Note: Descriptions are shown in the official language in which they were submitted.
: ~ :
W O 9t/13054 PCT/EP91/00296
7~5~) .
''Cycl~ y~ uted Glu~aramide Antihypertensive Agents"
This invention relates to a series of cycloalkyl-substituted
glutaramide derivatives which are antihypertensive agents having
utility in the treatment of various cardiovascular disorders,
including hypertension and heart failure.
According to the specification of our European patent
application 274234, we disclose certain cycloalkyl-substituted
glutaramide derivatives which are inhibitors of the zinc dependent
neutral endopeptidase E.C.3.4.24.11 (atriopeptidase) and which are
thereby able to potentiate the biological effects of atrial
natriuretic factor and in particular, are natriuretic,
antihypertensive and diuretic agents of value in the treatment of
various cardiovascular disorders. In our European patent
application no. 89308740.3 we describe compounds which are
inhibitors of the enzyme E.C.3.4.24.11 and, in addition, they are
also able to inhibit angiotensin converting enzyme (ACE~, a ;~
further enzyme which is involved in the control of blood pressureO
The compounds thus have a dual pharmacological action through
inhibiting two key enzymes involved in blood pressure control
which makes them particularly useful in the treatment of various
forms of hypertension and associated cardiovascular disorders,
e.g. congestive heart failure and glaucoma.
According to the present inyention there are provided further
related compounds having activity as atriopeptidase and ACE
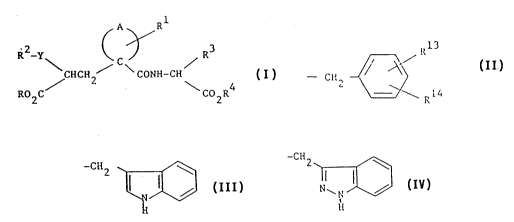
inhibitors of the formula:
, , , 1 ",
PLC 534
A Rl
~2 y ~ C ~ R3
/ CHCH2 / \ CONH-CH
RO2C \ C02R ~;
(I) :
wherein:
A completes a 5 or 6 membered carbocyclic ring which may be
saturated or monounsaturated;
Y is an alkylene group of from 1 to 9 carbon atoms which may be
straight or branched chain; ::
R is H or (Cl-C4)alkyl;
R and R are each independently H, (Cl-C6)alkyl, (C3-C7)
cycloalkyl, benzyl, or an alternative biolabile ester-forming
group;
R is hydroxy, (Cl-C6)alkoxy, hydroxy(C2-C6)alkoxy, (Cl-C6)alkyl-
S(O)n-, (Cl-C4)alkoxy(Cl-C6)alkoxy, (Cl-C~)alkyl-S(O)n-(Cl-C6)-
alkoxy, (Cl-C4)alkoxy(C2-C6)alkenyloxy, N3, (R )2N, (R )2N-(Cl-
C6)alkoxy, (R )2N-(C2-C6)alkenyloxy, heterocyclyl-Z-, hetero-
cyclyl(Cl-C4)alkyl-Z-, aryl-Z- or aryl(Cl-C4)alkyl-Z, wherein Z is
~ S()n or NR and R is H, (Cl-C4)alkyl or aryl(Cl-C4)alkyl;
or R is a group of the formula R7R CH- in which case Y may also :`
be a direct link and wherein R7 is (R5)2N(Cl-C4)alkyl, (Cl-C4)-
alkoxy(C2-C4)alkylaminomethyl, heterocyclyl(Cl-C4)alkyl or aryl
and R is (Cl-C6) alkoxy, (Cl-C4)alkoxy(C2-C6)alkoxy,
~ ..
PLC 534
hydroxy(C2-C6)alkoxy or hydroxy(C1-C6)alkyl; wherein aryl means
phenyl or naphthyl which may optionally be substituted with, one
or more OH, CN, CF3, (Cl-C4) alkyl, (C1-C4)alkoxy, halo, j ~:~
carbamoyl, aminosulphonyl, amino, mono or di(C1-C4 alkyl)amino,
~Cl-C4 alkanoyl)amino, amino(Cl-C4)alkyl, di(Cl-C4 alkyl)amino~
(Cl-C4)alkyl, or (Cl-C4) alkyl-S(O)n (Cl C4)alkyl groups a
heterocyclyl means a 5 or 6 membered nitrogen, oxygen or sulphur
containing heterocyclic group which may be saturated or
unsaturated and which may optionally include a further oxygen or
one to three nitrogen atoms in the ring and which may optionally
be benzofused or substituted with one or more haloJ (Cl-C4)alkyl,
hydroxy, carbamoyl, benzyl, oxo, amino or mono or di-(Cl-C4
alkyl)amino or (Cl-C4 alkanoyl)amino groups; :~
or R is a group of the formula R9Co- wherein R9 is a l-piperidine
or 1-piperazine group, either of which may optionally be .
substituted by OH, =O, (Cl-C4)alkyl or N(R5)2;
or R is a group of the formula R 0CONR6- wherein R6 is as
previously defined, and R is (R )2N, (Cl-C4)alkoxy(C2-C4~alkyl~
Rl R CH-, or substituted phenyl wherein the substituent is
(R )2N-(Cl-C4)alkyl, (Cl-C4)alkyl-S(0)n- or (Cl-C4)alkyl-S(O)n-
(Cl-C4)alkyl;
with the proviso that R2 is not NH2 or CH3NH when Y is CH2;
R is a group of the formula:
- C~2 ~ ~ ~ R14
r-7
'~4, '
~ , .: ; " , , , : :.: ,~
PLC 534
wherein Rl is H, halo, 4-OH, 4-(Cl-C6 alkoxy),
4-(C3-C7 cycloalkoxy) 4-(C2-C6 alkenyloxy), 4-[(Cl-C
alkoxy)carbonyloxyl, 4-[(C3-C7 cycloalkoxy)carbonyloxy], or
3-(Cl-C4 alkyl)SO2NH-; and R14 is H, (Cl-C4)alkyl, (Cl-C~)alkoxy, ' ~ -
(C2-C6) alkanoyl or halo; or R is a group of the formula:
-CH2 `~
H
: -
wherein said groups may optionally be substituted in the fused ~.
benzene ring by (Cl-C4)alkyl, (Cl-C4)alkoxy, OH, halo or CF3;
each R is H, (Cl-C6)alkyl, phenyl(Cl-C6)alkyl or the two gro~lps
R are taken together to form, with the nitrogen to which they are
attached, a pyrrolidinyl, piperidino, morpholino, piperazinyl or
N-(Cl-C4)alkyl-piperazinyl group;
R is (Cl-C4)alkyl-S(O) NH- or (Cl-C4)alkanoylamino and R is
(Cl-C4)alkyl-S(0) (Cl-C4)alkyl or morpholinomethyl or R and R
are both morpholinomethyl or (Cl-C4)alkoxy(Cl-C4)alkoxy(Cl-C4)-
alkyl;
n is 0, 1 or 2
and pharmaceutically acceptable salts thereof and bioprecursors
therefor.
. . ' : . . ' ', ' . '' ' , : ' ' ' :;: ' , ', ' ,'', ' ' ';; ' ' ' ,''' . "" i;' , 1.:. . '' ' . .~ . '
PLC 534 ~ I
In the above definition~ unless otherwise indicated, alkyl
groups having three or more carbon atoms may be straight or
branched chain. The term halo means fluoro, chloro, bromo or
iodo. When R includes a heterocyclic group, particular examples
of heterocycles include pyridyl, pyrazinyl, pyrirnidinyl,
pyridazinyl, pyrrolyl, piperidino, imidazolyl, pyrazolyl,
triazolyl, tetrazolyl, furanyl, tetrahydrofuranyl,
tetrahydropyranyl, dioxanyl, thienyl, oxazolyl, isoxazolyl,
thiazolyl, indolyl, isoindolinyl, quinolyl, quinoxalinyl, ;
quinazolinyl and benzimidazolyl, each being optionally substituted
as previously defined.
The compounds of formula (I) may contain several asymrnetric
centres and thus they can exist as enantiomers and diastereomers.
The invention includes both the separated individual isomers as
well as mixtures of isorners.
______ _ :
Wo gl/13054 2~7~5~ PCr/EP91/00 ~
The pharmaceutically acceptable salts of the compounds of
formula (I) containing ~n acidic centre are those formed with
bases which form non-toxic salts. Examples include the al~ali or
alkaline earth metal salts such as the sodium, potassium or
calcium salts or salts with amines such as diethy:Lamine.
Compounds having a basic centre can also form acid addition salts
with pharmaceutically acceptable aclds. Examples include the
hydrochloride, hydrobromide, sulphate or bisulphate, phosphate or
hydrogen phosphate, acetate, citrate, fumarate, gluconate,
lactate, maleate, succinate, tartrate tosylate and lauryl sulphate
salts.
The term bioprecursor in the above definition means a
pharmaceutically acceptab]e biologically degradable derivative of
the compound of formula (I) which, upon administration to an
animal or human being, is converted in the body to produce a
compound of the formula (I). Examples include biolabile ester
derivatives and amide or amino acid derivatives of the compounds
of formula I.
A preferred group of compounds of the formula (I) are those
wherein A is (CH2)4 and Rl is H, i.e. compounds of the formula
(II) wherein R, R , R and R are as previously defined for
formula (I):
~ WO9~/13054 . . P~f~ 296
~?-i?~ 3~S8
.
R2 y ~ / R3
CHCH CONH-CH
2 2 \ C2R
(II
Also preferred are those compounds of formulae (I) and (TI)
wherein R and R4 are both H (diacids) as well as biolabile mono
and di-ester derivatives thereof wherein one or both of R and R4
is a biolabile ester-forming group.
The term biolabile ester-forming group is well understood in
the art as meaning a group which provides an ester which can be
readily cleaved in the body to liberate the corresponding diacid
of formula (I) wherein R and R4 are both H. A number of such
ester groups are well known, for example in the penicillin area or
in the case of the ACE-inhibitor antihypertensive agents.
Ln the case of ehe compounds of for~ulae (I) and (II) such
biolabile pro-drug esters are particularly advantag,eous in
providing compounds of the formula (I) suitable for oral
administration. The suitability of any particular e~ster-forming
group can be assessed by conventional animal or in vitro enzyme
hydrolysis studies. Thus, desirably for optimum effect, the ester
should only be hydrolysed after abscrption. accordingly, the ester
should be resistant S hydrolysis by digestive enzymes before
. : , ~
,, ,. . . ... ... ~ .. . .... .
W O 9l/13054 PCT/~ D~
2~7~5~ ~ ~
absorption but should be readily hydrolyzed by, ror example,
gut-wall, plasma or liver enzymes. In this wav the active di~cid
is released into the bloodstream following oral absorption.
In addition to lower alkyl esters (particularly ethyl) and
benzyl esters, alternative biolabile esters include
alkanoyloxyalkyl esters, including alkyl, cycloalkyl and aryl
substituted derivatives thereof, aroyloxyalkyl esters, arylesters 9
aralkylesters, halo~lkyl esters and hydroxyalkyl esters including
ketal derivatives thereof, wherein said alkanoyl or alkyl groups
have from 1 to 18 carbon atoms and are branched or straight chain
and said aryl groups arè phenyl, naphthyl or indanyl optionally
substituted with one or more (Cl-C14)alkyl, (Cl-C14)alkoxy or
(C2-C14)alkoxycarbonyl groups or halo atoms.
Thus examples of R and R4 when they are biolabile ester
groups include ethylj indanyl, isopropyl, n-butyl, sec-butyl9
t-butyl, cyclohexyl, benzyl, phenethyl, phenpropyl9 acetonyl~ -
glyceryl, pivaloyloxymethyl, 5-(4-methyl-1,3-dioxolene-2-
onyl)methyl, cyclohexylmethyl, cyclohexylcarboxyethyl,
cyclohexylacetoxyethyl, propionyloxyisobutyl, hexanoyloxyethyl,
pentanoyloxyethyl, acetoxyethyl, acetoxyben~yl,
pentanoyloxybenæyl, cyclohexyloxycarbonyloxyethyl,
butyloxycarbonyloxyethyl, isobutyloxycarbonylethyl and
ethoxycarbonyloxyethyl.
In one preferred aspect of the invention, the group R is
4-hvdroxybenzyl and the carbon atom to which it is attached is of
(S) stereochemistry; the group N~lCH(R3)C02R being derived from
L-~yrosine. Also preferred are compounds wherein R3 is
4-metnoxybenzyl.
.. . . . ... ..
W O 91/13054 ~ 5~ PCTiEPgl~00296
The substituent R Y is preferably (Cl-C6)alkoxy(Cl-C4~alkyl
or (Cl-C4)alkoxy(Cl-C6)alkoxy(cl-C4~alkyl. Thus particular and
preferred examples include ethoxvetkyl and 2-(methoxvethoxy)-
propyl.
Also preferred are compounds where R Y is amino(C4-C9)alkyl.
A particular and preferred exa~ple of this type is 8-amino-
octyl.
In a further group of preferred compounds Y is CU2 and R isof formula Rl CONR - wherein R is H. Particularly preferred are
examples wherein RlO is substituted phenyl particularly when said
substituent is H2NCH2-, (CH3)2NCH2- or CH3S(O?nCH2- (wherein n is
O, 1 or 2).
Thus particular and preferred compounds of the invention
include-: -
N-[1-~2~S)-carboxy-4-(2-methoxyethoxy)pentylJ-l-cyclopentane-
carbonyl]-(S)-tyrosine,
N-~ 2(S)-carboxy-4-ethoxybutyl)-1-cyclopentanecarbonyl]-(S)- ~ '
tyrosine,
N-[1-(2(R,S)-carboxy-10-aminodecyl)-1-cyclopentanecarbonyl]-(S)-
tyrosine and
N-~ 2(S)-carboxy-3-(4-aminomethylbenzamido)propyl~-l-cyclo-
pentanecarbonyl]-(S)-tyrosine.
W O 91~13054 , P ~ /EPg1/00 ~
- 2~7~ ) :
' ~
The compounds of formula (L) are prepared by a number of I :
different processes:
a) One procedure involves the synthesis of a partially
protected cycloalkyl-substituted glutaric acid derivative which is
coupled to an amino acid ester derivative to give the desired
glutaramide. Any reactive groups in R and R3 may require
protection during the coupling step and such protecting groups are
removed in the final stage of the process.
The synthetic route is illustrated in the following reaction
scheme wherein A~ Y and R are as previously defined, R and R
are as defined for R and R with any reactive groups therein
protected if necessary and Rl and R are as defined for R and R
excluding H, or they are conventional carboxylic acid protecting
groups:
CHCU2 CO2~ + U2 \ 18
(III) I (IV)
A Rl
R2 _y ( C ~ R3'
CHCH2 / CONH-CH
2 \ CO2R 8
\~ ,
(l)'' ~,
.. . ..
W O 91/130~4 ~ 5~ PCT/EP91~00296
11 t
The reaction of the compounds of formula (TII) and (IV) is
achieved using conventional amide coupling techniques. Thus in
one process the reaction is achieved with the reactants dissolved
in an organic sol~ent, e.g. dichloromethane, using a diimide
condensing agont! for example l-ethyl-3-(dimethylaminopropyl)-
carbodiimide, or N,~'-dicyclohexylcarbodiimide, advantageously in
the presence of l-hydroxybenzotriazole and an organic base such as
N-methylmorpholine. The reaction is generally complete after a
period of from 12 to 24 hours at room temperature and the product
is then isolated by conventional procedures, i.e. by washing with
water or filtration to remove the urea biproduct and evaporation
of the solvent. The product may be further purified by
crystallisatlon or chromatography, if necessary.
The compounds of formula (V) include compounds of formula (I)
wherein R and R are Cl-C6 alkyl or benzyl.
The diesters of formula (V) are subsequently reacted to give
the monoester or diacid derivatives of formula (I) wherein one or
both of R and R4 are H. The conditions used will depend on the
precise nature of the groups R and R present in the compound
of formula (V) and a number of variations are possible. Thus for
example when both of Rl7 and Rl8 are benzyl, hydrogenation of the
product will yield the diacid of formula (I) wherein R and R are
both H. Alternatively if one of Rl7 and Rl8 is benzyl and the
other is alkyl, hydrogenation will yield a ~onoester product.
This can then be hydrolysed, if desired, to again yield the diacic
product. When Rl or Rl~ is t-butyl, treatment of the compound of
formula (~r) with trifluoroacetic acid or hydrogen
. . ; : .
WO 91/13054 ,j - ~ - P~/EY;~/0~5
2~?~7~5~ 12
chloride yields the corresponding acid. If some other carbo~ylic
acid protecting group is used for ~ or R then clearly
appropriate conditions for its removal must be e~ployed in the
final step to give the ester or diacid product of formula (f).
For example when R or R is trimethylsilylethyl it may be
removed by treatment with tetrabutylammonium fluoride. Any
protecting groups present in R and R must also be removed and
this may be performed concominantly with removal of protecting
groups present in R17 and R18 or as a
procedures appropriate to the particular protecting group
employed. Thus, for example when R contains a substituted or
protected amino group (for example a benzylamino, dibenzylamino,
benzyloxycarbonylamino or t-butyloxycarbonylamino group) the
compounds may be converted to the free amines by hydrogenation or
hydrolysis as appropriate.
.
(b) In an alternative process, compounds of the formula (I)
wherein R is R CONR - are prepared by a process which involves
reacting an amine of the formula:
~ A ~ Rl
R NH-~ ~ C J / R
CHCH CONH-CH
R170 C / 2 \ CO2R
(~Il)
I
W O 91/~3054 ~73~L~ ` PCT/EP~1/00296
13
wherein A, Y, R , R , R , Rl and R are as previously defined;
with a carboxylic acid or acid chloride of the formulae:
':
:
Rl0co~H or Rlcocl
~ .
wherein R10 is as previously defined, and wherein any reactive
groups therein are optionally protected, to yield for example a
compound of the formula: .
:
R6 A~ Rl
R C O N-Y \ C R3
CHCH2 CONH-CH
R 02C ~ RO 2R
(VII)
wherein Rl~ is as previously defined for R10 with any reactive
group.s therein optionally protected; and subsequently removing any
protecting groups, if present and hydrolysing the ester product to
yield the compounds of formula (I) wherein R and R are H. ~:
:: .
.
: . , ~,
W O 91/l3054 ' i PCT/EP91/O~?a.6
14
The reaction of the amine of formula (VI) and acid of formula
R C02H is achieved using conventional amide coupling techniques
a~ previously described. In the case where R1 is tR5)2N, the
acid cllloride may be prepared by first reacting the amine with
phosgene; subsequent reaction with the amine of formula (VI)
yields the urea products. Subsequent removal of protecting groups
is achieved using appropriate procedures as previously described.
The am1nes of formula (VI) are prepared following the same
procedure outlined in process (a) above but using an acid of
formula (III) wherein R? is a protected amine. Thus, in one
~ariant of this process, the coupling reaction with the amino acid
derivative is achieved using a compound of formula (III) wherein
R2 ls dibenzylamino or di(alpha-methylbenzyl)amino .
Hydrogenation of the coupled product of formula (V) gives the
amine of for~ula (VI) wherein R is H.
c) Certain compounds of formula (I) are best prepared by routes
which involve coupling a precursor stage to give a diester
derivative of formula (V) type, which is then subjected to a
chemical transformation reaction to give the particular R2 group
desired.
Thus for example the coupling process may be performed using
an acid of formula (III) wherein R is halo (e.g. bromo). This
may then be reacted with an amine of formula (R )2NH to yield the
compounds wherein R is (R )2N-, or with an azide to give
compounds wherein R- is N3. Subsequent reduction of the azide
group by catalytic hydrogenation, either on the protected prodvct
or on the deprotected diacid, gives the corresponding amine o~
formula (I) wherein R is NH2.
j
W O 91/130~4 ~ PCT/EP~/00296
-,~ s 15
In another variation, an acid of formula ~III) is used
wherein R Y- is 2-propenyl. Reaction of the coupled product of
formu]a (V~ with a (Cl-C6)alkanol or (Cl-C,)alkoxy(Cl-C6)alkanol
in the presence of mercuric acetate and potassium iodide yields
the 3-iodo-2-~(Cl-C6)alkoxy]- or 2-[(~l-c4~alkoxy(cl-c6)alk
propyl derivative, subsequent reduction (e.g. with tributyltin
hydride) yields the corresponding compound of formula (V) wherein
Y is propyl and R is ~Cl-C6)alkoxy or (Cl-C4)alkoxy(Cl-C6)alkoxy
attached at the 2-position. In a further variant of this process
the iodo intermediate can be reacted with a heterocyclic compound
(e.g. imidazole) to yield the compound of formula (V) wherein R
is of formula R R CH, R is heterocyclyl(Cl-C4)alkyl and R is
t~l-C6)alkoxy.
Compounds of the formula (I) wherein Z is present and is S(O)
or S(0)2 can naturally be prepared by oxidation of the
corresponding thio derivatives where Z is S. The oxidation may be
performed either on the protected diester formula (V) or on the
deprotected diacid product. Appropriate reagents will be well
known to those skilled in the art but, for example, metachloro-
peroxybenzoic acid in excess can be used to give the corresponding
sulphones (Z = SO2) or an equimolar amount of sodium metaperiodate
to give the sulphoxides (Z = SO).
Appropriate reagents and conditions for all of the above
transformations will be well known to those skilled in the art by
reference to standard text books and to the examples provided
hereafter. Other variations and possibilites for the convenient
svnthesis of the range of R substituents will also be evident to
those skilled in the art and the above are representacive of some
of the variations which are possible.
:
, . ,
;. . : ~.: ,
.
:, ' ' . ' ' ', ' ' ' ! ' , . . . : ' :
W O 91/13054 : PCT/EP91/00 ~
~7~
16
Compounds of che formula (I) wherein one or both of R and R
is a biolabile ester-forming group are prepared following similar
procedures to those outlined above using the appropria~te ester
group for R or R .
As well as removing any protecting group which may be present
in R , a number of chemical transformation reactions are possible
on the final mono-ester or diacid products as previously
described. In each case the product ~ay be obtained as the free
carboxylic acid or it may be neutralised with an appropriate base `
and isolated in salt form.
Appropriate coupling and protecting methods for all of the
above steps and alternative variations and procedures will be ~ell
known to those skilled in the art by reference to appropriate text
books and to the examples provided hereafter.
The starting spiro-substituted glutaric acid ~ono esters
of formula (III) may be prepared as described in our European
patent application 274234. The amino acid esters of formula (IV)
are generally known compounds which are either commercially
available or they may be prepared by standard methods in
accordance with literature precedents.
As previously mentioned, the compounds of the invention are
potent inhibitors of the neutral endopeptidase (E.C.3.4.24.ll).
This enzyme is involved in the breakdown of a nu~ber of peptide
hormones including 9 in particular the breakdown of atrial
natriuretic factor (ANF?. Thus, the compounds of the invention,
by preventing the degradation of ANF by endopeptidase
~.C.3.4.24. 11, can potentiate its biological effects and the
compounds are thus diuretic, natriuretic and antihypertensive
~ 9l/l3054 17 ~7q~5~ PCT/EP9l/00~96
agents of utility in a number of disorders incl.uding hypertension, ;~
heart failure, angina, renal insufficiency, premenstrual syndrome,
cyclical oedema, Menieres disease~ hyperaldosteroneism ~primary
and secondary) and hypercalciuria. In addition, because of their
ability to potentiate the effects of ANF the compounds have
utility in the treatment of glaucoma. As a further result of
their ability to inhibit the neutral endopeptidase E.C.3.4.24.11
the compounds of the invention may have activity in other
therapeutic areas including for example the treatment of asthma,
inflammation, pain, epilepsy, affective disorders, dementia and
geriatric confusion, obesity and gastrointestinal disorders
(especially diarrhoea and irritable bowel syndrome), the
modulation of gastric acid secretion and the treatment of
hyperreninaemia.
Activity agalnst neutral endopeptidase E.C.3.4.24.11 is
assessed using a procedure based on the assay described by J. T.
Gafford,.R. A. Skidgel, E. G. Erdos and L. B. Hersh, Biochemistry,
1983, 32, 3265-3271. The metbod involves determining the
concentration of compound required to reduce by 50~ the rate of
release of radiolabelled hippuric acid from hippuryl-L-
phenylalanyl-L-arginine by a neutral endopeptidase preparation
from rat kidney.
As previously mentioned, the compounds of the invention are
also inhibitors of angiotensin converting enzyme. As such they
are useful in tre~ting a further variety of conditions for which
A~E inhibitors are known to be useful including limitation of
ischaemic damage to the myocardium, protection of ~he kidney
against hyperfiltration damage, prevention or reversal of left
W O 91/13054 ~ PCr~EP~I/00
18
ventricular hypertrophy, memory enhancement, control of cognitive
function, dementia, and preventing reocclusion following coronory
angioplasty or coronory artery bypass surgery. Their activity
against this enzyme is assessed using a modified procedure based
on the assay described by Rohrbach, ~I.S., Anal. Biochem., 1978
84, 272. The metho~ involves determining the concentration af
compound required to reduce by 50% the extent of release of
radiolabelled hippuric acid from hi~puryl-L-histidyl-L-leucine by -~
angiotensin converting enzyme isolated from the rat kidney.
Inhibitory activity is also measured in vlvo following
intravenous injection to anaesthetised rats using the methods
described by I. L. Natoff et al, Journal of Pharmacological
Methods, 1981, 5, 305 and by D. M. Gross et al, J. Pharmacol. Exp. ;
Ther., 1981, 216, 552. The dose of inhibitor required to reduce
; the pressor response produced by intravenous injection of
angiotensin I (50 ng bolus) by 50% is determined.
The activity of the compounds as diuretic agents is
determined by measuring their ability to increase urine output and
sodium ion excretion in saline loaded conscious mice. In this
test, male ~ice (Charles River CDl, 22-28 g) are acclimatised and `-
starved overnight ln metabowls. The mice are dosed intravenously
via the tail vein, with the test compound dissolvéd in a volume of
saline solution equivalent to 2.5% of body weight. Urine samples
are collected each hour for two hours in pre-weighed tubes and
analysed for electrolyte concentration. Urine volume and sodium
ion concentration from the test animals are compared to a contro~
group which rec~ived only saline.
The antihypertensive activity of the compounds is evaluated
.. . . .
i : :
,C ~3~
0 91/13054 P ~ /EP91/a~296
19, ' ' - :"
by measuring the ~all in blood pressure following oral or
intravenous administration to salt depleted, diuretic primed,
spontaneously hypertensive rats, salt depleted renall~
hypertensi~re do~s, or DOCA/salt hypertensive rats.
For administration to man in the curative or prophylactic
treatment of hypertension, congestive heart failure or renal
insufficiency, oral dosages of the compounds will generally be in
the range of from 3-lS00 mg daily for an average adult patient t70
kg). Thus for a typical adult patient, individual tablets or
capsules contain from 1 to 500 mg of active compound, in a
suitable pharmaceutically acceptable vehicle or carrier for
administration singly, or i.n multiple doses, once or several times
a day. Dosages for intravenous administration would typically be
within the range 1 to 500 mg per single dose as required. In
practice the physician will determine the actual dosage which will
be most suitable for an individual patient and it will vary with
the age, weight and response of the particular patient. The abo~e
dosages are exemplary of the average case but there can, of
course, be individual instances where higher or lower dosage "
ranges are merited, and such are within the scope of this
invention.
For human use, the compounds of the formula (I) can be
administered alone, but will generally be administered in ;~
admixture with a pharmaceutical carrier selected with regard to
the intended route of administration and standard pharmaceutical
prac~ice. For example, they may be administered orally in ~he
form of tablets containing such excipients as starch or lactose,
- or in capsules or ovules either alone or in admixture with
.. . .: , . . .. , . . , .. ... :
~; :
. .
e~cipients, or in the form of elixirs or suspensions
containing flavouring or colouring agents. The may be
injected parenterally, for example, intravenously, intra-
muscularly or subcutaneously. For parenteral administra-
tion, they are best used in the form of a sterile aqueoussolution which may contain other substances, for example,
enough salts or glucose to make the solution isotonic with
blood.
The compounds may be co-administered with other agents
as may be beneficial for the control of blood pressure or
the treatment of cardiac conditions or renal insufficiencyO
Thus for example they may be co-administered with digitalis
or another cardiac-stimulant drug or with an alpha-blocker,
beta~blocker, exogenous ANF or with a potassium channel
activator or another diuretic agent as shall be determined
by the physician as appropriate to the particular patient
or disease state.
Thus in a further aspect the invention provides a
pharmaceutical composition comprising a compound of the
formula (I) or (II), or a pharmaceutically acceptable salt
thereof or bioprecursor therefor, together with a pharma-
ceutically acceptable diluent or carrier.
The invention also includes a compound of the formula
(I) or (II), or a pharmaceutically acceptable salt thereof
or bioprecursor therefor, for use in medicine, in particular
in the treatment of hypertension, congestive heart failure
or renal insufficiency in a human being.
The invention also extends to a commercial package
containing, as active pharmaceutical ingredient, a compound
of the invention, together with instructions for its use in
medicine in particular for the treatment of hypertension,
congestive heart failure or renal insufficiency in a human
being.
The preparation of the compounds of the invention and
of intermediates for use in their preparation is illustrated
by the
F~
~ ~ .
,
.. ...
~ 91/13054 ; ~ ~ PC~/EP91/00296
~1 '
following Examples. Thin layer chromatography was performed on
silica plates using the following solvent systems: ethyl acetate, ¦
hexane, l:l(ss l); ethyl acetate, hexane, 1:3(ss-~); methyl :
isobutyl ketone, water, acetic acid, 2:1:1,(ss 3); diethyl ether,
hexane, 3:7(ss 4); ethyl acetate(ss 5); diethyl ether(ss 6);
dichloromethane, methanol, acetic acid, 80:20:1(ss 7), diethyl :::
ether, hexane, l:l(ss 8), heXane, ethyl acetate, 4:1 (ss 9), or :~
n-butanol, acetic acid, water, 12:3:5 (ss 10)
.
W O 91/130~4 PCT/EP9D~0~ ~
` 2~7~5~ t~
22
EXAMPLE 1
_ _ _ _ ~
1-(2-t-Butoxycarbonyl___ ethoxybutyl)cyclo~ntane carbox lic acid
____. __ Y
A solution of t-butyl 3-(1-carboxycyclopentyl~propanoate (300
g, 12.38 mmol) in dry tetrahydrofuran (10 ~1) was added to a
stirred solution of lithium diisopropylamide (26 mmol) in a
mixture of hexane (10.4 ml) and tetrahydrofuran (45 ml) at -70C
under nitrogen. After 1 hour a solution of 2-iodo-1-ethoxvethane
(4.95 g, 24.8 m~ol) in dry tetrahydrofuran (10 ml) was added
maintaining the temperature at -70C. After an hour at that
temperature, the solution was allowed to warm up to room
temperature overnight and left for a further two days. The
mixture was then acidified to pEI 2 with 2N hydrochloric acid, and
extracted with ether. The organic extract was washed with brine,
dried (MgS04) and the solvent evaporated to give the crude acid
which was chromatographed on silica. Elution with increasing
proportions of ethyl acetate in hexane (1:4 to neat ethyl acetate)
gave an oil (2.5 g, 55%). Rf 0.15 hexane, ethyl acetate, 401)o
Found: C,65.10; H,9.36. C17H3005 requires C,64.94; H,9.62%.
EXAMP~ES 2-8
The following compounds were prepared by alkylating
l-carboxy-cyclopentylpropanoic acid t-butyl ester using the
appropriate bromo or iodocompound following the procedure of
Example 1.
~Y r~ ~
Bu02C / \/ \ C()~i
.; ~ . . ,, .......... .
. ~ ......... ~ .. .... . ; . .
W O 91/13054 PCT~EPgl/00~96 .
23
, ; :
Example R2~Y- For~ I Analysis ~
No isolated (Theoretical in brac~ets)
T.L.C. C H N
. _ .
2 / oil 64.43 8.82 :~.
o ~ _OCH2-Rf 0.3 (64.02 9.05)
/(ss 1),
__ . __. . . I
3 CH30(CH2)40(cH2)2 oil , 64.24 9.90 ?
Rf 0.45 ¦ (64.49 9.74) `~.
. (5sl) . .':
_ __ _ . ~ '
4 CH30CH2CH=CHCH20(CH2)2 oil 62.36 8.83
Rf 0.45 (62.62 8.95)
¦ (S5 1) (O. 2 mole CH2C12)
. ! !~ ~
Br(CH2)6- ¦ oil ~1 .
Rf 0.5 _
. I(ss 2)
; ' .
.
W O 9IJ130~4 PCr/EP9l/00 ~
~7~5~) ~
~4
6 ,, I oi~. 61.86 8.~q 3.36, ,
~ ¦ Rf 0.4 (61.79 8.93 2.80)~ :
BOC-- N ~-~C~z~ j
~ (ss 1) (Q.25 mole ~H2Cl~
7 Br(CH2)8- oil 58.08 8.60
. . Rf 0.9 t58.15 8.60)
. (ss 3)
: 8R19OCH2 ~ -O(CH2)2 oil 65.36 8,97
. . Rf 0.9 (65.20 9.19)
(ss 1) (0.5 mole H20)
_ ... ~ . . _ _._ .
BOC = t-butoxycarbonyl
R 9 = dimethyl-t-butylsilyl
EXAMPLE 9
1-[2(RS)-t-Butoxycarbonyl- _-azidodecyl~ cyclopentane carboxvlic
acid
Tetramethylguanidinium azide (0.7 g, 6 mmol) was added as a
solution in chloroform (10 ml) to a solution of 1-[2(RS)-t-
butoxycarbonyl-10-bromodecyl]-1-cyclopentane carboxylic acid (1.4
g, 3 mmol) in chloroform (10 ml). A few crystals of potassiu~
iodide were added and the resulting mixture was refluxed for 2
days. The cooled reactlon mixture was ehen diluted with
:~
~ O 91~13054 2~3~ 5~ ~ PCT/EP9l/00296
chloroform (20 ml~ and washed with water (2 x 20 ml), dried
(MgS04), filtered and evaporated. The residue was chromatographed
on silica gel using a gradient of ethyl acetate and hexane to give
the title compound as an oil (0.69 g, 54%). Found: C,63.84;
H,9.41; N,9.22. C2lH37N304 requires C,63.77; H,9.43; ~,10.62%o
Rf 0.9 (ethyl acetate).
` ~ .
EXAMPLE 10
acid
A mixture of 1-(2(R,S)-t-butoxycarbonyl-4-pentenyl)cyclo-
pentane carboxylic acid (EP-A-0274234, Example 44) (28.24 g, 0.1
mmol) and (+)- pseudoephedrin were recrystallised three times from
hexane to give a white crystaline solid (18.9 g, 42~3 m.p.
106.5-107.5C. 1~]DS+ 20.5 [~]26S + 70.7 (c = 1.07, methanol).
The above salt was dissolved in ether and washed with lN
hydrochloric aeid and saturated salt solution. Drying (MgS04) and
evaporation gave the requlred R-acid as an oil (ll.9 g). Rf 0,7 ~;
(ethyl acetate, hexane, 1:1). [~i~D~- 11.8, [dl~655 _ 36.5 (c =
1.03, methanol).
,'
XA~PLE ll
1-[2~ _)-t-Butoxycarbonyl-4-pentenyl]-1-cyclopentane carboxylic
acid benzyl ester
. . . _ _ . _
Anhydrous potassium carbonate (13.5 g, 98 mmol) was added in
one portion to a stirred and ice-cooled solution of 1-[2(RS)-t
butoxycarbonyl-4-pentenyl]-l-cyclopentane carhoxvlic acid (]0.95
g, 38 mmol~ and benzyl bromide (~.6 g, 38 mmol) in dry dimethyl-
formamide (30 ~Il). After 3 hours the reaction mixture was diluted
" :' , ' ' . ' : ' '~, : . ,' ' ' : : :, . ' :' : ' ' ' . :, . , : . ' . ' , ' ' : : ' : ' '
W O 91/13054 '. ; PCT/EP9~ ~
2~3~5~ `26
with ethyl ace~ate (100 ml) and water (100 mi). The or~anic phase
was separated and washed with further por~ions of water (1 x 20
ml) lM hydrochloric acid (5 x 20 ml), brine (1 x 10 ml), dried
(MgS04), and the solution filtered and evaporated to yield the -
title compound as an oil (14.14 g, 98%). This oil was used
without further purification. Rf 0.86 (ethylacetate, toluene~ ¦
1:4).
EXAMPLE 12
1-[2(RS)-t-Butoxycarbo y -4-oxobutyl~ yclopentane _arboxylic
a _d benzyl ester
Osmium tetroxide (56 mg, 0.2 mmol) was added as a 2.5% w/v
solution in t-butanol (2.25 ml) to a stirred solution of 1-[2(RS)-
t-bu~oxycarbonyl-4-pentenyl]l-cyclopentane carboxylic acid benzyl
ester (8.25 g, 22 mmol) in acetonitrile (60 ml) and water (10 ml)
at room te~perature. After 30 minutes, the black-brown solution
was treated with sodium metaperiodate (10 g, 47 mm:l) in one
portion. Stirring was continued for 18 hours and then the
suspended solid was filtered and washed with a small portion of
acetonitrile. The filtrate was evaporated and the dark residue
was chromatographed on silica gel eluting with a gradient of ethyl
acetate and hexane to yield the title compound as an oil (3.77 g,
45%). Rf 0.58 (diethyl ether, petrol, 1:1).
,, : ~. , ,~. :. . - ,. . . , : . ::
I;
0 91/13054 ~ PCT/E ~1/002g6
EXAMPLE 13
_-[2(~S)-t-Butoxycarbo_v1-3-carboxy~ opyll~l-cyclopentane
carboxylic acid benzyl ester
_ ..
Potassium permanganate (7.6 g, 48 mmol) as a solution in
water (48 ml) was added slowly to a stirred solution of l-[2(RS)-
t-butoxycarbonyl-4-oxobutyl]-1-cyclopentane carboxylic acid benzyl ~
ester (51 g, 12 mmol) in t-butanol (70 ml) and lM sodium ~;
dihydrogen phosphate buffer solution (20 ml). The resulting
mixture was stirred vigorously for 30 minutes. Excess oxidant was
destroyed by the addition of solid sodium metabisulphate t8 g).
The brown suspension was filtered, the filter pad being washed
with ethyl acetate (50 ml). The filtrate was separated into two
phases and the aqueous phase was extracted with ethylacetate (30
ml). The combined organic solutions were dried (MgS04), filtered
and evaporated to give the title compound as aD oil t4.52 g, 96%).
Rf 0.7 (ss 5j.
' '
EXAMPLE 14
N-~ 3-[4-(t-Butoxycarbonylaminomethyl)b~nzoylamino]-2(S)-t-
butoxycarbonylpropyl~-l-cyclopentanecarbonyl~-O~t-butyl-(S)-
osine-t-butyl ester
To an ice-cold solution of 4--t-butoxycarbonylaminomethyl-
benzoic acid (0.689 g, 2.74 mmol) in dry dichloromethane (30 ml)
was added l-hydroxybenztriazole (0.409 g, 3.02 mmol), l-ethyl-3-
(dimethylaminopropyl)-carbodiimide (0.684 g, 3.57 mmol~ and N-
methylmorpholine (0.360 g, 3.57 mmol) and the resulting solution
stirred at 0C for 30 minutes. N-[1-(3-Aminopropyl-2(S)-t-
butoxycarbonyl)-l-cyclopeneanecarbonyll-0-t-butyl-(S)-tyrosine
: ' ' . ',, . : .', . ' . ' . : ': ' ' ' ': ., ': . ' : `,; , ;.,: ': .: .,. ' ' :: :,' : ' : . :; ' : :. : ' ' ~ . ' , . ':: '
' ' ' : : : : ' '- ' ; . : ' ' ' : : : : : . .: . . : , " : : ,. .; ,:,' ' ', , .:,: ' . ,. , :
W O 91/13054 ~` i PCT/EP91/00 ~
2@7~5~ 28
t-butyl ester was ad~ed to this solution and stirred at room
temperature for 24 hours. The solvent was evaporated under
reduced pressure and the resultant oil dissolved in ethyl acetate
(~0 ml) and washed with 2M hydrochloric acid (50 ml), saturated
aqueous sodiu~ bicarbonate (2 ~ 5~ ~1) and saturated brine (50
~1), dried (MgS04) and the solu~ion fi].tered and evaporated to
yield the crude product as an oil. Chromatography on silica gel
using dichloromethaneldieehyl ether as eluent gave the title
compound (1.695 g, 79%) as a white foam. Rf 0.52
(dichloromethane, diethyl ether, 1:1). Found: C,67.52; H,8.47;
N,5.26. C44H65N309 requires C,67.75; H,8.40; N,5.39%
EXAMPI,ES 15-18
The following compounds were prepared following the procedure
of Example 14, using the appropriate carboxylic acid derivative of
formula R C02H.
~ ( 3)3 ..
R CONH ~ ~
(CH3)3CO,~C CO~H C02C(Cl'.3)3
W O 91/13054 2 ~ PCT/EP91/00296
29 ~
. _
Example¦ RlO¦ Form Analysis %
No isolated (Theoretical in brackets)
T.L.C. C H N
. _ '~ `
foam 69.21 8.70 5.65
(CH3)2NCH2- ~Rf 0.38(69.55 8.68 5.93) ~ ~;
~ (ss 3) `
16 foam 67.57 8.22 3.94
CH3SCH2- ~ (f505)5(67.25 8.08 4.02)
17 CH3S02NH foam 58.52 8.00 5.39
(S~CH-Rf 0.83(58.78 8.13 5.56)
CH3SCH2CH2(ss 5)
_ _ _
18 CH30CH2CH20C\H2 foam 64.70 8.89 3.69
CH-0.29 (64.37 8.96 3.66)
_ CH30CH2CHOCH2 _ . ~ :
,
.
W O 91/13054 ` ` PCr~EP9~0
EXAMP_E 19
N~ ~ thoxy-2-(t-butoxYcarbonyl)butyl~-cyc'opentarlecarbonyl]-
-0-methyl-(S)-t~rosine t-butvl ester
l-Ethyl-3-(3-dimethylaminopropyl)carbodiimi~e hydrochloride
(762 mg~ 3.98 mmol) was added to an ice cold stirred mixture of 1-
(2-t-butoxycarbonyl-4-ethoxybutyl)cyclopentane carboxylic acid
from Example 1 (626 mg, 1.99 mmol), 0-methyl-(S)-tyrosine t-butyl
ester (500 mg, 1.99 mmol), l-hydroxybenzotriazole (269 mg, 1.99
mmol) and N-methylmorpholine (0.44 ml, 3.98 mmol) in dry
methylenechloride (10 ml). After standing at room temperature for
16 hours the mixture was diluted with methylene chloride, washed
with water, dried (MgS04) and evaporated under reduced pressure to
give an oil. Chromatography on silica gel, eluting with a mixture
of hexane and ethyl acetate (4:1) gave the pure diester (975 mg;
89%) as an oil. Found: C,67.54; H,8.74; N,2.13. C31H49N07. 0025
H20 requires C,67.42; H,9.03; N,2.53%.
.... . . ., :
EXAMPLES 20-29
The following compounds were prepared as described above
using the appropriate cyclopentane carboxylic acid of Examples
1-6 and 8-10 in the coupling step together with the appropriate
amine of formula (IV).
~ ~lo
R2
(C~3)3co~c ~ ~ ~ C0~ ~ C0~?~
Examples 25 and 26 are the 2(R) isomers, Example 28 is the 2(~)
isomer.
2~3f.~.5~
WO 91tl3054 PCI`/EP91/00296
31
~ a7_ ~ r~ O ~0
a~z ~ ~ ~ ~r ~ ~ ~ co
C`l
U~ o '~
a o c~ c~ ~ u~ ~ o ~ o c~
~ 1 S ~ ~ ~ oo ~ c~l ~ m 1~ co
3 ~I co o al ~ oo o~ oo ~ o~ oo oo
o ~ ~ ~ o ~r ~ ~ O Lff ~ o ~ ~ e
.c ~ ~r _ ~ ~ ~ ~ o ~ ~ ~
E~ ~ ) -~o ~ I_ r~ ~ ~ ~ ~ c~ ~ ~ :,~_ `D ~D ~ ~ ~ ~ ~O ~O ~O ~D ~ ~D O .,
, _ ._
~ ~ ~ ~ u~
- ~ ~ -
O ~D O ~ O r. O ~ O ~ O
11 ll ll 1
~: ~ ~:; ~ P:: ~ '~
. _ .__ .._~ ._ . _
t l l ~ l ~ ' :; '
~ r-~ ~ ~ ~ ~ ~ ~.
~ s~ m~ ~ ~ I ~
_ , ~ .
o ,o I
D~ C~ m~ ~ ~
. _ _ ,, I -- -
'` ,IC~ ~ I ;~
._
x~z ~o 1~ 1'`' 1'~' 1
.
WO 91/130S4 ` . PCI~/EP91/0 1 :
7~5~ 32
1,
. ~ _.
~^ ~ ~
~ ~ C~l ~ ' ' '.~
~ ~ . ~ ,~ l ':
. oo CO . : ~
. ~o o ~o o , ;.
,
.. _ _ . ~``'
-;r u~ ~ a~ :'
In ,~ ~ ~ ~n
o~ o~o o~ o~
.
~ C~ ~ P~
--~
o ~ i ~.
i
+~
. ,,, o I o~; :
~ z~
l~
91J13054 '~? ~ PCT/EPg~/0~29
33
EXAMPLE 30
N-[1-~2(S)-t-Butoxvcarbonyl)-5-iodo-4-(2-methoxyethoxypentvl~-]-
cyclopentanecarbonyl]-0-t-butyl-(S)-tyrosine t-butyl ester
Mercuric acetate (662 mg, 2.08 mmol) was added to a stirred
solution of the diester from Example 26 (892 mg, 1.6 mmol) in
~-methoxyethanol (4 ml). After 20 hours at room tempera~ure, 2N
sodium hydroxide (3.6 ml) was added followed by potassium iodide
(2.9 g, 17.5 mmol) dissolve~ in water (3 ml~. The mixture was
diluted with water, extracted with diethyl ether and the extract
washed with water. Drying over MgS04 and evaporation gave an oil
which was dissolved in carbon tetrachloride. Iodine (406 mg, 1.6
mmol) was àdded and the solution stirred for six hours. The
mixture was then filtered, evaporated under reduced pressure and
the residue chroma~ographed on silica. Elut.ion with a mixture of
diethyl ether and hexane (4:6) gave the title product as an oil
(620 mg, 51%). Rf 0.35 (diethyl ether, hexane, 1:1).
EXAMPLE 31
N~ 2(S)-t-Butoxycarbony )=5-iodo-4-(2-methoxyethoxypen
-l-cyclopetanecarbonyl]-0-methyl-(S)-tyrosine _-but~ ester
The title compound was prepared from Example 25 using the
procedure of Example 30 above. The product was obtained as an oil ~;
(57% yield). Rf 0.28, (diethyl ether, hexane, 1:1). Found:
C,55.60; H,6.99; N,1.92. C33H52IN08 requires C,55.23; H,7.30;
N,1.95%.
W O 91/13054 ,~ ~ P ~ /EP9l/00
.~ 34
F,XAMP~E 32
N~ 2(S)-(t-~utoxYcarbonyl)-4-(2-methoxyethoxy)~entyl~-1-cyclo-
. _ _ . _ . . . .. . . ~ ~ _ . ... _ _ ., . .. . _ _ . _ . _ _ . _ .
~ent neca bonyl_-0-t-butyl-(S)-tyrosine t-butyl_ester
Tributyltin ~Iydride (0.5 ml, 1.86 mmol) was added to a
solution of the iodo compound from Example 30 (590 mg, 0.78 mmol)
in dry tetrahydrofuran (ln ml). After heating at 55C for 16
hours, the solution was diluted with diethyl ether, washed with
dilute potassium fluoride solution (x4) and then with saturated
salt solution. Drying (MgS04) and evaporation under reduced
pressure gave an oil which was chromatographed on silica. Elution
with a mixture of diethyl ether and hexane (1:1) gave the required
product as an oil (410 mg, 83%). Rf 0.32 (ether/he~ane 1:1).
Found: C,68.00; H,9.25; N,2.25. C36H~gN0~ requires C,68.21;
H,9.38; N,2.21%.
EXAMPLE 33
N-_l-~?(S)-(tert-Butoxycarbonyl)-4(2-methox ethoxy)pentyl~-
l-cyclo entanecarbonyl]-0-methyl-(S)-tyrosine tert-butyl ester
A solution of the iodo compound from Example 31, (430 mg, 0.6
mmol) in ethyl acetate (30 ml) containing triethylamine (67 mg,
0.66 mmol) was hydrogenated over 10~ palladium on charcoal at room
temperature at 50 p.s.i. (3.45 bar! pressure. The mi~ture was
filtered, the solvent evaporated and the residue chromatographed
on silica to give the title compound as an oil (300 mg, 85~) Rf
0.2 (diethyl ether, hexane, 1:1).
:- , :. ... . : . ..
~ W O 9l/13054 ~g~ PC~/EP~1/00296
................................................. ~
3~
EXAMPLE 34
__ .
N~ (S)-(t-Butoxycarbonyl)-5-(1-imidazolyl)-4-metho~ypentyl}-1-
cyclopentanecarbonyl]-0-methyl-(S)-tyrosine t-butyl ester
a) Mercuric acetate (797 mg, 2.5 mmol) was added to a solution
of the diester of Example 25 (1.14 g, 2.21 mmol) in dry methanol
(5 ml). After stirring for an hour at room temperature, 2N sodium
hydroxide (5.5 ml) was added followed by potassium iodide (1.83 g,
11.05 mmol) dissolved in water ~2.5 ml). Water was added and the
mixture extracted with ether. The organic extract was washed with
water, dried (MgS04) and evaporated to give an oil, which was
dissolved in carbon tetrachloride. Iodine (558 mg, 2.2 mmol) was
added and after stirring at 0C under nitrogen for 1.5 hours, the
solvent was evaporated under reduced pressure. The residue was
chromatographed on silica, eluting with a mixture of diethyl ether
and hexane (4:6) to give N~[1-~2(S~-(t~butoxycarbonyl)-5-iodo-4-
methoxypentyl~-l-cyclopentanecarbonyl~-(S)-0-methyl-tyrosine
t~butyl ester as an unstable oil (1.27 g, 85%). Rf 0.37 (diethyl
ether, hexane, 1:1) which was used directly for the next step.
b) A solution of the above iodide (620 mg, 0.92 mmol) and
imidazole (627 mg, 9.2 mmol) in acetonitrile (12 ml) was refluxed
for 24 hours. Evaporation of the solvent under reduced pressure
gave a gum ~hich was chromatographed on silica. Elution with a
mixture of ethanol and ethyl acetate (l:l9) gave the title product
as a gum. Rf 0.4 (ethanol, ethyl acetate, 1:9). Found: C,66.05;
H,8.22; N,6.80. C34H5lN307 r q
W O 91/13054 ` : PCTIEP91/00 ~
2~?73f~ 36
EXAMPLE 35
N-Fl-~8-Morpholino-2-(t-butoxycarbonyl~oct ~ -1 cyclopentane
, ,, ,. ., ... .. ~ . i
carboxy]-(S)-0-methv]tvrosine t-butyl eseer
. _ . _ _... ~ .. .,.. ... .... _ _ _ . _ _ _ I
The bromo compound from Example 23 (1.2 g, 1.88 mmol) and
morpholine (650 mg, 7.5 mmol) were heated at 50C in dimethyl-
formamide (lS ml) containing potassium iodide (120 mg). After 20
hours, water was added and the mixture extracted with ethyl
acetate. Washing with water, drying (MgS04) and evaporation gave
an oil (1.2 g) which was chromatographed on silica. Gradient
elution starting with hexane, ethyl acetate (7:3) progressing to
neat ethyl acetate and finally with ethyl acetate, ethanol, (95:5)
gave the required compound as a gum (1.0 g). Rf 0.3 (hexane,
diethyl ether, 1:1). Found: C,68.10; H,9.50; N,4.14. C37H60N207.
0.5 CH3C02C2H5 requires C,68.09; H,9.37; N,4.30~.
EX PLE 36
N-[1-2(S?- - utoxycarbonyl-3-(diethylaminocarbonylamino)propyl-
l-cyclopentanecarbonyl]-0-methyl-(S)-tyrosine t-butyl ester
. _
Phosgene (72 mg, 0.7 mmol) as a 12.5% w/v solution in toluene
(0.6 ml) was added in one portion to a stirred and cooled solution
of diethylamine (85 mg, 0.65 mmol) and N-methyl morpholine (73 mg,
0.7 mmol) in methylene chloride (10 ml). After stirring at 0C
for 2 hours, nitrogen was bubbled gently through the solution to
remove excess phosgene. To this solution at room temperature was
added more N-methylmorpholine (73 mg, 0.7 mmol) and
~-~1-(3-aminopropyl-2(S)-c-butoxycarbonyl)-l-cyclopenrane-
carbonyl]-0-e-butyl-(S)-tyrosine t-butyl ester (360 mg, 0.65 mmo])
~VO 91/l30 Y 2072~50 rcr/~ "~" ll
- 37 '~
as a solution in methylene chloride (3 ml). After stirring at
room temperature overnight, the solvent was evaporated and the , ~
residue was dissolved in ethyl acetate (15 ~1). This solution was ~ ;
washed with water (2 x 5 ml), lM hydrochloric acid (2 x 5 ml)~
saturated sodium bicarbonate solution (1 x 5 ml), brine (l x 5
ml), dried (MgS0.), filtered and evaporated. The residual oil was
chromatographed on silica gel using a gradient of methylene
chloride and methanol saturated with aqueous a~monia to give the
title compound as an oil (332 ~g, 82~). Rf 0.67 (ethyl acetate).
EXAMPLE 37
N-[1-(2(RS)-c-Butoxycarbonyl-3-~4-keto-1-piperidylcarbony~
propyl)-l-cyclopentanecarbonyl]-0 methyl-(S)-tyrosine t-butyl
ester
a) 4-Piperidone was coupled to 1-~2(RS)-t-butoxycarbonyl-3-
carboxypropyl]-l-cyclopentanecarboxylic acid benzyl ester (Example ~;
13) using the procedure of Example 14 to give 1-[2(RS)-t-butoxy-
carbonyl-3-(4-keto-1-piperidylcarbonyl~propyl]l-cyclopentane
carboxylic acid benzyl ester.
b) The above product was dissolved in an ethanol, water mixture
(9:1) and hydrogenated at room temperature under an atmosphere of
hydrogen (60 p.s.i., 4.1 bar) over 10~ palladium on carbon for 3
hours. The reaction mixture was filtered, and the filtrate
evaporated to dryness. The residue was azeotroped with
dichloromethane to yield 1-(2tRS)-t-butoxycarbonyl-3-(4-ke~o-l-
piperidylcarbonvl~propyl)-l-cyclopentane-carboxvlic acid as a
foam. Rf 0.21 (ethyl acetate). Found: C,62.42; H,8.02; N,3.67.
C34H50N208 requires C,62.97; H,8.19; N,3.67~.
; . . : : :. ,:. ::, - . , . ,. . '
W O 91/13054 : PCr/EP91/OD
~ 38
c) The above acid was coupled to 0-methyl-(S)tyrosine-t-butyl
ester following the procedure of Example 19 to yield the title
product as a foam, Rf 0.71 (echyl acetate). I
EXAMPLE 38
.. ... _ I
N~ (2(RS)-t-Butoxycarbony1-3-(4-hydroxy-1-piperi_inylcarbonyl)
propyl)-l-cyclopentanecarbo~]-0-methyl-(S)t~rosine t-bu yl ster t
lM Hydrochloric acid was added to a stirred ice~cooled
solution of N-[1-(2(RS)-t-butoxycarbonyl-3-(4-keto-1-piperidyl-
carbonyl)propyl)-l-cyclopentanecarbonyl] 0-methyl-(S)-tyrosine t-
butyl ester (400 mg, 0.6 mmol) in ethanol (3 ml~ and water (2 ~1) -
to adjust the pH of the solution to between 4 and 6. More ethanol
was added as necessary to maintain a homogeneous solution. To the
above solution was added sodium cyanoborohydride (45 mg, 0.7 m~ol)
in one portion. The resulting mixture was allowed to warm to room
temperature overnight. The ethanol was evaporated and the aqueous
residue was partitioned between ethyl acetate (20 ml) and water 20
(ml). The organic phase was separated and washed with water (1 x
lG ml), lM hydrochloric acid (2 x 10 ml), saturated sodium
bicarbonate solution (1 x 10 ml), brine (1 x 10 ml), dried (MgS04) ,
and filtered. The filtrate was e~aporated and the residue was
chromatographed on silica gel eluting with a gradient of ethyl
acetate and hexane to yield the title compound as a gum (160 mg,
38%). Rf 0.29 (ethyl acetate).
.. : ,, ,., ,, . ; ~ ~ . , : ::: :, ~ :: : .: . .
W O ~1/13054 PCTiEP91/00296
~i~73~50 - ~
39
_AMP~E 39
N-~1-(2(RS?-t-Butoxycarbonyl-3-(4-di~ethylamino-1-piperidyl-
carbonyl)propyl)-l-cyclopentanecarbonyl]-0-methyl-(S)tyrosine t-
:
butyl ester
Sodium cyanoborohydride (60 mg, 0.9 m~ol) was added in one
portion to a stirred and ice-cooled mixture of N-[1-(2(RS)-t-
butoxycarbonyl-3-(4-keto-1-piperidylcarbonyl)propyl)~l-cyclo- z
pentanecarbonyl]-0-methyl-(S)tyrosine t-butyl ester (550 mg, 0.9
mmol) from Example 37), anhydrous sodium acetate (730 mg, 9 mmol,
and dimethylamine hydrochloride (360 mg, 4.5 mmol) in dry methanol
(15 ml). The mixture was allowed to warm to room temperature.
After 24 hours, the solvent was evaporated and the residue
partitioned between ethyl acetate (50 ml) and water (30 ml). The
organic layer was separated and washed with water (1 x 10 ml),
saturated sodium bicarbonate solution (2 x 15 ml), brine (1 x 10
ml), dried (MgS04) and filtered. The filtrate was evaporated and
the crude residue was chromatographed on silica gel to yield the
title product as a gum (313 mg, 54%). Rf 0.32 (SS 3).
- EXAMPLE 40
N-[1-~2(S)-t-Butoxycarbonyl-3-(4-methanesulphonylmethylbenzoyl-
amino)propyl ~ -c
t-butyl ester
90% ~letachloroperoxybenzoic acid (0.29 g, 1.5 mmol) was added i:
in one portion to a stirred and ice-cooled solution of N-[l-
2(S)-t-butoxycarbonyl-3-(4-methylthiomethvlbenzoylamino)propyl
-l-cyclopentanecarbonyl]-0-t-butyl-(S)-tyrosine t-butyl ester.
(Example 1~, 0.38 g, 0.5 mmol~ in methylene chloride (10 ml). The
solution was allowed to warm to room temperature overnight. The
W O 9l/130S4 PC~tEP91/Ofl ~
;~7:~S~ 40
solvent was evaporated and the residue was dissolved in ethyl
acetate (20 ml). This solution was washed with saturated sodium
bicarbonate solution (2 x 10 ml), 10% aqueous sodium carbonate
solution (1 x 10 ml), brine (1 x 10 ml), dried (MgS04), filtered
and evaporated to give the title compound as a white foam (0.39 g,
100~). Found: C,63.21; H,7.46; N,3.58. C40H58N209S. H20
requires C,63.13; H,7.94; N,3.68%. Rf 0.66 (ethyl acetate).
EXAMP~E 41
N~ (2(S)-t-ButoxYcarbonyl-3-(2(S)-methanesul~honamido-4~methane-
sulphonylbutyrylamino?eropyl)-l-cyclopentanecarbonyl]-O-t-bu_yl-
(S)-tyroslne t-butyl ester
The title compound was prepared from Example 17 by oxidation
with metachloroperoxybenzoic acid following the procedure of
Example 40 and was obtained as a foam. Rf 0.63 (ethyl acetate).
Found: C,56.09; H,7.47; N,5.58. C H N S requires C,56.39; H,7~80;
N,5.33%.
EXAMPLE 42
N-~ 2-(t-Buto_ycarbonyl)-4-(4-hydroxymethylphenoxy)buty
cYclopentanecarbonylJ-O-methyl-(S)-tyrosine t-butyl ester
lM Tetrabutylammonium fluoride (2.16 ml) was added at 10C
under nitrogen to a stirred solution of the product of Example 29
(800 mg, 1.08 mmol) in tetrahydrofuran (10 ml). ~fter standing at
room temperature for 2~ days, the mixture was diluted with ethyl
acetate, washed with 0.5N hydrochloric acid followed by water,
dried (MgS04) and the solvent evaporated under reduced pressure.
The residue was chromatographed on silica, eluting with increasing
W O 91~3~S4 ~3~ PCr/EP91/O~g6
~ `. ` .
4 ~
proportions of ethyl ace~ate in hexane (from 3:7 to l:l) to gi~e a
clear oil (610 mg, 90%). Rf 0.4 (hexane, ethyl acetate, l:l).
Found: C,69.47; H,8.01; ~92.31. C36H5~N03 requires C,69 09;
H,8.21; N,2.24%.
EXAMPLE 43
N-[1-~2-(t-Butoxycarbony_)-4-(4-~bis-t-b_toxycarbonylaminome~hvl~-
phenoxy_butyl~-l-cyclopentanecarbonyl]-0-methyl-(S)-tyrosine
t-butyl ester
Di-t-butyliminodicarboxylate (333 mg, 1.53 mmol) and
triphenylphosphine (402 mg, 1.53 mmol) were added at 10C under
nitrogen to a stirred solution of the product of Example 42 (640
mg, 1.023 mmol) in tetrahydrofuran ~6 ml). Diethyldiazodi-
carboxylate (0.24 ml, 1.53 mmol) in tetrahydrofuran (2 ml~ was
slowly added dropwise and the mixture stirred for 20 hours at room
temperature. The mixture was then evaporated to dryoess, the
residue preabsorbed on silica and chromatographed. Elution with
increasing proportions of ethyl acetate in hexane (from 5:95 to
1:4~ gave the required product as a clear gum (166 mg, 20%) Rf
0.55. (hexane, ethyl acetate, 1:1). .
. ' I~ :: ; ' . . . . I ' . . ' " .' . ' . ' ' ' '
W O 91/13054 - PC~EP9~0 ~
2~ 5~ 42
EXAMPLE 44
N=E1-~2(S)-Carboxy-4-(2-methoxyethoxy)pentyl~-1-cyclopentane-
carbonyl]-(S)-tyrosine
Trifluoroacetic acid (5 ml) was added to an ice cold solution
of N-[1-~2(S)-t-buto~ycarbonyl)-4-(2-methoxyetho~y)pentyl~-cyclo~
pentanecarbonyl]-0-t-butyl-(S)-tyrosine t-butyl ester (from
Example 32, 390 mg, 0.62 mmol~ and anisole (998 mg, 9.2 mmol) in
dry methylenechloride (5 ml). After standing at 0C overnight the
solution was evaporated to dryness under reduced pressure and
dried azeotropically with toluene. The residual gum was dissolved
in ether and the product was extracted with lN sodium hydroxide
(10 ml). The basic extract was then acidified with concentrated
hydrochloric acid, saturated with salt and extracted with ethyl
acetate. Washing with brine, drying (MgS04) and evaporation gave
2 glass which was crushed to a white powder t275 mg). Rf 0.52 (ss
7). Found: C,61.22; H,7.52; N,2.99. C24H35N08, 0.1 CH3C02C2H5,
0.25 H20 requires Cj61.20; H,7.64; N,2.92%.
EXAMPLES 4 -53
The following compounds were prepared from the corresponding
0-methyl or 0-t-butyl tyrosine t-butyl ester, derivative as
2~?~5~
W O 91/~3054 ~CT/~P91/00296
43
appropriate by treat~ent with trifluoroacetic acid following the
procedure of Examp].e 44. Examples 49, 50, 52 and 53 were purified
by ion-exchange chromatography using Dowex AG 50W - X8 resin 2nd
eluting with 5% aqueous pyridine. Examples 51 and 52 are the 2S
isomers.
X A
~02C CO~H CO~H
WO 91/13il54 PCI`/EP91/00~
~ ~ ~J~ "~ 5!~ ) 44
_ ~ ~ O ~ A
._ ~ C~ ~ 0~C~ 1- ~- 1-
o Z ~ ~ ~ ~ C~l ~
~ .
h
r~ cr~ O ~ IC)~
~ O O 0~ ~S) CO C~ ~ V) ~ 00
) -- ~ ~D `D I_ ~ I~ I I I_
cr;: ~ o c~cJ~ u~ cO InU~
~1 ~ O ~ Il~~O U~ t~l
O O O 0~ ~ ~ ~ ~i (~ ~')
a) ~o ~ u~ u~ ~~D ~O ~O ~O ~D
~ ~. ~
`O ,,
1- 1~ 1_ :r'`l ,_
U~ ,_ l_
~) . o ~n o ~_ ~o ~ u
~ ~ ) E ~ E ~ v~ ~ ^ a) ~ ~_
E v . ~ ~ . ~ ~ t~ E . E ~ _, o
~ o ~ . o ~ . . :~ o :~ ~1 ~ U)
O ,~ . q~ O u O 00 ~ C~ r_ ~
O E~ ~ ~ 4~ ~ r~ .
1~ P: o ~ P~ ~ o o
. o ~ ~
I ~o ~ o 1~ :~ ~
1 1~
~ ~ ~ .
l 1~ '-'~ 1~ :~ .
~
i E~o u~ ~ ~ 0 llc~
ta æ ~ ~;r ~ ~ .~
X _ i i I ~ .
~WO 91/13054 45 ~?7~ pcr/Epgl~oo2g6
,,.`j. i ` .
~ , _~ ,_
~ oo ~ ~ U) C~ CO
o o oo , ~ ~ ~ ~
U~ U~ ~ C~ oo CO ~ .
o ~ CO ~ ~ ,o Co U~ ,~
~ CO ~ ~ ~ ~ ~D ~ ~ '
. , .
oo O ~ ~ a~ `D ~ I_
s~ ~ o o ~ ~ ~ ~ , .
~, ~
.
.
~n ~ r- . "~.
C`~ ~:r ~ 0~ ~ ~ u~ ~ ~
~ ~ o ~ ~ C4 ~ O O
o ~
i
~ o ~
1,, i I . I
W O 91/130~4 PC~/EP9l/0~ ~
i513 46
EXAMPLE 54
N~[1-(2(S)-_ r oxy-3-~ (S)-methanesulphonamido-4-methane-
sulphonyl-butyrylamino7propyl)-1-cyclopentanecarbonyl]-(S)-
tyrosine
.... ..
Hydror,en chloride gas was passed gent.Ly through a stirred and
ice-cooled solution of N-[1-(2(S)-t-butoxycarbonyl-3-i2(S)-
methanesulphonamido-4-methanesulphonylbutyrylamin~ propyl)-1-
cyclopentane-carbonyl]-0-t-butyl-(S)-tyrosine t-butyl ester (227
mg, 0.29 mmol) and anisole (470 mg, 4.3 mmol) in dry
dichloromethane (5 ml) until saturation was achieved. The
resulting solution was allowed to stand overnight at 0C. The
solvent and excess hydrogen chloride were evaporated under reduced
pressure and the residue was partitioned between ethyl acetate (19
ml) and saturated sodium bicarbonate solution (5 ml~. The organlc
phase was separated and extracted with more saturated sodium r
bicarbonate solution (2 x 5 ml). The combined aqueous extract~
were washed with ether (2 x 5 ml) and acidified to pH 3 with
concentrated hydrochloric aci.d in the presence of ethyl acetate
(15 ml). The acidic layer was separated and extracted with ethyl
acetate (5 ml). The combined organic solutions were dried
(MgS04), filtered and evaporated to give the title co~pound as a
white solid (140 mg, 78~). Found: C,47.32; H,5.86; N,6.39.
C25H37N3011S2. 0.75 H20 requires C,47.71; H,6.17; N,6.68X.
~3~S~)
91tl3054 : PCT/EP91/002g6
47 ,.
EXAMPLES 55-63
1'he following Examples were prepared from the corresponding
t-butyl protected compounds by treat~ent wi~h hydrogen chloride
following the procedure gi~ren in Example 54.
p~ 6
r~
2 COi~2 ~ CO A P,
Examples 55-58 and 62 are 2S isomers.
.:: : .: ; -
-
. , , :: . ::
WO 91/13054 48 PCr/EPgl~00~
Z ~7 3~ h ~ ~
_ _ .
U~ 0~ ~ ~D r- _~ ~OO
,~ ~ r~ ~ O u~ O O~r~
~ Z ~ ~ ~r ~ ~ . I~ r~
V~ D
.,1 1~ r~ u~ ~ Cl~ ~ oO 'D
c; ~ ~ a~ ot
I_ ~D ~ ~ Lr~ In ~D
~3
a~ ~ ~r u~ ~ ~- ~ O O
o 1~ ~ ~ o ~ ~ ~ ~
~ ~ ~ O ~ U~ ~D r~ I_
r ~ ~D ~ ~O ~D u) u~ u~ u~
. . . __~ ' ''
X G
_~ ~_ ~ ~ O
~ ~ ~ X ~) XC`I a~
O~ ~ U~ ~ u~ ~U)
. U~ ;) U~ ~ U7 ~ , .
~ C~ O~' ~ ~ ~ _, O ~ .'
e ~ ~i ~ ~ ,~ ~ ~, ~ ~~,~ ~ c~~ . :
O r~ ~11 0 ~ ~11 1~ ~ ~ 1~1 ~J .
O ~ O ~. Oul . O ~ . ~ O . `
O~ ~O S O u) ~O to O
_1 I r~ 11~ ~: C~ 4
~ O F~ O ~ 3 S c
o
. l I ~.
~ T
'1 'O O _ O
~ O ~ `
~ X~ . , .
O '_~
~)
_._____ ,, . . j
1.~ 1
I ~ o u~ ,~ ' co
X Z ,u~ U~
_ .
-, .. ." . ` -- . ., ~ , . ~ .. ~ . ..
~O 91/13054 49 ~?73~5~ PCI/EP91/03296
.. ' ... . .
_ ~ :'
~ ~ d` O
o~ o~ U~ U~ o~ oo . , ~
O~ O 00 ~ ' ~ ~ 1_ 1~ 1'O cr~ O CO ~ U~ `D ~O
co 1~ ~ ~ 1 ~ ~ ~o
. ~ r
u) ~ ~_ ~ ~ ~; r~ I~
O . ~ lO ~ ~ ~ .
. . _, I _~ :'' :~-
_ ~ _
_~ _ I O ' .~ .
Z U7 ~ Ul U~ ~ . : .
U~ UJ U~ ~U~ ~
t~ . ~ ~ ~~ ~3 ~ 3 _,
OO ~ O ~ O~ O~D ~.q ~
4~ . 4~ .4~ .~ . ~ .
,. o o o o ~ ~ C
~~ c~ ~ ~ ~
_
1~ 0~ ~.) i0
Z
Z Z r~ I
~ , 2~
t~ O o '~ ~ I
o
. ~ ~ ~ , ~ ~
i . . I
~. ., ... ..... . i. ; .. ' ., .. .. ;. ,. . .;";, .... , . ~
. WO 91/13054 '' ' ' ' , ' pcr/~
2~ ~ ?`~ 5 ~ ~7'~
EXAMPLE 64
_ . _ _ _ ,
N~ (2(S)-Carboxy-3-methoxvethoxypropyl)-1-cYclopentanecarbonyl1-
O-methyl-S)-tyrosine
An ice-cold solution of the diester from Example 28 (1.4 g)
and anisol (3.0g) in dry methylene chloride (20 ml) was saturated
with hydrogen chloride gas. After 30 minutes the mixture was
allowed to attain room temperature and after a further 30 minutes
the solvent was evaporated. The residue was chromatographed using ,
silica gel and eluting with dichloromethane, methanol, acetic acid
(95:5:0.5) to give the ethyl ester as an oil. This product was
dissolved in ethanol (2 ml) and dioxane (2 ml) and hydrolysed with
lN sodium hydroxide (6 ml). After standing at room temperature
for 2 hours ~ost of the solvent was evaporated under reduced
pressure, water was added, the mixture acidified with concentrated
hydrochloric acid and extracted with ethyl acetate. Washing with
water, drying (MgS04) and evaporation gave the title product as an
oil. Rf 0.18 (ss 7). Found: C,60.45; H,7.43; N,3.36. C23H33~08o `
0.25 H20 requires C,60.58; H,7.40; N,3.07%.
EXAMPEE 65
N~ (2(R,S)-Carboxyl-10-azidodecyl)-1-cyclopentanecarbonyl]-(S)-
tyrosine
The title compound was prepared from Example 27 by treatment
with hydrogen chloride followed by alkaline hydrolysis following th2
procedure of Exar;ple 64 above; the product was obtained as a gu~.
Rf 0.79, (ss 3).
~ 0 9~/130~4 2~73~ PC~/EP91/002~6
_AMPLE 66
N~ 2(R,S)-Carboxyl-10-aminodecyl)-1-cyclopentanecarbonyl~-(S)-
tyrosine
_ _ .
The title co~pound was prepared from the azido derivative of
Example 65 above by hydrogenation, following the procedure given
in Example 37 (part b). The product was obtained as a foam Rf
0.59, (ss 3). Found: C,64.10; H,8.49; N,4.73. C26FI40N2O6. 0.5
H20 requires C964.30; H,8.51; N,5.76%.
,;'
EXAMPLE 67
N-[1-~2(S)-Carboxy-3-(4-methanesulphinylmethylben~oylamino)-
propyl~l-cyclopentanecarbonyl]-(S)tyrosine
To a cooled solution of sodium metaperiodate (39.5~mg, 0.18
mmol) in water (2 ml) was added N-[1-~2(S)-carboxy-3-(4-methyl-
thiomethylbenzoylamino)propyl~-l-cyclopentanecarboxy]-(S)-tyrosine
from Example 56, (87 mg, 0.16 mmol) as a solution in methanol (9
ml). Methanol was added as needed to achieve a homogeneous
solution. The reaction mixture was allowed to stir at 0C
overnight. The methanol was evaporated and ~the aqueous residue ~ -
was diluted with water (5 ml) saturated with solid sodium chloride
and extracted with ethyl acetate (6 x 5 ml). The combined organic
extracts were dried ~MgS04), filtered and evaporated to give the
title compound as a white solid (71 mg, oOZ). Found: C,57.07;
C28H34N28S~ 0-25 CH2C12' H20 requires C,56.75;
H,6.15; N,4.69%. Rf 0~48 (ss 3).
' `: ' . .
, ' ' :, : ', ' ' . :' ", ;~' , ' ;,.,' ,'.' .' ' " . i `., ' :' '., ' '.'
W 0 91/l3054 . PCT/EP9l/0
~?~ 52
EXAMPLE 68
N-Ll-~2(S)-Carbo~y-5-~1-(4-methylpiperazinyl~-4-methoxypentvl~
cyclopentanecarbonyl]-0-methyl-tS)tyrosine
a) The procedure of Exa~ple 34 was followed but using N-methyl-
piperazine in step (b~ to give N~ 2(S)-tt-butoxycarbonyl)-5-
~(4-methylpiperazinyl)~-4-methoxypentyl~-1-cyclopentanecarbonyl]-0-
methyl-(S)-tyrosine t-butyl ester as a gum. Rf 0.19 (methanol~
dichloromethane, acetic acid, 10:90:1). Found: C,67.01; H,9.12;
N,6-42- C36H59N307 requires C,66.95; H~9.21; N~6.51%.
b) ~eprotection of the above diester with trifluoroacetic acid
following the procedure of Exa~ple 44, followed by ion-exchange -
chromatography gave the title diacid as a white powder. Rf O.lS
(ss-3). Found: C,62.68; H,8.37; N,7.76. C28H43N307 requires
C,63.02; H,8.12; N,7.87%.
~ ~ .
91~130S4 ~73~L~ PC~/E~9~ 6
53 ;~
PR~PARATION I
3~Methoxvethoxy-2-methoxyethoxymethyl ~roprionic acid
~,
a) To a solution of t-butyl 2-(bromethyl)acrylate (2.0 g, 9.00
mmol) in 2-methoxyethanol (30 ml) at room temperature was added,
in one portion, potassium carbonate (2.5 g, 18 mmol) and the
mixture stirred at room temperature for 72 hours. The reaction
was diluted wi;h distilled water (100 ml) and extracted with
dichloromethane (100 ml). The layers were separated and the
aqueous layer further extracted with dichloromethane (2 x 50 ml)~
The combined organic extracts were dried over magnesium sulphate~
filtered and concentrated under reduced pressure. The residue was
purified by chromatography on silica by eluting with ethylacetate/ -
hexane to give t-butyl-3-methoxyethoxy-2-methoxyethoxymethyl-
propionate as a yellow oil (1.947 g, 74%), Rf 0.33 (ethyl acetate/
hexane 1:1). Found: C,57.67; H,9.85. C14H2806 requires C,57051;
H,9.65%
.
b) t-Butyl-3-methoxyethoxy-2-methoxyethoxymethylproprionate
(1.60 g, 5.47 mmol) was dissolved in dichloromethane (20 ml) and
cooled to 0C. The solution was saturated with hydrogen chloride
and stirred for 3 hours at 0C. ~vaporation of the solvent under
reduced pressure gàve the title compound as an oil (1.292 8, v
l00%) Found: C 49.90; H,8.41. C]oH2006. 0.07 CH2C 2 q
C,49.93; H,8.38/o.
.,. .. . ,.- . , . - ~ - :
... : . .... .. ~, ...... : .. ... ... . .
W O 9It~3054 : P ~/~P~
~73~$~ 54
PREPARATION 2
_(2-Iodoethoxy?-methoxybutane
a) A solution of 4-methoxybutanol (3.64 g, 34.9 mmol) in dry
tetrahydrofuran (20 ml) was added dropwise under nitrogen to a
stirred suspension of sodium hydride (1.05 g, 34.9 mmol, 80%
dispersion in oil) in dry tetrahydrofuran (55 ml) keeping the
temperature at 10C. The resulting mixture was stirred at room
te~perature for 11~ hours, cooled to 10C and 1,3,2-dioxathiolane-
2,2-dioxide (4.77 g, 38.4 mmol) in dry tetrahydrofuran (30 ml) was
added dropwise. After a further two hours at room temperature9
water (0.63 ml, 34.9 mmol) was added followed carefully, with
cooling, by concentrated sulphuric acid. After a final 1 hour
stirring, solid sodium bicarbonate (7 g) was added with water (10
ml). The mixture was filtered, evaporated to a small volume
without heat at reduced pressure and the residue partitioned
between ethyl acetate and saturated salt solution. The organic
extract on washing with brine9 drying (MgS04) and evaporation ga~e
2-(4-methoxybutoxy)ethanol (4.54 g, 88%). Rf 0.25 (hexane,
ethylacetate, 1:1). Found: C,54.98; H,10.65. C7R1603. 0.25 H20
requires C,55.06; H,10.89%.
b) 4-Methylbenzenesulphonyl chloride (17.52 g, 91.90 mmol) was
added to an ice cooled stirred solution of the alcohol from
part a) (4.54 g, 30.63 mmol) and ~-methylmorpholine (10.44 ml,
94.96 mmoi) in dry meth~lene chloride ~100 ml). After standing at
O~C for 20 hours, the solvent was evaporated under reduced
pressure and the residue was chromatographed on silica eluting
with increasing proportions of ethyl acetate in hexane to give the
4-methylbenzenesulphonate as an oil (6.58 g, 71%).
c) Sodium iodide (7.25 g, 48~35 mmol) was added to a stirred
!
2~73~5q)
91/13054 PCT/EP9l~90296
~
solution of the above product (7.31 g, 24.17 mmol) in dimethyl-
acetamide (30 ml), and the mixture was maintained at 60-65~ for
for 20 hours. On cooling, water was added and the product
extracted with ether. The organic extract was washed succesivel~
with water, dilute sodium thiosulphate solution and water, dried
(~gS04) and evaporated to give the product as an oil (5.85 g,
94%). Rf 0.45 (hexane, ethyl acetate, 1:1). Found: C,32.23;
H,5.51. C7H15I02 requires C,32.56; ~1~5-86~-
'
PREPARATION 3
4-~2-Iodoethoxy)piperidine-l-carboxylic acid t-butyl ester
The title compound was prepared fro~ 4-hydroxypyridine-1-
carboxylic acid t-butyl ester following the procedure of
Preparation 2 and was obtained as a clear oil. Rf 0.4 (hexane,
ethyl acetate, 4:1). Found: C,40.94; H,6.20; N,3.93. Cl2H22IN0
requires: C,40.58; H,6.24; N,3.94%.
.: . - - :
PREPARATIO~ 4
1-(2-Iodoethoxy)-4-methoxy-but-2-ene
__. _ _
a) Dimethyl-t-butylsilyloxyethanol (5.29 g, 30 mmol) in
tetrahydrofuran (20 ml) was added dropwise under nitrogen at
10-15~C to a stirred suspension of sodium hydride (900 mg, 30
mmol, 80% dispersion in oil) in tetrahydrofuran (80 ml). On
stirring at room temperature for an hour l-bromo-4-methoxy-2-
butene (5.0 g, 30 mmol) in tetrahydrofuran (20 ml) was added
dropwise with ice cooling. The mixture was then stirred at room
te~perature overnight, cooled in ice and ether (100 ml) adde~,
followed carefully by water (100 ml). The organic phase was
~`~" ~
~ .~
W O 9~/13054 PC~tEP91/00
~ ~ ~ ? ~ 5 ~ 56
washed with water, dried (MgSO4) and evaporation gave the crude
product which was chronlacographed on silica. Elution with a
mixture of hexane and e.hyl acetate (9:1) gave a pale yellow
volatile oil (4.6 g, 59%).
lM Tetrabutylammonium fluoride in tetrahydrofuran (33.7 m~ol~
was added to a stirred ice cooled solution of the above product
(4.38 g, 16.83 mmol) in tetrahydrofuran (25 ml~. On stirring at
room temperature overnight, ether was added and the mixture was
washed with 0.SN hydrochloric acid and water. The aqueous
washings were saturated with salt and extracted with ethyl acetate
(3 x 100 ml). The combined organic extracts were washed with
saturated salt solution, dried (MgS04) and evaporation gave a
yellow oil (10.8 g). Chromatography on silica gel, eluting with a
mixture of hexane and ethyl acetate (1:1~ gave 2-(4-methoxy-2-
butenoxy)ethanol as an oil (1.59 g, 65%). Rf 0.14 (hexane, ethyl
acetate, 1~
b3 The above product was reacted with 4-methylbenzenesulphonyl
chloride followed by sodium iodide following the procedure of
Preparation l(b) to give the title compound as a pale yellow oil.
Rf 0.34 thexane, ethyl acetate, 1:1). Found: C,32.47; H,5.04.
C7H13O2f requires C,32.83; H,5.12%.
PREPARATION 5
_-C~!loromethoxytetra_ydropyran
4-Hydroxytetrahydropyran (8.0 g, 70.33 ~mol) was saturated
with hydrogen chloride gas at 0C, S-trioxane (3.24 g, 35.3 mmol)
was added with stirring and hydrogen chloride introduced for a
further 4 hours. The resulting solution was then diluted with a
O 91/130~4 ~3~5~ PCT/EP91/00296
57
1:1 mixture of ethet and hexane (150 ~1), dried over MgS04 and the
solvent evaporated under reduced pressure. Distillation gave a
clear oil, b.p. 100-110C/10 torr (13.3 pa) which was used
directly.
PREPARATION 6
1-(2-Iodoethoxy)-4-(dimethyl-t-butylsilyloxymethyl)benzene
a) Alpha,4-bis(Dimethyl-t-butylsilyloxy)toluene
Imidazole (49!34 g, 0.724 mmol) and dimethyl-t-butylsilyl chloride
(54.62 g, 0.362 mmol) were added successively under nitrogen at
15C to a stirred solution of 4-hydroxymethylphenol (15 g, 0.121
mmol) in dry di~ethylformamide (55 ml). On stirring at roo~
temperature for 24 hours, water was added and the mixture
extracted with ether. The extract was washed with water, dried
~MgS04) and evaporation gave an oil which was chromatographed on
silica gel. Elution with hexane and then a mixture of ether in
hexane (5:953 gave a clear oil. ~36.35 g, 85%).
b) 4-(Di~ethyl-t-butylsilyloxymethyl)phenol
lM Tetrabutylammoniu~ fluoride in tetrahydrofuran (76.85 ml) was
added dropwise at 0C to a stirred solution of the product from
part (a) (27.1 g, 76.8 mmol) in dry tetrahydrofuran (100 ml).
Af~er a further 30 minutes, ammonium chloride solution was added
and the mixture extracted with ether. The extract was washed with
water, dried (MgS04) and evaporated under reduced pressure.
Chromatography on silica gel eluting with increasing proportions
of ethyl acetate in hexane gave the required product as a clear
oil (4.15 g, 23~). Rf 0.4 (hexane, ethvl acetate, 4:1). Found:
C,64.26; H,9.57. C13H2202Si, 0-25 H20 requires C,64.2~; H,9-34%.
. ~
., . ~, .: .. . .: : `: ~
W O 91/13054 ~ : PCr/EP9~OQ ~ ~
~ ~,
2~?7~
c) 1-(2-Chloroethoxy)-4-~d methyl-t-butylsilyloxy-methyl)benzene
Sodium hydride (515 ~g, 17.6 ~mol, 80% dispersion in oil) was
added under nitrogan at room temperature to a stirred solution of
the phenol from part (b) (4.09 g, 17.16 mmol) in dry
dimethylformamide (30 ml). The mixture was heated at 60C for 30
minutes, cooled and 2-(4-methyl-ben~enesulphonyloxy)-1-chloro-
ethane (3.11 ml, 17.16 mmol) added in one portion. The mix~ure
was then reheated to 60C for 3 hours~ cooled, diluted with water
and extracted with ether. The organic extract was washed with
water, dried (MgS04) and solvent evaporated under reduced
pressure. Chromatography on silica gel, eluting with a mixture of
ethylacetate and hexane (1:9) gave the required compound as a waxy ~`
solid (2.48 g, 47~). Found: C,59.64; H,8.19. C15H25C102S
requires C,59.87; H, 8.37%.
d) 1-(2-Iodoethoxy)-4-(dimethyl-t-butylsilyloxymethyl)beni~ene
A mixture of sodium iodide (2.44 g, 16.28 mmol) and chlorocompound
from part (c) (2.45 g, 8.14 mmol) were refluxed in acetone for 3O5 1
days. The solvent was evaporated off and the residue partitioned
between ether and water, dried (MgS04) and evaporated under
reduced pressure. The residue was chromatographed on silica gel,
eluting with a mixture of ether and hexane (1:4) to give a clear
oil (2.71 g, 85%). Rf 0.8 (ethylacetate, hexane 1:4).
. ~ .
w~ 91/13054 z~73~ p~T/~ 29~ 1
I .
59
PREPARATION 7
N~ (3-Ami opropyl-2(S)-t-buto_ycarbonyl)-l=cyclopentane-
carbonyl]-O-t-butyl-(S)-tyrosine-t-butyl ester
a) To an ice cold solution of l-t2-t-butyloxycarbonyl-3-
dibenzylaminopropyl)-l-cyc10pentane carboxylic acid (12.7 g9 27
mmole) in dry dichloromethane (1~0 ml) was added l-hydroxy-
benztriazole (4.2 g, 31 mmole), and 1-ethyl-3-(dimethylamino-
propyl)-carbodiimide (7 g, 36 mmole) and the resulting solution
stirred at 0C for 30 minutes. To this solution was added
O-t-butyltyrosine t-butyl ester (8.4 g~ 28.6 mmole) and
N-methylmorpholine (5.25 g~ 52 mmole) and the solution allow~d to
stand overnight at room temperature. The solvent was evaporated
under reduced pressure and the ~esultant mobile oil was dissolved
in methylene chloride and washed with water (2 x ), 2M
hydrochloric acid and saturated aqueous sodium bicarbonate (1 x)
dried (MgSO4), and the solution filtered and evaporated to yield
the crude product as a gum. Recrystallisation from n-hexane gave
N-[1-(2-t-butyloxycarbonyl-3-dibenzylaminopropyl)-1-cyclopentane-
carbonyl]-0-t-butyl-(S)-tyrosine t-butyl ester as a solid (13 g,
69%), m.p. 82-87C. A further batch of material was obtained by
evaporation of the supernatant liquors and further
recrystallisation. Found: C,74.12; H,8.69; N,3~87. C45H62N206
requires C,74.34; H,8.59; N,3.85%.
.
:. : ~: ,. . :; :: :. .: .~ : : . : :: :
W O 91~13054 PC~/EP91/00
2~ i5~ ~,
.
b) ~-[1-(2-t-~utyloxycarbonyl-3-dibenzylaminopropyl)-1- ~ .
cyclopentanecarbonyl]-0-t-butyl-(S)-tyrosine t-butYl ester (~rom . .
part a), 19 g) was dissolved in an ethanol:water mixture ~8:1~ 300
r.~l) and hydrogenated under an at~osphere of hydrogen (60 p.s.i~
4.1 bar) at room te~perature, over 20% paIladiu~ hydroxide on
carbon (2 g). After 24 hours, the solution was filtered through a
solkafloc pad, and the filtrate evaporated to yield an oil which
crystallised. This was triturated with hexane, chilled and
filtered to yield the pure enantiomer title co~pound as a solid (6
g, 42%) m.p. 122-127C. Found: C,67.90: H,9.33; ~,5.08.
C31H50N206 requires C,68.09; H,g.22; N~5-12%-
.
.
,