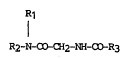Note: Descriptions are shown in the official language in which they were submitted.
y 207~3~ ~
DERIVE~ DE GLYCIN~DE, LE~R EsKE:PARAT:[CN
La présente invention ooncerne des dérivés de formule :
R1
I
R2-N-CO-CH2-NH-CO-R3 (I)
leurs procédés de préparation et les médicaments les oontenant.
Dans la formule (I),
-R1 représente :
. un radi~1 phényle ou phenyle substitué par un ou
plusieurs substituants choisis parmi les atomes d'h3logène et les
radicaux aIkyle, alooxy, alkylthio, nitro et cyano,
. une cha~ne -CH(R4)-CO-Rs dans laquelle R4 représente un
atome d'hydrogène, un radical alkyle, alcoxycarbonyle ou phényle
(éventuellement substitué par un ou plusieurs substituants choisis
parmi les atomes d'halogene et les ~adicaux alkyle, alcoxy,
alkylthio et nitro) et Rs rep~esente un radical alooxy,
cyclo ~ loxy (é~entuellement substitué par au m~ins un radical
alkyle), cycloalkyalkyloxy, p~énylalkyloxy, polyfluor~alkyloxy,
cinnamyloxy, un reste -NR6R7 dans l~quel R6 et R7 identiques ou
différents représentent un atome d'hydr~gene, un radical alkyle,
phényle (éventuellement substitué par un ou plusieurs substituants
choisis paGmi les a~omes d'halogène et les radicaux alkyle, alcoxy
et alkylthio), ~ycloalkylalkyle, cycloalkyle, indanyle,
phenylalkyle ou bien R6 et R7 forment ensemble avec l'atome d'a70te
auquel ils sont rattachés un héter~cycle mon~ ou polycyclique
saturé ou insaturé contenant 4 à 9 atomes de carb~nP et un ou
plusieurs héteroa~omes IO,N,S) et éventuellement substitué par un
ou plusieurs radioaux alkyle, alcoxy, alooxycarbonyle,
dialkylcar~amoyle, phényle cu en oombinaisan avec un ato~e de
cartane de l'héte~ocycle par un cycle spiromonocyclique à 4 ou 5
cha~^nons et oontenant éventuell~ent un ou plusieurs hét~roatcmes
(O,S,N),
2~3~
-~2 represente un radical pyridyle, isoquinolyle, ~yinDlyle,
quinoxalinyle (ces hétérocycles étant éventuellement substitués par
un cu plusieurs substituants choisis parmi les radicaux aIkyle,
phényle ou les at3nes d'halogène), alkyle, phénylalkyle, naphtyle,
tétrahydro-5,6,7,8 naphtyle, tétrahydrc-1,2,3,4 naphtyle,
alooxy bonylaIkyle ou cyloalkyle,
-R3 représente un radical phényle (éventuellement substitué par un
ou plusieurs substituants ch~isis panmi les atomes d'h2logène et
les radicaux alkyle, alooxy et alkylthio), naphtyle, indolyle,
quinolyle ou phénylamino dont le noyau phényle est éventuellement
substitué par un ou plusieurs substituants choisis parmi les atomes
d'halogène et les radicaux alkyle, alooxy, alkylthio, carboxy,
alooxycarb~nyle, hydroxy, nitro, amin~, acyle, cyano, sulfamoyle,
carbamDyle, benzoyle, phénylhy ~ éthyle, pi~ ridino, hydroxyiml-
noalkyle, alooxyiminoalkyle, hydroxy~ninLcarbooyle, alooxyamin~-
carbonyle, tétrazolyl-5, tétrazolyl-5 alkyle, trifluorométhylsul-
fon2mido, alkylsulfinyle, mQno ou polyhydkoxyaIkyle, sulfo,
-alk~O-CO~alk, -alk-COOX, -alk-0-alk, -alk'-CDOX, -C-alk-COOX,
-CH=CH-OOOX, -CC-COOX, -alk-SO3H, -CH=CH-alk', -C(=NOH)-COOX et
-S-alk-Coox
- X représente un atome d'hydrogene ou un r ~ cal alkyle,
- alk représente un radir~l alkyle ou alkylene,
- alk' représente~un radical hydrcxyaIkylène ou hydroxya ~ le.
Dans les défmiticns qui precèdent et celles qui serDnt
citées ci-après, sauf mention oontraire, les radicaux alkyle,
alkyl ~e et alooxy et les portions alkyle, aIkylène et alcoxy
ccntiennent 1 à 4 atc~es de ~ e en cha~^ne drDite ~u ~ fiée,
les radicaux et portions cycloalkyle oontiennent 3 à 6 atGmes de
carbcne et les radicaux acyle ocntienNent 2 à 4 atomes de carbcne.
Dans la formule (I), les a~ome~ d'halogen~ sont de
préerence des atomes de chlore, de brome ou de fluor.
Lorsgue R6 et R7 forment avec l'a~ome d'azote auquel ils
sant rattache~ un hetérocycle, celui-ci est de préference un cycle
piperidino (éventuellement substit~e par au m~ins u~ radlcal
aIkyle, ph'enyle, alcY~tcarbonyle eu dialkylcarbamoyle), un cycle
3 2~3~
perhydroazépinyl-1, tétrahydro-1,2,3,6 pyridyl-1,
tétrahydro-1,2,3,4 quinolyl-1, pyrr~lidinyl-1, dihydro-3,4 2H-
benzoxazine-1,4 yl-4, dihydro-3,4 2H-benzothiazine-1,4 yl-4,
N-alkyl tétrahydro-1,2,3,4 quinoxalinyl-1, perhydroquinolyl-1,
tétrahydrc-1,2,3,4 isoquinolyl-2, aza-8 spiro [4,5] décanyl-8,
phenyl-2 ou -3 pyrrolidinyl-1, æ a-8 dioxa-1,4 spiro [4,5]
décanyl-8, thiomorpholino (éventuellement substitué par au moins un
radical alkyle) ou indolinyl-1.
LÆS oompcsés de formule (I) comportant un ou plusieurs
centres asymétriques présentent des forrnes isomeres. Les racemiques
et les énantiomères de ces composés fcnt également partie de
1'invention.
Les oomposés de formule (I) pour lesquels R3 représente un
radical phénylamino dbnt le noyau phényle est éventuellement
substitué par un ou plusieurs substituants choisis parmi 1 s atomes
d'halogène et les radicaux alkyle, alooxy, alkylthio, nitro, acyle,
cyano, sulfamoyle, benzoyle, alooxyc~rkonyle, -alk-O-aIk,
tétrazolyl-S, tétrazolyl-5 aIkyle et trifluorométhylsulfonamido
peuvent être préparés par action d~un dérivé de fo¢nule :
R1
I
R2-N~CO~CH2-NH2 (II)
dans laquelle Rl et R2 ont les mêmes significations que dans la
formule (I), sur un isocyanate de foImule :
OCN-Rg (III)
dans laquelle R8 represente un radical phenyle éventuellement
substitué par un ou plusieurs substit ~ ts choisis ~ i l~s atomes
d'halogène et les radicaux alkyle, alooxy, aIkylthio, nitro, acyle,
cyano, sulfamcyle, benzDyle, alcoxycarbcnyle, -al~-O-alk,
tétrazolyl-5, tétrazDlyl-5 aIkyle et triflucmethylsul~cnamido.
Cbtte réaction s'ef~ectue ge'neralement au sein d'un solvant
4 2~7~3~
inerte tel ~ue le tétrahydrDfuranne, le dimethylformamide, un
solvant chloré (chloroforme, dichloro~1,2 éthane par exemple), un
solvant aromatique ~benzene, toluène par exe~ple), à une
te~pérature ~omprise entre 10C et la temperature d'ébullition du
solv3nt.
Les isocyanates de formule (III) peuvent être obtenus par
application ou 2daptation de la methcde décrite par R. RIcHrER et
coll., Ihe Chemistry of Cyanate and their thio derivatives,
S. PAT~I, part 2, Wiley New-Yor~ (1977).
Les dérivés de formule (II) peuvent être obtenus par
applicaticn ou adaptation de la méthode decrite par T. WqELAND et
coll.l Justus Liebigs Ann. Chem.t 613,84 (1958) ou par adaptatian
de la méthode de GABRIEL (M.S. GIBSCN et ooll., Ang~w. Chem. Int.
Ed., 7,919 (1968)) qui con~iste a faire reagir une hydrazine de
formLle :
H2N-NHRg (:[V)
dans laquelle Rg représente un atome d'hydrogène ou un radical
methyle, Æ un dérivé de formule :
o
R1 /!~ (V)
~/ .
o
dans laquelle R1 et R2 cnt les mêmes significations que dans la
formule ~I).
Cbtte réaction s'effectue de préference au se~n d'un solvant
inerte tel gu'un alcool (méthanol, éthanol par ~xemple) ou un
sDlvant chloré (chloroforme, dichlorcmethane par exemple), a une
2S temperature oomprise entre 0C et la temp#rature d'~bullition du
solvant.
T~C d'erivés de f~rmule (V1 peuvent être obtenus par action
d'un derivé ~e formule :
c3~
Hal-R1 (Vq~
dans laquelle Rl a les mêmes significations que dans la formule
(I), et Hal représente un atome d'halogène (de préference chlore ou
brcme) sur un dérive de formLle :
R2-NH-OO-CH2-N
~/ ~
I (VII)
o
dans laquelle X2 a les nêmes significations que dans la ormule
(I).
Cette réaction s'effectue géneralement au sein d'un solvant
m erte tel que l'acétonitrile, le dimethylformamide ou le tétra-
hydrofuranne, en présence d'une base telle qu'un hydrure de métalalcalin, à une température oomprise entre 0C et la température de
reflux du milieu réacticnnel.
Les dérives de formule (VII) peuvent être obtenus par action
d'une ~mine de formLle :
R2-NH2 (VIII)
dans laquelle R2~a les mêmes si~nificaticns que dans la formule
(I), sur le chlorure de phtalimldb-2 acetyle.
Cette reaction s'effectue gé ~ ement au seLn d'un solvant
inerte tel gu'un solvant chlore (chloroforme, dichloro-1,2 éthane
par exemple), en présence d'unQ base telle qu'une amine tertiaire
oomme une trialkylamine, un carbonate ou un bicarbonate de met
alcalin, à une ~ ture v~isine d~ 20C~
Le chloruxe de phtalimidb-2 acetyle peut être yxeparé par
application de la meth~de de'crite par W. GRASSM~NN et ooll., Chem
Eer., 83,2~4 (1950).
Les derives de formLl~ (V) peuvent egalement ~tre obtenus
par action d'une amine de ~or~ule o
6 2,07~35 ~
R~ -R2
dans laquelle R1 et R2 ont les m~mes significations que dans la
formule ~I) sur le chlorure de phtalimido-2 acetyle.
Cette reacticn s'effectue dans les oonditions décrites
précédemment pour la préparation des ~ de fcrmule (VII).
Les amines de formule (IX) sont oommercialisees ou peuvent
être obtenues par application ou adaptation des méthodes décrites
dans les exemples.
En particulier, ces oomposés peuvent être cbtenus par
hydrolyse d'un derivé de formLle :
COCF3 (X)
dans laquelle R1 et R2 ont les mêmes significaticns que dans la
formule (I).
Cette hydrolyse s'effectue généralement au m~yen d'une base
15 telle qu'un hydmxyde de métal alcalin (soude, potasse) ~u
l'hydroxyde d'a0mcnium au sein d'un solvant in~rte tel gue l'eau,
un alcool ou un mélange de ces solvants, à une temperature comprise
entre 20C et la temperature de reflux du m~lieu réactionnRl.
Les derives de formule (X) peuvent être obtenus par acticn
20 d'un dérivé de formule :
R2-NH~F3 (X~:)
dans laguelle R2 a les mêmes sign~fications que dans la fcrmwle (I)
sur un dérivé de fo~le (VI).
Cbt~te reactian s'effectue generalement au sein d'un ~olvant
inerte tal gue le tétrahydro~uranne, le dim'ethylformamide,
l'aoe tonitrile, en pré~ence d'une ba~e telle qu'un hydrure de metal
alcalin ou un carkonate de metal alcalin, à une temperature
oomprise entre 10~C et la temperature de reflux du milieu
reactionnel.
~0 Ies dérives de formule (XI) peuvent être obtenus par action
d'anhydride trifluoroacetique sur une amine de formule (VIII).
2~3~
- Cette réaction s'effectue géneralement au sein d'un solvant
inerte tel que la pyridine, a une temperature oo~prise entre -25C
et 10C.
Les composés de formule (I) pcur lesquels R3
représente un radical phénylamino dont le noyau phényle est
éventuellement substitué peuvent également être prépares par action
d'un dérivé de form~le :
R2-N-CO-CH2-NH-CC-N N (XII)
. l
dans laquelle R1 et R2 ont les mêmes significations que dans la
foroule (I), sur un dérivé de formule :
H2N-R10 (XIII)
dans laquelle R1o représente un raAlcal phényle éventuellement
sub6titue par un cu plusieurs substituants choisis parmi les atcmes
d'h2logene et les radicaux alkyle, alooxy, alkylthio, carboxy,
alooxycarbonyle, hydroxy, nitro, amino, acyle, cyano, sulfamoyle,
carbamoyle, benzoyle, phénylhydroxyméthyle, piperidino, hydroxy-
imlnoalkyle, alooxyimin~aIkyle, hydroxy~mincclrbonyle, alooxyamuno-
carbonyle, té ~ lyl-5, tétræolyl-5 alkyle, trifluorométhylsul-
fonamido, alkylsulfinyle, mono ou polyhydroxyalkyle, sulfo,
-alk-0-C0-alk, alk-aOoX, -al~-C-alk, -alk'-C00~ -C-aIk-OOQX
-CH--CH-COQX, -aIk-S03H, -CO-COCX, -~H=CH-alk', -C(=NoH)-COOX et
-S-alk-COOX.
Cette reaction s'eff~ctue g'ene'ralement au sein d'un solvant
inerte tel qye le tétrahydrofuranne, le diméthyl~ormamide/ un
solvant chloré cu un s~lvant aromatique, a une t ~ tu~e oomprise
entre 20C et la temperature d'e'bullition du sDlvant.
LRS anilinRs substituees de formule (XIII) sont
commercialisees cu peuvent être obtenucs par applicat~on ou
adaptation de 1~ méthode decrite par R. SCHR5IERt Mbthoden der
30 O~ganishen Chffnie, ~ Weil, Bar~ X/1, p 360.
~7~3.~
-; Les derives de formule (XII) peuYent être cbtenus par acticn
d'un dérivé de formwle (II) sur le N,N'-diimidazole carbonyle.
Cette réaction s'effectue généralement au ~ein d'un solvant
inerte tel que le tétrahydrofuranne, le d ~ thylformamide, un
solvant chloré cu un solvant aromatique, à une température oomprise
entre 20C et la temperature d'ebullition du solvant.
Les oompcsés de formule (I) pour lesquels R3 représente un
radical phénylamino dont le noyau phényle est éventuellement
s-~stitué peuvent également être préparés p r actian d'un dérivé de
formwle :
~1
R2-N ~ H2-Na) (XIV)
dans laquelle R1 et R2 ont les mêmes significations que dans la
formule (I), sur un dérivé de formule (XIII).
Cette reaction s'effectue genéralement au sein d'un solvant
inerte tel que le tétrahydrofuranne, le diméthylformamide, un
solvant chloré (chloroforme, dic~loro-1,2 éthane par exemple), un
solvant aromatique (benzene, toluene par exemple~, à une
temperature oomprise entre 10C et la temperature d'ebullition du
solvant.
T~c isocyanat~s de formule (XIV) peuvent être cbtenus par
action d'une amIne de fo¢mLle (IX) sur le chlorure
d'isocyanatoacétyle.
Cbtte réaction s'effectue géneralem~nt au sein d'un sol~ant
inerte tel qu'un éthe~ (éther diéthylique par exemple), un solvant
ammatique (benzène, boluene par exemple), ~n ~ c~ d'un
accepteur d'æide telle qy'une amlne conme la ~riéthyl2mine, la
pyridine, à une te~perature ocmprise entrs en~iron 5C eb 30C.
Les mmposes de formule ~I~ pour lesquRls R3 re~resente un
radir~l ph~nylamino éventu~llement sub*titue par un ou plusieurs
substituants ch~isis parmi les atomes d'halogene et les ~adica~x
alkyle, alccxy, alkylthio, nitro, acyle, CyanD, sulfam~yle,
benzoyle, alooxycartonyle, -alk-O-alk, ~'etrazolyl-5 aIkyle,
2 ~
té~razolyl-5 et trifluorométhylsul~cnamido peuvent égale~.ent être
prépares par action d'une amine de formule (IX~ sur un acide de
formule:
HOOC-CH2~ H-R3 (XV)
dans laquelle R3 a les mêmes significations que précéd ~ nt ou un
dérivé réactif de cet acide.
Lorsque l'on met en oeuvre l'acide, on opere en présence
d'un agent de condensation peptidique tel qu'un carbcdiimide ~par
exemple le dicyclohexylcarhodiimide) ou le N,N'-diimidazole
carbonyle, dans un solvant inerte tel qu'un éther ~tétrahydrofu-
ranne, dioxanne par exemple), un ~ de ~dime'thylfor~amide) cu un
solvant chloré ~chlorure de methylene, dichloro-1,2 éthane,
chlorDforme par exemple) à une temperature oomprise entne 0C et la
température de reflux du melange réactionnel.
Lorsque l'an met en oeuvre un derivé reactif de l'acide, il
est possible de faire réagir l'anhydride, un anhydride mixte, un
halogènure d'acide ou un ester (qui peut être choisi parmi le~
esters actives ou no,n de l'acide).
Qn opere alors soit en milieu organique, eventuellement en
présen oe d'un accepteur d'acide tel qu'une base crganique azotée
(triaIkylamine, pyrid~ne, diaza-1,8 bicyclo ~5.4.0] ur~e'ce`re-7 cu
diaza-1,5 bicyclo [4.3.03 nonène-5 pa~ exemple), dans ~ solvant
tel que cité ci-dessus, ou un meLange d~ ces solvants, à une
te ~ ture oomprise entre 0C et la temperature de reflux Zu
méLange reactic~nel, soit en milieu hydroorgFnique biphasigye en
presence dlune base alcaline ~u alcalino-terreuse (soude, potassP)
clu d'un c ~ te ou bicarbonate d'un metal alcalin ou
alcalino-terreux à une t ~ ture ocmprise ~ntre 0 et 40C.
Les acides de fc~mLle (XV) peuvent être ~bt~n~s par action
d'un isocyanate de ~c~mule (III) sur la glycine.
Cette reacticn s'effectue generalement en solutian ~queuse
en pr ~ d'une base telle gu'un bicarbonate de metal alcalin, à
une temperature v~i~ine de 20C.
Les ccmç06es de ~ormule (I3 pour lesqyels R3 représente un
207~35 7
~o
`~ radical phénylamino d~nt le noyau phényle est substitué par au
m~ins u~ radical carboxy, -aLk-COaH, -0-alk-aOCH, -alk'-aOOH,
-CH=CH-COOH, -CO-COQH, -S-alk-OOQX cu -C(=NCH)-OOOH ~euvent
e~alement être préparés par hydrolyse des esters co¢respcl lants.
Cette hydrolyse s'effectue généralement au moyen d'une base
telle que la so~de ou la potasse, au sein d'un solvant inerte tel
que le tétrahydrofuranne, le dioxanne, l'eau ou un mélange de ces
solvants, à une température oomprise entre 20C et 40C, ou au
moyen d'acide trifluoroacétique, au sein d'un solvant inerte tel
qu'un solvant chlore (dichlorométhane, chloroforme, dichloro-1,2
éthane par exemple), à une t~mpérature comprise entre 20C et la
température d'ébullition du solvant.
Les ocmpo6és de formule (I) pour lesquels R3 représente un
radical phényle éventuellement substitué, naphtyle, indolyle ou
qyinolyle peuvent être préparés par action d'un derivé de formule
(II) sur un aoide de formule :
.
HOOC-R3 (XVI)
dans laquelle R3 a les ~ significations que precedemment eu un
d~rivé réactif de oe t acide.
Cette réaction s'effectue dbu~ les co~diticns decrites
precedem~ent pcu~ la reacticn entre les amhnes de formule (IX) et
les acides de formule (XV).
Il est entendu pour l'hcmme du metier que, pcur la mise en
oeuYrs des procedes selon l' mventicn decrits preoedeiment, il peut
être ~ saire d'introdNire des gro~pes prot~cteurs des fonctions
amln~ af m d'eYiter des reactions secan~aires. Ce~ fcnctions
peuvent par exemple etre bloquees ~ous form~ db trifluorcmethyla-
cétamide puis reg ~ par action db méthan~l ammcniacal apres
avDlr mis en se~vre le pro~ede selon l'inventian.
Les enantiomeres des ocmposes de ~oGmule ~I) eontenant au
~oins un centre asymétrigue peuvent être obt~nus par de'dbublement
des race~iques par exemple par chromatographie sur oDlonne chirale
~elon W.H. PIR~LE et OD11.~ asyretric syn~esis, ~l.1, AC ~ ic
3 ~ ~
11
Press (1983~ cu par synthèse a partir des précurseurs chiraux.
Les composes de formLle (I) peuvent être purifiés par les
méthodes connues habituelles, par exemple par cristallisation,
chromatographie, extraction
Les composés de formule (I) présentent des propriétés
phaLmaoologiques intéressantes. Ces composés possèdent une forte
affinite pour les récepteurs de la cholécystokinlne (CCK) et de la
gastrine et sont don~ utiles dans le traitement et la prévention
des désordres liés à la CCK et à la gas~rine au niveau du systeme
nerveux et de l'a~pareil gastrDintestinal.
C'est ainsi que ces OQmpOses peuvent être utilises pour le
traitement ou la préventian des psychcses, des troubles anxieux, de
la maladie de Parkinson, de la diskinésie tardive, du syndrome du
colon irri~able, de la pancréatite aiguë, des ulceres et des
d~ardres de la motilité intestinale, de ~eItaines tumeurs de
1'o ~ ge inférieur, du colon et de l'intestin et comme
regulateur de l'appetit.
Ces cc~poses ant egalement un effet de pDtentialisation sur
1'activité ~ gé~ique des medicaments naroDtiques et n3n
naroDtiques.
L'affL~ité des oomposés de formule (I) pcur les recepteurs
CCK a été déterminée selon une technlque inspiree de oe lle de A.
SAIT~ et ooll. (J. Neuro. Chem., 37, 483-490 (1981)) au niveau du
cortex cérebral et au niveau du panc~eas.
D~ns ces tests, la CIso des ~ de ~o~mule (I) est
généralement e'gale ou inferieure a 1 000 nM.
Par ailleurs, il e~t connu gue les produits qyi reoonnais-
sent les récepteurs centraux de la CCK cnt une specificité similai-
re po~r les recepteurs de la gastrine dbn le tractus gastr4inte~-
tinal (BO~K et ooll., J. Mbd. Chem., 32, 16-23 (1989) ; REYFELD et
coll., Am. J. P~ysi~l., 240, G255-266 (1~81) ; EEINFELD et ooll.,
NbunoQeptides, 3, 411-427 (1g83)~.
Les oomposes de fo~ule (I) present~nt un tcxicité faible.
Leur DLso es~ generalement superieure à 40 m3/kg par v~ie sous
cutanee chez la ~ouris.
`` 2~7~3~
12
D'un intérêt particulier sont les composes de formwle (I)
pour lesquels R1 représente une cha~^ne -CH(R4)-CORs dans laquelle
R4 représente un atome d'hydrogène et Rs représente un radi~1
alcoxy et de préférence tert-butoxy, ou un reste -NR6R7 dans lequel
R6 représente un radical aLkyle et R7 représente un radical phényle
ou bien R6 et ~7 forment avec l'atome d'a~ote auquel ils sQnt
rattachés un radical tétrahydro-1,2,3,4 quinolyl-1, R2 représente
un radir~1 quinolyle ou lsoquinolyle et R3 représente un radical
phénylamino dont le noyau phényle est substitué par un ou plusieurs
substituant5 choisis parmi les radicaux alkyl~, carboxy,
hydroxyiminoalkyle cu hydroxyalkyle.
Sont particulièrement intéressants les composés suivants :
- ~(hydroxyimlnométhyl-3 phényl)-3 uréid~]-2 N-(qulnolyl-8)
N-[(tétrahydro-1,2,3,4 quinolyl-1) carbonylm~thyl] acétamide-(E)
- [[(méthyl-3 phényl3-3 uréido]-2 N [(tétrahydro-1,2,3,4
quinolyl-1)-2 cwo-2 éthyl] N-(quinolyl-8) acétamide
- acide {{N-(quinolyl-8) N-[(tétrahydro-1,2,3,4 quinolyl-1)-2 oxD-2
éthyl] carbam~ylméthyl}-3 uréido}-3 benzoïque
- {[(méthyl-3 phényl)-3 uréido]-2 N-(isoquinolyl-4) acétamido}-2
acétate de tert-butyle
- [(hydroxyméthyl-3 phényl)-3 uréido]-2 N-(quinolyl-8)
N-[(tétrahydro-1,2,3,4 quinolyl 1)-2 oxo-2 éthyl] acétamide
- [[(mé~hyl-3 phényl)-3 uréido]-2 N-(qum Dlyl 5) acétamidD]-2
acétate de tert-butyle
- ~(méthyl-3 phényl)-3 uréid~]-2 N-(quinolyl-8) acétamido]-2
acetate de tert-butyle
- {~[(hydroxy-1 éthyl)-3 ph~nyl]-3 uréidD~-2 N-(quinolyl-8)
acétamido}-2 acétate de tert-butyle
- {[(hydroxyméthyl-3 phényl)-3 uréido]-2 N-(isoquinolyl-4)
acétamido}-2 acétate de tert-butyle
- acide {[N-(N-methyl N-~hényl-carbamoylmethyl) N-(quinolyl-8)
carbam~ylmethyl~-3 uréido}-3 ben20ïque
- acide {[N-tert-butoxycarkonylmethyl N-(quinolyl 8)
carbamoylméthyl]-3 uréido}-3 ben~oïque
13 2-07~7
Les exemples suivants illustrent l'invention s~ns la
limiter.
ExEMæLE 1
A une solution de 6,1 g de [phtalimido-2 N-tpyridyl-3)
acétamido]-2 acétate de tert-butyle dans 90 cm3 de dichlorométhane
on ajoute, à une temperature v~isine de 0C, 2,13 g de
méthylhydrazine. Le mélange réactionnel est agité pendant 30 heures
à une temperature voisine de 20C puis pendbnt 1 heure au reflux.
Après refroidissement on ajoute 100 cm3 d'eau, a~ite et separe par
decantation la ph3se aqueuse qui est réextraite par 2 fois 60 cm3
de dichloronethane. Les phases organiques sont réunies, lavé~s par
2 fois 15 cm3 d'eau, sechées sur sulfate de magnesium, filtrées
Fuis ccncentrees à sec sous pression reduite (2,7 kPa) à 40C. an
obtient ainsi 3,75 g d'[a ~ 2 N-(pyridyl-3) acéta~ido]-2 acétate
de tert-butyle scus forme d'une huile jaune qui est dissaute dans
40 cm3 de tétrahydrDfuranne anhydre. ~n ajoute à oe tte ~olution,
1,9 g d'isocyanate de methyl-3 phenyle puis agite le melange
réactionnel pendant 1 heure à une température vDisine de 20C, et
oon~entre à sec sous pression re'duite (2,7 ~Pa) à 40C. Cn obtient
après recristallisation du residu dans l'acétate d'ét~yle, 2,25 g
de [[(methyl-3 ~henyl)-3 uréido]-2 N-(pyridyl-3) acétamido~-2
acétate de tert-butyle fondant à 173C.
Le [phtalimido-2 N-(pyridyl-33 acetamido]~2 acétate de
tert-butyle peut être prépare de la mani*re ~uivante : à une
solution de 11,4 g de phtalimidb-2 N-(pyridyl-3) asetamide dans 100
cm3 de tétrahydIDfuranne ~nhydre maint~nue sous abmosphere d'arg3n,
0 ajcute à une tempera~ure v~isine de 10~C, 2,13 g d'une
suspension huileuse (50 % en poids) d'hydrure de sDdium et agite la
suspensicn obt~nue pendant 1 heure à une temperabure ~isine de
20C.Cn aj~ute alors une sDlution de 9,5 g de b~cmoac'etate de
tert-butyle dEns 20 cm3 de tatrahydr~fura~ne anhydre et l'on
pour~uit l'ayitation peMdant 3 heures à une temperature voisine de
2 ~ 7
14
20C puis pendant 45 minutes à une température voisine de 40C. Le
mélange réactionnel est ensuite refroidi à une terperature v~isine
de 0C puis versé dans un mélange de 0C de 150 cm3 d'eau et de 200
cm3 d'acétate d'éthyle. La phase aqueuse est séparée par
décantation et réextraite par 2 fois 40 cm3 d'acétate d'éthyle. Les
phases organlques sont réunies, lavées par 3 fois 50 cma d'eau,
séchées sNr sulfate de magnésium, filtrées puis concentrées à sec
sous pressicn réduite (2,7 ~Pa) à 40C. On obtient ainsi, après
recristallisation dans l'acétate d'éthyle, 7,8 g de [phtalimido-2
10 N-(pyridyl-3) acétamido]-2 acétate de tert-butyle fondant à 154C.
Le phtalimldb-2 N-(pyridyl-3~ acétamide peut être préparé de
la manière suivante : à une solution de 4,7 g d'amins-3 pyridine
dans 80 cm3 de dichlorcmethane maintenue saus abmosphère d'argon,
on ajoute 5,6 g de ~riéthylamine puis, en maintenant la temperature
au voisinage de 20~, une soluticn de 11,2 g de chlorure de
phtalimido-2 acétyle dans 70 cm3 de dichlorcmethane. La solution
obtenue est agitee pendan~ 3 heures à une temperature voisine de
20C puis additicnnée de 300 cm3 d'eau. Le solide ~on~é est séparé
par fil~ration,lavé par 3 fois 10 cm3 d'oxyde de diissp~q~yle puis
par 3 fois 20 cm3 d'eau et seché à l'air. La phase onganique du
filtrat est separée par décantatian, lavée par 2 fois 20 cm3 d'e u,
se'chée sur sulfate de magnesium, filtrée puis ocncentrée à ~ec sous
pression réduite`(2,7 kPa) à 40C. Le solide obtenu est réuni au
précéd~nt et l'e ~ le est recristallisé dans l' ~ tate d'éthyle.
On obtient ainsi 11,4 g de phtalinidb-2 N-~pyridyl-3) acétamide
fondant à 229C.
Le chlorure de phtalimidk-2 acétyle peut être preparé selon
la neth~de decrite par ~. GRASSM~NN et ~oll., R~rO, 83,244 (1950).
~E ~
Eh cperant d'une maniere ~nalogue à celle d~srite a
l'exemple 1, mais à partir de 9,3 g de tPhtalimidb-2 N~(quinolyl-8)
acetamido~-2 acetate de tert-butyle, de 2,9 g de methyIhydrazine et
2~3.-~7
de 2,8 g d'isocyanate de méthyl-3 phényle, an bbtient après
recristallisation dans l'aoetate d'éthyle, 5,6 g de t[(~éthyl-3
phényl)-3 uréido]-2 N-(guinolyl 8) acétamido]-2 acétate de tert-bu-
tyle fcndant à 131C.
Le [phtalimido-2 N-(quin~lyl-8) acétamido]-2 acétate de
tert-butyle peut être préparé d'une manière analogue à celle
décrite à l'exemple 1 pour la préparatian du lphtalimidc-2 N-(pyri-
dyl-3) acétamido]-2 acétamide, mais à partir de 12,3 g de phtalimi-
db-2 N-(qu~nolyl-8) acétamide, de 2 9 d'une suspensian huileuse (50
~ en poids) d'hydrure de sodium et de 8,8 ~ de brcmoacétate de
tert-butyle. On obtient ainsi, apres recristallisaticn dans
l'acétate d'éthyle, 12,3 g de [phtalimidb-2 N -(quin~lyl-8) aceta-
mid~]-2 acétate de tert-butyle fondant à 196C.
Le phtalimido-2 N-(qyinolyl~8) acétamide peut être préparé
d'une manière analogue à celle decrite à l'exemple 1 pour la
préparatian du phtalimidb-2 N-(pyridyl-3) acéthmide, nais à partir
de 5,8 g d'h ~ 8 qyinoléine, de 4,4 g de triéthylamine et de
8,9 g de chlorure de phtalimidb-2 ~ tyle. On obtient ainsi 4,3 g
de phtalimidc-2 N-(quin~lyl-8) acetamide f ~ nt à 224C.
EXEMPLE 3
En ~ an~ d'une maniere analogue à oelle decrite à
l'exemple 1, mais à partir de 3,45 g de [phtalimidb-2 N-(pyridyl-2~
acéta~ido]~2 acétate de tert-butyle, de 1,2 g de méthylhydrazine et
de 1,15 g d'isocyanate de methyl-3 ph~nyle~ on cbtient après
recristallisatiQn dans un melange d'cxyae de diiscproQyle et
d'acetate- d'éthyle (95-5 en volumes), 0,45 9 de [[~methyl-3
ph'enyl)-3 uréidb]-2 N-(pyridyl-2) acetamido]-2 acetate de
tert-butyle fondant à 112C.
Le [phtalimidb-2 N-(pyridyl-2) acet3midD]-2 acetate de
tert-butyle peut être prepare d'une maniere analogue à celle
decrite à l'exemple 1 pcur la preparaticn du [phtallmido-2
N-(pyridyl-3) aoe tamid~]-2 asetamide, mais à part~r de 4,3 g de
phtalimido-2 N-(pyridyl-2~ acetamide, de 0,8 g d'une suspension
16 2~
huileuse (50 % en poids) d'hydrure de sodium et de 3,5 g de
brcmcacétate de tert-butyle. Qn bbtient ainsi, après recristal-
lisati~n dans l'acétate d'éthyle, 3,5 g de [phtalimido-2
N-(pyridyl-2) acétamido]-2-acétate de tert-butyle fondant à 120C.
Le phtalimido-2 N-(pyridyl-2) acétamide peut être préparé
d'une manlère analogue à celle décrite à l'exemple 1 pou~- la
préparati~n du phtalimidc-2 N-(pyridyl-3) acétamide, mais à partir
de 2,4 g d'amino-2 pyridine, de 2,8 g de triéthylamine et de 5,6 g
de chlorure de phtalimidb-2 acétyle. ~n obtient ainsi 4,3 g de
phtalimido-2 N-(pyridyl-2)-acétamide fondbnt à 193C.
A une solution de 1,6 g d'[~mino-2 N-(isoquin~lyl-8) aceta-
mido]-2 acetate de tert-kutyle dans 20 om3 de tetrahydr~furanne, on
ajoute, à une temperature voisine de 20C, 0,72 g d'isocyanate de
méthyl-3 p ényle. La solution obtenue est agitée pendant 4 heures à
une temperature voisine de 20C puis ooncentrée à sec s~us pression
réduite (2,7 kPa) à 40C. L'huile residuelle est purifié par
chrcma ~ ie s~r 150 g de silice (0,063-0,2 mm) contenus dans
une colcnne de 2 cm de dlamètre [éluant : cyclbhexane-aoetate
d'éthyle (75-25 en vDlumes)] en recueillant des fractions de 20
cma. Les fractions ne cvntenant que le produit cherché sont réunies
et ~ .centrées à sec sous pression reduite (2,7 kPa) à 40C. On
obtient apres recristallisation dans l'acétate d'éthyle, 0,85 g de
ll(methy1-3 phényl)-3 ureido]-2 N-(isoguin3lyl 8~ acétamido]-2
acetate de tert-kutyle ~ondant à 170C.
L'[amino-2 N-(isDquinolyl-8 acetami~o]-2 a~etate de
tert-butyle peut être ~ paré d'une whnlere analogue à oelle
decrite à l'exemæle 1 pour la ~ tion de l'[aminc~2
N-lpyridyl-3) acetamido]-2 acetate de tert-butyle, m~i8 à partir de
2,6 g de [phtalimidb-2 N-(isoquinolyl 8) asetamid~1-2 asetate de
tert-butyle et de 0,93 g d'hydrate d'hydrazine. on obtient ainsi
1,7 g d'[amino-2 N-(isoquinolyl-8) acetamido]-2 acetate de
tert-butyle sous forme d'une huile utilise~ telle quelle dbns le~
17 207~3~
syntheses ultérieures.
Le [phtalimido-2 N-(isoquin~lyl-~) acétamido]-2 acétate de
tert-butyle peut être préparé d'une manière analogue à celle
décrite à l'exemple 1 pour la préparation du [phtalimido-2
N-(pyridyl-3) acétamido]-2 acétate de tert-butyle, mais à partir de
4,3 g de phtalim~idb-2 N-(iso~uinolyl-8~ acétamide, de 0,76 g d'une
suspension huileuse (50 % en poids~ d'hydrure de sodium et de
2,65 g de bromoacétate de tert-butyle. On obtient ainsi 2,6 g de
[phtalimido-2 N-(isoquinolyl-8) acétamido]-2 acétate de tert-butyle
sous forme d'une huile utilisee telle quelle dans les synthèses
ultérieures.
Le phtalimidb-2 N-(isoqyinolyl-8) acétamide peut être
préparé d'une manière analogue à celle decrite à l'exemple 1 pour
la préparatiGn du phtalimido-2 N-(pyridyl 3)-acéta~ide, mais à
partir de 3,4 g d'amino-8 iscquinoléine, de 2,63 g de
triéthylamine, et de 5,75 g de chlorure de phtalimidb-2 acétyle. On
bbtient ainsi 4,3 g de phtalimidc-2 N-(is ~ lyl-8) acétamide.
EXEMPLE 5
En o~erant d'une manière analogue à celle décrite a
l'exemple 4, mai~ à partir de 3,3 g d'[amino-2 N-(qyinolyl-5)
acétamido]-2 acétate de tert-butyle et de 1,37 g d'isocyanate de
méthyl-3 phényle, on obtient apres recristalli~ation dans l'cxyde
de diisspropyle, 3,9 g de t[~méthyl~3 phe'nyl)-3 uréido]-2
N-(quin~lyl-5) ~cétamldo]-2 a oe tate de tert-butyle fcndant à 121C.
L'[amin~-2 N-(qyinolyl-5) acetamid~]-2 acétate de
tert-butyle peut être pré ~ d'une manière analogue a celle
decrite à l'exemple 1 pcur la ~ paration de l'[ ~ 2
N-(pyridyl-3) acétami~o]~2 aoe tate de tert-butyle, mais à partir de
5,5 g de ~phtalimido-2 N~(quin~lyl-5) acétamido]-2 acétate de
tert-butyle et de 1,85 g d'hydrate d'hydrazine. Qn obti~nt aLnsi
3,3 q d'[amino-2 N-(quinDlyl-5) acetamido]-2 aceta~2 de t~rt-butyle
sous forme d'une huile utilisee telle quelle dans les s~ntheses
ulterieures.
18 ~0~3~7
Le [phtalimidb-2 N-~qyinolyl-5) acétamido]-2 acétate de
tert-butyle peut être préparé d'une manière analogue à celle
décrite à l'exemple 1 pour la préparation du [phtalimido-2
N-(pyridyl-3) aoe tamido]-2 acétate de tert-butyle, mais à partir de
8 g de phtalimido-2 N-(quinolyl-5) acétamide, de 1,16 g d'une
suspension huileuse (50 % en poids) d'hydrure de ~odium et de 4,7 g
de bromoacétate de tert-butyle. Qn obtient ainsi, après recristal-
lisaticn dans l'oxyde de diisopropyle, 5,5 g de [phtalimido-2
N-tquinolyl-5) acetamido]-2 acétate de tert-butyle fondant à 168C.
Le phtalimidb-2 N-(quinolyl-5) aoe tamide peut être préparé
d'une manière analogue à oelle decrite à l'exenple 1 pcur la
préparation du phtalimidb-2 N-(pyridyl-3) acétamide, mais à partir
de 3,6 g d'amu`nc~5 quinoléine, de 2,8 g de triéthylamine et de
5,6 g de chlorure de phtalimidk-2 acétyle. Cn obtient ainsi 8 g de
ph~alimidb-2 N-(quinolyl-5) acétamide.
EXEMPLE 6
En ~ t d'une manlère analogue à celle décrite à
l'exemple 4, m~is à partir de 1,95 g d'~amino-2 N-(isoquinolyl-5)
acétamid~]-2 acétate de tert-butyle et de O,82 g d'iso~yanate de
methyl-3 phenyle, on obtient ap¢es recristallisaticn dans l'oxyde
de diiso~ropyle,~ 1,3 g de [[(methyl-3 phenyl)-3 ureid~-2
N-(isoquinolyl-5) acétamido]-2 a oetate de tert-butyle fondant à
188C.
L'[amino~2 N-(isoquinDlyl-5~ acetamido~-2 acetate de
tert-butyle peut être préparé d'une maniere analogue à celle
decrite à l'ex0mple 1 pour la préparation de l'[amino-2
N-(pyridyl-3) aCetami~Dl-2 asetate de tert-butyle, mais à partir de
3,65 9 de [p talimidb-2 N-(isDquinDlyl-5) acetamid~]-2 acetate de
tert-butyle et de 1,23 g d!hydrate d'hydrazine. Cn obti~nt ainsi
1,95 g d'1aminc-2 N-(isoquin~lyl-5) a¢etamiZD]-2 asetate de
tert-butyle sous forme d'une huile utilisee telle guelle dans les
synth`esEs ultérieures.
Le [phtal~midb-2 N-(iscquinDlyl-53 acetamidD]-2 acetate de
2~ 3S7
19
tert-butyle peut être préparé d'une manière analogue à celle
decrite à l'exemple 1 pcur la préparation du [phtalimido-2
N-(pyridyl-3) acétamido]-2 acétate de tert-butyle, mais à partir de
6,15 g de phtalimidc-2 N-(isoquinolyl-5) acétamide, de 1,07 g d'une
suspension huileuse (50 % en poids) d'hydrure de sodium et de 3,6 g
de broacétate de tert-butyle. ~n obtient ainsi, après recristal-
lisation dans l'acétate d'éthyle, 3,65 g de [phtalimidc-2 N-(iso-
quinolyl-5) acétamido]-2 acétate de tert-butyle fondant à 176C.
Le phtalimido-2 N-(isoquinolyl-5) acétamide peut être
préparé d'une maniere analogue à celle decrite à l'exemple 1 pour
la préparatian du phtalimido-2 N-(pyridyl-3) acétamide, mais à
partir de 3,6 g d'amino-5 isoqu~noléine, de 2,8 g de triéthylamLne
et de 5,6 g de chlo¢ure de phtalimido-2 acétyle. ~n bbtient ainsi
6,2 g de p~talimidb-2 N-(isoquinDlyl-5) acétamide.
EXEMPLE ~
En operant d'une naniere analogue à celle décrite à
l'exemple 4, mai~ à partir de 1,~ g d'1amino-2 N-(phényl-4
qyinolyl-8) acéta~ido]-2 acétate de tert-butyle, et de 0,67 g
d'isocyanate de methyl-3 phenyle, on obtient apres recristal-
lisation dans l'acétonitrile, 0,95 g de l[(methyl-3 ~ nyl)-3
uréido]-2 N-(phényl-4 guin~lyl-8) acétamido~-2 ac2tate de
t OE t-butyle ~ondant a 228C.
L'[aminc-2 N-(phenyl-4 qyin~lyl-8) acet2mido]-2 acétate de
$ert-butyle peut et~e p ~ d'un0 maniere an~logue à celle
decrite à l'exemple 1 pour la préparation de l'~amino-2
N-(pyridyl-3) acetamid4]-2 acétate de tert-butyle, mais à partir de
2,5 g de [phtal ~ db-2 N-(p~enyl-4 qyinolyl-8) a oe tamid~]-2 acetate
de teIt-butyle, et de 0,77 g d'hydrate d'hydrazine. ~n obtient
ainsi 2 g d'lamino~2 N-(phe'nyl-4 quinolyl-8 acetamid~]-2 acetate de
tert-butyle SGUS forme d'une hulle utilise~ telle qu~lle dans les
syntheses ultérieures.
Le [phtalimidb-2 N-(phenyl-4 gyinDlyl-8) acetamid~]-2
2~7~3~7
acetate de tert-butyle peut être préparé d'un manière analogue a
celle décrite à l'exemple 1 pour la préparation du tPhtalimidk-2
N-(pyridyl-3~ acétamido~-2 acétate de tert-butyle, mais à partir de
4,9 g de phtalimidb-2 N-(phényl-4 quinolyl-8~ acétamide, de 0,7 g
d'une susFensiGn huileuse (50 ~ en poids) d'hydrure de sodium et de
2,45 g de brom~acétate de tert-butyle. On obtlent ainsi 2,6 g de
[phtalimidc-2 N-(phényl-4 quinolyl-8) acétamido]-2 acétate de
tert-butyle sous fonme d'une huile utilisée telle quelle dans les
synthèses ulterieures.
Le phtalim~do-2 N-(phényl-4 qyinolyl-8) acétamide peut être
préparé d'une manière an310gue à celle décrite à l'exemple 1 pour
la préparation du phtalimido-2 N-(p~ridyl-3) acétamide, mais à
partir de 5 g d'amino-8 phényl-4 quinDléine, de 3,05 g de
triéthylamine et de 5,4 g de chlorure de phtalimidb-2 a oe tyle. On
obtient ainsi 5 g de phtal ~idb-2 N-(phényl-4 quinDlyl-8) ace~amide
fondant à 200C.
EXEMæLE 8
A une solution de 2,7 g de N,N'-carbonyldiimidazole dans 40
cm3 de dichloro-1,2 éthane, on aj~ute à une temperature vDisine de
20C, une solution de 4,8 g d'[amino-2 N-(quin~lyl-8) acétamid~]-2
acétate de tert-butyle dans 50 cm3 de dichioro-1,2 éthane et ~ite
le mélange pendant 2 heures à une temperature voisine de 20C. On
ajoute alors une solution de 2,1 g d'(hydrsxy-1 ~thyl)-3 aniline
dans 50 cm3 de dichlorc-1,2 éthane et l'on chauffe au reflux du
solvant pendant 6 heures. Le melange reacticnnel est ensuite
refroidi a une t ~ ture voisine de 10C puis additionne de 100
cma d'éau. La phase aqueuse est Bépanee par d'ec~ntation puis
reextraite par 2 fois 80 cm3 de dichloromethane. Les phases
crganig~es ~ont reunies, lavees par 50 cm3 d'eau, puis par 100 cm3
d'une s31ution aqueuse 0,5N d'acide chlorhydrigue et par 2 fois 80
om3 d'eau, sechees sur sulfate de magnesium, filtrees puis
soncentre'es à sec ~ous pression reduite (2,7kPa~ à 40C. L'huile
obtenue est purifiée par chromatDgraphie sur 150 g de silice
21 20~3~7
. (0,063-0,2mm) contenus dans une colonne de 2,5 cm de dia~ètre
[éluant : dichlorométhane-methan~l (98-2 en volumes)] en
recueillant des fractions de 30 cm3. Les fractions ne oontenant que
le produit cherché sont réunies et oonoentrées à sec sous pression
réduite (2,7kPa) à 40C. Qn bbtient après recristallisaticn dans
l'acétonitrile, 0,4 g d'{{[(hydroxy-1 éthyl)-3 phényl]-3 uréido}-2
N-(quinolyl-8) acétamido)-2 acétate de tert-butyle fondant à 200C.
L'[amino-2 N-(quinolyl-8) acétamido]-2 acétate de
tert-butyle peut être préparé de la manière suivante : à une
solution de lS g de [phtalimido-2 N-(quinolyl-8) acétamido]-2
acétate de t~rt-butyle dans 300 cm3 de méthan~l, on ajoute 4,8 g
d'hydrate d'hydrazine. Le mélange réactionnel est agité pendant 4
heures à une température voisine de 20C puis oancentré à sec sous
pression réduite (2,7kPa) à 40~C. L'huile résiduelle est disssute
dans 400 cm3 d'acéta}e d'éthyle et la solut~on bbtenue est
additionnée de 100 cm3 d'eau. La phase aqueuse est séparée æar
décantation puis réextraite par 2 fois 50 cm3 d'acétate d'éthyle.
Les phases organiques sant réunies, lavées pOEr 2 fois 60 cm3 d'eau,
séchées sur sulfate de magnésium, filtrees puis concentrées à sec
sous pression redlite (2,7kPa) à 40C. Qn obtient ainsi 10,2 g
d'[amino-2 N-(quin3lyl-8) acétamido]-2 acétate de tert-butyle sous
forme d'une huile utillsee telle quelle dans les synthèses
ultérieures.
EXB~ g
A une solution de 2,9 g d'[amlnc~2 N-(chlorc-S
isoqulnolyl-8) acetamid~]-2 acétate de tert-butyle dans 30 cm3 de
tétrahydr~furanne anhydre, an ajoute, à une t ~ ture v~isLne de
20C, 1,2 g d'is wyanate de methyl-3 phenyle. La sDlution obtenue
est agitee pendant 4 heures à une temperature vDisine de 20C puis
ooncentrée à sec scus pre~sion r~duite (2,7kPa) à 40Co ~'hulle
résidNelle est purifiée par chroma ~ e s~r 75 g de silioe
(0,063-0,2mm) oontenus dans une ~olonne de 2 cm de diametre
[éluant : dichloLomethane-mét~a~Dl (98-2 en v~lumes)] en
207~3~7
~2
recueillant des fracticns de 20 cm3. ~es fractions ne contenant que
le produit cherché sont réunies et concentrees à sec sous pression
reduite (2,7kPa) à 40C. On obtient ainsi, après recristallisation
dans l'acétate d'éthyle, 2,1 g de {N-(chloro-5 iscquinolyl-8)[(me-
5 thyl-3 phényl)-3 uréido]-2 acétamido)-2 aoétate de tert-butyle
fondant à 160C.
L'[amino-2 N-(chlorc-5 isoqyinolyl-8) acétamido]-2 acétate
de tert-butyle peut être préparé de la manière suivante : à une
solution de 4,0 ~ de [N-(chloro-5 isoquinolyl-8) phtalimido-2
10 acétamido]-2 acétate de tert-bu *le dans 80 cma de méthanol, an
ajoute 0,54 g d'hydrate d'hydrazine. Le mélange réactionnel est
chauffé à reflux pendant 3 heures puis ooncentré à sec sous
pression réduite (2,7kPa) à 40C. L'huile résiduelle est dissoute
dans 150 cm3 d'acétate d'éthyle et la solution obtenue est
additionnée de 50 cm3 d'eau. La phase aqueuse est séparée par
décantation puis reextraite par 2 fois 30 cm3 d'acétate d'éthyle.
Les phases organiques sont réunies, lavees par 3 fois 15 cm3 d'eau,
sechées sur sulfate de magnésium, filtrées puis oonoentrées à sec
sous pression réduite (2,7kPa) à 40C. on obtient ainsi 3,0 g
d'[amino-2 N-(chloro-5 isoquinolyl-8) acétamido]-2 aoétate de
tert-butyle sous forme d'une huile utilisee telle qyelle dans les
syntheses ultérieures.
Ie ~N-(chloro-5 isoquinolyl-8) phtalimidk-2 acétamido]-2
acétate de tert-butyle peut être prépare de la naniere ~uivante : à
une soluticn de 7,8 g de N-(chloro-5 isoquinolyl-8) phtalimidk-2
acétamide dans 150 cm3 de tétrahydrofuranne anhydre maintçnue sous
atmcKphère d'argon, cn ajoute, à une t ~ ture v~isine de 10C,
1.,3 g d'une ~ sion huileuse (50% en poids) d'~ ~ e de sodium
et agite la ~ sion obtenue pendant 2 hRu~es à une temperature
vDisine de 20C. Qn ajoute alors une solution de 5,S g de
brcmoacétate de tert-butyle dans 10 cm3 de te'trahydrofuranne
anhydre et l'on pcursuit l'agitation p~ndant 15 heuIes à une
temperature voisine de 20C. Le melange reactionnel est ensuite
versé dans 50 cm3 d'eau, le sDlvant est éllmlne par evapo¢ati~n
scus p¢essicn re'duite (2,7kPa) à 405C, et l'an ajoute un melange
207~3~7
23
-
de 500 cm3 d'acétate d'éthyle et de 100 cm3 d'eau. La phase aqueuse
est séparée par décantation puis réextraite Rar 2 fois 100 cm3
d'acétate d'éthyle. Les phases organiques sont réunies, lavées par
3 fois 80 cm3 d'eau, séchées sur sulfate de magnésium, filtrées
puis concentrees à sec sous pression réduite (2,7kPa) à 40C.
L'huile obtenue est purifiée par chrcmatographie sur 200 g de
silice (0,063-0,2mm) oontenus dans une colonne de 2,5 cm de
diamètre [éluant : dichlorométhane-méthanol (98-2 en volumes)] en
recueillant des fractions de 30 cn3~ Les fractions ne ccntenant que
le produit cherché sont réunies et oonoentrées à sec sous pression
réduite (2,7kPa) à 40C. ~n obtient après recristallisaticn dans
l'acétate d'éthyle, 4,7 g de [N-(chloro-5 isoqulnolyl-8)
phtalimido-2 acétamido]-2 acétate de tert-butyle fcndant à 200C.
Le N-(chloro-5 isoquin~lyl-8~ phtalimidb-2 acétamide peut
être préparé de la maniere suivante : à une solution de 5,4 g
d'amino-8 chloro-5 isoquinoléine et de 3 g de triéthylamine dans
150 cm3 de dichlorométhane maintenue sous at ~ ere d'argan, on
ajoute en maintenant la temperature au voisinage de 20C, une
soluticn de 7,4 g de chlorure de phtallmido-2 acetyle dans 50 cm3
de dichlo ~ thane. La solution obtenue est agitée pændant 4 heures
à une te~perature voisine de 20C, puis additionnee de 50 om3
d'eau. Le produit insoluble est sepane par filtration, lavé par 2
fois 25 cm3 d'oxyde de dii~cprcpyle puis par 5 fois 50 om3 d'eau et
seche à l'air. On bbtient ainsi 7,9 g de N-(chloro-5
i ~ olyl-8) phtalimidb-2 acetamide.
L'~mm o-8 chlo¢o-5 isoquin~léine peut etre prqparé ælon la
méthode decrite par J.H.H. FRAENXEL, E. SCH~oED~R, Iiebigs. Ann.
Ch~m., 396, 53-75 (1~13).
E~eLE 10
En operant d'une manière analogue à oelle decrite à l'exem-
ple 9, mais a partir dR 2,6 g d'laminc-2 N-(quinoKalinyl-5~ acéta-
midD~-2 acétate de tert-butyle et de 1,2 g d'isocyanate de methyl-3
phfinyle, cn obtient après necristallisatio~ dans l'acetate d'ethy-
24 2~7~3~
le, 1,1 g de {[(méthyl-3 phenyl-3) uréido]-2 N-(quinoxalinyl-5)
acétamido}-2 acétate de tert-butyle fcndant à 132C.
L'[aminc-2 N-(quinoxalinyl-5) acétamido]-2 acétate de
tert-butyle peut être préparé d'une manière analogue a celle
décrite à l'exemple 9, pour la préparation de l'[amino-2 N-(chlo-
ro-5 is ~ inolyl-8) acétamido]-2 acétate de tert-butyle, mais à
partir de 7,4 g de [phtalimidc-2 N-(quinoxalinyl-5) acétamido]-2
acétate de tert-butyle et de 1,7 g d'hydrate d'hydrazine. On
obtient ainsi 2,6 g d'[aminc-2 N-(quinoxalinyl-5) acetamido]-2
acétate de tert-butyle sous ~orme d'une huile utilisée telle quelle
dans les synthèses ultérieures.
Le [phtalimido-2 N-(quinoxalinyl-5~ acétamido]-2 acétate de
tert-butyle peut être préparé d'une naniène analogue à celle
décrite à l'exemple 3, pcur la préparation du [phtalimidc-2
N-~chloro-5 i ~ nolyl-8) aoe tamido]-2 acétate de tert-butyle,
mais à partir de 8,7 g de phtallmido-2 N-tquinoxalinyl-5)
aoe tamide, de 1,3 g d'une suspension huileuse (50% en poids)
d'hydrure de ~ ium et de 5,1 g de bromoacétate de tert-butyle. On
obtient ainsi 7,4 g de [phtalimido-2 N-(quinoxalinyl-5)
acétamido]-2 acétate de tert-butyle fondant à 195C.
Le phtalimidb-2 N-(quinoxalinyl-5) acéta~ide peut être
préparé d'une mam ère analogue à celle decrite à l'eKemple 9 pour
la préparation ~ du phtalimidb-2 N-(chloro-S isoquin~lyl-8)
acétamide, mais à partir de 5,6 g d'amlnc-5 quinoxaline, ~e 3,9 g
de triéthylamine et de 8,6 g de chlorure de phtalimidb-2 acétyle.
On bbtient ainsi 10,8 g de p talimidb-2 N-(guinoxalinyl-5
acét ~ de fcndant à 261C.
L'aminc-5 qyincxaline peut être ~ pare selon la methode
decrite par PLATT B. C., 5H~P T.M, J. Chem. Soc., 2129-2134
(19~8).
EXE~PLE 11
A ule solution de 0,41 g d'~hydrcxy-1 éthyl)-3 aniline d3ns
30 cm3 de tétrahydrDfuranne anhydke, maintenue sous abmosphexe
2~3~7
d'argon, on ajoute, à une température voisine de 20C, 3 g de
N-méthyl N-phényl [N-(qyinolyl-8) isocyanatoacétamido]-2 acétamide.
La solution obtenue est agitée pendant 16 heures à une température
voisine de 20C puis concentrée à sec sous pression réduite
5 (2,7kPa) à 40C. L'huile résiduelle est purifiée pa~
chromatographie sur 75 g de silice (O,063-0,2mm) contenus dans une
colonne de 2,5 cm de diamètre [éluant : dichloraméthane-méthanol
(98-2 en volumes)] en recueillant des fractions de 20 cm3. Les
fractions ne contenant que le produit cherché sont réunies et
concentrées à sec sous pressicn réduite (2,7kPa) à 40C. On abtient
après recristallisation daT~ l'acétate d'othyle, 0,80 g
d'{{[(hydroxy-1 éthyl)-3 phényl]-3 uréido)-2 N-(quinolyl-8)
acétamido}-2 N-méthyl N-phényl-acétamide fcndant à 188C.
Le N-méthyl N-phényl lN-(quinolyl-8) isocyanatoa oetamid~]-2
acétamide peut être préparé de la maniere suivante : à une solution
de 1,6 g de chlor~re d'isccyanatoacétyle dans 25 cm3 d'éther
diéthylique anhydre maintenue scus atmosphere d'argcn, on ajcute en
maintenant la t ~ ture au vDisiT~ge de 5C, une solution de 2,9
g de N-méthyl N-phenyl (quinolyl-8) aminc-2 acetamide et de 0,8 g
de pyrid me dans 200 cm3 d'éther diéthylique anhydre. Le mélange
reactionnel est agité pendant 4 heures à une t~mperature voisine de
20C puis le produit insDluble est separé par filtratian et lavé
par 3 fois 10 cm3 de tétrahydrDfuranne anhydre. Les filtrats sont
réunis et ooncentre's à sec sou~ p~ession re'duite (2,7kPa) à 40C.
On bbtient ainsi 2,3 g de N-méthyl N-ph'enyl tN-¦qyinDlyl-8)
isccyanatoacétamidb]-2 acétamide sous forme d'une huile utilisée
telle qyelle dans les synthèses ult2rieures.
Le chlorure d'isocyanatoace'tyle peut être préparé selon la
méthode ~ite par Y06HIO IW~KU~A, KEIKICHI UND, S~NhM KANG, J.
30 Qrg. ~m., 30, 1158-1161 (196S).
Le N-methyl N-p~yl (quin~lyl-8) amin~2 ace;tamide peut
être préparé de la maniere suivante: à une solutil ~le 2,4 g de
N~éthyl N-ph~yl [N-(quir~olyl-8) triflu~oaoetamidc)~-2 aoetamide
dans 40 cn3 d'éthanol, on ajoute, à une temp#rature Y2isLne de
35 20C, 6,2 03 d'une solution aqueuse 2N d'hyd~de de sodiun. Le
`~ 2~3~7
26
mélan~e reactionnel est chauffé à reflux pendant 30 minutes puis
1'éthanol est élinané p æ évaporaticn sous pression réduite
(2,7kPa) à 40C. Le résidu est agité avec 50 cm3 d'eau et le
prcduit insoluble est séparé par filtration puis recristallisé dans
l'éthan~l. on obtient ainsi 1,6 g de N-méthyl N-phényl ~quinolyl-8)
amino-2 acétamide fondant à 169C.
Le N-methyl N-phényl [N-(quin~lyl-8) trifluoroacétamido]-2
acétamide peut être préparé de la manière suivante : à une solution
de 1,2 g de trifluoroaoe tylamino-8 quinolé~ne dans 20 cm3 de
tétrahydr~furanne anhydre maintenue sous atmcsphère d'argon, on
ajcute, à une température voisine de 10C, 0,25 g d'une SUSpenSiGn
huileuse (50% en poids) d'hydrure de sodium et agite la suspension
cbtenue pendant 30 minutes à une te~perature voisine de 20C. On
ajoute alors une solution de 1,B g de bromc~2 N-methyl
l5 N-phényl-acétamlde dans 15 cm3 de tétrahydrofuranne anhydre et cn
chauffe à reflux pendant 5 heures. Le mélange réactio~nel est
ensuite refroidi à une temperature ve~isine de 20C et verse dans un
mélange de 25 cm3 d'eau et 50 cm3 d'acétate d'é~hyle. La phase
aqueuse est séparee par décantatian et reextraite par 2 fois 25 cm3
d'aoetate d'éthyle. Les phases o¢gz m ques sont réunles, la~ées par
3 fois 20 cm3 d'eau, sechées sur sulfate de magnesium, filtrées
puis ocncentrees à sec SGUS p¢essicn reduite (2,7kPa) à 40C.
L'huile cbtenue æ t purifiee par chromat ~ e sur 75 g de silice
(0,063-0,2mm) oonten~s dans une colonne de 2,5 om de diametre
léluant : dichloIométhane~ en rYcueillant des fracticns de 10 cm3.
LRS ~ actions ne oontenant que le ~ it cherch'e sont réunies et
ooncentrees à sec scus pression re'duite (2,7kPa) à 40C. On cbtient
ainsi 1,7 g de N-methyl N-phenyl [N-( ~ lyl-8)
trifluoroace~amido]-2 acétamide ~ous forme d'une hu~l~ utilisée
telle guelle dbns les synth`ese ulte'ri~ures.
Le bromc-2 N-méthyl N-phe'nyl-acetamide peut 2tre préparé de
la maniere suivantR: à une solution ds 10,7 g de ~hméthylaniline
dans 65 am3 de dichlorcmethane, on ajcute ~uccessivem~nt, à une
temperature voisine db -5C, 11,1 g de triéthylamdne et une
~olution de 29,4 g db ~rnmure de bromo20etyle dans 10 am3 de
2~7~3~
27
dichlorométhane. La suspension est agitée pendant 2 heures à une
temperature voisine de 20C puis additionnee de 25 cm3 d'eau. La
phase aqueuse est séparée par décantation et ree~traite par 2 ~ois
15 cm3 de dichloromethane. Les phases organiques sont réunies,
lavées par 3 fois 25 cm3 d'eau, séchées sur sulfate de magnésium,
filtréPs puis ocncentrées à sec sous pression reduite (2,7kPa) à
40C. L'huile résiduelle est additi~nnée de 100 cm3 d'éther
diéthylique anhydre ; le prcduit insoluble est séparé par
filtration et lavé par 3 fois 15 cm3 d'éther diéthylique. Les
filtrats sont réunis et concentres à sec sous pression réduite
(2,7kPa) à 40C. On bbtient ainsi 20,5 g de brcno-2 N-méthyl
N-phényl-acétamide sous forme d'une huile utilisée telle quelle
dans les synthèse ultérieures.
La trifluoroacetylamino-8 quin~léine peut être préparee de
la manière suivante : à une s~luticn de 2,9 g d'amino-8 qyinDléine
dans 25 cm3 de pyridine, on a~c~te à une temperature vQisine de
-20C, 4,2 g d'anhydride trifluoroacetique. Le mélange réactionnel
est a~ité pendant 30 minutes à une température v~isine de -20C,
puis pendant 1 heure à une temperature voisine de 0C et versé dans
150 cm3 d'eau refroidie à une temperature voisine de 0C. Le
produit insoluble est sépare par filtratian, lavé par 5 fois 10 cm3
d'eau et seché à l'air. On obtient ainsi 4,7 g de
trifluaroacétyl2mino-8 quin~léine ~cndant à 84C.
~ 12
En opeIant d'une manière analogue à celle decrite à l'exem-
ple 11, mais à partir de 0,37 g d'hy ~ éthyl-3 an1line e~ de 1,1
g de N-rethyl N-phenyl [N~(qu~nDlyl-5) isocyanatoacetamido]-2
acet~n~de, cn obtient a~res recristallisaticrl dans l'aoetate
d'ethyle, 0,80 ~ d'{1lhydroxymethyl-3 ph'enyl)-3 ureido]-2 N-(qui-
n~lyi-5) acetamido~-2 N-methyl N-ph~nyl-acetamide ~ond~nt à 205C.
Le N-methyl N-phe'nyl [N-(quinDlyl-5) isocyanatoaoetami~D]-2
aoetamide peut être prepare d'u~e maniere analogue à r~lle decrite
à l'ex~le 11 pa~r la preparaticn du N~thyl N-p~ényl
2~7~3~7
28
[N-(quin41yl-8) isocyanatoa oetamido~-2 acét ~ de, mais à partir de
1,7 g de chlorure d'isocyanatoacétyle, de 3,5 g de N-méthyl
N-phényl (quinolyl-5) amino-2 acétamide et d0 0,96 g de pyridine.
On obtient ainsi 2,3 g de N-méthyl N-phényl lN-(qulnolyl-5)
isocyanatoacétamido]-2 acétamide sous forme d'une huile utilisée
telle quelle dans les synthèses ultérieures.
Le N-méthyl N-phényl (quinDlyl-5) amino-2 acétamide peut
être prépare de la manière suivante : a une solution de 6,2 g de
N-méthyl N-phenyl ~N-(quinDlyl-5) trifluoroacétamido]-2 acétamide
dans 20 cm3 d'éthanol, Gn ajoute, à une t~mperature Y~isine de
20C, 16 cm3 d'une solution aqueuse 2N d'hydroxyde de sodium. Le
mélange reactionnel est agité pendant 1 heure à une temæerature
voisine de 20C puis l'éthanol est élimuné par évaporaticn scus
pression réduite (2,7kPa) à 40C. Le residu est agité avec 75 cm3
d'eau et le produit insoluble est separé par filtration puis
recristallisé dans l'éthanDl. ~n obtient ainsi 3,6 g de N-méthyl
N-phényl (qyi~olyl-5~ amino-2 acétamide f ~ nt à 165C.
Le N-méthyl N-p ~nyl [N-(quinolyl-5) trifluoroacetamid~D]-2
acetamide peut être préparé de la manière suivante : à une solutian
de 4 g de trifluoroacétylamino-5 qyinoléine dans 60 cm3 de
tétrahydrofuranne anhydre maintenue scus atmo~ph`ere d'argon, on
ajoute, à une temperature ~DiSine de 0C, 1,0 g d'une suspensicn
huileuse (50% en pDids) d'hydrure de sodium et agite la suspension
obtenue ~ nt 30 mlnutes à une tempera~ure voisine de 0C. On
ajout2 alors une solutisn d~ 5,7 g de brcmo-2 N-methyl
N-phényl-acétamide dans 30 cm3 ~e tetrahydrofuranne anhydre et
chauffe à reflux, sous agitation pendant 5 heures. Le mélange
r~ticnnel est ensuite refrDidi à une t~ture voisine de 20C
et verse dans un melan3e de 30 cm3 d'eau ~t de 200 cm3 d'acétate
d'éthyle. La phase 2queuse est séparee par ~ecantation et
r'eextraite par 2 fois 20 cm3 d'acetate d'éthyle. Les phases
organiqyes sont reu m ~s, lavees par 5 fois 25 cm3 d'eau, sech~es
sur sulfate de ~agnesium, filtrees puis oonoentre'~s à sec sous
pression reduite (2,7kPa) a 40C. L'huile obt~nue est purifiee par
chro~atographie sur 125 g de æilioe ~0,063-0,2 mm) contenus dans
2~3~ ~
29
une oolonne de 3 cm de diamètre (éluant : dichlorométhane) en
recueillant des fractions de 20 cm3. Les fractions ne contenant que
le produit cherché sont réunies et concentrées à sec sous pression
réduite (2,7kPa) a 40C. Cn bbti~nt ainsi 6,2 g de N-méthyl
N-phényl [N-(quinolyl-5) trifluoroacétamido]-2 acétamide sous forme
d'une huile utilisée telle quelle dans les synthèses ultérieures.
La trifluorcacétylamino-5 qui~oléine peut être préparée de
la manière suivante : à une solution de 2,9 g d'amino-5 quinoléine
dans 25 om3 de pyridine anhydre, on ajoute à une temperature
voisi~e de -20C, 4,2 g d'anhydride trifluoroacétique. Le mélange
réacticnnel est agité pendbnt 30 mlnutes à une temperature voisine
de -20C, puis pendant 1 heure à une temperature voisine de 0C et
versé dans 200 cm3 d'eau refroidie à une temperature voisine de
0C. Le pr~duit insoluble est séparé par filtration et seché à
l'air. On obtient ainsi 4,7 g de trifluoroacétyl ~ 5 ~ oléine
fcndant à 124C.
ExEMæLE 13
En oper~nt d'une manière analogue à oelle d'ecrite à l'exem-
ple 11, nais à partir de 0,31 g d'hydroxymethyl-3 aniline et de
1,0 g de N-methyl N-phenyl [N-( ~ lyl-8) isDcyanatoacetamido]-2
acetamide, on obtient apres ~ecristallisation dans l'aoe tanitrile,
0,47 g d'{[(hydroxymethyl-3 phenyl)-3 uréido]-2 N-(guinDlyl-B)
acetamido}-2 N-méthyl N-phenyl-acetamide fondbnt à 190C.
EXEMæLE 14
Uhe sDlutian de 1,5 ~ d'~[~imddazDlyl-1) caobcxamido~-2
N-(isoquin~lyl-4) acetamido}-2 acet~te de tert-butyle et de 0,9 g
d'al~oDl aminc-3 b~nzylique dbns 25 cm3 de ~Dlu~ne est chauffee au
reflux pendant 24 heures, puls le nili~u reactio~nel est cGn~entré
à æ c sous pression reduite (2,~kPa) à 70C. L~ residu ~st traité
~0 par 150 cm3 d'acétate d'é~hyle et la svlution obtenue est lavee par
100 cm3 d'eau, sech~e sur sulfate de magnesium, filtree
2~3~7
puis concentrée à sec sous pression reduite (2,7kPa) à 70C. Le
produit obtenu est c~romatographié sur 30 ~ de silice
(O,063-0,200mm) contenus dans une oolonne de 1,8 om de diamètre
(éluant : dichlorométhane-méthanDl (98-2 en v~lumes)) en
5 recueillant des fractions de 15 cm3. Les fractions 54 à 60 sont
réunies et ooncentrées à sec sous pression réduite (2,7kPa) à 60C.
On obtient, après recristallisation dans l'oxyde de diisopropyle,
0,17 g d'{[(hydroxyméthyl-3 phényl)-3 uxéido]-2 N-(isoquinolyl-4)
acétamido)-2 acétate de tert-butyle fondant à 153C.
L'{[(imidazolyl-1) carboNamid~]-2 N-(isoqyinolyl-4) acetami-
do}-2 acétate de tert-butyle peut être préparé de la manière
suivante : une solution de 2,7 g d'[amino-2 N-(isoquinolyl-4)
acétamido]-2 acétate de tert-butyle et de 1,4 g de N,N'-diimidazole
carbonyle dans 25 cm3 de tétrahydrD ~ ne est agitée pendant 24
15 heures à une temçerature voisine de 20C. Le produit insoluble est
séparé ~ filtration et lavé par 5 cm3 de tétrahydro ~ ne
anhydre. On obtient ainsi 1,4 g d'{l(imudazolyl-1) caIto~in~d~]-2
N-(isoquinDlyl-4) acétamidD}-2 acétate de tert-butyle fcndant à
130C.
L'~ ~ 2 N-(isoguinDlyl-4] aoetamido]-2 acétate de
tert-butyle peut être préparé de la manière suivante : à une
sDlution de ~,9 g de ~phtalimidb-2 N-(isoquinDlyl-4) acétamido]-2
acétate de tert-butyle dans 100 cm3 de methanol on ajoute à une
temperature vDisine de 20C, 1,3 g d'hydrate d'hydrazine. Le
mélange reactionnel est agité pendant 24 h~ures à une temperature
de 20C, puis on ajoute 30 cm3 d'~au. Le n~lange est ooncentré sous
pressicn ~eduite (2,7kPa) à 60C, puls dilue par 100 cm3 d'eau et
extrait ~ 2 fois 100 am3 de dichlo ~ thane. Le~ phases scnt
réunies, la~ée~ par 100 cm3 d'eau, s'echees ~ur ~ulfate de
~agnesium, filtrees puis ccncentre'es à sec sous pression r~duite
(2,7kPa) à 50C. on obtient ainsi 2,7g d'[amino-2 N-(isoquinolyl-4)
acétamid~]-2 acetate de tert-butyle sous for~e d'une ~uile utilisee
telle quelle dans les synthe`ses ulterieures.
Le [phtalimido-2 N-(isDquin~lyl-4) aoe~amid~-2 acétate de
~5 tert-butyle peut être p~eparé de la ma m ere suivante : à une
2~3~7
31
suspension de 8 g de phtalimido-2 N-(isoquinolyl-4) acétamide dans
150 cm3 de tétrahydrDfuranne anhydre naintenue sc~s atmosphère
d'azote, cn ajoute à une temperature voisine de 20C, 0,73 g d'une
suspension huileuse (50% en poids) d'hydrure de sc~ium. La
suspensicn cbtenue est agitée pendant 3 heures à oe tte temperature.
On ajoute 4,7 g de bromaoetate de tert-butyle et l'cn poursuit
l'agitation pendant 18 heures à une température voisine de 20C. an
ajoute alors 10 cm3 d'eau puis an verse le mélange réacticnnel dans
400 cm3 d'eau. On extrait par 2 fois 250 cm3 d'acétate d'éthyle.
Les phases organiques sont réunies, lavées par 100 cm3 d'eau,
séchees sur sulfate de magnésium, filtrées et ocncentrées à sec
sc~s pressicn réduite (2,7kPa) à 70C. Cn cbtient ainsi, après
recristallisaticn dbns l'oxyde de diiscpropyle, 6,1 g d~
[phtalimido-2 N-(isoquinolyl-4) acétamido]-2 acetate de tert-butyle
fcndant à 183C.
Le phtallmido-2 N-(iscquinolyl-4) acétamide peut être
préparé de la manière suivante : à une suspensicn de 7,0 g
d'amino-4 iscquln~léine dans 80 cm3 de dichlc~cmethane cn ajoute
4,85 g de triéthyl~mine puis une solutic~ de 10,9 g de chlorure de
20 phtalmidt-2 acétyle dans 70 cm3 de dichlorcméthane. La suspension
obtenue ~st agitée pendant 18 heures à une température voisine de
20C puis v~rsee dans 600 cm3 d'eau. Le solide formé est separé par
filtration, lavé par 3 fois 100 cm3 d'eau. On cbtient ainsi 8 g de
phtalimidb-2 N-(isoquinolyl-4) acetamide ~ondant à 260C.
EXEMPLE 15
En operant oomme à l'exemple 11 mals à partir de 2,9 g de
N-méthyl N-Fhenyl [N-(quin~lyl-8) isocyanatoaoetamido]-2 acetamide
et ds 1,0 g d'amino-3 benzcate d'ethyle, on cbtient après
recristallisation dbns l'isop ~ ol, 1,05 g de ~[N-(N-méthyl
N-phényl-carbam4ylmethyl) N-(quinolyl-8) carbamoylméthyl~-3 uréi-
do}-3 bP~nzoate d'éthyle fsndant à 173~C.
~7~
32
EXEMPLE 16
Une solution de 0,54 g de {[N-(N-methyl N-phényl-carba~oyl-
methyl) N-(quinolyl-8) car~am~ylméthyl]-3 uréido}-3 bKnzoate d'é-
thyle dans 10 cm3 d'une solution aqueuse 0,1 N d'hydroxyde de
sodium, 10 cm3 de dioxanne-1,4 et 10 cm3 de tétrahydrofuranne est
agitée à une temperature voisi~e de 20 C pendant 46 heures. Le
~ilieu réactionnel est cancentré jusqu'à un volu~e de 10 cm3 sous
pression réduite (2,7 kPa) à une température voisine de 45C. Le
résidu obtenu est séparé par filtration et traité par 10 cm3 d'une
solution aqueuse 0,lN d'hydrcxyde de scdium puis 10 cm3 d'eau. Les
phases aqueuses sont réunies, l~vees par 2 fols 10 cm3 d'acétate
d'éthyle, acidifiées par 2 om3 d'une solution aqueuse 1N d'acide
chlorhydrique jusqu'à un pH voisin de 3 et extraites par 3 fois 20
om3 de dichlor~méthane. Les extraits organiques sont réunis et
lavés par 25 cm3 d'une solution aqueuse saturee de chloru~e de
scdium. Le solide est filtré, lavé par 2 fois 5 cm3 de
dichlorcméthane, remis ~n suspension d2ns 30 cm3 d'eau, filtré et
rincé p2r 3 fois lp c~3 d'eau. On obtient ainsi 0,26 g d'acide
{[N-(N-méthyl N~phényl-carbamoylmethyl) N-(qyinolyl-B) carbamoyl-
methyl]-3 uréid~}-3 benzoïque fondant à lBO~C.
EXEMPLE 17
En ~ t d'une maniere analogue à oelle d~crlte à l'ex~m-
ple 11 mais à partir de 2,0 g d'isocy~natc-2 N-(quinDlyl-8) N-[(te-
trahydro-1,2,3,4 qyinolyl-1)-2 oYo-2 éthyl] acetamide et de 0,61 g
d'alcool ~ 3 benzyliqye, on ~btient 0,13 g d'l(hydroxymethyl-3
phényl)-3 uréido]-2 N-(qulnolyl-8) N~[(tétrahydro-1,2,3,4 quino-
lyl~ 2 oxo-2 éthyl] acetamide ~ond2nt à 125C.
L'isocyanabo-2 N-(quin~lyl-8~ N~l(tetrahydro-1,2,3,4 quinc-
lyl-1)-2 oxo-2 éthyl] aoe tamlde peut être prepQIe d'une manière
analogue a celle decrite à l'exemple 11 pour la preparation Zu
N-methyl N-phenyl [M-(quinolyl-8) isocyanatoacetamido]-2 acetamide,
mais à partir de 2,0 g de ~hlorure d'isocyanatoa oetyle, dR 5,3 g de
3 ~ ~
33
[(quinolyl-8) amins-2 acetyl]-1 tétrahydro-1,2,3,4 quinoléine. ~n
obtient ainsi 6,6 ~ d'isocyanato-2 N-tquinolyl-8) N-~(tétrahy-
dro-1,2,3,4 quinolyl-1)-2 owo-2 éthyl] acétamide sous forme d'une
huile utilisée telle quelle dans les synthèses ultérieures.
La [(quinolyl-8) amino-2 acétyl]-1 tetrahydro-1,2,3,4
quinoléine peut être préparée d'une manière analogue à celle
décrite à l'exemple 11 pour la préparation du N-méthyl N-phenyl
(quinolyl-8) amlno-2 acétamide, mais à partir de 65 cm3 d'une
solution aqueuse 2N d'hydroxyde de sodium et de 27 g de
N-(quinolyl-8) N-[(tétrahydro~1,2,3,4 quinolyl-1)-2 oxo-2 éthyl]
trifluor~acétamade. On sbtient ainsi 17 g de [(guinolyl-8) aminc-2
acétyl~-1 tétrahydro-1,2,3,4 quinDlélne fondant à 134C.
Le N-(qyinolyl-8) N-[(tétrahydro-1,2,3,4 qyinolyl-1)-2 oxo-2
éthyl] trifluoroacétamide peut être préparé de la manière
suivante : à ~ne solution de 21 g de trifluoroacétylamino-8 qyino-
léine dbns 210 cma de N,N-dime'thylformamide, on ajoute s~ccessi-
vement 12 g de carbonate de pDtassium puis une sDlution de 22 g de
bromacétyl-1 tétrahydro-1,2,3,4 qyin~léine dans 80 cm3 de N,N-dime-
thylformamide. La suspension ainsi obtenue est agitée pendbnt 18
heures à une temperature voisine de 20C. Le mélange réactionnel
est alors versé dans 11 d'eau et on extrait par 3 fois 500 cm3
d'oxyde de diéthyle. Les ph2ses organiques sont réunies, séchées
sur sulfate de nagnesium, filtrées puis ccncentrees à sec sous
pression reduite (2,7kPa) à 50C. Qn obtlent ainsi, après
recristallisation dans l'oxyde de dii ~ yle, 22,6 g de
N-(quin~lyl~8) N-[(tétrahy ~ 1,2,3,4 qyin2lyl~ 2 kKD-2 éthyl]
trifluoroacétamide ~o~dant à 132C.
La bromaoetyl-1 tétrahydro-1,2,3,4 quinoléine peut etre
préparee d'une maniere analogu~ à celle decrite à l'~xe~ple 11 psur
~0 la p¢épOEration du brcmc-2 N-methyl N- pényl-acetamide, mais à
partir de 40 g de tét~ahydro-1,2,3,4 quinaleine et de 64 g de
bromure de krsmcacetyle. on obtient ainsi 67 g de brcmoacetyl-1
tétrahydrc-1,2,3,4 ~uinoléine SQU5 fo~me d' w huile utilisee telle
quelle dans les synth`eses ulterieures.
2~7~7
34
EXEM2LE 18
En o~erank d'une manière analogue à oelle décrite à
l'exemple 11 mais à partir de 4,5 g d'isocyanato-2 N-(quinolyl-8)
N-[tétrahyd m-1,2,3,4 quinolyl-1)-2 oxc-2 éthyl] acétamide et 1,9 g
d'amin~-3 benzoate d'éthyle, on obtient 0,7 g de {{N-(quinolyl-8)
N-[(tétrahydro-1,2,3,4 qyinolyl-1)-2 oxc-2 éthyl] bamoylme-
thyl~-3 uréido}-3 b~nzoate d'éthyle fondant à 140C.
EXEMPLE 19
Une solution de 2,7 g d'[amino-2 N-(guinolyl-8) acétamido]-2
acétate de tert-butyle et de 1,5 g de N,N'-diimidazole bonyle
dans 30 cm3 de dichloro-1,2 éthane est agitée à une température
voisine de 20 C pendant 2 heures et 30 minutes. Cn ajoute ensuite
1,1 9 d'alcool amlno-3 benzylique et on chauffe le mélange à reflux
pendant 8 heures. Le milieu réacticnnel est ensulte refroidi à une
temperature voisine de 20C et dilué par 150 cm3 de
dichlor~methane. La ~ e o~ganique est lavée par 2 fois 80 cm3
d'eau, sechée sur sulfate de magnesium, filtrée et ooncent~ee à sec
sous pression réduite (2,7kPa) à 60~C. Ie residu cbtenu est
chromatographié sur 90 g de sili oe (0,063-0,200mm) oontenus dans
~0 une o~lonne de 2,2 am de diamètre (~luant : dichlo-
rométhane-méthanol (99,5-0,5 en volumes)) en recueillant des
fractlons de 10 c~a. Les fractions 40 à 60 sont réunies et ooncen-
trees à sec sou~ pression r'edNite (2,7kPa) à 60C. ~n obti~nt,
après recristallisation dans l'acetate d'éthyle, 0,65 g de
{[N-tert-butoxycarbonylmethyl N-(quinDlyl-8~ carbamcylmethyl]-3
uréido)-3 benzoate d'éthyle fondant à 200C.
En ~ t d'une maniere analogue à ~elle decrite à
l'exemple 19 nais à p~rtir db 2,7 g d'~amino-2 X~(quinDlyl-8)
acetamido]-2 acetate de tert-butyle, de 1,5 g de N,N'- diimidazole
2~43;~J
carbonyle et de 1,1 g d'alcool amunc-3 benzylique, on obtient 0,85
g d'{[(hydroxymethyl-3 phenyl)-3 uréido]-2 N-(quinolyl-8)
acétamido}-2 acétate de tert-butyle f ~ nt à 115C.
EXEMPLE 21
En opérant oomme à l'exemple 16, mais à partir de 1,0 g de
{{N-(quin dyl-8) N-[(tétrahydro-1,2,3,4 quinolyl-1)-2 oxo-2 éthyl]
carbamoylmethyl}-3 uréido}-3 ben2oate d'éthyle et de 18 cm3 d'une
solution aqueuse 0,1 N d'hydraxyde de sodium, on bbtient 0,81 g
d'acide {{N-(qui~olyl-8) N-[(tétrahydro-1,2,3,4 quinolyl-1)-2 oxo-2
éthyll ~arbam~ylm'ethyl}-3 uréido}-3 benzoïque f~ndant à 164C.
EXEMPLE 2~
En o~érant comme à l'exemple 11, mais à partir de 2,75 g
d'isocyana~c-2 N-( ~ lyl-8) N-[(tétrahydro-1,2,3,4 quinolyl-1)-2
owo-2 éthyl] acétamide et de 0,93 g d'am~no-3 benzaldoxi~e, on
Gbtient 0,76 g d'[(hydroxyiminométhyl-3 phényl)-3 uréiaD~-2
N-(quinolyl-8) N-[(tétrahydrc-1,2,3,4 quinolyl-1) carbGnylméthyl]
acétamide-(E) fondant à 221C.
L'amino-3 benzaldoxime peut être préparée selon la méthode
décrite par S. Gabriel, Chem. Ber., 16, 1997 (1883).
EXEMæLE 23
En operant ccmme à l'exemple 16, nzis à partir de 0,4 g de
{[N-tert-butDxycarbonylméthyl N-(quinolyl-8) carbamoylmethyl]-3
uréid~}-3 benzoate de méthyle et de 5,8 Qm3 d'une solution aqueuse
0,1N d'hydroxyde de sodium, on obtient 0,04 g d'acide
{[N-tert-butoxycarbanylmethyl N-(quinolyl-8) carbamoylmethyl]-3
uréidD}-3 benzoïque fondant à 180C.
Ie {[N-tert-butoxycarbonylmethyl N-( ~ lyl-8) carba-
moylméthyl]-3 uréidD}-3 benzoate de méthyle peut être préparé d'une
maniere analogue à celle decrite à l'exemple 11 pour la préparation
2 ~ J 7
36
de l'{{[(hydroxy-1 éthyl~-3 phénylj-3 uréid3}-2 N-(quin~lyl-8)
acétamido}-2 N-methyl N-phényl-acetamide, mais à partir de 4,2 g de
N-tert-butoxy bcnylmethyl isocyanato-2 N-(quinolyl-8) acétamide
et de 1,9 g d'aminc-3 benzoate de méthyle~ Gn obtient ainsi 1,8 g
de {[N-tert-butoxycarbQnylméthyl N-(quinolyl-8) carbamoylméthyl~-3
uréido}-3 benzoate de méthyle fondant 96C.
Le N-tert-butoxy ~ nylmethyl isccyanato-2 N-(quinolyl-8)
acétamide peut être préparé d'une manière analogue à celle décrite
à l'exemple 11 pour la préparation du N-methyl N-phényl
~N-(quinolyl-8) isocyanatoaoe tamido]-2 acétamide, mais à partir de
3,2 g d'amino-2 (quinolyl-8) acétate de tert-butyle, de 1,5 g de
chlorure d'isocyanatoacétyl~ et de 1,0 g de pyridine. On bbtient
ainsi 4,4 g de N-tert-butoxycarbanylméthyl isocyanato-2
N-(qyinolyl-8) acétamide sous forme d'une huile utilisée telle
quelle dans les synthèses ulterieures.
L'amino-2 (quinolyl-8) acétate de tert-butyle peut être
préparé d'une maniere analogue à celle décrite à l'exemple 11, pour
la préparation du N-methyl N-phenyl (quinolyl-8) amuno-2 acétamide,
mais à partir de 18,7 g de [N-(quinolyl-8) trifluoroacétamido]-2
acetate de tert-hutyle et de 53 cm3 d'une ~olution aqueuse 2N
d'hydroxyde de sodium. On obtient ain~i 3,5 g d'aminc-2
(quinolyl-8) acétate de tert-butyle fondant à 62C.
Le [N-(quinolyl-8) trifluoroacetami~o~-2 acetate de
tert-butyle peut être préparé de la nani~re ~uivante : à une
soluticn de 15 g de trifluoroacetylamlno-8 quinoléine dan~ 150 om3
de diméthylformam~de anhydre, on ajoute, à une t ~ t~re vDisLne
de 20C, 8,6 g de carbonate de potassium puis u~e sDlution de 12 g
de ~nomoaoe tate de tert-butyle dans 50 cma de dim~thylf ~ de
anhydre. Le melange reactionnel est a~ité pendant 17 heures à une
temperature voisine de 20C puis ver~e d2ns un litre d'eau. La pha-
se a4ueu~e est extraite par 3 fois 250 om3 d'acetate d'éthyle. Les
phases organiques sont r~wnies, lavees par 100 oma d'une solution
agueuse saturee en chlorure de sodium, ~echees sur sulfate de
magnésium, filtrees puis ccncentre'es a sec scus pression reduite
(2,7kPa~ à 40C. Le ~olide cbtenu est mis en suspensian dans 30 cm3
3 ,3 ~
31
d'oKyde de diiscpropyle et le produit insDluble est séparé parfiltraticn, lavé par 2 fois 30 cm3 de pentane Fuls séché sous
pression réduite (50kPa) à 25C~ On obtient ainsi 18,7 g de
lN-(quin41yl-8) trifluoroacétamido~-2 acétate de tert-butyle
fondant à 98C.
EXEMPLE 24
A une solution de 1,07 g de methyl-3 aniline dans 25 cm3 de
tétrahydrofuranne anhydre, maintenue svus atmosphère d'argon, on
ajcute, à une temperature voisine de 20C, 2,3 g d'isocyanato-2
N-[(diméthyl-3,3 piperidlnv)-2 owo-2 éthyl] N-(quinolyl-81
acétamide. La solution bbtenue est agitée pendant 2 heures à une
te ~ ature voisine de 20C puis ooncentrée a sec saus pression
réduite (2,7kPa) à 40C. L'huile résiduelle est purifiée par
chramatographie sur 75 g de silice (0,063-0,2mm) oontenus dans une
oolonne de 2,5 om de diamètre ~eluant : dichlorométhane-méthan~l
t98-2 en volumes)] en recueillant des fractions de 20 om3. Les
fractions ne contenant que le produit cherché sont réunies e~
concentrées à sec sous pression re'duite (2,~kPa~ à 40C. On obtient
après recristallisation dans l'aoétcnitrile, 1,2 g de [(méthyl-3
phényl)-3 uré.ido]-2 N-[(dlme'thyl-3,3 piperidlno)-2 oxc-2 éthyl]
N-(qyinolyl-8) acétamide fondant à 180C.
L'isocyanato-2 N-[(dim'ethyl-3,3 piperidinD~-2 C~D~2 éthyl]
N-(quinolyl-8) acétamide peut être préparé de la maniere suivante :
à une solution de 0,84 g de chlorure d'isocyanatoaoe tyle dans 25
om3 d'éther diéthyliqu~ maintenue sous atmosph`ere d'argan, on
ajoute en maintenant la t ~ ture au voisinage de 5C, une
solution de 1,8 g de ~(dim'ethyl-3,3 piperidlno)-2 owo-2 éthyl~
amino-8 qum oléi~e et de 0,48 g ds pyridine dbns 25 om3 de dio~anne
anhydre. Le mélan~e reactial~el est agite pendant 2 heures à une
t ~ ture vDisine de 20C puis le produit lnsDluble est séparé
par filtraticn et lavé par 2 fois 5 cn3 de diox3nne anhydre. Les
filtrats sont Yeunis et concentrés à sec ~ous p¢ession r'eduite
(2,7kPa) à 409C. Cn obtient ainsi 2,35 g d'isocyanato-2
-` 2~7~3)~
38
N-[(dlmethyl-3,3 pipéridino)-2 owo-2 éthyl] N-(quinolyl-8)
acétamide sous forme d'une huile utilisée telle ~uelle dans les
syntheses ulterieures.
La [(diméthyl-3,3 pipéridino)-2 CKo-2 éthyl] amino-8
quinolélne peut etre préparee de la nanière suivante : à une
solution de 3 g de N-[~diméthyl-3,3 pipéridino)-2 oxo-2 éthyl]
N-(quinolyl-8) trifluoroacétamide dbns 50 om3 d'éthanol, on ajoute,
à une temperature voisine de 20C, 7,5 cm3 d'une solution aqueuse
2N d'hydroxyde de sodium. Le mélange reactionnel est agité pendant
3 heures à une température voisine de 20C puis l'éthanol est
éliminé par évaporation ~ous pression reduite (2,7kPa) à 40C. L2
résidu est additionné d'un mélange de 50 cm3 d'eau et de 150 cm3
d'acétate d'éthyle. La phase aqueuse est séparée par décantati~n
puis réextraite par 2 fois 25 cm3 d'acétate d'éthyle. Les phases
organiques sont réunies, lavees p2r 3 fois 150 cm3 d'eau, séchées
sur sulfate de magnésium, filtrees puis ooncentrées à sec scus
pression réduite (2,7kPa) à 40C. On obtient ainsi 2,1 g de
[(dimethyl-3,3 piperidino)-2 ~Ko-2 éthyl] amin~-8 quinoléine scus
~orme d'une huile utilisee telle quell~ dans les synthèses
ulterieures.
Le N [(diméthyl-3,3 piperidino)-2 oxo-2 éthyl]
N-(quinolyl-8) trlfluaroacétamide peut être préparé de la maniere
suivante : à une sDlutic~ de 2,4 g db trifluorDaoetylaminc-8
quinolé me dans 40 cm3 de tétrahydrofuranne anhydre ma mtenue sous
atmosphere d'argcn, on ajoute, à une t ~ ture voisine de 10C,
0,6 g d'une suspension huileuse (50% en poids) d'hydrure de scdium
et agite la suspension obtenue pendant 30 minutes à une t ~ ture
voisine de 20C. on ajoute alors une sDlution de 3,5 g de bromo-2
1dime'thY1-3,3 piperidinD)-1 éthanone-1 dans 30 cm3 de
tétrahydrofuranne anhydre ~t chauffe à reflux , sous agitaticn
pendant 5 heures. Ie melange reacticnnçl est ensulte refroidi à une
temperature vDisine de 20C et versé dæns un mElan9e de 25 cn3
d'eau et de 100 cm3 d'aoe ~ate d'ethyle. La phase aqueuse est
separée par decantation et re'extIaite par 2 fois 25 om3 d'aoétate
35 d'éthyle. LRS phases organiqyes sont reunies, lavees par 3 fois 20
cm3 d'eau, se'chees sur su~fate de magnesiu~., filtr~es puis
39
concentrees à sec sous pression r~duite (2,7kPa) à 40C. L'huile
bbtenue est purifiée par chromatographie sur 100 g de silic2
(0,063-0,200mm) contenus dans une oolonne de 2,5 cm de diamètre
[éluant : dichlorcméthane-méthanDl ~98-2 en volumes)] en
recueillant des fractions de 20 cm3. Les fractions ne oontenant que
le produit cherché sont réum es et concentrées à sec s~us pression
réduite (2,7kPa) à 40C. On obtient ainsi 3,0 g de N-[(diméthyl-3,3
pipéridino)-2 oxc-2 éthyl] N-(quinolyl-8) trifluoroacétamide sous
forme d'une huile utilisée telle quelle dans les synthèses
10 Ultérieures~
La brcmo-2 (dime~hyl-3,3 piperidino)-1 éthanone-1 peut être
préparée de la manière suivante : à une solution de 11,3 g de
dimethyl-3,3 piperidine dans 65 cm3 de dichlorométhane, cn ajoute
successivement, à une t~mperature voisine de -5C, 11,1 g de
triéthylamine et une solution de 20,4 g de bromure de bromoacétyle
dans 10 cm3 de dichlorometh~ne. La suspension o~tenue est agitée
pendant 2 heures à une temperature voisine de 20C puis ~itionnee
de 25 cm3 d'eau. La phase aqueuse est séparée par décantation et
réextraite par 2 fois 15 cm3 de dichlorométhane. Les phases
20 ~rganiques scnt réunies, lavees par 3 fois 25 om3 d'eau, sechées
sur sulfate de magnésium, filtrees puis ccncentrées à sec ~ous
pression reduite (2,7kPa) à 40C. L'huile residuelle est
additionnée de 1~0 cm3 d'éther diéthylique anhydre ; le produit
insoluble est séparé par filtration et lavé par 3 fois 15 cm3
25 d'éther diéthylique. LRS filtrats sont réuni~ et ~oncentres à sec
sous pression reduite (2,7kPa) à 40C. On obtient ainsi 6,5 g de
brcmo-2 ~diméthyl-3,3 piperidino)-1 éthanone-1 ~ous forme d'une
h~ le utilisée telle quelle dbns les synthe`se~ ulterireures.
La tri1 ~ oetylamino-8 guinoléine pe~t être preparée de
30 la maniere suivante : à une solutis~ de 2,9 g d'amino-B quinolé me
dans 25 cn3 ~e pyridine, on ajcute à une t ~ ture voisine de
-20C, 4,2 g d'anhydride trifluoroacetique. Le me'lange reacticnnel
est agité pe~dant 3Q minutes à ~ne temperature voisine de -20C,
puis pen~ant 1 heure à une temperature vDisine de 0C et versé dans
35 150 cm3 d'eau refroidie à une tempe'rature Yoisine de 0C. Le
2074~r~
~o
produit insoluble est séparé par filtration, lavé par 5 fois 10 om3
d'eau et 6eche à l'air. On ~bt~ent ainsi 4,7 g de
trifluoroacétylamino-8 qyinoléine fondant à 84C.
EXEMæLE 25
.
A une solution de 0,56 g de méthylthio-3 aniline dans 10 cm3
de tétrahydrofuranne anhydre, maintenue sous atm~sphere d'argon, on
ajoute, à une t ~ ature v~isine de 20C, 1,5 g de N-nethyl
N-phenyl [N-(quinolyl-8) isocyanatoacétamido]-2 acétamide. La
solution obtenue est agitée pendant 16 heu¢es à une temperature
voisine de 20C puis ooncentree a sec sous pression reduite
(2,7kPa) à 40C. Cn obtient ainsi après recristallisation dans
l'acétonitrile, 0,65 ~ de N-méthyl [[(méthylthio-3 phényl)-3
~réid~]-2 N-(qym olyl-8) acétamido]-2 N-~henyl-acetamide fondant à
174C.
Le N-methyl N-phényl [N-(quinolyl-8) isccyanatoacétamido]-2
acétamude peut être préparé de la manière suivante : à une solution
de 1,6 g de chlcrure d'isocyanatoacétyle dans 25 cm3 d'éther
diéthylique anhydre maintenue sous atmosphère d'argon, o~ ajoute en
maintenant la temperature au voisinage de 5C, une solution de 2,9
g de N-méthyl N-pbenyl (quinolyl-8) amino-2 acétamide et de 0,8 g
de pyridine dans 200 cm3 d'éther diéthylique anhydre. Le mélan~e
réactionnel est ~gité pendant 4 heures à une temperature vDisine de
20C puis le produit insQluble est sépare par filtration et lavé
par 3 fois 10 cm3 de tetrahydrofuranne anhydre. LÆS filtrats sont
réunis et concentrés à s~c sous Fressicn ~eduite ~2,7kPa) à 40C.
On obtient ainsi 2,3 g de N ~ thyl N-phenyl tN-( ~ lyl-8)
isocyanatDa oetamldol-2 acétamide sous forme d'une huile utilisee
telle quelle dbns les synthe`ses ultérieures.
Le N-methyl N- p enyl (quinolyl-8) amlno-2 ace~amide peut
être prépaxé de la maniere æui~ante : à une sDlution de 2,4 g de
N-mRthyl N-phenyl [N-(qui~Dlyl-8 trifluorc~cetamido]-2 acetamide
dans 40 om3 d'éthanol, on ajoute, à une temQerature vDisine de
20C, 6,2 cm3 d'une solutlon aqueuse ZN d'hydroxyde de ~cdium. Le
- 20~3~
41
mélange réactionnel est agité à reflux pendant 30 minutes puis
1'éthanol est éliminé par évaporation sous pression réduite
(2,7kPa) à 40C. Le résidu est agité avec 50 cm3 d'eau et le
produit est séparé par filtratian pu$s recristall$sé dans
l'éthanol. on Gbtient ainsi 1,6 g de N-méthyl N-phényl (quinolyl-8)
amino-2 acétamide fondant à 169C.
Le N-méthyl N-phényl [N-(qyinolyl-8) trifluoroacétamido-2
acétamide peut être préparé de la man$ere suivante : à une solutian
de 1,2 9 trifluoroacétylamino-8 quinoléine dans 20 cm3 de tétrahy-
drofuranne anhydre maintenue sous atmosphère d'argon, on ajoute à
une température voisine de 10C, 0,25 g d'une suspension huileuse
(50~ en poids) d'hydrure de sDdium et agite la suspension cbtenue
pendant 30 minutes à une temperature voisine de 20C. On ajoute
alors une solution de 1,8 g de brcmo-2 N-méthyl N-phényl-acétamide
dans 15 cm3 de tétrahydrofuranne anhydre et chauffe à reflux, scus
agitation pendant 5 heures. Le mélange réactionnel est ensuite
refroidi à une temperature voi~ine de 20C et versé dans un mélange
de 25 cm3 d'eau et de 50 cm3 d'acetate d'éthyle. La phase aqyeuse
est sépar~ par d ~ tation et re'extraite par 2 fois 25 cm3 d'ace-
tate d'éthyle. Les phases organigues sont réunies, lavées par 3foi5 20 cm3 d'eau, sechées sur sulfate de ma ~ ium, filtré~s puis
concentrées à sec sous pression ~ ite (2,7 kPa) à ~0C. L'huile
obtenue est purifiée par chromatDgraphie sur 75 g de silice
(0,063-0,2mm) contenus dans une oolonne de 2,5 om de diametre
(eluant : dichlorcmethane) en recueill~nt des fractions de 10 cm3.
IRS fracticns ne c~ntenant que le produit cherch'e sont reunies et
conoentrees à sec ~ous pression r~duite (2,7kPa) à 40DC. Qn cbtient
ainsi, 1,7 g de ~-methyl N-phfinyl [N-(guinolyl-8)
trifluo¢nacétamido]-2 acé~amdde sous ~orme d'une huile utilisée
telle quelle dans les synth`ese~ ulterieures.
1e brcmo-2 N-méthyl N-phenyl-acetamdde peut être preparé de
la nanière suivante : à une s~lution de 10,7 g de ~me'thyl-aniline
dans 65 cm3 de dlchlorcméthane, on ajaute sucoessivement, à une
temperature voisine de -5C, 11,1 g de triéthylamine et une
solution de 20,4 g ~e bromure de bromoacetyle dans 10 cm3 de
~7~
42
dichloromethane. La suspensian obtenue est agitée pendant 2 heures
à une température voisine de 20C puis additiannée de 25 cm3 d'eau.
La phase aqueuse est séparée par décantation et reextraite par 2
fois 15 cm3 de dichlorométhane. Les phases organiques sont réunies,
lavées par 3 fois 25 cm3 d'eau, séchées sur sulfate de magnésium,
filtrées puis concentrées à sec sous pression réduite (2,7kPa) à
40C. L'huile résiduelle est additionnée de 100 cm3 d'éther
diéthylique anhydre ; le produit insoluble est séparé par
filtration et lave par 3 fois 15 cm3 d'éther diéthylique. Les
filtrats sont réunis et ooncentrés à sec sous pression réduite
(2,7kPa) à 40C. On obtient ainsi 20,5 g de brommo-2 N-méthyl
N-phényl-acétamide sous forme d'une huile ut~lisée telle quelle
dans les synthèses ulterieures.
EXEMPLE 26
A une solution de 0,8 g d'[amino-2 N-(quinolyl-5)
acétamido]-2 N-méthyl N-phényl-acétamide dans 70 cm3 de
tétrahydrofuranne anhydre, on ajoute, à une te ~ ture voisine de
20C, 0,34 g d'isocyanate de méthyl-3 phényle. La solution obtenue
est agitée pendant 3 heures à une temæerature voisine de 20C puis
cGncentrée à sec sous pression réduite (2,7kPa) à 40C. L'huile
résiduelle est p~rifiée par chromatographie sur 60 g de silice
(0,063-0,2mm) contenus dans une colonne de 2om de diamètre
(éluant : acétate d'éthyle) en necueillant des fractions de 20 cm3.
LRS fractions ne oontenant que le prvduit cherche' sont réunies et
ooncentrees à sec sous pression reduite (2,7kPa) à 40C. an sbtient
zprès rec~istallisation dbns l'acétate d'~thyle, 0,67 g de N-~éthyl
[[(methyl-3 phenyl)-3 ureido~-2 N-(quinQlyl-5) acetamido~-2
N-phényl acétamide fondant à 206C.
L'1amino-2 N-(qyinDlyl-5) acétamid~-2 N-methyl N~phenyl
acetamide peut être pré ~ db 1~ mani~re sui~ante : à une soluticn
de 2,3 g de N-méthyl N~phe'nyl ~pht limidb-2 N~(~yinolyl-5)
acetamido]-~ ace~amide dans 100 cm3 de me~hanal, cn ajoute 0,72 9
d'hydrate d'hydrazine. Le me'lange reacticnnel est a3ité pendant 3
~71~1 3^j~
43
heures à une temperature voisine de 20C pUi5 ooncentré à sec sous
pression re'duite (2,7kPa) à 40C. On obtient ainsi 1,3 g d'[a ~ 2
N-(quin~lyl-5) acé~amido]-2 N-methyl N-phényl-acétamide fondant à
157C.
Le N-méthyl N-phényl [phtalimido-2 N-~quinolyl-5)
acétamido]-2 acétamide peut être préparé de la manière suivante : à
une solution de 5 g de phtalimidb-2 N-(quinolyl-5) acétamide dans
100 om3 de tétrahydrofuranne anhydre maintenue SGUS atmosphère
d'argon, on ajoute, à une temæerature voisine de 10C, 0,72 g d'une
suspension huileuse (50% en poids) d'hydrure de scdium et agite la
suspensicn ohtenue pendant 1 heure à une temperature voisine de
20C. Qn ajoute alors une solution de 5,1 g de brcmc-2 N-méthyl
N-phényl-acétamide dans 100 cm3 de tétrahydrDfuranne anhydre et
1'on poursuit l'agitaticn pendbnt 3 heures à une température
voisine de 20C. Le mélange réacticnnel est ensuite versé dans un
mélange refroidi à une température voisine de 0C, de 60 cm3 d'eau
et de 200 om3 d'acétate d'éthyle. La phase aqueuse est séparée par
decantation et reextraite p~r 2 fois 20 cm3 d'acétate d'éthyle. Les
phases organiques sont réunies, lavées par 3 ~ois 50 om3 d'eau ,
se'chées sur sulfate de magnesium, filtrees puis concentre'es à sec
sous pressian re'duite (2,7kPa) à 4~C. an obtient ainsi, après
recristallisation dans l'acétate d'éthyle , 3,4 g de N-méthyl
N-phényl [phtalimido-2 N~quinolyl-5) aoet ~ do~-2 acétamide
fondant à 208C.
Le phtalimudb-2 N-(qyin~lyl-5) ~ tamide peut être préparé
de la nanlère suivante : à une solution db 4,3 g d'aminc-5
qulnoléine dans 60 cm3 de dichloromethane maintenue sçus at ~ ere
d'argon,. ~n ajoute 3,9 9 de triéthylamine pui~ en maintenant la
temperature au v~isinage de 20C, une ~olution de 8,1 g de chlorure
de phtalimldb-2 acetyle dbns 60 am~ de dichloromethane. La
~uspensian obtenue est agitee pendant 1 ~eure à une tempe'rature
v~isine de 20C. Le solide f~rmé est sépare par filtration, lavé
par 3 fois 50 cm3 d'eau et se'che' à l'air. Qn obti~nt ainsi, 9,9 g
de phtallmldb-2 N-~quin~lyl-5) acetamide.
2~7~3~7
~4
EXEMPLE 27
A une solution de 1,8 g de N,N'-carbcnyldiimldazole dans 25
om3 de dichloro-1,2 éthane, cn ajoute à une temperature voisine de
20C, une soluticn de 3,2 g d'[amino-2 N-(quinolyl-8) acétamido]-2
acétate de tert-butyle dans 30 ~m3 de dichloro -1,2 éthane et agite
la solutiGn bbtenue pendant 2 he.ures à une température voisine de
20C. On ajoute alors une solution de 1,4 g de méthylthio-3 aniline
dans 10 cm3 de dichlorc-1,2 éthane et l'cn poursuit l'agitatian au
reflux du solvant pendant 5 heures. Le mélange réactionnel est
ensuite refroidi à une température voisine de 10C puis additionné
de 100 cm3 d'eau. La phase aqueuse est séparée par decantation puis
rée~traite par 2 fois 80 cma de dichloromethane. LRS phases
organiques ænt réunies, lavées par 50 om3 d'eau, puis par 100 cm3
d'une solution aqueuse 0,5 ~ d'acide chlorhydrique et par 2 fois 80
cm3 d'eau, sechées sur sulfate de nagnésium, filtrées puis
ooncentr es à sec scus pression reduite (2,7kPa~ à 40C. L'hulle
obtenue est purifiée par chromatographie sur 200 g de silice
(0,063-0,2mm) oontenus dans une oDlonne de 2,5 cm de diamètre
[éluant : dichlonométhane-methanol (98-2 en vDlumes)] en
recueillant des fractions de 30 cm3. Les fractions ne oontenant que
le produit cherché sont xe~ni~s et ccncentrees à sec sous pression
réduite (2,7kPa) à 40C. On obtient après recristallisation dans
l'acétonitrile, 3,0 g de [t(méthylthio-3 phenyl)-3 uréldo]-2
N-(quinolyl-8) acét~mido]-2 acetate de tert-butyle fcndant à 165C.
EXEMPLE 28
En oQerant d'une ~ ere analogue a celle decrite a
l'exemple 24, mais à partir de 4,4 g de me~hyl-3 aniline et de 4 g
d'i ~ anato-~ N-[oxo-2 (pyrrolidinyl-1)-2 éthyl] N-(quinolyl-8)
acé~amide, cn obtient apres recristallisaticn dans l'a~étonitrile,
0,7 g de [(methyl-3 phenyl~~3 uréid~]-2 N-1OKQ-2 (pyrTolidinyl-1)-2
éthyl] N-(quinDlyl-8) acetanide fc~dant a 227C.
~07~3.~J~
L'isocyanato-2 N-[owo-2 (pyrrolidinyl-1)-2 éthyl]
N-(quinolyl-8) acétamide peut être préparé d'une m~nière analogue à
celle décrite à l'exemple 24 pour la préparation du N-[diméthyl-3,3
pipéridino)-2 owo-2 éthyl] isocyanato-2 N-(quinolyl-8) acétamide,
mais à partir de 0,65 g de chlorure d'isocyanatoa oe tyle, de 1,1 g
de [oxo-2 (pyrrolidinyl-1~-2 éthyl] amino-8 quinoléina et de 0,34 g
de pyridine. On obtient ainsi 1,4 g d'isocyanato-2 N-1oxo-2
(pyrrolidinyl-1)-2 éthyl] N-(quinolyl-8) acétamide sous forme d'une
huile utilisée telle quelle dans les synthèses ultérieures.
L'[(owc-2 pyrrolidinyl-1)-2 éthyl~ amino-8 quinoléine peut
être préparée d'une manière analogue à celle décrite à l'exemple 24
pour la préparation de la [(dimethyl-3,3 piperidino)-2 oxo-2 éthyl]
amino-8 quinoléine, mais à paItir de 1,9 g de N-[oxo-2
~pyrrolidinyl-1)-2 éthyl3 N~(quinolyl-8) trifluoroacétamide et de
S,8 cm3 d'une soluticn aqueuse 2N d'hydroxyde de sodium. ~n obtient
ainsi, 1,1 g d'[owo-2 (pyrrDiidinyl-1)-2 éthyl] amino-8 quinoléine
fondant à 178C.
Le N-[owo-2 (pyrrolidinyl-1)-2 éthyl] N-(quinolyl-8)
triflu!oroacétamide peut être préparé d'une maniere analogue à celle
decrite à l'exemple 24 pour la préparatian de la N-[(diméthyl-3,3
pipéridino)-2 oxo-2 éthyl] N-(quinolyl-8) trifl~oroacétamide, mais
à partir de 2,5 g de trifluoroacetylaminc-2 quinoléine, de 0,6 g
d'une suspension huileuse (50~ en poids) d'hydrure de sodium et de
2,4 g de bromo-2 ~pyr~olidinyl-1)-1 éthanane-1. on obtient ainsi
1,9 g de N-[oxo-2 (pyrrolidinyl-1)-2 éth~l] N-(quinolyl-8)
trifluoroacétamide sous ~crme d'u~e huile utilisée telle qyelle
dans les synthe`ses ulterieunes.
Ls`brcmo-2 (pyrrolidinyl-1)-1 éthancne-1 peut être préparee
d'une manière analogue a celle decrite à l'exemple 24 pour la
30 P¢ ~ tion de la b¢cmc-2 (dimethyl-3,3 pipéridino)-l éthanone,
mais à partir de 7,1 g de pyrrolidine, de 11,1 g de ~riéthylamine
et de 20,4 g de b¢omure de bronoacétyle. on obtient ainsi 9,1 g de
bro00-2 (pyrrDlidinyl-1)-1 éthanane-1 s~us f~rme d'une huile
utilisee telle quelle dans l~s synth`eses ultérieures.
46 2~3~7
EXEMPLE 29
A une solution de 0,6 g d'[aminc~2 N-(qyinolyl-7)
acétmido]-2 acétate de tert-butyle dans 15 cm3 de tétrahydrofuranne
anhydre, cn ajoute, à une température voisine de 20C, 0,4 g
d'isocyanate de méthyl-3 phényle. ~ soluticn obtenue est agitée
pendant 3 heures à une température voisine de 20C puis concentrée
à sec sous pression réduite (2,7kPa) à 40C. L'huile résiduelle est
purifiée par chrcmatographie sur 40 g de silice (0,063-0,2mm)
contenus dans une colonne de 2 om de diamètre (éluant : acétate
d'éthyle) en recueillant des fractions de 20 cm3. Les fractions ne
contenant que le produit cherché sont réunies et ooncentrées à sec
sous pression réduite (2,7kPa) à 40C. On obtient ainsi après
recristsllisation dans l'acétate d'éthyle, 0,37 g de [[(méthyl-3
phényl)-3 uréido]-2 N-(quinolyl-7~ acétamido]-2 acétate de
tert-butyle fondant à 165C.
L'[amino-2 N-(quinolyl-7) acétamido]-2 acétate de
tert-butyle peut être préparé de la manière sulvant : à une
solution de 1,3 g de [phtalimido-2 N-(quinDlyl-7) acétamido]-2
acétate de tert-butyle dan~ 40 om3 de methanDl, on ajoute 0,6 g
d'hyd~ate d'hydIazine. Le melange reactionnel est chauffé à reflux
pendant 3 heures pui~ ocncentre à sec sous pression réduite (2,7
kPa) à 40C. L'hulle résiduelle est di5scute dans 50 cm3 d'eau. La
phase aqueuse est separee par decantaticn puis reextraite par 2
fois 30 cm3 d'acétste d'éthyle. L~s pha~es organiques scnt réunies,
lavees par 3 fois 15 cm3 d'eau, sechees sur sulfate de magnesium,
filtrees puis ooncentrées à sec 50U5 pres~ion redNite (2,7kPa) à
40C. On bbtient ainsi 0,65 g d'~amino-2 N-(quinolyl~7)
acétamido]-2 acetate de telt-butyle sous foxme d'une huile utilisee
telle quelle dbns les synthe`ses ulterieures.
le [phtalimidb-2 N~(~uinDlyl-7) acé~amido~-2 acetate de
tert-butyle peut être prepare de la nanlere suivante : a une
~olution de 3,4 g de phtalimidb-2 N-(quinolyl-7) aoetanide dans 100
cm3 de tétrahydr~furanne anhydre maintenue sous atmc~pbe`re d'argon,
on ajoute, à une temp'erature voisine de 10C, 0,53 g d'une
47 2~7~3~
suspension huileuse (50% en poids) d'hydrure de sodium et agite la
suspension obten~e pendant 2 heures à une temperature voisine de
20C. On ajoute alors une solution de 2,1 g de brcmoacétate de
tert-butyle dans 10 cm3 de tétrahydrofuranne anhydre et l'on
poursuit l'agitation penaant 16 heures à une températue voisine de
20C. Le mélange réactionnel est ensuite versé dans 50 cm3 d'eau,
le solvant est éliminé par évaporation sous pression réduite
(2,7kPa) à 40C, et l'on rajoute un mélange de 100 cm3 d'acétate
d'éthyle et de 50 cm3 d'eau. La phase aqueuse est séparée par
décantation puis réextraite par 2 fois 100 om3 d'acétate d'éthyle.
Les phases organiques sont réunies, lavées par 3 fois 80 cm3 d'eau,
séchées sur sulfate de magnésium, filtées puis concentrées à sec
sous pression reduite (2,7kPa) à 40C. On obtient ainsi après
recristallisation dans l'acétate d'éthyle, 1,4 g de [phtalimido-2
N-(quinolyl-7) acétamido]-2 acétate de tert-butyle fondant à 213C.
Le phtalimido-2 N-(quinolyl-7) acétamide peut être préparé
de la manière suivante : à une solution de 3,0 g d'amins-7
quinoléine dans 80 cm3 de dichlorcmethane maintenue scus atmosphère
d'argon, on ajoute 2,1 g de triéthylamine puis en maintenant la0 temperature au voisinage de 20C, une solution de 4,7 g de chlorure
de phtalimidc-2 acétyle dans 50 cm3 de dlchloromethane. La solution
obtenue est a~itée pendant 4 heures à une temperature voisine de
20C, puis additionnee de 50 cm3 d'eau. Le solide forme est séparé
par filLL~tion, lavé par 2 fois 25 cm3 d'oxyde de diisopropyle puis
par 4 fois 50 cm3 d'eau et séche à l'air. On abtien,t ainsi 3,5 g de
phtalimldb-2 N-(quinolyl-7) acétamlde ~ondant à 259C.
L'amlno-7 qulnoléine peut être préparée selcn la methode
decrite p~r F. LINSKER et R.L. EV~NS., J. Amer. Chem. Soc., 68,
149-150 (1946).
EXEMæLE 30
En cperant d'une manière analogue à celle decrite à
l'exemple 29, mais à partir de 3,0 g d'[aminc-2 N-~quinDlyl-fi)
acetamido]-2 acetate de tert-butyle et de 1,06 g d'isocyanate de
~7~3~7
48
méthyl-3 phényle, on obtient apres recristallisation dans l'oxyde
de diiscpropyle, 0,9 g de [~(méthyl-3 phényl)-3 uréido]-2
N-(quinolyl-6~ acétamido]-2 a~étate de tert-butyle fondant à 105C.
L'[amlno-2 N-(quinolyl-6) acétamido]-2 acétate de
tert-butyle peut être préparé d'une manière analogue à celle
décrite à l'exemple 29 pour la préparation de l'~amino-2
N-(quinolyl-7) acétamido~-2 acétate de tOE t-butyle, mais à partir
de 3,3 g de [phtalimidb-2 N-(quinolyl-6) acétamido~-2 acétate de
tert-butyle et de 1,2 g d'hydrate d'hydrazine. On obtient ainsi 3,0
g d'[amino-2 N-(quinolyl-6) acétamido~-2 acétate de tert-butyle
sous forme d'une huile utilisée telle qyelle dans les synthèses
ultérieures.
~ e [phtalimido-2 N-(quinolyl-6) acétamid~]-2 acétate de
tert-butyle peut être préparé d'une manière analogue à celle
décrite à l'exemple 29 pour la p¢éparatian du [phtalimidç-2
N-(quinolyl-7) acétamid~]-2 acétate de tert-butyle, mais à partir
de 9,7 g de ~ talimidb-2 N-~quinolyl-6) acétamide, de 1,55 g d'une
~ sicn huileuse ~50% en poids) d'hydrure de sodium et de 6,05 g
de bromoacétate de tert-butyle. On obtient ainsi, 3,4 g de
[ph~alimidb-2 N-(guinolyl-6 acétamido]-2 ac~tate de tert-butyle.
Le phtalimidb-2 N-(quinoly-6) acétamide peut être préparé
d'une manière analogue à celle decrite à 1'exemple 29 pour la
préparation du phtalimidb-2 N-(qui~olyl~7) acétamide, mais à partir
de 4,35 g d'aminc-6 quinoléine, de 3,3 ~ de triéthylamine et de
7,2 y de chlorure ~e Fhtalimido-2 acétyle. On dbtient ainsi 9,8
de ptalimldk-2 N-(qyinDlyl-6) acétamide.
EXEMPLE.31
En ~ t d'une nanlere analogue à celle décrite à
l'exemple 24, nals à partix de 0,64 g de methyl-3 aniline et de 2,5
9 d'isocyanato-2 N-[tétrahydro-1,2,3,4 quinolyl-1)-2 oKo-2 ethyl]
N-(quinolyl-8) acetamide, cn cbtient apres recristallisation dans
l'acetonitrile, 1,6 g de [[(methyl-3 phenyl)-3 uréido]-2
N-[(t~trahydro-1,2,3,4 quinolyl-1)-2 oxc~2 éthyl] N-(guinDlyl-8)
- 2~74~57
49
acetanlde fondant à 187~C.
L'isocyanato-2 N-[(tétrahydro-1,2,3,4 qyinolyl-1)-2 oxo-2
éthyl] N-(quinoly-8) acétamide peut etre prépare d'une manière
analogue à celle décrite à l'exemple 24 pcur la préparation de
l'isocyanato-2 N-[(diméthyl-3,3 piperidino)-2 owo-2 éthyl]
N-(quinolyl-B) acétamide, mals a partir de 0,85 g de chlorure
d'isocyanatoacétyle, de 1,9 g de E (tétrahydro-1,2,3,4 quinolyl-1)-2
oKc-2 éthyl] amino-8 quinoleine et de 0,48 g de pyridine. On
obtient ainsi 2,6 g d'isocyanato-2 N-l(tétrahydro-1,2,3,4
quinolyl-1)-2 oxo-2 éthyl] N-(quinolyl-8) acétamide sous forme
d'une huile utilisée telle quelle dans les synthèses ultérieures.
La [~tétrahydro-1,2,3,4 quinolyl-13-2 oxc-2 éthyl] amino-8
quinoléine peut être préparee d'une manière analogue à celle
decrite à l'exemple 24 pour la préparation de la ~(diméthyl-3,3
pipéridino)-2 oxo-2 éthyl] amlnc-8 quinoléine, mals a p ~ ir de
2,8 g de N-tquinolyl-8) N-[(tetrahydro-1,2,3,4 quinDlyl-1)-2 oxo-2
éthyl] trifluoroacétamide et de 6,7 cm3 d'une solution aqueu3e ZN
d'hydroKyde de ~odium. On obtient ainsi, apres recristallisaticn
dans l'acétate d'éthyle, 2,0 g de l(tétxahydrc-1,2,3,4
quin~lyl-1)-2 oxo-2 éthyl~ aminc-8 quinDléine fondant à 128C.
Le N-(~uinoly-8) N-[(tétrahydro-1,2,3,4 qyin3lyl-1)-2 owo-2
éthyl] trifluoroacetamide peut être préparé d'une manière d ogue
à celle décritè à l'~xemple 24 pour la preparation du
N-l(diméthyl-3,3 piperidinD)-2 oxo-2 éthyl] N~(quinolyl-8)
trifluoroacétamide, mais à partir de 2,4 g de
trifluoroacétylamino~8 quin~léine, de 0,6 g d'une suspensicn
huileuse (50% en p~ids) d'hydrure de sodium et de 3,75 g de b¢onc-2
(tétrahy ~ 1,2,3,4 quinolyl-1)-1 éthanone-1. on obtient ainsi 2,8
g de N-(quinolyl-8) N-[(tétrahydrc-1,2,3,4 quinolyl-1) 2 oxD~2
30 ethyl] trifluoroacetamide sous forme d'une hNlle utili~ee telle
qyelle dans les synth`eses ulter~eures.
La brons-2 (tétrahydxc-1,2,3,4 qyinDlyl-1) éthancne peut
être préparee d'une mzniere analogue à celle decribe à l'exemple 24
pour la preparation de la b~omo-2 (dime'thyl-3,3 piperidino~-1
35 éthanone-1, mais a partir de 13,3 g de tétrahydro-1,2,3,4
`-- 2~7~3~
quinoléine, de 11,1 g de triéthylanine et de 20,4 g de brcmNre de
brcmoacétyle. On obti~nt ainsi 20,3 g de brcmo-2
(tétrahydro-1,2,3,4 quin~lyl-1)-1 éthanone-1 scus forme d'une
huile utilisee telle quelle dans les synthèses ultérieures.
EXEMæLE 32
En opèr,mt d'une manière analogue à celle décrite à
l'exemple 24, mais à partir de 0,43 g de méthyl-3 aniline et de 1,5
g d'isccyanato-2 N-(piperidino-2 oxc-2 éthyl) N-(quinolyl-8)
acétamide, on obtient après recristallisation dans l'acetonitrile,
0,8 g de [~methyl-3 phenyl)-3 uréido]-2 N-(pipéridino-2 cxs-2
éthyl) N-(quinDlyl-8) acetamide fondant à 183C.
L'isocyanato-2 N-(pipéridino-2 oxc-2 éthyl) N-(quinolyl-8)
acétamide peut être préparé d'une manière analcgue à celle décrite
à l'exemple 24 pour la préparation de l'isocyanato-2
N-[(dimethyl-3,3 pipéridino)-2 cxc-2 éthyll N-( ~ olyl-8
acétamide, mais à partir de 0,75 g de chlo~ure d'isocyanatoacetyle,
de 1,4 g de (pipéridino~2 oxc-2 éthyl) amino-8 qy~noléine et de
0,42 g de pyridine. ~n obtient ainsi 1,5 g d'isocyanatc-2
N-(piperidino-2 owo-2 éthyl) ~-(quinolyl-8) acétamide sous forme
d'une huile utilisee telle quelle dbns les syntheses ulterieures.
La (pi ~ idinc-2 oxc-2 éthyl) aminc-8 quinDléine peut etre
préparée d'une manière analogue à celle decrite à l'exemple 24 pour
la préparation de la [(dimethyl-3,3 pipéridiro)-2 oxc-2 éthyl]
amino-8 quin~léine, n2~.s à partir de 2,55 g de N-[(pi ~ idino-2
oxo-2 éthyl)] N-(quinolyl-8) trif~uoroacétamide et de 7,U cm3 d'~e
solution ~queuse ZN d'hydnoKyde de sodium. on obtient ainsi, apres
recristallisation dar~ l'aoe tate ~'éthyle, 1,5 g de (piperidino-2
oxc-2 ethyl~ amino-8 quinoléine fc~dant à 135C.
Le N~[(pi ~ idinc-2 CXD-2 éthyl~] N-(quinolyl 8)
trifl ~ tamide p~ut être prépare d'une n~niere ~nalogue à celle
decrite ~ l'exemple 24 pour la préparation du ~-~(dim'ethyl-3,3
piperidino)-2 owo-2 éthyl] N-(qyin~lyl-8) trifluoroacétamide, mais
à partir ~e 2, 4 g de trifl~tylarnin~8 quin~léir~, de 0, 6 g
51 20~3~ ~
d'une suspension huileuse (50% en poids) d'hydrure de sodium et de
3,1 g de ~romc-2 piperidinc-1 é ~ ne-1. on Gbtient ainsi 2,55 g
de N-l(pipéridinc-2 oxo-2 éthyl)~ N-(quinolyl-8) trifluoroacétamide
sous fonme d'une huile utilisee telle quelle dans les synthèses
ulterieures.
La bromo-2 pipéridino-1 éthanone-1 peut être préparée d'une
manière analogue à celle décrite a l'exemple 24 pour la préparation
de la bromo-2 (diméthyl-3,3 piperidino)-1 é ~ ne-1, mais à partir
de 8,5 g de piperidine, de 11,1 g de triéthyl2mine et de 20,4 g de
bromure de brcmoacétyle. On obtient ainsi 7 g de bromc-2
piperidinc-1 éthanone-1 sous forme d'une huile.
EXEMPLE 33
On opere d'une maniere analogue a celle décrite à l'exemple
1, mais à partir de 2,~5 g de [phtalimido-2 N-(iscquinolyl-4)
acétamido]-2 acétate de tert-butyle, de 0,75 g d'hydrate
d'hydrazine et de 0,68 g d'isocyanate de methyl 3 pényle. Le
produit brut obtenu est chroma ~ ié sur 25 g de silice
(0,063-0,2mm) oontenus dans une colonne de 1,6 cm de diamètre
(éluant : dichloromethane3 en recueillant des fractions de 10 cm3.
LRS fractions 48 à 97 sont réunies et ~ ntrees à sec sous
presssion réduite (2,7kPa) à 50C. Cn ~btient, après
recristallisation dans l'éther de petrole, 1,35 g de {[(méthyl-3
phenyl)-3 uréid~]-2 N-(isDquin3lyl-4) acetamido}-2 acétate de
tert-butyle fordant à 100C.
Le [phtalimidb-2 N-(isoqyinolyl-4) acetamidD~-2 acétate de
tert-butyle peut être bbtenu d'une manière a~alogue à oelle décrite
à l'exemple 1 pcur la preparation dN [phtalimidb-2 N~(pyridyl-3)
acetami~3]-2 acétate de tert-butyle, mais à partir de 1,3 g de
N-(isDquinolyl-4~ phtalimidb-2 acetamide, de 0,24 g d'une
suspensicn huileu~e (50% en poids) d'hydrure de sodium et de 0,78 ~
de bromoaoetate de tert-butyle. On bbtient ainsi, 0,85 g de
[phtalimido-2 N-(isoquinDlyl-4) acetamid3]-2 acetate de tert-butyle
scus form~ d'une resine utilisee ~elle quelle dans les synthèses
2 ~
52
ultérieures.
Le phtalimido-2 N-(isoquin~lyl-4) acétamide peut être
préparé d'une manière analogue à celle décrite à l'exemple 1 Fcur
la préparation du phtalimidc-2 N-(pyridyl-3) acétamide, mais à
partir de 1,45 g d'amino-4 isoquinDléine, de 1,0 g de triéthylamine
et de 2,25 ~ de chlorure de phtalimidk-2 acétyle. On obtient ainsi
1,6 g de phtalimido-2 N-(is~quinolyl-4) acétamide fondant au
dessus de 260C.
EXEMPLE 34
En opérant d'une maniere analogue à celle décrite à
l'exemple 1, mais à partir de 4,45 g d'[amino-2 N-(quin~lyl-8)
acétamido]-2 acétate de tert-butyle et de 1,37 g d'isocyanate de
methoxy-3 phenyle, on obtient 1,6 g de {[(methoxy-3 ~hényl)-3
uréido]-2 N-(qyinolyl-8) acétamido)-2 acétate de tert-butyle
fcndant à 149~C.
EXEMPLE 35
A une sDlution de 1,7 g de mé~hyl-3 aniline dans 100 cm3 de
tétrahydrofuranne anhydre, maintenue sous abmosphe`re d'argon, an
ajoute, à une tè ~ ature v~i5ine de 20C, 3,5 g de N-benzyl
N-(isocyanatoacétyl) glycinate de tert-butyle. La solution obtenue
est agitee pendant 3 heures à une temperature voisi~e de 20~C ~ s
concentree à sec sous pression reduite (2,7 kPa) à 40C. on obtient
après recristallisation du residu dans l'aoetate d'ethyle, 2,2 g de
1[(méthyl-3 phe'nyl)-3 uréido~-2 N-benzyl-acétamid~]-2 acetate de
tert-butyle ~ondant à 117C.
Le N-benzyl N-(isocyanatoac~tyl) glycinate de tert-butyle
peut être preparé de la manie~e suivante : à ~ne solution ~e 1~2 g
de chlarure d'isocyanatoac~tyle dbns 50 om3 d'eth~r dié~hylique
anhydre maintenue ~ous atmosph`ere d'argon, on ajoute en maintenant
la tempexature au vDisinage de 10C, une solution de 2,21 g de
kenzylamino-2 acétate de tert-butyle et de 0,8 g de pyridine dans
7~3~,~
53
25 om3 d'ether diéthylique anhydre. Le mélange réactionnel est
agité pendant 2 heures à une température voisine de 20C puis le
produit insoluble est séparé par filtration et lavé par 15 cm3
d'éther diéthyliqye anhydre. I~ filtrats sant réunis et cancentrés
à sec sous pressi~n redulte (2,7 kPa) à 40C. On abtient ainsi
3,5 g de N-benzyl N-(isocyanatDacétyl) ylycinate de tert-butyle
sous forme d'une huile utilisée telle quelle dans les synthèses
ultérieures
.
Le benzylamino-2 acé~ate d& tert-butyle peut être préparé de
la manière suivante : à une solution de 21,4 g de benzylamine dans
200 om3 d'acétonitrile, on ajoute 19,5 g de ~romocétate de
tert-butyle. La solution obtenue est chauffée à reflux ~ ant 4
heures. Après refroidissement, le produit insoluble est séparé par
filtration et lavé par 3 fois 20 om3 d'acetGnitrile. Les filtrats
scnt réunis et ooncentres à sec scus pressian reduite (2,7 KPa) à
40C. On abtient ainsi 44,7 g de benzylamino-2 acetate de tert-
butyle sous forme d'une huile utilis e telle quelle dans les
syntheses ultérieures.
LR chlorure d'isocyanatoacétyle peut être préparé selon la
méthode decrite par Y0SHIO I~KURA, KEIKICHI UNO et SANWM KANG, J.
Org. Chem., 30, 1158 (1965).
EXEMPLE 36
A une solution de 6 g de [phtalimidb-2 N-~phenyl-1 éthyl)
acetamido]-2 aoe tate de tert-butyle dbns 100 cm3 de methanol cn
ajoute, à une temperature voisine de 20C, 2,17 g d'hydrate
d'hydrazine. Le mélange réactionnel est agite pendbnt 3 heures a
une température voisine de 20C puis ocncentre' à sec ~ ~ pression
re'duite (2,7 kPa) à 40Co L'huile ~ idNelle e~t dissoute dans
50 cm3 d'acetate d'éthyle et la solution obtenue est additicnnee de
50 cm3 d'eau. La phase agueuse est separee par de'ca~tati~n et
ree~traite par 2 fois 25 om3 d'acetate d'éthyle. Ies phases
oryaniques sont reunies, lavees par 2 ois 15 cm3 d'eau, seshees
sur sulfate de magnesium, filtrees puis ooncentrees à sec
-" 2~3~
~4
scus pression reduite (2,7 kPa) à 40C. Le résidu est dissous dans
30 cm3 de tétrahydrofuranne et on ajoute à cette solution 1,89 g
d'isocyanate de méthyl-3 phényle. La solution obtenue est agitée
pendant 1 heure à une temperature voisine de 20C puis concentrée à
sec scus pression réduite (2,7 ~Pa) à 40C. Le résidu est purifié
par chromatographie sur 150 g de silice (0,063-0,2 mm) contenus
dbns une colonne de 2 cm de diamètre [éluant : cyclohexane-acétate
d'éthyle (50-50 en volumes)] e~ recueillant des fractions de
20 cm3. Les fractions ne contenant quP le produit cherché sont
réunies et concentrées à sec sous pression réduite (2,7 kPa) à
40C. On obtiPnt après recristallisation dans l'oxyde de
diisopropyle, 1,7 g de [[(méthyl-3 Fhényl)-3 uréido]-2 N-(phényl-1
éthyl) acétamido]-2 acétate de tert-butyle fondant à 120C.
Le [phtalimidc-2 N-(phenyl-1 éthyl) acétamido]-2 acétate de
tert-butyle peut être préparé de la manière suivante : à une
solution de 4,2 g de (phényl-1 éthyl)aminc-2 acétate de tert-butyle
dans 60 cm3 de dichloro-1,2 éthane, maintenue sous atmosphère
d'argcn, cn ajoute 2,15 g de triéthylamlne puis goutte à goutte, à
une température de 20C, une solution de 4 g de chlorure de
phtalimido-2 acétyle dans 20 cm3 de dichloro-1,2 éthane. La
soluti~n obtenue est agitée pendant 3 heures à une temperature
voisine de 20C, puis additionnee de 50 cm3 d'eau. La phase aqueuse
est séparée par decantation puis r~extraite par 2 fois 50 cm3 de
dichlorc-1,2 ét~ne. LRS phases organiques sont réunies, lavées par
2 fois 10 cm3 d'eau, sechées sur sulfate de magnesium, filtrées
puis ccncentrées à sec scus pression réduite t2,7 kPa) à 40C. On
obti~nt ainsi, après recr1stallisaticn dans l'oxyde de
diisoprDp~le, 6 g de [phtallmidb-2 N-(phényl-1 éthyl) acé~am~do]-2
acétate de tert-butyle fcndbnt à 131C.
Le (phenyl-1 éthyl)amm o-2 acétate de tert-butyle peut être
préparé de la manière suivante : à une solution de 6,1 g de
phenyl-1 éthylamine dans 60 om3 d'acétonitrile, on ajoute a une
température vois me de 20C, 2,1 g d'hydrogénocarbonate de ~cdium
puis 4,9 g de brcmoacétate de tert-butyle. Le melange ~eactionnel
est chauffé à reflux pendant 48 heures puis
7~7
refroidi à une température voisine de 20C. L'insoluble est séparé
par filtration et lavé par 50 cm3 d'acétonitrile. IRS filtrats scnt
réunis et concentrés à sec scus pression réduite (2,7 kPa) à 40C.
On obtient ainsi 5,5 g de (phényl-1 éthyl)amlnc-2 acétate de
5 tert-butyle sous forme d'une huile utilisée telle quelle dans les
synthèses ultérieures.
EXEMYLE 37
A une suspension de 2,6 g d'acide [(méthyl-3 phényl)-3
uréido]-2 acétique dans 230 om3 de dichloro-1,2 éthane, maintenue
10 sous atmosphère d'argon, an ajoute, à une température voisine de
20C, 2 g d'isopropylaminL-2 acétate de tert-butyle. Le melange
réactionnel est chauffé sous agitation jusgu'au refl~x du solvant.
Qn ajGute alors, goutte à gcutte, en maintenant le reflux, 2,12 g
de chlorure de sulfinyle. L'addition terminee, on poursuit le
ohauffage à reflux pendant 10 minutes puis le melange reactionnel
est refroidi à une temperature v~isine de 10C et v~rsé dans une
solution de 15 g d'hydrogenocarbcnate de sodium dans 300 cm3 d'eau.
La phase aqueuse est séparée par decantation et re'ex*raite par 2
fois 50 cm3 de dichloro-1,2 éthane. Les phases organlques sont
20 réunies, lavees par 3 fois 20 cn3 d'eau, sechees sur sulfate de
magnesium filtrée~ puis concentrees à sec scus pression réduite
(2,7 kPa) à 40~C. Le résidu est purifié par chromatographie sur 200
g de silice (0,063-0,2 mm) oontenus dans une oolcnne de 3,5 om de
diamètre [éluant : cyclohexane-acetate d'éthyle (50-50 en Yolumes)]
25 en recueillant des fractions de 30 cm3. LRS ~ractions ne oontenant
que le p~odNit ch ~ é sont réunies et ssncentrees à ~ec scus
pressicn reduite (2,7 kPa) à 40C. On obtient apr~
recristallisation dans l'acétate d'ethyle 0,8 g de ~[(méthyl-3
phenyl)-3 u¢eido]-2 N~isoproQy1-acetamid~]-2 aoeta~e de tert-butyle
30 fcndbnt a 143C.
L'isopropylamino-~ acetate de tert-butyle peut être preparé
de la maniere suivante : à une sDlution de 4,4 g d~isopropylamine
dans 60 cm3 d'acetsnitrile, on a~oute 7,3 g de brcmoaoe t~te de
56 2~3~7
tert-butyle. La soluticn obtenue est agitée a reflux pendant 4
heures. Après refroidissement, le produit insoluble est séparé par
filtration et lavé par 30 cm3 d'acétonitrile. Les filtrats sont
réunis et ooncentrés à sec sous pressicn réduite (2,7 kPa) à 40C.
L'huile residuelle est dissoute dans 150 om3 de dichloromét~ne et
la solution est lavée par 4 fois 15 om3 d'eau. La phase organique
est séchée sur sulfate de magnésium, filtrée puis ccncentrée à sec
sous pression réduite (2,7 kPaj à 40C. On obtient ainsi 4,1 g
d'isopropylaminc-2 acétate de tert-butyle scus forme d'une huile
utilisée telle quelle dans les synthèses ultérieures.
L'acide [(méthyl-3 phényl)-3 uréido]-2 acétique peut être
préparé de la mam ère suivante : à une solution de 7,5 g de glycine
et de 8,4 g d'hydrogenoc~rtcnate de sodium dans 250 cm3 d'eau, on
ajoute, à une temperature ~isine de 20C, 13,3 g d'isocyanate de
méthyl-3 phényle. Le mélange réactionnel est agité pendant 18
heures à une temperature voisine de 20C puis le produit insoluble
est separé par filtration et lavé par 2 fois 30 cm3 d'eau puis par
2 fois 30 om3 d'acétate d'éthyle. Les filtrats sont réunls, la
phase aqueuse est séparée par decantation et acidifiée par une
solution aqueuse d'acide chlorydrique 5N jusqu'à un p~ voisin de 1.
Le solide formé est sépare par filtration, lave par 2 fois 30 cm3
d'eau puis par 2 fois 30 om3 d'acétate d'éthyle et s'eché à l'air.
On obtient ainsi` 16,3 g d'acide l(n~thyl-3 phenyl)-3 uréido]-2
acétique fondant à 225C.
EXEMPLE 38
A une solution de B g d'[amino-2 N-(naphtyl-1) acetamido]-2
acétate de tert-~utyle dans 100 om3 de tétrahydrD~uranne anhydre,
on ajoute, à w t ~ ture v~isine de 20C, 0,6 g d'isocranate de
méthyl-3 phenyle. La sDlution bbtenue est agitee pendant 4 he~res à
une temperature voisine de 20C puis ooncentree à sec scus pression
réduite (2,7 kPa) à 40C. L'huile residuelle est purifiée par
chromatographie sur 400 g de silioe ~0,063-0,2 mm) contenus dbns
une oDlcnne de 3 om de diametre [éluant : cyclohe~ane-acétate
57 2 ~ 7
d'éthyle (75-25 en Yolumes3] en recueillant des fractions de
20 om3. Les fracticns ne contenant gue le produit cherché scnt
réunies et ccncentrées à sec sous pression xéduite (2,7 kPa) à
40C. On obtient après recristallisation dans l'acétcnitrile, 3,4 g
de [[(méthyl-3 phényl)-3 uréido]-2 N-(naphtyl-1) acétamido]-2
acétate de tert-butyle fondant à 156C.
L'aminc-2 N-(naphtyl-1) acétamido]-2 acétate de tert-butyle
peut être preparé de la mam ère suivante : à une solution de 8,9 g
de [N-(naphtyl-1) phtalimido-2 acétamido]-2 acétate de tert-butyle
dans 100 cm3 de méthanol, cn ajoute 3 g d'hydrate d'hydrazine. Le
mélange réactionnel est agité pendant 3 heures à une température
voisine de 20C puis oonoentré à sec sous pression reduite
(2,7 kPa) a 40C. Le résidu est agité avec 400 cm3 d'éther
diéthylique et le produit insDluble est ~éparé par filtraticn. Le
filtrat est concentré à sec scus pression ~eduite (2,7 kPa) à 40C.
On obtient ainsi 8 g d'[amino-2 N-(naphtyl-1) aoe tamido]-2 acétate
de tert-butyle sous forme d'u~e h~ile utilisée telle quelle ~ s
les syntheses ultérieures.
Le [N-(naphtyl-1) phtalimidb-2 aoétamido]-~ acetate de
tert-butyle peut être preparé de la msniere ~uivante : à une
solution de 14,9 g de N-(naphtyl-1) phtalimidD-2 acetamide dans
150 om3 de tétrahydrofuranne anhydre malntenu~ sous atmosphere
d'argon, on ajoute, à une t ~ ture voisine de 10C, 2,4 g d'un~
suspension huileuse ~50 % en pDids) d'hydrure de sodium et agite la
25 suspension cbtenue pendant 1 hRure à une te ~ ature vDisine de
20C. On ajoute alors une solution de 9,75 g de bromoacétate de
tert-butyle dans 40 cm3 de tétrahydro~uranne anhydre et l'an
poursuit l'agitation ~ ant 3 heures à une t ~ ture vDisin de
20C. Le mél~nge reactionnel est ensuite vexse dans un melange
30 r froidi à une tempe~ature voisine de 0C, de 80 cm3 d'eau et de
800 c~3 d'acetate d'éthyle. La phase aqueuse est separee Far
decantatiGn et ~ ite par 2 fois 80 cm3 d'~cetate d'~thyle. LÆS
phases organlques sont reunies, lavees par 3 fois 100 am3 d'eau,
sechées sur sulfate de magnesium, filtrees puis ooncentre'es à sec
35 scus pression reduite (2,7 kPa) à 40C. On obtient ainsi 20 g de
2~r,,l~:3~7
58
~N-(naphtyl-1) phtalimidb-2 aoétamido]-2 acétate de tert-butyle
sous forme d'une huile utilisée telle quelle dans les synthèses
ultérieures.
Le N-(naphtyl-l) phtalimido-2 acétamide peut être préparé
de la manière suivante : à une solution de 7,1S g de naphtylamine-l
dans 100 cm3 de dichlorométhane maintenue sous atmo6phère d'argon,
on ajoute 5,56 g de triéthylamine puis en maintenant la temperature
au voisinage de 20C, une solution de 11,2 g de chlorure de
phtalimidb-2 acétyle dans 75 cm3 de dichlorométhane. La soluticn
cbtenue est agitée pendant 3 heures à une température voisine de
20C puis additicnnée de 75 om3 d'eau. Le solide formé est séparé
par filtration, lavé par 3 fois 15 om3 de dichlorométhane puis par
3 fois 30 cm3 d'eau et seché à l'air~ La phase organique du filtrat
est séparée par décantatian, lavee par 2 fois 30 om3 d'eau, sechée
sur sulfate de magnésium, filtrée puis ooncentrée à sec sous
pression reduite (2,7 kPa~ à 40~. Le solide bbtenu est réuni au
précédent. On obtient ainsi 16 g de N-(naphtyl-1) pht~limidc-2
acetamide.
Le chlorure de phtalimidb-2 acétyle peut être préparé selon
la methode décidee par W. GæAS5MANN et CD11., Eer., 83, 244 ~1950).
EXEMPLE 39
A une ~olution de 8,9 g de [phtalimido-2 N-(tert-butoxycar-
bonylmethyl) acétamidD~-2 acetate de tert-butyle dans 100 cm3 de
dichloromethane, cn ajoute, à une temperature v~i~Lne de 0C, 2,8 g
de méthylhydkazine. Le melange reactionnel est agité pendant 30
heures à une t ~ ture ~DiS~ne de 20C pui~ pendant 1 heure au
reflux. Apres refrDidissement, on ajoute 100 om3 d'eau, agite et
sép~re par d~cantation la phase aqueuse qui est re'~xtraite par 2
fois 80 ~n3 de dichlorométhane. Les phases organi~ues sont reunies,
lavees par 2 fois 15 om3 d'eau, ~e'chees sur ~ulfabe de magnesium,
filtrees pUls ooncentrees à sec SGUs pression reduite (2,7 kPa) à
40C. L'huile ainsi cbtenue est dissoute dans 60 am3 de
tétrahydrDfuranne anhydre. Qn a~oute à oe tte ~Dlutian 2,7 g
2~7~ 7
59
d'isocyanate de méthyl-3 phényle puis agite le mélange réactionnel
pendant 1 heure à une temperatuL-e voisine de 20C et ocncentre à
sec sous pression réduite (2,7 kPa) à 40C. Qn obtient 9 g d'une
huile jaune que l'an purifie par chromatographie sur 250 g de
silice (0,063-0,2 mm) ccntenus dans une colonne de 4 cm de diamètre
[éluant : cyclohexane-acétate d'éthyle (50-50 en volumes)] en
recueillant des fractions de 40 cm3. Les fracti~ns ne contenant que
le produit cherché sont réunies et concentrees à sec sous pression
réduite (2,7 kPa) à 40C. Le résidu obtenu est purifié par une
seo~nde chramatographie sur 75 g de silioe (0,063-0,2 mm) contenus
dbns une col~.e de 2 cm de diamètre [éluant : cyclohexane acétate
d'éthyle (60-40 en volumes~] en recueillant des fractions de 15
om3. Les fracticns ne contenant que le produit cherché s~nt réunies
et aQncentrées à sec sous pression réduite (2,7 kPa) à 40C. Qn
obtient après deux recrictallisations successives dans un mélange
d'oxyde de diisoprcpyle et d'acétate d'éthyle (90-10 en volumes),
2 g de [~méthyl-3 phényl)-3 uréido]-2 N,N-bis(tert-b~to-
xycarbcnylm~thyl) acétamide fondant à 119C.
Le [phtalimldo-2 N-(tert-butoxycarbonylméthyl~ acétamido~-2
acétate de tert-butyle peut etre préparé de la manière suivante : à
une solution de 7,9 g de glycinate de tert-butyle dbns 40 cm3
d'acétonitrile malntenue scus atmosphère d'argcn, on ajoute, à une
temperature ~oisine de 20C, 2,5 g d'hydrogenccarLonate de sDdium,
puis en maintenant la t ~ ture aux envircns de 20C, une
solution de 5,85 g de bromoacétate de tert-butyle dans 80 cm3
d'aoetonitrile. Le melange reactionnel est agite pendant 2 heures a
une temperature voisin~ de 20C puis pendant 1 heure à une
temperature de 40C. Le p¢oduit est sépare par filtration et laYé
par 15 cm3 d'acétonitrile. Le~ fil~rats sont reNnis et concentrés à
sec scus pression reduite (2,7 kPa) à 40C. Le residu est agité
avec 100 om3 d'oxyde de diisoproæyle, le p¢oduit in~oluble est
~eparé par filtration et le filtrat est conoentre à sec scus
pression reduite (2,7 kPa) à 40C. L'huil~ residuelle est dissoute
dans 120 cm3 de dichloro-1,2 éthane et à la solution ainsi obtenue
oQ ajGute 2,5 g d'hydro~enocDrbcnate de sodium, puls en naintenant
`- 2~7~
la temperature au voisinage de 20C, une solution de 6,7 g de
chlorure de phtalimido-2 acétyle dans 20 om3 de dichloro-1,2
éthane. La suspension bbtenue est agitée pendant 16 heures à une
temperature vois.ine de 20C, puis additionnée de 50 om3 d'eau. La
phase aqyeuse est séparée par décantation puls reextraite p~r 2
fois 100 om3 de dichloro-1,2 éthane. Les phases or3aniques sont
réunies, lavees par 2 fois 10 om3 d'eau, se'chées par sulfate de
magnésium, filtrees puis oonoentrees à ~ec scus pression réduite
(2,7 kPa) a 40C. on obtient, après r~cristallisation dans
l'acétate d'éthyle, 8,9 g de [phtalimido-2 N-(tert-butoxycarbanyl-
méthyl~ acetamido]-2 acetate de tert-butyle fondant à 150C.
EXEMPLE 40
En operant d'une manière analogue à oe lle décrite à
l'exemple 38 mais à partir de 6,4 g d'[amuna-2
N-(tétrahydro-1,2,3,4 naphtyl) acétamido]-2 acetate de tert-butyle
et de 2,26 g d'isocyanate de méthyl-3 phenyle, on cbtient après
recristallisatian dans l'oxyde de diisopropyl~, 3,8 g de
~[(m~thyl-3 phényl)-3 uréido]-2 N-(tétrahydro-1,2,3,4 naphtyl-5)
acétamido]-2 acétate de tert-butyle fGndant à 146C~
L'[~minc-2 N-(tétrahydro-1,2,3,4 naphtyl-5) acétamid3]-2
acétate de tert-butyle peut être préparé d'une maniere analogue a
celle décrite à l'exemple 38 pour la preparation de 1'[ ~ 2
N-(naphtyl-1) acé ~ ido]-2 acetate de tert butyle, nais à partir de
7,9 g de [phtalimidb-2 N-(tétrahydro-1,2,3,4 naph~yl-5) acétamldo]
-2 acétate de tert-butyle et de 2,7 g d'hydrate d'hydra2ine. On
obtient a~nsi 6,5 g d'[amino-2 N-(tétrahydro- 1,2,3,4 naphtyl-5)
acétamido~-2 acétate de tert-butyle s3us or~e d'u~e huile utilisee
telle yuelle dans les synth`ees ultérieur~s.
~ e [phtalimidc-2 N-ltétrahydro-1,2,3,4 naphtyl-~) aceta-
n~d~]-2 acetate de tert-butyle peut etre prepare d'une m2 m ere
analogue à oelle decrite à l'~xemple 38 pour la prepOE~a~lon du
[N-(naphtyl-1) phtalimido-2 acetamido]-2 acetate de tert-butyle,
2 ~ J ~
61
mais à partir de 10 g de phtalimido-2 N-(tétrahydro-1,2,3,4
naphtyl-5) acetamide, de 1,6 g d'une suspensiGn huileuse (50 % en
poids) d'hydrure de sodium et de 6,05 g de bromoacétate de tert-
butyle. On bbtient ainsi, après recristallisation dans l'oxyde de
diisopropyle, 9 g de [phtalimido-2 N-(tétrahydro-1,2,3,4 naphtyl-5)
acétamido]-2 acétate de tert-~utyle faldant a 144C.
Le phtalimidb-2 N-(tétrahydro-1,2,3,4 naphtyl-5) acétamide
peut etre prépare d'une manière analogue à oelle décrite à
l'exemple 38 pour la préparation du N-(naphtyl-1) phtalimidk-2
acétamide, mais à partir de 5,9 g de tétrahydro-1,2,3,4
naphtylamlne-5, de 4,4 g de triéthylamine et de 9,6 g de chlor~re
de phtalimido~2 acétyle. ~n obtient ainsi 13 g de phtalimido-2
N-(tétrahydro-1,2,3,4 naphtyl-5) acétamide.
EXEMPLE 41
A une solution de 5,5 g de (N-cyclcprcpyl phtalimadb-2
acetamido)-2 acétate de tert-butyle dans 80 cm3 de méthanol on
ajoute, à une température voisine de 20C, 2,3 g d'hydrate
d~hydrt 7ine. Le ~élange réactionnel est agité pendant 1 heure à
cette te ~ ature. ~'insc,luble forme est élimine pa~ filtration et
lavé par 2 fois 20 cm3 d'ether daéthyligue puis le filtrat est
concentré à sec sous pression ~eduite (2,7kPa) à 40C. L'huile
résiduelle est additicn~ee d'un mélange de 40 cm3 d'eau et de 40
cm3 d'acétate d'éthyle. La phase aqueuse est ~ ee par
décantation puis reextraite par 2 fois 20 cm3 d'acetate d'éthyle.
Les phases organiques sont réunies, lavees par ~ Poi~ 20 cm3 d'eau,
~echées sur sulfate de magnesium, filtrees puis c ~ trEes à sec
s~us pression reduite ~2,7kPa) à 40C. On ob~i~nt ainsi 3,2 g
d'(amino-2 N-cyclopropyl-acétamid~-2 asetate de tert-butyle sous
~onme d'une huile jaune qui est ~issoute dans 50 cm3 de
30 tétrahydrD~uranne anhydke. On ajcute a cette ~oluticn , 1,9 g
d'isccyanate de ~éthyl-3 ~ yle pu1s a~ite le melange ~eactionnel
pendant 1 heure à une tzmperature voisine de 20C et concentre à
sec ~ous pression ~eduite (2,7kPa) à 40C. On obtient
20~3~
62
ainsi après recristallisaticn du résidu dans l'acétate d'éthyle/
2,0 g de N-cycloprcpyl ~(méthyl-3 phényl)-3 uréido]-2 acétamido-2
acétate de tert-butyle fondant à 128C.
Le (N-cyclopropyl phtalimido-2 acétamido)-2 acétate de
tert-butyle peut être préparé de la manière suivante : à une
solution de 4,1 g de cyclGpluyylamino-2 acétate de tert-butyle dans
50 cm3 de dichloromethane, maintenue scus atmcsph~re d'argon, on
ajoute 2,9 g de triéthylamine puis goutte à goutte, à une
température voisine de 20C, une solution de 5,4 g de chlorure de
phtalimidb-2 acétyle dans 50 cm3 de dichlorométhane. La solution
obtenue est agitée Fendant 1 heure à une températu¢e voisine de
20C, puis additionnée de 50 cm3 d'eau. La phase aqueuse est
séparée par décantation puis r~extraite par 2 fois 25 cm3 de
di~hlorométhane. Les phases organiques sont réuniPs~ lavées par 2
fois 20 cm3 d'eau, séchées sur sulfate de magnésium, filtrées puis
concentrées à se~ sous pression réduite (2,7 kPa) à 40C. Cn
obtient ainsi, après recrist llisation dans l'acétate d'éthyle,
5,5 g de (N-cyclûpropyl phtalimido-2 acetamido)-2 acétate de
tert-butyle fondant à 167C.
Le cyclcpropylaminc-2 acétate de tert butyle peut être
préparé de la manière suivante : à une solution de 2,9 g de
cycloprcpylamine dans 100 cm3 d'acétonitrile on ajoute, à une
temperature Y~iSine de 20C, 2,1 g d'hydrogenoc~rbcnate de sodium,
puis une s~lution de 4,9 g de bromoacétate de tert-butyle dans 50
om3 d'a~etonitrile. Le melange r~actionnel est chauffé à reflux
pendant 4 heures puis refroidi à une te ~ ture v~isine de 20C.
Le produit insoluble est separé par filtLaticn et lavé par 50 cm3
d'aoe tonitrile et les fil~rats reunis sont oonoentres à sec scus
pression redNlte (2,7kPa) à 40C. ~'huile residuelle est
additionn~e d'un m'elange de 50 ~m3 d'eau et de 100 cm3 d'acetate
d'éthyle pUlS la phase ~ ~s~ ~ par decantation et
reextraite par 2 fois 25 cm3 d'acetate d'éthyle. LRS phases
organiques sont reunies, lavees par 2 fois 20 cm3 d'eau, sechées
sur sulfate de magnesium, filtre'es puis ocncentre'es a sec SGUS
pressicn reduite (2,7kPa) à 40C. On bbtient ainsi 4,1 g de
` 2~3~
63
cyclc~ropylamino-2 acétate de tert-butyle scus forme d'une huile
utilisée telle quelle dans les synthèses ultérieures.
EXEMæLE 42
A une solu~ion de 7,6 g d'[amino-2 N-(tétrahydro-1,2,3,4
napthyl-1) acétamido]-2 acétate de tert-butyle dans 15 om3 de
tétrahydrofuranne anhydre, on ajoute, à une temperature v~isine de
20C, 3,1 g d'isocyanate de méthyl~3 phényle. La solution obtenue
est agitée pendant 16 heures à une te~perature voisine de 20C puis
ooncentrée à sec sous pression reduite (2,7kPa) à 40C. L'huile
residuelle est pulifiee par chrcmatographie sur 450 g de silice
(0,063-0,2mm) OQntenuS dans une oolonne de S cm de diamètre
téluant : acétate d'éthyle) en recueillant des fractions de 20 cm3.
Les fractians ne oontenant que le produit cherché sont réunies et
ooncentrées à sec sous pression réduite (2,7kPa) à 40C. Cn obtient
1~ après recristallisation dbns l'acétate d'éthyle, 3,7 g de
[[(méthyl-3 phényl)-3 uréido~-2 N-(tétrahydro-1,2,3,4 naphtyl-1)
acétamido]-2 acétate de telt-butyle fondant à 144C
L'~amino-2 N-(tétrahydrc-1,2,3,4 naphtyl-1) acétamid~]-2
acétate de tert-butyle peut être prépar~ de la manière suivante : à
une solution de 9,5 g de ~phtalimido-2 N-(tétrahydro-1,2,3,4
naphtyl-1) acéta~ido]-2 acétate de tert-butyle dans 150 cm3 de
méthanol, on ajoute 3,2 g d'hydrate d'hydkazine. ~e mélange
réactisnnel est agité pendant 3 heures à une temperature voisine de
20C pulS ooncentre à sec sous pressicn r'eduite (2,~kPa) à 40C. Le
residu est txaité par 200 cm3 d'éther diéthylique puis le produit
insDluble est se ~ par filtraticn, lavé par 2 fois 100 cm3
d'acetate d'éthyle et les filtrats sont reunis et oonoentres à sec
sous pression re'duite (2,7kPa) à 40C. Qn cbtient ainsi 6,8 g
d'tamuno-2 N-(tetrahydro-1,2,3,4 naph~y1-13 aoetamido~-2 acétate de
tert-~utyle scus forme d'une huile utilis~e telle quelle dbns les
synth`eses uat~rieures.
~e [p tallmldb-2 N-(tétrahydro-1,2,3,4 naphtyl-1)
acetamidD]-2 acetate dde tert-butyle peut être preparé de la
2 ~
64
manière suivante : à une solution de 7,2 g de ~tétrahydro-1,2,3,4
naphtyl-1) aminc-2 acétate de tert-butyle dans ~0 cm3 de
dichloro-1,2 éthane, maintenue sous atmosphère d'argon a une
temperature voisine de 20C cn ajoute 3,4 g de triéthylamlne puis,
une solution de 6,8 g de chlorure de phtalimido-2 acétyle dans 60
cm3 de dichloro-1,2 éthane. La solution obtenue est agitée pendant
16 heures à une température voisine de 20C, puis additionnée de
150 cm3 d'eau. La phase aqueuse est séparée par décantation puis
réextraite par 2 fois 50 cma de dichloro-1,2 éthane. Les phases
organiques sont réunies, lavées par 2 fois 10 cm3 d'eau, séchees
sur sulfate de magnésium, filtrees puis o~.centrées à sec scus
pression reduite (2,7kPa~ à 40C. Cn cbtient ainsi, après
r istalllsaton dans l'acétate d'ékhyle, 10,1 g de [phtalimldo-2
N-(tétrahydro-1,2,3,4 naphtyl-1) acetamidol-2 acétate de
tert-butyle fondbnt à 176C.
Le (tétrahydro-1,2,3,4 napthyl-1) amino-2 acétate de
tert-butyle peut être préparé de la manière suivante : à une
solutian de 14,2 g de (tétrahydrc-1,2,3,4 naphtyl-1) amlne dans 100
om3 d'acétonitrile on ajoute, à une temperature v~isine de 20C,
4,2 g d'hydrogenoc1rbcnate de sodiu~, puis une sDlutian de 9,8 g de
~romoacétate de tert-butyle dans 50 cm3 d' æétonitrile. Le mélanye
réactionnel est chauffé à reflux pendant 4 heures puis refr~idi à
une temperatllre v~is me de 20C. Le prcduit insoluble est séparé
par filt~atian et lave par 50 cm3 d'acétonitrile puds le filtrat
est o~ncentré à sec sous pressicn re'duite (2,7kPa) à 40~C. Le
residu est purifié par ~ atographie sur 700 g de silice
(0,063-0,200mm) ocntenus dbns une oolonne d~ Som de diametre
~éluant cyclbhexane-acetate d'éthyle (70-30 en vDlumes)]en
recueillant des fractions de 50 cm3. LRS fractions ne oontenant que
30 le produit ~herch'e sont réunies et ooncentrees à sec scus pressicn
reduite (2,7kPa) à 40C. on cbtient alnsi 7,3 g de
(tétrahydro-1,2,3,4 naphtyl~ mino-2 acetate de tert-~utyle sous
forme d'une huile utilisee telle quelle dans les syntheses
ultérieures.
- ~7~3i~
EXEMPLE 4~
En opérant d'une manière analogue à celle décrite à
l'exemple 35, mais à partir de 0,54 g de méthyl-3 aniline et de 1,5
g de N-cyclchexyl N-(isocyanatoacétyl) glyclnate de tert-butyle, on
abtient après recristallisation dans l'oxyde de diisopropyle,
0,53 g de [~(méthyl-3 phényl)-3 uréido]-2 N-(cyclohexyl)
acétamido]-2 acétate de tert-butyle fondant à 7~C.
Le N-(isocyanatoacétyl) N-cyclahexyl glycinate de
tert-butyie peut être preparé d'une manière analogue à celle
1o décrite à l'exemple 35 p~ur la préparation du N-ben2yl
N~(isocyanatoacétyl) glycinate de tert-butyle, mais à partir de
1,2 g de chlorure d'i~ocyanatoacétyle, de 2,13 g de
cyclahexylamino-2 acétate de tert-~utyle et de 0,8 g de pyridine.
On obtient ainsi 1,5 g de N-(isccyanatoacetyl~ N-cyclahexyl
glycinate de tert-butyle fcndant à 135C.
Le cyclohexylamino-2 acétate de tert-butyle peut être
préparé de la maniere suivante : a une solutiGn de 19,8 g de
cyclohexylamine dans 100 cm3 d'acétonitrile, on ajoute 19,5 g de
bromcacétate de tert-butyle. La solution ~btenue est chauffée à
reflux pendant 4 heures. Apr~s refro~dissement, le produit
insoluble est ~p~ré par ilt~atian et lavé par 3 fois 30 cm3
d'acétonitrile ~ s les filtrats sont ré~is et ~Dncentres à sec
sous pression reduite (2,7kPa) à 40C. L'huile résiduelle est
purifiée par chromatographie ~ur 350 9 de silioe (0,063-0,200mm)
25 oontenus dans une colonne de 3,5 am de diametre [éluant
cyclohexane-acetate d'é~hyle (70-30 en vDlumæs~ en recueillant des
fractions de 20 cm3. ~es fractions ne ~ont~nant qye le ~ it
cherché sont reunies et concentrées à s~c ~ous pression re'duite
(2,7kPa) à 40PC. on bbtient ainsi 11,8 g de cyclohexyla~ino-2
acétate de tert-butyle sous fonme d'une huile utilisee telle quelle
dans les synth`ese5 ulterieures.
E)~ 4~
On opere d'une manière ~nalogue a oelle decrite à l'exemple
207~3~ ~
66
11, m is a partir de 0,5 9 de N-methyl N-phényl [N-(quinolyl-8)
isocyanatoacétamido]-2 acétamide et de 0,15 g de méthyl-3 aniline.
On cbtient, apres recristallisaticn dans l'acétonitrile, 0,25 g de
{[(methyl-3 phényl)-3 uréido]-2 N-(gyinolyl-B) acétamido}-2
5 N-méthyl N-phényl-acétamide fondant à 206C.
La présente invention oonce~ne également les medicaments
constitués par au moins un oomposé de formule (I) à l'état pur ou
s~us fonme d'une ccmposition dans laquelle il est associé à tDUt
autre produit pharmaceutiquement oompatible, pouvant être inerte ou
physiologiquement actif. Les medicaments selon l'invention peuvent
être employés par voie orale, parentérale, rectale ou topique.
Cbmme compcsitions solides pour administration orale,
peuvent être utilises des oomprimes, des pilules, des poudres
(capsules de gélatine, cachets) ou des gIanules. Dans ces oomposi-
15 tions, le principe actif selon l'inventian est mélangé à un cuplusieurs diluants inertes, tels que amidcn, cellulose, saodharose,
lactose ou silioe. ( es oanp~sitions peuvent également ~nprendre
des substanoes autres que les diluants, par ex~nple un cu plusieurs
lubrifiants tels que le stearate de magnesiun ~u le talc, un
20 oolorant, un enr~age (dragées) ou un verrLis.
C~nme oanp~sitions liquides E~r aBn$nistraticn o~ale, on
peut utiliser des solutils, des supensicrls, des ~lsicns, des
sirops et des élixirs pila:rn~oeutiquement aoceptables caltenant des
diluants inertes tels que l'eau, l'éthar~ol, le glyc~rol, les huiles
ve~étales ou l'huile de paraffine. C~s oar~ siti~s peuvent
ocmprendre des substances autres qu~ les diluants, par exemple des
produits mouillants, ~duloorants, épaissi~sants, aramatisants ou
stabilisants.
Les ccnpositicns stériles pour admdnistxation ~arenterale,
peuvent être de préference des soluticns aqueuses ~u des soluticns
non aqueuses, dbs suspensions ou des e'mulsions. Cbmme solYant ou
vehicule, on peut employer l'eau, le propyleneglyool, un polyéthy-
lèneglycol, des huiles vegé~ales, ~n particulier l'huile d'olive,
des esters o¢ganiques injectables, par exemple l'oleate dléthyle ou
d'autres solvants organiques convenables. Ces ocmpositisns peuvent
egalement oontenir des adjuvants, en particulier des agents mouil-
3 ~ 7
67
lants, isotonisants, emulsifiants, dispersants et st~bilisants. Lastérilisation peut se faire de plusieurs façons, par exemple par
filtration aseptisante, en incorporant à la oompcsition des agents
stérilisants, par irradiation ou par chauffage. Elles peuvent
egalement être préparées sous forme de oompositions solides stéri-
les qui peuvent être disso~tes au moment de l'emploi dans un milieu
stérile injectable.
Les compositions pour a~ministratian rectale sont les
suppositoires ou les capsules rectales qui contiennent, outre le
produit actif, des excipients tels que le beurre de cacao, des
glycérides seml-synthétiques cu des polyéthylèneglycols.
Les ccmpositions pour administration topique peuvent être
par exemple des cremes, des pommades, des lotions, des collyres,
des oollutoires, des gouttes nasales ou des aer~sols.
En thérapeutique humaine, les composés selcn l'invention
sont particulièrement utiles dans le traitement et la prévention
des desordres liés à la CCK et à la gastrine au niveau du systeme
nerveux et de 1'appareil gastrointestinal. Ces ccmposés peuvent
donc être utilisés dans le traitement et la prévention des
psychoses, des trcubles anxieux, de la maladie de Parkinsan, de la
diskinésie tardive, du syndrcme du colon irritable, de la
pancréatite qiguë, des llcères, des déscrdres de la motilité
intestinale, de ~oertaines tumeurs de l'oescphage inferieur, du
colon et de l'intestin, comme potentialisateur de l'activité
analgésique des me'dic3ments analgésiques narootiques et n~n
narcotiques et oomme r ~ ateur de l'appetit.
LRS dbses dépendent de 1'effet recherche, de la durée du
traitemeni et de la v~ie d'administration utili ~ ; elles sont
géneralement ~ rises entre 0,05 g et 1 g par jour par Yoie orale
pcur un adulte av2c des dbses unitaires allant de 10 m~ à 500 mg de
substanoe active.
D'une façon ge'ne'rale, le medecLn determinera la pcsologie
appropriee en fonction de l'âge, du poids et de bous les autr~s
facteurs propres au sujet a traiter.
Les exemples suivants illustrent des oompositions selon
2 ~ 7
68
1'invention :
EXEMPLE A
On prépare, selon la technique habituelle, des oomprimés
dosés à 50 mg de produit actif ayant la ocTposition suivante :
- ~[(méthyl-3 phényl)-3 uréido]-2 N-(isoquinolyl-4)
acétamido} 2 acétate de tert-butyle ~ 50 mg
- lactose ........................................... 104 mg
- cellulose .......................................... 40 mg
- polyvidone ......................................... 10 mg
- cartoxymethylamidon sDdique ....................... 22 mg
- talc .............................................. 10 mg
- staarate de magnésium .............................. 2 mg
- silice oolloïdale .................................. 2 mg
- mélange d'hydrcxyméthylcellulase, glycérine, oxyde
de titane (72-3,5-24,5) ............... q.s.p. 1 oomprime
pelliculé texminé à 245 mg
EXEMPLE B
On prépare, selon la technique habituelle, des ~élules
dbsées à 50 mg de p~oduit actif ayant la oompoæiticn suivante :
- ~{[(hydrcxy-1 éthyl)-3 phenyl]-3 ureid~}-2 N~(quin~lyl-8~
acétamido}-2 ac~tate de tert-butyle ............... 50 mg
- cellulose ......................................... 1B mg
- lactase ........................................... 55 mg
- silice oolloïdale .................................. 1 ~g
~5 - corboYym~thylamldan ~dique ....................... 10 mg
- talc .............................................. 10 mg
. - stearate de magnesium ............................ ,. 1 mg
EXEMPLE C
On prépare une solution injectable conten2nt 10 mg de
prcduit actif ayant la oomposition suivante :
~` 2~3~
69
- t(hydIoxyimdnométhyl-3 phenyl)-3 uréido3-2 N-(quin4lyl-83
N-l~tétrahydro-1,2,3,4 quinolyl-1) carbonylme'thyl~
acétamide-(E) ....................................... 10 mg
- acide benzoïque ..................................... 80 mg
- alccol benzylique ................................ 0,06 cm3
- benzoate de ~odium .................................. 80 mg
- éthan~l à 95 % .................................... 0,4 om3
- hydroxyde de sodium ................................. 24 mg
- propylène glyool .................................. 1,6 om3
- eau .......................................... q.s.p. 4 om3