Note: Descriptions are shown in the official language in which they were submitted.
WO 91 / 10734 PCT/CA91 /00009
'~ ~.~ ~:~ j~
i
~N~RO~s E~~BeTR r,'~ ~P~F~ fw~~T~ asirn~sls GE11~E
Mu~~T~oNS ~T ~~~ot~s ~os~~yo~rs o~ r~~ ~~rE
FaEa~n ~F T$E ~~avl~~oN
The present invention relates generally to the
cystic fibrosis (CF) gene, and, more particularly to the
identification, isolation and cloning of the DNA sequence
corresponding to mutants of the CF gene, as well as their
transcripts, gene products and genetic information at
exon/intron boundaries. The, present invention also
relates to methods of screening for and detection of eF
carriers, CF diagnosis, prenatal CF screening and
diagnosis, and gene therapy utilizing recombinant
technologies and drug therapy using the information
derived from the DNA, protein, and the metabolic function
of the protein.
83~rCRGRO1~1D of 'fF~IE Tl~EId'1°TON
Cystic fibrosis (CF) is the most common severe
autosomal recessive genetic disorder in the Caucasian
population. It affects approximately 1 in 2000 live
births in Narth America (Boat et al, The Metabniir. ua~is
of Tnhgrited DiRp~~r~
6th ed, pp 2649-2680, McGraw Hill,
NY (1989)). Approximately 1 in 20 persons are carriers of
the disease.
Although the disease was first described in the late
193o~s, the basic defect remains unknown. The major
symptoms of cystic fibrosis include chronic pulmanary
disease, pancreatic exocrine insufficiency, and elevated
sweat electrolyte levels. The symptoms are consistent
with cystic fibrosis being an exocrine disorder.
3o Although recent advances have been made in the analysis
of ion transport across the apical membrane of the
epithelium of CF patient cells, it is not clear that the
abnormal regulation of chloride channels represents the
primary defect in the disease. Given the lack of
understanding of the molecular mechanism of the disease,
an alternative approach has therefore been taken in an
attempt to understand the nature of the molecular defect
'WO 91/1a734 PC.T/~CA911OQ009
2
through direct cloning of the responsible gene on the
basis of its chromosomal location.
However, there is no cleax phenotype that directs an
approach to the exact nature of the genetic basis of the
disease, or that allows for an identification of the
cystic fibrosis gene. The nature of the CF defect in
relation to the population genetics data has not been
readily apparent. Both the prevalence of the disease and
the clinical heterogeneity have been explained by several
different mechanisms: high mutation rate,
heterozygote advantage, genetic drift, multiple loci; and
reproductive compensation.
Many of the hypotheses can not be tested due to the
lack of knowledge of the'basic defect. Therefore,
alternative approaches to the determination and
characterization of the CF gene have focused on an
attempt to identify the location of the gene by genetic
analysis.
Linkage analysis of the CF gene to antigsnic and
protein markers was attempted in the 1950'x, but no
posit'ave results were obtained [Steinberg et al ~T.
FIum<, Genet. 8: 162-176, (I956); St~inberg and Morton Am.
J. Hum. Genet 8: 177-189, (1956); Goodchild et al J. Med.
G a 7: 417-419, 1976.
More recently, it has become possible to use RFLP's
to facilitate linkage analysis. The first linkage of an
RFLP marker to the CF gene was disclosed in 1985 [Tsui et
al. ciencg 230: 1054-1057, 1985) in which linkage was
found between °the CF gene and an uncharacterized marker
DOCRI-917. The association was found in an analysis of
39 families with affected CF children. This showed that
although the chromosomal location had not been
established; the location of the disease gene had been
narrowed to about 1% of the human genome, or about 30
million nucleotide base pairs.
The chromosomal location caf the DOCRI-917 probe: was
established using rodent-human hybrid cell lines
CA 02073441 2000-12-07
3
containing different human chromosome complements. It was shown that DOCR1-
917 (and therefore the CF gene) maps to human chromosome 7.
Further physical and genetic linkage studies were pursued in an attempt to
pinpoint the location of the CF gene. Zengerling et al [Am. J. Hum. Genet. 40:
228-
236 (1987)] describe the use of human-mouse somatic cell hybrids to obtain a
more
detailed physical relationship between the CF gene and the markers known to be
linked with it. This publication shows that the CF gene can be assigned to
either the
distal region of band q22 or the proximal region of band q31 on chromosome 7.
Rommens et al [Am. J. Hum. Genet. 43: 645-663, (1988)] give a detailed
discussion of the isolation of many new 7q31 probes. The approach outlined led
to the
isolation of two new probes, D7S122 and D7S340, which are close to each other.
Pulsed field gel electrophoresis mapping indicates that these two RFLP markers
are
between two markers known to flank the CF gene, MET [White, R., Woodward S.,
Leppert M., et al. Nature 318: 382-384, (1985)] and D7S8 [Wainwright, B. J.,
Scambler, P. J., and J. Schmidtke, Nature 318: 384-385 (1985)], therefore in
the CF
gene region. The discovery of these markers provides a starting point for
chromosome walking and jumping.
Estivill et al, [Nature 326: 840-845(1987)] disclose that a candidate cDNA
gene was located and partially characterized. This however, does not teach the
correct location of the CF gene. The reference discloses a candidate cDNA gene
downstream of a CpG island, which are undermethylated GC nucleotide-rich
regions
upstream of many vertebrate genes. The chromosomal localization of the
candidate
locus is identified as the XV2C region. However, that actual region does not
include
!V0 91/10731 PC1'/CA91l00009
4
A major difficulty in identifying the CF gene has
been the lack of cytologically detectable chromosome
rearrangements or deletions, which greatly facilitated
all previous successes in the cloning of human disease
genes by knowledge of map position.
Such rearrangements and deletions could be observed
cytologically and as a result, a physical location on a
particular chromosome could be correlated with the
particular disease. Further, this cytological location
could be correlated with a molecular ,location based on
known relationship between publicly available DNA probes
and cytologically visible alterations in the chromosomes.
Knowledge of the molecular location of the gene for a
particular disease would allow cloning and sequencing of
that gene by routine procedures, particularly when the
gene product is known and Toning success can be
confirmed by immunoassay of e}:pression products of the
cloned genes,
Tn contrast, neither the cytological location nor
the gene product of the gene for cystic fibrosis was
known in the prior art. With the recent identification
of MET and D7S8, markers which flanked the CF gene but
did nit pinpoint its molecular location, the present
inventars devised various novel gene cloning strategies
to approach the CF gene in accordance with the present
invention. The methods employed in these strategies
include chromosome jumping from the flanking markers,
cloning of DrIA fragments from a defined physical region
with the use of pulsed field gel electrophoresis, a
combination of somatic cell hybrid and molecular Cloning
techniques designed to isolate 1~NA fragments from
undermethylated CpG islands near CF, chromosome
microclissection and cloning, and saturation cloning of a
large number of DNA markers from the 7q31 region. By
means of these novel strategies, the present inventors
were able to identify the gene responsible for cystic
CA 02073441 2000-12-07
fibrosis where the prior art was uncertaW or, even m one case, wrong.
The application of these genetic and molecular cloning strategies has allowed
5 the isolation and cDNA cloning of the cystic fibrosis gene on the basis of
its
chromosomal location, without the benefit of genomic rearrangements to point
the
way. The identification of the normal and mutant forms of the CF gene and gene
products has allowed for the development of screening and diagnostic tests for
CF
utilizing nucleic acid probes and antibodies to the gene product. Through
interaction
with the defective gene product and the pathway in which this gene product is
involved, therapy through normal gene product supplementation and gene
manipulation and delivery are now made possible.
The gene involved in the cystic fibrosis disease process, hereinafter the "CF
gene" and its functional equivalents, has been identified, isolated and cDNA
cloned,
and its transcripts and gene products identified and sequenced. A three base
pair
deletion leading to the omission of a phenylalanine residue in the gene
product has
been determined to correspond to the mutations of the CF gene in approximately
70%
of the patients affected with CF, with different mutations involved in most if
not all
the remaining cases. This subject matter is disclosed in co-pending United
States
patent 5,776,677.
Specific aspects of the invention defined in these U.S. patent applications
are
discussed in journal articles by the inventors, namely, Science (1989) 245 No.
4922
pp 1066-1073 and Science (198~~) 245 No. 4922 pp 1073-1080. These journal
articles
discuss the CFTR gene sequence and the mutation claimed in the U.S. patent
W~ 91/10734 ~.~ ~ ~ (1 ~~ PC.°f/CA91/00009
6
With the identification and sequencing of the mutant
gene and its gene product, nucleic acid probes and
antibodies raised to the mutant gene product can be used
in a variety of hybridization and immunological assays to
screen for and detect the presence of either the
defective CF gene or gene product. Assay kits for such
screening and diagnosis can also be provided. The
genetic information derived from the intron/exon
boundaries is also very useful in various screening and
to diagnosis procedures.
Patient therapy through supplementation with the
normal gene product, whose production can be amplified
using genetic and recombinant techniques, or its
functional equivalent, is now also possible. Correction
or modification of the defective gene product through
drug treatment moans is now possible. In addition,
cystic fibrosis can be cured or controlled through gene
therapy by correcting the gene defect ,~ situ or using
recombinant or other vehiclet~ to deliver a DNA sequence
capable of expression of the normal gene product to the
cells of the patient.
According to another aspect of the invention, a
purified mutant CF gene comprises a DNA sequence encoding
an amino acid sequence for a protein where the pxotein,
when expressed in cells of the human body, is associated
with altered cell function which correlates with the
genetic disease cystic fibrosis.
According to another aspect of the invention, a
purified RNA molecule comprises an RNA sequence
corresponding to the above DNA sequence.
According to another aspect of the invention, a DNA
molecule comprises a cDNA molecule corresponding to the
above DNA sequence.
According to another aspect of the invention, a.DNA
anolecule comprises a DNA sequence encoding mutant CFTR
polypeptide having the sequenc.~a according to the
following Figure 1 for amino acid residue positions 1 to
W~O 91/10734 PCT>CA91100009
7 '~~~.y~~'~~i
w.~ L _
1480 as further characterized by a nucleotide sequence
variants resulting in deletion or alteration of amino
acids or residue positions 85, 148, 378, 455, 493, 507,
542, 549, 551, 560, 563, 574, 1077 arid 1092.
According to another aspect of the invention, a DNA
molecule comprises an intronless DNA sequence encoding a
mutant CFTR polypeptide having the sequence according to
FIgure 1 for DNA sequence positions Z to 4575 and,
further characterized by nucleotide sequence variants
resulting in deletion or alteration of DNA at DNA
sequence positions 129, 556, 621+1, 713.+1, 1717-1 and
3659.
According to anather aspect of the invention, a DNA
molecule comprises a cDNA molecule corresponding to the
above DNA sequence.
According to another aspect of the invention, the
cDNA.molecule comprises a DNA sequence selected from the
group consisting of:
(a) DNA sequences which correspond to the mutant
2o DNA sequence selected from the group of mutant amino acid
positions Of 85, 148, 178, 455, 493, 507, 542, 549, 551,
560, 563, 574, 1077 and 109? and mutant pNA seqnece
positions 129, 556, 621+1, 711+1, 1717-1 and 3659 and
which encode, on expression, for mutant CFTR polypeptide;
(b) DNA sequences which correspond to a fragment of
the selected mutant DNA sequence, including at least
twenty nucleotides;
(c) DNA sequences which comprise at least twenty
nucleotides and encode a fragm~:nt of the selected mutant
3o CFTR protein amino acid sequence;
(d) DNA sequences encoding an epitope encoded by at
least eighteen sequential nucleotides in the selected
mutant DNA sequence.
According to another aspect of the invention, a DNA
sequence selected from the group consisting of:
WO 91/10734 ~ ~ PC"~'/CA91/0~009
s
(a) DNA sequences which correspond to portions of
DNA sequences of boundaries of exons/introns of the
genomic CF gene;
(b) DNA sequences of at least eighteen sequential
nucleotides at boundaries of exons/introns of the genomic
CF gene depicted in Figure 18; and
(c) DNA sequences of at least eighteen sequential
nucleotides of intron portions of the genomic CF gene of
Figure 18.
1o According to another aspect of the invention, a
purified nucleic acid probe comprises a DNA or ~tNA
nucleotide sequence corresponding to the above noted
selected DNA sequences of groups (a) to (c).
According to another aspect of the invention,
purified RNA molecule comprising RNA sequence corres-
ponds to the mutant DNA sequence selected from the group
of mutant protein positions consisting of 85, 148, 178,
455, 493, 507, 542, 549,, 551, 560, 563, 574, 1077 and
1092 and of mutant DNA sequen~a positions consisting of
129, 556, 621+1, 711+1, 1717-1 and 3659.
A purified nucleic acid probe comprising a DNA or
RNA nucleotide sequence corresponding to the mutant
sequences of the above recited group.
According to an~ther aspect of the invention, a
recombinant cloning vector comprising the DNA sequences
of the mutant DNA and fragments thereof selected from the
group of mutant protein positions consisting of 85, 148,
178, 455, 493, 507, 542, 549, 551, 563, 574, 1077 arid
1092 and selected from the group of mutant DNA sequence
positions consisting of 129, 556, 621+1, 711+1, 1717-1
and 3659 is provided. The vec~t:or, according to an aspect
of this invention, is operatively linked to an expression
control sequence in the recombinant DNA molecule so that
the selected mutant DNA sequences for the mutant CFTR
polypeptide can be expressed. The expression control
sequence is selected from the w~roup consisting of
sequences that control the exp~:ession of genes of
wo 9mo~~a Pcre~a~gnoooo9
L~~r~~~,~e~i.3'.
prokaryotic or eukaryotic cells and their viruses and
combinations thereof.
According to another aspect of the inventian, a
method for producing a mutant CFTR polypeptide comprises
the steps of:
(a) culturing a host cell transfected with the
recombinant vector for the mutant DNA sequence in a
medium and under conditions favorable for expression of
the mutant CFTR polypeptide selected from the group of
mutant CFTR polypeptides at mutant protein positions 85,
148, 1?$, 455, 493, 507, 542, 549, 551, 560, 563, 574,
1077 and 1092 and mutant DNA sequence positions 129, 556,
621+1, 711-1 1717-1 and .3659; and
(b) isolating the expressed mutant CFTR
polypeptide.
According to another aspect of the invention, a
purified protein of human cell membrane origin comprises
an amino acid sequence ~:ncoded by the mutant DNA
sequences selected from the group of mutant protein
positions of 85, 148, 178, 455, 493, 507, 542, 549, 551,
560, 563, 574, 1077 and 1092 and from the group of mutant
DNA sequence positions 129, 556, 621+1, 711+1, 1717-1 and
3659 where the protein, when present in human cell
membrane, is associated with cell function which causes
the genetic disease cystic fibrosis.
According tc~ another aspect of the invention, a
method is provided for screening a subject to determine
if the subject is a CF carrier or a CF patient comprising
the steps of providing a biological sample of the subject
3o to be screened and providing an assay for detecting in
the biological sazaple, the presence of at least a member
from the group consisting of:
(a) mutant CF gene selected from the group of
mutant protein positions 85, 14$, 17$, 455,
493, 507, 542, 549, 551, 560, 563, 574, ~L077
and 1092 and from the group of mutant DNA
W~ 91 /10734 PCf/CrS91 /00009
~~'~'~4~
sequence positions 129, 556, 621+1, 711°~1,
1717-1 and 3659;
(b) mutant CF gene products and mixtures thereof;
(c) DNA sequences which correspond to portions of
5 DNA sequences of boundaries of exons/introns of the
genomic CF gene;
(d) DNA sequences of at least eighteen sequential
nucleotides at boundaries of exons/introns of the genomic
CF gene depicted in Figure 18; and
10 (e) DNA sequences of at least eighteen sequential
nucleotides of intron portions of the genomic CF gene of
Figure 18.
According to another aspect of the invention, a kit
for assaying for the presence of a CF gene by immunoassay
techniques comprises:
(a) an antibody which specifically binds to a gene
product of the mutant DNA sequence selected from the
group of mutant protein positions 85, 148, 178, 455, 493,
507, 542, 549, 551, 560, 563, 574, 1077 and 1092 and from
the group of mutant DNA sequence positions 129, 556,
621+1, 711+1, 1717-1 and 3659;
(b) reagent means for detecting the binding of the
antibody to the gene product; and
(c) the antibody and reagent means each being
present in amounts effective to perform the immunoassay.
According to another aspect of the invention, a kit
fer assaying for the presence of a mutant CF gene by
hybridization technique comprises:
(a) an oligonucleotide probe which specifically.
binds to the mutant CF gene having a mutz~tion at a
protein position selected from the group consisting of
85, 148, 178, 455, 493, 507 , 542, 549, 551, 560, 563,
574, 1077 and 1092 or having a mutation at a DNA sequence
position selected from the group consisting of 129, 556,
621+1, 711+1, 1717-1 and 3659;
(b) reagent means for detecting the hybridization
of the oligonucleotide probe to the mutant CF gene; and
WO 91/1073 PC'T/CA91/00009
11
~, ~ '°7. c..~ i ,
(c) the probe and reagent means eac2i"~ing present
in amounts effective to perform the hybridization assay.
According to another aspect of the invention, ari
animal comprises an heterologous cell system. The cell
system includes a recombinant cloning vector which
includes the recombinant DNA sequence corresponding to
the mutant DNA sequence which induces cystic fibrosis
symptoms in the animal.
According to anpther aspect of the invention, in a
polymerase chain reaction to amplify a selected axon of a
cDNA sequence of Figure Z., the use of oligonucleotide
primers from intron portions near the 5~ and 3'
boundaries of the selected axon of Figure 18.
BRTBF DBBCRIP°$°aoN of B'~~' lpR1',WIN~;B
Figure 1 is the nucleotide sequence of the CF gene
and the amine acid sequence of the GFTR protein amino
acid sequence with n indicating mutations at the 507 and
508 protein positions.
Figure 2 is a restriction map of the CF gene and the
2o schematic strategy used to chromosome walk and jump to
the gene.
Figure 3 depicts the physical map of the region
including and surrounding the CF gene generated by pulsed
field gen electrophoresis. panels A, 8, C, and D shoWi
hybridization data for the restriction enzymes Sal I, Xho
I, Sfi T, and Nae I, respectively generated by
representative genomic and cDNA probes which span the
region. The deduced physical maps for each restriction
enzyme is shown below each panel. A composite map of the
entire MET-D7S8 interval is shown in panel E (J. M.
Rommens et al., Am. J. Hum. Genet. 45:932-947., 1990).
The open boxed segment indicates the portion cloned by
chromosome walking and jumping, and the filled arrow
indicates the gortion covered by the CF' transcript.
Figures 4A, 4B and 4C show the detection of
conserved nucleotide sequences by cross-species
hybridization.
wo ~m1o~34 1PCT/~CA97J00009
Figure 4D is a restriction map of overlapping
segments of probes E4.3 and H1.6.
Figure 5 is an RNA blot hybridization analysis using
genomic and cDNA probes. Hybridization to RNA of: A-
fibroblast with cDNA probe ~-2; B-trachea (from
unafflicted and CF patient individuals), pancreas, liver,
HL6o cell line and brain with genomic probe CF16; C-T84
cell line with cDNA probe 10-1.
Figure 6 is the methylation status of the E4.3
1o cloned region at the 5~ end of the CF gene.
Figure ? is a restriction map of the CFTR cDNA
showing alignment of the cDNA to the genomic DNA
fragments.
Figure 8 is an RNA gel blot analysis depicting
hybridization by a portion of the CFTR cDNA (alone 10-1)
to a 6.5 kb mRNA transcript in various human tissues.
Figure 9 is a DNA blot hybridization analysis
depicting hybridization by the CFTR,cDNA clones to
genomic DNA digested with EcoRI and Hind III.
Figure 10 is a primer extension experiment
characterizing the 5~ and 3~ ends of the CFTR cDNA.
Figure 12 is a hydropathy profile and shows
predicted secondary structures of CFTR.
Figure Z2 is a dot matrix analysis of internal
homologies in the predicted CFTR polypeptide.
Figure 13 is a schematic model of the predicted CFTR
protein.
Figure 14 is a schematic diagram of the restriction
fragment length polymorphisms (RFLP~s) closely linked to
the CF gene where the inverted triangle indicates the
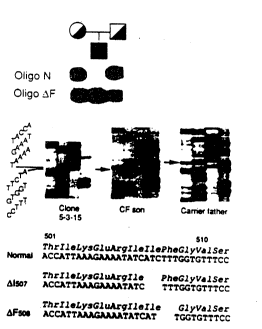
location of the F508 3 base pair deletion.
Figure 15 represents alignment of the most conserved
segments of the extended NBFs of CFTR with comparable
regions of other proteins.
Figure 16 is the DNA sequence around the F50~
deletion.
WO 91/107341 PCIf/CA91/00009
r .:
I~ ;.
13
Figure 17 is a representation of the nucleotide
sequencing gel showing the DNA sequence at the F508
deletion.
Figure 18 is the nucleotide sequence of the portions
of introns and complete axons of the genomic CF gene for
27 axons identified and numbered sequentially as 1
through 24 with additional ex~ns 6a, 6b, 14a, 14b and
17a, 17b of cDNA sequence of Figure l;
Figure 13 shows the results of amplification of
l0 genomic DNA using intron oligonucleotides bounding axon
10e
Figure 20 shows the separation by gel
electrophoresis of the amplified genomic DNA products of
a CF family; and
Figure ~1 is a restriction mapping of cloned intron
and exan portions of genomic DNA which introns and exans
are identified in Figure 18.
~1~~AI~~D DE~CR~~'~IC1Y~ OF 'f81; kktEFERIt~D 1,~1~ODIl~k'NTg
,~,s. D~%F~Rt~'f~~I~Y~
In order to facilitate review of the various
embodiments of the invention and an understanding of
various elements and constituents used in,making the
invention and using same, the following definitian of
terms used in the invention description is as follows:
eF - cystic fibrosis
CF carrier - a person in apparent health whose
chromosomes contain a mutant CF gene that may be
transmitted to that personls offspring.
CF patient - a person who carries a mutant CF gene
on each chromosome, such that they exhibit the clinical
symptoms of cystic fibrosis.
CF gene - the gene whale mutant forms are associated
with the disease cystic fibrosis. This definition is
understood to include the various sequence polymorphisms
that exist, wherein nucleotide substitutions in the gene
sequence do not affect. the essential function of the gene
product. This term primarily relates to an isolated
WO 9111074 pC f/CA91 /00009
?~~3~~~~.
coding sequence, but can also include some or all of the
flanking regulatory elements and/or introns.
Genomic CF gene - the CF gene which includes
flanking regulatory elements and/or introns at baundaries
of crone of the CF gene.
CF - PI - cystic fibrosis pancreatic insufficient,
the major clinical subgroup of cystic fibrosis patients,
characterized by insufficient pancreatic exocrine
function.
CF - pS - cystic fibrosis pancreatic sufficient, a
clinical subgroup of cystic fibrasis patients with
sufficient pancreatic exocrine function for normal
digestion of food.
CFTR ~ cystic fibrosis transmembrane conductance
regulator protein, encoded by the CF gene. This
t3efbr~ition includes the protein as isolated from human or
animal sources, as produced by recombinant organisms, and
as chemically or enzymatically synthesized. This
definition is understood.ta include the various
polymorphic forms of the protein wherein amino acid
substitutions in the variable regions of the sequence
does not affect the essential functioning of the protein,
or its hydropathic profile or secondary or tertiary
structure.
DNA - standard nomenclature is used to identify the
bases.
Intronless DNA - a piece of DNA lacking internal
non-coding segments, for example, cDNA.
IRP locus sequence - (protooncogene int-1 related),
a gene located near the CF gene.
Mutant CFTR - a protein that is highly analagous to
CFTR in terms of primary, secondary, and tertiary
structure, but wherein a small number of amino acid
substitutions and/or deletions and/or insertions result
in impairment of its essential function, so that
organisms whose epithelial sells express mutant CFTR
CA 02073441 2000-12-07
rather than CFTR demonstrate the symptoms of cystic fibrosis.
mCF - a mouse gene orthologous to the human CF gene
5 NBFs - nucleotide (ATP) binding folds
ORF - open reading frame
PCR - polymerase chain reaction
Protein - standard single letter nomenclature is used to identify the amino
acids
10 R-domain - a highly charged cytoplasmic domain of the CFTR protein
RSV - Rous Sarcoma Virus
SAP - surfactant protein
RFLP - restriction .fragment length polymorphism
507 mutant CF gene - the; C'.F gene which includes a DNA base pair mutation
15 at the 506 or 507 protein position of the cDNA of the CF gene
507 mutant DNA sequence - equivalent meaning to the
507 mutant CF gene
507 mutant CFTR protein or mutant CFTR protein amino acid sequence, or
mutant CFTR polypeptide - the mutant CFTR protein wherein an amino acid
deletion
occurs at the isoleucine 506 or 507 protein position of the CFTR.
Protein position means amino acid residue position.
2. ISOLATING THE CF GENE
Using chromosome walking, jumping, and cDNA hybridization, DNA
sequences encompassing > 500 kilobase pairs (kb) have been isolated from a
region
on the long arm of human chromosome 7 containing the cystic fibrosis (CF)
gene.
This technique is disclosed in detail in the aforementioned United States
Patent
5,776,677. For purposes of convenience in understanding and isolating the CF
gene
and identifying other mutations, such as at the 85, 148, 1178, 455, 493, 507,
542, 549,
560, 563, 574, 1077 and 1092 amino acid residue positions, the technique is
reiterated
here. Several transcribed sequences and conserved segments have been
identified in
this region. One of these corresponds
WO 91/10734 P(.'T/CA91/00009
Z6
to the Cc, gene and spans approximately 250 kb of genomic
DNA. bverlapping complementary DNA (cDNA) clones have
been isolated from epithelial cell libraries with a
genomic DNA segment containing a portion of the cystic
S fibrosis gene. The nucleotide sequence of the isolated
cDNA is shown in Figures 1 through 18. In each row of
the respective sequences the lower row is a list by
standard nomenclature of the nucleotide sequence. The
upper row in each respective row of sequences is standard
single letter nomenclature for the amino acid
corresponding to the respective codon.
Accordingly, the isolation of the CF gene provided a
cDNA molecule comprising a DNA sequence selected from the
group consisting of:
(a) DNA sequences which correspond to the DNA
sequence of Figure 1 from amino acid residue position 1
to po~:2.tion 1480;
(b) DNA sequences encoding normal CFTR polypeptide
having the sequence according to Figure 1 for amino acid
2o residue positions from 1 to 1480;
(c) DNA sequences which correspond to a fragment of
the sequence of Figure 1 including at least 16 sequential
nucleotides between amino acid residue positions 1 and
1480;
(d) DNA sequences which comprise at least 16
nucleotides and encode a fraganent of the amino acid
sequence of Figure 1; and
(e) DNA sequences encoding an epitope encoded by at
least 18 sequential nucleotides in the sequence of Figure
1 between amino acid residue positions 1 and 1480.
According to this invention, the isolation of other
mutations in the CF gene alsa provides a cDNA molecule
comprising a DNA sequence selected from the group
consisting of:
a) DNA sequences which correspond to the DNA
sequence encodinglmutant CFTR polypeptide characterized
by cystic fibrosis-associated activity inhuman
dvo 9~rio~~a PcrrcA9~roooo9
17 v. ~
rd~ ' 4.3':'t ~.f
epithelial cells, or the DNA sequence of Figure 1 for the
amino acid residue positions 1 to 1480 yet further
characterized by a base pair mutation which results in
the deletion of or a change for an amino acid at residue
positions 85, 148, 178, 455, 493, 507, 542, 549, 551,
560, 563, 574, 1077 and 1092;
b) DNA sequences which correspond to fragments of
the mutant portion of the sequence of paragraph a) and
which include at least sixteen nucleotides;
c) DNA sequences which comprise at least sixteen
nucleotides and encode a fragment of the amino said
sequence encoded for by the mutant portion of the DNA
sequence of paragraph a); and
d) DNA sequences encoding an epitope encoded by at
least 18 sequential nucleotides in the mutant poz°tion of
the sequence of the DNA of paragraph a).
Transcripts of approximately 6,500 nucleotides in
size are detectable in tissues affected in patients with
CF. Hosed upon the isolated nucleotide sequence, the
2o predicted protein consists of two similar regions, each
containing a first domain having properties consistent
with membrane association and a second domain believed to
be involved in ATP binding.
A 3 by deletion which results in the omission of a
phenylalanine residue at the center of the first
predicted nucleotide binding domain (amino acid position
508 of the CF gene product) was detected in CF patients.
This mutation in the normal DNA sequence of Figure 1
corresponds to approximately 70% of the mutations in
3o cystic fibrosis patients. Extended haplotype data based
on DNA markers closely linked to the putative disease
gene suggest that the remainder of the CF mutant gene
pool consists of multiple, different mutations. This is
now exemplified by this invention at, for example, the
506 ar 507 protein position. A small set of these latter
mutant alleles (approximately 8%) may confer residual
W~ 91/1073a 1'CT/CA91/00009
18
pancreatic exocrine function in a subgroup of patients
who are pancreatic sufficient.
2.1 CH120MOBO1~IE W~,7GkCyNB ND ~'~MMpING
Large amounts of the DNA surrounding the D7S122 and
D75340 linkage regions of Rommens et al supra were
searched for candidate gene sequences. In addition to
conventional chromosome walking methods, chromosome
jumping techniques were employed to accelerate the search
process. From each jump endpoint a new bidirectional
l0 walk could be initiated. Sequential walks halted by
"unclonable" regians often encountered in the mammalian
genome could be circumvented by chromosome jumping.
The chramosome jumping library used has been
described previously [Collins et al, S~,ience_ 235, 1046
(1987);W anuzzi et al, Am J ~,ium Genet 44, 695
(1989)]. The original library was prepared from a
preparative pulsed field gel, and was intended to contain
partial EcoRl fragments of 70 - 130 kb; subsequent
experience with this library indicates that smaller
fragments were also represented, and jumpsizes of 25 -
110 kb have been found. The library was plated on sup
host 1!1C1061 and screened by standard techniques,
[Maniatis et al]. Positive clones~were subcloned into
pBRo23Ava and the beginning and end of the jump
identified by EcoRi and Ava 1 digestion, as described in
COllinS, Genome anallrsiso ~, taraCtiCa~ ar~nrnanh ~~~,,
London, 1988), pp. 73-94) For each clone, a fragment
from the end of the jump was checked to confirm its
location on chromosome 7. The contiguous chromosome
region covered by chromosome walking and jumping was
abaut 250 kb. Direction of the jumps was biased by
careful choice of probes, as described by Collins et al
and Ianuzzi et al, supra. The entire region cloned,
including the sequences isolated with the use of the CF
gene cDNA, is approximately 500 kb.
The schematic representation of the chromosome
walking and jumping strategy is illustrated in Figure 2.
iV0 91 / 10734 PCT/Cf191 /00009
'' ~ '~ '~ ,i;~ ')
19
CF gene axons are indicated by Roman numerals in this
Fire. Horizontal lines above the map indicate walk
steps whereas the arcs above the map indicate jump steps.
The Figure proceeds from left to right in each of six
tiers with the direction of ends toward 7cen and 7qter as
indicated. The restriction map for the enzymes EcoRI,
HindIII, and BamHT is shown above the solid line,
spanning the entire cloned region. Restriction sites
indicated with arrows rather than vertical lines indicate
l0 sites which have not been unequivocally positioned.
Additional restriction sites for other enzymes are shown
below the line. Gaps .in the cloned region are indicated
bY ~~. These occur only in the portion detected by eDNA
clones of the CF transcript. These gaps are unlikely to
be large based on pulsed field mapping of the region.
The walking clones, as indicated by horizontal~arrows
above the map, have the direction of the straw indicating
the walking progress obtained with each clone. Cosmid
clones begin with the letter c; all other clones are
phage. Cosmid CF26 proved to be a chimera; the dashed
portion is derived from a different genomic fragment on
another chromosome. Roman numerals I through XXIV
indicate the location of axons of the CF gene. The
horizontal boxes shown above the line are probes used
during the experiments. Three of the probes'represent
independent subcloning of fragments previously identified
to detect polymorphisms in this region: H2.3A corresponds
to probe XV2C (X. EstiVill et al, at , 326: 840
(1987)), probe E1 corresponds to KM19 (Estivill, supra),
and probe E4.1 corresponds to Mp6d.9 (X. Estivill et al.
Am. J. Hum Ggnet 44 ,704 (1989)), G~2 is a subfragment
of E6 which detects a transcribed sequence. 8161, 8159,
and 8160 are synthetic oligonucleotides constructed from
parts of the IRP locus sequence [B. J. Wainwright et al,
O J., 7: 1743 (1988)], indicating the location of this
transcript on the genomic map.
CA 02073441 2000-12-07
As the two independently isolated DNA markers, D7S 122 (pH131) and
D7S340 (TM58), were only approximately 10 kb apart (Figure 2), the walks and
5 jumps were essentially initiated from a single point. The direction of
walking and
jumping with respect to MET and D7S8 was then established with the crossing of
several rare-cutting restriction endonuclease recognition sites (such as those
for Xho I,
Nru I and Not I, see Figure 2) and with reference to the long range physical
map of
A.M. Poustka, et al, Genomics 2, 337 (1988); M.L. Drumm et al. Genomics 2, 346
10 (1988). The pulsed field mapping data also revealed that the Not I site
identified by
the inventors of the present invention (see Figure 2, position 113 kb)
corresponded to
the one previously found associated with the IRP locus (Estivill et al 1987,
supra).
Since subsequent genetic studies showed that CF was most likely located
between
IRP and D7S8 [M. Farrall et al, Arn. J. Hum. Genet. 43, 471 (1988), B.S. Kerem
et al.
15 Am. J. Hum. Genet. 44, 827 (1989)], the walking and jumping effort was
continued
exclusively towards cloning of this interval. It is appreciated, however, that
other
coding regions, as identified in Figure 2, for example, G-2, CF14 and CF16,
were
located and extensively investigated. Such extensive investigations of these
other
regions revealed that they were not the CF gene based on genetic data and
sequence
20 analysis. Given the lack of knowledge of the location of the CF gene and
its
characteristics, the extensive and time consuming examination of the nearby
presumptive coding regions did not advance the direction of search for the CF
gene.
However, these investigations were necessary in order to rule out the
possibility of the
CF gene being in those regions.
Three regions in the 280 kb segment were found not to be readily recoverable
in the amplified genomic libraries initially used. These less clonable regions
WO 91/10734 PCT/CA91/00009
21 ~~~~~~~'~'t
were 1~cated near the DNA segments H2.3A and ?C.6, and
just~beyond cosmid cW44, at,positions 75-100 kb, 205-225
kb, and z75-285 kb in Figure 2, respectively. The
recombinant clones near H2.3A were found to be very
unstable with dramatic rearrangements after only a few
passages of bacterial culture. To fill in the resulting
gaps, primary walking libraries were constructed using
special host-vector systems which have been reported to
allow propagation of unstable sequences [A. R. Wyman, L.
B. Wolfe, D. Botstein, _proc. Nat Acad Sci ~r a ~ g2~
2880 (1985); K. F. Wertman, A. R. Wyman, D. Botstein,
Gene 49, 253 (1986); A. R. Wyman, K. F. Wertman, D.
Barker, c. Helms, W. H. Petri, Gene, 49, 263 (1966)),
Although the region near cosmid cW44 remains to be
recovered, the region near X.6 was successfully rescued
with these libraries.
COZdBTRDC'~T~At OF l3gNO~trr r °~~~~or~a
Genomic libraries were constructed after procedures
described in Manatis, et al, Nalecu~ar Clon~tng~
Laby g4~ (Cold Spring Harbor Laboratory, Cold
Spring Harbor, New Yark 1982) and are listed in Table i.
This includes eight phage libraries, one of which was
provided by T. Maniatis [Fritsch et al, Ce , 19:959
(1980)]; the ~°est were constructed as part of this work
according to procedures described in l~aniatis et al,
su~~a. Four phage libraries were cloned in aDASH
(commercially available from Stratagene) and three in
aF~X (commercially available from Stratagene), with
vector arms provided by the manufacturer. One aDASH
library was constructed from Sau 3A-partially digested
DNA from a human-hamster hybrid containing human
chromosome 7 (4AF/102/K015) [Rommens et al Am. ~' Hum
a et 43, 4 (1988)], and other libraries from partial
Sau3A, total BamHI, or total EcoRI digestion of human
peripheral blood or lymphoblastoid DNA. To avoid loss of
unstable sequences, five of the phage libraries were
propagated on the recombination-deficient hosts DH1316
CA 02073441 2000-12-07
22
(recD-), CES 200 (recBC-;) [Wyman et al, supra , Wertman et al supra, Wyman et
al
supra]; or TAP90 [Patterson et al Nucleic Acids Res. 15:6298 (1987)]. Three
cosmid
libraries were then constructed. In one the vector pCV 108 [Lau et al Proc.
Natl.
Acad. Sci USA 80:5225 (1983)] was used to clone partially digested (Sau 3A)
DNA
from 4AF/102/KO15 [Rommens et al Am.J. Hum. Genet. 43:4 (1988)]. A second
cosmid library was prepared by cloning partially digested (Mbo I) human
lymphoblastoid DNA into the vector pWE-IL2R, prepared by inserting the RSV
(Rous Sarcoma Virus) promoter-driven cDNA for the interleukin-2 receptor a-
chain
(supplied by M. Fordis and B. Howard) in place of the neo-resistance gene of
pWElS
[Wahi et al Proc. Natl. Acad. Sci. USA 84:2160 (1987)]. An additional partial
Mbo I
cosmid library was prepared in the vector pWE-1L2-Sal, created by inserting a
Sal I
linker into the Bam HI cloning site of pWE-EL2R. This allows the use of the
partial
fill-in technique to ligate Sal I and Mbo I ends, preventing tandem insertions
[Zabarovsky --et al Gene 42:19 (1986)]. Cosmid libraries were propagated in E.
coli
host strains DH1 or 490A [M. Steinmetz, A. Winoto, K. Minard, L. Hood, Cell
28,
WO 91110734 Pt.°TlCA91/00009
23
TABLK 1
C3EP1~~i:~t~ x,lrB CBS
Vectr.~r Source of human DNA ~,gst ~om plexitxRef
~ Charon lIaeII/Alul-partially LE392 1 106 Lawn
x
4A digested total human (amplified) et
al
liver DNA 1980
pCV108 Sau3a-gartially digested DK1 3 106
x
DNA from 4AF/K015 (amplified)
adash Sau3A-partially digested LE392 1 106
x
DNA from 4AF/K015 (amplified)
adash Sau3A-partially digested DB13161.5 x
106
total human peripheral
blood DNA
adash BamFiI-digested total DB1316 1.5 x
106
human peripheral blood
DNA
adash EcoRI-gartially digested DB1316S 10~
x
total human peripheral
blood DNA
aFIX Mbol-partially digested LE392 1.5 x
10g
human lymphvblastoid DNA
3o aFlx Mbol-partially digested cE2oo 1.2 x
lob
human lymphoblastoid DNA
aFIX MboI-partially digested TAp90 1.3 x
106
human lymphoblastoid DNA
pWE-IL2R P3boI-partially digested 490A 5 lOs
x
human lymphoblastoid DNA
pWE-IL2R- ~lbol-partially digested 490A 1.2 x
106
Sal human.lymphoblastoid DNA
ACh3A EcoRI-partially digested MC10613 lOg
x
collins
nlac (24-110
kb)
et al
(nu human lymphoblastoid DNA
mping) supra
a
d
Iannuzzi
et al
supra
w~ 91/10734 IPCT/CA91/00009
24
Three of the phage libraries were propagated and
amplified in ~. co ' bacterial strain LE392. Four
subsequent libraries were plated on the recombination-
deficient hosts DB1316 (recD'~ or CES200 (rec BC's [Wyman
1985, su ra; Wertman 1986, su ; and Wyman 1986, sera]
or in one case TAP90 [T. A. Patterson and NI. Dean, Nucleic
Acids Research 15, 6298 (1987)].
Single copy DNA segments (free of repetitive
elements) near the ends of each phage or cosmid insert
were purified and used as probes for library screening to
isolate overlapping DNA fragments by standard procedures.
(Maniatis, et al, sutra).
1-2 x 106 phage clones were plated on 25-30 150 mm
petri dishes with the appropriate indicator bacterial
host and incubated at 37°C for 10-16 hr. Duplicate
"lifts" were prepared for each plate with nitrocellulose
~;~:s nylon membranes, prehybridized and hybridized under
conditions described [ltommens et al, 1988, supra].
Probes were labelled with 32P to a specific activity of >5
x 10a cpm/~cg using the random priming procedure (A. P.
Feinberg and ~. Vogelstein, glp,a~l. l~iochem. 132, 6
(1983)]. The cosmid library was spread on ampicillin-
containing plates and screened in a similar manner.
DNA probes which gave high background signals could
often be used more successfully by preannealing the
boiled probe with 250 ~.g/ml sheared denatured placental
DNA for 60 minutes prior to adding the probe to the
hybridization bag.
For each walk step, the identity of the cloned DNA
fragment was determined by hybridization with a somatic
cell hybrid panel to confirm its chromosomal location,
and by restriction mapping and Southern blot analysis to
confirm its colinearity with the genome.
The total combined cloned region of the genomic DNA
sequences isolated and the overlapping cDNA clones,
extended >500 kb. To ensure that the DNA segments
isolated by the chromosome walking and jumping procedures
dV0 91!10734 PCT/CA91/00009
~5
were colinear with the genomic sequence, each segment was
examined bys
(a) hybridization analysis with human-rodent somatic
hybrid cell lines to confirm chromosome 7 localization,
(b) pulsed field gel electrophoresis, and
(c) comparison of the restriction map of the cloned
DNA to that of the genomic DNA.
Accordingly, single copy human DNA sequences were
isolated from each recombinant phage and cosmid clone and
used as probes in each of these hybridization analyses as
performed by the procedure of Maniatis, et al supra>
While the majority of phage and cosmid isolates
represented correct walk and jump clones, a few resulted
from cloning artifacts or cross-hybridizing sequences
fram other regions in the human genome, or from the
hamster genome in cases where the libraries were derived
from a human-hamster hybrid cell line. Confirmation of
correct localizatian was particularly important for
clones isolated by chromosome jumping. Many jump clones
were considered and resulted in non-conclusive
information leading the direction of investigation away
from the gene.
~s.~, CDNFIRM14,R~'~oTJ p~ R~gt~ s~~~~aTr~Tn~ MAP
s ,~. ya
Further confirmation of the overall physical map of
the overlapping clones was obtained by long rmnge
restriction mapping analysis with the use ~f pulsed field
gel electrophoresis (J. M. Rommens, et al. Am. J. ~Ium
G net in press, A. M. Poustka et al, 1988, supra M.L.
Drumm et al, 1988 su ra).
Figures 3A to 3E illustrates the findings of the
long range restriction mapping study, where a schematic
representation of the region is given in Panel E. DNA
from the human-hamster cell line 4AF/102/K015 was
digested t~ith the enzymes (A) Sal.I, (H) Xho I, (C) Sfi I
and (D) Nae I, separated by pulsed field gel
electrophoresis, and transferred to zetaprobe~' (BioRad).
For each enzyme a single blot was sequentially hybridized
CA 02073441 2000-12-07
26
with the probes indicated below each of the panels of Figure A to D, with
stripping of
the blot between hybridizations. The symbols for each enzyme of Figure 3E are:
A,
Nae I; B, Bss HII; F. Sfi I; L, Sal I; M, Mlu I; N, Not I; R, Nru I; and X,
Xho 1. C
corresponds to the compression zone region of the gel. DNA preparations,
restnction
digestion, and crossed field gel electrophoresis methods have been described ~
The
gels in Figure 3 were run in 0.5X TBE at 7 volts/cm for 20 hours with
switching
linearly ramped from 10-40 seconds for (A), (B), and (C), and at 8 volts/cm
for 20
hours with switching ramped linearly from 50-150 seconds for (D). Schematic
interpretations of the hybridization pattern are given below each panel.
Fragment
lengths are in kilobases and were sized by comparison to oligomerized
bacteriophage
7~DNA and Saccharomyces cerevisiae chromosomes.
H4.0, J44, EG1.4 are genomic probes generated from the walking and jumping
experiments (see Figure 2). J30 has been isolated by four consecutive jumps
from
D7S8 (Collins et al, 1987, supra; Ianuzzi et al, 1989, supra; M. Dean, et al,
submitted
for publication). 10-l, B.75, and CE1.5/1.0 are cDNA probes which cover
different
regions of the CF transcript: 10-1 contains exons I -VI, B.75 contains exons V
- XII,
and CE1.5/1.0 contains exons XII - XXIV. Shown in Figure 3E is a composite map
of the entire MET - D7S8 interval. The open boxed region indicates the segment
cloned by walking and jumping, and the closed arrow portion indicates the
region
covered by the CF transcript. The CpG-rich region associated with the D7S23
locus
(Estivill et al, 1987, supra) is at the Not I site shown in parentheses. This
and other
sites shown in parentheses or square brackets do not cut in 4AF/102/K015, but
have
__ . . , . , , ,_,,__~ __",.____
WO 91/10734 PCT/CA93/00009
27 ~~v~~~c'3~'t.a
d.
..~ o xn~rrrx~~cA~rTorr ~~ cF
used on the findings of long range restriction
mapping detailed above it was detenained that the entire
CF gene is contained on a 38o kb Sal I fragment.
Alignment of the restriction sites derived from pulsed
field gel analysis to those identified in the partially
overlapping genomic DNA clones revealed that the size of
the CF gene was approximately 250 kb.
The mast informative restriction enzyme that served
to align the map of the cloned DNA fragments and the long
range restriction map was Xho I; all of the 9 Xho 1 sites
identified with the recombinant DNA clones appeared to be
susceptible to at least partial cleavage in genomic DNA
(compare :asps in Figures Z and 2). Furthermore,
hybridization analysis with probes derived frown the 3~
en~? of the CF gene identified 2 Sfil sites and confirmed
the position of an anticipated Nae I site.
These findings further supported the canclusion that
the DNA, segments isolated by the chromosome walking arid
jumping procedures were colinear with the genuine
sequence.
CRxTRR%1~1 F'oR '~i~RN'~,'TIa''r'G~Trn~
A positive result based on one or more of the
following criteria suggested that a cloned DNA segment
may contain candidate gene sequences:
(a) detection of cross-hybridizing sequences in
other species (as many genes show evolutionary
conservation),
(b) identification of CpG islands, which often mark
the 5~, end of vertebrate genes [A. P. bird, Natu,~g, 32Z~
209 (1986); M. Gardiner-Garden and M. Frommer, J~o~,
Biol. 196, 261 (1987)],
(c) examination of possible mRNA transcripts in
tissues affected in CF patients,
(d) isolation of corresponding cDNA sequences,
(e) identification.of open reading frames by direct
sequencing of cloned DNA segments.
WO 9]/10734 P('('/Cp9]/00009
-~;~c~~.
28
Cross-species hybridization showed strong sequence
conservation between human and bovine DNA when CF'14, E4.3
and H1.6 were used as probes, the results of which are
shown in Figures 4A, 4B and 4C.
Human, bovine, mouse, hamster, and chicken genomic
DNAs were digested with Eco RI (R), Hind III (H), and Pst
I (P), electrophoresed, and blotted to Zetabind~'
(BioRad). The hybridization procedures of Rommens et al,
1988, su ra, were used with the most stringent wash at
55°C, 0.2X SSC, and 0.1% SDS. The probes used for
hybridization, in Figure 4, included: (A) entire cosmid
Cfl4, (B) E4.3, (C) H1.6. In the schematic of Figure
(D), the shaded region indicates the area of cross-
species conservation.
The fact that different subsets of bands were
detected in bovine DNA with these two overlapping DNA
segments (H1.6 and E4.3) suggested that the conserved
sequences were located at the boundaries of the
overlapped region (Figure 4(D)). ~nThen these DNA segments
were used to detect RNAwtranscripts from a variety of
tissues, no hybridization signal was detected. In an
attempt to understand the cross-hybridizing region and to
identify possible open reading frames, the DNA sequences
of the entire H1.6 and part of the E4.3 fragment were
determined: The results showed that, except for a long
stretch of CG-rich sequence containing the recognition
sites for two restriction enzymes (Bss HI,I and Sac II),
often found associated with undermethylated CpG islands,
there were only short open reading frames which could not
easily explain the strong cross-species hybridization
signals.
To examine the methylation status of this highly
CpG-rich region revealed by sequencing, genomic DNA
samples prepared from fibroblasts and lymphoblasts were
digested with the restriction enzymes Hpa II and Msp I
and analyzed by gel blot hybridization. The enzyme Hpa
IT cuts the DNA sequence 5~-CCGG-3' only when the second
CA 02073441 2000-12-07
29
cytosine is unmethylated, whereas Msp I cuts this sequence regardless of the
state of
methylation. Small DNA fragments were generated by both enzymes, indicating
that
this CpG-rich region is indeed undermethylated in genomic DNA. The gel-blot
hybridization with the E4.3 segment (Figure 6) reveals very small hybridizing
fragments with both enzymes, indicating the presence of a hypomethylated CpG
island.
The above results strongly suggest the presence of a coding region at this
locus. Two DNA segments (E4.3 and H1.6) which detected cross-species
hybridization signals from this area were used as probes to screen cDNA
libraries
made from several tissues and cell types.
cDNA libraries from cultured epithelial cells were prepared as follows. Sweat
gland cells derived from a non-C',F individual and from a CF patient were
grown to
first passage as described [G. Collie et al, In Vitro Cell. Dev. Biol. 21,
592,1985].
The presence of outwardly rectifying channels was confirmed in these cells
(J.A.
Tabcharani, T.J. Jensen, J.R. Riordan, J.W. Hanrahan, J. Memb. Biol., in
press) but
the CF cells were insensitive to activation by cyclic AMP (T.J. Jensen, J.W.
Hanrahan, J.A. Tabcharani, H. Buchwald and J.R. Riordan, Pediatric
Pulmonolo~y,
Supplement 2, 100, 1988). RNA was isolated from them by the method of J.M.
Chirgwin et al (Biochemistry 18, 5294, 1979). Poly A+RNA was selected (H. Aviv
and P. Leder, Proc. Natl. Acad. Sci. USA 69, 1408, 1972) and used as template
for the
synthesis of cDNA with oligo (dT) 12-18 as a primer. The second strand was
synthesized according to Gubler and Hoffman (Gene 25, 263, 1983). This was
methylated with Eco RI methylase and ends were made flush with T4 DNA
polymerase. Phosphorylated Edo RI linkers were ligated to the cDNA and
restricted
with Eco RI. Removal of excess linkers and partial size fractionation was
achieved
by BiogelTM A-50 chromatography. The cDNAs were then ligated into the Edo RI
site of the commercially
Wl7 91/10734 PCTlCA91/00009
available lamdba zAP. Recombinant were packaged and
propagated ~in ~ cozy HH4. Portions of the packaging
mixes.were amplified and the remainder retained for
screening prior to amplification. The same procedures
were used to construct a library from RNA isolated from
preconfluent cultures of the T-84 colonic carcinoma cell
line (Dharmsathaphorn, K. et al. Am. ,T. Phvs~ni. 246,
6204, 1984). The numbers of independent recombinant in
the three libraries were: 2 x 106 for the non-CF sweat
l0 gland cells, 4..5 x 106 for the CF sweat gland cells and
3.2 x 106 from T-84 cells. These phages were plated at
50,000 per 15 cm plate and plaque lifts made using nylon
membranes (Biodyne) and probed with DNA fragments
labelled with 'aP using DNA polymerise I and a random
mixture of oligonucleotides as primer. Hybridization
conditions were according to G.M. Wahl and S.L. Herger
(Meth Enzymol. 152,415, 1987). Bluescript'~ plasmids
were rescued from plaque purified clones by excision with
M13 helper phage. The lung and pancreas libraries were
purchased from Clontech Lab Inc. with reported sizes of
1.4 x 106 and 1.7 x 106 independent clones.
After screening 7 different libraries each
containing 1 x lOs - 5 x 106 independent clones, 1 single
clone (identified as l0-1) was isolated with H1.6 Eram a
cDNA library made from the cultured sweat gland
epithelial cells of an unaffected (non~CF) individual.
DNA sequencing analysis showed that probe 10-1
contained an insert of 920 by in size and one potential,
long open reading frame (oRF). Since one end of the
3o sequence shared perfect sequence identity with H1.6, it
was concluded that the cDNA clone was probably derived
from this region. The DNA sequence in common was,
however, only 113 by long (see Figures 1 and 7). As
detailed below, this sequence in fact corresponded to the
5~-most exon of the putative CF gene. The short sequence
overlap thus explained,the weak hybridization signals in
library screening and inability tee detect transcripts in
W~ 91/1fl734 Pt.'T/CA91/flfl009
r
31. ~.r~ s~e~~~~!i
RNA gel-blot analysis. In addition, the orientation of
the transcription unit was tentatively established on the
basis of alignment of the genomic DNA sequence with the
presumptive ORF of 10-1.
since the corresponding transcript was estimated to
be approximately 6500 nucleotides in length by RNA gel~-
blot hybridization experiments, further cDNA library
screening was required in order to clone the remainder of
the coding region. As a result of several successive
to screenings with eDNA libraries generated from the colonic
carcinoma cell line T84, normal and GF sweat gland cells,
pancreas and adult lungs, 18 additional clones were
isalated (Figure 7, as subsequently discussed in greater
detail). DNA sequence analysis revealed that none of
these cDNA clones corresponded to the length of the
observed transcript, but it was possible to derive a
censensus sequence based on overlapping regions.
Additional cDNA clones corresponding to the 5' and 3'
ends of the transcript were derived from 5' and 3'
primer-extension experiments. Together, these clones
span a total of about 6.1 Icb and contain an ORF capable
of encoding a polypeptide of 1480 amino acid residues
(Figure 1).
It was unusual to observe that most of the cDNA
atones isolated here contained sequence insertions at
various locations of the restriction map of Figure 7.
The map details the genomic structure of the CF gene.
Exon/intrc~n boundaries are given where all cDNA clones
isolated are schematically represented on the upper half
of the figure. Many of these extra sequences clearly
corresponded to intron regions reversely transcribed
during the construction of the cDNA, as revealed upon
alignment with genomic DNA sequences.
Since the number of recombinant cDNA clones for the
CF gene detected in the library screening was much less
than would have been expected from the abundance of
transcript estimated from RNA hybridization experiments,
w0 91/10734 P(.°T/CA91/00009
32
it seemed probable that the clones that contained
aberrant structures were preferentially retained while
the proper clones were lost during propagation.
Consistent with this interpretation, poor growth was
observed for the majority of the recombinant clones
isolated in this study, regardless of the vector used.
The procedures used to obtain the 5° and 3° ends of
the cDNA were similar to those described (M. Frohman et
al, Proc. Nat. Aca~Sci, tTSA, 85, 8998-9002, 1988). Fox
the 5r end clones, total pancreas and T84 poly A -~ RNA
samples were reverse transcribed using a primer, (10b),
which is specific to exon 2 similarly as has been
described for the primer extension reaction except that
radioactive tracer was included in the reaction. The
fractions collected from an agarase bead column of the
first strand synthesis were assayed by polymerase chain
reac~Lion (PCR) of eluted fractions. The oligonucleotides
used were within the 10-1 sequence (245 nucleotides
apart) just 5° of the extension primer. The earliest
fractions yielding PCR product were pooled and
concentrated by evaporation and subsequently tailed with
terminal deoxynucleotidyl transferase (BRL Labs.) and
dATP as recommended by the supplier (BRL Labs). A second
strand synthesis was then carried out with Taq Polymerase
(Cetus, AmpliTaq~') using an oligonucleotide containing a
tailed linker.sequence 5°CGGAATTCTCGAGATC(T)123°.
amplification by an anchored (PCR) experiment using
the linker. sequence and a primer just internal to the
extension primer which possessed the Eco RI restriction
site at its 5° end was then carried out. Following
restriction with the enzymes Eco RI and Bgl II and
agarose gel purification size selected products were
cloned into the plasmid Bluescript KS available from
Stratagene by standard procedures (Maniatis et al,
supra). Essentially all of the recovered clones
contained inserts of less than 350 nucleotides. To
abtain the 3° end clones, first strand cDNA was prepared
W~ 91/1073q QCT/CA91/00009
33 a
~~v~~.., ~;
with reverse transcription of 2 ag T04 poly A ~- RNA using
the tailed linker oligonucleotide previously described
with conditions similar to those of the primer extension.
Amplification by PCR was then carried c;ut with the linker
oligonucleotide and three different oligonucleotides
corresponding to known sequences of clone T16-4.5. A
preparative scale reaction (2 x 100 u1) was carried out
with one of these oligonucleotides with the sequence
5'ATGAAGTCCAAGGATTTAG3'.
This oligonucleotide is approximately 70 nucleotides
upstream of a Hind III site within the known sequence of
T16-4.5. Restriction of the PCR product with Hind III
and Xho 1 was followed by agarose gel purification to
size select a band at 1.0-1.4 kb. This product was then
cloned into the plasmid Hluescript KS available from
stratagene. Approximately 20~ of the obtained clones
hybridized to the 3' end portion of T16-4.5. 10/10 of
plasmids isolated from these clones had identical
restriction maps with insert sizes of approx. 1.2 kl~.
All of the PCR reactions were carried out for 30 cycles
in buffer suggested by an enzyme supplier.
An extension primer positioned 157 nt from the 5'end
of 10-1 clone was used to identify the start point of the
putative CF transcript. The primer was end labelled with
-~(32P]ATP at 5000 Curies/mole and T4 polynucleotide kinase
and purified by spun column gel filtration. The
radiolab~led primer was then annealed with 4-5 ug poly A
-~ RNA prepared from T-84 colonic carcinoma cells in 2X
reverse transcriptase buffer for 2 hrs. at 60'C.
3o Following dilution and addition of AMV reverse
transcriptase (Life Sciences, Inc.y incubation at 41'C
proceeded for 1 hour. The sample was then adjusted to
0.4M Na~H and 20 mM EDTA, and finally neutralized, with
NH~OAc, pH 4.6, phenol extracted, ethanol precipitated,
redissolved in buffer with formamide, and analyzed on a
polyacrylamide sequencing gel. Details of these methods
wo ~mo73a racricA~rioooo9
~~ ii 34
have been described (Meth. EnzVmol. 152, 1987, Ed. S.L.
Bexger, A.R. ICimmel, Academic Press, N.Y.).
Results of the primer extension experiment using an
extension oligonucleo~:ide primer starting 157 nucleotides
from the 5~ end of 10-1 is shown in Panel A of Figure 10.
End labelled X174 bacteriophage digested with Hae III
(BRL Labs) is used as size marker. Two major products
are observed at 216 and 100 nucleotides. The sequence
corresponding to 10o nucleotides in 10-1 corresponds to a
ZO very GC rich sequence (11/12] suggesting that this could
be a reverse,transcriptase pause site. The 5~ anchored
PCR results are shown in panel B of Figure 10. The 1.4%
agarose gel shown on the left was blotted and transferred
to Zetaprobe~° membrane (Bio-Rad Lab). DNA gel blot
hybridization with radiolabeled l0-1 is shown on the
right. The 5~ extension products are seen to vary in
size from 170-280 nt with the major product at about 200
nucleotides. The PCR control lane shows a fragment of
145 nucleotides. Tt was obtained by using the test
oligomers within the 10-1 sequence. The size markers
shown correspond to sizes of 154, 220210, 298, 344, 394
nucleotides (lkb ladder purchased from BRL Lab).
The schematic shown below Panel B of Figure 10
outlines the procedure to obtain double stranded cDNA
used for the amplification and cloning to generate the
clones PA3-5 and TB2-7 shown in Figure 7. The anchored
PCR experiments to characterize the 3~end are shown in
panel. C. As depicted in the schematic below Figure 10C,
three primers whose relative position to each other were
known were used for amplification with reversed
transcribed T84 RNA as described. These products were
separated on a 1% agarose gel and blotted onto nylon
membrane as described above. DNA-blot hybridization with
the 3~ portion of the T16-4.5 clone yielded bands of
sizes that corresponded to the distance between the
specific oligomer ~used.and the 3~end of the transcript.
These bands in lanes 1, 2a and 3 are shown schematically
WO 91 / 1073d PCT/CA91 /00009
3 5 ~ i~ ~~ ~ ~ r'~ ~.a.
below Panel C in Figure 10. The band in lane 3 is weak
as only 60 nucleotides of this segment overlaps with the
probe used. Also indicated in the schematic and as shown
in the lane 2b is the product generated by restriction of
the anchored PCR product to facilitate cloning to
generate the THZ-d clone shown in Figure 7,
DNA-blot hybridization analysis of genomic DNA
digested with EcoRI and HindIII enzymes probed with
portions of cDNAs spanning the entire transcript suggest
that the gene contains at least 26 axons numbered as
Roman numerals I through XXVI (see Fi.c~ure 9), These
correspond to the numbers 1 through 26 shown in Figure 7.
The size of each band is given in kb.
In Figure 7, open boxes indicate approximate
positions of the 24 axons which have been.identified by
the isolation of X22 clones from the screening of cDNA
libraries and from anchored PCR experiments designed to
clone the 5~ and 3~ ends. The lengths in kb of the Eco
RI genomic fragments detected by each axon is also
2o indicated. The hatched boxes in Figure 7 indicate the
presence of intron sequences and the stippled boxes
indicate other sequences. Depicted in the lower left by
the closed box is the relative position of the clone H1.6
used to detect the first cDNA clone 10-1 from among 106
phage of the nox~al sweat gland library. As shown in
Figures 4(D) and 7, the genomic alone H1.6 partially
overlaps with an EcoRI fragment of X1.3 kb. All of the
cDNA clones shown were hybridized to genomic DNA and/or
were fins restriction mapped. Examples of the
restriction sites occurring within the eDNAs and in the
corresponding gendmic fragments are indicated,
With reference to Figure 9, the hybridization
analysis includes probes; i.e., cDNA clones 10-1 for
panel A, T16-1 (3o portion) for panel B, T16~4.5 (central
portion) for panel C and T16-4.5 (3~ end portion) for
panel D. In panel~A of,Figure 9, the cDNA probe 10-1
detects the genomic bands for axons I through VI. The 3'
WO 91/10734 PCTlCA91/00009
36
portion of T16-5. generated by NruI restriction detects
axons IV through XIIT as shown in Panel B. This probe
partially overlaps with 10-1. Panels C and D,
respectively, show genomic bands detected by the central
and 3' end EcoRI fragments of the clone T16-4.5. Two
EcoRI sites occur within the cDNA sequence and split
axons XIII and XIX. As indicated by the axons in.
parentheses, two genomic EcoRI bands correspond to each
of these exans. Cross hybridization to other genomic
1o fragments was observed. These bands, indicated by N, are
not of chromosome 7 origin as they did not appear in
human-hamster hybrids containing human chromosome 7. The
faint band in panel D indicated by XI in brackets is
believed to be caused by the cross-hybridization of
sequences due to internal homology with the cDNA.
Since l0-~ detected a strong band on gel blot
hybridization of RNA from the T-8~ colonic carcinoma cell
line, this cDNA was used to screen the library
constructed from that source. Fifteen positives were
obtained fro~a which clones T6, T6/20, T11, T16-1 and T13-
1 were purified and sequenced. Rescreening of the same
library with a 0.75 kb Bam HI-Eco RI fragment from the 3f
end of T16-1 yielded T16-4.5. A l.8kb EcoRI fragment
from the 3' end of T16-4.5 yielded T8-B3 and Tl2a, the
latter of which contained a polyadenylation signal and
tail. Simultaneously a human lung cDNA library was
screened; many clones were isolated including those shown
here with the prefix 'GDL'. A pancreas library was also
screened, yielding clone CDp,TS.
To obtain copies of this transcript from a CF
patient, a cDNA library from RNA of sweat gland
epithelial cells from a patient was screened with the
0.75 kb Sam HI - Eco RI fragment from the 3' end of T16-1
and clones C16-1 and C1-1/5, which covered all but axon
1, were isolated. These two clones both exhibit a 3 by
deletion in axon 10 which is not present in any other
clone containing that axon. Several clones, including
'WO 91/1073A PCT/CA91/00009
37 ~~r~~'k~~~
CDLS28-1 from the lung library and T6J20 and T13-1
isolated from T84 were derived from partially processed
transcripts. This was confirmed by genomic hybridization
and by sequencing across the exon-intron boundaries for
each clone. T11 also contained additional sequence at
each end. T16-4.5 contained a small insertion near the
boundary between axons l0 and 11 that did not correspond
to intron sequence. Clones CDLS16A, lla and 13a from the
lung library also contained extraneous sequences of
unknown origin. The clone C16-1 also contained a short
insertion corresponding to a portion of the 7-transposon
of E. coli; this element was not detected in the other
clones. The 5' clones PA3-5, generated from pancreas RNA
and-TB2-7 generated from T84 RNA using the anchored PCR
technique have identical sequences except for a single
nucleotide difference in length at the 5' end as'shown in
Figure 1. The 3' clone, THZ-4 obtained from T84 RNA
contains the 3' sequence of the transcript in concordance
with the.genomic sequence of this region.
A combined sequence representing the presumptive
coding region of the CF gene was generated from
overlapping cDNA clones. Since most of the eDNA clones
were apparently derived from unprocessed transcripts,
further studies were performed to ensure the authenticity
of the combined sequence. Each cDNA clone was first
tested for localization to chromosome 7 by hybridization
analysis with a human-hamster somatic cell hybrid
containing a single human chromosome 7 and by pulsed
field gel electrophoresis. Fine restriction enzyme
mapping was also performed for each clone. While
overlapping regions were clearly identifiable far most of
the clones, many contained regions of unique restriction
patterns:
To further characterize these cDNA clones, they were
used as probes in gel hybridization experiments with
EcoRI -or HindIIZ-digested human genomic DNA. As shown in
Figure 9, five to six different restriction fragments
wo 9a~ao~3a Pcricn9aiomoo~
~~~~.~~a~~ ~ 3s
could be detected with the 10-1 cDNA and a similar number
of'fragments with ather cDNA clones, suggesting the
presence of multiple axons for the putative CF gene. The
hybridization studies also identified those cDNA clones
with unprocessed intron sequences as they showed
preferential hybridization to a subset of genomic DNA
fragments. For the confirmed cDNA clones, their
corresponding genomic DNA segments were isolated and the
axons and exon/intron boundaries sequenced. As indicated
in Figure 7, at least 27 exoris have been identified which
includes split axons 6a, 6b, 14a, 14b and 17a, 17b.
Based on this information and the results of physical
mapping experiments, the gene locus was estimated to span
250 kb on chromosome 7.
2.6 THE BEOaJEIZCE
Figure 1 shows the nucleotide sequence of the cloned
cD.~JA encoding CFTR together with the deduced amino acid
sequence. The first base position corresponds to the
first nucleotide in the 5' extension clone PA3-5 which is
one nucleotide longer than TB2-7. Arrows indicate
position of transcription initiation site by primer
extension analysis. Nucleotide 6129 is followed by a
poly(dA) tract. Positions of axon junctions are
indicated by vertical lines. Potential membrane-spanning
segments were ascertained using the algorithm of
Eisenberg et al J. Mol. Bio~ 179:125 (1984). Potential
membrane-spanning segments as analyzed and shown in
Figure 11 are enclosed in boxes of Figure 1. In Figure
11, the mean hydropathy index [Kyte and Doolittle, J.
Molec. Biol. 157: 105, (1982)] of 9 residue peptides is
plotted against the amino acid number. The corresponding
positions of features of secondary structure predicted
according to Garnier et al, [J. Molec a3so~ 157, 1s5
(1982)] are indicated in the lower panel. Amino acids
comprising putative ATP-binding folds are underlined in
Figure 1. Possible sites of phosphorylation by protein
kinases A (PKA) or C (PKC) are indicated by open and
WO 91 / 9 0734 PCT/CA91 /00009
3~ ~G~ ~~~~~t
closed circles, respectively. The open triangle is over
the 3bp (CTT) which are deleted in CF (see discussion
below). The cDNA clones in Figure 1 were sequenced by
the dideoxy chain termination method employing 35S
labelled nucleotides by the Dupont Genesis 2000'
automatic DNA sequencer.
The combined cDNA sequence spans 6129 bass pairs
excluding the poly(A) tail at the end of the 3'
untranslated region and it contains an ORF capable of
encoding a polypeptide of 1480 amin~ acids (Figure 1).
An ATG (AUG) triplet is present at the beginning of this
ORF (base position 133-X135). Since the nucleotide
sequence surrounding this colon (5'pAGACCCA-3') has
the proposed features of the consensus sequence (CC)
Z5 A/GCCAUGG(G) of an eukaryotic translation initiation site
with a highly conserved A at the -3 position, it is
highly probable that this AUG~corresponds to the first
msthionine colon for the putative polypeptide.
To obtain the sequence corresponding to the 5' end
of the transcript, a primer-extension experiment was
performed, as described earlier. As shown in Figure 10A,
a primer extension product of approximately 216
nucleotides could be observed suggesting that the 5' end
of the transcript initiated approximately 60 nucleotides
upstream of the end of cDNA clone 10-1. A modified
polymerase chain reaction (anchored PCR) was then used to
facilitate cloning of the 5'-end sequences (Figure 10b).
Two independent 5'-extension clones, one from pancreas
and the ether from T84 RNA, were characterized by DNA
30. sequencing and were found to differ by only 1 base in
length, indicating the most probable initiatian site for
the transcript as shown in Figure 1.
Since most of the initial cDNA clones did not
contain a polyA tail indicative of the end of a mRNA,
anchored PCR was also applied to the 3' end of the
transcript (Frohman et. al, 1988, s_.upra). Three 3'-
extension oligonucleotides were made to the terminal
wo 9li~o~3a PCfl~A91/00009
~~,~'~~i~i: ; 40
portion of the cDNA clone T16-4.5. As shown in Figure
lOc, 3 PCR products of different sizes were Obtained.
All were consistent with the interpretation that 'the end
of the transcript was approximately 1.2 kb downstream of
the HindIII site at nucleotide position 5027 (see Figure
1). The DNA sequence derived from representative clones
was in agreement with that of the T84 cI3NA cline Tl2a
(see Figure 1 and ~) and the sequence of the
corresponding 2.3 kb EcoRI genomlc fragment.
lo 3 ~ O P2OIrFii."OL~R CaFI3dETTP~:Q n~ Visa
~ . ~. sxT~s o~ EAg~eEBSi_oN
To visualize the transcript for the putative CF
gene, RNA gel blot hybridization experiments were
performed with the 10-l cDNA as probe. The RNA
hybridization results are shown in Figure 8.
RNA samples were prepared from tissue samples
obtained fram surgical pathology or at autopsy according
to methods previously described (A. M. Kimmel, S.L.
Bergen eds. Meth. Fn'~~.no~. 152, 1987)._ Formaldehyde
2o gels were transferred onto nylon membranes (zetaprobe TM~
BioRad Lab): The membranes were then hybridized with DNA
probes labeled to high specific activity by the random
priming method (A. P. Feinberg and B. Vogelstein, Anal,
Bioi chum 132, 6, 1983) according to previously published
procedures (J. Rommens et al, Am. J. Hum Gent 43, 645-
663, 1988). Figure 8 shows hybridization by the cDNA
clone 10-1 to a 6.5kb transcript -in the tissues
indicated. Total RNA (10 ~cg) of each tissue, and Poly A+
RNA (1 fag) of the T84 colonic carcinoma cell line were
separated on a 1% formaldehyde gel, The positions of the
28S and 18S rRNA bands are indicated. Arrows indicate
'the position of transcripts. Sizing was established by
comparison to standard RNA markers (BRL Labs). HL60 is a
human promyelocytic leukemia cell line, and T84 is a
human colon cancer cell line.
Analysis reveals a prominent band of approximately
6.5 kb in size in T84 cells. Similar, strong
iY~ 91 / I 0734 PC°T/CA91 /00009
~;
~~ 1 ~ ~ .~ e~ ~t cs ~:..
hybridization signals were also detected in pancreas and
primary cultures of cells from nasal polyps, suggesting
that the mature mRNA of the putative CF gene is
approximately 6.5 kb. Minor hybridization signals,
probably representing degradation products, were detected
at the lower size ranges but they varied between
different experiments. Identical results were obtained
with other cBNA clones as probes. Based on the
hybridization band intensity and comparison with those
detected for other transcripts under identical
experimental conditions, it was estimated that the
putative CF transcripts constituted approximately 0.01
of total mRNA in X84 cells.
A number of other tissues were also surveyed by RNA
gel blot hybridization analysis in an attempt to
correlate the expression pattern of the 10-I gene hnd the
pathology of CF. As shown in Figure 8, transcripts, all
of identical size, were found in lung, colon, sweat
glands (cultured epithelial cellsj, placenta, liver, and
parotid gland but the signal intensities in these tissues
varied among different preparations and were generally
weaker than that detected in the pancreas and nasal
polyps. Tntensity varied among different preparations,
for example, hybridization in kidney was not detected in
the preparation shown in Figure 8, but can be discerned
in subsequent repeated assays. No hybridization signals
cauld be discerned in the brain or adrenal gland (Figure
8j, nor in skin fibroblast and lymphoblast cell lines.
Tn summary, expression of the CF gene appeared to
occur in many of the tissues examined, with higher levels
in those tissues severely affected in CF. While this
epithelial tissue-specific expression pattern is in good
agreement with the disease pathology, no significant
difference has been detected in the amount or size of
transcripts from CF and control tissues, consistent with
the assumption that CF mutations are subtle changes at
the nucleotide level.
CVO 91/10734 PCTlCA91100009
42
3.2 TFI~ 3'~~OR CF' ~IUTA.T,~~~1
Figure 16 shows the DNA sequence at the F508
deletion. On the left, the reverse complement of the
sequence from base position 1649-1664 of the normal
sequence (as derived from the cDNA clone T16). The
nucleotide sequence is displayed as the output (in
arbitrary fluorescence intensity units, y-axis) plotted
against time (x-axis) for each of the 2 photomultiplier
tubes' (P2~T~1 and ,~2) of a Dupont Genesis 2000TM DNA
l0 analysis system. The corresponding nucleotide sequence
is shown underneath. On the right is the same region
from a mutant sequence (as derived from the cDNA clone
C16). Double-stranded plasmid DNA templates were
prepared by the alkaline lysis procedure. Five ~Sg of
plasmid DNA and 75 ng of oligonucleotide primer were used
in each sequencing reaction according to the protocol
recommended by Dupont except that the annealing was done
at 45°C for 3o min and that the elongation/termination
step was for l0 min at 42°C. The unincorporated
fluorescent nucleatides were remaved by precipitation of
the DNA sequencing reaction product with ethanol in the
presence of 2.5 M ammonium acetate at pH 7.0 and rinsed
one time with 70~ ethanol. The primer used for the T16-1
sequencing was a specific oligonucleotide
5'GTTGGCATGCTTTGATGACGCTTC3° spanning base position
1708 - 1731 and that far C16-1 was the universal primer
SK far the Bluescript vectar (Stratagene).
Figure 17 also shows the DNA sequence araund the
F508 deletion, as determined by manual sequencing. The
normal sequence from base pasition 1726-1651 (from cDNA
T16-1) is shown beside the CF sequence (from cDNA C16-1).
The left panel shows the sequences from the coding
strands obtained witty the B primer
(5'GTTTTCCTGGATTATGCCTGGCAC3') and the right panel those
from the opposite strand with the D primer
(5'GTTGGCATGCTTTGATGACGCTTC3'). The brackets indicate
the three nucleotides in the normal that are absent in CF
~'O 91/10734 PCT/CA91/00009
4 3 ~ ~~ ~~ ~ $ (~ y;
(arrowheads). Sequencing was performed as described in
F. Sanger, S. Nicklen, A. R. Coulsen, Proc. Nat. Acad.
sci. u. s. A. ?~: 5463 (i9??~.
The extensive genetic and physical mapping data have
directed molecular cloning studies to focus on a sanall
segment of DNA on chromosome 7. Because of the lack of
chromosome deletions and rearrangements in CF and the
lack of a well-developed functional assay for the CF gene
product, the identification of the CF gene required a
detailed characterization of the locus itself and
comparison between the CF and normal (N) alleles.
Random, phenotypically normal, individuals could not be
included as controls in the comparison due to the high
frequency of symptomless carriers in the population. As
a result, only parents of CF patients, each of whom by
definition carries an N and a CF chromosome, were
suitable for the analysis. Moreover, because of the
strong allelic association observed between CF and some
of the closely linked DNA markers, it was necessary to
exclude the possibility that sequence differences
detected between N and CF were polymorphisms associated
with the disease locus.
3.3 IDENT~FI~~fJL"ION OF RFLPs AND FAMIh'X sT~TD~Es
To determine the relationship of each of the DNA
segments isolated from the chromosome walking and jumping
experiments to CF, restriction fragment length
polymorphisms (RFLPs) were identified and used to study
families where crossover events had previously been
detected between CF and other flanking DNA markers. As
shown in Figure 14, a total of 18 RFLPs were detected in
the 500 kb region; 1? of them (from E6 to CE1.0) listed
in Table 2; some of 'them correspond to markers previously
reported.
Five of the RFLPs, namely 10-1X.6, T6/20, H1.3 and
CE1.0, were identified with cDNA and genomic DNA probes
derived from the putative CF gene. The RFLP data are
presented in Table 2, with markers in the MET and D?S8
W~ 91 /10734 PCTlCA91 /00009
k
as
regions included for comparison. The physical distances
between these markers as well as their relationship to
the M>;T and D7S8 regions are shown in Figure 14.
!'!'O 91 > I 074 PCr/~~91 /00009
45 ~?~~"('~e".:..
hJ 'J ,n h~ :Y ~:C
Q1
~ CY fIF ~ d~
~
4Y ~" f7 QJ
f.3, f7 .N N
.E.1
~ ~
~.I jJ1p .Ca C9 d) Gl N
r1 '~1 '~w lI1 lC ~ 01
~ ~ 3 ~ '~ CO
~ CD
W w j"..0l a CO
01
~r~ r"~ .~ ~ ~
4"'i ~ .~ Pd
P'~
G: f'7 C9'r Ra '3 0.7 b'
4l 1d N IO ~-'
h~ e!1 .x",
'~-~
O 1S 41 ~0
O O O
'a ~ O O O
O !9 tt~
eD r'1 et
A,' O O O
O
OD lt1 t~1 01 O N ' P
N t~ c1' eh N tD ~I
(yb CO Ot afi G1 tC1 CO CO N
..~
N !t1 h srl V' M lh eh
N
~ .i~,x
1 v
t0 C ~D CD N t0 tP1 O ~l' ~0
W !\ v0 1C sN P er cr c1
ri
H H H H
W ~
H H H
I
~ G ~
~ ~ W
W~ 91/10734 PC'I"/C~,91100009
46
W .-.
~
i tb
b O r
rI
x
~ l
i O
N . r
~ .- ~
.ri
O
w
H
i.,~
" _
r-4 .EJ~ ~ N r-1 CO N
, CO N bd l'
.~ .'Y
C9
~' ~ !A n.G ibl N rI tCl
t0 ~ lCJ
x ",~
~.J t~
A1
iJ
w
N
r7 C?~' f~7 ~C M A
OJ ~"' ~U G7 W
h ~ PO
ri
O r.~ M
,.
.~ ri
O O O O
O
y. M CO '0'
~ ~
e ~ ~ O
.
O p ~ o
p
v
.,.~
O
f
r !P1 M d'M ep !L1 1G
e-!
N
H
O ~-i ~-1 c0cV N Cv
~l' !n 00
, rdCO N M M M lL1
~ ~' ~ - t0
er CO
M ~ O O C N
N -1
v-1 f"1
N
H
H
H~, W b H H
1'~ .~ ~' a ' f
H x ,r f
1 H
x
U
d1, 3 x W
~-'
WHO 91/0734 PC'I'/CA91/00009
~ y~r3~,a 1
~ o
'
~ so .P.,
Pa ~ ~,
~> 4
~
~ ~
w N v ro.~
W ~ ~ ~
~
b
~ ~
d-~
i.i p
~C CI t0 C7 Caa U t;
4! N
CA CO e~ t~ '"i
'i
r~ r~ r.~ N
ri
C O O O O O
~ On0 ~ ~ O
CO O1
~ p o O O O O
U ~ n 1ritC1
tp 01 e-1 y0 !I1In 01 p W o n-1
N
W
E.,
r' N Oi tT 10 N N l~ CO O ~Y h
N 10 t~ f"1 ~,'N ll~f") P'1 V' a9 1D
~ ~ + r7
~ N Ln
N '1 l1 tryf1
r1 , ~ pd N p r-1 t0 r1 r1 ~D
~ , ~, N n ~
U
x ~ ~
~ U
""~ ~p .
~ , 0~
N 00 -1 ~
W W ~
h :~ W v h
WO 91/10734 PCt'/C/191l00009
48
~.a
N QI
h'~
H lf1 ri CO
ir. iG .~.
tLt tO
CJ ~
U cn h
~ .~
.
co
o~
~
cr~3b
;
td'S at' Ifi M N cap
N !87 ri O p O
O O O O O p
H M h H h
01 V7 6C 'd' rd N
a
n
r~
.~
Q
v 10 ~O r! tp 00 !~ 10 h f1 N ~'i C1 M
N
W
a
H
r~ r1 h lt1 N !L'!i1 00 CO N at' 1") tG
O
t1
'f-
~ N f'-1ef'ra tt h N 00
a
o o ~ . v -t' v v ~ o .
M ri CIO et'N r1 t~l e1 t-1 lp sr r'1
'
F-I
M
H
b N U
b b
x ~ x
~e o . o ,..,
M N M . H
1 w . ri N
~ W ra M
~ x U
WO 91/10734 PCT/C~.91i00009
49
N y.a
H
r1
U
~ O
O
0
r~
O
N
O
O
U ~ t~
M t°1
N
G~7
a
H
u~
~o
H
H
Gr
d1
N
p7
w0 93/10734 PCT/CA9i/00009
P~IO~'~8 FOR 'f~IBTW 2
(a) The number of N and CF-PT (CF with pancreatic
insufficiency) chromosomes were derived from the
parents in the families used in linkage analysis
5 [Tsui et al, Cold Spring Harbor Sump Ouant Biol
51:325 (1986)].
(b) Standardized association (A), which is less
influenced by the fluctuation of DNA marker allele
Z~ distribution among the N chromosomes, is used here
for the comparison Yule~s association coefficient
A=(ad-bc)/(ad+bc), where a, b, c, and d are the
number of N chromosomes with DNA marker allele l, CF
with 1, N with 2,. and CF with 2 respectively.
15 Relative risk can be calculated using the
relatianship RR = (I~A)/(1-A) or its reverse.
Allelic association (*), calgulated according to A.
Chakravarti et al, Am. J Hum cP~e~ 36:1239,
20 (3.984) assuming the frequency of 0.02 for CF
chromosomes in the population is included for,
comparison.
Because of the small number of recombinant families
25 available for the analysis; as was expected from the
close distance between the markers studied and CF, and
the possibility of misdiagnosis, alternative approaohes
were neoessary in further fine mapping of the CF gene.
3.4 L ~h C A88oCIA2~~eat
3a Allelic association (linkage disequilibrium) has
been detected for many closely linked DNA markers. While
the utility of using allelic association for measuring
genetic distance is uncertain, an overall correlation has
been observed between CF and the flanking DNA markers. A
35 strong association with CF was noted for the closer DNA
markers, D7S23 and D7S122, whereas little or no
WO 91 /10734 PCT/CA91I0000~
~~~~.~~tt !~.~
~ i-a .r _d '.... .....
association was detected for the more distant markers
MET, D7S8 or D7S424 (see Figure 1).
As shown in Table 2, the degree of association
between DNA markers and CF (as measured by the Yule's
5 association coefficient) increased from 0.35 for metes and
0.17 for J32 to 0.91 for 10-1X.6 (only CF-PI patient
families were used in the analysis as they appeared to be
genetically more homogeneous than CF-PS). The
association coefficients appeared to be rather constant
over the 300 kb from EG1.4 to H1.3; the fluctuation
detected at several locations, most notably at H2.3A,
E4.1 and T6/20, were probably due to the variation in the
allelic distribution among the N chro2~osomes (see Table
2). These data are therefore consistent with the result
from the study of recombinant families (see Figure 14).
A similar conclusion could also be made by inspection of
the extended DNA marker haplotypes associated with the CF
chromosomes (see below). However, the strong allelic
association detected over the large physical distance
between EGl.4~and Fil.3 did not allow further refined
mapping of the CF gene. Since J44 was the last genomic
DNA clone isolated by chromosome walking and jumping
before a eDNA clone was identified, the strong allelic
association detected for the JG2E1-J44 interval prompted
us to search for candidate gene sequences over this
entire interval. It is of interest to note that the
highest degree of allelic. association was, in fact,
detected between CF and the 2 RFLPs detected by 10-1X.'6,
a region near the major CF mutation.
Table 3 shows pairwise allelic association between
DNA markers closely linked to CF. The average number of
chromosomes used in these calculations was 75-80 and only
chromosomes from CF-PI families were used in scaring CF
chromosomes. Similar results were obtained when Yule's
standardized association (A) was used.
W~ 91/10734 PCT/CA97/00009
:y '-2e ii ? 3
i,a .~ CI cJ a ~~ _L
52
~ ~'° u' as r
r r
r ~ ~ r. r
~ ~~ ~ ~ d d s~ cd ~ ~ ~ ~ ~q F'3 '~ 'c~ '~3 r a. ,~ ,
d ~ sa ~ ~ d ~ sa o c~
r r
r r 1 O
n b ~ ~ ~ A ~ ~ ~ O o ~ o ~ ~ 6~ O C3 la ~ ~
h ~p
~ ey n '~ '~, '~ e~ e°'4 ~ ~t r? ~ ~! N ~ ~ o °~ r
r r
o ti ~ o ~ o ~ ~ o o e9 0 0 0 0 ~ o o d ~ o
O r ~ O ~ S ~ r P ~ ~ ~ 1 ~ O
C O O S~ d O d ~ d ~ t~ ~ ~ o o d d ~ O ~ ~ Ci
'r° r a ~r :~ ~ ° ~ ~ tr ~r ~ t~ s°~ s~ c~ '.~ 'u~ . '~
tad r
~ o 0 o d ~ ~ ~ d ~ d o d d o r~ d d o
8 ~ ~ ~ ~ ~ ~ ~ ~ ~ o ~ $ ~ ~ ~.' ~ 1 r o
~ d d ~ ~ 0 0 o d d d o d d d ~ d d ~ ~ o
o o e' h .-
o .
3C ~ ~ ~ ~ d d ~ d o d ~ d d d d ~ d ~ ci
~ ~ ~ ~ d d d o d d d ~ ~ ~ o ... d o rS o 0 0
8 =a~P'~~~~~i~~~c~~ 1 ~~: ~~~'yc~e
-i o d d d d d ~ ~ ~ d d ~ ~ ~ ~ d o ~ d
~~1~', 8~~~~3'c3~ oneo. ~a*°. ~~~~~~~ ~'r°
o ~ ~i ~ d a d o o a o o d d ~ d d ~ ~ ~ r3 0
d~~~o~dddooa °~~~~~~~A~~
~O( ~ ~ ~ r ~ ~ ~ ~ ~ ~ a ~ ~ ~ ~ ~ ~ ~ ~ ~ r0 p
d ai d d d d d d d ~ d d r d d d d d ~ ea C 0
o " ~' $ ~-
0 0 ~ ~ o ~ ~ O ~ r r
a.. d d d d d d d d o o ~ .. o o d d o d o d o
U
o d d d d d d d o 0 0 0~ 0 0 0 o d d o ~ o
o ~ d ~ ~ ~ d ~ ~ ~ ~o o d o ~ d ~ ~ v
c'~ g ~ °~ ~ ~ ~ '~ ~ ~ ~ o
1- .0 0 0 o d d o o d d o d d d o o d ~ d
~~
~o~~~
~~~~,~~~~~a~~gA~~~~~~~0
~dd~~ dddddod~dddoodd~d~
t~ ~ r O , ~ ~ ~ O n ~ w r P p O
r0 ~ ~ s~ ~ o o O O O ~ ~ d O d d O ~ d d d O
1 10
0 0 0 o r
I-° O d ~ d o o d d O d ~ d ~ d o O O o O d ~ O
~ ~ ~ Q
D 1 ~ 8. ~ 8 O d d d d d d O ~ ~ ~ Gi C O O
0 ~ ~ ~ ~ r n ~ ~1' r
o O ~ d o O O O O d O O d O d d O ~ C ~ ~ CA
a
LU w '~P et r ~' ~ ,~ t0 ~ ~ ~ ~ r
r
r P
S~LId~S~1,1~0,ti.d~ ~~
S S'T !'T '!' !~ E T.
wo 9W lo~3a PCT/CA91/000~9
53 ~~;~~'':z'x,'
Strong allelic association was else detected among
subgroups of RFLFs on bath the CF and N chromosomes. As
shown in Table 3, the DNA markers that are physically
close to each other generally appeared to have strong
association with each other. For example, strong (in
some cases almost complete) allelic association was
detected between adjacent markers E6 and E7, between
pH131 and W3D1.4~between the AccT and HaeTTT polymorphic
sites detected by 10-1X.6 and amongst EG1.4, JG2E1,
E2.6(E.9), E2.8 and E4.1. The two groups of distal
markers in the MET and D?S8 region also showed some
degree of linkage disequilibrium among themselves but
they showed little association with markers from E6 to
CE1.0, c~nsistent with the distant locations for MET and
D7S8. On the other hand, the lack of association between
DNA markers that are physically close may indicate the
pres~:nca of recombination hot spots. Examples of these
potential hot spots are the region between E'T and pH131,
around H2.3A, between J44 and the regions covered by the
pr~bes 10-1X.6 and T6/20 (see Figure 14). These regions,
containing frequent recombination breakpoints, were
useful in the subsequent analysis of extended haplotype
data for the CF region.
3 a 5 ~iA~~!1'YPL ANA.LYBTB
Extended haplotypes based on 23 DNA markers were
generated for the CF and N chr~mosomes in the collection
of families prewiausly used for linkage analysis.
Assuming recombination between chromosomes of different
haplotypes, it was possible to construct several lineages
of the observed CF chromosomes and, also, to predict the
location of the disease locus.
To obtain further information useful for
understanding the nature of different CF mutations, the
F508 deletion data were correlated with the extended DNA
i'VO 91110734 P~H'/CA91/00009
54
marl~er haplotypes. As shown in Table 4, five major
groups of 1d and CF haplotypes could be defined by the
RFLPs within or izmnediately adjacent to the putative CF
gene (regions 6-~8) .
WO 91/10734 PCT/Ch91/00009
ss ~~~~;~,. ~
.................. .....
................................
a . a . . . ~ . ~ . . . . . . . . . . . . . . . . . . . . . . . .
rl......s..s,~.~v..o~vs.a.o'.1~mv.p.~asn.1
S v N
0
w ~ .a . .a ~o wo ri ~s .a . .~ ,.~ ~ ,.a re ..i ev re ~ Pa Pe ~ r.e . .., .
,~ ry ~ ra .i
U
tt8
~r
~I~C~~~Z~cDOSU~CWC ~.e~d~~6~~~C~o~f~~m~acAl~t~oA~9~2cOCa
t~ s~~~ ~ ~e~'~.~afRbCaC~ ~~'al~~~ ~~aC~CaCaE~C~BWC~~~~t~
pa
I~IiC °~~&'~'~0.'~~e'~'~~'~~~~oCili~', ~9~'b'~4'~~~'~'. ~~C~~V'~'~
H ,,fir
~i°BsCe~aE~~~'.odd.~~'~'~6'~~6'4°~rC~'~'.iC~uCieCoC~'.a'.i&eGA~Ca
CG
I~i ~ ~ ~ .1 ° ~ ° ~ ~ ~ ~ ~~l ~ ~r ~ ~ ~ v n o1'
V A d ~ ~ ~ ~ V V
Q
v
N
WO 91!10734 . PCTlCA91/OOOU9
56
p'. . . .p . , p ,r-1r-lpppr7 pp r~1 . . . .Mr-7prlpn-~1 .r-lr4r-!
~ a ,..0 , s , a . a . a . . . . . . . p . . p a . o . a . . p
. . a p ,./ a r~p.'~ . p a . a a , a a a n ,../ p a ~ a . a . ~ . s . a
. . . . . . a a s o . a a a a o n a ~ a a s a . . , s s a . , a
. p . . . a a a s . . a , a . . . . ~j .s . , a . , a a . ~ . s.
~ GC ~ ~ t~ a tp GD 0~ CO 65 ~ ~ ~ U ,~ r~ f~ m QE fta ~p s~ ~ ~ a6 61 V G ~
~$ L7
U
~~wC~d~E~a6 aaaaaaaa ~U UU~UUUUC9uUtlCduU
ACS. ~~aC~!~~~ ~i~'~~°aCiC ~ ~ ~E~C ~CaCa6~~'A6 ~w,~A ~nC~~laC
WCdaCeC~Q ~.'~4'a&p6~6W6aC ~~,' mmmmmmmmmf4mmmm
~d ~ ~BAC~~e6.6~d uUUUClu6Ji6 A:~ ~t'~ ~t~m6Dmi~6~itD ~~m~
W 6~C m~f$m~ODfttW CU oDmmtn~IB~IDtAfi~00s0ft1m09
a II~~ a IPa ~ ~ ~ ~ ~ ~ ~ ~ .r ~ ~ ~ ~ m ~ ~ ~ ~ ~ ~ ~ ~ m ~ m
a
mu uuuuuu uuu~aamam mu .e~~cvmcrue~uuuuuu
~eca ~w~aa~m mmd~w~maa~ am cg .m~oaat~a,~.~mcamoa
I
..
.a O ro M
e.. .. r H
SSTfT TEST
WrD 91110734 ~J ~ :~~ a ~ ~ ~ p~,T/CA91/0000'9
57
.~a .-t ev ra ..r ~ .-o .~ r.~ .-o Pa r.r r.o r1 P1 :v ..1 0.r ~ e.a a~ . ..~
Pa ~ ..a . ,.~ .~ . re re Pa
a a . . a o a . a a s a . a o a a s . a . r.1 . a s . . s a r4 a a s
a a a a . a . a . a . . s a s . a a . . . . a . . . r/ a . o a . a
ucscaucaca.uuuucsut~uuuucauueau~vu~~u~uc,3uuu
a
CD ~ W C9 W ~ R~ m SD ~9 6~ ~ i9 ~i CA ~l 8p C4 ~ ~d CO G9 ~ 61i 0~ l~ ~ ~i GO
to ~ OG Qi
a a v ~ ~. ~. ~ IAe ~. ~ ifa ps a oe ' ~ ~ a ~ .
U U
outs .uu~e~~c~saaaan~acauuuuuo'~auaaaoc~uuuaac~msa
~~. ~I~~~~a~~I~rbi~~,~~~~~~ V ID~~~~~~~1P~ e~.
WO 91/1073a PCT/CA91/04049
~~~ ~~Zl~~
58
P
,.~ep . . . vi CV rC H RV .4 rd . o-a n-C
o e-1 r8 ri . r~1 .~1
tV r4 .1
.y . . . . . a . . ,.~ a . ~.,yN
a a . rd o . . o .
~O r1 r-1 . . . . . . N . n . . .
. N r1 . . . ..~ .
.
. . . p . . s . .. a . . . a a . p . o .
. . . .
. . . p ,.~. . . s . . . . o, . . N . . .
. . . o
6.7t~ a Ca 6'Oa~~~Ca~60~a~6'If~G.iDa ~ .SD
mra a .c .eaaa~aaa.saa~~ea~a aaa uuc~
as a oa as asaoasoaasaocamsamma,easaasaaeo a,m .
.
aaao as a vuuuuuuuvuuuc~uuu uuu W uu
,~aa a. ~eaaaaaaaa~aaaaaa ~aa~m aaa
~~ ~ ~ ~.caaaa.savaaaaa.ea ea~a, aaa
a . . ca aomm . . saaa~ma .aaaaaoaa aaaa
.a
s~u o a auuuaa~mucoaaa~~auuo~ae~aa uuca
as oa m asaaaa~aaeaoa~s~~a~aaaa.A a.aaa
_ ._ _
l7 V N .i7
b
v N v N
v
~JS'~~'~~J'E ET
WO 91/10734 ~ ;~ p' P01'/CA9I/00009
~)
l~
~~
,
~
~
F~~ J
.. ~:
5 9 .
. . .-r a us rr ..~ Pa e~ o
,~ .a ra .r .a .-o
.a .i ~.
. . >~,. . . ~, ~, . . . ,
. . . . . . p
O rirl.sa. .. N .....e p N N
O , a s . p , s . s a
. . a . , a p
a p . a . . p , s a se ~ ~
. . . . . , p
(~ fed ~6 ~ f~ ~C iD ~ ~ ~ ~ .
1~ f!d O ~' Gi
~
cau a >uuuu vv ~c~eauucs~'
ca ~~~~s~t ~ comoo .~e~~ ,
c.~
Ui~ CD UUUtd G7 ODOo~,140f4~G~
C1
. t4 G0 . f~D U .C eE 09 tii
f4 0D G G4 ~C iC
~
oC a1 ~ GO CD 04 ~G ~C' k7 ~1
~ fa 00 EA ek', eP
R7
sDW alC9RJP~ AC GAG~fW C~ ~
>R'
xca cruooc~u uu ucsuuauca
.ea a~~cm~aaaaaix maaaacom~c~
~ ..
a m
"' m m .. ~
~u ' o
~
~
. r H
o ..
a ~
~
5 ~ ~'~ '~E S T
i~'O 91110734 FGf/CA91 /~~DOU9
TAELE 4 (continued)
(a) The extended haplotype data are derived from the CF
families used in previous linkage studies (see footnote
(a) of Table 3) with additional CF-PS families collected
5 subsequently (Kerem et a1, Am. J. GeDet. 44:827 (1989)).
The data are shown in groups (regions) to reduce space.
The regions are assigned primarily according to pairwise
association data shown in Table 4 with regions 6-8
spanning the putative CF locus (the F508) deletion is
l0 between regions 6 and 7). A dash (-) is shown at the
region where the haplotype has not been deter~ainsd due to
incomplete data or inability to establish phase.
Alternative haplotype assignments are also given where
date are incomplete. Unclassified includes those
15 chromosomes with more than 3 unknown assignments. The
hap~,o-~ype definitions for each of the 9 regions are:
Region 1- metD metD mates
Banl
2o A = 1 1 1
B = 2 1 2
C g 1 1 2
D = 2 2 1
E = 1 2
25 F = 2 Z 1
G = 2 2 2
Region 2- E6 E7 pHI31 W3D1.~
30 Taal Taa
A = 1 2 2 2
s = 2 Z
C = 1 2 1 1
35 D = 2 1 2 2
E = 2 ~ 2 2 1
F = 2 2 1 1
WO 91/10734 PCT/CA91/00009
67.
G = 1 2 1 2
H = 1 1 2 2
Region 3- H2.3A
Taqz_
A = 1
B = 2
Region 4- EC1.4 EG1.4 JG2E1
~,~n~~x Ba~.x ~Stz
A = 1 1 2
B = 2 2 1
C = 2 2 2
D = 1 l 1
E = 1 2 1
~0
Region 5- E2.6 E2.8 E4.1
NCA MspI
A a 2 1 2
B ~ 1 2 1
C = 2 2 2
Region 6- J44 10-1X.610-1X.6
3 t1 Xbal cc HaeIII
A = 1 2 1
B = 2 1 2
C = 1 1 2
D = 1 2 2
E = 2 2 2
F = 2 2 1
ii~0 91!30734 ~'Cd'/CA91100009
r ~,~~~~~ ~ 62
RegJ.oI1 7° T6~20
MS1~I
A = 1
B = 2
Region 8- H1.3 CE 1.0
co eI
to A = 2 1
B= 1 2
c= 1 1
D = 2 2
Region 9- J32 J3.11 J29
Sac NisnI PvuII
A = 1 1 1
B = 2 2 2
C = 2 1 2
D = 2 2 1
E = 2 1 1
(b) Plumber of chromosomes scored in each class:
CF-PI(F) = CF chromosomes from CF-PI patients with
the F508 deletion;
CF-PS(F) = CF chromosomes from CF-PS patients with
the F508 deletion;
CF-PI = Other CF chromosomes from CF-PI patients;
CF-PS = Other CF chromosomes from CF-PS patients;
N = Normal chromosomes derived from carrier parents
dV~ 91 / 10734 PC'1!'/CA.91 /00009
63 ~:::~f ~ ~ ~)
tJ i1 ~ t'
It was apparent that most recombinations between
haplotypes occurred between regions Z and 2 and between
regions 8 and 9, again in good agreement with the
relatively long physical distance between these regions.
Other, less frequent, breakpaints were noted between
short distance intervals and they generally corresponded
to the hot spots identified by pairwise allelic
association studies as shown above. It is of interest to
note that the F508 deletion associated almost exclusively
1C with Group I, the most frequent CF haplotype, supporting
the position that this deletion constitutes the major
mutation in CF. I4lore important, while the F508 deletion
was detected in 89~ (62/70) of the CF chromosomes with
the AA haplotype (corresponding to the two regions, 6 and
7) flanking the deletion, it was not was found in the 14
N chromosomes within the same group (~2 = 47.3, p <10'~).
The ~'S08 deletion was therefore not a sequence
polymorphism associated with the core of the Group I
haplotype (see Table 5).
Together, the results of the oligonucleotide
hybridization study and the haplotype analysis support
the fact that the gene locus described here is the CF
gene and that the 3 by (F508) deletion is the most common
mutation.in CF.
3.6 INTRONjEXON HOONDhRIER
The entire genomic CF gene includes all of the
regulatory genetic information as well as intron genetic
information which is splicad out in the expression of the
CF gene. Portions of the introns at the intron/exon
boundaries for the exons of the CF gene are very helpful
in lacating mutations in the CF gene, as they permit PCR
analysis from genomic DNA. Genomic DNA can be obtained
from any tissue including leukocytes from blood. Such
intron information can be employed in PCR analysis for
purposes of CF screening which will be discussed in more
detail in a later section. As set out in Figure 18 with
the headings "Exon 1 through Exon 24", there are portions
6V0 91/10734 ~'C1'/CA91100009
s4
of the bounding introns in particular those that flank
the axons which are essential for PCR axon amplification.
Further assistance in interpreting the information
of Figure 18 is provided in Figure 21. Genomic DNA
clones containing the coding region of the CFTR gene axe
provided. As is apparent from Figure 21, there are
considerable gaps between the clones of the axons which
indicates the gaps in the intron portions between the
axons of Figure 18. These gaps in the intron portions
to are indicated by 'o.,,e~. In Figure 21, the clones were
mapped using different restriction endonucleases (AccI,A;
AvaI,W; BamFiI,B; BgIII,G; BssIiI,Y; EcoRV,V; FspI,F;
HinCII,C; FIIndIII,H; Kpn,K; NCOI,.T; PstI,P; PVUII,U;
SmaI,M; SacI,S; SspI,E; StyI,T; XbaI,X; XhoI,O). Iri
Figure 21, the axons are represented by boxed regions.
The ~apen boxes indicate non-coding portions of the axons,
whereas closed boxes indicate Boding portions. The
probable positions of the axons within the genomic DNA
are also indicated by their relevative positions. The
arrows above the boxes mark the location of the
oligonucleotides used as sequencing primers in the PCR
amplification of the genomic DNA. The numbers provided
beneath the restriction map represent the size of the
restriction fragments in kb.
Tn sequencing the intron portions, it has been
determined that there are at least 27 axons instead of
the previously reported 24 axons in applicants'
aforementioned co-pending applications. Exons 6, 14 and
17, as previously reported, are Bound to be in segments
and are now named axons 6a, 6b, axons 14a, 14b and axons
17a, 17b.
The in~tron portions, which have been used in PCR
amplification, are identified in the following Table 5
and underlined in Figure 18. The portions identified by
the arrows are selected, but it is understood that other
portions of the intron,sequences are also useful in the
PCR amplification technique. For example, for axon 10
vy0 91/10734 PCf/CA,91/00009
65 ~~'j~ ~~b~!
a.
the relevant genetic information which is preferd~ed in
PCR is noted by reference to the 5° and 3' ends of the
sequence. The intron section is identified with an "i".
Hence in Table 5 for axon 2, the preferred portions are
identified by 2i-5 and 2i-3 and similarly for axons 3
through 24. For axon 1, the selected portions include
the sequence GGA...AAA for 8115-B and ACA...GTG for IOD.
For axon 13, portions are identified by two sets: 13i-5
and C1-lm and %13B-5 and 13i-3A. (This axon (13) is
to large and most practical to be completed in two
sections). C1-1M and %13B-5 are from axon sequences.
The specific conditions for PCR amplification of
indivisual axons are summarized in the following Table 6
and are discussed in more detail hereinafter with respect
to the procedure explained in R.K. Saiki et al, Sci nce
23p:1350 (1965).
These oligonucleotides, as derived from the intron
sequence, assist in amplifying by PCR the respective
axon, thereby providing for analysis for I~NA sequence
alterations corresponding to aautations of the CF gene.
The mutations can be revealed by either direct sequence
determination of the PCR products or sequencing the
products eloped in plasmid vectors. The amplified axon
can also be analyzed by use of gel electrophoresis in the
manner to be further described. Tt has been found that
tlae sections of the intron for each respective axon are
of sufficient length to work particularly well with PCR
technique to provide for amplification of the relevant
axon.
wc~ 9aiao7~a ~cr,cA~aio~o9
~,~~~~~~i.'~
~'ASr~ s
ss
Oligonuchttdes useQ for amplification of CF gene axons by 1?CR
Exon 1'CR primers; 5'-> 3' Amplired produce (bp)
1 , GGAGTTCACTCACCTAAA (Ells-B) 933
ACACGCCCTCCTCTI°TCGTG (lOD)
2 CCAAATCTGTATGGAGACCA (2i-~ 3?8
TATGTTGCCCAGGCTGGTAT (2i~3)
3 CTTGGGTTAATC't'CtrThGGA (3i~5) 3pg
ATTCACCAGAT'i'1'CGTAGT!C (3i-3)
4 TCACATATC~G?'ATGA(4I-5) 438
TT'GTACCAGC'I~CACTAG~fA (4i-3)
ATTTCTGCCTAGATGCTGGG (Si-,5~ 395
AACTCCGCC'I°T"TCCAGTl'GT (5i-3)
~ TTAGTGTGC1°CAGAACCACG (bAi-~ 385
CTATGaCATAGAGCAGTCC1~ (tSAi-3)
6'b TGGAATGAGTCTGTACAGCG (6Ci-5) 41?
GAGGTGGAAGTCTACCATGA (6Ci-3)
? AGACCATGCTCAGATC'ITCCAT (7i-5) 410
GCAAAGTTCATTAGAACTGATC (7i-3)
' 8 TGAATCCrAGTGCTTGGCAA (8i~~ 3S9
TCGCCATTAGGATGAAATCC (8I-3)
9 TAA'IY~GATCATGGGCCATGT (9i-5) ~p
ACA~AATGTGGTGGA (9i-3)
GCAGAGTACCTGAAACAGGA (l0i-5) 491
CAT'TCACAGTAGCTTAG'CCA (l0i-3)
11 CAAL'11GTGOTTAAAGCAATAG'lY3T (a li~3) 425
GCACAGATTCTCiAGTAACCATAAT (1li~3)
12 GTGAA'hCpA7T3TGGTCiACCA (12i-5) 426
C~T'ITAGCATOAGGC(3G1° (121.3)
I3 (a) TGCTAAAATACGA~ACATATTGCA (13i-5) 52g
A'd~.°I'CrGI°AC'I'AAGCiACA(3 (Cl-1M)
(b) TCAATCCAATCAAC'PCTATACGAA (X138-5) 49?
TACACCTTATCCTAATCCTATGAT (13i-3A)
14a AAAAGG1'ATi3CCAL'n31'1'AAG (l4Ai-5) 511
GTATACATCOa',AAAC1°ATCT (l4Ai-3) .
14b GAACALXTAGTACAGCT~1' (idii;i~5) 449
AAC'lY~'1~GGGC'rCleAf3TGAT (14BI-3)
GTGCATG~CZ°!'C1'AA'lYiCA (15I-5) 485
AAGGCACaITGt~CTC'1°GTtiCA (15i~3)
16 CAGAGAAATTGGT~r °TACT (16i-~ 5?p
A'I'~."1'AAAGGA1'~T (16i~3)
1?s ' CAA,'PC3TGCACA'hCiTAt~C'TA (1?AI-5) 5?9
TGTACACCAAC'1°GTGGTAAG (1?Ai-3)
1?b TTCAAAGAATGGCACCAGTGT (17BI-5) 463
ATAACGTATAGAATGCAGCA (1?Bl-3)
18 GTAGATGCTGTC1ATGAAG'TQ (18i~5) 451
AGTraGCTATGTATGAGAAGG (18i~3)
19 GQOCGACAAATAACCAAGTGA (19I-5) ~ 454
GCTAACACATTGCT1'CAGGC'T (19i~3)
GG"1T,'AGGATTGAAAGTGTGCA (1~OI-5) 4?3
C'd'ATGAGAAAACTGCAChCiGA (?Gi-3)
21 AATGTTCACAA~iGGACTCCA (21i-~ 4?7
GTACCTGT1'GC'1'CCA (21i-3)
~ AAAACAAGA (22i-5) 562
TGTCAt7CATGAAGCAGGCAT (22i~3)
23 AGC'nGAT'hC,iTGCGTAAG'GC'1' (23i-:~ 4~p
TAAAGCT~CiGATATG (23i-3)
24 GGACACAGCAGTTAMTGTO (24i-5) 5~
ACTATTGCCAGGAAGCCATT (2aii-3)
-~ ~-~ ~' E .
igd0 91/10734 PC~i'/C~9i100009
67
TA LE 6
Z'hennal
cycle
Exon BufferInitial DenatuaationAnnealingExtensionFrltal
o
denaturatian extension
~elte~nPtitx~eJterraPtimcJtetnptime/te~ti~mp
3-5 ,ba A 6 4 C 30 secf9430 sec/351 asiM2T taiN72 C
,bb, (1.5) C C C
?-10,
12,
14a,
i6.
17b,
18-24
l B 6 tnirv9430 s~c/9430 s~/552,5 7 minl72 C
C C C min/12
C
2,11 B 6 mini9430 sr~19430 s~1521 mit~i27 tnir~!'2
C C C C C
13a A(1.7~6 mits/9430 secl9430 sec/542.5 ? minll2 C
C C C minf72
C
13b A(1.T~6 miN'9430 serf9430 seeP522.5 7 mir~J2 C
C C C miN72
c
14b $ 6 4 C 30 secl9430. soc/561 mitt/72T t~ninPl2
C C C C
1Ta A(1.5)6 X94 30 sec~430 secl361 X72 7 ~in!!2 C
C C C C
(a) Buffet' A(13): . ,~ buffer with l.SusMI wig~Cla
Buffes A(1.T~: .~ bu~fea with l.TSmhd MgCIZ
But~'or B: 67 tarM lrfs-HC7 pH 8.8. 6.7 mM MgCI~ 16.6 mM (NfIayaSt~4, 0.67uM
EDT" ,
ld~ $-m~aptoethaaol,1T0 ug/asl BSA.1096 DMSO, 1.5 mIrf of each dN'TP's
~. ~ tx'~~r ~ Cc~rrfu ir~S : to ~, dh 'frr s pry g, 3 (~,~~~G)
50 ~ ~t KCQ
~; Dol ~~(w j~,v) ~Q,~i ~.
l~.z~,M dNTPs.
dNTPm =3eexynLCleotide t~ithosnhatpa
US~ITII~'~ SEAT
fVO 93/10734 F'CT/Cf9,91/00009
Fr Y _.
,~~~~"~~.~ 68
3.~ CF' MZJ~'~1'I°TOIds ~ AZS~6 O'7~3 Z~507
The association of the F508 deletion with 1 common
and 1. rare CF haplotype provided further insight into, the
number of mutational events that could contribute to the
present patient population, Based on the extensive
haplotype data, the original chromosome in which the F508
deletion occurred is likely to carry the haplotype -
AAAAAAA- (Group Ia), as defined in Table 4. The other
Group I CF chromosomes carrying the deletion are probably
recombination products derived from the original
chromosome. If the CF chromosomes in each haplotype
group are considered to be derived Pram the same origin,
only 3-4 additional mutational events would be predicted
(see Table 4). However, since many of the CF chromosomes
Z5 in the same group are markedly different from each other,
further subdivision within each group is possible. As a
result, a higher number of independent mutational events
could be considered and the data suggest that at least 7
additional, putative mutations also contribute to the CF-
PI phenotype (see Table 3). The mutations leading to the
CF-PS subgroup are probably more heterogeneous.
The 7 additional CF-PI mutations are represented by
the haplotypes: -CAAAAAA- (Group Ib), -CABCAAD- (Group
I~), ___BBBAC- (Group IIa), -cASBaAB- Group va).
Although the molecular defect in each of these mutations
has yet to be defined, it is clear that none of these
mutations severely affect the region c~rresponding to the
oligonucleotic~e binding sites used in the
PCR/hybridization experiment.
One CF chromosome hydridizing to the ~F508-ASO
probe, however, has been found to associate with a
different haplotype (group IIIa). It appeared that the
~F508 should have occurred in both haplotypes, but with
the discovery of aI507, it is discovered that it is not.
Instead, the 0F508 is in group Ia, whereas the ~I50? is
in group IIIa. None of the other CF nor the normal
chromosomes of this haplotype group (IIIa)have shown
WO 91 / 10734 PCT/Cr191 /00009
n .~ r a Il
:f l'~ .~ r'4 ~1
hybridization to the mutant (~F508) ASO [B. Kerem et al,
Science 245:1073 (1989)x. In view of the group Ia and
IIIa haplotypes being distinctly different from each
other, the mutations harbored by these two groups of CF
chromosomes must have originated independently. To
investigate the molecular nature of the mutation in this
group IIIa CF chromosome, we further characterized the
region of interest through amplification of the genomic
DNA from an individual carrying the chromosome IIIa by
the polymerase chain reaction (PCR).
These polymerase chains reactions (PCR) were
performed according to the procedure of R.K. Saiki et al
Science 230:1350 (1985). A specific DNA segment of 491
by including axon l0 of the CF gene was amplified with
the use of the oligonucleotide primers 10i-5 (5'-
GCAGAGTACCTGAAACAGGA-3') and 10i°3
(5'CATTCACAGTAGCTTACCCA-3')located in the 5' and 3'
flanking regions, respectively, as shown in Figure 18 and
itemzied in Table 5. Eoth oligonucleotides were
2o purchased from the FiBC DNA Biotechnology Service Center
(Toronto). Approximately 500 ng of genomic DNA from
cultured lymphoblastoid cell lines of the parents and the
CF child of Family 5 were used in each reaction. The DNA
samples were denatured at 94°C for 30 sec., primers
annealed at 55°C for 30 sec., and extended at 72°C for 50
sea. (with 0.5 unit of Taq polymerise, Perkin-
Elmer/Cetus, Norwalk, CT) for 30 cycles and a final
extension period of 7 min. in a Perkin-Elmer/Cetus DNA
Thermal Cycler. Reaction conditions for PCR
amplification of other axons are set out in Table 6.
Hydridization analysis of the PCR products from
three individuals of Family 5 of group ~IIa was
performed. The carrier mother and father are represented
by a half-filled circle and square, respectively, and the
affected son is a filled square in Figure 19a. The
conditions for hybridizaton and washing have been
previously described (Kerem et al, su~a). There is a
I~VO 91/10734 1'CTJCA91J00009
f '~l~le)~a~~ j 70
relatively weak signal in the father°s PCR product with
the mutant (oligo 0F508) probe. In Figure 19b, DNA
sequence analysis of the clone 5-3-15 and the PCR
products from the affected son and the carrier father are
shown. The arrow in the center panel indicates the
presence of both A and T nucleotide residue in the same
position; the arrow in the right panel indicates the
points of divergence between the normal and the 0I507
sequence. The sequence ladders shown are derived from
the reverse-complements as will be described later.
Figure 19c shows the DNA sequences and their
corresponding amino acid sequences of the normal, aI507,
and 0F508 alleles spanning the mutation sites are shown.
With reference to Figure 19a, the PCR-amplified DNA from
the carrier father, who contributed the group IIIa CF
dhr~mosome to the affected son, hybridized less
efficiently with the eF508 ASO than that from the mother
who parried the group Ia CF chromosome. The difference
became apparent when the hybridization signals were
compared to that with the normal As0 probe. This result
therefore indicated that the mutation carried by the
group IIIa CF chromosome might not be identical to eF508.
To define the nucleotide sequence corresponding to
the mutant allele on this chromosome, the PCR-amplified
product of the father°s DNA was excised from a
polyacrylamide-electrophoretic gel and cloned into a
sequencing vector.
The general procedures for DNA isolatian and
purification for purposes of cloning into a sequencing
vector are described in J. Sambrook, E.F. Fritsch, T.
Maniatis, Molecular Cloni_na~ A I,a a~r~ry gq~~nual, 2nd ed.
(Cold Spring Harbor Press, N.Y. 1989). The two
homoduplexes generated by PCR amplification of the
paternal DNA were purified from a 5% non-denaturing
polyacrylamide gel (30:1 acrylamide:bis-acrylamide). The
appropriate bands were visualized by staining with
ethidium bromide, excised and elwted in TE (10 mM Tris-
4f~ 91110734 PC1'/~A91/00009
71 ~~ ~j~~~~~:d
HClp 1mM EDTAa pH 7,5j for 2 to 12 hours at room
temperature. The DNA solution was sequentially treated
with Tris-equilibrated phenol, phenol/CHC13 and CHClj.
The DNA samples were concentrated by precipitation in
ethanol and resuspension in TE, incubated with T4
polynucleotide kinase in the presence of ATP, and ligated
into diphosphorylated, blunt-ended Bluescript KS~° vector
(Stratagene, San Diego, CA). Clones containing amplified
product generated from the normal parental chromosome
l0 were identified by hybridization with the aligonucleotide
N as described in Kerem et al supra.
Clones cantaining the mutant sequence were
identified by their failure to hybridize to the normal
ASO (Kerem et al, supra), One clone, 5~3-15 was isolated
and its DNA sequence determined. The general protocol
for sequencing cloned DNA is essentially as described
[3,Rs ~t~,ordan et al, c' a 24511066 (1989)] with the
use of an U.S. Biochemicals Sequenase~' kit. To verify the
sequence and to exclude any errors introduced by DNA
poly~nerase during PCR, the DNA sequences for the PCR
products from the father and one of the affected children
were also determined directly without cloning.
This procedure was accomplished by denaturing 2
pmoles of gel-purified double=stranded PCIt product in 0.2
M NaOH/0.2 mM EDTA (5 min. at room temperature),
neutralized by adding 0.1 v~lume of 2 M ammonium acetate
(pH 5.4) and precipitated with 2.5 volumes of ethanol
at -70°C for 10 yin. After washing with 70~ ethanol, the
DNA pellet was dried and redissolved in a sequencing
reaction buffer containing 4 pmoles of the
oligonucleotide primer 10i-3 of Figure 18, dithiothreitol
(8.3 mM) and (a-35S]-dATP (0.8 ACM, 1000 Ci/mmole). The
mixture was incubated at 37~C for 20 min., following
which 2 ~sl of labelling mix, as included in the
Sequenase" Kit and then 2 units of Sequenase enzyme were
added. Aliquotes~of the reaction mixture (3.5 ~1) were
transferred, without delay, to tubes each containing 2.5
CA 02073441 2000-12-07
72
~1 of ddGTP, ddATP, ddTTP and ddCTP solutions (U.S. Biochemicals SequenaseTM
kit) and the reactions were stopped by addition of the stop solution.
The DNA sequence for this mutant allele is shown in Figure 19b. The data
derived from the cloned DNA and direct sequencing of the PCR products of the
affected child and the father are all consistent with a 3 by deletion when
compared to
the normal sequence (Figure 19c). The deletion of this 3 by (ATC) at the 1506
or
1507 position results in the loss of an isoleucine residue from the putative
CFTR,
within the same ATP-binding domain where OF508 resides, but it is not evident
whether this deleted amino acid corresponds to the position 506 or 507. Since
the 506
and 507 positions are repeats, it is at present impossible to determine in
which
position the 3 by deletion occurs. For convenience in later discussions,
however, we
refer to this deletion as oI507.
The fact that the 0:1507 and OF508 mutations occur in the same region of the
presumptive ATP-binding domain of CFTR is surprising. Although the entire
sequence of 01507 allele has not been examined, as has been done for OF508,
the
strategic location of the deletion argues that it is the responsible mutation
for this
allele. This argument is further supported by the observation that this
alteration was
not detected in any of the normal chromosomes studied to date (Kerem et al, su
ra .
The identification of a second single amino acid deletion in the ATP-binding
domain
of CFTR also provides information about the structure and function of this
protein.
Since deletion of either the phenylalanine residue at position 508 or
isoleucine at
position DI507 is sufficient to affect the function of CFTR such that it
causes CF
disease, it is suggested that these; residues are involved in the folding of
the protein
but not directly in the binding of ATP. That is, the length of the peptide is
probably
.~~.,ra..~a aL..~ aL.. ....a....7 .....,......, ....:.-7
'W~ 91/10734 PCT/CA91/00009
~~.~.)'~ l; .3
~d Cf ~a
73
res;dues in this region. In support of this hypothesis,
it has been found that the phenylalanine residue can be
replaced by a serine and that isoleucine at position 506
with valine, without apparent loss of function of CFTR.
When the nucleotide sequence of dT507 is compared to
that of aF5O8 at the ASO-hybridizing region, it was noted
that the difference between the two alleles was only an A
-~ T change (Figure 19c). This subtle difference thus
explained the cross-hybridization of the dF508-ASO to
to 0I507. These results therefore exemplified the
importance of careful examination of both parental
chromosomes in performing ASO-based genetic diagnosis.
it has bean determined that the eF5o8 and nT507 mutations
can be distinguished by increasing the stringency of
oligonucleotide hybridization condition or by detecting
the unique mobility of the heteroduplexes formed between
each of these sequences and the normal DNA on a
polyacrylaminde gel. The stringency of hybridization can
be increased lay using a washing temperature at ~5°C
instead of the prior 39°C in the presence of 2XSSC (1XSSC
- 25,0 mM NaCL and 15 mM Na citrate).
Identification of the ~I507 and eF508 alleles by
polyacrylamide gel electrophoresis is shown in Figure 20.
The PCR products were prepared from the three family
members and separated on a 5~ polyacrylamide gel as
described above. A DNA sample from a known heterozygous
nF508 carrier is included for comparison. With reference
to Figure 20, the banding pattern of the PCIt-amplified
genomic DNA from the father, who is the carrier of nI507,
is clearly distinguishable from that of the mother, who
is of the type of carriers with the aF508 mutation. In
this gel electrophoresis test, there were actually three
individuals (the carrier father and the two affected sons
in Family 5) who carried the ~iI507 deletion. Since they
all belong to the same family, they only represent one
single CF chromos~me in our population analysis [Kerem et
al, supra] The two patients who also inherited the nF508
WO 9111a73~t &'C'1'/CA91/00009
~~ ~ '~3~.y y
74
mutation from their mother showed typical symptoms of CF
with pancreatic insufficiency. The father of this family
was the only parent who carries this QI507 mutation; no
other CF parents showed reduced hybridization intensity
signal with the eF508 mutant oligonucleotide probe or a
peculiar heteroduplex pattern for the PCR product (as
defined above) in the retrospective study. In addition,
two representatives of the group IIIb and one of the
group IIIc CF chromosomes from our collection [Kerem et
l0 a1, sutra] were sequenced, but none were found to contain
eI507. Since the electrophoresis technique eliminates
the need for probe-labelling and hybridization, it may
prove to be the method of choice for detecting carriers
in a large population scale [J. M. Rommens et al, Am. J.
Hum. Genet. 46:395-396 (1990)x.
The present data also indicate that there is a
strict correlation between DNA marker haplotype and
mutation in CF. The ~F508 deletion is the most common CF
mutation that odcurred on a group Ia chromosome
background [Kerem et.al, sutara~. The pI507 mutation is,
however, rare in the CF population; the one group~IIIa CF
chromosome carrying this deletion is the only example in
our studied population (1/219). Since the group III
haplotype is relatively common among the normal
chromosomes (17/198), the ~I507 deletion probably
occurred recently. Additional studies with larger
populations of different geographic and ethnic
backgrounds should provide further insight in
understanding the origins of these mutations.
3 0 3~8 ~?DITIONAL C~ MtIT~TTCNB
Following the above procedures, other mutations in
the CF gene have been identified. The following brief
description of each identified mutation is based on the
previously described procedures for locating the mutation
involving use of PCR procedures. The mutations are given
short form names. The numbering used in these
abbreviations refers to either the DNA sequence or the.
r3'0 g1!10734 PC1'/CA91/OtD009
75 ~~~u mLi~
amino acid sequence position of the mutation depending on
the type of mutation. For example, splice mutations and
frameshift mutations are defined using the DNA sequence
position. Most otrar mutations derive their nomenclature
from the amino acid residue position. The description of
each mutation clarifies the nomenclature in any event.
For example, mutations 6542X, Q493X, 3659 dal C, 556
dal A result in shortened polypeptides significantly
different from the single amino acid deletions or
alteration. 6542X and Q493X involve a pnlypeptide
including on the first 541 and 493 amino acid residues,
respectively, of the normal 1480 amino acid polypeptide.
3659 dal C and 556 dal A also involve shortened versions
and will include additional amino acid residues.
Mutation 711+1G -~ T and 171?-1G ~ A are predicted to lead
to polypeptides which cannot b~ as of yet exactly
def3.ned. They probably do lead to shortened polypeptides
but could contain additional amino acids. DNA sequence
encoding these mutant polypeptides will now probely
contain intros sec,~aence from the normal gene or possible
deleted axons.
3 ~ 8 . 0 ~i,~'ATLQ~T.$ ~H ~~OTJ 1
In the 1296 -~ C mutation, there is a single basepair
change of G to C at nucleotide 129 of the cDNA sequence
of Figure 1. The PCI2 product for amplifying genomic DNA
containing this mutation is derived from the 8115-B and
lOD primers as set out in Table 5. The genomic DNA is
amplified as per the conditions of Table 6.
~~z~N~ zr~ ~~oN ~
The G85E mutation in exon 3 involves a G to A
transition at nucleotide position 386. It is detected in
family X26, a French Canadian family classified as PI.
This predicted Gly to Glu amino acid change is associated
with a group IIb haplotype. The mutation destroys a
F3inf1 site. The PCTd product derived from the 3i-5 and
3i-3 primers, as per conditions of Table 6, is cleaved by
this enzyme into 3 fragment, 172, 105 and 32 bp,
WO 91/1073~t PCT/CA91/00009
76
resp~ctivelyP for the normal sequence; a fragment of 277
by would be present for the mutant sequence. We analyzed
54 CF chromosomes, 8 from group II, and 50 normal
chromosomes, 44 from group II, and did not find another
example of G85E.
~.~.a a~~~~xxoN~ xN ~xoN ~
556 dal A is a frameshift mutation in axon 4 in a
single CF chromosome (Toronto family X17, GM1076). There
is a deletion of A at nucleotide position 556. This
mutation is associated with Group IIIb haplotype and is
not found in 31 other CF chromosomes (9 from IIIb) and 30
N chromosomes (16 from IIIb). The muation creates a BglI
1 enzyme cleavage site. The PCR primers are 4i-5 and 4i-
3 (see Table 5) where the enzyme cuts the mutant PCR
product (437 bp) into 2 fragments of 287 and x,50 by in
size.
The I148T mutation in axon 4 involves a T to C
basepair transition at nucleotide position 575. This
results in an Ile to Thr change at amino acid position
a48 of Figure Z. The PCR product used in amplifying
genomic DNA containing this mutation uses primers 4i-5
and 4i-3 as set out in Table 5. The reaction conditions
for amplyfing the genomic DNA are set out in Table .6.
3.8.3 M~TATxtaNS ',,~i O~
In mutation G178R the Gly to Arg missense mutation
in axon 5 is due to a G t~ A change at nucleotide
position 664. The mutation is found on the mother s CF
chromosome in family X50; the other mutation in this
family is OF508. primers 5i-5 and 5i-3 were used for
amplifying genomic DNA as outlined in Tables 5 and 6.
a. a ~t~r~~xoNS xN ~xo~a~ ~
A mutation in axon 9 is a change of alanine (GCG) to
glutamic acid (GAG) at amino acid position 455
(A455 -~ E). Two of the 38 non-eF508 CF chromosomes
examined carries this mutation; both of them are from
patients of a French-Canadian origin, which we have
identified in our work as familie;~ X27 and X53, and they
VV~ 91/~~?734 PCT/CA91/OU009
'.
7 7 .x .
belong to haplotype group Ib. The mutation is detectable
by allele-specific oligonucleotide (ASO) hybridization
with PCR-amplified genomic DNA sequence. The PCR primers
are 91-5 (5'-TAATGGATCATGGGC~:ATGT-3') and 9i-3 (5'-
ACAGTGTTGAATGTGGTGCA-3') for amplifying genomic DNA under
the conditions of Table 6. The ASOs are 5'-
GTTGTTGGCGGTTGCT-3~ for the normal allele and 5'-
GTTGTTGGAGGTTGCT-3' for the mutant. The oliganucleotide
hybridization is as described in Kerem et al (1989) su ra
at 37°C and the washings are done twice with SXSSC for 10
min each at room temperature followed by twice with 2 X
SSC for 30 min each at 52°C. Although the alanine at
position 455 (A1a455) is not present in all ATP-binding
folds across species, it is present in all known members
of the P-glycoprotein family, the protein most similar to
CFTR. Further, A455 -~ E is believed to be a mutation
xat3ier than a sequence polymorphism because the change is
not found in 16 non-oF5o8 CF chromosomes and three normal
chromosomes carrying the same group I haplotype.
2 0 3 . $ . 5 MDT~S'C'I~1~T8 IId EXON ~( O
In the Q493X mutation G1n493 (CAG) is changed into a
stop codon (TAG) in Toronto family ~'9 (nucleotide
position 1609 C -~ T). The muation occurs on a CF
chromasome with haplotype IIIbe it is not found in 28
normal chromosomes (15 of which belong to 11b) nor in 33
other CF chromosomes (5 of which IIIb). The mutation can
be detected by allele-specific PCR, with 10i-5 as the
common PCR primer, 5'-GGCATAATCCAGGAAAACTG-3' for the
normal sequence and 5'-GGCATAATCCAGGAAAACTA-3' for the
mutant allele. The PCR condition is 6 min at 94°
followed by cycles of 30 sec at 94°, 30 sec at 57° and 90
sec at 72°, with 100 ng of each primer and -40o ng
genomic DNA. The primers 9i-3 and 9i-5 may be used for
internal PCR control as they share the same reaction
condition.
3.8.6 MUTATIONS IN E9COIV 11
WO 9,1/10734 P~1'1CA91100009
In mu~catian G542X the glycine radon (GGA) at amino
acid position 542 is changed to a stop colon (TGA) (G542
Stop). The single chromosome carrying this mutation is
of Ashkenazic Jewish origin (family A) and has the B
haplotype (XV2C allele 1; KM.19 allele 2). The mutant
sequence can be detected by hybridization analysis with
allele-speoi~ic oligonucleotides (ASOs) on genomic DNA
amplified under conditions of Table 6 by PCR with the
11i-5 and 11i-3 oligonucleotide primers. The normal ASO
is 5'-ACCTTCTCCAAGAACT-3° and the mutant ASO, 5°-
ACCTTCTCAAAGAACT-3°. The oligonucleotide hybridization
condition is as described in Kerem et al (1989) su ra and
the washing conditions are twice in 5 x SSC for 10 min.
each at room temperature followed by twice in 2 X SSC for
30 min. each at 45°C. The mutation is not detected in 52
other non-ef508 CF chromosomes, 11 of which are of Jewish
vr~,c~a.n (three hays a B haplotype), nor in 13 normal
chrom~somes.
In mutation S549R, the highly conserved serine
residue of the nucleotide binding domain at position 549
is changed to arginine (5549 -~ R);,the colon change is
AGT -~ AGG. The CP.chromosome with this mutation is
carried by a non-Ashkenazic Jewish pateitn from Morocco
(family B). The chromosome also has the B haplotype.
Detection of this mutation may be achieved by ASO
hybridization or allele-specific PCR. In the ASO
hybridization procedure, the genomic DNA sequence is
first amplified under conditions of Table 6 by PCR with
the 11i-5 and 11i-3 oligonucleotides; the ASO for the
normal sequence is 5°-ACACTGAGTGGAGGTC-3' and that for
the mutant is 5°-ACACTGAGGGGAGGTC. The oligonucleotide
hybridization condition is as described by Kerem et al
(1989) supra and the washings are done twice in 5 x SSC
for 10 min. each at room temperature followed by twice in
2 x SSC for 30 min. eachat 56°C. In th.e allele-specific
PCR amplification, the,oligonucleotide primer for the
normal sequence is 5'TGCTCGTTGACCTCCA-3', that for the
WO 91/10731! ~ n ,7 ~ ;Q .~ :; PCTlCA91/00009
7~
mutant is 5°TGCTCGTTGACCTCCC-3° and that fir the common,
outside sequence is 11i-5. The reaction is performed
With 500 ng of genomie DNA, 100 ng of each of the
oligonucleotides and 0.5 unit of Taq polymerase. The DNA
template is first denatured by heating at 94°C for 6
min., followed by 30 cycles of 94° for 30 sec, 55° for 30
sec and 72° for 60 sec. The reaction is completed by a 6
min heating at ?2° for 7 min. This 5549 -» R mutation is
not present in 52 other non-oF508 CF chromosomes, 11 of
which are of Jewish origin (three have a B haplotype),
nor in 13 normal chromosomes.
In the S549I mutation there is an AGT~ATT change
(nucleotide position 1778 G-~T) which represent the third
mutation involving this amino acid colon resulting in a
loss of the Ddel s3.te. We have only one example who is
of Arabic origin and is sequenced; no other Ddel-
resistant chromosome is found in 5 other Arabic CF, 21
Jewish CF, 41 Canadian CF, and 13 Canadian normal
chromosomes.
In mutation R560T the arginine (AAG) at amino acid
position 560 is changed to threonine (AAC). The
individual carrying this mutation (R560 -~ T) is from a
family we have identified in our work as family X32 and
the chromosome is marked by haplotype IIIb. The mutation
creates a MaeII site which cleaves the PCR product of
axon 11 (generated with primers !1i-5 and !1i-3 under
oonditions of Table 6) into two fragments of 214 and 204
by in size. None of the 36 non-~F508 CF chromosomes
(seven of which have haplotype IIIb) or 23 normal
chromosomes (16 have haplotype IIIb) carried this
sequence alteration. The 8560 w T mutation is also not
present on eight CF chromosomes with the ~F508 mutation.
In mutation G551,D glycine (G) at amino acid position
551 is changed to asgartic acid (D). 6551 is a highly
conserved residue within the ATP-binding fold. The
corresponding colon change is from GGT to GAT. The
6551-~D change is found in 2 of our families (~'1, X38)
'1'~~ 91110734 PC1'/CA91/00009
~3~~~~~'-,~~~~ ' ~0
with pancreatic .insufficient (PI) CF patients and 1
family (#54) with a pancreatic sufficient (PS) patient.
The other CF chromosomes in family #i and X38 carry the
AF508 mutation and that in family #54 is unknown. Based
on our °°severe and mild mutation°' hypothesis (Iterem et
al. 1989), this mutation is expected to be a "severe"
one. All 3 chromosomes carrying this mutation belong to
Group IIIb. This 6551-~D substitution does not represent
a sequence polymorphism because the change is not
detected in 35 other CF chromosomes without the eF508
deletion (5 of them from group IIIb) and 19 normal
chromosomes (including 5 from.group IIIb). To detect
this mutation, the genomic DNA region may be amplified
under conditions of Table 6 by PCR with primers 11i-5
(5'-CAACTGTGGTTAAAGCAATAGTGT-3') and 11i--3 (5~-
GCACAGATTCTGAGTAACCATAAT-3') and examined for the
presence of a Mbol (Sau3A) site created by nucleotide
change; the uncut (normal) form is 419 by in length and
the digestion products (from the mutant form) are 241 and
178 bp.
.3 ~ ~ . 7 ~f»T~ I~1 EI~Id 3.2 '
Zn the Y563N mutation a T to A change is detected at
nucleotide position 1820 in exon 12. This switch would
result in a change from Tyr to Asn at amino acid position
563. It is found in a single family with 2 PS patients
but the mutation in the other chromosome is unknown. We
think Y563N is probably a missense mutation because (1)
the T to A change is not found in 59 other CF
chromosomes, with 8 having the same haplotype (IIa) and
30 having vF508; and (2) this alteration is not found in
54 normal chromosomes, with 39 having the 11a haploytype.
Unfortunately, the amino acid bhange is not drastic.
enough to permit a strong argument. This putative
mutation can be detected by ASO hybridization with a
normal (5°-AGCAGTATACAAAGATGC-3') and a mutant (5~-
AGCAGTAAACAAAGATGC-3~) oligonucleotide probe. The
washing condition is 54°C with 2xSSC.
WO 91/10734 PCT/CA91/~D0009
cy~ w1 :a .S .;
~a ~ d c7 ' ~' ~:.:
81
In the P574H mutation the C at nucleotide pasi,~ion
1853 is changed to A. Although the amino acid Pro at
this position is not highly conserved across different
ATP-binding folds, c change to His could be a drastic
substitution. This change is not detected in 52 ether CF
chromosomes nor 15 normal chromosomes, 4 of which have
the same group IV haplotype. Based on these arguments,
we believe P574H is a mutatian. To detect this putative
mutation, one may use the following ASOs: 5'-
l0 GACTCTCCTTTTGGA-3' far the normal and 5°-GACTCTCATTTTGGA-
3° for the mutant. Washing should be dune at 47~ in
2xSSC.
In the L1077P mutation, the T at nucleotide position
3362 is changed to C. This results in a change of the
amino acid Leu to Faro at amino position 1.077 in Figure 1.
As with the other mutations in this axon, the genomic DNA
is amplified by use of the primers of Table 5; namely
l7bi-5 and 1?bi-3. The reaction conditions in amplifying
the genamic DNA are set out in Table 6.
The Y1092x mutation involves a change of C at
nucleatide p~sition 3408 to A. This would result in
protein synthesis termination at amino position 1092.
Hence the amino acid Tyr is not present in the truncated
polypeptide. As with the above procedures, the primers
used in amplifying this mutation are i7bi-3 and l7bi-3.
3 s ~ W ~~a~N~ Z~ ~r~~~ Z
3659 dal C is a frameshift mutation in axon 19 in a
single CF chramasome (Taronta family ~2); deletian of C
at nucleatide position 3659 or 3960; haplotype IIa; not
present in 5? non-aF508 CF chromosomes (7 from IIa) and
50 N chromosomes (43 from IIa); the deletion may be
detected by PCR with a common oligonucleatide primer 191-
5 (see Table 5) and 2 ASO primers, HSCB (5°-
GTATGGTTTGGTTGACTT GG-3°) far the normal and HSC9 (5°-
GTATGGTTTGGTTGACTTGT-3°) for the mutant allele; the PCR
condition is as usual except the annealing temperature is
at 60~C to improve specificity.
W~ 91/10734 PCI'/C/e91/000~9
~flr~~~.~~~.~ 82
3.8.9 ,'MtJ°x°A!~°xCNB IN Il~fiTRON 4
In the 621 + 1G -~ T mutation there is a single by
change affecting the splice site (GT -~ TT) at the 3' end
of axon 4; this mutation is detected in 5 French-Canadian
CF chromosomes (one each in Toronto families X22, 23, 26,
36 and 53) but not in 33 other CF chromosomes (18 from
the same group, group I) and 29 N chromosomes (13 from
group I); the mutation creates a Msel site; genomic DNA
may be amplified by the 2 intron primers, 4i-5 adn 4i-3,
20 and cut with Msel to distinguish the normal and mutant
a11e1es; the normal would give 4 fragments of 33, 35, 7Z
and 298 by in size; the 298 by fragment in the mutant is
cleaved by the enzyme to give a 54 and 244 by fragments.
3.8. AO ~QTATIO~TB IN INTRON ;Z
In the 711 + 1.G ~ T mutation this G to T switch
occurs at the splice junction after axon 5. The mutation
is found ~n the mother's CF chromosome in family ,~22, a
French Canadian family from Chicoutimi. The other
mutation in this family is 621+iG -~ T.
2 0 ~QTA~'gONB IN INI,~_ON 10
In the 9.73.7-1G -~ A mutation a putative splice
mutation is found in front of axon 7.1. This mutation is
located at the last nucleotide of the intron before axon
11. The mutation may be detected with the following
ASO's> normal ~~ 5'-TTTGGTAATAGGACATCTCC-3'; mutant ASO =
5'-TTTGGTAATAAGACATGTCC-3'. The washing conditions afar
hybridization are 5xSSC twice for 10 min at room temp,
2xSSC twice for 30 min at 47° for the mutant and 2xSSC
twice to 30 min at 48° for the normal ASO. We have only
1 single example from an Arabic patient and there is no
haploytpe data. The mutation is not found in 5 other
Arabic, 21 Jewish, and 41 Canadian CF chromosomes, nor in
13 normal chromosomes.
3.9 DNA SRQtTLNCR 1~OL5tMORP~fIBM~
Nucleotide position Amino acid change
1540 (A or G) Met or Val
1716 (G or A) no change (Glu)
2694 (T or G) no change (Thr)
W(~ 91J10734 PCTJCA91l00009
8 3 r.. ~S 9 _s .,..
356 (G or A) Arg or Gln
A polymorphism is detected at nucleotide position 1540-
the ~. residue can be substituted by G, changing the
corresponding amino acid from Met to ~7a1. At postion
2694- the T residue can be a G; although it does not
change the encoded amino acid. The polymorphism may be
detected by restriction enzymes AvaII or Sau9GI. These
changes are present in the normal population and show
good correlation with haploytpes but not in GF disease.
There can be a G to A change for the last nucleotide
of exan 10 (nucleotide position 1716). We think that
this nucleotide substitution is a sequence polymorphism
because (a) it does not alter the amino acid, (b) it is
unlikely to cause a splicing defect and (c) it occurs on
some normal chromosomes. In two Canadian families, this
rare allele is found associated with haplotype IIIb.
fihe more common mucleotide at 356 (G) is found to be
changed to A in the father's normal chromosome in family
~'~4. The amino acid changes from Arg to Gln.
2 0 ~ C,E~ ~~tMP~I~J
As discussed~~aith respect to the DNA sequence of
Figure 1, analysis of the sequence of the overlapping
cDNA clones predicted an unprocessed polypeptide of 1480
amino acids with a molecular mass of 166,138 daltons. As
later described, due to polymorphisms in the protein, the
molecular weight of the protein can vary due to possible
substitutions or deletion of certain amino acids. The
molecular weight will also change due to the addition of
carbohydrate units to form a glycoprotein. It is also
3d understood that the functional protein in the cell will
Wa 9i/IU7~t i'C'T/CA91/00409
84
....'
be aimiiar to the unprocessed polypeptide, but may be
modified due to cell metabolism.
Accordingly, purified normal CFTR polypeptide is
characterized by a molecular weight of about 170,000
daltons and having epithelial cell transmembrane ion
conductance activity. The normal CFTR polypeptide, which
is substantially free of other human proteins, is encoded
by the aforementioned DNA sequences and according to one
embodiment, that of Figure 1. Such polypeptide displays
the immunological or biological activity of normal CFTR
polypeptide. As will be later discussed, the CFTR
polypeptide and fragments thereof may be made by chemical
or enzymatic peptide synthesis or expressed in an
appropriate cultured cell system. The invention provides
purified 507 mutant CFTR polypeptide which is
characterized by cystic fibrosis-associated activity in
human epithelial cells. Such 507 mutant cFTR
polypeptide, as substantially free. of other human
proteins, can be encoded by the 50T mutant DNA sequence.
2 0 ~ ST130~°iJRB O~ t:F'fR
The most characteristic feature of the predicted
protein is the presence of two repeated motifs, each of
which consists of a set of amino acid residues capable of
spanning the membrane several times followed by sequence
resembling consensus nucleotide (ATP)-binding folds
(NBFs) (Figures 11, 12 and 15). These characteristics
are remarkably similar tc those of the mammalian
multidrug resistant P-glycoprotein and a number of other
membrane-associated proteins, thus implying that the
predicted CF gene product is likely to be involved in the
transport of substances (:tons) across the membrane and is
probably a member of a membrane protein super family.
Figure 13 is a schematic modal of the predicted CFTR
protein. xn Figure 13, cylinders indicate membrane
spanning helices, hatched spheres indicate NBFs. The
stippled sphere is~the polar R-domain. The 6 membrane
spanning helices in each half of the molecule are
w~ ~~rio7~a Pcrrc~.~nroooo9
;~ ~ ~ ~~ S : j :,
depicted as cylinders. The inner cytoplasmically
oriented FdBFs are shown as hatched spheres with slots to
indicate the means of entry by the nucleotide. The large
polar R-domain which links the two halves is represented
by an stippled sphere. Charged individual amino acids
within the transmembrane segments and on the R-domain
surface are depicted as small circles containing the
charge sign. Net charges on the internal and external
loops joining the membrane cylinders and on regions of
the NBFs are contained in open squares, Bites for
phosphorylation by protein kinases A or C are shown by
closed and open triangles respectively. K,R,H,D, and E
are standard nomenclature fox the amino acids, lysine,
arginine, histidine, aspartic acid and glutamic acid
respectively.
Each of the predicted membrane-associated regions of
the CFTR protein consists of 6 highly hydrophobic
serpents capable of spanning a lipid bilayer according to
the algorithms of Kyte and Doolittle and of Garnier et al
2g (J. Mal. Bio' 1Z0, 97 (1978) (Figure 13). The membrane-
associated regions are each followed by a large
hydrophilic region containing the NBFs. Based on
sequence alignment with other known nucleotide binding
proteins, each of the putative NBFs in CFTR comprises at
least 15o residues (Figure 13). The 3 by d~letion at
position 507 as detected in CF patients is located
between the 2 most highly conserved segments of the first
NBF in CFTR. The amino acid sequence identity between
the region surrounding the isoleucine deletion and the
30. corresponding regions of a number of other proteins
suggests that this region is of functional importance
(Figure 15), A hydrophobic amino acid, usually one with
an aromatic aide chain, is present in most of these
proteins at the position corresponding to I507 of the
CFTR protein. It is understood that amino acid
polymorphi~ms may exist as a result of DNA polymorphisms.
Similarly, mutations at the other positions in the
WO 91/10734 PCT/CA91/00009
~;
86
protein suggested that corresponding regions of the
protein'are also of functional importance. such
additional mutations include substitutions of:
i) Glu for Gly at amino acid position 85;
ii) Thr for Ile at amino acid position 148;
iii) Arg for Gly at amino acid position l?8;
iv) Glu for ALA at amino position 455;
v) stop colon for Gln at amino acid portion 493;
vi) stop colon for Gly at amino acid position 542;
vii) Arg for Ser or Ile for Ser at amino acid
position 549;
viii) Asp for Gly at amino acid position 551;
ix) Thr for Arg at amino acid position 560;
x) Asn for Tyr at amino acid position 563;
xi) His for Pro at amino acid position 574;
xii) Pro for Leu at amino acid position 1077;
xiii) Stop colon for Tyr at amino acid position
1092.
Figure 15 shows alignment of the 3 most conserved
segments of the extended NBF's of CFTR with comparable
regions of other proteins. These 3 segments consist of
residues 433-473, 488-513, and 542-584 of the N-terminal
half and 1219-1259, 1277-1302, and 1340-1382~of the C-
terminal half of CFTR. The heavy overlining points out
the regions of greatest similarity. Additional general
homology can be seen even without the introduction of
gaps.
Despite the overall symmetry in the structure of the
protein and the sequence conservation of the NBFs,
sequence homology between the two halves of the predicted
CFTR protein is modest. This is demonstrated in Figure
12, where amino acids 1-1480 are represented on each
axis. Lines on either side of the identity diagonal
indicate the positions of internal similarities.
Therefore, while four sets of internal sequence identity
can be detected as shown in Figure 12, using the Dayhoff
scoring matrix as applied by Lawrence et aZ. [C. B.
°~~°f IJTT.
WO 91/10734 PC'd'/CA97/00009
~~i~ a a
87
Lawrence, D. ~. Goldman, and R. T.. Hood, Bull Math liol
48, 569 (1986)], three of these are only apparent at low
threshold settings for standard deviation. The strongest
identity is between sequences at the carboxyl ends of the
NBFs. Of the 66 residues aligned 27% are identical and
another 11% are functionally similar. The overall weak
internal homology is in contrast to the much higher
degree (>70%) in P~glycoprotein for which a gene
duplication hypothesis has been proposed (taros et al,
Ce 47, 371, 1986, C. Chen et al, C~11 47, 381, 1986,
Gerlach et al, Nature, 324, 485, 1986, taros et al, Mol.
Celt. Biol. 8, 2770, 1988). The lack of conservation in
the relative positions of the axon-intron boundaries may
argue against such a model far CFTR (Figure 2).
15. Since there is apparently no signal-peptide sequence
at the amino-terminus of CFTR, 'the highly charged
hydrophilic segment preceding the first transmembrane
sequence is probably oriented in the cytoplasm. Each of
the 2 sets of hydrophobic helices are expected to form 3
transversing loops across the membrane and little
sequence of the entire protein is expected to be exposed
to the exterior aurtace, except the region between
transmembrane segment 7 and 8. It is of interest to note
that the latter region contains two potential sites for
N-linked glycosylation.
Each of the membrane-associated regions is followed
by a NBF as indicated above. xn addition, a highly
charged cytoplasmic domain can be identified in the
middle of the predicted CFTR polypeptide, linking the 2
halves of the protein. This domain, named the R-domain,
is operationally defined by a single large axon in which
69 of the Z41 amino acids are polar residues arranged in
alternating clusters of positive and negative charges.
Moreover, 9 of the 10 consensus sequences required for
phosphosphorylation by protein kinase A (PxA), and, 7 of
the potential substrate sites for protein kinase C (PRC)
found in CFTR are located in this axon.
bV~ 91/10734 P~'/CA91/00009
'J s.~ s . n .
~~ ~ ~l~~L a . s8
FuzaCTlC~la of CFfiR
Properties of CFTR can be derived Pram comparison to
other membrane-associated proteins Figure 15). In
addition to the overall structural similarity with the
mammalian P-glycoprotein, each of the two predicted
domains in CFTR also shows remarkable resemblance to the
single domain structure of hemolysin B of ,~ Coli and the
product of the White gene of Drosophila. These fatter
proteins are involved in the transport of the lytic
2o peptide of the hemolysin system and of eye pigment
molecules, respectively. The vitamin B12 transport
system of ,~,,. cc~li, BtuD and MbpX which is a liverwort
chloroplast gene whose function is unknown also have a
similar structural motif. Furthermore, the CFTR protein
Z5 shares structural similarity witty several of the
periplasmic solute transport systems of gram negative
bacteria where the transmembrane region and the ATP-
binding folds are contained in separate proteins which
function in concert with a third substrate-binding
20 polypeptide.
The overall structural arrangement of the
transmembrane domains in CFTR is similar to sesreral
cation channel proteins and some canon-translocating
ATPases as well.as the recently described adenylate
25 cyclase of bovine brain. The functional significance of
this topological classification, consisting of C
transmembrane damains, remains speculative.
Short regions of sequence identity have also been
detected between the putative transmembrane regions of
30 CFTR and other membrane-spanning proteins.
Interestingly, there are also sequences, 18 amino acids
in length situated approximately 50 residues from the
carboxyl terminus of CFTR and the ref serine/threonine
kinase protooncogene of Xeng~ous ~a~yis which are
35 identical at 12 of these positions.
Finally, an amino. acid sequence identity (10/13
conserved residues) has been noted between a hydrophilic
~'O 91/10734 ~ ~;;'y ~ ~ _~ ~ PCT/CA91/U0009
89
segment (position 701-713) within the highly charged R-
domain of CFTR and a region immediately preceding the
first transmembrane loop of the sodium channels in both
rat brain and eel. The charged R-domain of CFTR is not
shared with the topologically closely related R-
glycoprotein; the 241 amino acid linking-peptide is
apparently the major difference between the two proteins.
In summary, features of the primary structure of the
CFTR protein indicate its possession of properties
to suitable to participation in the regulation and control
of ion. transport in the epithelial cells of tissues
affected in.CF. Secure attachment to the membrane in two
regions serve to position its three major intracellular
domains (nucleotide-binding folds 1 and 2 and the R-
domain) near the cytoplasmic surface of the cell membrane
where they can modulate ion movement through channels
formed either by CFTR transmembrane segments themselves
or by other me~abrane proteins.
In view of the genetic data, the tissue-specificity,
and the predicted properties of the CFTR protein, it is
reasonable to conclude that CFTR is directly responsible
for CF. It, however, remains unclear how CFTR is
involved in the regulation of ion conductance across the
apical membrane of epithelial cells.
It is possible that CFTR serves as an ion channel
itself. As depicted in Figure 13, 10 of the 12
transmembrane regions contain one or more amino acids
with charged'side chains, a property similar to the brain
sodium channel and the GABA receptor chloride channel
subunits, where charged residues.are present in 4 of the
6, and 3 of the 4, respective membrane-associated domains
per subunit or repeat unit. The amphipathic nature of
these transmembrane segments is believed to contribute to
the channel-forming capacity of these molecules.
Alternatively, CFTR may not be an ion channel but instead
serve to regulate ion channel activities. In support of
the latter assumption, none of the purified polypeptides
IJS~'~T1'HET
WO 91J1073a fC'f/CA91/00009
'J ~1 9 ~ t~ (. ~3 90
h:
from trachea and kidney that are capable of
reconstituting chloride channels in lipid membranes
[Landry et al, Science 224:1469 (3.989)) appear to be CFTR
if judged on the basis of the molecular mass.
In either case, the presence of ATP-binding domains
in CFTR suggests that ATP hydrolysis is directly involved
and required for the transport function. The high
density of phosphorylation sites for PKA and PKG and the
clusters of charged residues in the R-domain may both
l0 serve to regulate this activity. The deletion of a
phenylalanine residue in the N5F may prevent proper
binding of ATP or the conformational change which this
normally elicits and consequently result in the observed
insensitivity to activation by PKA- or PKC-mediated
phosphorylation of the CF apical chloride canductance
pathway. Since the predicted protein contains several
doma~.n,s and belongs to a family of proteins which
frequently function as parts of mufti-component molecular
systems, CFTR may also participate in epithelial tissue
functions of activity or regulation not related to ion
transport.
With the isolated CF gene (cDNA) now in hand it is
possible to define the basic biochemical defect in CF and
to further elucidate the control of ion transport
pathways in epithelial cells in general. Piost important,
knowledge gained thus far from the predicted structure of
CFTR.together with the additional information from
studies of the protein itself provide a basis far the
development of improved means of treatment of the
disease, In such studies, antibodies have been raised to
the CFTR protein as later described.
S . 0 CF' SCRE~INIIdG
5~1 DNA HABED D%AGNOSIS
Given the knowledge of the 85, 148, 178, 455, 493,
507, 542, 549, 551, 560, 563, 574, 1077 and 1092 amino
acid position mutations and the nucleotide sequence
varients at DNA sequence positions 129, 556, 621+1,
SI~TI'~!J°~h~ET.
iV0 91/10734 PC'T/CA9y/00009
r~ :~
~'~ :1 ~3
91 ø' ,. c~ 's '., ?
711-1, 1717°1 and 3659 as disclosed herein, carrier
screening and prenatal diagnosis can be carried out as
follows.
The high risk population for cystic fibrosis is
Caucasians. For example, each Caucasian woman andjar man
of child°bearing age would be screened to determine if
she or he was a carrier (approximately a 5~ probability
for each individual). If both are carriers, they are a
couple at risk for a cystic fibrosis child. ~aoh child
l0 of the at risk couple has a 25% chance of being affected
with cystic fibrosis. The procedure for determining
carrier status using the probes disclosed herein is as
follows.
For purposes of brevity, the discussion on screening
by use of one of the selected mutations is directed to
the I507 mutation. It is understood that screening can
also be accomplished using one of the other mutations or
using several of the mutations in ~ screening pr~cess or
mutation detection process of this section on CF
2o screening involving DNA diagnosis and mutation detection.
one major application of the DNA sequence
information of the normal and 507 mutant CF gene is in
the area of genetic testing, carrier detection and
prenatal diagnosis. Individuals carrying mutatians in
the CF gene (disease carrier or patients) may be detected
at the DNA level with the use of a variety of techniques.
The genomic DNA used for the diagnosis may be obtained
from body cells, such as those present in peripheral
blood, urine, saliva, tissue biopsy, surgical specimen
34 and autopsy material. The DNA may be used directly for
detection of specific sequence or may be amplified
enzymatically ,~,n vitro by using FCR [Saiki et al. Science
230: 1350-1353, (1985), Saiki et al. to a 324: 163-3.66
(1986) prior to analysis. RNA or its cDNA form may also
be used for the same purpose. Recent reviews of this
subject have been presented by Casket', [science 236:
ST~'~ TE SHEEN'
WO 91/1073x1 PC.T/CA91/00009
1
J
92
1223-8 (1989) and by Landegren et al (Science 242: 229-
237 (7.989) ].
The detection of specific DNA sequence may be
achieved by methods such as hybridization using specific
oligonucleotides [Wallace et al. Cold Spring Harbour
Symp. Ouant. Riol. 51: 257-261 (1986)], direct DNA
sequencing [Church and Gilbert, Proc. Nat. Aced. Sci. U.
S. A. 81: 1991-1995 (1988)], the use of restriction
enzymes (Flavell et al. Ce 15: 25 (1978), Geever et al
Frdc. Nato Acad. Scl,. 'U. S. A. 78: 5081 (1981)],
discrimination on the basis of electrophoretic mobility
in gels with denaturing reagent (Myers and Maniatis, Cold
Spring Harbour Sym. guant. Biol. 51: 275-284 (1986)),
RNase protection (Myers, R. M., Larin, J., and T.
Maniatis Science 230: 1242 (1985)), chemical cleavage
(Cotton et al Proc. Nat. Aced. Sci. U. S. A. 85: 4397-
4401, (2985)) and the ligase-mediated detection procedure
[Landegren et al sc'ence 241:1077 (1988)].
Oligonucleotides specific to normal or mutant
sequences are chemically synthesized using commercially
available machines, labelled radioactively with isotopes
(such as 3zP) ar non-radioactively (with tags such as
biotin (Ward and Langer et al. Proc. Nat. Ac~,d. Sci. U.
S. A. 78: 6633-6657 (1981)), and hybridized to individual
DNA samples immobilized on membranes or other solid
supports by dot-blot or transfer from gels after
electrophoresis. The presence or absence of these
specific sequences are visualized by methods such as
autoradiography or fluorometric (Landegren et al, 1989,
supra) or colorimetric reactions (Gebeyehu et a. Nucleic
Acids Research 1.5: 4513-4534 (198?)): An embodiment of
this oligonucleotide screening method has been applied in
the detection of the I507 deletion as described herein.
Sequence differences between normal and mutants may
be revealed by the direct DNA sequencing method of Church
and Gilbert (su~r~,). Cloned DNA segments may be used~as
probes to detect specific DNA segments. The sensitivity
SSTi~'T'E ET
W() 91/10734 1"CTlCA91/00009
93
~d V ~~ C.) '~ ' t
of this method is greatly enhanced when combined with PCR
[Wrichnik et al, Nucleic Ac?d~ ~~e 15:529-542 (1987);
Worig et al, I,~'~~g 330:384-386 (1987)i StOflet et al,
'e c 239:491-494 (1988)x. In the latter procedure, a
sequencing primer which lies within the amplified
sequence is used with double-stranded PCR product or
single-stranded template generated by a modified PCR.
The sequence determination is performed by conventional
procedures with radiolabeled nucleotides or b~ automatic
sequencing procedures with fluorescent-tags.
sequence alterations may occasionally generate
fortuitous restriction enzyme recognition sites which are
revealed by the use of appropriate enzyme digestion
followed by Eonventional gel-blot hybridization
(Southern, J. M01. 8in1 98: 503 (1975)). DNA fragments
carrying the site (either normal or mutant) are detected
bf their reduction in size or increase of corresponding
restriction fragment numbers. Genomic DNA samples may
also be amplified by PCR prior to treatment with the
appropriate restriction enzyme; fragments of different
sizes are then visualized under W light in the presence
of ethidium bromide after gel electrophoresis.
Genetic testing based on DNA sequence differences
may be achieved by detection of alteration in
electrophoretic mobility of DNA fragments in gels with or
without denaturing reagent. Small sequence deletions and
insertions can be visualized by high resolution gel
electrophoresis. For example, the PCR product with the 3
by deletion is clearly distinguishable from the normal
sequence on an 8% non-denaturing polyacrylamide gel. DNA
fragments of different sequence compositions may be
distinguished on denaturing formamidQ gradient gel in
which the mobilities of different DNA fragments are
retarded in the gel at different positions according to
their specific l~partial-melting~o temperatures (Myers,
su~ra)~ In addition, sequence alterations, in particular
small deletions, may be~detected as changes in the
WO 91/10734 PCT/CA91/00009
94
migration pattern of DNA heteroduplexea in non-denaturing
gel electrophoresis, as haws been detected for the 3 by
(T507) mutation and in other experimental systems
N [Nagamine et al, Am. ~. Hum. Genet, 45:337-339 (1989)).
Alternatively, a method of detecting a mutation
comprising a single base substitution or other small
change could be based on differential primer length in a
PCR. For example, one invariant primer could be used in
addition to a primer specific for a mutation. Ths PCR
products of the normal and mutant genes can then be
differentially detected in acrylamide gels.
Sequence changes at specific locations may also be
revealed by nuclease protection assays, such as RNase
(Myers, supra) and S1 protection (Berk, A. J., and P. A.
Sharps Proc. Nat: Aced. Sci. t1: S. A 75: 1274 (1978)),
the chemical cleavage method (Cotton, ) or the
ligase-mediated detection procedure (Landegren ).
Tn addition to conventional gel-electrophoresis and
blot-hybridization methods, DNA fragments may also be
visualized by methods where the individual DNA samples
are not immobilized on membranes. the probe and target
sequences may be both in solution or the probe sequence
may be immobilized [Saiki et al, Proc. Natl. Act
Wig, 86:6230-6234 (1989)). A variety of detection
~25 methods, such as autoradiography involving radioisotopes,
direct detection of radioactive,decay (in the presence or
absence ~f scintillant), spectrophotometry involving
colorigenic reactions and f.luorometry involving
fluorogenic reactions, may be used to identify specific
individual genotypes.
Sinee more than one mutation is anticipated in the
CF gene such as I507 and F508, a multiples system is an
ideal protocol for screening CF carriers and detection of
specific mutations. For example, a FCR with multiple,
specific oligonucleotide primers and hybridization
probes, may be used to, identify all possible mutations at
the same time (Chamberlain et al. Nua~eic Acids Research
wo ~ar~o73a pcz~rcA9~roooo~
9 5 ~e
16: 1141-1155 (1988)). 'The procedure may invoave
immobilized sequence-specific oligonucleotides probes
(Saiki et al, supra).
~'2 DFTE(:'fIN(3 TNN Cx' 507 MUTl~'J~"Ic72d
These detection methods may be applied to prenatal
diagnosis using amniotic fluid cells, chorionic villi
biopsy or sorting fetal cells from maternal circulation.
The test for CF carriers in the population may be
incorporated as an essential component in a broad-scale
genetic testing program for common diseases.
According to an embodiment of the invention, the
portion of the DNA segment that is informative for a
mutation, such as the mutation according to this
embodiment, that is, the portion that immediately
surrounds the 2507 deletion, can then be amplified by
u~,ing standard PCR techniques [as reviewed in Landegren,
Ulf, Robert Kaiser, C. Thomas Casket', and Leroy Hood, DNA
Diagnostics - Molecular Techniques and Automation, in
Science 242: 229-237 (1988)x. It is contemplated that
the portion of the DNA segment which is used may be a
single DNA segment or a mixture of different DNA
segments. A detailed description of this technique now
follows.
A specific region of genomic DNA from the person or
fetus is to be screened. Such specific region is defined
by the oligonucleotide primers C16B
(5'GTTTTCCTGGATTATGCCTGGCAC3') and C16D
(5'GTTGGCATGCTTTGATGACGCTTC3') or as shown in Figure Z8
by primers 101-5 and 10i-3. The specific regions using
10i-5 and 10i-3 were amplified by the polymerase chain
reaction (PCR). 200-400 ng of genomic DNA, from either
cultured lymphoblasts or peripheral blood samples of CF
individuals and their parents, were used in each PCR with
the oligonucleotides primers indicated above. The
,oliganucleotides were purified with Oligonucleotide
Purification Cartridges'" (Applied Biosy~tems) or NENSORB'~
PRBP columns (Dupont) with procedures recommended by the
WiD 91/10734 PCTlCA91J00009
9
suppliers. The primers were annealed at 55°C for 30 sec,
extended at 72'C for 60 sec (with 2 units of Taq DNA
polymerase) and denatured at 94'C for 60 sec, for 30
cycles with a final cycle of 7 min for extension in a
Perkin-Elmer/Cetus automatic thermocycler with a Step-
Cycle program (transition setting at 1.5 min). Portions
of the PCR products were separated by electrophoresis on
1.4~ agarose gels, transferred to Zetabind"'; (Biorad)
membrane according to standard procedures.
The normal and 0I507 oligonucleotid~ probes of
Figure 19 (l0 ng each) are labeled separately with 10
units of T4 polynucleotide kinase (Pharmacia) in a 10 ~1
reaction containing 5o mM Tris-HC1 (pH7.6}, 10 mM MgCl2,
0.5 mM dithiothreitol, l0 mrii spermidine, 1 mM EDTA and
30-40 ~eCi of ~r[3ap~ - ATp for 20-30 min at 37°C, The
unincorporated radionucleotides were removed with a
Sephadex G-25 column before use. The hybridization
conditions were as described previously (J.M. Rommens et
al Am. J. Hug- Genet. 43,645 (1988}) except that the
temperature can be 37'C. The membranes are washed twice
at room temperature with 5xSSC and twice at 39°C with 2 x
SSC (1 x SSC = 150 mM NaCl and 15 mM Na citrate}.
Autoradiography is performed at room temperature
overnight. Autoradiographs are developed to show the
hybridization results of genomic DNA with the 2 specific
oligonucleotide probes, Probe C normal detects the
normal DNA sequence and probe C pI507 detects the mutant
sequence.
Genomic DNA sample from each family member can, as
explained, be amplified by the .polymerase chain reaction
using the intron sequences of Figure 18 and the products
separated by electropharesis on a 1.4% agarose gel and
then transferred to Zetabind (Biorad) membrane according
to standard procedures. The 3bp deletion of eI507 can be
revealed by a very convenient polyacrylamide gel
electrophoresis procedure. When the PCR products
generated by the above-mentioned 10i-5 and 10i-3 primers
SUST~T TE SKEET.
W~O 91/1073a PC'f/CA91/00009
~ 11 ~d~ 7
97 ,~~,9 ~J~_ø. :.
are applied to an 5~ polyacrylamide gel, sleetrophoresed
f~r 3 hrs at 20V/cm in a 90mM Tris-borate buffer (pFi
8.3j, DNA fragments of a different mobility are clearly
detectable for individuals without the 3 by deletion,
heterozygous or homozygous for the deletion.
As already explained with respect to Figure 20, the
PCR amplified genomic DNA can be subjected to gel
electrophoresis to identify the 3 by deletion. As shown
in Figure 20, in the four lanes the first lane is a
control with a normal/oF508 deletion. The next lane is
the father with a normal/0I507 deletion. The third lane
is the mother with a normal/oF508 deletion and the fourth
lane is the child with a eF508/aI50? deletion. The
homoduplexes show up as solid bands across the base of
each lane. In lanes 2 and 3, the two heteroduplexes show
up very clealy as two spaced apart bands. In lane 2, the
father's bI507 mutation shows up very clearly, whereas in
the fourth lane, the child with the adjacent 507, 508
mutations, there is no distinguishable heteroduplexes.
Hence the showing is at the homoduplex line. Since the
father in lane 2 and the mother in lane 3 show
heteroduplex banding and the child does not, indicates
either the child is normal or is a patient. This can be
father checked i~ needed, such as in embryoic analysis by
mixing the 507 and 508 probes to determine the presence
of the dI507 and 0F508 mutations.
Simihar alteration in gel mobility for
heteraduplexes formed during PCR has also been reported
for experimental systems where small deletions are
involved (Nagamine et al sux~ra). These mobility shifts
may be used in general as the basis for the non-
radioactive genetic careening tests.
~sr~, CF' 8~$NI3llf~ hROaR.nMB
It is appreciated that approximately 1% of the
carriers can be detected using the specific 0I507 probes
of this particular embodiment of the invention. Thus, if
an individual tested is not a carrier using the aI5o7
WO 91/10734 P~f/CA91100009
c'~ r.. a ;;
~,4~ ~~:~m . 98
probes, their carrier status can not be excluded, they
may carry same other mutation, such as the oF508 as
previously noted. However, if both the individual and
the spouse of the individual tested are a carrier for the
eI507 mutation, it. can be stated with certainty that they
are an at risk couple. The sequence of the gene as
disclased herein is an essential prerequisite for the
determination of the other mutations.
Prenatal diagnosis is a logical extension of carrier
screening. A couple can be identified as at risk for
having a cystic fibrosis child in one of two ways: if
they already have a cystic fibrosis child, they are both,
by definition, obligate carriers of the defective CF'TR
gene, and each subsecguent child has a 25% chance of being
affected with cystic fibrosis. A major advantage of the
present invention eliminates the need for family pedigree
analysis, whereas, according to this invention, a gene
mutation screening program as outlined above or other
similar method can be used to identify a genetic mutation
that leads to a protein with altered function. This is
not dependent on prior ascertainment of the family
through an affected child. Fetal DNA samples, for
example, can be obtained, as previously mentioned, from
amniotic fluid cells and chorionic villi specimens.
Amplification by standard PCR techniques can then be
performed on this template DNA.
If b~th parents are shown to be carriers with the
0I507 deletion., the interpretation of the results would
be the following. If there is hybridization of the fetal
DNA to the normal probe, the fetus will not be affected
with. cystic fibrosis, although it may be a CF carrier
(50% probability for each fetus of an at risk couple). If
the fetal DNA hybridizes only to the aI507 deletion probe
arid not to the normal probe, the fetus will be affected
with cystic fibrosis.
It is appreciated, that for this and other mutations
in the CF gene, a range of different specific procedures
1V0 91 /10734 PC~'/CA91 /00009
99 n'v'j''L''i.:B
can be used to provide a complete diagnosis for all
potential CF carriers or patients. A complete
description of these procedures is later described.
The invention therefore provides a method and kit
for determining if a subject is a CF carrier or CF
patient. In summary, the screening method comprises the
steps of:
providing a biological sample of the subject to be
screened; and providing an assay for detecting in the
biological sample, the presence of at least a member from
the group consisting of a 507 mutant CF gene, 507 mutant
CF gene products and mixtures thereof.
The method may be further characterised by including
at least one more nucleotide probe which is a different
DNA sequence fragment of, for example, the DNA of Figure
1, o~' a different DNA sequence fragment of human
chromosome 7 and.located to either side of the DNA
sequence of Figure 1. In this respect, the DNA fragments
of the intron portions of Figure .2 are useful in further
confirming the presence of the mutation. Unique aspects
of the introns at the axon boundaries may be relied upon
in screening procedures to further confirm the presence
of the mutation at the T507 position or othe mutant
positions.
A kit, according to an embodiment of the invention,
suitable for use in the screening technique and for
assaying for the presence of the mutant CF gene by an
immunoassay comprises:
(a) an antibody which specifically binds to a gene
product of the mutant CF gene having a mutation at one of
the positions c~f 85, 148, 178, 455, 493, 507, 542, 549,
551, 560, 563, 574, 1077 and 1092;
(b) reagent means for detecting the binding of the
antibody to the gene product; and
(c) the antibody and reagent means each being
present in amounts effective to perform the immunoassay.
~J ~'~ I'T '~ S E E'~
~~ 91/10734 PCT/CA91/00009
300
c ~~ 't
h~ kit for assaying for the presence for the mutant
CF gene may also be provided by hybridization technidues.
The kit comprises:
(a) an oligonucleotide probe which specifically
binds to the mutant CF gene having a mutation at one of
the positions 85, 148, 178, 455, 493, 507, 542, 549, 551,
560, 563, 574, 1077 and 1092;
reagent means for detecting the hybridization
of the oligonucleotide probe to the mutant CF gene; and
(c) the probe and reagent means sash being present
in amounts effective to perform the hybridization assay.
'---_ ~~s 3~o~T ~~rT~
As mentioned, antibodies t~ epitopes within the
mutant CFTR protein at positions 85, 148, x.78, 455, 493,
507, 542, 549, 551, 560, 563, 574, 1077 and 1092 are
raised to provide extensive information on the
chara~cter.istics of the mutant protein and othex° valuable
information which includes:
1. The antibodies can be used to provide another
2o technicjue in detecting any of the other CF mutations
which result in the synthesis of a protein with an
altered size.
2~ Antibodies to distinct domains of the mutant
protein can be used to determine the topological _
arrangement of the protein in the cell membrane.
This provides informa~tian on segments of the protein
which are accessible to externally added modulating
agents for purposes of drug therapy:
3. ~'he structure-function relationships of
3o portions of the protein can be examined using
specific antibodies. For example, it is possible to
introduce into cells antibodies recognizing each of
the charged cytoplasmic loops which join the
transmembrane sec;uences as well as portions of the
nucleotide binding folds and the R-domain, The
influence o! these antibodies on functional
Parameters of the protein provide insight into cell
WO 91/1073 IPCT/CA91/00009
~i
101 ~~'~~~C'~.w
regulatory mechanisms and potentially suggest means
of modulating the activity of the defective protein
in a CF patient.
4. Antibodies with the appropriate avidity also
enable immunoprecipitation and immuno-affinity
purification of the protein. Immunoprecipitation
will facilitate characterization of synthesis and
post translational modificatian including ATP
binding and pt~osphorylation. Purification will be
required for studies of protein structure and for
reconstitution of its function, as well as protein
based. therapy.
In order to prepare the antibodies, fusion proteins
contaaning~defined portions of anyone of the mutant CFTR
polypeptides can be synthesized in bacteria by expression
of corresponding mutant DNA sequence in a suitable
cloning vehicle. Smaller peptide may be synthesized
chemically. The fusian proteins can be purified, for
example, by affinity chromatography on gluta~thione-
agarose and the peptides coupled to a carrier protein
(he~aocyanin), mixed with Freund~s adjuvant ahd injected
into rabbits. JFollowing booster injections at bi-weekly
intervals, the rabbits are bled and sera isolated. The
developed polyclonal antibodies in the sera gay then be
combined with the fusion proteins. Immunablots are then
firmed by staining with, for e~cample, alkaline-
phosphatase conjugated second antibody in accordance with
the procedure of Make et al, ~nal,,~Hiochem. 136:175
X1984).
Thus, it is possible to raise polyclonal antibodies
specific for both fusion proteins containing portions of
the mutant CFTR protein and peptides corresponding to
short segments of its sequence. Similarly, mice can be
injected with KLH conjugates of peptides to initiate the
production of anonoclonal antibodies to corresponding
seglnentB ~f Alutant C.~''TR prCtein.
lVO 91/10734 PCT/CA~1/00009
X02
~~ ~i ~( !~ .~s for the generation of monoclonal antibodies,
immunogens for the raising of monoclonal antibodies
(mAbs) to the mutant CFTR protein are bacterial fusion
proteins [smith et al, 67:31 (1986)j containing
portions of the cF~t polypeptide or synthetic peptides
corresponding to short (12 to 25 amino acids in length)
segments of the mutant sequence. The essential
methodology is that of Kohler arid i~Iilsteln [N~tuj~ 256:
495 (1975)j.
Balb/c mace are immunized by intraperitoneal
injection with 500 ~cg of pure fusion protein or synthetic
peptide in incomplete Freund~s adjuvant. A second
injection is given after 14 days, a third after 21 days
and a fourth after 2s days. Individual animals so
immunized are sacrificed one, two and four weeks
following the final injection. Spleens are removed,
their cells dissociated, collected and fused with Sp2/0-
Agl4 myeloma cells according to Defter et al, Somati,g
~'~~1 Genet ~a 3:231 (197'7). The fusion mixture is
distributed in culture ~aedium selective for the
pr~pagation of fused cells which are grown until they are
about 25~ confluent. At this time, culture supernatants
are tested for the presence of antibodies reacting with a
particular CFTR antigen. An alkaline phosphatase
labelled anti-mouse second antibody is then used for
detection of positives. Cells from positive culture
wells acre than expanded in culture, their supernatants
collected for further testing and the cells stored deep
fx°ozen in crycaprotectant-containing medium. To obtain
large quantities of a mAb, producer cells are injected
into the peritoneum at 5 x 106 cells par animal, and
ascites fluid is obtained. Purification is by
chromotography on Protein G- or Protein A-agarose
according to Ey et al, Im_munochem;~i,-~. 15:429 (1977),
' Reactivity of these mAbs with the mutant CFTR
protein can be confirmed by polyacrylamide gel
electrophoresis of membranes isolated from epithelial
'0~0 91 / 10734 ~'CT/Cr~91 /00009
103 ~~a T~ a c7' ':i
cells in which it is expressed and immunoblotted [Towbin
et al, P~~g. t3atl. Acad. Sci. USA 76:4350 (1979) ] .
In addition to the use of monoclonal antibodies
specific for the particular mutant domain of the CFTR
protein to probe their individual functions, other mAbs,
which can distinguish between the normal and mutant forms
of CFTR protein, are used to detect the mutant protein in
epithelial cell samples obtained from patients, such as
nasal mucosa biopsy ~~brushings" [ R. De-Lough and J.
Rutland, 3. C7,in.~at ol. 42, 613 (1989) ] or stein biopsy
specimens containing sweat glands.
Antibodies capable of this distinction are obtained
by differentially screening hybridomas from paired sets
of mice immunized with a peptide containing, for example,
the isoleucirae at amino acid position 507 (e. g.
GTIKENI,~FGVSY) or a peptide which is identical except for
the: absence of 1507 (GTIKENIFGVSY). mAbs capable of
recognizing the other mutant forms of CFTR protein
present in patients in addition or instead of 1507
2o deletion are obtained using similar monoclonal antibody
production strategies.
antibodies to normal and CF versions of CFTR protein
and of segments thereof are used in diagnostically
immunocytochemical and immunofluorescence light
microscopy and immunoelectron microscopy to demonstrate
the tissue, cellular and subce11u1ar distribution of CFTR
within the organs of CF patients, carriers and non-CF
individuals.
l~ntibodies are used to therapeutically modulate by
3o promoting the activity of the CFTR protein in CF patients
and in cells of CF patients. Possible modes of such
modulation might involve stimulation due to cross-linking
of CFTR protein molecules with multivalent antibodies in
analogy with stimulation of some cell surface membrane
receptors, such as the insulin receptor [O~Brien et al,
EurQ. ~o~, B,~o,).. Organ. J. 6:4003 (1987) ], epidermal
growth factor receptor [Schreiber et a1, J. Biol. Chem.
CVO 91 / 1 X734 PCT/CA91 /00009
~d ~ ~; ix ~_
104
25s:s46 (1983), and T-cell receptor-associated molecules
such as CD4 (veillette et al ~latt~tre, 338 0 257 (1989) ] ,
Antibodies are used to direct the delivery of
therapeutic agents to the cells which, eXpress defective
cFTR protein in cF. For this purpose, the antibodies are
incorporated into a vehicle such a;s a liposome [Matthay
et al, Cancer Res. 46:4904 (a986), which carries the
therapeutic agent such as a drug or the normal gene.
~.5 RfZP ANA~XSIS
to DNA diagnosis is currently being used to assess
whether a fetus will be barn with cystic fibrosis, but
historically this has only been done after a particular
set of parents has already had one cystic fibrosis child
which identifies them as obligate carriers, However, in
combination with carrier detection as outlined above, DNA
'diagnosis fox all pregnancies of carrier couples will be
pe~sible. If the parents have already had a cystic
fibrosis child, an extended haplotype analysis can be
done an the fetus and thus the percentage of false
positive or false negative will be greatly reduced. If
the parents have not already had an affected child and
the DNA diagnosis on.the fetus is being performed on the
basis of carrier detection, haplotype analysis can still
be performed.
Although it has been thought for many years that
there is a great deal of clinical heterogen~aty in the
cystic fibrosis disease, it is now emerging that there
are two general categories, called pancreatic sufficiency
(CF-PS) and pancreatic insufficiency (CF-PI). If the
mutations related to these disease categories are well
characterized, one can associate a particular mutation
with a clinical phenotype of the disease. This allows
changes in the treatment of ~ach patient. Thus the
nature of the mutation will to a certain extent predict
the prognosis of the patient and indicate a specific
treatment.
W~ 91110734 P~'/~A9111)0009
l05
~o~~c~ ~xo~oo~ o~ cx~~TC~ ~~~~os~~
The postulate that CFTR may regulate the activity of
ion channels, particularly the outwardly rectifying C1
channel implicated as the functional defect in CF, can be
tested by the injection and translation of full length in
vitro transcribed CFTR mRNA in Xenopus oocytes. The
ensuing changes in ion currents across the oocyte
membrane can be measured as the potential is clamped at a
fixed value. CFTR may regulate endogenous oocyte
channels or it may be necessary to also introduce
epithelial cell RNA to direct the translation of channel
proteins. LTse of mRNA coding for normal and for mutant
CFTR, as provided by this invention, makes these
experiments possible.
i5 ~ther modes of expression in heterologous cell
system also facilitate dissection of structure-function
relationships. The complete CFTR DNA sequence ligated
into a plasmid expression vector is used to transfect
cells so that its influence on ion transport can be
assessed. Plasmid expression vectors containing part of
the normal CFTR sequence along with portions of modified
sequence at selected sites can be used in vitro
mutagenesis experiments performed in order to identify
those portions of the CFTR protein which are crucial for
regulatory function.
~s.,& EXPItE~~IOP1 !~~ 'fRE ~ItJTA..~'9' DNFv 8 nrgv~rng
The mutant DNA sequence can be manipulated in
studies to understand the expression of the gene and its
product, and, to achieve production of large quantities
of the protein for functional analysis, antibody
production, and patient therapy. The changes in the
sequence may or may not alter the expression pattern in
terms of relative quantities, tissue-specificity and
functional properties. The partial or full-length cDNA
sequences, which encode for the subject protein,
unmodified or modified, may be ligated to bacterial
expression vectoxs such as the pRTT (Nilsson et al. EMB_O
wo ~mo7~ pcri~A~lioooo~
~ ~'~ ~ !.~ C~ 106
,~, 4: 1075-1080 (1985)), pGEX (Smith and Johnson,
67: 31-40 (1988)) or pATIi (8pindler et al. J. V x-01. 49:
132-141 (1984)) plasmids which can be introduced into ~.
coli cells for production of the corresponding proteins
which may be isolated in accordance with the previously
discussed protein purification procedures. The DNA
sequence can also be transferred from its existing
context to other cloning vehicles, such as other
plasmids, bacteriophages, cosmids, animal virus, yeast
to artificial chromosomes (YAC)(Burke et al. Saie re 236;
8os-812, (1987)), somatic cells, and ether simple or
complex organisms, such as bacteria, fungi (Timberlake
and Marshall, ,~ fence 244: 1313~-1317 (1989),
invertebrates, plants (Gasser and Fraley, a a 244:
1293 (1989), and pigs (Pursel et al. ~_ience 244: 1281-
128s (19s9)).
. For expression in mammalian cells, the cDNA sequence
may be ligated to heterologous promoters, such as the
simian virus (SV) 40, promoter in the pSV2 vector
[Mulligan and Berg, DoE, Nat, Acad ~~t yran,
?8:2072-
2076 (1981)] and introduced into cells, such as monkey
CCS-1 cells [Gluzanan, C~1~,, 23:17x-182 (1981)J, t4
achieve transient or long-term expression. The stable
integration of the chimeric gene construct may be
2S maintained in mammalian sells by biochemical selection,
such as neomycin [Southern and Berg, J. Mol, Aooln.
Genet. 1:327-341 (1982):] and mycophoenolic acid (Mulligan
and Berg,
DNA sequences can be manipulated with standard
procedures such as restriction enzyme digestion, fill-in
with DNA polymerase, deletion by exonuclease, extension
by terminal deoxynucleotide transferase, ligation of
synthetic or cloned DNA sequences, site-directed
sequence-alteration via single-stranded bacteriophage
intermediate or with the use of specific ol'igonucleotides
in combination with PCR.
'WO 91/10734 PC.T/CA91/OO~Dlf9
107 ~~J~C~~~/~ ~l.x
The cDNA sequence (or portions derived from it), or
a mini gene (a cDNA with an intron and its own promoter)
is introduced into eukaryotic expression vectors by
conventional techniques. These vectors are designed to
permit the transcription of the cDNA in eukaryotic cells
by providing regulatory sequences that initiate and
enhance the transcription of the cDNA and ensure its
proper splicing and polyadenylation. Vectors containing
the promoter and enhancer regions of the simian virus
~o (sv)4o or long terminal repeat (LTR) of. the Rous Sarcoma
virus and polyadenylation and splicing signal from SV 40
are readily available [Mulligan et al Proc. Natl Acad
Sci. LISA 78:1078-2076, (1981); GOrmari et al ProC Nail
ACad. Sci tiaA 79: 6777-6781 (1982)j. Alternatively, the
CFTR endogenous promoter may be used. The level of
expression of the cDNA can be manipulated with this type
of vector, either by using promaters tPaat have different
activities (for ea~ample, the baculovirus pAC373 can
express cDNAs at high levels in ;~,. ~runaincells [M.
2o D. Summers and G. E. Smith in, Genetically Altered
viruses and the Environment (B. Fields, et al, eds.) vol.
22 no 3i9-328, Cold Spring Harbour Laboratory Press, Cold
Spring Harbour, New York, 1985] or by usang vectors that
contain promoters amenable to modulation, for example the
glucocorticoid-responsive promoter from the mouse mammary
tumor virus [I~e et al, P~ature 294:228 (1982)J. The
expression of the cDNA can be monitored in the recipient
cells 24 to 72 hours after introduction (transient
expression).
In addition, some vectors contain selectable markers
[such as the ,gp~, [Mulligan et Berg supra] or neo
[Southern and Berg ~. Mol. Aptaln Gp~p~ ~,:327-3d1 (1982)]
bacterial genes that permit isolation of cells, by
chemical selection, that have stable, long term
expression of the vectors (and therefore the cDNA) in the
recipient cell. The vectors can be maintained in the
cells as episomal, freely.replicating entities by using
WO 91/10734 PCT/CA91/00009
v 1 108
~~ ~~~~i~?
regulatory elements of viruses such as papilloma [Server
et al Mol. Cell lBiol~ 8:486 (1981)] or Epstein-Barr
(Sugden et al MOI. Cei_t yiol 5:410 (1985)].
Alternatively, one can also produce cell lines that have
integrated the vector into genomic DNA. Both of these
types of cell lines produce the gene product on a
continuous basis. One can also produce cell lines that
have amplified the number of copies of the vector (and
therefore of the cDNA as well) to create cell lines that
can produce high levels of the gene product [Alt et al.
J~ Bio1 them 253: 1357 (1978)].
The transfer of DNA into eukaryotic, in particular
human or other mammalian cells is now a conventional
technique. The vectors are introduced into the recipient
cells as pure DNA (transfection) by, for example,
precipitation with calcium phosphate [Graham and vender
Eb, Virolocrv 52:466 (1973) or strontium phosphate [Brash
et al Mol. Cell Bio, 7:2013 (1987)], electroporation
[Neumann et al EMBO J 1:841 (1982)J, lipofect~.on [Felgner
~r-i rmn
et al Pros Nato Aged 84.7413 (1987)J, DEAF
dextran [McCuthan et al ~. Nat_'! Can_ce~r Tns~_ 41;351
1968)J, microinjection [Mueller et al Cellq15:579 1978)],
protoplast fusion [Schafner, Proc Natl AGE Sci tten
72:2163) or pellet guns [Klein et al, Na~u~g 327: 70
(1987)]. Alternatively, the cDNA can be introduced by
infection with virus vectors: Systems are developed that
use, for example, retroviruses [Bernstein et al. ~en~t~,,~
F~ n ina 7: 235, (1985)), adenoviruses [Ahmad et al T~
,Vi~rol 5'1:267 (1986)) or Herpes virus [Spaete et al Cell
30:29 (1982)].
These eukaryotic expression systems can be used for
many studies of the mutant CF gene and the mutant CFTR
product,.such as at protein positions 85, 148, 178, 455,
493, 507, 542, 549, 551, 560, 563, 574, 1,077 and 1092.
These include, for example: (1) determination that the
gene is properly expressed and that all post-
translational modifications necessary for full biological
!v0 91110734 PCT/CA91/00009
109 ~3~~~~~1~~.~,
activity have been properly completed (2) identify
regulatory elements located in the 5o region of the CF
gene and their role in the tissue~ or temporal-regulation
of the expression of the CF gene (3) production of large
amounts of the normal protein for isolation and
purification (4) to use cells expressing the CFTR protein
as an assay system for antibodies generated against the
cFTR protein or an assay system to test the effectiveness
of drugs, (5) study the function of the normal complete
to protein, specific portions of the protein, or of
naturally occurring or artificially produced mutant
proteins. Naturally occurring mutant proteins exist in
patients with CF while artificially produced mutant
protein can be designed by site directed sequence
1S alterations. These latter studies can probe the function
of any desired amino acid residue in the protein by
mutating the nucleotides coding far that amino acid.
Using the above techniques, the expression vectors
containing the mutant CF gene sequence or fragments
2o thereof can be introduced into human cells, mammalian
cells from other species ar non-mammalian sells as
desired. The choice of cell is determined by the purpose
of the treatment. For example, one can use monkey COS
cells ~Gluzman, Cell 23:175 (1981),, that produce high
25 levels of the SV4o T antigen and-permit the replication
of vectors containing the SV40 origin of replication, can
be used to show that the vector can express the protein
product, since function is not required. Similar
treatment could be performed with Chinese hamster ovary
30 (CH~) or moues NZH 3T3 fibroblasts or with human
fibroblasts or lymphoblasts.
The recombinant cloning vector, according to this
invention, then comprises the selected DNA of the DNA
sequences of this invention for expression in a suitable
35 hash The DNA is operatively linked in the vector to an
expression contral.sequence in the recombinant DNA
molecule so that normal CFTR polypeptide can be
WO 91!10734 P(.°TlCA91/00009
~a~~ .~~,'~C~ ~ mo
expressed. The expression control sequence may be
selected from the group consisting of sequences 'that
control the expression of genes of prokaryotic or
eukaryotic cells and their viruses and combinations
thereof. The expression control sequence may be
specifically selected from the group consisting of the
~ system, the t~ system, the system, the ~c
system, major operator and promoter regions of phage
lambda, the control region of id coat protein, the early
1.o and late promoters of SV40, promoters derived from
polyoma, adenovirus, retrovirus, baculavirus and simian
virus, the promoter for 3-phosphoglycerate kinase, the
promoters of yeast acid phosphatase, the promoter of the
yeast alpha-mating factors and combinations thereof,
as The host cell, which may be transfected with the
vector of this invention, may be selected from the group
consisting of ~, coli, pseudomonas, Saci_Wuc sub 'l y
~~ci pus s~earothe moph9lus or other bacili; other
bacteria; yeast; fungi; insect; mouse or other animal;
20 or plant hosts; or human tissue cells.
It is appreciated that for the mutant DNA sequence
similar systems are employed to express and produce the
mutant product.
z!ROT~zN ~mN~~~orr ~orrszn~~~r_Tnu~
25 To study the function of the mutant CFTR protein, it
is preferable to use epithelial cells as recipients,
since proper functional expression may require the
presence of other pathways or gene products that are only
expressed in such cells. Cells that can be used include,
30 for example, human epithelial cell lines such as T84
(ATCC ,~CRL 248) or FANC-1 (ATCC ,~ CLL 1459), or the T43
immortalised CF nasal epithelium cell line [Jettan et al,
,~.ence (1989) and primary [Yanhoskes et al. Ann. Rev.
Res~a. Dis 132: 1281 (1985)) or transformed [Scholte et
35 al. Exc. Cell. Res 182: 559(1989)] human nasal polyp or
airways cells, pancreatic cells [Harris and Coleman ~,
Cell. Sci 87: 695 (298?),, or sweat gland cells [Collie
iy0 91/10734 1'CT/CA91/00009
111 ~~~'~~~u~(a:y_
et al. ,fin Vi~ro 21s 597 (1985)] derived from normal or CF
subjects. The CF cells can be used to test far the
functional activity of mutant CF genes. current
functional assays available include the study of the
movement of anions (C1 or I) across cell membranes as a
function of stimulation of cells by agents that raise
intracellular AMP levels and activate chloride channels
[Stutto et al. g~,Qc. Nat. Aced. Sci. U. S. .A. 82: 6677
(1985)]. Other assays include the measurement of changes
to in cellular potentials by patch clamping of whole cells
or of isolated membranes [Frizzell et a1. Science 233:
558 (1986), Welsch and Liedtke 322: X67 (1986)]or
the study of ion fluxes in epithelial sheets of confluent
cells [Widdicombe et al. Proc. Nat. Aced. Sci. 82: 6167
(1985)]. Alternatively, RNA made from the CF gene could
be injected into ~,~nogus oocytes. The oocyte will
translate RNA into protein and allow its study. As other
more specific assays are developed these can also be used
in the study of transfected mutant CFTR protein function.
~~pomain-switching~~ experiments between aautant CFTR
and the human multidrug resistance P-glycoprotein can
also be performed to further the study of the mutant CFTR
protein. In these experiments, plasmid expression vectors
are constructed by routine techniques from fragments of
the mutant CFTR sequence and fragments of the sequence of
P-glycoprotein ligated together by DNA ligase so that a
protein containing the respective portions of these two
proteins will be synthesized by a host cell transfected
with the plasmid. The latter approach has the advantage
3o that many experimental parameters associated with
multidrug resistance can be measured. hence, it is now
possible to assess the ability of segments of mutant CFT~t
to influence these parameters.
These studies of the influence of mutant CFTR on ion
transport will serve to bring the field of epithelial
transport into the molecular arena.
'WO 91/10734 PCT/CA91/00009
~~~~I~ y ~ 112
_',~L'P~IaB
It is understood that the major aim of the various
biochemical studies using the compositions of this
invention is the development of therapies to circumvent
or overcome the CF defect, using both the pharmacological
and the "gene-therapy" approaches.
In the pharmacological appraach, drugs which
circumvent or overcome the CF defect are sought.
Initially, compounds may be tested essentially at random,
and screening systems are required to discriminate among
many candidate compounds. This invention provides host
cell systems, expressing various of the mutant CF genes,
which are particularly well suited for use as first level
screening systems. Preferably, a cell culture system
using mammalian cells (most preferably human cells)
transfected with an expression vector comprising a DNA
sequence coding for CFTR protein captaining a CF-
generating mutation, for example the 1507 deletion, is
used in the screening process. Candidate drugs are
20, tested by incubating the cells in the presence of the
candidate drug and measuring those cellular functions
dependent on CFTR, especially by measuring ion currents
~rhere the transmembrane potential is clamped at a fixed
value. To accommodate tha large number of assays,
however, more convenient assays are based, for example,
on the use of ion-sensitive fluorescent dyes. To detect
changes in C1''on concentration SPA or its analogues are
useful.
Altera~atively, a cell-free system could be used.
Purified CFTR could be reconstituted into articifial
membranes and drugs oould be screened in a cell-free
assay [A1-Aqwatt, ,S,~ience, (1989)x.
At the second level, animal testing is required. It
is possible to develop a model of CF by interfering with
the normal expression of the counterpart of the CF gene
in an animal such~as the moue. The "knock-out" of this
gene by introducing a mutant form of it into the germ
WO 91/10734 1'C'f/CA91/00009
l~~ ~, L ~ ~ ~ ~i
line of animals will provide a strain of animals with cF-
like syndromes. This enables testing of drugs which
showed a promise in the first level cell-based screen.
As further knowledge is gained about the nature of
the protein and its function, it will be possible to
predict structures of proteins or other compounds that
interact with the CFTR protein. That in turn will allow
for certain predictions to be made about potential drugs
that will interact with this protein and have some effect
~o on the treatment of the patients. Ultimately such drugs
may be designed and synthesized chemically on the basis
of structures predicted to be required to interact with
domains of CFTR. This approach is reviewed in Capsey and
Delvatte, a
Stockton press, ldew York, x.988. These potential drugs
must also be tested in the screening system.
PI~oT~Ild REPr:a~~at~am ~ror~ru!naw
Treatment of CF can be performed by replacing the
defective gratein with normal protein, by modulating the
2o function of the defective protein or by modifying another
step in the pathway in which CFTR participates in order
to correct the physiological abnormality.
To be able to replace the defective protein with the
normal version, one must have reasonably large amounts of
pure CFTR protein. Pure protein can be obtained as
described earlier dram cultured cell systems. Delivery
of the protein to the affected airways tissue will
require its packaging in lipid-containing vesicles that
facilitate the incorporation a! the protein into the cell
membrane. It may also be feasible to use vehicles that
incorporate proteins such as surfactant protein, such as
SAP(Val) or sAP(Phe) that performs this function
naturally, at least for lung alveolar cells. (PCT patent
Application WO/8803~.70, Whitsett et a1, May 7, 1888 and
PCT patent Application W089/04327, Benson et al, May 18,
1989). The CFTR-containing vesicles are introduced into
WCa 91/10734 I'C~'/C~91/00009
~~~~~~~.i 114
the airways by inhalation or irrigation, techniques that
are currently used in CF treatment (Boat et al, supxa).
6.3.2 DISUGf '~'~I~PX
Modulation of CFTR function can be accomplished by
the use of therapeutic agents (drugs). These can be
identified by random approaches using a screening program
in which their effectiveness in modulating the defective
CFTR protein is monitored ,~ vitro. Screening programs
can use cultured cell systems in which the defective CFTR
protein is expressed. Alternatively, drugs can be
designed to modulate CFTR activity from knowledge of the
structure and function correlations of CFTR protein and
from knowledge of the specific defect in the CFTR mutant
protein (Capsey and Dalvatte, ). It is possible
that the mutant CFTR protein will require a different
drug for specific modulation. It, will then be necessary
to identify the specific mutations) in each CF patient
before initiating drug therapy.
Drugs can be designed to interact with different
aspects ~f CFTR protein structure or function. For
example, a drug (or antibody) can bind to a structural
fold of the protein to correct a defective structure.
Alternatively, a drug might bind to a specific functional
residue and increase its affinity for a substrate or
cofactor. Since it is known that members of the class
of proteins to which CFTR has structural homology can
interact, bind and transport a variety of drugs, it is
reasonable to expect that drug-related therapies may be
effective in treatment of CF.
A third mechanism for enhancing the activity of an
effective drug would be to modulate the production or the
stability of CFTR inside the cell. This increase in the
amount of CFTR could compensate for its defective
function.
Drug therapy can also be used to compensate for the
defective CFTR function by interactions with other
components of the physiological or biochemical pathway
w~ 9aiao~~ pcricA~noooo9
'~~~t~.l~.
necessary for the expression of the CFTIt function. These
'interactions can lead to increases or decreases in the
activity of these ancillary proteins. The methods for the
identification of these drugs would be similar to those
described above for GFTR-related drugs.
2n other genetic disorders, it has been possible to
correct for the consequences of altered or missing normal
functions by use of dietary modifications. This has
taken the form of removal of metabolites, as in the case
to of phenylketonuria, where phen~lalanine is removed from
the diet i.n the first five years of life to prevent
mental retardation, or by the addition of larger amounts
of metabolites to the diet, as in the case of adenosine
deaminase deficiency where the functional correction of
the activity of the enzyme can be produced by the
addition of the enzyme to the diet. Thus, once the
details of the CFTR function have been elucidated and the
~aWic defect in CF has been defined, therapy may be
achieved by dietary manipulations.
2~ The second potential therapeutic approach is so-
called "gene-therapy" in which normal copies cf the CF
gene errs introduced in to patients so as to successfully
code far normal protein in the key epithelial cells of
affected tissues. It is most crucial to attempt to
achieve this with the airway epithelial cells of the
respiratory tract. Ths CF gene is delivered to these
cells fn form in which it can betaken up and code for
sufficient protein to provide regulatory function. As a
result, "the patient s quality and length of life will be
greatly extended. tTltimately, of course, the aim is to
deliver the gene to all affected tissues.
One appraach to therapy of CF is to insert a normal
version of the CF gene into the airway epithelium of
affected patients. It is important to note that the
respiratory system is the primary cause of mordibity and
mortality in CF; while pancreatic disease is a major
WO 91/10734 PCT1CA91/(~09
15.6
feature, it is relatively well treated today with enzyme
supplementation. Thus, somatic cell gene therapy (for a
review, see T. Friedmann, a ee 244:1275 (1989))
targeting the airway would alleviate the most severe
problems associated with CF.
A. Retroy~ra7 Vectors. Retroviruses have been
considered the preferred vector for experiments in
somatic gene therapy, with a high efficiency of infection
and stable integration and expression [orkin et al Pr~a.
20 Med. Genet 7:130, (1988}J. A possible drawback is that
cell division is necessary for retroviral integration, so
that the targeted cells in the airway may have to be
nudged into the call cycle prior to retroviral infection,
perhaps by chemical means. The full length CF gene cDNA
can be cloned into a retroviral vector and driven from
either its endogenous promoter or from the retroviral LRT
(Long teruinal repeat). Exgression of levels of the
normal protein as low as 1o% of the endogenous mutant
protein in CF patients would be expected to be
2o beneficial, since this is a recessive disease. Delivery
of the virus could be accomplished by aerosol or
instillation into the trachea.
B. other ~ira~ vo.a~,.ao Other delivery systems
which can be utilized include adeno-associated virus
[AAV, McLaughlin et al, J. Viroi 62:1963 (1988)],
vaccinia, virus [doss st al Annu. ev '~ > >nol, 5:3~s,
1987)1, bovine gegil1oma virus [Rasmussen et al, J~gt ode
], 139:642 (1987)] or ~e:aber of the hergesv3rus
group such as Epstein-Barr virus (Margolskee et al ~tol.
3o Cel1.B~o~ 8:2937 (1988),. Though much would need to be
learned about their basic biology, the idea of using a
viral vector with natural trogism for the respiratory
track (e. g. respiratory syncytial virus, echovirus,
Coxsackie virus, etc.) is possible.
C. Non_-vira~ Gene Tran~fp.-, ether methods of
inserting the CF gene into respiratory epithelium may
also be productive; many of these are lower efficiency
WO 91/1074 ~'~Ci°/CA9IlOOOU9
11? ~~~ 3
and would potentially require infection ,~ vitro,
selection of tr~nsfectants, and reimplantation. Thus
would include calcium phosphate, DEAE dextran,
electroporation, and protoplast fusion. A particularly
attractive idea is the use of liposome, which might be
possible to carry out ,~ vivo (Ostro, ~ioo~ spores, Marcel-
Dekker, 198?]. Synthetic cationic lipids such as DOTrIA
[Felger et al pros N~. t1 AC~~ ~ni TTc~ 84:?413 (198?) ]
may increase the efficiency and ease of carrying out this
approach.
s . ~, c~ Artzr~ ~oaELa
The creation of a mouse or other animal model for CF
will be crucial to understanding the disease and for
testing of possible therapies (for general review of
creating animal models, see Erickson, Am. ,? Hum G n t
43:582 (1988)]. Currently no animal model of the CF
exists, The evolutionary conservation of the CF gene (as
demonstrated by the cross-species hybridization blots for
E4.3 and H1.6), as is Shawn in Figure 4, indicate that an
orthologous gene exists in the mouse (hereafter to be
denoted mCF, and its corresponding protein as mCFTR), and
this wall be possible to clon~ in mouse genomic and cDNA
libraries using the human CF gene probes. It is expected
that the generation of a specific mutation in the mouse
gene analogous to the T50? mutation will be most optimum
to reproduce the phenotype, though complete inactivation
of the mCFTR gene will also be a useful mutant to
generate.
'A~ d~utagL, esis Inactivation of the mCF gene can
be achieved by chemical [e. g. Johnson et al pros, Hats,
Acad, Sci. USA 78:3138 (1981)] or 7(-ray mutagenesis [Popp
et al J. Mol. Bln~_ 12?:141 (19?9)] of mouse gametes,
followed by fertilization. Offspring heterozygous fox
inactivation of mCFTR can then be identified by Southern
blotting to demonstrate loss of one allele by dosage, or
failure to inherit one parental allele if an RFLP marker
is being assessed. This approach has previously been
WO 91/10734 PCg'/~A91/00009
~~~~~~~ C
ma
successfully used to identigy mouse mutants for a-globin
[~hltney et al ~~OC Nat3 $r~a~ Cyi U~"A 77 v a.o8'~
(1980)), phenylalanine hydroxylase [McDonald et al
Pediatr Res 23:63 (1988)), and carbonic anhydrase II
[Lewis et al pr c. Natl Acad ~r~~TCA 85~1c362, (198g)),
B. TransA mutant version og CFTR or mouse
CFTR can be inserted into the mouse germ line using now
standard techniques of oocyte injection [Camper, a ds
in Genetics (1988)l; alternatively, if it is desirable to
1o inactivate or replmce the endogenous mCF gene, the
homologous recombination system using embryonic stem (ES)
cells [Capecchi, 'ence 244:1288 (1989)] may be applied.
1. oocvte _rni ion placing one or more copies
of the normal or mutant mCF gene at a random location in
the mouse germline can be accomplished by microinjection
of the pronucleus of a just-fertilized mouse oocyte,
followed by reimplantation into a pseudo-pregnant foster
mother. The liveborn mice can then be screened for
integrants using mnalysis of trail DIdA for the presence of
2o human CF' gene sequences. The same protocol cmn be used
to insert a mutmnt mCF gene. To generate m mouse model,
one would want to plmce this trmnsgene in m mouse
background where the endogenous mCF gene has been
inactivated, either by mutagenesi.s (see above ) or by
homologous recombination (see,below). The transgene can
be eithers m) a complete genomic .sequence, though the
size of this (mbout 250 kb) would require that it be
injected as a yeast artificial chromosome or a~ chromosome
frmgment; b) m cDNA with either the natural promoter or a
heterologous promoter; c) a °~minigene" contmining all of
the coding region and various other elements such ms
introns, promoter, and 3° flanking elements found to be
necessmry for optimum expression.
2. Retrovira~ Tnt~er ton o
This mlternative involves inserting the CFTR or mCF.gene
into a retroviral 'vector and directly infecting mouse
embroyos at early stages of development generating a
W('D 91 /10734 PtT/CA91 /00009
~ l9 ~ ~e~ ~,~ ~ ~ /_~
chimera [Soriano et al Gel1 46:~.g (1986)]. At least some
of these will lead to germline transmission.
3. ~$ CellS and Hnmnlnnnnc yo.. 1.t ~~ The
embryonic stem cell approach (Capecchi, and
s Capecchi, Trends Genet 5:70 (1989)] allows the
possibility of performing gene transfer and then
screening the resulting totipotent cells to identify the
rare homologous recombination events. c7nce identified,
these can be used to generate chimeras by injection of
1o mouse blastocysts, and a proportion oz the resulting mice
will show germline transmission from the recombinant
line. There are several ways this could be useful in the
generation of a mouse model for CF:
a) Inactivation of the mCF gene can be conveniently
15 accomplished by designing a DNA fragment which contains
sequences from a mCFTR axon flanking a selectable marker
such as neo. Homologous recombination will lead to
insertion of the neo sequences in the middle of an axon,
inactivating mCFTR. The homologous recombination events
20 (usually about 1 in 1000) can be recognized from the
heterologous ones by DNA analysis of individual clones
[usually using PCR, Kim et al ~cleic Acids Res 16:8887
(1888), ~oyner et al Nature 338:153 (1989); Zimmer et al
sutra, p. 150] ox by using a negative selection against
25 the heterologous events [such as the use of an HSV ,TK
gene at the end of the construct,.followed by the
gancyclovir selection, Mansour et al, Nature 336x348
(1988)]. This inactivated mCFTR mouse can then be used
to introduce a mutant CF gene or mCF gene containing, for
30 example, the 1507 abnormality or any other desired
mutation.
b) It is possible that specific mutants of mCFTR
cDNA be created in one step. For example, one can make a
construct containing mCF intron 9 sequences at the 5~
35 end, a selectable n~o gene in the middle, and intro 9 +
axon 10 (containing the mouse version of the I507
mutation) at the 3~ end. A homologous recombination
WO 91/10734 Pt.'T/CA91/00009
~~~~~c~x'' ~ 120
event would lead to the insertion of the nip gene in
intron 9 and the replacement of axon 10 with the mutant
version.
c) zf the presence of the selectable neo marker in
s the intron altered expresson of the mCF gene, it would be
possible to excise it in a second hoaaologous
recombination step.
d) zt is also possible to create anutations in the
mouse germline by injecting oligonucleotides containing
1o the mutation of interest and screening the resulting
cells by pCR.
This embodiment of the invention has considered
primarily a mouse model for cystic fibrosis. Figure 4
shows cross-species hybridization not only to mouse DNA,
but also to bovine, hamster and chicken DNA. Thus, it is
contemplated that an orthologous gene will exist in many
other species also. Tt is thus contemplated that it will
be possible to generate other animal models using similar
technology.
20 Although preferred embodiments of the invention have
been described herein in detail, it will be understand by
those skilled in the art that variatione~ may be made
thereto without departing from the spirit of the
invention or the scope of the appended claims.