoc. 199, 549 ~1967)],
for about 2 to 10 days, preferably for 3 to 5 days, whereby
the monoclonal antibody can be obtained from the culture
solution. The antibody can further be obtained by
intraperitoneally inoculating mammals with the hybridoma
cells, thereby proliferating the cells and then collecting
:
WO91t09875 PCT/JP90/01726
- 13 - ,~r,~h~ ~
the ascites. In the case o a mouse, for example, about l X
104 to l X 107, preferably 5 X 105 to 2 X 106 of the
hybridoma cells are intraperitoneally inoculated into a
mouse such as BALB/c preliminarily inoculated with mineral
oil and the like, and the ascites is collected after about 7
to 20 days, preferably after about lO to 14 days. The
monoclonal antibody formed and accumulated in the ascites
can be easily isolated as pure immunoglobulin by ammonium
sulfate fractionation, DEAE-cellulose column chromatography
or the like.
The monoclonal antibodies of ~he present invention have
high binding sensitivity not only with the immunogen
peptides, but also with the bFGF proteins, and further
exhibit the neutralizing activity to the bFGF proteins.
As a result of this high binding sensitivity, the
monoclonal antibodies of the present invention are very
useful as reagents for assaying the bFGF proteins and for
purifying the bFGF proteins. ;
The bFGF proteins can be assayed to 20 pg/ml by the
20 assays using the monoc}onal antibodies of the present !~
invention. The ability to assay such an extremely small
amount of bFGF in vivo is an important development in the
art. As diseases induced by overproduction of bFGF, tumors
are mentioned. For the tumors, there are the case that
cancer cells proliferate directly by bFGF and the case that
vascular endothelial cells react with bFGF produced from the ;~
tumors to proliferate and induce n-w formed blood vessels
" ~:
~: .
w ~ n9B75 Pcr
- 14 -
which supply nutritive substances to the tumors, which
results in enlarged tumor masses. In these cases, these
diseases can be anticipated by measuring the amount of the
bFGF overproduced. Since bFGF easily adheres to the inner
walls of blood vessels, it is therefore important to have a
detecting or determining assay method as sensitive as
possible.
Also in treating traumas and burns~ bFGF is considered
to improve the symptoms thereof effectively. In this case,
it is more effective to use a mutein in which at least one
cysteine residue of the bFGF is substituted for a serine
residue and which is higher in stability than the bFGF.
When the mutein is used as a therapeutic drug, the
monoclonal antibodies of the present invention can be used
as means for tracing the amount of the mutein in vivo,
because the monoclonal antibodies also have high binding
sensitivity with the mutein in which at least one cysteine
residue is substituted for a serine residue.
As described above, many tumors are considered to be
proliferated by bFGF in bodies. The monoclonal antibodies
of the present invention inactivate the bFGF in vivo and
exhibit antitumor activity, because of their strong
immuno-neutralizing activity. }n this case, the monoclonal
antibodies are administered in an amount of about 100 ~g/kg
to 10 mg/kg. These monoclonal antibodies are also effective
for treatment of enlarged tumors following angiogenesis.
Examples of the methads for detecting or assaying the
., . , . . . .
, -
,. , ~ :
. . - . :
WOgl/0987~ P~T/JP90/01726
- 15 -
~ ; J'-~J 7 ~
bFGF proteins include an immunoassay for assaying the bFGF
proteins by using an anti-bFGF antibody supported on a
carrier and a conjugate ob~ained by combining an anti-bFGF
antibody directly with a labeling agent, the anti-bFGF
antibody differing from the antibody held on the carrier in
an antigen determinant.
The carriers on which the antibody is held in the
above-mentioned assay include, for example, gel particles
such as agarose gels [for example, Sepharose 4B and`
Sepharose 6B (Pharmacia Fine Chemical, Sweden)~, dextran
gels [for example, Sephadex G-75, Sephadex G-lO0 and
Sephadex G-200 (Pharmacia Fine Chemical, Sweden)] and
polyacrylamide gels [for example, Biogel P-30, Biogel P-60
and Biogel P-lO0 (Bio RAD Laboratories, U.S.A.)~; cellulose
particles such as Avicel tAsahi Chemical Industry, Japan)
and ion exchange cellulose (for example, diethylaminoethyl
: cellulose and carboxymethyl cellulose); physical adsorbents
such as glass (for example, glass balls, glass rods,
aminoalkyl glass balls and aminoalkyl glass rods), silicone
pieces, styrenic resins (for example, polystyrene balls and
polystyrene particles) and plates for immunoassay ~for
example, Nunc, Denmark); and ion exchange resins such as
weakly acidic cation exchange resins lfor example, Amberlite
IRC- 50 ~Rohm & Haas, U.S.A.) and Zeocurve 226 ~Permutit,
West ~ermany)], and weakly basic anion exchange resins [for
example, Amberlite IR-4B and Dowex (Dow Chemical, U.S.A.)].
In order to couple the antibody onto the carrier,
,
~ `'`
WO91/09875 PcT/Jp9o/o172s
~ t~ ? ~ 16 - -
methods k~own in th~ art are applied. Examples of such
methods include the cyanogen bromide method and the
glutaraldehyde method which are described in Metabolism, 8,
696 (1971). As a simpler method, the antibody may be
physically adsorbed on the surface of the carrier.
The labeling agents with which the antibodies are
combined include radioisotopes, enzymes, fluorescent
substances and luminous substances. However, it is
preferred to use the enzymes. As the enzymes, which are
preferably stable and high in specific activity, there can
be used peroxidases, alkaline phosphatases, ~-D-
galactosidases, glucose oxidases and the like. In
particular, peroxidases is preferably used. Peroxidases of
various origins can be used. Examples of such peroxidases
include peroxidases obtained from horseradishes, pineapples,
figs, sweet potatoes, broad beans and cone. In particular,
horseradish peroxidase (HRP) extracted from horseradishes is
preferable.
In combining peroxidase with the antibody, the thiol
group of Fab~ as the antibody molecule is utilized, and
peroxidase into which a maleimide group is preliminarily
introduced is conveniently used.
When a maleimide group is introduced into peroxidase,
it can be introduced through an amino group of peroxidase.
For this purpose, N-succinimidyl-maleimide-carboxylate
derivatives can be used. N-~y-maleimidobutyloXy)succinimide
~hereinafter also briefly referred to as GMBS) is preferably
' . r , ` . . ' . ' .. :., .. .. 1~ . ; ': ' ` . ' ~ . , ' ~ . ' . '.. ' ' . .. ' '
WO91J09X75 PCT/JP90/~17~6
- 17 ~ t'~ ~`, - ?
used. A certain group may therefore intervene between the
maleimide group and perxidase.
G~BS is reacted with peroxidase in a buffer solution
having a pH of 6 to 8 at about 10 to 50C for about 10
minutes to about 24 hours. The buffer solutions include,
for example, 0.1 M phosphate buffer (pH 7.0). The
maleimidated peroxidase thus obtained can be purified, for
example, by gel chromatography. Examples of carriers used
in the gel chromatography include Sephadex G-25 (Pharmacia
Fine Chemical, Sweden) and Biogel P-2 (Bio RAD Laboratories,
U.5.A.).
The maleimidated peroxidase can be reacted with the
antibody molecule in a buffer solution at about 0 to 40C
for about 1 to 48 hours. The buffer solutions include, for `;
example, 0.1 M phosphate buffer (p~ 6.0) containing 5 mM
sodium ethylenediaminetetraacetate. The peroxidase labeled
antibody thus obtained can be purified, for example, by gel
chromatography. Examples of carriers used in the gel
chromatography include Sephadex G-25 ~Pharmacia Fine
Chemical, Sweden) and Biogel P-2 (Bio RAD Laboratories,
U.S.A.).
A thiol group may be introduced into peroxidase to ~;
allow it to react with the maleimidated antibody molecule.
Enzymes other than peroxidases can be directly combined ;~
with the antibodies similarly to the methods of combining
peroxidases, and known methods which achieve such combining
include body fluids or, for example, the glutaraldehyde
:
``~. , ,~, , "' ' ,- "., ~ ' . ',, , " ", !~
` ~ ~.' ': . ; ' ' ' . . , . : '' ,' ' ' ., . ' , ' ' ' ' . "``' . '
WO91/0~87~ PCT/JP90/~1726
~ 18 -
method, the periodic acid method and the water-soluble
carbodiimide method.
Test samples used in the assay system of the present
invention include humors such as urine, serum, plasma and
cerebrospinal fluid, extracts of animal cells, and culture
supernatants thereof.
As an example of the assays of the present invention, a
case is hereinafter described in detail in which peroxidase
is used as the labeling agent, but the present invention is
not limited to peroxidase.
(1) First, a test sample containing the bFGF protein to
be assayed is added to the antibody held on a carrier to
conduct antigen-antibody reaction, and then the conjugate of
the peroxidase with the anti-bFGF protein antibody obtained
above is added thereto, followed by reaction.
(2) The substrate of the peroxidase is added to the
reaction product obtained in (l), and then the absorbance or
the fluorescent intensity of the resulting substance is
measured, thereby knowing the enzyme activity of the above
reaction product.
(3) The procedures described in ~l) and t2) are
preliminarily carried out for the standard solution of the
bFGF protein of a known amount to prepare a standard curve
showing the relation between the amount of the bFGF protein
and the absorbance or the fluorescent intensity thereof.
t4) The absorbance or the fluorescent intensity
obtained for the test sample containing the bFGF protein of
WO~1/098~ PCT/JP90/Ot726
- 19 - P~ r~ 7,~ ~.
an unknown amount is applied to the standard curve to
determine the amount of the bFGF protein in the test sample.
In order to purify the bFGF protein, the purified
antibody of the present invention is coupled with a suitable ~-~
carrier such as activated agarose gel beads according to
conventional methods, and packed in a column. Then, a
sample containing the crude bFGF protein, such as a culture
supernatant or disrupted cells, is loaded onto the antibody
affinity column to allow the sample to be adsorbed thereby,
followed by washing. Then, elution is carried out with a
chaotropic reagent such as potassium thiocyanate ~KSCN) or
under such acescent conditions that the bFGF is not
inactivated. Thus, the bFGF protein can be efficiently ~ ;
purified.
The antibody column can be prepared by coupling the
monoclonal antibody of the present invention, which is, for ``
example, purified from ascites or other humors inoculated `~
with the hybridoma cells, with an appropriate carrier.
Any carrier may be used as long as the bFGF protein is
20 specifically efficiently adsorbed thereby after coupling and ~`
suitable elution is thereafter possible. Examples of such
carriers include agarose gels, cellulose and acrylamide
polymers. By way of example, polyacrylamide gel beads in
which primary amines of the proteins are activated so as to
be easily combinable, such as Af~i-Gel 10 ~Bio RAD), are
conveniently used according to the following method. The
antibody is reacted with Affi-Gel 10 in a buffer solution
.. , : . ,- ,~ ,:: ::. :.. . ., , . , . . . . , : . ,
w09l~0987s PCT/JP90/~1726
- 20
such as a bicarbonate solution having a concentration of
about 0.001 to l M, preferably about 0.1 M. The r~action is
conducted at about 0 to 20C at a broad p~ range for about
10 minutes to about 24 hours, preferably at about 4C at a
pH of about 3 to lO for about 4 hours. With respect to the
mixing ratio of the antibody to Affi-Gel lO, the larger
amount of antibody is mixed with Affi-Gel 10, the larger
amount of antibody becomes combined therewith, within the
range up to about 50 mg of antibody per 1 ml of Affi-Gel 10.
The antibody may ~herefore be mixed with Affi-Gel lO in any
ratio within this range. However, about lO to 30 mg of the
antibody is conveniently used, considering the combining
efficiency and the purification efficiency in affinity
chromatography. The antibody-carrier combined material thus
formed is thoroughly washed with the buffer solution used
for the reaction. Then, residual unreacted active groups
are blocked by allowing the washed material to stand for
several days, by adding a compound containing a primary
amine such as ethanolamine-hydrochloric acid or glycine
~ thereto to a final concentration of about 0.05 to 0.10 M,
followed by reaction at about 4C for about 1 to 4 hours, or
by reacting a protein such as 1 to 5% bovine serum albumin
~SA) therewith at 4C overnight. ~he combined material
thus treated is packed in an appropriate column to form the
antibody column.
In purifying a sample with the above antibody column,
the bFGF protein-containing sample is dissolved in a buffer
WO91/09875 PCT/JP90/~1726
- 21 -
7~?
solution having a pH around neutrality such as phosphate
buffer or Tris-hydrochloric acid buffer, followed by
adsorption by the antibody column. Then, the column is
washed with the same buf~er, and then the bFGF protein is
S eluted. As eluents, the following solutions are commonly
used: weakly acidic solutions such as acetic acid solutions,
solutions containing polyethylene glycol, solutions
containing peptides more easily combinable with the antibody
than the sample, high concentration salt solutions and their ~;
combined solutions. Solutions which do not so promote the
degradation of the bFGF protein are preferred.
Eluents are neutralized with buffer solutions by
conventional methods. The above purification procedure can
be repeated again as needed.
15Thus, the substantially pure bFGF protein substantially
free from pyrogens and endotoxins can be obtained. The
substantially pure bFGF protein of the present invention
contains the bPGF protein in a concentration of 90% tw/w) or
more, and preferably in a concentration of 95~ (w/w) or
more.
The monoclonal antibodies of the present invention
immuno-neutralize the biological activity of ~he bFGF
proteins at low concentrations and have high binding
sensitivity with the bFGF proteins, so that they can be used
as therapeutic drugs for treatment of diseases such as
cancer, and as reagents for assaying the bFGF proteins.
Mouse 3H3 cells obtained in Example 2-~4) described
.: .- .... . :: , - : : : . : , .. . - . - -, : . - . . .: ,.... . .. . .
,;,;,: : ' ~ ' : : :: : . : . : , '. ' ' :: : .:: .:: : : : . : , :: : ' : : . ., :: . : . :: :, : , , :,: . .
wosl/~98~5 PCT/JP9~/01726
- 22 -
~ r~
below was deposited with the Institute for Fermentation,
Osaka, Japan tIFO) under the accession number IFO 50216 on
November 10, 1989. The above cells were also deposited with
the Fermentation Research Institute, Agency of Industrial
Science and Technology, Ministry of International Trade and
Industry, Japan (FRI) under the accession number FERM BP-
2658 on November 14, 1989.
Anti-bFGF monoclonal antibodies MAbl2, MAb52 and MAb98
described in the following Examples can be produced by the
methods described in Hybridoma, 8, 209-221 (1989) and
European Patent Publication No. 288,687 the disclosures of
which are herein incorporated by reference. The recognition
site of MAbl2 is included in the amino acid sequence of ~rom
position 1 ~the N-terminus) to position 9 of bFGF, and the
recognition sites of MAbS2 and MAb98 are included in the
amino acid sequence of from position 14 to position 4~ of
bFGF. Recombinant human aFGF which was produced by the
methods described in Reference Example 1 was used.
Recombinant human bFGF (rhbFGF) which was produced by
the methods described in Iwane et al., Biophys. Biochem.
~ Res. Commun. 146, 470 ~1987) and European Patent Publication
; No. 237,966 was used.
rhbFGF mutein CS23 which was produced using
transformant Escherichia coli MM294/pT~762 (IFO 14613, FERM
BP-1645) by the methods described in Sano et al., BioPhys.
Biochem. Res. Commun. 151, 701 (1988) and European Patent
Publication No. 281,822 was used. The above E. coli
wosl/09~7s PC~/JP90~01726
- 23 -
MM294/pTB762 was deposited with IFO under the accession
number IF0 14613 on May 27, 1987. This transformant was
also deposited with FRI under the accession number FERM
P-9409 on June 11, 1987. This deposit was converted to the
deposit under the ~udapest Treaty and the transformant is
stored at the YRI under the accession number FERM BP-1645.
Escherichia coli MM294(DE3)/LysS,pTB762 which is
. produced in Reference Example 1 was deposited with IF0 under
the accession number IF0 14936 on September 12, 1989. This
transformant was also deposited with FRI under the accession
number FERM BP-2599 on September 20, 1989.
The following examples are given to illustrate
embodiments of the present invention as it is presently
preferred to practice. It will be understood that the
examples are illustrative, and that the invention is not to
be considered as restricted except as indicated in the
appended claims.
Examples
Reference Example 1 (Preparation of Recombinant Human aFGF)
Human aFGF was produced by the ~ollowing method, with
reference to the methods described in BiotechnoloqY, 5, 960
~1987) and ICU5 Short Report, vol. 8, Advances in Gene
Technology; Protein ~ngineering and Production, Proceedings
of the 1988 Miami Bio/Technology Winter Symposium, page 11~,
IRL Press.
(a) Construction o~ Expression Plasmid
Plasmid pTB917 obtained by incorporating chemically
.
~,
, ...... . . . .
WO9l/~9875 . pCT/JPgo/01726
~ 24 -
synthesized human aFGF DNA (Fig. 1) into pUC18 [Methods in
Enzymoloqy 101, 20-78 (1983)] was digested with BspMI, and
the cleaved sites were changed to flush ends by reaction
with DNA polymerase large ~ragment, followed by digestion
with BamHI to produce a 0.45-kb DNA fragment.
As a vector DNA, there was used pET3c [F. W. Studier et
al., J. Mol. Biol., 189, 113-130 (1986)] carrying a ~10
promoter for a T7 phage. pET3c was cleaved with NdeI and
the termini thereof were made flush by treatment with large
fragment. Then, an NcoI linker, 5'-CCATGG-3', was ligated
thereto with T4 DNA ligase. The resulting plasmid was
cleaved with NcoI, and the cleaved ites were made flush with
DNA polymerase large fragments, followed by cleavage with
Bam~I to remove the sequence of S10. Then, the above
0.45-kb blunt BspMI-BamHI fragment was incorporated
thereinto with T4 DNA ligase to obtain pTB975 (Fig. 2).
~b) Expression of Human aFGF cDNA in E. coli
~Phage DE3 ~F. W. Studier et al., J. Mol. Biol. 189,
113-130 (1986)~ in which an RNA polymerase gene of the T7
phage was incorporated into E. coli strain MM294 was
lysogenized, and plasmid pLysS [F. W. Studier et al., J.
Mol. Biol. 189, 113-130 ~1986)1 having a lysozyme gene of
the T7 phage was further introduced thereinto to prepare E.
coli MM294~DE3)/pLysS. This strain was transformed with
pTB975 to give E. coli MM294(DE3)/pLysS, pTB975~IFO 14936,
FERM BP-2599).
This strain was cultivated in a medium containing 35
WO9l/~9875 PCT/3P9~/Ot726
- 25 ~
~g/ml of ampicillin and 10 ~g/ml oE chloramphenicol. When
the turbidity reached 170 Kletts, isopropyl-~-D-
thiogalactoside (IPTG) was added thereto to a final
concentration of 0.5 mM, and cultivation was further
continued for 3 hours. The cells were collected by
centrifugation and washed with ice-cooled PBS. Then, the
cells were collected again and stored at -20C until their `
use.
(c) Purification of Human aFGF
The cells collected from a one liter culture were
suspended in 100 ml of an ice-cooled solution [10 mM
Tris-HCl tpH 7.4), 10 mM EDTA, 0.6 M NaCl, 10% sucrose, 0.25
mM PMSF], and egg white lysozyme was added thereto to a
concentration of 0.5 mg/ml. The mixture was allowed to~
stand in ice for 1 hour, followed by incubation at 37C for
5 minutes. Then, the product was subjected to
ultrasonication twice under ice cooling for 20 seconds, and
then centrifuged at 18 Krpm at 4C for 30 minutes ~Sorvall)
to obtain a supernatant. This supernatant was mixed with
200 ml of an ice-cooled solution [20 mM Tris-HCl ~pH 7.4), 1
mM EDTAI~ and the mixture was loaded onto a heparin
Sepharose column (2.5 cm diameter X 4 cm) equilibrated with
a buffer 120 mM Tris-HCl (pH 7.4), 1 mM EDTA]. The column
was washed with 150 ml ~f a solution ~20 mM Tris-HCl (pH
7.4), 1 mM EDTA, 1.5 M NaCl], and then the protein was
eluted with an eluting solution ~20 mM Tris-HCl (pH7.4~, 1
mM EDTA, 1.5 M NaCl]. The eluate was fractionated into 6 ml
WO91/098~5 PCT/JPgO/01726
- 26 -
t ~ ., ~J ~
portions and OD28~ was monitored to collect the second peak
fraction (8th to 11th, the total amount: 24 ml) ~Fig. 3).
Twenty-two ml of the eluate was mixed with an equal volume
of solution 20 mM Tris HCl (pH 7.4), 1 mM EDTA, 2 M
(NH4)2SO~), and the mixture was loaded onto a phenyl
Sepharose column (2.5 cm diameter X 8 cm) equilibrated with
a buffer [20 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1 M (~H4)2S04]
(flow rate: 0.5 ml/min). The column was washed with 20 ml
of the same buffer, and eluted with a linear gradient of 0
to l M ammonium sulfate tflow rate: 0.5 ml/min, gradient
time: 200 minutes). The eluted fractions 40 to 55 ~Fig. 4)
were collected as purified human aFGF.
(d) Reverse-Phase C4 HPLC
A l.2 mg/ml solution of the purified human aFGF was
mixed with 0.25 ml of 0.1% trifluoroacetic acid (TFA), and
the mixture was applied to a reverse-phase C4 column
lVYDAC). The column was eluted with a linear gradient of 0
to 90% acetonitrile in 0.1% TFA to examine an elution
pattern. The flow rate was l ml/min and the gradient time
was 60 minutes (Fig. 5).
le) Bioloqical Activity
The activity of the human aFGF was determined by
assaying the incorporation of ~3H] thymLdine as an
indication o~ the DNA synthesis induction of mouse BALB/c3T3
cells (ATCC No. CRL 6587) according to the method of Sasada
et al. [Sasada et al., Mol. Cell. Biol. 8, 588-594 11988)].
When the test sample was added, a heparin lSigma, Grade I)
WO91/09B7S PCT/JP90~01726
- 27 ~ , 7( ~
solution was incorporated in the medium and the test sample
as required.
Reference Example 2 ~ .
(a) Construction of Plasmid pTB1000 for ExPression of Human
bFGF Mutein
Plasmid pTB762, obtainable from E. coli MM294tDE3)/
Lys,pTB672 (IFO 14936, FERM BP-2599), and described in
Japanese Patent Unexamined Publication (Laid-Open) ~o.
2-193/1990 (corresponding to European Patent Publication No.
281,822) was cleaved with restriction enzymes EcoRI and
BamHI to obtain a 0.38-kb DNA fragment containing a region
coding for human bFGF mutein CS23. Additionally, plasmid
pTB503 described in ~apanese Patent Unexamined Publication
. (Laid-Open) No. 62- 175182/1987 (corresponding to
15 EP-225,701) was cleaved with ClaI and EcoRI to obtain a 1.7-
kb DNA fragment containing an LTR of a mouse leukemia virus
(MuLV) region, an SV40-derived promoter and a splicing
region and a leader sequence of human interleukin 2.
Further, plasmid pTB675 described in Japanese Patent
~ Unexamined Publication (Laid-Open) No. 2-193/1990
~corresponding to EP-281,822) was cleaved with ClaI and
BamHI to obtain a 3.4-kb DNA fragment containing a region
coding for the C-terminal side of the human bFGF, a
3'-untranslated region thereof, a pLasmid pBR322-derived
ampicillin-re5lstant gene and a replicator in E. coli.
These three DNA fragments were ligated using T4 DNA ligase
to obtain plasmid pTB1000 (Fig. 13).
. ~ "".
~ -
~091/09~75 PCT/JP90/Ot726
- 28 -
(b) Transformation of Mouse BA~B/c3T3 Cell and Establishment
of Transformed Cell
Mouse BALB/c3T3 clone A31 cells ~T. ~akunaga et al.
Science, 209, 505-507 (1980)] were seeded on a 6 cm
diameter dish for tissue culture having DMEM medium
[Dulbecco et al. Viroloqy, 8, 396 (1959)] containing 10~
calf serum. The next day, the medium was exchanged for the
same medium. After 4 hours, 10 ~g or 1 ~g of plasmid
pTB1000 obtaine~ in the above (1) was transfected by the
calcium phosphate method ~Graham et al., ViroloqY, 52, 456
(1973)]. After transfection, cultivation was continued in
DMEM medium containing 5% calf serum. After about 3 weeks,
the formed focuses (focus forming frequency: 10 to 20/~g
DNA) were transferred into a new dish to cultivate them, and
cloning was further conducted by the limiting dilution
method. For the three transformed cell clone strains
K1000-Fl, K1000-F2 and K1000-3 thus obtainedi the FGF
activity of the culture solution and a cell extract was
assayed as D~A synthesis-promoting action to the A31 cells
in a still state. The results are shown in the following
table.
FGF activity ~ng FGF equivalent/dish)
CellCulture solution Cell extract
~ , .
R1000-Fl 0.09
K1000-F2 0.16 11.3
X1000-F3 0.09 4.5
A31 ~ 0.05 < 0.2
Each of these transformed cells showed the form of a
.; .
W091/09875 PCT/JP90/01726
- 29 ~ ) !; `'l~'7?
malignant cell gland and formed colonies on a soft a~ar
plate.
Example 1 (Immunization)
BALB/c mice ~female, 8 weeks old) were
intraperitoneally injected with 50 ~g of antigen rhbFGF
mutein CS23 (a mutein in which each of Cys residues at
- positions 70 and 88 of human bFGF were substituted for a Ser
residue) which was dissolved in Freund's complete adjuvant
(Difco). Two weeks later, the mice were intraperitoneally
given again 50 ~g of antigen rhbFGF mutein CS23 dissolved in
0.4 ml of Freund's complete adjuvant. Further 2 weeks
later, the mice were additionally immunized with 50 ~g of
antigen rhbFGF mutein CS23 dissolved in 0.4 ml of Freund's
incomplete adjuvant. Two weeks after the additional
immunization, S0 ~g of rhbFGF mutein CS23 dissolved in
physiological saline was inoculated into the caudal veins of
the mice.
ExamPle 2
(1) Cell Fusion
Three days after the final inoculation, the spleens
were removed from the mice immunized in Example 1 to obtain
cells to be used for cell fusion. These cells were
suspended in IH medium.
Mouse myeloma cells SP2/0-AG14 ~ATCC ~o. CRL 1581) were
subcultured in DMEM medium containing 10% fetal calf serum
under an atmosphere of 5% carbon dioxide and 95% air.
Cell fusion was carried out in accordance with the
-~ , : - . , : . " .. .
W~91/0987~ PCT/3P90/~1726
- 30 -
method established by Kohler and Milstein [G. Kohler and C.
Milstein, Nature, 256, 995 (1975)]. The above myeloma cells
(2 X 107 cells) were mixed with the immunized lymphocytes
(1.5 X 108 cells) obtained by the above method, and the
mixture was centrifused. Then, 1 ml of a 45% solution of
polyethylene glycol 6000 (hereinafter referred to as PEG
6000) in IH medium was dropwise added thereto. The PEG 6000
solution was preheated to 37C and slowly added for 1
minute.
Then, 1 ml of IH medium was added for 1 minute, 1 ml
for 1 minute and 8 ml for 3 minutes. The solution was
thereafter centrifuged at room temperature at 1,000 rpm for
5 minutes to remove a supernatant. The resulting cell
precipitate was suspended in 30 ml of IH medium containing
20% calf serum. The suspension was seeded on a 96-well
microtiter plate (~unc) in an amount of 100 ~l/well. One
day later, IH medium (containing 20% calf serum)
supplemented with HAT (1 X 10 4 M hepoxanthine, 4 X 10 7 M
aminopterin, 1.6 X 10 5 M thymidine) was added to the
microtiter plate in an amount of 100 ~l/well. The IH medium
supplemented with HAT is hereinafter referred to as HAT
medium. Further every 3 days, one-half the amount of the
medium was exchanged for HAT medium. The cells which thus
grew were hybrid cells.
(2) Screening of Antibody-Producing Cells
A fixing buffer [0.1 M sodium hydrogencarbonate (pH
9.6), 0.02~ sodium azide] containing 200 ng/ml of rhbFGF
, :
; - - :.,:.-. .-. .; . .
WO9l/~9875 - 31 - PCr/JP90/~1726
mutein cs23 was added in an amount of 100 ~l/well to a 96-
well polystyrene microtiter plate (Nunc). After 2 hours,
the microtiter plate was washed with a rinsing liquid (0.05
Tween 20, physiological phosphate buffer), and then 100 ~1
of the combined solution of 50 ~1 of the culture supernatant
and S0 ~1 of a buffer for dilution (0.05 M Tris-HCl buffer
pH 8.01, 1 mM magnesium chloride, 0.15 M sodium chloride,
0.05% Tween 20, 0.02% sodium azide, 0.3% gelatin) was added
to the microtiter plate. After 2 hours, the culture
10 supernatant was washed with a rinsing liquid, followed by - .
addition of the alkaline phosphatase-labeled anti-mouse IgG
goat antibody ~Bio RAD) as the second antibody. After 2
hours, the second antibody was washed with a rinsing liquid,
and then coloring reaction was conducted by ~dding a
}5 reaction substrate ~ELISA method). By this method, the
rhbFGF mutein CS23 combining activity was observed in 4
wells.
~3) Screening of anti-bFGF Immuno-Neutralizing
Antibody-Producing Cells
Human umbilical vein-derived vascular endothe}ial cells
was suspended in GI~ medium ~commercially available from .`
Wako Pure Chemical Industries, Ltd. Japan; a mammalian
serum-derived composition for animal cell cultivation,
produced by subjecting mammalian serum to purifying
treatment including an inactivation process for cantaminant
microorganisms and a salting-out/desalting process; cf. U.S.
Patent No. 4,654,304) containing 2.5~ feta} calf serum,
.
WOgl/0~875 PCT/JP90/01726
~? ~ 3 2
and the suspension was seeded in an amount of 100 ~l/well
(2,000 cells/well) to a 96-well microtiter plate. The next
day, GIT culture solution cont~ining various concentrations
of hybridoma culture supernatants, 4 ng/ml rhbFGF and 2.5%
fetal calf serum was added in an amount of 100 ~l/well, and
cultivation was conducted at 37C under an atmosphere of 5
C2 and 7% 2 for 3 days. After 3 days, the culture
solution was removed, and then GIT culture solution
containing 1 mg/ml MTT (4,5-dimethyl-2-thiazolyl-
2,5-diphenyl-2H-tetrazolium bromide) and 2.5% fetal calf
serum was added in an amount of 100 ~l/well. After
cultivation was carried out at 37C under an atmosphere of
5% C2 and 7% 2 for 4 hours, 10% sodium dodecyl sulfate
~SDS) was added in an amount of 100 ~l/ml. After 4 hours,
the absorbance at 590 nm was measured with a spectro-
photometer for 96 wells ~Titertek) ~MTT method). By this
method, the strong neutralizing activity was observed in one
well.
(4) Cloning of Hybrid Cells
The cells in this well were spread to 0.5 cell per well
on a 96-well microtiter plate on which mouse thymocytes had
preliminarily been spread as vegetative cells, and cloning
was carried out. As a result, hybridoma mouse 3H3 cells
~IFO 50216, FERM BP-2658) were obtained.
The cloned cells were stored in liquid nitrogen, adding
dimethyl sulfoxide ~DMSO) to IH medium containing 20% calf
serum to a concentration of 10%.
... .. . ...
WOgl/09875 PCT/JP90/01726
- 3
2r~ ,7~
Example 3 ~Immunoglobulin Class of Monoclonal Antibody)
The culture supernatant of 3H3 cells obtained in
Example 2-(3) were reacted with various immunoglobulin
samples by a subclass detecting kit (Rio RAD). The results
5 are shown in Table 1.
Table 1
Immunoglobulin sample 3H3 antibody
IgGl +
` I G
IgG2b
IgG3
IgM
IgA
:~ ....
In Table 1, "+" indicates that the reaction is
positive, and "-" indicates that the reaction is negative.
Table 1 shows that the antibody in the culture
2~ supernatant o~ 3H3 cells belonss to IgGl in the
i immunoglobulin class.
Example 4 (Purification of Monoclonal Antibody from
Culture Supernatant and ~scites)
~1) Purification from Culture Supexnatant
The combined solution of the culture supernatant of
mouse 3H3 cells and a combining buffer [3 M sodium chloride,
1.5 8 glycine ~pH 8.7)] in a 1:1 ratio was loaded onto a
,;,
. :
WO91/0~7~ PCT/JP90/01726
- 34 -
~ . . i; ~.. , ~ ,.
Protein A column. After washing with a combining buffer,
elution was carried out with an elution buffer [0.1 M citric
acid (pH 5)]. To the eluate was added 1 M Tris (pH a.0) to
neutralize it, followed by dialysis against physiological
phosphate buffer.
The IgG amount of the samples was determined according ~`
to the method described in Example 2-t2) in the following
manner. Various dilutions of mouse IgG whose concentration
was known and the 3H3 antibody were fixed on a 96-well
polystyrene microtiter plate with a fixing buffer. After 2
hours, the alkaline phosphatase- labeled anti-mouse IgG goat
antibody (Bio RAD) was added thereto. After 2 hours,
coloring reaction was conducted by adding a reaction
substrate (ELISA method). With respect to mouse IgGl, a
determination curve was drawn, and the IgG amount of the
samples was determined based on this curve, whereby a 60
~g/ml solution of the 3H3 antibody was prepared.
Fig. 6 shows the results of the antibody titer to
rhbFGF mutein CS23 measured for the monoclonal antibody thus
purified, according to the method described in Example 2-
(2). One ~g/ml of the rabbit anti-bFGF polyclonal antibody
was fixed and rhbFGF mutein CS23 was added thereto. Then, 1
~ig/ml of the 3H3 antibody was added and 1 ~g/ml of the
alkaline phosphatase-labeled anti-mouse IgG goat antibody
25 Wa5 further added thereto. By this method, rhbFGF mutein
CS23 could be detected up to 3 ng/ml.
WO91/09X~5 PCT/JP9~/017~6
- 35 -
~ r~
(2) Purification from Ascites
Further, the antibody was purified from ascites. The
mouse 3H3 cell strain was injected illtO the mice (Balb/c).
IgG was purified from the ascites according to conventional
- 5 methods. Namely, 5 ml of the ascites was subjected to salt
precipitation using a 45~ saturated solution of ammonium
sulfate, and the precipitate was dissolved in borate buffer
(BBS, pH 8.5) containing 0.15 M NaC1, followed by dialysis
against BBS at 4C for 20 hours. The dialyzed solution was
applied to a DE-50 column (1 cm diameter X 60 cm, Whatman,
Great Britain), and the column was eluted with a linear
gradient of 0.1 to 0.35 M NaCl in 0.1 M phosphate buffer ~pH
; 8.0), whereby 7 mg of monoclonal antibody 3H3 was obtained
from 5 ml of the ascites.
Example 5 (Determination of Antigen Recognition Site)
The antigen recognition site of the 3H3 antibody whose
antibody titer was measured in Example 4 was examined by
competitive binding inhibition experiments. As competitive
substances, there were used human aFGF, rhbFGF, rhbFGF
mutein CS23, synthetic peptides pep 1: Pro-Ala-Leu-Pro-Glu-
Asp-Gly-Gly-Ser-Tyr [the peptide in which Tyr was added to
the C-terminus of N-terminal amino acids 2 to 10 of human
bFGF, ~3L~ Y ~ , 10, 309-317 ~1985)] and pep 2:
Leu-Pro-Met-Ser-Ala- Lys-Ser (corresponding to amino acids
141 to 147, refer to European Patent Publication No.
288,687) and heparin sodium.
~O 91/09875 PCr/.lP90/0~726
-- 36 --
The synthetic peptides, heparin sodium, human aFGF,
rhbFGF and rhbFGF mutein CS23 were diluted with the buffer
for dilution used in Example 2-~2) to a concentration of 100
~g/ml. When the synthetic peptides, heparin sodium, human
aFGF, rhbFGF and rhbFGF mutein CS23 were used as the
competitive substances, 100 ng/ml of the 3H3 antibody and
the competitive substance were suspended, and the suspension
was maintained at 37C for 60 minutes.
The amount of antibody not com~ined in this solution
was assayed by the EIA shown in Example 2-(2). The results
are shown in Table 2.
Table 2
3H3 antibody
Human bFGF +
Human aFGF
CS23 +
pepl
pep2
Heparin sodium
In Table 2, "~" indicates that the competitive
inhibition occurred, and "-" indicates that the competitive
inhibition did not occur. From these results, the
recognition site of the 3H3 antibody is considered to be the
peptide of 10th to 141st amino acids of human bFGF molecule
' :. '
- .. ~ . . , .:, . . . . .... . .. .. . .
WO9l/0~87s PCT/~P90/01726
- 37 ~ j7~
and binding site of human bFGF to human bFGF receptor or its
adjacent site, other than the heparin combining site, or a
region adjacent thereto.
Example 6 (Examination of Immuno-Neutralizing Action)
As to the 3H3 antibody purified by the method of
Example 4(1), the human bFGF-immunoneutralizing activity
against rhbFGF was studied by the MTT method of Example
2-(3) (Fig. 7). Namely, the activity was assayed by 3-day
proliferation in tbe presence of 2 ng/ml of bFGF of the
human umbilical vein endo~helial ~H W E) cell. The
proliferation inhibitory effect is shown by the absorbance
at OD-590 nm in case of adding the monoclonal antibodies
where the value is estimated as 100~ both in the presence of
the bFGF and in the absence of the monoclonal antibody.
Referring to Fig. 7, - o - indicates the results of the 3H3
antibody, and - ~ - indicates the results of normal mouse
IgG. The line of 44~ indicates the number of cells when the
bFGF was not added. When the 3H3 antibody was added, 50
ng/ml thereof hindered the proliferation of the HUVE cells
to the number of cells in the absence of the bFGF. The
normal mouse IgG did not exhibit the proliferation
inhibitory effect.
The assay was further changed to study the
proliferation inhibition effect of the HUVE cells. Namely,
the HUVE cells were seeded on a 24-well Linbro plate in an
amount o~ l X 104 cells/well ~the culture conditions
were37he same as with Example 2-t3)). The next day, 2 ng/ml
of the bFGF and the 3H3 antibody were added thereto,
~, .. . ., ... . ~ .. . . .
' ' ,. . . ' . . . '. . ., ' ' ' ' ~ . . ., ' '.' , . , ` , ,
WOgl/09X7~ PCT/JP9~/Ot726
- 38 -
followed by cultivation for 3 days and for 5 days. Then,
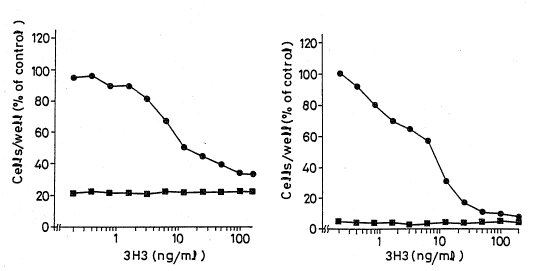
the number of cells was measured by using a Coulter counter
(Coulter Electronics, Inc.) for each case. Referring to
Figs. 8(A) and 8(B), Fig. 8(A) shows the results when
cultivation was carried out for 3 days, and Fig. 8(B) shows
the results when cultivation was carried out for 5 days.
The number of cells when the 3H3 antibody was added was
expressed in percentage, taking the number of cells when the
bFGF was added and the 3H3 antibody was not added as 100%.
In both of Fig. 8(A) and Fig. 8(B), - o - and - ~ - indicate
the effect of the 3H3 antibody in the presence of 2 ng/ml of
the bFGF and in the absence of the bFGF, respectively. The
IC50 value of the proliferation hindrance (the amount of the
antibody which hinders 50~ of cell proliferation when the
antibody is not added) shows 13.2 and 7.6 ng/ml in Fig. 8(A)
and Fig. 8(B), respéctively. Further, the 3H3 antibody did
not affect the number of cells in the absence of the bFGF.
It is clear from the above data that the 3H3 antibody has
strong immuno-neutralizing action to the biological activity
of the bFGF.
Example 7 (Preparation of Horseradish Peroxidase-Labeled 3H3
Antibody)
The purified 3H3 antibody (7 mg/ml) was dialyzed
against 0.1 M acetate buffer (pEI 4~5) containing 0.1 M NaCl
at 4C for 20 hours, followed by addition of pepsin (0.1 mg)
(Sigma, U.S.A.). Then, digestion was carried out at 37C
for 8 hours. The solution was adjusted to pH 8 with l M
i`
., : . ., . , . . ,: : . ~
wo~l/09875 PCT/JP90/01726
- 39 -
~ ,7`~
Tris to terminate the reaction. The resulting solutlon was
placed on a column of Ultrogel AcA44 (IBF, France) and
eluted with 0.02 M borate buffer ~pH 8.0) containing 0.15 M
NaCl to obtain F(ab')2. The solution containing F(ab')2 was
concentrated to 1 ml. Thenl the concentrated solution was
dialyzed against 0.1 M phosphate buffer (pH 6.0) at 4C for
20 hours, and 0.1 ml of a solution ~0.2 M mercaptoethyl-
amine, 5 mM EDTA, 0.1 M phosphate buffer (pH 6.0)] was
added, followed by reduction at 37C for 90 minutes. The
reaction solution was placed on a Sephadex G-25 fine column
(1 cm diameter X 60 cm, Pharmacia Fine Chemical, Sweden) and
eluted with an eluent ~5 mM EDTA, 0.1 M phosphate buffer (pH
6.0)] to obtain an Fab' fraction.
Additionally, 10 mg of horseradish peroxidase (HRP,
Behringer Manheim, West Germany) was dissolved in 1.5 ml of
0.1 M phosphate buffer (pH 7.0), and 3.5 mg of
maleimidobutyloxy)succinimide ~GMBS) was dissolved in
100 ~1 of N,N-dimethylformamide ~DMF). The solution of GMBS
in DMF was added to the HRP solution, followed by stirring
at 30C for 60 minutes. Then, the resulting solution was
placed on the Sephadex G-25 fine column ~1.2 cm diameter X
60 cm) and eluted with 0.1 M phosphate buffer ~pH 7.0) to
obtain maleimide group- introduced HRP ~maleimidated HRP).
Fab' and maleimidated HRP were mixed with each other to a
mol ratio of 1:1, followed by reaction at 4C for 20 hours.
The reaction solution was applied to an ultrogel AcA44
column and eluted with 0.1 M phosphate buffer ~pH 7.0) to
uosl/os~75 PCT/JP~0/01726
~ q ~ - 40 -
obtain an enzyme- labeled antibody (3H3-HRP).
Example 8 (preparation of Antibody-Sensitized Plate)
MAbl2 or a 50-50 mixture of MAb52 and MAb98 was diluted
with 0.1 M carbonate buf~er (pH 9.6) to a concentration of
10 ~g/ml. The resulting solution was poured in an amount of
100 ~1/well into an immunoplate for EIA ~Maxisoap: Nunc,
Denmark), followed by standing at 4C overnight to sensitize
or fix the antibody to the plate. After washing the plate
with 0.01 M phosphate buffer (pH 7.0) containing 0.15 M
NaCl, 0.01 M phosphate buffer (pH 7.0) containing 0.1~ BSA
was poured into each
well. Then, the plate was stored in a chilled place until
its use.
Example 9 (Assay of Human bFGF)
1. Preparation of Calibration Curve
A. Reaqents
~1) Enzyme-labeled antibody obtained in Example 7,
(2) Antibody-sensitized microtiter plate obtained in
Example 8,
(3) Recombinant human bFGF ~rhbFGF): 0-50 ng/ml,
~4) Buffer AE0.02 M phosphate buffer (pH 7.0)
containing 0.15 M NaCl],
Bufer B~0.02 M phosphate buffer ~pH 7.Q) containing
25% Blockace ~blocking agent prepared from milk protein)
(Dainippon Pharmaceutical), 0.15 M NaCl],
~ 5) Peroxidase substrate solution ~sodium citrate
buffer ~pH 5.5) containing 0.02~ hydrogen peroxide and 0.15%
W091/09875 PCT/JPgO/01726
f. ~ r~
o-phenylenediamine],
Enzyme reaction-terminating solution(2N sulfuric acid).
B. Assay
Into each well of the antibody-sensitized plate
obtained in Example 8, 100 ~1 of a solution of the bFGF (0.1
to lOOOpg/ml) in buffer B was added, followed by reaction at
4C for 24 hours. After washing each well with buffer A,
100 ~1 of an enzyme- labeled antibody solution diluted 200
times with buffer B was added to each well, followed by
further reaction at 25C for 2 hours. Each well was washed
with buffer A, and 100 ~1 of the peroxidase substrate
solution was added thereto, followed by reaction at 25C for
30 minutes. Then, 100 ~1 of the enzyme reaction-terminating
solution was added thereto to terminate reaction. The
absorbance at 492 nm was thereafter measured by using an
automatic colorimeter for microtiter plates (MTP-32.
Corona). Fig. 9 shows the relationship between the
concentration of the hbFGF and the absorbance. Referring to
Fig. 9, - o - and - ~ - indicate the relationship between
the concentration of the hbFGF and the absorbance when the
microtiter plate sensitized with MAbl2 was used and when the
microtiter plate sensitized with the 50-50 mixture of MAbS2
and MAb98 was used, respectively. It was revealed that 20
pg/ml of the bFGF protein could be detected, when the plate
was sensitized with MAbl2.
2. bFGF Immunoassay Kit and Assay of bFGF
The amount of the bFGF in the test samples was assayed
Wo9l/09x7s ~T/JP90/01726
~ 2 -
by using the following bFGF immunoassay kit according to the
following procedure:
A. Reagents
(1) Enzyme labeled antibody (3H3 antibody-HRP) obtained
in Example 7,
(2) Antibody-sensitized microtiter plate obtained in
Example 8,
(3) Recombinant human bFGF (rhbFGF): 0-50 ng/ml,
(4) Buffer A C0.02 M phosphate buffer ~pH 7.0)
containing 0.15 M NaCl],
Buffer B~0.02 M phosphate buffer (pH 7.0) containing
25% Blockace, 0.15 M NaCl],
~S) o-Phenylenediamine,
(6) Buffer D used for dissolution of o-phenylenediamine
10.1 M citrate buffer (pH 5.5) containing 0.02
hydrogenperoxide and 0.005% thimerosal],
~7) Enzyme reaction-terminating solution (2N sulfuric
: acid),
B. Ass~y ~ .
Each well of the antibody-sensitized plate obtained in
Example 8 was washed with buffer A, and 100 ~1 of a standard
solution of the bFGF in buffer B or a solution of the test
sample diluted with bufer B was poured thereinto, ollowed
by reaction at 4C for 24 hours. After washing each well
25 with buffer A, 100 ~1 of an enzyme-labeled antibody solution ~:
diluted 200 times with buffer B was added to each well,
followed by further reaction at 25C for 2 hours. After
:: .
wn ~1/0987~
PCT/JP9OtOt726
washing each well with buffer A, 100 ~1 of a 0.15~ solution
of o-phenylenediamine in buffer D was added thereto,
followed by reaction at 25C for 30 minutes. Then, 100 ~1
of the enzyme reaction-terminating solution was added
thereto to terminate reaction. The absorbance at 492 nm was
thereafter measured by using an automatic colorimeter for
microtiter plate ~MTP-32. Corona). The calibration curve of
the standard bFGF was prepared, and the bFGF concentration
was determined from the absorbance obtained for the test
10 samples.
Example 10 (Influence of Heparin in hbFGF Assay System)
In Example 9-1, heparin was added to buffer B to a
concentration of 0, 1, 10 or 100 ~g/ml, when the standard
hbFGF was diluted. Then, the concentration of the hbFGF was
assayed in the manner of Example 4-(1). As a result, there
were observed no considerable changes in the standard curves
with the presence of heparin, as shown in Figs. 10(1) and
10(2).
Referring to Figs. 10(1) and 10(2), Fig. 10~1)
indicates the results when MAbl2 was fixed, and Fig. lOt2)
indicates the results when the 50-50 mixture of MAb52 and
MAb98 was ~ixed. Further in each igure, - -, - o ~
- and - ~ - indicate the results when heparin concentrations
are 0, 1, 10 and 100 ~g/ml, respectively.
5 Example 11 ~Reactivity of Acid-Modified hbFGF in hbFGF Assay
Systems)
A 20 ~1 solution of the hbFGF (in 20 mM Tris-HCl buffer
WO9l/09875 PCT/JP90/01726
- 44 -
~" . ~.'`_J ~);
(pH 7.4) containing hbFGF at a concentration of 200~g/ml and
containing 1 M NaC1 was added in an amount of 20 ~1 to laO
~1 of 1 M acetate buffer, followed by incubation at 25C for
0, 1, 3 or 10 minutes. 400 ~1 of 1 M Tris was added thereto
to neutralize it, and ~hen the resulting solution was
diluted with buffer B used in Example 9-1. Then,
calibration curves were prepared for respective incubation
times according to the method of Example 9-1, as shown in
Figs. 11(1) and 11(2). Referring to Figs. 11(1) and 11(2),
Fig. 11(1) indicates the results when MAbl2 was solidified,
and Fig. 11(2) indicates the results when the 50-50 mixture
of MAb52 and MAb98 was solidified. Further in each figure,
- O -, - O -, - a - and - ~ indicate the results when the
hbFGF was incubated at pH 4 for 0, 1, 3 and 10 minutes,
respectively. Figs. 11(1) and 11~2) show that the hbFGF
modified at pH 4 is reduced in reactivity to about 1~ by
incubation for 10 minutes in each assay system. The assay
systems of Example 9 are assay systems by which only the
native hbFGF can be assayed.
Example 12 (Assay of bFGF in Various Cells)
A375 (human melanoma), A431 ~human sguamous cell
carcinoma)~ A549 ~human lung cancer), SK-Hepl ~human hepatic
cancer) ~all o them were supplied by ATCC, U~S.A.) and
human umbilical vein endothelial cells were each scraped in
an amount of 2 to 6 X 107 cells by a cell scraper. The
cells were washed twice with a phosphate buffered saline
~PBS, pH 7.2) containing 0.15 M NaCl, and then 0.6 ml of PBS
'~.
W~gl/0~75 PC~/JP~0/~1726
.
- 45 - ~r ~ ?
was added thereto, followed by ultrasonication under ice
cooling for 25 seconds. After centrifugation at 15,000 rpm,
the concentration of the bFGF in the supernatant was
determined according to the method of Example 9-2.
The results are shown in Table 3. In Table 3, A shows
the results when MAbl2 was fixed, and ~ shows the results
when the 50-50 mixture of MAb52 and MAb98 was fixed.
Table 3 Amount of bFGF in Cell
~' .
Amount of bFGF
: A B
~ame of Cell(pg/106 cells)(ng/106 cells)
A375 1.4 4.0
A431 1.5 0.5
A549 8.4 8.5
SK-Hepl 4 . 0 11. 7
HUVE 11 . 7 0 . 8 4
From Table 3r it is observed that the difference in
measured amount of bFGF between the A assay system and the B
assay system is great. From this fact, the possibility is
considered that the N-terminus of the bFGF is cleaved during
extraction operation, or that the ~-terminus of the actually
produced bFGF is cleaved or masked.
Example 13 (Antitumor Effect to KlO00 Tumor)
The KlO00-Fl cells obtained in Reference Example 2 were
~.
wosl/0987s PCT/JP90/01726
- 46 -
sub ~ a~o~ impLanted in an amount of 3 X lO6 cells/mouse
into nude BALB/c mice. Three days after implantation, the
3H3 antibody obtained in Example 4 and nonimmune mouse IgG
were given to the caudal veins in a dose of 200 ~g/mouse/day
continuously for 5 days. After implantation, the diameter
of the tumors was measured, and the volume thereof ~=0.5 X
(major axis) X (minor axis)2] was calculated. The results
are shown in Fig. 12. Referring to Fig. 12, - ~ -, - 4 -
and - ~ - indicate the results for an untreated control
group, a nonimmune mouse IgG-given group and a 3H3
antibody-given group, respectively. The 3H3 antibody
exhibited antitumor effect against the RlO00 tumor (14 days
after implantation, the 3H3 antibody-given group is
decreased in tumor volume to 34% of that of the untreated
control group). The antitumor effect of nonimmune mouse IgG
was not observed. ;
,', ~';'~
,:
., . , ' :
` ,
' '~
~
.
.:
~ '
;~,
~91/0987~ PCT/JP90/017Z6
47
Incorporated by refere~ce
~ature 249, 123 ~1974)
Biophys. Res. Commun. 151, 701 (1988)
European Patent Publication No. 281,822
FEBS Letters 213, 189 ~1987)
Biophys. Res. Commun. 146, 470 (1987)
European Patent Publication No. 237,966
J. Immun. Method 80, 55 tl985)
Nature 256, 495 (1975)
J. Am. Med. Assoc. 199, 549 (1967)
Metabolism, 8, 696 (1971)
Hybridoma, 8, 209-221 (1989)
European Patent Publication No. 288,687
Biotechnology, 5, 960 (1987)
ICUS Short Report, vol. 8, Advances in Gene Technology;
Protein Engineering and Production, Proceedings of the
1988 Miami Bio/Technology Winter Symposium, page 110, IRL
Press
J. Mol. Biol., 189, 113-130 (1986)
Mol. Cell. Biol. 8, 588-594 ~1988)
Methods in Enzymology 101, 20-78 (1983)
~apanese Patent Unexamined Publication tLaid-Open) No.
2-193/1990 ~European Patent Publication No. 281,822)
Japanese Patent Unexamined Publication (Laid-Open) No. 62-
175182/1987 (EP-225,701)
Science, 209, 505-507 (1980)
Virology, 8, 396 (1959)
Virology, 52, 456 (1973)
U.S.P. No. 4,654,304
Regulatory Peptides, 10, 309-317 (1985)