Note: Descriptions are shown in the official language in which they were submitted.
DEMANDES OU BREVE~S VOLUMINEUX
LA PRÉSENTE PART1E-DE CEI IE DEMANDE OU CE BREVET
COMPREND PLUS D'UN TOME.
CECI EST L,E TOME ~ DE 2
NOTE: Pour les tomes additionels, veuillez c~ntacter le Bureau canadien des
brevets
,2 ~ 7 ~ 7
JUI\IIBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE
THAN ONE VOLUME
THIS IS VOLUME L_ OF
NOTE: ~or additional volumes please c~ntact the Canadian Patent Office
WO91/11~ PCT/USgl/~X~7
C~,gO~ q - V 3
2375627
TITI~ OF T~F. T~V~ ON
ANGIOTENSIN II ANTAGONISTS INCORPORATING A
SUBSTIlul~D BENZYL F.T .F.M~T
BACRGRO~ND OF T~ I NV~;~l I ON
The present application is a
continuation-in-part of copending application Serial
No. 479,786 filed on ~ebruary 13, 1990.
The Renin-angiotensin sy~tem (RAS) plays a
central role in the regulation of normal blood
pressure and seems to be critically in~olved in
hypertension development and maintenance as well as
congestive heart failure. Angiotensin II (A II), is
an octapeptide hormone produced mainly in the blood
during the cleavage of angiotensin I by angiotensin
converting enzyme (ACE) localized on the endothelium
of blood vessels of lung. ~idney, and many other
organs. It is the end product of the renin-
angiotensin system (RAS) and is a powerful arterial
vasoconstrictor that exerts its action by interacting
with specific receptors present on cell membranes.
WO91/11~ PCT/US91/009~7
2a75~27
One of the possible modes of controlling the RAS is c
angiotensin II receptor antagonism. Several peptide
analogs of A II are ~nown to inhibit the effect of
this hormone by competitively bloc~ing the receptors,
5 but their e~perimental and clinical applications have
been limited by partial agonist activity and lack of
oral absorption tM. Antonaccio. Cl in . ~Yp.
~y~ertens. A4, 27-46 (1982); D. ~. P. Streeten and
G. H. Anderson, Jr. - ~ndbook of ~ypertension,
10 Clinical Pharmacolo~y of Antihy~erte~sive Dr--es, ed.
A. E. Doyle, Vol. ~, pp. 246-271, Elsevier Science
Publisher, Amsterdam, The Netherlands, 1984].
Recently, several non-peptide compounds have
been described as A II antagonists. Illustrative of
15 such compounds are those disclosed in U.S. Patents
4,207,324; 4,340,598; 4,576,958; 4,582,847; and
4,880,804 and in European Patent Applications
028,834; 245,637; 253,310; and 291,969; and in
articles by A.T. Chiu, et al. [Fur. J. Pharm. F.yp.
Therap, 157, 13-21 (1988)] and by P.C. Wong, ~ ~1-
~J. Ph~rm. ~ Therap, 247, 1-7(1988)~. All of the
U.S. Patents, European Patent Applications 028,834
and 253,310 and the two articles disclose substituted
imidazole compounds which are generally bonded
through a lower al~yl bridge to a substituted
phenyl. European Patent Application 24S,637
discloses derivati~es of 4,5,6,7-tetrahydro-2~-
imidazo~4,5-c~-pyridine-6-carboxylic acid and analogs
thereof as antihypertensive agents.
None of the compounds disclosed within this
application or in any US Patent, ~uropean
Applications or literature publication are of the
WO91/11~9 PCT/US91/oogs7
i
2 0 7 ~ 6 ~ 7
-- 3
type containing su~stituted heterocycles bonded
through an al~yl bridge to a novel ~ubstituted phenyl
of the type disclosed herein. The imidazole-5-,6-,
and 7-fused heterocycles ha~e been disclosed in
5 earlier U.S. Patent applications focusing on the
heterocyclic fragment of the antagonist design. The
serial numbers of these applications are 351,508;
358,971; 375,6~5; 360,673; 375,217; and 386,328 and
are hereby incorporated by reference.
- 8RIEF DESCRIPTION OF T~ V~N110N
This invention is directed to substituted
heterocycles attached through a methylene bridge to
novel substituted phenyl derivatives to give
compounds of the Formula I, which are angiotensin II
antagonists and are useful in the treatment of
hypertension and congestive heart failure.
Specifically, the compounds of this invention contain
a heterocyclic moiety which is substituted at the
specified positions and to which a methylene bridge
connecting a novel substituted phenyl group as
defined by the lower portion of Formula I, is
attached. Additionally, pharmaceutically acceptable
compositions of these novel compounds, as the sole
therapeutically acti~e ingredient and in combination
with diuretics and other antihypertensive agents,
including beta blockers, angiotensin converting
enzyme inhibitors, calcium channel blockers or a
combination thereof are disclo8ed and claimed.
~urther, methods of treating hypertension and
congestive heart failure are described and claimed.
.
~ WO91/11999 PCT/USgl/00957
207~627
The compounds of this invention have central
nervous system (CNS) activity. They are useful in
the treatment of cognitive dysfunctions including
Alzheimer's disease, amnesia and ~enile dementia.
These compounds also have anxiolytic and
antidepressant properties and are therefore, useful
in the relief of symptoms of anxiety and tension and
in the treatment of patients with depressed or
dysphoric mental states.
WO91/11~ PCT/US91/009S7
- .
207~6~7
D~TAITFn ~FSC~TPTTON OF T~ ~hvr;~llON
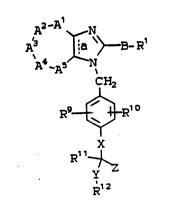
Thi~ invention relates to compounds of the
general Formula I:
-A1
S ~ E-R'
A -A5
CH2
R9~1 o
~
, R~
~12
Rl is:
(a) (Cl-C6)-alkyl, (C2-C6)-alkenyl or
(C2-C6)-alkynyl each of which is
unsubstituted or substituted with a
substituent selected from the group
consisting of:
i) aryl as defined below in Rl(b),
ii) (C3-C7)-cycloalkyl,
iii) Cl, Br, I, F,
2s i~) 0~,
v) NH2.
vi) N~(cl-c4)-al~yl~
~ii) N~(Cl-C4)-alkyl)]2,
~Ji i i ) NHS02R2,
ix) CF3,
x) COOR2, or
xi) S02N~R2a; and
; WO91/11~ PCT/US91/009~7
2075627
-
(b) aryl, wherein aryl i 8 defined as phenyl or
naphthyl and is unsubstituted, mono- or
disubstituted with ~ubstituents ~elected
from the group consi~ting of:
5i) Cl, Br, I, F,
ii) (Cl-C4)-alkyl~
iii ) (Cl-C4)-alko~cy,
i'~) N02
v) CF3
10~i) S02NR2aR2a
vii) (Cl-C4)-alkylthio,
~iii) hydroxy,
ix) amino,
x) (C3-C7)-cycloalkyl,
15xi) (C3-C10)-alkenyl; and
(c) heteroaryl, wherein heteroaryl is defined as
a 5- or 6-membered heteroaromatic moiety,
which can contain one or two members
selected from the group consisting of N, O,
20S and wherein the heteroaryl is
unsubstituted, mono- or disubstituted with
substituents selected from the group
consisting of:
i) Cl, ~r, I, F,
ii) OH,
iii) SH,
iv) N0
v) (Cl-C4)-alkyl,
vi ) (C2-C4)-al~enyl,
30~ii) (C2-C4)-alkynyl,
viii) (Cl-C4)-al~oxy, or
i~) CF3, or
(d) perfluoro-(Cl-C4)-al~yl; and
WO91/11999 PCT/US91/~ss7
~ 2~75S~7
_Al-A2-A3-A4-A~
when A4 and A5 are absent, then -Al-A2-A3- is:
~22
(a) -C-G-C=,
R22
R,22
lo (b) =N-G-C=
R22
(c) =C-G-N=
R22 R22
(d) =C-N-C=
R23
(e) =N-G-N=
R123
(f) =N-N-N=
R23
(g) ~N-N-C=
R22
R22
(h) =C-N-N=
R23
wherein ~ represents a single bond in these
definitions of A in structure I but, hereafter
will represent a double bond.
wogl/llggg 207~627
PCT/US91/00957
R23
(i) -N-C=N-,
R22
R22
(j) -N=C-N-,
R23
R23 R22
(k) -N-N=C-,
R22
(1) -S-C=CH-,
15(m) -CH=C-S-,
R22 .
(n) -C=N-N-,
R23
R22
(o) -G-C=N-,
R22
25(p) -N=C-G-,
R22
(q) -C=N-G-,
R22
(r) -G-N=C-,
WO 91/119~ ~ , ~ t ~ PCT/US91/00957
2075~27
R22 R23
(8) -C=C-N-,
R22
R22
(t) -C=C-G-,
R22
R23 R22
R22
R22
(v) -G-C=C-.
R22
R23
(w) -N=N-N-,
R23
(x) -N-N=N-,
R24 R24
(y ) --C--O--C--,
0
~ /
- (z) --C--C--O--,
O
R24 R24
(ba) 0
r~ wO 9~ g~ 2 0 7 S 6 2 7 PCT/US91/~9~7
, .
-- 10 --
R24 R24
- (bb) -C-O-C-,
o
O R24 R24
(bc) -C-N-C-,
R23
R24 R24 o
(bd) -C- N-C-,
when A4 is present and A5 is absent, then
_Al_A2_A3_A4_ represents:
R4 R4
(be) -C=C-C=C-,
R4 R4
R4 R4
(bf) -C=C -C=N-,
R4
R4 R4
(bg) -N=C- C=C-,
R
R4 ,R4
(bh) -C=C- N=C-,
WO 91/11999 PCI/US91/00957
2 0 7~ S2 7
11
R14 R~4
(bi ) -C=N--C-C-,
R~
( b j ) -C=C--N=N-,
R4
( bk ) -N=N C , 4
R4
( bl ) -C=N--N=C-,
R4
R4
( bm ) -N=C--C=N-,
R4
R4
( bn ) -N=N-N=C-,
2 5 ( bo ) -C=N-N=N-,
R4
( bp ) -NsN-C=N-,
R4
( bq ) -N=C-N=N-,
Wo g~ g99 2 ~ 7 ~ 6 2 7
PCT/US91/~957
, .. = ~
R4 R4
(br) -N=C- N=C-,
R4 R4
(bs) -C=N -C=N-,
O O
(bt) -~C-N -C-N-,
R5 R5
R5 R5
(bu) -N-C - N-C-,
O O
R4 o
(bv) -C=C- C-N-,
R4 R5
R5 R4
(bw) -N-C-C=N-,
o
,R4 R5
(~x) -N=C-C-N-,
2s 0
R4 o
(by) -C=C-C-N-,
R4 R5
,R4 R5
(bz) -C=C-N-C-,
R4 ol
PCT/US91/00957
WO 91111999
~ 2075627
_ 13 -
R5 R4
(ca~ -~-C-C=C-.
o R4
(cb) -~-N-C=C-,
R5 R4
R5
(cc)-N-C-N=N-,
(cd) -N=N-C-N-,
(ce) -C-N-N=N-,
R4
(cf)-Cl-N-C=N-,
R4
(cg)-N=C-N-C-,
0 R5
(ch)-C-N-N-C~-,
? i . wo 9l/llg99 2 0 7 5 6 2 7 PCI/US91/00957
.~ ' - .
-- 14 --
O O
(ci) -C-N=N-C-,
R5 O
( c j ) -N-C-C-N-,
O R5
R6a R6a R6a R5a
( ck ) -C --C --C --N-,
R6a R6a R6a
R5a R6a R6a R6a
- ( cl ) -N --C --C --C-,
R6a R6a R6a
R6a R6a R~
( cm ) -C --C --C-N-,
R6a R6a 0
20 -
R6a R6a R5
( cn ) -C --C --N-C-,
~.6a R6a b
R6a o R6a
( c o ) -C --~-N --C-,
R6a R5 R6a
O R4
3 o ( cp ) -C-C=C-N-,
R4 R5
WO91/11999 PCT/US91/~957
2 Q 7~ 62 7
O R6a R6a R5a
(cq) -C- C - C - N-,
R6a R6a
R6a R6a R5 R6a
(cr) -C -C -N -C-,
R6a R6a R6a
when A4 and A5 are present, then -Al-A2-A3-A4-A~-
are:
R6 R7 R8
(cs) -N-C-C -D-C-,
O O
O R7 R8 R6
(ct) -C-D--C--C-N-,
O
(cu) -C-N -C -C=N-,
~6 '2a
R6 R7 R8
(c~) -N-C- C-C=N-, or
O R2 a
R7 R8
(cw) -C-N- C- C~2-T-; and
R6
WO 91/11999
~ : ` ` ` 2 0 7 ~ 6 2 7 PCT/US91/00957
- 16 -
B is:
(a) a ~ingle bond,
(b) -S(O)n(CR2)~-, or
(c) -0-; and
n is 0 to 2; and
s is 0 to 5; and
lo D is
(a) -0-, or
(b) -N(R6)-; and
G is:
(a) -0-, or
(b) ~S(O)n; and
T is -S-, -0- or -N(R20)-; and
R2 is:
~a) E, or
(b) (Cl-C6)-alkyl; and
R2a iE::
(a) R2,
(b) C~2-aryl, or
(c) aryl; and
R4 ~roups are independently:
(a) ~,
WO91/11~9 PCT/US91/~s~7
- 207~627
(b) (Cl-C6)-alkyl, (C2-C6)-alkenyl, or
(C2-C6>-al~ynyl, each of which i~
unsubætituted or substituted with:
i) OH,
ii) (Cl-C4)-alkoxy,
iii) C02R2,
iv) OCOR2,
~) CON~R2a
~i ) CON(R2a)2,
~ii) N(R2a)C(=O)R2
~iii) N~2.
ix) (Cl-C4)-alkylamino,
x) dit(Cl-C4)-alkyl]amino,
xi ) -S-(Cl-C4)-alkyl,
xii) aryl,
xiii) heteroaryl,
(c) Cl, Br, I, F,
(d) CF3,
(e) CO2R2a,
(f) C(=O)N(R2a)2, or
(g) -C(=O)-aryl,
(h) (C3-C7)-cycloalkyl,
( i ) -oR24,
( j ) -S~ ,
2s (k) -S(O)n~(Cl-C4)-alkyl,
(1) -SO3~,
(m) -NR2R21
(n) -NR2C(=o)R21
(O) -NR2CooR21
-
WO91/11~9 PCT/USgl/009~7
207~627
`. ~:
.:
(P) -S02NR2aR2a
,.-`. (q) -N02,
(r) -NHS02-(Cl-C4)-al~yl, or
(s) when R4 groups are on adjacent carbon atoms
~5 - they may join to form a phenyl ring; and
R5 is:
(a) X, or
(b) (Cl-C6)-alkyl or (C2-C6)-alkenyl, optionally
substituted with:
i) hydroxy, or
ii) (Cl-C4)-alkoxy; and
R5a is
(a) R5, or
(b) (Cl-C4)-acyl; and
R6 is:
(a) ~,
(b) (Cl-C6)-alkyl, or
(c) (Cl-C6)-alkyl substituted with hydroxy;
and
R6a is:
(a) R6, or
(b) (Cl-C6)-alkyl substituted with:
i ) co2~2,
i i.) CON~IR2,
iii) CON(R2)2; and
WO91/11~ PCT/US91/~ss7
: ; i` 2~7~627
- 19 -
R7 and R8 are independently:
(a) H,
(b) (Cl-C6)-alkyl, (C2-C6)-alkenyl or
(C2-C6)-alkynyl unsubstituted or ~ubstituted
with a substituent selected from the group
consisting of:
i) hydroxy,
ii) (Cl-C4)-alkoxy,
iii) (Cl-C4)-alkylthio,
iv) amino,
v) (Cl-C4)-alkylamino,
vi~ di(Cl-C4)-alkylamino,
vii) carboxy,
viii) carboxamido,
ix) C02R2a,
x) OC(O)R2a, or
xi) ~uanidino,
(c) phenyl or phenyl-(Cl-C4)-alkyl, wherein the
phenyl group is unsubstituted or substituted
with a member selected from the group
consisting of:
i) hydroxy,
ii) Cl, Br, I, F,
iii ) (Cl-C4)-alkyl,
iv) (Cl-C4)-alkoxy,
(d) imidazolyl-(Cl-C4)-alkyl, or
(e) indolyl-(Cl-C4)-alkyl; and
WO91/11~9 PCT/US91/OOgs7
- 207S627
... . .
- 20 -
R9 and ~10 are independently:
(a) H,
(b) (Cl-C6)-alkyl, unsubstituted or substituted
with (C3-C7)-cycloalkyl,
(c) (C2-C6)-alkenyl,
(d) (C2-C6)-alkynyl,
(e) Cl, Br, F, I,
(f) (Cl-C6)-alko~y,
(g) when R9 and R10 are on adjacent carbons,
lo they can be joined to form a phenyl ring,
(h) perfluoro-(Cl-C6)-alkyl,
(i) (C3-C7)-cycloalkyl, unsubstituted or
substituted with (Cl-C6)-alkyl,
(j) aryl; and
X is:
(a) -0-,
(b) ~S(O)n~,
(c) _NR13_
(d) -CH20-,
(e) -CH2S(O)n~
(f) -CH2NR13 -,
(g) -0CH2-,
(h) -NR13CH2-,
(i) ~S(O)nCH2-,
(j ) -CH2-,
(k) -(C~2)2-'
(1) single bond, or
(m) -C~=j wherein Y and R12 are absent forming a
-C=C- bridge to the carbon bearing Z and
Rll; and
WO 91/11~ 2 ~ 7 5 ~ 2 7 PCT/US91/~957
Y is:
(a) single bond,
(b) -o_,
(c) ~S()n~.
(d) -NR13_, or
(e) -CH2-; and
Except that X ~nd Y are not defined in such a way
that the carbon atom to which Z i6 attached also
simultaneously is bonded to two heteroatoms (0, N, S,
SO . SO2 )
Rll and R12 are independently:
(a) ~,
(b) (Cl-C6)-alkyl, unsubstituted or ~ubstituted
with a substituent selected from the group
consisting of:
(i) aryl, or
(ii) (C3-C7)-cycloalkyl,
(c) aryl, unsubstituted or substituted with 1 to
5 substitutents selected from the group
consisting of:
i) Cl, Br, I, F,
ii ) (cl-c4)-alkyl,
iii) ~(Cl-Cs)-alkenyl]C~2-,
iv) ~ (Cl-C5 )-alkynyl]C}I2-,
v) (Cl-C4)-alkoxy,
vi) (Cl-C4)-alkylthio,
~i i ) -N02 ~
viii) -CF3.
ix) -C02R2a, or
x ) --O~I,
~ WO 9~ 2 0 7 5 6 2 7 PCT/US91/009S7
113/VJC45 - 22 -
(d) aryl-(Cl-C2)-alkyl. un6ubstituted or
~ubstituted with 1 to 5 sub6titutent6
selected from the group con~isting of:
i) Cl, ~r, I, F,
ii) (Cl-C4)-alkyl,
iii) ~(Cl-C5)-alkenyl]CH2-,
iv) [(cl-c5)-alkynyl]cH
v) ( Cl-C4 )-alkoxy,
vi) (Cl-C4)-alkylthio,
vii) -N02,
viii) -CF3,
ix) -C02~2a, or
x) -OH, or
(e) (C3-C7)-cycloalkyl; and
R13 is:
(a) H,
(b) (Cl-C6)-alkyl,
(c) aryl,
(d) aryl-(Cl-C6)-alkyl-(C=O)-,
(e) (Cl-C6)-alkyl-(C=O)-,
(f) t(C2-Cs)-alkenyl~CH2-
~(g) ~(C2-C5)-alkynyl]CH2-, or
(h) aryl-CH2-; and
Z i6:
(a) -CO2H,
(b) -CO2-(Cl-C6)-,alkyl,
(c) -tetrazol-5-yl,
(d) -CO-NH(tetrazol-5-yl)
(e) -CON~-S02-aryl,
WO91/11~ PCT/US91/~s7
2075627
(f) -CONH-SO2-(Cl-Cg)-alkyl, wherein the alkyl
group is un~ubstituted or substituted with a
substituent selected from the group
consi~ting of: -O~. -SH, -O(Cl-C4)-alkyl,
-S-(Cl-C4)-al~yl, -CF3, Cl, Br, F, I, -NO2,
-C02E- -C2-(Cl-C4)-alkyl, -N~2,
-NHt(Cl-C4)-alkyl], or -Nt(Cl-C4)-alkyl]2,
(g) -CON~-SO2-perfluoro-(Cl-C4)-alkyl,
(h) -CONH-SO2-heteroaryl,
(i~ -CON~S02NR2aR2a,
( j ) -S02NHCO-aryl,
(k) -SO2NHCO-(Cl-Cg)-alkyl~ wherein the alkyl
group is unsubstituted or substituted with a
substituent selected from the group
consisting of: -OH, -S~, -O(Cl-C4)-alkyl,
-S-(Cl-C4)-alkyl, -CF3, Cl, Br, F, I, -NO2,
-C02H~ ~co2-(cl-c4)-alkyl~ -N~2,
-NHt(Cl-C4)-alkyl], or -Nt(Cl-C4)-alkyl]2,
(1) -S02NHCO-(Cl-C4)-perfluoroalkyl,
(m) -S02N~CO-heteroaryl,
(n) -SO2CONR2aR2a,
(o) -PO(0~)2 ~
(p) -PO(OR2)2, or
(q) -PO(OH)(OR2); and
2~
R20 is:
(a) ~.
(b) (Cl-C6)-alkyl,
(c) allyl,
(d) (C3-C6)-cycloalkyl,
(e) (Cl-C4)-acyl,
(f) benzyl, or
(g) phenyl; and
WO 91/11999 ') ~
V ~ J U ~ ~ PCI`/US91/00957
-- 24 --
R21 iS:
(a) H, or
(b) (Cl-C4)-alkyl, unsubstituted or sub~tituted
with:
i) NR2,
i i ) NH r ( cl-c4 ) -alkyl],
iii) N~(Cl-C4)-alkyl]2,
iv) C02~.
v) CO2(Cl-C4)-alkyl,
~i) OH,
vii) SO3H, or
viii) SO2NH2; and
22
R ~roups are independently:
(a) H,
(b) (Cl-C6)-alkyl, (C2-C6)-alkenyl or
(C2-C6)-alkynyl each of which is
unsubstituted or substituted with a
substituent selected from the group
consisting of: (C3-C7)-cycloalkyl, Cl, Br,
I, F, OH, -NH2, -N~t(Cl-C4)-alkyl~,
-N[~Cl-C4)-alkyl~2, -NRSo2R25, -Co2R25,
(Cl-C4)-alkoxyl, (Cl-C4)-alkylthio,
(Cl-C4)-acyl, or C(=O)NH2,
(c) aryl,
(d) substituted aryl in which the substituents
are V or W, as defined below,
(e) aryl-(Cl-C4)-alkyl, which can be substituted
with V or W as defined below,
(f) Cl, Br, I, F,
(g) hydroxyl,
(h) amino,
W091/11~9 PCT/US91/00957
2Q75627
- 25 -
( i ) N~[ (Cl-C4)-alkyl],
(j) N[(Cl-C4)-al~Yl]2
(k) (Cl-C6)-alkogy,
(1) CF3,
(m) Go2R25,
(n) C(=o)N(R25)2,
(o) N(R25)-C(=o)R25
(p) (Cl-C4)-alkylsulfonyl,
(q) (Cl-C4)-alkylsulfinyl, or
(r) (Cl-C4)-alkylthio; and
R23 iS
(a) H,
(b) (Cl-C6)-al~Yl. (C2-C6)-alkenyl or
(C2-C6)-alkynyl each of which is
unsubstituted or substituted with a
substituent selected from the group
consisting of: (C3-C7)-cycloalkyl, Cl, Br,
I, ~, OH, -NH2, -N~(Cl-C4)-alkyl],
-Nt(Cl-C4)-alkyl]2. -N~So2R25, -Co2R25,
(Cl-C4)-al~oxyl, (Cl-C4)-alkylthio,
(Cl-C4)-acyl, or C(=O)NH2,
(c) -C(=o)R25
(d) -Co2R25,
2s (e) aryl, which is unsubstituted or substituted
with substituents V or W,
(f) aryl-(Cl-C4)-alkyl. which is unsubstituted
or substituted with V or W; and
R24 groups are independently:
(a) H,
(b) (Cl-C6)-alkyl. (C2-C6)-al~enyl or
(C2-C6)-alkynyl. each of which i8
unsubstituted or su~stituted with a
substituent selected from the group
WO91/11~ PCT/US91/ooss7
207~62~
. .
- 26 -
consisting of: (C3-C7)-cycloalkyl, Cl, Br,
I, F, 0~, -N~2. -N~[(Cl-C4)-alkyl~,
-N[(Cl-C4)-alkyl]2, -N~So2R25, -Co2R25,
(Cl-C4)-alkoxyl, (Cl-C4)-alkylthio,
(Cl-C4)-acyl, or C(=O)N~2,
(c) aryl or aryl-(Cl-C4)-alkyl which is
unsubstituted or substituted with V or W
V and W are each independently selected from:
(a) ~,
(b) (Cl-C5)-alkoxy,
(c) (Cl-C5)-alkyl,
(d) hydroxy,
(e) -S(O)n(Cl-C5)-alkyl,
lS (f) -CN,
(g) -N02~
(h) _NR2R2a
(i) ~(Cl-C5)-alkyl]-NR2R2a,
( j ) -C02R2a,
(k) -CO(Cl-C5)-alkyl,
(1) CF3,
(m) I, ~r, Cl, F
(n) hydroxy-(Cl-C4)-alkyl-,
(o) carboxy-(Cl-C4)-alkyl-,
2~ (p) -tetrazol-S-yl,
(q) -N~-S02CF3, or
(r) aryl; and
R25 iS: .
(a) E,
(b) (Cl-C6)-alkyl,
(c) aryl, or
(d) aryl-(Cl-Cs)-alkyl; and
the pharmaceutically acceptable ~alts thereof,
WO91~11999 PCT/US91/~9~7
,. .:
- 2075627
Wherein a preferred embodiment is when:
R~
(a) (Cl-C6)-alkyl or (C2-C6)-alkenyl or
(~2-C6)-alkynyl each of which i8
unsubstituted or substituted with a
substituent selected from the group
consisting of:
i) (Cl-C4)-alkylthio,
ii ) (Cl-C4)-alkoxy,
iii) CF3,
iv) CF2-CF3, or
v) (C3-C5)-cycloalkyl,
(b) perfluoro-(Cl-C4)-alkyl, or
- (c) (C3-C5)-cycloalkyl; and
-Al-A2-A3-A4-A5- is:
when A4 and A5 are absent, then -Al-A2-A3- is:
R122
(a) =C-G-C-,
R22 R122
(b) =C-N-C=
R23
wo 9~ g99 2 0 7 S 6 ~ ~ PCT/US91/~957
- 28 -
(c) =N-S-N=,
wherein ~ represent~ a ~ingle bond in these
definitions of -Al-A2-A3- in structure Ia but,
hereafter will represent a double bond.
~23
(d) -N-C=N-,
R22
R22
(e) -N=C-N-,
R23
~23 ~22
(f) -N-N=C-,
R22
(g) -C=N-N-
~23
R22
(h) -G-C=N-
R22
(i) -N=C-G-
R22
(j) -~C=N-G-
PCrIUS91100957
WO 91/llffl
. .
. .
21)7S627
-- 29 --
R22
(k ) -G-N=C-
. ~22
)C C G
R22
R22
G C C
R24 R24
(n ) -C--O-C-
(o) --C--O--C--
o
( p ) -C-~--C-
R24 R24 o
( q ) --C--~--C--
R
WO91/11999 2 0 ~ 5 6 2 7 PCT/USgl/U99S7
- 30 -
when A4 is present and A5 is absent, then
-Al-A2-A3-A4- represents:
R4 R4
(r) -C=C- C=C~-,
R4 R4
R4 R4
(s) -C=C- C=N-,
R4
R4 R4
(t) -C=C -~=C-,
R4
R14 ,R4
(u) -C=N-C=C-,
R4
~4 R4
(~) -N=C-C=C-,
R4
(w) -IC=C -NCN-,
R4
R4
(x ) -~ C=C-,
R4
WO91/11999 PCT/US91/009~7
207~627 `:~
- 113/VJC45 - 31 -
(y) -N=C-N=C-,
R4
(z) -C=N-C=N-,
R4
R4
(aa) R4
O O
(ab)
R5 R5
(ac) -N-C-N-C-,
0 0
R4 0
(ad) -C=C- C-N-,
l4 `5
~R5 R~4
(ae) -N-C-C=N-,
-
WO91/11999 pcT/us9l/oo9s7
r !~ 2 0 7 5 6 27
:
- 32 -
R4 R5
(af) -C=C-N-C-,
R4 o
R6a R6a R6a R5a
(ag)-C - C - C - N-,
. R6a R6a R6a
R6a R6a R5
lo (ah)-C - C - C-N-,
R6a R6a 0
R6a R6a R5
(ai)-C - C - N-C-,
R6a R6a 0
,R6 R,8
(aj) -N-C-CH-D-C-,
ll ll
O O
O R8
(ak) -C-N-CH-C=N-, or
R6 R2a
R8 R6
(al)-C-D-CH-C-N-; and
O O
B is:
(a) ~ingle bond,
(b) -S-, or
(c) -0-; and
WO91/11 ffl PCT/US91/009~7
~ - 207562~
113/VJC4~ - 33 -
n is 0, 1, or 2; and
D is
~a) -O-, or
(b) -N(R6)-; and
G is:
(a) -O-, or
(b) ~S()n.
R2 is:
(a) H, or
(b) (C1-C6)-alkyl; and
R2a is:
(a) R2,
(b) benzyl, or
(c) phenyl; and
R4 groups are independently:
(a) H,
(b) (Cl-C6)-alkyl, which is unsubstituted or
substituted with:
i) OH,
ii) C02R2,
iii) N~2.
iv) (Cl-C4)-alkylamino,
. v) dit(Cl-C4)-alkyl]amino,
(c) C1, Br, I, F,
(d) CF3,
(e) C02R2a.
. WO91/11~ PCTJUS91/009s7
: . ,
- 2075627
113/VJC45 - 34 -
(f) C(=O)NR2aR2a
(g) -C(=0)-aryl,
(h) -oR24,
(i) -S-(Cl-C4)-alkyl,
(j) -Nt(cl-c4)-alkyl~
(k) -N~C(=O)(Cl-C4)-alkyl,
(1) -N~COO(Cl-C4)-alkyl,
(m) -S02N~(Cl-C4)-alkyl,
(n) -N02,
1 0 ( O ) -NHS02CH3,
(p) (C3-C7)-cycloalkyl, or
(q) when R4 groups are on adjacent carbon atoms
they may join to form a phenyl ring; and
~5 is:
(a) H, or
(b) (Cl-C6)-alkyl, which i5 unsubstituted or
substituted with: hydroxyl, or C02R2; and
~5a is
(a) H,
(b) (Cl-C4)-alkyl, or
(c) (Cl-C4)-acyl; and
R6 is:
(a) H, or
(b) (Cl-C6)-alkyl; and
R6a is:
(a) H, or
(b) (Cl-C4)-alkyl; and
WO91/11~ pcT/us9l/~ss7
.
2075627
- 35 -
R8 is:
(a) H,
(b) (Cl-C6)-alkyl,
(c) which is unsubstituted or substituted with a
- 5 substituent selected from the group
. consisting of:
i) hydroxy,
iii) (Cl-C4)-alkylthio,
iv) amino,
lo vii) carboxyl,
viii) carboxamido,
ix) C02R2a,
x) OC(O)R2a, or
xi) guanidino,
~d) phenyl,
(e) benzyl,
(f) 4-hydroxybenzyl,
(g) 4-imidazolylmethyl, or
(h) 3-indolylmethyl; and
R9 and R10 are independently:
~a) H,
(b) (Cl-C6)-alkyl, unsubstituted or substituted
with (C3-C7)cycloalkyl,
(c) (C2-C6)-alkenyl,
(d) (C2-C6)-alkynyl,
(e) Cl, Br, F, I,
(f) (Cl-C6)-al~oxy, or
(g) when R9 and R10 are on adjacent carbons,
they can be joined to form a phenyl ring,
(h) perfluoro-(Cl-C6)-alkyl,
(i) (C3-C7) cycloalkyl, which is unsubstituted
- or ~ubstituted with (Cl-C6)-alkyl, or
(j) aryl; and
WO91/11~9 ~ PCT/US91/~9s7
6~
.'~
- 36 -
X is:
(a) -0-,
(b) ~S()n~.
(c) _N~13_
(d) -C~2O-,
(e) -C~2S(O)n.
(f) -CH2NR13 -,
(~) --OC~2-,
(h) -NR13C~2-,
(i) -S(O)nCH2-
( j ) _C~2_ ,
(~) -(C~2)2-~
(1) single bond, or
(m) =(C~)-, wherein Y and R12 are absent formin~
a -C=C- bridge to the carbon bearing Z and
Rll; and
Y is:
(a) single bond,
(b) _o_,
( c ) --C~2---
(d) ~S(O)n~~ or
(e) _NR13_; and
E~cept that ~ and Y are DQ~ defined in such a way
that the carbon atom to which Z is attached also
simultaneously is bonded to two heteroatoms (O, N, S,
SO . S02 )
Rll and R12 are independently:
(a) ~,
(b) (Cl-C6)-alkyl, unsubstituted or substituted
with:
i) aryl, or
ii) (C3-C~)-cycloalkyl,
WO91/11~ PCT/US91/~s~7
207S62~
- 37 -
(c) aryl, unsubstituted or substituted with 1 to
5 substitutents selected from the group
consisting of:
i) Cl, Br, I, F,
5ii) (Cl-C4)-alkyl,
i i i ), [ (Cl-C5 )-al~enyl]C~2-,
iv) [(Cl-C5)-alkynyl]CH2-,
v) (Cl-C4)-alko~y, or
vi) (Cl-C4)-alkylthio. and
(d) aryl-(Cl-C2)-al~yl, unsubstituted or
æubstituted with 1 to 5 substitutents
selected from the group consiæting of:
i) Cl, Br, I, ~,
ii ) (Cl-C4)-alkyl,
iii) t(Cl-c5)-alkenyl]c~2-~
iv) t(Cl-C5)-alkynyl]C~2-,
~) (Cl-C4)-alko~y, or
~i) (Cl-C4)-alkylthio, and
(e) (C3-C7)-cycloalkyl; and
R13 is:
(a) ~,
(b) (Cl-C6)-alkyl,
(c) aryl,
(d) aryl-(Cl-C6~-alkyl-(C=0)-, or
(e) (Cl-C6)-alkyl-(C=O)-; and
z is:
(a) -C02~,
(b) -CO2-(Cl-C6)-alkyl,
(c) -tetrazol-5-yl,
(d) -CO-NH(tetrazol-5-yl),
(e) -CONH-502-aryl,
WO 91/11~9 2 0~ 5 62~ PCT/US91/~Q9~7
- 38 -
(f) -CON~-SO2-(Cl-C4)-alkyl,
(g) -CONH-SO2-perfluoro-(Cl-C4)-alkyl,
(h) -CONH-S02-heteroaryl, wherein heteroaryl is
defined as a 5 or 6 membered aromatic ring
containing one or two heteroatoms selected
from the group consisting of O, N, or S;
( i ) -CON~S02NR2aR2a,
(j) -S02N~ICO-aryl,
(k) -SO2N~CO-(Cl-C6)-alkyl,
(1) -S02N~CO-(Cl-C4)-perfluoroal~cyl,
(m) -S02N~CO-heteroaryl, wherein heteroaryl is
defined as a 5 or 6 membered aromatic ring
containing one or two heteroatoms Eelected
from the group consisting of O, N, or S,
(n) -S02NHCONR2aR2a,
(o) -PO(0~)2 ~
(p) -PO(OR2)2, or
(q) -PO(OH)(OR2); and
R22 groups are independently:
(a) E,
(b) (Cl-C6)-alkyl, (C2-C6)-al~enyl or
(C2-C6)-alkynyl each of which is
unsubstituted or substituted with a
substituent selected from the group
consisting of: (C3-C7)-cycloalkyl, Cl, Br, I, F,
-0~, -NH[(Cl-C4)-alkyl]. -Nt(Cl-C4)-alkyl]2,
-CO2R25, or C(=o)NHt(cl-c4)-alkyl)]~
(c) aryl or aryl-(Cl-C4)-alkyl, which is
unsubstituted or substituted with a
substituent selected from the group
consistin~ of: H, Br, Cl, I, F,
(cl-c4)-alkyl, hydroxy-(Cl-C4)-alkyl,
(cl-c4j-al~oxy~ or Co2R25.
WO91/11~ PCT/US91/~gs7
- 2075S27
113/VJC45 _ 39 _
(d) Cl, Br, I, ~,
(e) amino,
(f) N~[(Cl-C4)-alkyl],
(g) Nt(Cl-C4)-alkYl]2~ -
(h) (cl-c6)-alkoxy~
( i ) Co2R25,
(j) C(=o)N(R25)2, or
(k) (Cl-C4)-alkylthio; and
R23 is:
(a) E,
(b) (Cl-C6)-alkyl. which is unsubstituted or
substituted with a substituent selected from
the group consisting of: -OH, -N~2,
-Nt(Cl-C4)-al~Yl~2~ or -Co2R25~
(c) aryl or aryl-(Cl-C4)-alkyl, which is
unsubstituted or ~ubstituted with: Br, Cl,
F, I, (Cl-C4)-alkyl, hydroxy-(Cl-C4)-alkyl,
Co2R25, CoR25, or So2R25,
R24 groups are independently:
(a) H,
(b) (Cl-C6)-alkyl. which is unsubstituted or
substituted with a substituent selected from
2s the group consisting of: Cl, Br, I, F,
-Co2R25, hydroxy-(Cl-C4)-alkyl, or
(Cl-C4)-acyl, or
(c) aryl or aryl-(Cl-C4)-alkyl; and
R25 is:
(a) H,
(b) (Cl-C6)-alkyl,
(c) aryl, or
(d) aryl-(Cl-C5)-alkyl; and
the pharmaceutically acceptable 5alt thereof.
WO 91/11999 Pcr/uS9l/~ngs7
`i 7~6~
,~ .
- 40 -
Wherein a more preferred embodiment of the in~ention
is when:
~1 is:
(a) (Cl-C~)-alkyl (C2-C6)-alkenyl or
. (C2-C6)-alkynyl, each of which is
unsubstituted or ~ubstituted with a
substituent selected from the group
consisting of:
i ) (Cl-C4)-alkylthio,
ii) (Cl-C4)-alkoxy,
iii) C~3,
iv) CF2CF3, or
~) (C3-C5)-cycloal~yl, or
(b) perfluoro-(Cl-C4)-alkyl; and
-Al-A2-A3-A4-A5- is:
when A4 and A5 are absent, then -Al-A2-A3- is:
R23
(a) -N-C=N-,
R22
RZ2
(b) -C=N-N-
,, R23
R22
(c) -G-C=N-
R22
(d) -N=C-&-
~. WO91/11999 -~ PCT/US91/oogs7
!
20756~7
- 41 -
R22
(e) -~=N-G-
R22
(f) -C=C-G-
R22
when A4 is present and A5 is absent, then
-Al-A2-A3-A4- represents:
R4 R4
( g ) -C=C--C=C-,
R4 R4
R4 R4
(h) -C=C- C=N-,
R4
(i) -C=N-C=N-,
R4 0
(j) -C=C- C-N-,
R4 R5
R6a R6a R6a R5a
(k) -C - C - C - N-,
R6a R6a R6a
WO91/11~9 PCT/USgl/~gs7
~ 20756~ ~
- 42 -
When Al-A2-A3-A4-A~ are all present:
R6 R8 o
(1)-N-C- C~-N-~-, or
0 R
(m) R8 R6
-C-N - CX-C-N-; and
16
B is a single bond; and
G is -O-, or -S-; and
R2 is:
(a) ~,
(b) (Cl-C6)-alkyl; and
R2a is:
(a) R2,
(b) benzyl, or
(c) phenyl; and
R4 groups are independently:
(a) E,
(b) (Cl-C6)-al~yl, which is unsubstituted or
substituted with:
i) O~
i i ) C02R2a,
iii) N~2~
iv) (Cl-C4)-al~ylamino.
v) dit(Cl-C4)-al~yl~amino,
WO91/11999 PCT/US91/~957
' ~
207362~
- 43 -
(c) Cl, Br, I, F,
(d) CF3,
(e) CO2R2a,
(f) c(=o)NR2aR2a
(g) (C3-C7)-cycloalkyl;
(h) -C(=O)-aryl,
(i) -oR24,
(j) -N[(Cl-c4)-alkyl]2~
(k) -NHC(=O)(Cl-C4)-alkyl,
(1) -NHCO2(Cl-C4)-alkyl,
(m) -SO2NH-(Cl-C4)-alkyl,
(n) -S02NH-aryl,
(o) -N02,
( p ) -NHS02CH3,
R5 is:
(a) H, or
(b) (Cl-C6)-alkyl, unsubstituted or substituted
with: hydroxyl, or CO2R2; and
R5a is
(a) H,
(b) (Cl-C4)-alkyl, or
(c) (Cl-C4)-acyl; and
R6 is:
(a) H, or
(b) (Cl-C6)-alkyl; and
R6a is:
(a) H, or
(b) (Cl-C4)-alkyl; and
.
~ WO 91/11999 2 0 7 5 6 27 Pcr~usgl/~q~s7
- 44 -
~8 is:
(a) H,
(b) (Cl-C6)-alkyl, which is unsubstituted or
substituted with a ~ub~tituent selected from
the group consi~ting of:
i) hydroxy,
ii) (Cl-C4)-alkoxyl,
iii) ~mino,
iv) carboxyl,
~) carboxamido,
vi ) C02R2a,
vii) OC(O)R2a, or
viii) guanidino,
(c) phenyl,
lS (d) benzyl,
(e) 4-hydroxybenzyl,
(f) 4-imidazolylmethyl, or
(g) 3-indolylmethyl; and
R9 and R10 are independently:
(a) H,
(b) (Cl-C6)-alkyl, unsubstituted or substituted
with (C3-C7)cycloalkyl,
(c) (C2-C6)-alkenyl,
(d) (C2-C6)-alkynyl,
(e) Cl, Br, F, I,
(f) (Cl-C6)-alkoxy, or
(g) when R9 and R10 are on adjacent carbons,
they can be joined to form an aryl ring,
(h) perfluoro-(Cl-C6)-al~yl,
(i) (C3-C7)-cycloalkyl, unsubstituted or
su~stituted with (Cl-C6)-alkyl, or
(j) aryL; and
.~ WO91/11~9 PCT/US91/00957
2075~27
_ 45 -
X is:
(a) -0-,
(b) -S()n~~
(c) _NR13_
5 (d) -C~20-,
(e) ~CH2S(O)n,
(f) -C~2NR13 -,
(g) -OC~I2-,
(h) -NR13CH2-,
(i) -S(O)nC}~z-,
( j ) -CH2- ,
(k) -(C~2)2-~
(1) single bond, or
(m) -C~=, wherein Y and R12 are absent forming a
-C=C- bridge to the carbon bearing Z and
Rll; and
Y is:
(a) single bond,
(b) -0_,
( c ) --C~2- ~
(d) ~S(O)n~, or
(e) -NR13-; and
Except that X and Y are nQ~ defined in such a way
that the carbon atom to which Z is attached also
simultaneously is bonded to two heteroatoms (0, N, S,
SO . SO2 ) -
Rll and R12 are independently: -
(a) ~.
(b) (Cl-C6)-al~yl. unsubstituted or substituted
with:
i) aryl, or
ii) (C3-C7)-CYcloal~yl,
WO91/11~9 7 PCT/US91/~gs7
2 0 ~ 5 6 2
. ~ , .
- 46 -
(c) aryl,
(d) aryl-(Cl-C2)-al~yl, or
(e) (C3-C7)-cycloalkyl; snd
R13 i~
(a) ~,
(b) (Cl-C6)-alkyl,
(c) aryl,
(d) aryl-(Cl-C6)-al~yl-(C=0)-, or
lo (e) (Cl-C6)-alkyl-(C=0)-; and
Z is:
(~) -C02~.
(b) -C02-(Cl-C6)-alkyl,
(c) -tetrazol-5-yl,
(d) -C0-N~(tetrazol-5-yl),
(e) -CON~-502-aryl,
(f) -CON~-S02-(Cl-C4)-alkyl,
(g) -CON~-S02-perfluoro-(Cl-C4)-alkyl,
(h) -CON~-S02-heteroaryl, where in heteroaryl i6
a ~ or 6 membered aromatic ring containing
one or two heteroatoms selected from the
group consi6ting of 0, N, or S,
( i ) -CON~S02NR2aR2a,
(j) -S02N~CO-aryl,
(k) -S02N~C0-(Cl-C4)-alkyl,
(1) -S02N~CO-(Cl-C4)-perfluoroalkyl,
(m) -S02N~CO-heteroaryl. where in heteroaryl is
a 5 or 6 membered aromatic ring containing
one or two heteroatoms 6elected from the
group consi6ting of 0, N, or S,
(n) -SO2N~coNR2a~2a
(o) -PO(O~I)2 ~
(p) _PO(OR2)2, or
(q) -Po(O~(OR2); and
WO91/1l ffl . PCT/US91/oogs7
2~75627
- 47 -
R22 groups are independently:
(a) H,
(b) (Cl-C6)-alkyl, (C2-C6)-alkenyl or
(C2-C6)-al~ynyl each of which is
unsubstituted or substituted with a
- substituent selected from the group
consisting of: (C3-C7)-cycloalkyl, Cl, Br,
I, F, -0~, -N~t(cl-c4)-alkyl]~
-Nt(cl-c4)-alkyl32~ -C02R or
C(=O)N~t(Cl-C4)-alkyl)].
(c) aryl or aryl-(Cl-C4)-al~yl, which is
unsubstituted or substituted with a
substituent selected from the group
consisting of: Br, Cl, I, F, (Cl-C4)-alkyl,
hydroxy-(Cl-C4)-alkyl, (Cl-C4)-alkoxy, or
Co2R25,
(d) Cl, Br, I, F,
(e) amino,
(f) NH[(Cl-C4)-alkyl],
(g) Nt(Cl-c4)-alkYl]2
(h) (Cl-C6)-alkoxy.
( i ) Co2R25,
(j) C(=o)N(R25)2, or
(k) (Cl-C4)-alkylthio; and
R23 is:
(a) H,
(b) (Cl-C6)-alkyl. which is unsubstituted or
substituted with a substituent selected from
the group consisting of: -0~, -N~2,
-Nt(Cl-C4)-alkyl]2, or -Co2R25,
WO91/ll ffl
2 0 7 S 6 2 7 PCr/US91/OO~S7
. .
- 48 -
(c) aryl or aryl-(Cl-C4)-alkyl, ~hich i~
unsubstituted or substituted with: Br, Cl,
~, I, (Cl-C4)-alkyl. hydroxy-(Cl-C4)-alkyl,
Co2R25, CoR25, or So2R25,
R~4 groups are independently:
(a) E,
(b) (Cl-C6)-al~yl, which i8 unsubstituted or
substituted with a ~ubstituent selected from
lo the group consisting of: Cl, Br, I, F,
-Co2R25, hydroxy-(Cl-C4)-alkyl, or
(Cl-C4)-acyl, or
(c) aryl or aryl-(Cl-C4)-al~yl; and
R25 is:
(a) ~,
(b) (Cl-C6)-alkyl,
(c) aryl, or
(d) aryl-(Cl-C5)-alkyl;
or a pharmaceutically acceptable salt thereof.
WO91~11~9 pcT/us9l/ooss7
,
: .
207~S27
- 49 -
The al~yl substitutents recited above denote
straight and branched chain hydrocarbons of the
length specified such as methyl, ethyl, isopropyl,
isobutyl, neopentyl, isopentyl, etc.
The alkenyl and alkynyl substituents tenote al~yl
groups as described above which are modified 80 that
each contains a carbon to carbon double bond or
triple bond, respectively, ~uch as vinyl, allyl and
2-butenyl.
Cycloal~yl tenotes rings composed of 3 to 8
methylene groups, each which may be substituted or
unsubstituted with other hydrocarbon substituents,
and include for e~ample cyclopropyl, cyclopentyl,
cyclohexyl and 4-methylcyclohe~yl.
The alkoxy substituent represents an alkyl group
as described above attached through an o~ygen bridge.
The aryl substituent recited above represents
phenyl or naphthyl.
The heteroaryl substituent recited above
represents any 5- or 6-membered aromatic ring
containing from one to three heteroatoms selected
from the group consisting of nitrogen, o~ygen, and
sulfur, for example, pyridyl, thienyl, furyl,
imidazolyl, and thiazolyl.
WO 91/11~9 2 0 7 5 6 2 7 PCT/US91/0~57
! ~
-50-
- Compounds of the present in~ention illu~trative of
- ~ubclasses of Formula I are:
5,5_F~S~n T~TDA7.oT~F~s:
2-~utyl-1-t4-(1-carboxy-1-phenyl)metho~yphenyl~methyl-
1,4-dihydro-4-methylimidazot4,5-d]imidazole
1-t4-(1-Carboxy-l-phenyl)methoxyphenyl3methyl-1,4-
dihydro-2-ethyl-4-methylimidazo[4.5-d]imidazole
1-t4-(1-Carboxy-1-(2-chlorophenyl))metho~yphenyl]-
methyl-1,4-dihydro-2-ethyl-4-methylimidazot4,5-d]-
imidazole
.
l-t4-((1-Carboxy-1-(2-chlorophenyl))methoxy)-3-propyl-
phenyl]methyl-1,4-dihydro-2-ethyl-4-methylimidazo-
t4.5-d]imidazole
l-t4-(1-Carboxy-1-(2-isopropylphenyl))methoxyphenyl]-
methyl-1,4-dihydro-2-ethyl-4-methylimidazot4,5-d]-
imidazole
l-t4-(1-Carboxy-1-(2-trifluromethylphenyl))methoxy-
phenyl]methyl-1,4-dihydro-2-ethyl-4-methylimidazo-
t4.5-d]imidazole
l-t4-((1-Carboxy-l-phenyl)methoxy)-3-propylphenyl]-
methyl-1,4-dihydro-2-ethyl-4-methylimidazot4,5-d]-
imidazole
WO91/11~ pcT/us9l/~ss7
207~627
1-[4-(l-Carboxy-l-(l-naphthyl))metho~yphenyl]methyl-
1,4-dihydro-2-ethyl-4-methylimidazot4.5-d~imidazole
1-[4-~-(1-Carboxy-1-(2-chlorophenyl))methylamino-
phenyl3methyl-1,4-dihydro-2-ethyl-4-methylimidazo-
[4,5-d]imid~zole
l-t4-N-(l-Carboxy-1-(2-chlorophenyl))methyl-N-ethyl-
aminophenyl]methyl-1.4-dihydro-2-ethyl-4-methyl-
imidazot4,5-d]imidazole
l-t4-(1-Carboxy-1-(2-chlorophenyl))methylthiophenyl~-
methyl-1,4-dihydro-2-ethyl-4-methylimidazo[4,5-d]-
imidazole
l-t4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-1,4-
dihydro-5-hydroxymethyl-4-methyl-2-propyl-imidazo-
t4.5-d]imidazole
2-Butyl-l-t4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
1,4-dihydro-4-ethyl-5-methylimidazot4,5-d]imidazole
2-Butyl-l-t4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
1,6-dihydro-6-methylimidazo~4,5-d]imitazole
Z-butyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
l~-thieno[3,4-d]imidazole
l-t4-(1-CarboXy-l-phenyl)methoxg-phenyl~methyl-2-
propyl-lE-furo[3,4-d]imidazole
2-.~utyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
4-methyl-1~-thieno[3,4-d]imidazole
WO91/11~ PCT/US91/oogs7
.. ; , _
-~- 2075627 ---
. ~ .
l-t4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-4-ethyl-
- 2-propyl-lR-thieno~3.4-d~imidazOle-~,5-diogide
2-Butyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl~methyl-
1,5-dihydro-5-methylpyrrolot3,4-d]imidazole
2-Butyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
1,5-dihydro-4,5-dimethylpyrrolo[3,4-d]imidazole
2-Butyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
1,5-dihydro-4-ethyl-5-methylSul~onylpyrrolot3,4-d~-
imidazole
3-t4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-6-methyl
-5-hydroxymethyl-2-propyl-3R-thienot2.3-d~imidazole
2-Rutyl-5-carboxy-3-t4-(1-carboxy-1-phenyl)methoxy-
phenyl]methyl-6-methyl-3H-thienot2,3-d]imidazole
2-Butyl-5-carbomethoxy-3-[4~ carboxy-1-phenyl)-
methoxyphenyl3methyl-6-methyl-3R-thienot2,3-d]-
imidazole
5-Carbomethoxy-3-t4-(1-carboxy-1-phenyl)methoxyphenyl`~
methyl-2-ethyl-6-methyl-3R-thienot2,3-d]imidazole
5-Carbomethoxy-3-[4-(1-carboxy-1-(2-chlorophenyl))-
methoxyphenyl~methyl-2-ethyl-6-methyl-3R-thieno-
~2,3-d]imidazole
3D
5-Carbomethoxy-3-[4-(1-carboxy-1-(2-isopropylphenyl))-
methoxyphenyl3methyl-2-ethyl-6-methyl-3E-thieno-
t2.3-d]imidazole
WO91/11~9 PCT/US91/oogs7
207562~
-53-
5-Carbomethoxy-3-~4~ carboxy-1-(1-naphthyl))-
methoxyphenyl~methyl-2-ethyl-6-methyl-3~-thieno-
t2.3-d~imidazole
5-Carbomethoxy-3-t4-(1-carboxy-1-(2-methoxyphenyl))-
methoxyphenyl]methyl-2-ethyl-6-methyl-3~-thieno-
[2,3-d]imidazole
5-Carbomethoxy-3-t4-(1-carboxy-1-(2-methoxyphenyl))-
lo methylthiophenyl]methyl-2-ethyl-6-methyl-3~-thieno-
t2.3-d]imidazole
5-Carbomethoxy-3-[4-N-(l-carboxy-1-(2-metho~yphenyl))-
methylaminophenyl]methyl-2-ethyl-6-methyl-3~-thieno-
lS t2.3-d]imidazole
6-[4-(1-carboxy-l-phenyl)methoxyphenyl~methyl-1,6-
dihydro-1.3-dimethyl-5-propylimidazot4.5-c~pyrazole
5-Butyl-6-t4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
2-hydroxymethyl-6~-imidazo[4,5-d]thiazole
5-Butyl-6-t4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
2-phenyl-6~-imidazot4,5-d~thiazole
5-Butyl-6-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
2-(2-chloro)phenyl-6~-imidazot4,5-d]thiazole
6-t4-(1-Carboxy-l-phenyl)metho~yphenyl]methyl-5-ethyl-
2-phenyl-6~-imidazo~4,5-d]thiazole
WO91/11~9 PCT/US91/00957
,t. , _ .
2075627 `~
114/VJC46 -54-
6-t4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-2-(2-
`- chloro)phenyl-5-ethyl-6H-imidazot4.5-d~thiazole
6-[4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-2-(2-
chloro)phenyl-5-ethyl-6H-imidazot4,5-d]oxazole
6-[4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-3-methyl
-5-butyl-6H-imidazot4,5-d]isothiazole
6-t4-(1-Carboxy-l-phenyl)metho~yphenyl]methyl-5-ethyl-
3-methyl-6~-imidazo[4,5-d~isothiazole
6-[4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl3-
methyl-5-ethyl-3-methyl-6H-imidazo[4.5-d]i60thiazole
6-[4-(1-Carboxy-1-(2-ethylphenyl))methoxyphenyl]methyl
-5-ethyl-3-methyl-6H-imidazot4.5-d]isothiazole
6-t4-(1-Carboxy-1-(2-trifluromethylphenyl))methoxyphen
yl]methyl-5-ethyl-3-methyl-6H-imidazo[4,5-d]-
isothiazole
6-t4-(1-Carboxy-1-(2-nitrophenyl))methoxyphenyl]methyl
-5-ethyl-3-methyl-6H-imidazot4,5-d~isothiazole
6-t4-N-(l-Carbo~y-1-(2-chlorophenyl))methylaminophenyl
]methyl-5-ethyl-3-methyl-6~-imidazot4~5-d]isothiazole
6-t4-(1-Carbo~Y-1-(2-chlorophenyl))methylthiophenyl]-
methyl-5-ethyl-3-methyl-6H-imidazo[4~5-d]isothiazole
WO91/11~ PCT/US91/~957
2D7562~
-55-
6-t4-(1-Carboxy-1-(2-chlorophenyl))methylsulfonyl-
phenyl]methyl-5-ethyl-3-methyl-6~-imidazot4,5-d]-
isothiazole
6-t4-(1-Carboxy-l-(l-naphthyl))metho~yphenyl3methyl-
5-ethyl-3-methyl-6E-imidazo[4,5-d]isothiazole
6-[4-(1-Carboxy-l-phenyl)metho~yphenyl]methyl-3,5-
diethyl-6H-imidazot4,5-d]isothiazole
2-Butyl-1,4-dihydro-4-methyl-1-[4-(1-(tetrazol-5-yl)-
l-phenyl)methoxyphenyl]methylimidazo[4,5-d]imidazole
1,4-Dihydro-2-ethyl-4-methyl-1-t4-(1-(tetrazol-5-yl)-
1-phenyl)methoxyphenyl]methylimidazot4,5-d]imidazole
1,4-Dihydro-2-ethyl-4-methyl-1-t4-(1-(tetrazol-5-yl)-
1-(2-chloro)phenyl)methoxyphenyl]methylimidazot4,5-d]-
imidazole
1,4-Dihydro-2-ethyl-4-methyl-1-[4-(1-(tetrazol-5-yl)-
1-(2-isopropyl)phenyl)methoxyphenyl]methylimidazo-
t4,5-d]imidazole
1,4-Dihydro-2-ethyl-4-methyl-1-t4-(1-(tetrazol-5-yl)-
1-(2-trifluromethyl)phenyl)methoxyphenyl]methyl-
imidazot4,5-d]imidazole
1,4-Dihydro-2-ethyl-4-methyl-1-t4-(1-(tetrazol-5-yl)-
1-(1-naphthyl))methoxyphenyl]methylimidazo[4,5-d]-
q imidazole
WO91/11~9 PCT/US91/oogs7
A ~' 2 0 7 5 6
' ~ ,I,,. ., ~ ~ ;,
1,4-Dihydro-2-ethyl-4-methyl-1-[4-N-(l-(tetrazol-5-
yl)-l-(2-chlorophenyl))methylaminophenyl]methyl-
- imidazo[4,5-d~imidazole
1,4-Dihydro-2-ethyl-4-methyl-1-t4-N-(l-(tetrazol-5-
yl)-l-(2-chloro)phenyl)methyl-N-ethylaminophenyl]-
methylimidazo[4,5-d]imidazole
1,4-Dihydro-2-ethyl-4-methyl-1-t4-(1-(tetrazol-5-yl)-
lo 1-(2-chlorophenyl))methylthiophenyl]methylimidazo-
t4,5-d]imidazole
1,4-Dihydro-5-hydroxymethyl-4-methyl-2-propyl-1-t4-(1-
(tetrazol-5-yl)-1-phenyl)methoxyphenyl]methylimidazo-
t4~5-d]imidazole
2-Butyl-1,4-dihydro-4-ethyl-5-methyl-1-t4-(1-
(tetrazol-5-yl)-1-phenyl)methoxyphenyl~methylimidazo-
t4.5-d]imidazole
2-Butyl-1,6-dihydro-6-methyl-1-t4-(1-(tetrazol-5-yl)-
l-phenyl)methoxyphenyl~methylimidazo[4,5-d]imidazole
2-Butyl-l-t4-(l-(tetrazol-5-yl)-1-phenyl)methoxy-
phenyl]methyl-lH-thienot3,4-d]imidazole
2-Propyl-1-[4-(1-(tetrazol-5-yl)-1-phenyl)methoxy-
phenyl]methyl-lH-furot3,4-d]imidazole
2-Butyl-4-methyl-1-t4-(1-(tetrazol-5-yl)-1-phenyl)-
methoxyphenyl]methyl-lH-thienot3.4-d]imidazole
WO91/11~ - PCT/USgl/009~7
~`- ` 207~627
114/VJC46 -57-
4-Ethyl-2-propyl-l-t4-(l-(tetraZol-5-yl)-l-phen
- metho~yphenyl]methyl-lH-thieno[3,4-d~imidazole-5,5-
dioxide
5 2-Butyl-1,5-dihydro-5-methyl-1-t4-(1-(tetrazol-5-yl)-
l-phenyl)methoxyphenyl]~ethylpyrrolot3,4-d]imidazole
2-Butyl-1,5-dihydro-4,5-dimethyl-1-t4-(1-(tetrazol-5-
yl)-l-phenyl)methoxyphenyl~methylpyrrolo[3,4-d]-
10 imidazole
2-Butyl-1,5-dihydro-4-ethyl-5-methylsulfonyl-1-[4-(1-
(tetrazol-5-yl)-1-phenyl)methoxyphenyl]methylpyrrolo-
~3,4-d]imidazole
5-~ydroxymethyl-6-methyl-2-propyl-3-t4-(1-(tetrazol-5-
yl)-l-phenyl)methoxyphenyl]methyl-3H-thieno~2,3-d]-
imidazole
2-Butyl-5-carboxy-6-methyl-3-[4-(1-(tetrazol-5-yl)-1-
phenyl)methoxyphenyl]methyl-3H-thieno[2,3-d]imidazole
2-Butyl-5-carbomethoxy-6-methyl-3-t4-(1-(tetrazol-5-
yl)-l-phenyl)methoxyphenyl]methyl-3H-thienot2,3-d]-
2s imidazole
5-Carbomethoxy-2-ethyl-6-methyl-3-[4-(1-(tetrazol-5-
yl)-l-phenyl)methoxYphenyl~methyl-3H-thieno~2,3-d~-
imidazole
5-carbomethoxy-2-ethyl-6-methyl-3-t4-(l-(tetrazol-5
yl)-1-(2-chloro)phenyl)methoxyphenyl]methyl-3H-
thieno~2,3-d]imidazole
WO 91/11~ 2 0 7 5 6 2 7 PCT/US91/ ~ 7
5-Carbomethoxy-2-ethyl-6-methyl-3-[4-(1-(tetrazol-5-
yl)-1-(2-isopropylphenyl))methoxyphenyl3methyl-3H-
thienot2,3-d]imidazole
5-Carbomethoxy-2-ethyl-6-methyl-3-t4-(1-(tetrazol-5-
yl)-1-(1-naphthyl))methoxyphenyl]methyl-3~-thieno-
t2.3-d]imidazole
5-Carbomethoxy-2-ethyl-6-methyl-3-t4-(1-(tetrazol-5-
lo yl)-1-(2-methoxyphenyl))methoxyphenyl]methyl-3~-
thienot2,3-d]imidazole
5-Carbomethoxy-2-ethyl-6-methyl-3-[4-(1-(tetrazol-5-
yl)-1-(2-methoxyphenyl~)methylthiophenyl]methyl-3H-
thienot2,3-d]imidazole
5-Carbomethoxy-2-ethyl-6-methyl-3-t4-N-(l-(tetrazol-5-
yl)-l-(2-methoxyphenyl))methylaminophenyl]methyl-3H-
thienot2,3-d]imidazole
1,6-dihydro-1,3-dimethyl-5-propyl-6-t4-(l-(tetrazol-5-
yl)-l-phenyl)methoxyphenyl]methyl-imidazot4,5-c]-
pyrazole
5-Butyl-2-hydroxymethyl-6-t4-(1-(tetrazol-5-yl)-1-
phenyl)methoxyphenyl]methyl-6~-imidazot4~5-d]thiazole
5-Butyl-2-phenyl-6-t4-(1-(tetrazol-5-yl)-1-phenyl)-
methoxyphenyl]methyl-6~-imidazot4~5-d3thiazole
5-Butyl-2-(2-chloro)phenyl-6-t4-(1-(tetrazol-5-yl)-l-
phenyl)methoxyphenyl]methyl-6~-imidazot4,5-d]~hiazole
WO 91/11999
PCT/US91/00957
,' '; ' ~
207~27
-59-
5-Ethyl-2-phenyl-6-[4-(1-(tetrazol-5-yl)-1-phenyl~-
methoxyphenyl~methyl-6~-imidazo~4.5-d]thiazole
2-(2-Chloro)phenyl-5-ethyl-6-[4-(1-(tetrazol-5-yl)-1-
phenyl)methoxyphenyl]methyl-6H-imidazot4,5-d]thiazole
2-(2-Chloro)phenyl-5-ethyl-6-t4-(1-(tetrazol-5-yl)-1-
phenyl)methoxyphenyl]methyl-6H-imidazo[4,5-d]oxazole
3-Methyl-5-butyl-6-~4-(1-(tetrazol-5-yl)-1-phenyl)-
methoxyphenyl]methyl-6H-imidazot4,5-d~isothiazole
5-Ethyl-3-methyl-6-t4-(1-(tetrazol-5-yl)-1-phenyl)-
methoxyphenyl]methyl-6H-imidazot4,5-d]isothiazole
5-Ethyl-3-methyl-6-t4-(1-(tetrazol-5-yl)-1-(2-chloro-
phenyl))methoxyphenyl]methyl-6H-imidazot4.5-d]-
isothiazole
5-Ethyl-3-methyl-6-t4-(1-(tetrazol-5-yl)-1-(2-ethyl-
phenyl))methoxyphenyl]methyl-6H-imidazot4,5-d]-
isothiazole
5-Ethyl-3-methyl-6-t4-(1-(tetrazol-5-yl)-1-
(2-trifluoromethylphenyl))methoxyphenyl]methyl-6~-
imidazot4,5-d]isothiazole
- 5-Ethyl-3-methyl-6-t4-(1-(tetrazol-5-yl)-1-
(2-nitrophenyl))methOXYPhenyl]methyl-6~I-imidazo-
t4.5-d]isothiazole
WO 91/11999 Pcr/us9l/oo
207~627 ~`
-60-
5-Ethyl-3-methyl-6-t4-N-(l-(tetrazol-5-yl)-1-(2-
chlorophenyl))methylsminOphenYl]methyl-6~-imidazo-
[4,5-d]isothiazole
- ~ S-Ethyl-3-methyl-6-t4-(1-(tetrazol-5-yl)-1-(2-chloro- phenyl))methylthiophenyl]methyl-6~-imidazo[4,5-d]-
isothiazole
~-Ethyl-3-methyl-6-t4-(1-(tetrazol-5-yl)-1-(2-chloro-
phenyl))methylsulfonylphenyl~methyl-6H-imidazot4,~-d~-
isothiazole
5-~thyl-3-methyl-6-[4-(1-(tetrazol-5-yl)-1-(1-
naphthyl)methoxyphenyl]methyl-6~-imidazo~4,5-d]-
lS isothiazole
3,5-Diethyl-6-~4-(1-(tetrazol-5-yl)-1-phenyl)methoxy-
phenyl]methyl-6~-imidazo[4,5-d]isothiazole
2-Butyl-l-t4-(l-((N-phenylsulfonyl)carboxamido)-l-
phenyl)methoxyphenyl]methyl-1,4-dihydro-4-methyl-
imidazot4,5-d]imidazole
2-Butyl-l-t4-(1-((N-methylsulfonyl)carboxamido)-l-
phenyl)methoxyphenyl~methyl-1.4-dihydro-4-methyl-
imidazo[4,~-d]imidazole
2-Butyl-1-~4-(1-((N-trifluoromethylsulfonyl)-
carboxamido)-l-phenyl)methoxyphenyl~methyl-l,4-
3~ dihydro-4-methylimidazo[4.5-d]imidazole
WO91/11~ . PCT/US91/00957
2075627
2-Butyl-l-t4-(1-((N-phenyl~ulfonyl)carboxamido)-l-
phenyl)methoxyphenyl~methyl-1,5-dihydro-5-methyl-
pyrrolo~3,4-d]imidazole
2-Butyl-l-t4-(1-((N-trifluoromethylsulfonyl)-
carboxamido)-l-phenyl)methoxyphenyl]methyl-1,5-
dihydro-5-methylpyrrolo~3,4-d]imidazole
2-(2-Chloro)phenyl-5-ethyl-6-[4-(1-((N-i~opropyl-
sulfonyl)carboxamido)-l-phenyl)methoxyphenyl]methyl-
6H-imidazo~4,5-d]oxazole
BF.~TMTn~0l.~S:
2-~utyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
benzimidazole
2-Butyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
7-methylbenzimidazole
l-t4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-7-methyl
-2-propylbenzimidazole
1-[4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-5,7-
dimethyl-2-ethylbenzimidazole
1-[4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethylbenzimidazole
1-[4-(l-Carboxy-l-(2-methylphenyl))methoxyphenyl]_
methyl-5,7-dimethyl-2-ethylbenzimidazole
WO9lJ11~ 2 0 7 5 6 2 7 PCT/USsl/onss7
1-[4-(1-Carboxy-l-(2.6-dichlorophenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethylbenzimidazole
l-t4-(1-Carboxy-1-(2-nitrophenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethylbenzimidazole
l-t4-(l-Carboxy-1-(2-trifluromethylphenyl))methoxy-
phenyl~methyl-5,7-dimethyl-2-ethylbenzimidazole
1o 1-[4-(1-Carboxy-1-(2-ethoxyphenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethylbenzimidazole
1-[4-(1-Carboxy-l-(l-naphthyl))methoxyphenyl]methyl-
5,7-dimethyl-2-ethylbenzimidazole
1-[4-(1-Carboxy-1-(2.6-dimethylphenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethylbenzimidazole
1-[4-(N-(l-Carboxy-1-(2-chlorophenyl))methyl)amino-
phenyl3methyl-5.7-dimethyl-2-ethylbenzimidazole
1-[4-(N-(l-Carboxy-1-(2-chlorophenyl))methyl-N-
methyl)aminophenyl]methyl-5,7-dimethyl-2-
ethylbenzimidazole
l-t4-(N-(l-carboxy-l-(2-chlorophenyl))methyl-N-acetyl)
aminophenyl]methyl-5.7-dimethyl-2-ethylbenzimidazole
l-t4-(1-Carboxy-1-(2-chlorophenyl))methylthiophenyl]-
methyl-5,7-dimethyl-2-ethylbenzimidazole
WO91/11~ PCT/US91/009S7
20~627
-63-
l-t4-(1-Carboxy-1-(2-chlorophenyl))methylsulfinyl-
phenyl]methyl-5,7-dimethyl-2-ethylbenzimidazole
1-~4-(1-Carboxy-1-(2-chlorophenyl))methyl~ulfonyl-
phenyl]methyl-5,7-dimethyl-2-ethylbenzimidazole
1-~4-(1-Carboxy-1-(2-chlorophenyl))metho~y-3-methyl-
phenyl3methyl-5,7-dimethyl-2-ethylbenzimidazole
l-t4-(1-Carboxy-1-(2-chlorophenyl))methoxy-3-ethyl-
phenyl]methyl-5,7-dimethyl-2-ethylbenzimidazole
1-~4-(1-Carboxy-1-(2-chlorophenyl))methoxy-3-propyl-
phenyl~methyl-5,7-dimethyl-2-ethylbenzimidazole
l-t4-(1-Carboxy-1-(2-chlorophenyl))methoxy-3-chloro-
phenyl]methyl-5,7-dimethyl-2-ethylbenzimidazole
1-~4-(1-Carboxy-1-(2-chlorophenyl))methoxy-3,5-
dichlorophenyl]methyl-5,7-dimethyl-2-ethyl-
benzimidazole
1-~4-(1-Carboxy-1-(2-chlorophenyl))metho~yphenyl3-
methyl-2-ethyl-S-hydroxymethyl-7-methylbenzimidazole
5-Carboxy-l-t4-(1-carbo~y-1-(2-chlorophenyl))metho~y-
phenyl]methyl-2-ethyl-7-methylbenzimidazole
5-Carbo~ethoxy-1-[4-(1-carboxy-1-(2-chlorophenyl))-
methoxyphenyl3methyl-2-ethyl-7-methylbenzimidazole
WO 91/11~9 2 0 7 5 6 2 7 PCT/US91/00957
-64-
l-t4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-2,5-diethyl-7-methylbenzimidazole
l-t4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl3-
methyl-2,5;7-triethylbenzimidazole
1-[4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-2-ethyl-4,5,7-trimethylbenzimidazole
1-t4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl~-
methyl-5,7-dimethyl-2-ethyl-4-hydroxybenzimidazole
5,7-Dimethyl-2-ethyl-1-t4-(1-(tetrazol-5-yl)-1-
(2-chlorophenyl))methoxyphenyl]methylbenzimidazole
5,7-Dimethyl-2-ethyl-1-t4-(1-(tetrazol-5-yl)-1-
(2-methylphenyl))methoxyphenyl]methylbenzimidazole
5,7-Dimethyl-2-ethyl-1-t4-(1-(tetrazol-5-yl)-1-
- 20 (2-ethoxyphenyl))methoxyphenyl~methylbenzimidazole
5,7-Dimethyl-2-ethyl-1- r 4-(1-(tetrazol-5-yl)-1-
(2-trifluoromethylphenyl))methoxy-3-methoxyphenyl]-
methylbenzimidazole
5,7-Dimethyl-~-ethyl-l-t4-N-(l-(tetrazol-5-yl)-1-
(2-chlorophenyl))methylaminophenyl]methylbenzimidazole
5,7-Dimethyl-2-ethyl-1-t4-N-(l-(tetrazol-5-yl)-1-
(2-chlorophenyl))methyl-N-methylaminophenyl~methyl-
benzimidazole
WO91/11~9 PCT/US91/009~7
2 0 7 ~ 6 2 7
5,7-Dimethyl-2-ethyl-1-[4-(1-(tetrazol-5-yl)-1-(2-
chlorophenyl))methylthiophenyl]methylbenzimidazole
5,7-Dimethyl-2-ethyl-1-t4-(l-(tetrazol-5-yl)-1-
(2-chlorophenyl)~methylsulfinylphenyl]methyl-
benzimidazole
5,7-Dimethyl-2-ethyl-1-t4-(1-(tetrazol-5-yl)-1-
(2-chlorophenyl))methylsulfonylphenyl]methyl-
benzimidazole
2-Ethyl-5-hydroxymethyl-7-methyl-1-[4-(1-(tetrazol-5-
yl)-l-(2-chlorophenyl))methoxyphenyl~methyl-
benzimidazole
5-Carboxy-7-methyl-2-ethyl-1-[4-(1-(tetrazol-5-yl)-1-
(2-chlorophenyl))methoxyphenyl]methylbenzimidazole
5-Carbomethoxy-2-ethyl-7-methyl-1-t4-(l-(tetrazol-5-
yl)-1-(2-chlorophenyl))methoxyphenyl~methyl-
benzimidazole
2,5-Diethyl-7-methyl-1-[4-(1-(tetrazol-5-yl)-1-(2-
chlorophenyl))methoxyphenyl]methylbenzimidazole
1-[4-(1-(Tetrazol-5-yl)-1-(2-chlorophenyl))methoxy-
phenyl~methyl-2.5.7-triethylbenzimidazole
2-Ethyl-1-[4-(1-(tetrazol-5-yl)-1-(2-chlorophenyl))-
methoxyphenyl~methyl-4,5.7-trimethylbenzimidazole
WO91/11~ PCT/US91/009s7
2075627
-66-
5,7-Dimethyl-2-ethyl-4-hydroxy-1-[4-tl-(tetrazol-5-yl)
-1-(2-chlorophenyl))methoxyphenyl]methylbenzimidazole
5,7-Dimethyl-2-ethyl-1-r4-(1-(N-Phenyl~ulfonyl)-
carboxamido-1-(2-chlorophenyl))metho~yphenyl]methyl-
benzimidazole
5,7-Dimethyl-2-ethyl-1-~4-(1-(~-trifluoromethyl-
sulfonyl)carboxamido-l-(2-chlorophenyl))methoxyphenyl]
methylbenzimidazole
5,7-Dimethyl-2-ethyl-1-[4-(1-(N-methylsulfonyl)-
carboxamido-l-(2-chlorophenyl))methoxyphenyl]methyl-
benzimidazole
TMID~0PY~TDT~F5
2-Butyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl~methyl-
7-methyl-3H-imidazo[4,5-b]pyridine
3-t4-(1-Carboxy-l-phenyl)methoxyphenyl~methyl-7-methyl
-2-propyl-3H-imidazo[4,5-b]pyridine
3-t4-(1-Carboxy-l-phenyl)methoxyphenyl~methyl-5,7-
dimethyl-2-ethyl-3~-imidazo[4,5-b]pyridine
3-[4-(1-Carboxy-1-(2-chlorophenyl)~methoxyphenyl]-
methyl-7-methyl-2-propyl-3H-imidazo[4,5-b]pyridine
3-[4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b~pyridine
WO91/11~ PCT/US91/~gs7
v~ ~ 2a75~27
3-~4-(1-Carboxy-1-(3-chlorophenyl))methoxyphenyl3-
methyl-7-methyl-2-propyl-3H-imidazot4,5-b]pyridine
3-[4-(1-Carboxy-1-(2,6-dichlorophenyl))metho yphenyl]-
methyl-7-methyl-2-propyl-3H-imidazo~4,5-b3pyridine
3-t4-(1-Carboxy-1-(4-chlorophenyl))methoxyphenyl~-
methyl-7-methyl-2-propyl-3H-imidazot4,5-b]pyridine
10 3-t4-(1-Carboxy-1-(2-bro~ophenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-[4-(1-Carboxy-1-(2-methylphenyl))methoxyphenyl]-
methyl-7-methyl-2-propyl-3~-imidazot4,5-b]pyridine
3-t4~ Carboxy-1-(2-methylphenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-t4-(1-Carboxy-1-(2-methoxyphenyl))metho~yphenyl]-
20 methyl-7-methyl-2-propyl-3~-imidazot4,5-b~pyridine
3-t4-(1-Carboxy-1-(2-methoxyphenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3~-imidazot4,5-b]pyridine
2s 3-t4-(1-Carboxy-1-(2-etho~yphenyl))methoxyphenyl]-
methyl-7-methyl-2-propyl-3H-imidazo~4,5-b]pyridine
3-[4-(1-Carboxy-l-(2-ethoxyphenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-[4-(1-Carboxy-1-(2-(1-hexyloxy)phenyl))methoxy-
phenyl]methyl-7-methyl-2-propyl-3H-imidazo-
[4.5-b]pyridine
WO 91/11999 . ; PCI/US91/OQ,n. 'i7
~ : i
2 07 ~ 6 2~
-68-
3-~4-(1-Carboxy-1-(2-(1-he~yloxy)phenyl))methoxy-
phenyl]methyl-5 ! 7-dimethyl-2-ethyl-3~-imidazo-
[4.5-b]pyridine -
3-[4-(l-Carboxy-1-(2~6-dichlorophenyl))meth
phenyl~methyl-5,7-dimethyl-2-ethyl-3~-imidazo-
[4,5-b]pyridine
3-~4-(1-Carboxy-1-(2-nitrophenyl))methoxyphenyl3-
methyl-7-methyl-2-propyl-3H-imidazo~4,5-b]pyridine
3-[4-(1-Carboxy-1-(2-nitrophenyl))methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b3pyridine
3-t4-(1-Carboxy)-1-(2-carboxyphenyl)methoxyphenyl3-
methyl-7-methyl-2-propyl-3~-imidazot4,5-b]pyridine
3-[4-(1-Carboxy)-1-(2-carboxyphenyl)methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3~-imidazot4,5-b]pyridine
3-t4-(1-Carboxy-1-(2-trifluoromethylphenyl))metho~y-
phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazot4,5-b]-
pyridine
3-t4-(1-Carboxy-l-phenyl)methoxy-3-chlorophenyl]methyl
-7-methyl-2-propyl-3~-imidazot4,5-b]pyridine
3-[4-(1-Carboxy-l-phenyl)methoxy-3-chlorophenyl]methyl
-5,7-dimethyl-2-ethyl-3~-imidazot4,5-b~pyridine
3-~4-((1-Carboxy-l-phenyl)methoxy)-2-chlorophenyl3-
methyl-5~7-dimethyl-2-ethyl-3~-imidazot4~5-b]pyridine
WO91/11~ PCT/US91/00957
. .
2~5627
-69-
3-t4-(1-Carboxy-1-(2-chlorophenyl))methoxy-3-chloro-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazo-
t4.5-b]pyridine
3-t3-Benzoyl-4-~(1-carboxy-1-phenyl)methoxy)phenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-t3-AcetYl-4-((l-carbo~y-l-phenyl)methoxy)phenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b~pyridine
3-[4-((1-Carboxy-l-phenyl)methoxy)-3-methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-[4-((1-Carboxy-l-phenyl)methoxy)-3-ethoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-t3-tert-Butyl-4-((1-carboxy-1-phenyl)methoxy)phenyl]
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-t4-((1-Carboxy-l-phenyl)methoxy)-3-methylphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-t4-((1-Carboxy-1-(2-methylphenyl))methoxy)-3-chloro-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazo-
t4.5-b]pyridine
3-t4-((1-Carboxy-l-phenyl)methoxy)-3.5-dichlorophenyl~
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4.5-b]pyridine
30 3-t4-(1-Carbo~y-1-(2-Chlorophenyl))metho~y-3,5-
dichlorophenyl~methyl-5,7-dimethyl-2-ethyl-3H-
imidazo~4,5-b]pyridine
WO 91/11999 Pcr/ussl/nnss7
2075627
7 0
3-t4-((1-Carboxy-l-phenyl)methoxy)-3-chloro-5-methoxy-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazo~4,5-~]-
pyridine
3-t4-((1-Carboxy-l-phenyl)methoxy)-3-(prop-2-enyl)-
phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazo[4,5-b]-
pyridine
3-[4-((1-Carboxy-l-phenyl)methoxy)-3-propylphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]pyridine
3-t4-((1-Carboxy-1-(2-methylphenyl))methoxy)-3-propyl-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]-
pyridine
3-[4-((1-Carboxy-1-(2-chlorophenyl))methoxy)-3-propyl-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]-
pyridine
20- 3-t4-((l-Carboxy-1-(4-chlorophenyl))methoxy)-3-propyl-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]-
pyridine
3-t4-((1-Carboxy-1-(2-methoxyphenyl))methoxy)-3-propyl
phenyl3methyl-5,7-dimethyl-2-ethyl-3~-imidazo[4,5-b]-
pyridine
3-t4-((l-Carboxy-1-(2,5-dibromo-3,4-dimetho~yphenyl))-
methoxy)-3-propylphenyl3methyl-5.7-dimethyl-2-ethyl-
3H-imidazot4,5-b]pyridine
WO 91/11999
PCT/US91/009~7
2D75627
3-[4-((1-Carboxy-1-(3,4-dimethoxyphenyl))methoxy)-3-
propylphenyl]methyl-5.7-dimethyl-2-ethyl-3~-imidazo-
[4,5-b~pyridine
3-{4-(1-Carboxy-l-phenyl)methoxy-3-propylphenyl~-
methyl-5-carbomethoy -2-ethyl-7-methyl-3E-imidazo-
[4,5-b]pyridine
3-[4-(1-Carboxy-l-phenyl)methoxy-3-propylphenyl3-
methyl-5-carboxy-~-ethyl-7-methyl-3H-imidazo[4,5-b]-
pyridine
3-[4-(1-Carboxy-l-phenyl)methoxy-3-propylphenyl]-
methyl-5-carbobenzyloxy-2-ethyl-7-methyl-3~-imidazo-
t4.5-b]pyridine
3-[4-((1-Carboxy-l-phenyl)methoxy)-3-(1-methyl-
cyclohex-l-yl)phenyl]methyl-5.7-dimethyl-2-ethyl-
3H-imidazo[4,5-b]pyridine
3-[4-((1-Carboxy-l-phenyl)methoxy)-3,5-dipropylphenyl~
methyl-5,7-dimethyl-2-ethyl-3~-imidazo[4,5-b]pyridine
3-[4-((1-Carboxy-1-(2-methoxyphenyl))methoxy)-3,5-
2s dipropylphenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazo[
4,5-b]pyridine
3-[4-((1-Carboxy-1-(2.5-dibromo-3,4-dimethoxyphenyl))-
methoxy)-3.5-dipropylphenyl]methyl-5.7-dimethyl-
2-ethyl-3~-imidazo[4,5-b]pyridine
" WO 91/11999
2 0 7 5 6 2 7 Pcr/ usg 1 /Or ~Q9~;7
-
3-[4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-2-ethyl-5-hydroxymethyl-7-methyl-3H-imidazo-
t4.5-b]pyridine
5-Carboxy-3-t4-(1-carbo~y-1-(2-chlorophenyl))methoxy-
phenyl]methyl-2-ethyl-7 methyl-3~-imidazot4,5-b]-
pyridine
5-Carbomethoxy-3-t4-(1-carboxy-1-(2-chlorophenyl~)-
1o methoxyphenyl]methyl-2-ethyl-7-methyl-3~-imidazo-
t4.5-b]pyridine
5-Carbomethoxy-3-t4-(1-carboxy-1-(2-chlorophenyl))-
methoxyphenyl]methyl-2-ethyl-7-trifluromethyl-3H-imida
zot4~5-b]pyridine
3-t4-(1-Carboxy-l-cyclohexyl)methoxyphenyl]methyl-7-
methyl-2-propyl-3H-imidazo[4,5-b]pyridine
3-[4-(1-Carboxy-l-propyl)methoxyphenyl]methyl-7-
methyl-2-propyl-3H-imidazot4,5-b]pyridine
3-t4-((1-Carboxy-1-(3-phenyl)propyloxy)phenyl]methyl-
5,7-dimethyl-2-ethyl-3~-imidazot4,5-b]pyridine
3-t4-((1-Carboxy-1-(2-phenyl)ethoxy)phenyl]methyl-5,7-
dimethyl-2-ethyl-3~-imidazot4.5-b]pyridine
3-[4-(1-carboxy-1-phenoxy)methylphenyl]methyl-7-
methyl-2-propyl-3~-imidazo[4,5-b]pyridine
WO91/ll ffl PCT/US91/009~7
- 2~75627
- (z)-3-t(4-((2-Carboxy-2-phenyl)ethylenyl)phenyl)-
methyl]-7-methyl-2-propyl-3H-imidazot4,5-b]pyridine
3-t(4-((2-Carboxy-2-phenyl)ethyl)phenyl)methyl3-7-
methyl-2-propyl-3~-imidazo[4,5-b]pyridine
3-[4-(1-Carboxy-l-methyl-l-phenyl)methoxyphenyl]-
methyl-7-methyl-2-propyl-3H-imidazo[4,5-b]pyridine
3-t4-(1-Carboxy-l-(naphth-l-yl))methoxyphenyl]methyl-
7-methyl-2-propyl-3~-imidazot4,5-b]pyridine
3-~4-(1-Carboxy-1-(3-methylnaphth-2-yl))methoxy-
phenyl]methyl-7-methyl-2-propyl-3~-imidazo[4,5-b]-
15 pyridine
3-t4-N-(l-Carboxy-l-phenyl)methyl)-N-methylamino-
phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazo-
t4,5-b]pyridine
3-[4-N-(l-Carboxy-l-phenyl)methyl)-N-ethylaminophenyl]
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4.5-b]pyridine
3-[4-N-(l-Carboxy-l-phenyl)methyl)-N-propylamino-
phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazo-
t4,5-b]pyridine
3-t4-N-(l-Carboxy-l-phenyl)methyl)aminophenyl]methyl-
5,7-dimethyl-2-ethY1-3~-imidaZot4~5-b]pyridine
3-t4-N-(l-Carboxy-l-phenyl)methyl)-N-allylaminophenyl]
methyl-5~7-dimethyl-2-ethyl-3~-imidazot4~5-b]pyridine
WO91/11~ PCT/USgl/00957
207 S 627 ~-:
.
-74-
3-[4-N-(l-Carboxy-l-phenyl)methyl)-N-i~o-butylamino-
phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazo-
[4,5-b]pyridine
3-[4-N-(l-Carboxy-l-phenyl)methyl)-N-cyclopropyl-
methylaminophenyl]methyl-5,7-dimethyl-2-ethyl-3H-
imidazot4,5-b]pyridine
3-[4-N-(l-Carbogy-l-phenyl)methyl)-N-sec-butylamino-
lo phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazo-
[4,5-b]pyridine
3-~4-N-(l-Carboxy-l-phenyl)methyl)-N-iso-propylamino-
phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazo~4,5-b]-
pyridine
3-[4-N-(l-(Tetrazol-5-yl)-1-phenyl)methyl)-N-methyl-
aminophenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazo-
[4,5-b]pyridine
2-Butyl-7-methyl-1-[4-(1-(tetrazol-5-yl)-1-phenyl)-
methoxyphenyl]methyl-3H-imidazo[4.5-b]pyridine
7-Methyl-2-propyl-3-[4-(1-(~etrazol-5-yl)-1-phenyl)-
methoxyphenyl3methyl-3H-imidazot4.5-b~pyridine
7-Methyl-2-propyl-3-[4-(1-(tetraZol-5-yl)-1-(2-chloro-
phenyl)methoxyphenyl3methyl-3H-imidazo[4.5-b3pyridine
7-Methyl-2-propyl-3-[4-(2-phenyl-2-(tetrazol-5-yl)-
ethyl~phenyl~methyl-3H-imidazo[ 4 . 5-b] pyr i d ine
WO91/11~ PCT/US91/00957
207S62~
5,7-Dimethyl-2-ethyl-3-[4-(1-(tetrazol-5-yl)-1-
phenyl)methoxyphenyl]methyl-3~-imidazot4,5-b]pyridine
5,7-Dimethyl-2-ethyl-3-[4-(1-(tetrazol-5-yl)-1-(2-
chlorophenyl))methoxyphenyl]methyl-3~-imidazot4,5-b]-
pyridine
5,7-Dimethyl-2-ethyl-3-t4-(1-(tetrazol-5-yl)-1-(2-
methylphenyl))methoxyphenyl]methyl-3E-imidazot4,5-b]-
10 pyridine
5,7-Dimethyl-2-ethyl-3-t4-(1-(tetrazol-5-yl)-1-(2-
etho~yphenyl))methoxyphenyl]methyl-3~-imidazo-
[4,5-b]pyridine
~,7-Dimethyl-2-ethyl-3-t4-(1-(tetrazol-5-yl)-1-(2,6-
dichlorophenyl))methoxyphenyl3methyl-3~-imidazo-
t4.5-b]pyridine
20 5,7-Dimethyl-2-ethyl-3-t4-(1-(tetrazol-5-yl)-1-
(2-nitrophenyl))methoxyphenyl]methyl-3H-imidazo-
t4,5-b]pyridine
5,7-Dimethyl-2-ethyl-3-t4-(1-(tetrazol-5-yl)-1-(2-
trifluoromethylphenyl))metho~yphenyl]methyl-3~-
imidazot4,5-b]pyridine
5,7-Dimethyl-2-ethYl-3-t4-(1-(tetraZol-5-yl)-1-
(2-chlorophenyl))methoxy-3-chlorophenyl]methyl-3~-
imidazot4,5-b]pyridine
WO91/11~9 PCT/US91/O~gs7
2075627
: - ~
5,7-Dimethyl-2-ethyl-3-t4-(1-(tetrazOl-5-yl)-1-
(2-chlorophenyl))metho~y-3.5-dichlorophenyl~methyl-3H-
imidazo[4,5-b]pyridine
2-~thyl-5-hydro~ymethyl-7-methyl-3-t4-(1-(tetrazol-5-
yl)-1-(2-chlorophenyl))metho~yphenyl]methyl-3H-
imidazot4,5-b]pyridine
5-Carbo2y-2-ethyl-7-methyl-3-t4-(l-(tetrazol-5-yl)-l-
1o (2-chlorophenyl))methoxyphenyl]methyl-3H-imidazo-
t4.5-b]pyridine
5-Carbmethoxy-2-ethyl-7-methyl-3-t4-(1-(tetrazol-5-yl)
-1-(2-chlorophenyl))methoxyphenyl~methyl-3H-imidazo-
t4~5-b]pyridine
5-Carbmethoxy-2-ethyl-3-t4-(1-(tetrazol-5-yl)-1-(2-
chlorophenyl))metho2yphenyl]methyl-7-trifluromethyl-
3H-imidazot4,5-b]pyridine
5,7-Dimethyl-2-ethyl-3-t4-(1-((N-phenylsulfonyl)-
carboxamido)-1-(2-chloro)phenyl)methoxyphenyl]methyl_
3H-imidazot4,5-b]pyridine
5,7-Dimethyl-2-ethyl-3-t4-(1-((N-methylsulfonyl)-
carbo~amido)-1-(2-Chloro)phenyl)metho2yphenyl]methyl-
3H-imidazo~4,5-b]pyridine
5,7-Dimethyl-2-ethyl-3-[4-(1-((N-trifluoromethyl-
sulfonyl)carboxamido)-l-(2-chloro)phenyl)meth
phenyl]methyl-3H-imidazot4,5-b]pyridine
WO91/11~9 PCT/US91/009~7
2075S2~
3-[4-(l-(~ydroxymetho~ypho~phoryl)-l-(2-methylphenyl))
methoxyphenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazo-
t4,5-b]pyridine
p~T~FS:
9-[4-(1-Carboxy-l-phenyl)methoxyphenyl~methyl-6-
methyl-8-propylpurine
9-t4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl3-
methyl-6-methyl-8-propylpurine
4,6-Dimethyl-8-ethyl-9-[4-(1-carboxy-1-~2-chloro-
phenyl))methoxyphenyl3methylpurine
9-t4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-8-ethyl-6-methylpurine
9-t4-(1-Carboxy-1-(2-chlorophenyl))methoxy-3-propyl-
phenyl]methyl-8-ethyl-6-methylpurine
9-~4-(1-Carboxy-l-phenyl)methoxy-3-propylphenyl]-
methyl-8-ethyl-6-methylpurine
9-t4-(l-Carboxy-l-(2-chlorophenyl))methoxyphenyl3-
methyl-4-dimethylamino-8-ethyl-6-methylpurine
9-t4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-8-ethyl-6-methyl-4-(morpholin-4-yl)purine
9-[4-(l-carboxy-l-(2-chlorophenyl))methoxyphenyl]
methyl-8-ethyl-6-methyl-4-methylaminOpurine
. WO91/11~ PCT/US91/~Q957
2075627 ---
-78-
9-t4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-4,8-diethyl-6-methylpurine
9-r4-(1-Carboethoxy-1-(2-chlorophenyl))methoy phenyl]- methyl-4,8-diethyl-6-methylpurine
9-~4-(1-Carboxy-1-(2-chlorophenyl))metho~yphenyl]-
methyl-4,8-diethyl-6-trifluoromethylpurine
10 9-t4-(l-carboxy-l-(2-trifluoromethylphenyl))meth
phenyl~methyl-8-ethyl-6-methyl-4-methylaminopurine
6-Methyl-8-propyl-9-t4-(l-(tetrazol-5-yl)-1-phenyl)-
methoxyphenyl]methylpurine
6-Methyl-8-propyl-9-t4-(1-(tetrazol-5-yl)-1-(2-chloro-
phenyl))methoxyphenyl]methylpurine
4,6-Dimethyl-8-ethyl-9-t4-(1-(tetrazol-5-yl)-1-(2-
chlorophenyl))metho~yphenyl]methylpurine
8-Ethyl-6-methyl-9-t4-(1-(tetrazol-5-yl)-1-(2-chloro-
phenyl))methoxyphenyl]methylpurine
4-DimethylaminO-8-ethyl-6-methyl-9-t4-(1-(tetrazol-5-
yl)-l-(2-chlorophenyl))methoxyphenyl~methylpurine
8-Ethyl-6-methyl-4-(morpholin-4-yl)-9-t4-(1-(tetrazol-
5-yl)-1-(2-chlorophenyl))metho~yphenyl]methylpurine
8-Ethyl-6-methyl-4-methylamino-9-t4-(1-(tetrazol-5-
yl)-1-(2-chlorophenyl))methoxyphenyl]methylpurine
WO91/11~ PCT/US91/~gs7
2075627
-79-
4,8-Diethyl-6-methyl-9-t4-(1-(tetrazol-5-yl)-1-(2-
chlorophenyl))metho~yphenyl3methylpurine
4,8-Diethyl-6-methyl-9-t4-(l-(tetrazol-5-yl)-1-(2-
S chlorophenyl))methoxyphenyl]methylpurine
4,8-Diethyl-9-t4-(1-(tetrazol-5-yl)-1-(2-chloro-
phenyl))methoxyphenyl3methyl-6-trifluoromethylpurine
8-Ethyl-6-methyl-4-methylamino-9-t4-(1-(tetrazol-5-
yl)-1-(2-trifluoromethylphenyl))methoxyphenyl]methyl-
purine
4,6-Dimethyl-8-ethyl-9-~4-(1-(N-phenylsulfonyl)-
carboxamido-1-(2-chlorophenyl))metho~yphenyl]-
methylpurine
4,6-Dimethyl-8-ethyl-9-~4-(1-(N-methylsulfonyl~-
carboxamido-1-(2-chlorophenyl))methoxyphenyl~methyl-
purine
4,6-Dimethyl-8-ethyl-9-[4-(1-(N-trifluoromethyl-
sulfonyl)carboxamido-l-(2-chlorophenyl))methoxy-
phenyl]methylpurine
4,6-Dimethyl-8-ethyl-9-t4-(1-(N-acetyl)sulfonamido-l-
(2-chlorophenyl))methoxyphenyl]methylpurine
4,6-Dimethyl-8-ethyl-9-t4-(1-(N-benzoyl)sulfonamido-l-
(2-chlorophenyl))methoxyphenyl]methylpurine
WO91/11~ PCT/US91/~gs7
2 0 ~ 5 6 2 ~ ~ ?
-80-
4,6-Dimethyl-8-ethyl-9-t4-(1-(N-pyrimidin-2-yl)-
6ulfonamido-1-(2-chlorophenyl))methoxyphenyl]methyl-
purine
- 5 5~7-FusT~n TMTD~OT.T.S
2-Butyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
1,4,6,7-tetrahydroimidazot4,5-e]tl,4]diazepine-5,8-
dione
2-Butyl-l-t4-(1-carboxy-1-(2-chlorophenyl))methoxy-
phenyl]methyl-1,4,6,7-tetrahydroimidazot4,5-e]~1,4]-
diazepine-5,8-dione
2-Butyl-l-t4-((1-carboxy-1-phenyl)methoxy)-3-propyl-
phenyl]methyl-1,4,6,7-tetrahydroimidazot4,5-e]tl,4]-
diazepine-5,8-dione
2-Butyl-l-t4-((1-carboxy-1-(2-chlorophenyl))methox)-
3-propylyphenyl]methyl-1,4,6,7-tetrahydroimidazot4,5-e
]tl.4]diazepine-5.8-dione
WO91/11~9 PCT/US91/00957
~ ~ ",
2075S~7
--81--
RAT. I~ )S ~OR ~ RATTON OF cn~ Jr.~s OF
G~F:~AT. ~'-Ol~-u I ~ T
The methods described in P~T T AND PA~T TI
below illustrate the preparation of angiotensin II
antagonists of Formula I. There are several general
approaches to the synthesis of antagonists of Formula
I, and it is taken as a general principle that.one or
another method may be more readily applicable for the
preparation of a given antagonist; some of the
approaches illustrated below may not be seadily
applicable for the preparation of certain antagonists
of Formula I.
It should be recognized that antagonists of
Formula I consist of a heterocyclic component
designated above by formula I and a substituted
benzyl substitutent which i6 attached to the
heterocyclic component at a nitrogen atom. Thus, two
generally applicable approaches to antagonists of
formula I are these:
1. A heterocycle, designated above with
Formula I is prepared as described in P~T T below.
Then the heterocycle is alkylated at a nitrogen atom
with a substituted benzyl halide or pseudohalide
gi~ing an alkylated heterocycle. In the Schemes
below, this alkylating agent is often designated as
"Ar-CH2Q where Q is a halide (-Cl,Br,I) or
pseudohalide (-OMs, OTs, OTf). In some cases,
WO91/11~ PCT/US91/~ss7
207 5 62~
alkylation may take place at more than one nitrogen
atom of the heterocycle, and in these cases,
separation by fractional crystallization or by
chromotographic methods may be necessary for
isolation of the desired product. In some cases, the
alkylation step produces a fully-assembled antagonist
of Formula I, e~cept that functional groups in the
alkylating agent or in the heterocycle may be present
in protected form and require deprotection steps to
be carried out to complete the synthesis. In other
cases, the alkylation is carried out with a
substituted benzylic halide or pseudohalide
("Ar-C~2Q"), but here the alkylation step is followed
by subsequent steps which are required to assemble
the substituted benzyl element of the antagonist of
Formula I. The alkylation steps and subsequent
steps used to prepare antagonists of Formula I, are
described in P~T TI below.
2- In another approach to antagonists of
Formula I, a substituted benzyl element is introduced
at the beginning of, or during the preparation of the
heterocyclic element. Routes of this type are
illustrated in Part II below. In most caseæ where
this general approach is used, the substituted benzyl
component which is introduced during the synthesis of
the heterocycle must be subjected to further
synthetic transformations in order to complete the
synthesis-of the antagonist of Formula I. In the
Schemes shown below in PART TI, this substituted
benzyl component is designated as "-C~2Ar," and is
usually introduced by an alkylation step with a
WO91/11~ PCT/US91/00957
207~627
-83-
- substituted benzyl halide or pseudohalide designated
ArC~2-Q (where Q i~, for e~ample, Cl, Br, I, ~, OTs,
or OMs). Substituted benzyl halides or pseudohalites
which are useful in the preparation of al~ylated
heterocycles described in PA~T T are illustrated by
those listed below in Table 1. In cases where these
benzylic halides, or pseudohalides are not
commercially available, they are prepared as
described in Part II below or by ~tandard methods of
lo organic synthesis. Subseguent steps which may be
required to complete the synthesis of antagonists of
Formula I are desribed in P~T TT below.
The compounds of this invention maybe
resolved using techniques ~nown in the art. The
diastereomeric salts or esters of the enantiomer~ are
separated and the desired compound is the more active
stereoisomer. The compounds of this invention, their
pharmaceutically acceptable ~alts and their prodrug
forms are included within the ~cope of this invention.
WO 91/11999 ~ 0~ 5 6 27 Pcr/US9l/oo9~7
--84--
Tabl e
CH2Br CHzBr CH2Br
(~ ~CH3
OCH2Ph OCH2Ph OCH2Ph
CH2Br CH2Br CH2Br
Cl ~ Cl J~C1 ~H3
OCH2Ph OCH2Ph OCH2Ph
CH2Br CH2Br CH2Br
CN CO2CH3 CH20TBD~;
CH2Br CH2Br CH20Ts
[~
S CH2 Ph N2 CH2 OTBD~;
WO91/11~9 PCT/US91/00957
.
2~7562~
Abbreviations used in the schemes and examples are
listed in Table 3.
T~ble 3
s
Re~ents
NBS N-bromosuccinimide
AIBN Azo(bis) isobutyronitrile
10 DDQ Dichlorodicyanoquinone
Ac20 acetic anhydride
TEA triethylamine
DMAP 4-dimethylaminopyridine
PPh3 triphenylphosphine
lS TFA trifluroacetic acid
TMS-Cl trimethylsilyl chloride
Im imidazole
AcSK potassium thioacetate
p-TsOH p-toluenesulfonic acid
20 FMOC-Cl 9-Fluorenylmethyloxycarbonyl
chloride
Solvents:
25 DMF dimethylformamide
HOAc (AcO~) acetic acid
EtOAc (EtAc) ethyl acetate
~ex hexane
T~F tetrahydrofuran
30 DMSO dimethylsulfoxide
MeOH methanol
iPrO~ isopropanol
WO91/11~ PCT/USsl/~ss7
~ 20~6~7
-86-
Others:
rt room temperature
TBDMS t-butyldimethyl~ilyl
5 OTf OS02CF3
. Ph phenyl
FAB-MS Fast atom bombardment mass
~pectroscopy
NOE Nuclear Overhauser Effect
10 SiO2 ~ilica gel
trityl triphenylmethyl
Bn benzyl
PART I: Preparation of the heterocycles ~hown in
Formula
A. Preparation of the imidazo-5,6-and 7-membered
fused heterocycles
The preparations of the imidazo-S-, 6-,
7-membered heterocycles are described in the Schemes
below. There are ~everal routes which have been used
to prepare these 8ystems. The first route involve6,
2~ in the final ~teps, the closure of the imidazole
ring. In other ca6e6 it ha6 been advantageou~ to do
the imidazole ring closure early in the ~ynthe~is and
generate the fused 5-, 6-, or 7- membered ring in the
final steps.
WO91/11~9 PCT/US91/ooss7
! ~' : 1 2 0 7 5 S 2 7
--87--
~MTD~0-5~ n F~SF:n ~TF~OCYC~-~S
A~ r \ ~ B-R
`A3 ~
CH2 Ar
The compounds of Formula I, wherein
-Al-A2-A3- represents a 3 ~tom seguence defined by
(a) - (dd) in the Detailed Description of the
Invention to generate the imidazo-5-fused 6ystem, can
be synthesized using the reactions and techniques
described herein below. The reactions are performed
in a solvent appropriate to the reagents and
materials employed and suitable for the
transformation being effected. It is understood by
those skilled in the art of organic synthesis that
the functionality present on the heterocycle and
other parts of the structure 8hould be consi6tent
with the chemical transformation6 proposed.
Depending upon the reactions and techniques employed,
25 - thi~ may involve changing the order of sy~thetic
steps, the use of required protecting groups followed
by deprotection. and, depending upon the particular
imidazo-fused 5-mem~ered heterocycle being formed,
the use of different strategie6 may be employed
regarding the cyclization 6teps and the particular
starting material utilized.
WO91/11~9 PCT/US91/00957
- 2075~:27
The ~tarting material~ for preparing the
compound~ of this invention are dependent upon the
nature of the heterocycle being formed. In many
caser the heterocycles can be prepared either from a
~uitably functionalized 5-membcred heterocycle by a
ring closure ~tep which gives an imida20 fused
bicyclic ring system (see Reaction Schemes I-l to
I-4) or by starting with a suitably functionalized
substituted imidazole and ring closing to give an
imidazo-fused 5-membered rin~ heterocycle (see
- Reaction Schemes 5-7). The particular route chosen
depends upon the nature of the bicyclic ring system
being formed and the availability of starting
material~.
lS One approach (Reaction Scheme I-l) start~
with monocyclic derivatives bearing vicinal carbon-
bound nitro and amino functions, such as 2 which are
often readily a~ailable, or can be prepared from the
mono nitro deri~atives 2 by reaction with
hydro~ylamine. Compounts ~ may be reduced by any one
of several methods, including catalytic hydrogenation
or reaction with SnC12 to give the diamino deri~ative
3. Such deri~atives are often unstable and can be
ring-closed to the imidazo fused heterocycle 4 (E=
single bond) by reaction with ~n appropriate
carbogylic acid, nitrile, imidate ester, thioimidate
ester, amidine, or orthoformate, either neat, or in a
- solvent sppropriate and compatible with the starting
materials and reagent8, ruch as polyphosphoric acid,
ethanol, methanol, hydrocarbon 801~entr and with a
catalytic amount of acid if required.
WO91/11~ pcTlus9l/~9s7
207SS27
-89-
Another pos6ible approach, not ~hown in
Reaction Scheme I-l, to compounds having the general
6tructure 4 (B= single bond) from 3 involve6 the
reaction of ~ with an appropriate altehyde in the
presence of an osidizing agent such as CuII,
nitrobenzene, or dicyanodichloroguinone (DDQ) to give
heterocycles such as 4.
Ring closure of the vicinal diamino
heterocycles 3, to give deri~atives 6uch as ~ can be
lo effected by treatment with reagent6 such as CS2,
CSC12, COC12j N~2CON~2, alkyl chloroformate6, dialkyl
carbonates, or potassium cyanate in the presence of
bases ~uch as ~0~ or K2CO3. Another potential route
to S (B= O) involves the use of a vicinal amino
carboxylate such as ~ or ~ which can be converted
to ~ via a Curtius or ~ofmann rearrangement on
suitable derivatives of ~a/~ such as scyl azites,
hydroxyamides or N-haloamide6. The bicyclic
derivatives ~ can be alkylated under the appropriate
conditions with al~yl halides, alkyl mesylates, alkyl
tosylates, trialkyloxonium 6alt6 or with diazomethane
to afford compounds of type 4 (B= O or S).
Another approach to 4 (~= single bond) which
has been used for example when Al-A2-A3- together are
-N=C(C~3)-N(C~3)- 6tart6 from ~ and utilize6
acylation of the amino function with an acyl chloride
or anhydride to give the nitro amido compound 7.
WO 91/11999 PCI/US91/009~7
207~627 .
--90--
~CTTON St~$~MF. T-1
H2N~ 2CXAl or NXA' 2
HO2C H2NA~ Hofr~nn 3
6a ~b rrent H 5
CSC12 ~
ot he r R' - Q
~ reagent
H2NXA3 H NXA3 ~ NXA3
1 5 ¦ R1 B- COC1 /~i d
or ~
o H XC)A2 [ H~ O H X)A2 , Na~
R1-~C-N A R1-B-C-N
2 ArCi~2
,
~NXAl 2 ArC~ DH ,~XOA~ [ H~ ~COA~ R
H A ~ Ar~N A or ot~er
H ~ r-ag~nt
lo tl
--- __
Q = a rultable l-avlng group ruoh or Cl. 9r. I. I~ yl or O-to~y
WO91/11~ pcT/us9l/~ss7
2075~27
Reduction of the nitro group leads to ~ and this can
be ring closed to g via cyclodehydration, by heating
and/or acid catalysis.
The imidazo fused heterocycle g can then be
- 5 al~ylated with ArC~2-Q (where Q is a suitable leaving
group such as Cl, Br, I, O-mesyl, or O-tosyl) in one
of several ways. One way is initially to form the
alkali metal salt of 4 by using M~ (where M is Li, Na
or ~) in anhydrous dimethylformamide (DMF) or by
treating 4 with a-metal alkoxide such as sodium or
potassium methoxide, etho~ide or t-butoxide in an
appropriate alcohol such as methanol, ethanol or
t-butanol as solvent. The alkylation is then carried
out by dissolving the above-mentioned salt of 4 in an
anhydrous aprotic solvent such as DMF, dimethylsul-
foxide (DMSO) or tetrahydrofuran (T~F) and reacting
it with the al~ylating agent ArC~2-Q (preparation of
ArC~2-Q is described hereinbelow) at 20-C to reflux
temperature of the solvent for 1 to 24 hours.
If the substituents on the heterocyclic ring
system result in an unsymmetrical heterocycle, then
the alkylation may produce a mixture of regioisomers.
These regioisomers pOSseSs distinct physico-chemical
and biological properties and in most cases can be
separated and purified using conv-ntional separation
techniques such as chromatography and/or
crystallization. In those ca8es where the separation
of regiosomers is difficult by conventional methods,
the mi~ture can be transformed into suitable
derivatives that are more amen~ble to the usual
separation methods. The identification of the
indi~idual regioi80mer8 can be made u~ing Nuclear
WO 91/11999 2 ~ ~ 5 6 ~ 7 PCT/USsl/009s7
-92-
Overhauser Effect (NOE) NMR methods, 13C NMR methods
(e.g. vicinal 13C-l~ coupling constant6) or by single
crystal ~-ray crystallography.
It should be noted that the relative amounts
of the regioisomers formed in the al~ylation reaction
can be influenced by several factors including the
nature of the base used (while the al~ali metal salt
of the hete~ocycle is generally used, the
regioisomeric ratio can be altered in some instances
lo by using the heterocycle in the presence of a weaker
base such as triethylamine, diisopropylethylamine,
potassium carbonate or sodium bicarbonate) and the
nature of the solvent used in the reaction.
An alternative approach, also shown in
Reaction Scheme I-l, starts with the mono nitro
derivative 9. This can be reacted with a substituted
hydroxylamine bearing the ArCH2 side chain, to give
lQ Reduction of the nitro function gives the
vicinal substituted tiamine 11 which can be ring
closed in the usual fashion. This allows for
regioselective introduction of the Ar-C~2 side chain
into the bicyclic system.
Another approach to the compounds of this
invention is shown in Reaction Scheme I-2. In this
instance, a monoamino heterocycle such a6 1~ can be
acylated under 8tandard condition6 to give the amido
derivative 1~. Thi8 amido compound can be reduced
with LiA1~4 in a anhydrou6 801~ent 8uch as '1~ or
WO91/11~ PCT/US91/oogs7
.
20756~7
-93-
Et20 to the al~ylamino derivative 4 which can then
be nitro~ated with isoamyl nitrite to give 1~- Such
al~ylamino nitroso derivatives particularly, for
example, when Al-A2-A3- together are
-C(C~3)~N-N(CH3)-, undergo cyclodehytration when
heated in pyridine to give the imidazo fused bicyclic
heterocycle 4. The conversion of g to 1 can be
carried out as described in Reaction Scheme I-l.
Alternatively, it should be possible to al~ylate 1
prior to ring closure to give the bis-N-al~ylated
derivative 1~ which can then be ~ubjected to
cyclodehydration in hot pyridine to give the blocked
1- Separation of any regioisomer~ that may be formed
can be effected by using conventional chromatographic
methods.
An alternative approach to the synthesi~ of
1~ might utilize al~ylation of the monoal~ylated
side-chain 14 with ArCH2-Q. This would provide the
bi~-N-alkylated derivative 11 which can be nitrosated
with isoamyl nitrite to give 1~-
An additional approach to the compounts ofthis invention i6 shown in Reaction Scheme I-3. In
thi~ approach the starting material is a vicinal
bromo nitro heterocycle 1~ which is reacted with an
appropriate mono al~ylamine to give 12- Ring clo~ure
can be effectet u8ing MeO~/NaO~ with heating to give
the N-o~ide derivative 2Q which can be reduced with
either triethylpho8phine. TiC12 or Si2C16 to give the
imidazo-fuset bicyclic heterocycle 4. This can be
converted to 1 in the u8ual fashion as described
above in Reaction Scheme I-l.
Wo g~ g~ 2 0 7 ~ PCT/US91/00957
-94_
Alternatively, 1~ can be reacted with the
appropriate dialkylamine to give 21 (which can also
be prepared by alkylation of 12 with ArC~2-Q under
the appropriate conditions). Ring closure of 21 in a
fashion similar to that described above in the
conversion of 12 to ~Q is followed by reduction and
separation of the products to give 1.
Another approach to compounds of the Formula
1 (particularly where -Al-A2-A3- together are
-C(R7a)=C(R7b)-S-) is shown in Reaction Scheme I-4.
In these instances, the starting material is the
substituted heterocycle 23 which can be alkylated
with ArC~2-Q to gi~e 2~. Treatment with
hydrazine/nitrous acid and a refluxing alcohol (HORl)
gives rise to ~ ~ia a Curtius rearrangement of the
intermediate acyl azide. The cyclization of 2~ can
be accomplished with polyphosphoric acid or other
acidic catalyst to provide 1-
WO 91/11999 PCI/US91/00957
-9S~- 2a7aS27
y~F~cTI 01~ S C~FMF T--2
X A2 X~ A2
12 H 13
H 1 ~ONO ON
R - CH2- N, R' - CH2- N A
H H \ pyridine
14 15
1. NaH/D~ 1. NaH/DMF
2. ArCH2-Q 2. ArCH2-Q
HXA~ 2 ~f NXA' 2 -<NX )
R - CH2- N R1 _ CH2- N A3 H A3
2 0 CH2Ar CH2Ar
16 \ / 4
/ 1 . Na H /DM~
1. pyridin6. ~ 2. ArCH2-Q
N Al
R~ 3A2
CH~Ar
WO 91/11999 PCr/US91/00957
207~627
--96--
- ~F~CTTON S~MF T_3
XOA ~XAA N~oH R ~NXdAA or ; R--
\ R- - CI~
C~2Ar ~ ~D~ 2. ArCH2-Q
0 \ 12. Ar-~-Q
CH~r ~ICH,Ar CH,A
Zl Z2
WO91/ll ffl ~ PCT/US91/009s7
'
2l17~;S27
-97-
Re~ctio~ S ch ~m e I-4
CH302C _~ CH302C _}~
~ 21. N~H~D~ 1O~A2 . N2H4,
~ 2. ArCH2-Q ~1 02CN~--A
H 1 3 . ~ HOR
CH2 Ar
23 24
H
R OzC ~ o ` 2 ~ ~ O ~ 2
CH2Ar
C H2 Ar
- The required heterocyclic precursors for
these transformations which are shown in Reaction
Schemes 1-4 may be prepared by adaptations of
literature procedures. A listing of representative
precursors to these imidazo fused bicyclic
heterocycles, along with literature references to
their preparations i6 shoWn below in Table 4.
WO 91/11~9 2 0 7 5 6 2 7 PCT/US91/~957
- ; ~;. , ,
-
-98-
TABLE 4
St ruct ure Nane ~ef erence
H__N 5-an~no-1- J. Che~ Soc.,
H ~ethyl- 2028 (1948)
H2N~-N ln~dazole
CH
H~_N~ 5-a~lno- Can. J. Re~.,
H2N~N in~dazole 19~3 296 (1941)
H
N 5-an~no-1.2- J. Che~ Soc,
~CH3 di~ethyl- 164S (1954)
H2N N in~dazole
CH3
H2N~_N 4-an~no-5- J. Het. Che~
~ ~CH3 nltro-1,2- 6, 53 (1969)
N02 I dim3t hyl-
CH3 in~dazole
NO2~ - ~ 5-an~no-4- Ibid.
~ ~CH3 4-nitro-1,2-
H2N I dinet hyl-
lnidazole
CH3
CH3
H~ 5-an~no-1,3- US. Patent
~ ,N dimethyl- 3,646,059
H2N N, pyrazole
CH3
H CH3
5-an~no-3- Ibld.
Il ,N net hyl-
H2N Nl pyrazole
Ph J. Gen. Chem
H2N~ 4,5-diamino- (USSR) 32.
Ll ,N 1 -'nethyl- 1898 (1962)
- N 3 - phe nyl -
CH3 pyrazole
SUBSTITUTE SHEET
WO 91/11999 PCI'/US91/00957
,. ; - ~,
- 99 ~ 2~75627
TAE~LE 4 ~ cont ) .
StructurQ Nar~ Reference
CH3 3-nitro-1, 5_ J. Gen. Chem
~,N-CH3 di~Tet hyl~ SR) 50,
~N pyrazole 21 06 ( 1 980)
H2N ~ 3, 4-dla~no- Ibid.
l_ ,N-CH3 1 . 5 - di m3 t hyl -
H2N--N pyrazole
2 CH3 4-nitro-1, 3- J. GerL Chorn
N~ dirrethyl- ( U~SR) 50,
H pyrazole 2106 (1980)
CH3
~3 5-arr~no-3- J. Het. Chem,
¦1 `,N rrethyl- 10, 181 (1973)
H2N~ oxazole
H2N CH3 4, 5-dlamLno- J. Het. Chem,
~,N 3-n~thyl- 10, 181 (1973)
H2N is oxazole
H 4- ~ut yr - Eur. J. Med.
~H amido- Chem 14 105
C3H2CONH--N t hiazole ( 1 979)
02N~S 5-nitro-2- J. Org. Chem,
CH3 rrethyl- 33 2545 (1968)
H --N t hiazole
H S 4- nit ro- Ibid.
3 XN>CH3 2 - ne t hyl -
NOz
SUBSTITUTE SHEET
WO 91/11999 PCI~/US91/00957
2075627
-- 100 --
TABLE 4 ( cont ) .
Structure Nar~e Re~erence
HzN 4-a~noi~o-J. Chem Soc.,
J~N thlazole 306 (1959)
H
CH3 4-arTiLno- Ibld.
H2 N~ 3 - iTe t hyl -
~--S i5 ot hiazole
CH3 5-amLno- Ibid.
~N 3 - r~e t hyl -
~ is ot hiazole
H2N ~
lS O2N~ 4-nitro- Ibid.
H2N isothiazole
2 ~, N 3, 4-dialTino- Liebigs Ann.
1- ~S2 1, 2, 5-thia- Cheln 4. 337
H2N' N diazole 1, 1- (1988)
N
H2N\~ 4, 5-dla~no- IZV. Akad. Nauk
H N~N' 1 H- 1 - ~enzyl- SSRR, Ser Khim
~3 1, 2, 3-triazole 11- 2633 (19
H2N~3 2, 3-dian~no- Arch, Pharm
H2N~S thlophene (Wbinheim), 314,
567 ( 1 981 )
SUB~ 111 lJT~ SHEEr
WO 91/11999 PCI`/US91/00957
2075~27
-lOOA-
TABLE 4 (cont).
Structure Nar~ ReFerence
- H2N~ ~ 1-benzyl-3. 4- Liebeigs P~nrL
J~N-CH2~ dia~no - 2 H- Chen~ 18 3 , 142 4
o pyrrol-2-one ( 1978)
N-CH~h~ 4. 5-diamLno-3_ Japanese Patent
H2N~ ( \=/ [ (phenylnethyl- 53/109527
~co ene) arr~no~ - 2H-
H2N^N pyrrol-2-one
0
H2N~ 2, 3-diarriLno- Chem ~er.,
¦I NH ~aleimLde 116, 2591
H2~ ( 1983)
H2 N~
I_,S 3, 4-dialT~no- ~3ull Soc.
H2N~ thiophene Chem Fr., 5-6,
pt. 2, 1 53 ( 1 983)
H2NrN 3, 4-dia~no- J. Het. Chem,
H2NJ--N 1, Z, 5-thia- 13, 13 (1976)
diazole
SUBSTITUTE SHEEr
WO91/11~ PCT/US91/009~7
- 2075S27 -~-
--101-- .
In certain ca~es due to the nature of the
heterocycle being prepared and to the availability of
starting materials, it may be advantageous to prepare
~ome of the compounds of this invention from a
~uitably functionalized imidazole ring by ring
closing to give compounds of Formula 1. Some
~pecific examples ~re shown in Reaction Schemes I-5
to I-7. Thus, Reaction Scheme 6 shows an approach to
the preparation of the ~ubstituted regioisomers of
lE- and 3~- thienot2,3-~imidazoles. The substituted
imidazole ~ can be readily al~ylated in the fa6hion
described earlier by using Na~ in DMF, followed by
treatment of the anion ~o formed with the al~ylating
agent ArC~2-Q to give a separable mixture of the
regioisomers ~1~ and 21~. These can be independently
converted to ~etones 2~ and ~, respectively, ~ia
oxidation with a suitable oxidizing agent ~uch as
MnO2 to the aldehyde, followed by reaction with an
appropriate ~rignard reagent to give the ~econdary
alcohol which is further oxidized with ~nO2 to ~, k.
These isomers can then be independently converted to
the corresponding thienot2,3-~]imidazoles ~2 by
treatment with a thioglycolic acid ester and the
appropriate al~oxide in the appropriate reflu~ing
alcohol (i.e., ~ gives the 3~-thieno-
t2,3-~]imidazole ~2~ and 2~ gives the lE-thieno-
[2,3-~]imidazole regioisomer 29b). Saponification of
2~ gives the carbo~ylic acid 33a~. Other
conversions possible with the ?9A.b regioisomers
include
WO91/11~ pcT/us9l/~ss7
207~S27
-102-
reduction with LiA1~4 to the alcohol 30~.b,
saponification followed by tecarbo~ylation to give
31~.b, and conver~ion of the al~yl carbo~ylate to a
~etone uith an al~yl lithium reagent to give ~?A.b.
In addition, the intermediate 2~ can be utilized as
a precursor to the 1,6-dihydro-imitazo-
t4,5-~3pyrazole, series by cyclization with a
~ubstituted hydrazine deri~ative, to give ~4a.
Similarly, ~ can be converted to the
lo imidazot4,5-~]pyrazole 1,4-dihydro sesie~ 34b.
WO 91/11999 PCI'/US91/00957
2075G27
--103-
~F.~CTI ON S t~MF. T - 5
R' ~ . R~ + R' --<N3~;
2. ArC~z-Q I~~
C~2 Ar C ~2 Ar
26 27b 27a
-- 1. MnO2
2 R22~ r
1 0 / 1 . MnOz
N / 2 R22-~7E3r
R'-<~ / 3. MnO2
C R22-
Ar ~ 2
2Bb N C-R2
R --<l~
J
Ar
2Ba
WO 91/11999
Pcr/us9l/oo9s7
2075627
--1 04--
CTTON S(~FMF T-5 CON ' T
2Bo ~ R'
~;C~,~R~, Ar J R~
N~O~
~DR~ 34
R~J R~
N~0!1 ~C
29
30~
/ \ LlOH H~O
~,~ 1. R~Ll \~F, ~0
R~2-
2 0 F' ~<~ R' ~,p~ F' ~4~ CC~i3
31~ 0
Ar J
32
Slrrll-rly 2~b c~n b cont~ rt-d ~ nto t~
r-glol-sn-riC 29b 30b, 31 b 32b, 33b nd 3~b
~ h-log-n ( Cl, ar F, I)
Ip~ yl, ~u~tltut--d lkyl ryl or
rub~tltut-d oryl
R~J = alkyl or ub~ t ~t ut d al kyl
R2J ~ olkyl or ~tltut~d olkyl
WO 91/11~ 2 0 7 5 6 2 7 PCT/US91/~9s7
.
-105-
Similarly, 2~ can be converted into the
regioisomeric 22~, 30b, 31b, ~Zb, ~, and
Ra - halogen (Cl, Br, F, I)
R22a - ~, alkyl, substituted alkyl, aryl or
substituted aryl
R23 = al~yl or substituted al~yl.
R25 _ alkyl or substituted alkyl.
A similar cyclization using hydro~ylamine
can be used to give the 6~-imidazor4,5-d]isoxazole
~eries (starting from ~) or the 4~-imidazot4,5-d]-
isoxazole series (starting from 2~
Preparation of the lactone derivatives 36A.b
can probably be accomplished from the regioisomers
35a.b as shown in Reaction Scheme I-6. These
tertiary alcohol 6tarting materials can be obtained
from respective ketones such as 28a.b (6ee Reaction
Scheme I-5) by reaction with an appropriate Grignard
reagent, followed by protection of the alcohol with
t-butyldimethylsilylchloride. Thus, lithiation of
35A.b can be carried out with ~-BuLi and the lithio
derivative reacted with methyl chloroformate. The
intermediate 60 formed can be treated with acid to
remove the ~-butyldimethylsilyl protecting group snd
to effect cyclization to the lactone. Deblocking can
be effected as described earlier in Reaction Scheme 1.
A possible route to the substituted
1,5-dihydropyrrolo~3,4-~]imidazole 41 is shown in
Reaction Scheme I-7. Thus, the functionalized
WO 91/11999 PCI/US91/009s~
2075627
-106-
imidazole ~7 (or its regioisomer) prepared as
describet in European Patent Application 2~3,310 can
be treated with tosyl chloride in pyridine to give
the 0-tosylate which can then be converted to the
aminomethyl derivatlve ~ia a Gabriel synthe~is
(displacement of tosylate with potassium phthalimide,
followed by de-phthaloylation with hydrazine). This
intermediate can be bloc~ed by treatment with
1,2-bis(chlorotimethylsilyl)ethane to give the
intermediate 38. Lithiation and sub~equent
formylation i8 accomplished by treatment with
butyllithium and DMF to give the intermediate,~
which can cyclize under acid cataly~is to the
pyrrolot3,4-~]imidazole derivative 40. Compound 40
then treated with an alkyl, acyl or ~ulphonyl
halide to block the pyrrole ring nitrogen.
Deblocking with acid under the conditions described
earlier gives rise to the required 41.
With regard to the preparation of
derivatives containing the furot2~3-~]imidazole
heterocycle (i.e., compounds of Formula 1 where
Al-A2-A3- to~ether are -C(R22)~C(R22)-0- and
-0-C(R22)zC(R22~-) these can be prepared by
alkylation of the appropriate
furo~2~3-~]imidazole tprepared as described in Chem.
Pap., 40, 675(1986)] using the general procedures
shown in Reaction Scheme I-l for the conver~ion of 4
to 1-
WO 91/11999 2 d 7 5 6 2 7 PCT/US91/00957
-107-
REACTIQN SCEEME I-6
RZ4b R22
R24bM3E~r N~- S i- C~ cH3)
- 2. T~DMS-Cl, Br
in~dazole J
Ar
35a
1. tBuLi R1 ~/ ~ C
2. ClCO2CH3 N
3. H~ J O
4. deblock ~r
36a
1 R24bMg~r N ~ Br CH /CH3
- 2. TBDM5-Cl. N ~ -S1-C(CH3)3
inidazole J I RZ2~ -
Ar Ra4b
35b
1. tBuLi N
2. ClCO2CH3 R 4
3- ~ J R
4. deblbck
Ar
36b
SUB~ ~ JTE SHEE~
W O 91/11999 ' ~ PC~r/US91/U0957
-107A- 2~75~27
R24a = aryl, substituted aryl, alkyl or substituted
alkyl
Ar = subs. phenyl Ar* = deprotected Ar
TBDMS = t-butyldimethylsilyl.
SU~ JTE SHEEl-
WO 91/11999 2 0 7 5 ~ 2 7 PCI`/US91/00957
. . ~
-- 108 _
RI:ACTION S(~ I-7
1 . Tos yl chloride/
pyridine \/
,~zOH O~ R~ ]
Ar O , Ar
3 . Nz H~
37 38
4 . S i S i ` kt 3 N
Cl Cl
1 . BULi /DMF ~ N~\NH2
. N--\CHO
39
N~ ,. ba s e
Ar J 2 R23 Q Ar
41 40
R23 = alkyl, acyl, or alkyls ulphonyl.
SUBSTITUTE S~IEET
WO91/11~9 PCT/US91/Oogs7
2075627
-lO9-
RF:~l'IITDA7:0~ FS
A2-A ~ N
H
(Formula I, wherein A5 is a single bond and
-Al-A2-A3-A4- is as defined by (r) above)
The compounds of Formula I wherein
(-Al-A2-A3-A4-) is a 4-atom sequence as defined in
the General Description of the inYention can be
synthesized using the reactions and techniques
described herein below. The reactions are performed
in a solvent appropriate to the reagents and materials
employed and suitable for the transformation being
effected. It is understood by those s~illed in the
art of organic synthesi6 that the functionality
present on the benzimidazole and other parts of the
structure should be consi6tent with the chemical
transformations proposed. Depending upon the
reactions and techniques employed, this may involYe
changing the order of Bynthetic step~, use of required
W091/11~9 . 2 0 7 S ~ ~ 7 PCT/US91/00957
--110- ,
protecting groups followed by deprotection, and
acti~ation of the benzylic po~ition of the alkylating
- agent~ used to enable alkylation at the nitrogen on
. the imidazole part of benzimidazoles.
I3F~CT~ON SC~t~: T--8
R b ~ -Rl . B"
(Q = I, ~r, OMs, OT~)
R3b R3~ R3b R3C
R'~ R~
R~b CH2~ ~-b CH2Ar
3a 3b
WO 91/11999
2 0 7 ~ S 2 7
..
--111--
As shown in Reaction Scheme I-8, compounds
of Formula (~ can be prepared by carrying out direct
al~ylation of al~ali-metal 8alt of benzimidazole (1)
(preparation of benzimidazole6 are described in
Reaction Schemes I-9 to I-12) using appropriately
protected benzyl halide, tosylate (OTs) or mesylate
(OMs) derivatives (2). The salt is prepared
preferably u~ing M~ (where M is lithium, sodium or
potassium) in anhydrous dimethylformamide (DMF), or
~Y treating it with metal alko~ide such as sodium or
potassium methoxide, ethoxide or t-buto~ide in an
appropriate alcohol such a~ methanol, ethanol or
t-butanol as the solvent. The al~ylation is
generally carried-out by di~solving the metal salt of
benzimidazole in a dipolar aprotic ~olvent such a~
DMF or dimethylsulfoxide (DMSO) and reacting it with
the al~ylating agent at 20C to reflux temperature of
the ~olvent for 1-24 hour~.
If substituents on the benzene ring result
in an unsymmetrical benzimidazole, the al~ylation may
produce a mi~ture of two regioisomers a~ products,
which may be represented by formulas ~ and ~.
These regioisomers possess distinct physico-chemical
and biological properties and in most cases can be
~eparated and purified by using conventional
separation techniques ~uch as chromatography (flash
column chromatograPhy, medium-pressure liquid
chromatography. high pre8sure liquid chromatography
(~PLC) and/or crystallization- In thoRe cases where
separation of regioi~omers i6 difficult by
conventional techniques, the mi~ture can be
transformed into suitable derivatives that can be
WO 91/11~ 2 0 7 ~ 6 ~ 7 PCT/US91/009~7
-112-
separated by usual separation methods. The
structural assignments of the isomers can be made
using proton NMR, Nuclear Overhauser Effect (NOE)
e~periments or X-ray crystallography.
w
~ v ~ zl;~;
~ K 3
D la
~;
15 ~ ~ 8,~ ~
c \
~: v \
O
r~
~" \ };
~ o~ rl
~p~ a D
SUBSTITUTE SHEE~
WO91/11~ PCT/US91/~ss7
.
207~62~
-113-
The starting benzimidazoles can be readily
prepared by any of the standart procetures described
in the literature tP. N. Preston, Ch~mistry of
~eterocyclic Co~Donnds, Vol. 40, part I, pp. 1-286
(1981) and references cited therein]. Se~eral
alternative routes to obtain benzimidazoles are
outlined in Reaction Scheme I-9. The most widely used
starting material, o-phenylenediamines (11). can be
readily prepared from the corresponding o-nitro-
I0 aniline (lQ) using standard reducti~e procedures suchas metal-acid reduction or catalytic reduction. The
substituted or un~ubstituted 11 can then be treated
with an appropriate imidate hydrochloride (1~) to
form corresponding benzimidazoles (l~).
Alternatively, the reaction of carboxylic acids (1~)
with o-phenylenediamines in the pre~ence of
polyphosphoric acid (PPA) is also effective in
producing benzimidazoles (1~). Benzimidazoles (ll)
can also be prepared from o-phenylenediamines and
aldehyde (1~) using cupric salt as an oxidant ~R.
Weidenhagen, Ch~m. Per., ~, 2263 (1936)].
` - 20~627
WO 91/11999 - - ~ PCI/US91100957
-- 114 --
R~;ACTION SCHEME I-lO
R4~ R3b ~1
(R= H, 0~ Cl) (R1= aryl, heteroaryl)
18 19
R3 b R3~
R4~ -~ +R1 _ C- NH- COOEt o r
~2 Et OH
4b 20
R
R3b R3a
2 5 R4~-~
R4 H
21
SUBST~TUTE St1EET
WO 91/11999 ; - 2 0 7 5 6 ~ 7 pcr/US91/009~7
~, . .
- - -114A-
REACTION ~CHEME I-10 cont ' d
[~ }
S, 170, 1 Oh R4 H
22
. 30
SUBSTITUTE SHEET
wo 9~ 2 0 7 5 6 2 7 - PCT/US91/~957
-115-
Although some benzimidazoles having aryl and
heteroaryl groups at the 2 po~ition can be prepared
using the methods described in Reaction Scheme I-9,
Reaction Scheme I-10 outlines methods which are more
~uitable for the cynthesis of this class of
compounds. N'-aryl-N-hydroxyamidines (l~; R=O~) are
cyclized under mild conditions using benzene-
sulfonyl chloride in pyridine or triethyl~mine to
give 12 in good yield ~M. W. Partridge and E. A.
Turner, J. Chem. Soc., 2086 (1958)]. Parent amidines
(1~; R=~) ;an also be oxidized with ~odium
hypochlorite under basic conditions to form 12 ~V. J.
Grenda, R. E. Jones, G. Gal and M. Sletzinger, ~.
- Or~. Chem., ~Q, 259, (1965)].
l~ Alternatively, a~ ~hown in Reaction Scheme
I-10, o-phenylenediamines (ll) can be reacted with
N-ethoxycarbonylthioamides (2Q) to give 2-substituted
benzimidazoles (21) in excellent yields. This method
avoids the use of acidic catalysts. The reagents
(~Q) are easily obtained in one ~tep from etho~y-
carbonyl isothiocyanate and 6imple aromatic or
heterocyclic compounds or al~ylmagnesium halides ~B.
~eorge and E. P. Papadopoulos., J. Or~. Chem ., ~1 .
3233(1976); E. P. Papadopoulos., J. Or~. Chem., gl,
962(1976)]. ~eterocyclic compounds containing
reactive methyl groups (e.g., 2-picoline) can also be
reacted with o-phenylenediamines in the presence of
sulfur at elevated temperatures to give 2-heteroaryl
benzimidazoles (~
wo 9l/llg99 2 ~ 7 ~ ~ ~ 7 PCI/US91/009~7
,
--116--
REACTION SC~IEME I-l 1
3~
~' R3~ H
R4 ~ z 4 R4~CNH
23 25
(W and Y= Cl, NH2 or N~N P~13
R3
R3~ R3b
R3b~ o ~ R'-O~Na~ ~Cl
27 ( Rl = C~ -C,5 alkyl) 26
~ + CS~ ~ R~.~SH
R3~
R3 b ~CN~)~ Rl
- 29
SUBSTITUTE SHEET
WO 91/11~ 2 0 7 5 6 2 7 PCT/US91/0~9s7
, ~ ...
-
-117-
As outlined in Reaction Scheme I-ll,
benzimidazole~ containin~ 2-al~osy and thioalkyl
substituent6 (27 and 22) can be prepared from the
corresponding benzimidazolone~ (2~) or benzimidazol-
ethione~ (2~). Benzimidazolone~ are conve~iently
prepared from o-phenylenediamines and pho~gene or
urea t~. Hofmann, ~Imidazole and its Deri~ati~e~,
Part 1," Wiley-Interscience, New ~or~, 1953, pp.
285-291]. Carbonate e~ters, diethylpyrocarbonate,
lo N,N-carbonyldiimidazole and N,N-diethylcarbamyl
chloride may al~o be used in this reaction. The
reaction of phosgene i~ apparently facilitated by the
use of N,N'-bis-trimethylsilYl (TMS) deri~ati~e (2~)
instead of the parent diamine tL. Bir~hofer, H. P.
guhlthau, and A. Ritter, Chem. Ber., 2~. 2810 (1960)].
WO 91/11999 ` PCI/US91/009~7
2~7~627
--118--
REACTION SCHEME I -12
R3a
R3 b ~,~NHz PPA
R4~ J~ + Cl-(CH2)n-COOH
1 1
R3
R3 b ~$~N R1 S ~ Na
R4b H Cl
R3~
( n= 1 - 3 R3 b ~_N
R1= alkyl, alkyl- R4~ ~--N> (~n
aryl and alkyl- R4b H S--
het eroaryl)
/RlQ
R4 ~ / (Q = I ~r, O~i, OT~)
R4b H
SUBSTITUTE SHEET
.!, 'WO 9 ~ 999 2 ~ 7 5 6 2 ;7 Pcr/ us g 1/00957
:
.
--119-
As described in Reaction Scheme I-12,
2-al~ylthioal~yl substituted benzimidazole~ (~1) can
be prepared from the reaction of RS-M (where M is
sodium, potassium or lithium) with 2-chloroal~yl
benzimidazoleE (~Q). 2-Chloroal~yl benzimidazoles
(~Q) can ~e conveniently prepared from the diamines
and the chloroal~yl carboxylic acid~ using PPA tW.
~nobloch, Ch~m Ber., 21. 2557 (1958)].
Alternatively, compound ~1 can also be prepared f~om
the readily available 2-thioal~yl derivative (~) tE.
S. Milner, S. Snyder, and M. M. Joullie, J. Ch~m.
~, 4151 (1964)].
TMT DAZ0- 6-FusFT) ~TF~OCYC~ F S
A2,A ~ N
~A4
CH2Ar
(FORMULA I, wherein A5 i~ a ~ingle bond and
-Al-A2-A3-A4- is a 4-atom sequence as defined in the
Detailed Description of the Invention)
WO91/11~ PCT/US91/00957
, ,
2075627
~ -120-
The compounds of ~ormula I, wherein
-Al-A2-A3-A4- are defined by (a~) to (bw) in the
Detailed De~cription of the invention can be
~ynthesized using the reactions and technique6
5 de~cribed herein below. The reaction~ are performed
in a solvent appropriate to the reagent~ and materials
employed and suitable for the transformation being
effected. It i~ under~tood ~y those s~illet in the
art of organic synthesis that the functionality
lO present on the heterocycie and in the reactants being
employed should be consistent with the chemic`al
transformations being conducted. Depending upon the
reactions and techniques employed, optimal yields may
require changing the order of ~ynthetic ~teps or use
15 f protecting groups followed by deprotection.
As shown in Reaction Scheme 1, compounds of
~ormula I can be prepared by carrying-out direct
alkylation of al~ali-metal salts of heterocycles (1)
(preparation of heterocycles are described in
20 Reaction Schemes 3-6) using appropriately protected
benzyl halide, tosylate (OTs) or mesylate (OMs)
derivatives (~). The salt is prepared preferably
using M~ (where M is lithium, ~odium or potassium) in
anhydrous dimethylformamide (DMF), or by treating it
25 with a metal al~oxide ~uch as ~odium or pota~sium
methoxide. etho~ide or t-buto~ide in an appropriate
alcohol such as methanol, ethanol or t-butanol as the
solvent. The al~ylation is generally carried-out by
di~sol~ing the metal salt of the heterocycle in a
30 dipolar aprotic sol~ent such as DM~ or
dimethylsulfO~ide (DMSO) and reacting it with the
alkylating agent at 20'C to reflux temperature of the
~ol~ent for 1-24 hour~.
-
WO 91/11~ 2 0 7 S 6 2 7 PCT/US91/00957
-121-
If ~ubstitucnts and/or the heteroatom
positions in the si~ membered ring are not
symmetrically di~posed, the al~ylation on the
imidazole nitrogen(s) generally produces a mi~ture of
two regioisomers as product~ arising from Nl and N3
al~ylation. These regioisomers I and Ia posses6
distinct physico-chemical and biological propcrties
and in most cases can be separated and purified by
using conventional separation techniques ~uch as
chromatography (flash column chromatography,
medium-pressure liquid chromatography, high
performance liquid chromatography) and/or
crystallization. In those cases where separation of
regioisomers is difficult by c~n~entional techniques,
the mixture can be transformed into ~uitable
derivati~es that can be separated by the above
separation methods. The structural assignments of
the isomers can be made using Nuclear Overhauser
~ffect (NOE), 1~-l3c coupled NMR e~periments or ~-ray
~rystallography.
When there is potential for alkylation of
the 6-membered heterocyclic ring, this can be a~oided
by the use of suitable protecting groups.
The heterocycles of type (1) can be prepared
by any of the standard procedures described in the
literature tJ.A. Montgomery and J.A. Secri~t III in
"Comprehensi~e Eeterocyclic Chemistry,~l Vol. 5, A.R.
Katritsky and C.W. Rees Eds., Pergamon Press 1984; pp
567-597 and 631-656 and references cited therein].
As shown in Reaction Scheme I-13, the most widely
used starting materials are si~ member heterocyclic
~icinal diamines (9). Fused imidazoles (lO) can be
- - - - - - - - - - - -
WO91/11~9 pcT/us9l/~9s7
,
2075~27
-122-
prepared by condensation of (9) with an appropriate
- carboxylic acid, nitrile, imidate ester, or
orthoester, either neat, or in a sol~ent appropriate
and compatible uith the starting material~ and
reagents, such as polyphosphoric acid, ethanol,
methanol, hydrocarbon solvent~, and with a catalytic
amount of acid if required. Oxidation of an imine
formed by reaction of diamine (9) with an appropriate
aldehyde using oxitants such as Cu (II), nitrobenzene,
lo or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)
also affords heterocycles (lQ)- Aminoamides (11. W =
H) or diamides (11, W R6CO) can be converted to
- fused imidazole~ (lQ) by heating neat, or at an
elevated temperature in a solvent such as ~ylene
under acidic or neutral conditions.
- Ealogenation of the imidazot4,5-b~pyridine
rin~ at the 6-position can be accomplished using Br2,
or N-bromosucCinimide. ~alogenation of the 7-position
can be accomplished by reaction of the corresponding
imidazopyridine-4-oxide (prepared by reaction of the
imidazopyridine with peracids such a~ m-chloroperben-
zoic acid) with POC13. When the 7-position i~
substituted other than hydrogen halogenation at the
5-position of the 4(N)-oxide precursor occurs on
treatment with POC13. Chlorides may be substituted
by bromides or iodides by treatment with either ~Br
or ~I, respecti~ely, in a sol~ent ~uch as EOAc.
WO91/11~ pcT/us9l/oo9s7
2075627 ~-
-123-
2-Al~yl-imidazot4,5-b]pyridines can be
~ubstituted at the 5, 6, or 7 po~itions by di~place-
ment of a halogen at that po~ition by nucleophile~
~uch as cyanide (followed by hydrolysis to obtain
carbo~ylic acids), amine~, copper alkosides, trial~yl-
phosphite~, and thiolate~. Also, sub6titution of the
halogens, in particular bromides or iotide~, can be
- accompli~hed by reaction with a coupling partner ~uch
as alkylzinc or arylzinc halide~, or monoalkylaryl-
phosphonites in the pre~ence of an appropriate metal
catalyst ~uch as nickel, palladium, ruthenium, or
platinum. In cases where the di~placement of a
halogen is luggish or otherwi~e complicated due to
- an acidic proton, the imidazopyridine may be protected
at the 1, 3, or 4 positions by benzyl or other
arylmethyl groups.
7-Methyl-2-propylimidazot4,5-b]pyridine-5-
carbo~ylic acid or the 2-ethyl analog i~ prepared
from 7-methyl-2-propylimidazot4,5-b]pyridine or the
2-ethyl analog by treatment with m-chloro-
peroxybenzoic acid to obtain the N-oxide which i~
then treated with POC13 to gi~e 5-chloro-7-
methyl-2-propylimidaZO-t4,5-b]pyridine or 2-ethyl
analog . The chloride i8 then e2changed for a
bromide by seaction of 5-chloro-7-methyl-
2-propylimidazot4,5-b]Pyridine or the 2-ethyl analog
with B r in acetic acid. The re~ulting
5-bromo-7-methyl-2-propylimidazot4~5-b]- pyridine or
2-ethyl analog is treated with Na~ in DMF followed by
benzyl bromide to o~tain 3-benzyl-5-bromo_
7-methyl-2-propylimitaZOt4.5-b]Pyridine or it~
corre~ponding 2-ethyl analog which is in turn treated
.
WO91/11~9 PCT/US91/00957
2 0 7 ~ 6 ~ 7
-124-
with CuCN in hot pyridine to obtain 3-benzyl-
5-cyano-7-methyl-2-propylimidazot4,5-b]pysidine or
the corresponding 2-ethyl analog. The cyano compound
is hydrolyzed to 3-benzyl-7-methyl-2-propylimldazo-
~4,~-b]pyridine-~-carbo~ylic acid or the
corresponding 2-ethyl analog by treatment with
E2S04-H20. Thi~ acid i8 e~terified by reaction with
MeOH-~Cl. The benzyl group is removet by
hydrogenation at 1 atm. in MeO~-ECl solution using
Pd(0~)2 as catalyst. This compound can be alkylated
as described earlier and the product methyl ester is
converted to the carboxylic acid by treatment with
hydroxide.
As ~hown in Reaction Scheme I-14, methods of
~reparing heterocycles of types 1~ and 1~ involve
treatment of diamines (9) with reagents such as urea,
phosgene, potassium cyanate, al~yl chloroformates,
dialkylcarbonates, or carbon di~ulfide in the
presence of bases such as potassium hydroxide or
potassium car~onate. Amino acid5 14 or 1~ can be
converted to 1~ via Curtius or ~offman rearrangement
of suitable derivatives such 8S acyl azides, hydroxy-
amides, or N-haloamides. ~icyclic compounds of type
(16, E = ~ulfur or o~ygen) are formed fsom 1~ ~y
reaction under neutral or ba~ic conditions with al~yl
halides, alkyl mesylates, al~yl tosylates, trial~yl-
oxonium ~alts, or with an appropriate diazoal~ane.
Compounds of type (16; B - o~y~en or sulfur) are
prepared by di~placement reactions u~ing alkogides or
alkyl mecaptides with chloro intermediate~ a~
indicated.
WO 91/11~ 2 0 7 5 6 2 7 PCT/US9l/~S7
-125-
Diamines of type 2 can be prepared by a wide
variety of methods such as hydroly8is of bis-amides
or amino amides, reduction of dinitro or aminonitro
or hydrazino or azido groups, displacement of
heteroaromatic halites or al~osy or thio or al~ylthio
or hydroxy or al~yl sulfonyl groups with ammonia or
amines, or rearrangement of ~cyl azides or amides or
acids (Curtius, Eoffman, or Schmidt rearrangements).
[A.S. Tomcufcik, L.~. Star~er in "~eterocyclic
Compounds, Pyridine and it's Deri~atives~' Pt 3, ~.
Klingsberg Ed., Wiley Interscience, 1962, pp S9-62,
and references cited therein; T. Na~agome in
"~eterocyclic Compounds, Pyridazines" Vol. 28, R.N.
Castle, ~d., Wiley Interscience, 1973, pp 597-601,
lS and references cited therein; "Heterocyclic
Compounds, The Pyrimidines" Vol. 1~, D.J. Brown Ed.,
Wiley Interscience 1985, pp 299-325; F, Schipper, and
A.R. Day J ~m Che~. Soc. (1952) 74, 350;
"Comprehensive ~eterocyclic Chemistry," Vol. 5, A.R.
2D Katritsky and C.W. Rees Eds., Pergamon Press 1984; pp
567-S97 and 631-656 and references cited therein].
In cases wherein heterocycles of type lQ or
1~ are not easily prepared from their corresponding
diamines, or when these diamines cannot be prepared
then alternati~e routes invol~ing fusion of the six
member heterocycle onto an 8ppropriately substituted
imidazole, are used. Two of these routes are
illustrated in Reaction Scheme I-15. ~or example,
imidazo[4,S-d]~1,2,33triazines (18) are
preferentially prepared ~y treatment of amino
carboxamido imidazoles (11) with sodium nitrite in
~queous acid. Precursor imidazoles (17) are prepared
WO91/11~ PCT/US91/009~7
.,, ~,-
~736~7 ~:
-126-
~y tegradation of an appropriately su~stituted
~anthine or by condensation of an appropriate imidate
ester uith aminocyanoacetamide. Imidazo~4,5-b3-
pyridazines (2Q) can be prepared from imitazodi-
carbo~ylate esters (1~) by treatment with hydrazine.
Oxidation of (~Q) gives pyridazinediones (21)- The
oxygen(s) in (~Q) or (21) can be con~erted to other
functionalities such as halides or thiones, which are
themsel~es precursors for the synthesis of more
lo ela~orate systems ["Comprehensi~e Eeterocyclic
Chemistry," Vol. 5, A.R. Katritsky and C.W. Rees
Eds., Pergamon Press 1984; pp 567-597 and 631-656 and
references cited therein].
2~
WO 91/11999 Pcr/uss~ s7
2 ~ 7 ~ 6 2 7
--127--
cTToN SL~ MF T_l~
H2 ~ ~A2 Rl C02 H~PPA ,~ R~
H2N ",A A~t~rn~te M~thod~ N A~'A3
9 H 10
X I heat~lnort rolvent R~
Rl C0~7H A~A or ne~t ~ ~,A3
11 H 10
AltGrn~t-~ r~genes ~nd re~ction condltlons:
Rl - CN, PPA
R~ - C- C oC2 Hs) . C2~sH
N~r ~1
2 0 Rl C( OCH3 ) 3, t ol ue ne, H~
RlCHO. C2~0~L CU(OCH3)2
~. WO 91/11999 PCI/US91/00957
2D 7562 7
-12B-
~FP~CT~ON S~ T_14
XA 13CS?/~ H~3~ Al
g H 12
2 XA ~A2 Cl 2 C= O~ e ~ `A2
H2N 4 ( NH2) 2C O, He~t ~ ~
X13cr X ;l3 1) 5O~l2~ N~N3. HO~ A
12 or 13 ~ R1Q N3~A~,A
H 1 6
12K~C13 Cl~ A3 R OH or ' 16
H 12
WO 91/11999 2 {) 7 S ~ 2 7 PCI`/US91/01"'~7
. .
--12 9-
CTION St~MF: T_l 5
II,N~)--8-R Nl ~
~,N~,O-BCl N
ll H
~ 7
C~OC~ E~ Rl
o
o ¦ DDQ
R
o H
As ~hown in Reaction Scheme I-16 amino
imidazole esters and amides are ~ersatile
intermediates for the preparation of purines. This
scheme 81so illustrates the synthesis of the
6-membered hetçrocyclic ring after the alkylating
agent 2 has been reacted with a suitably substituted
imidazole to afford ~ os 24.
WO91~11~9 PCT/USgl/009~7
~^ ~
207~;S27
-130-
~CTION S~F.MF. I-16
~-N=C-O ~
z2 23
10
R~- NH- CONH~
O O
1S ~c-oNH2 ~ ~ ~NH
R'-~ ~ ~ R'-~
NH2 R~-C(Oc2~)3 J
ArJ AC20 or D~F, Ar
24 ~ 25
The preparation of reduced forms of hetero-
cycles can be achieved by catalytic reduction, or by
synthesis from a suitable imidazole precursor. ~or
example. hi~tidine ant derivatives thereof react with
formaldehyde to afford partially ~aturated
imidazo(4!5-c) pyridines [cf. Neuberger, A. Biochem,
J., (1944), ~8, 309].
WO 91/11999 PCI/US91/00~57
2~75627 i ~ `
--13 1--
IMTD~7:o-7-~sFM~RF~T~ FIJSF:n ~ 0CYCIF:S
- A2-Al
A'3 r ~
S A'~_A~ --N
C~2
X
R~ ~z
y
R12
Compounds of FORMULA I, wherein -Al-A2-A3-A4-A5-
represents a 5-atom sequence as defined
in the Detailed Description of the Invention are
prepared as described below in Schemes I-17 to I-23.
S~M~ I-17
R'-B ~ - N H ~-A
(7) ArJ (l )
~ WO91/11999 PCT/US91/00957
2~75627
-132-
The imidazoles (7) reguired in al~ylation
Scheme I-17 can be prepared by a number of methods
well ~nown in the literature including those
de~cribed in EP0 publication 253,310. A useful
method of generating compound (7) wherein A* and A**
are NH2 and COM~R6 or Co2R7 i8 illustrated ln Scheme
I-18.
wo 9~ g99 - 2 0 7 5 6 2 ~ PCI/US91/0~457
--133--
Sch~me T_l 8
C--N
R C-N HClH R'-C-OEt CONH, '~J~H2
B I CONH2
C-N H
32-CH
l 0 C2 Et
N--~H2
R'~
N--\
I CO2Et
R~ NH, . Et O~l
R1 ~/ 3~
N
CONHR~
2 5 H
- W091/11999 PCT/US91/~ss7
2075~2~
-134_
The 6ynthesis of intermediate (l) wherein A*
i6 N~R6 and A** is CON~R6 or C02R7 (R7- ethyl) and B
is a single bond can be accomplished by the
al~ylation of the cyanoamidine (13) with a benzylic
halide or pseudohalide repre6ented a~ "ArC~2-Q" where
Q is a leaving group as outlined in Scheme I-l9.
WO 91/119g9 2 0 7 5 6 2 7 Pcr/US9~ 7
--135--
Sch~me T_1 9
NH N-C_N
R' - C- OEt + NH2C- N Et OH, R' - C- OEt
1 2
N-C=N
12 + NH2CH2CO2Et ~ R'-C
N-C-N
1 3 t ArCH2Q _ R'-C~CH2CO2Et
CH2Ar
1 4
~ WO91/11~9 l ~ PCT/US91/00957
20756~7
-136-
Sch~me T-l9 tCont'd)
~2 NH2
/~ R~NH2
N~IH,/~O,Et ~ R'~ON~
DMF C~Ar c~2
16
R~ I N--(
N~ H/DMF ~Oz Et
CH2 ~
Cyanoamidine (13) is prepared according to the
methods described by Edenhofer, ~ . Chim ~ , 58,
2192(1975).
Al~ylation of cyanoamidine 1~ may require an
alkylating reagent which incorporates functional
groups. In ~uch cases, protecting groups may be
required for these functional groups during the
al~ylation step. For egample, a carbosyl group can
be conviently protected a6 a t-butyl ester and a
tetrazole group as an N-trityl derivative. The
al~ylated cyanomidine (14) is purified by silica gel
chromatography as is the ring-closed product (15).
Conver~ion of (15) to amide (16) is accomplished by
heating the ester with R6N~2 in an inert solvent such
as ethanol. Compound (15) can be al~ylated on the
amino moiety using a small e~cess of Rl-I in DMF in
the presence of Na~.
WO 91~11999 2 0 7 5 S ~ 7 PCr/US91/Qn~57
-137-
Compounds of Formula (7) wherein A* and A**
are either Cl and C~20~ or C~2OE and Cl respectively
are also useful intermediates, and their preparations
are described in ~P0 publication 253,310. The
primary alcohol (C~20~) moiety in their al~ylation
products (18) can be o~idized directly to the
corresponding -C02C~3 ester groups using MnO2 in the
presence of NaCN and acetic acid in methanol as
illustrated in Scheme I-20.
Sche~e T-20
Cl Cl
S~ ~hO2. N~CN ~
R'~ OH HDA~, MEOR, B/~ 2C~3
~H2 Ar CH2 Ar
1~ 19
- C}~OH CO~CH3
R'-~ hO2. ~CN, R' ~3~1
C~ Ar~)A~ H CH2 Ar
2~ 21
2S Compounds (19) and (21) from Scheme I-20 can
be further converted to thiol compounds as
illustrated by the methodology of Scheme I-21.
Scheme I-21 also illustrates an alternate route to
amino compounds (22) which involves azide
displacement of Cl followed by hydrogenation.
~ WO91/11~9 ~ ~ PCT/US91/00957
i 20 ~5S2 7
-138-
Sch~me
CO2cH3 R C~CH~
R1 ~C 1 )CH,C_SH. N(Et~
-B ~ 2)~aOCH3 ~OH R'_B~;H
CH2 Ar
CH2 Ar
21 23
C2CH3 COZCH3
N~( 1 ?N~N3, DMF ~
~ N~C1 2)H2, Pd/C ,~2
CH2 Ar CH2 Ar
21 22
Formation of the products (I) wherein the
fused ring A represented by Al-A2-A3-A4-A5 contains D
= NR6 is carried out from intermediates (1) wherein
A* and A** are (M~R6, COM~R6) or (C0N~K6, NHR6)
2~ re~pectively by treatin~ (1) in DMF with
Q-C(o)-C(R7)(R8)-Q in the pre~ence of a tertiary
amine such as triethylamine. Q i5 a lea~ing group
which preferably is a halo group. When A* or A** is
Co2R7 and R7 = E then Y in the resultant products i~
oxygen. The transformations illu5trated in Scheme
I-22 with intermediate (16) are analogous to
transformatiOnS which can be employed to synthesize
similarly ~ubstituted benzodiazepine~.
WO 91/11999 2 0 ~ ~ 6 2 7 PCI~US91/~4~57
--13 9-
Sch~me ~ 2
Cl-C-C~R )~-Cl
16 24
R~
c02CH3 R~ ~ / ~7
1 5 /~( 1 ) Cbz - N- C~ R7 ~ ( R8 ) _ COOH /~
R1 ~--~32 DCC ~ R1 _ B~
CH2 Ar 2 ~ HE r /H0Ac J H
3)~ Ar 25
Scheme ~-22 also pro~ides an alternate route
to generate products (I) wherein ~ is NR6. In this
sequence N-protected amino acids are used to acylate
intermediate (1) wherein A* or A** is NR6, by
employing either an acyl halide, or a standard
carboxyl acti~ating reagent such a~ dicyclohexyl-
carbodimide (DCC) or (benzotriazol-l-yl)o~ytri~-
(dimethylamino) phosphonium hexafluorophosphate
(~OP). The N-protecting group of the amino acid ~uch
as the carbobenzyloxy (Cbz), t-buto~ycarbonyl (t-ROC)
or the fluorenylethylmetho2ylo~ycarbonyl (FMOC) group
~ WO91/11~9 PCT/US91/009~7
2075~7
.
-140-
is removed according to standard peptide synthetic
conditions. The final ring forming ctep is made by
heating this intermetiate in an alcoholic solvent or
by ~aponifying the imidazole Co2R7 group to yield a
carbo~ylic acid which iB treated with a carboxyl
activating reagent such as DCC or polyphosphoric acid.
Following the methodology of the above
described transformation of Scheme I-22, if A* and
A** are S~ and Co2R7, as illustrated by intermediate
lo (23), and the ring forming reagent is
~NR6C(R7)(R8)C~2Cl, it is possible to form products
of formula (I) wherein the Al-A2-A3-A4-A5 element is
-CON(R6)-C(R7)(R8)-C~2S
-SC~2C(R7)(R8)-N(R6)-Co-
-CoN(R6)-C(R14)(Rl5)-CH2S-
-SCII2C (R14 ) (R15 )-N(R16 )CO-
The preparation of products of Formula I,
wherein _Al_A2_A3_A4_A5 is -coN(R6)-c(R7)(R8)-c(R9)=
N- is carried out in Scheme I-23. The final ring
closure, which involves a dehydration to yield an
amine, can be assisted by heating in the presence of
molecular sieves and acetic acid in an inert solvent
such as dioxane. or by employing polyphosphoric acid
2~ as the dehydrating agent.
WO 91/11999 PCr/US91/00957
2~7S~27 ^
--141--
.,
Sche-ne T_?3
CO2C~
l )o~, F~ Chlorido
4~ 2)H2~C-CR )(R~)CCoEt)2-R7
ArJ 22 3)Plp~rldlne
o R7 R~
I I \ / o I ,~,-
C- NH- C ~ ~N~/~
R' - B~N~2 ~ R' -
CH2Ar ~ 27
26
WO91/11~ PCT/US91/00957
20756~7
- 142 -
PART II: Preparation of Qubstituted benzyl
derivatives of the general Formula I.
Preparation of compounds of Formula I starting from
the heterocycles or benzyl-sub~tituted heterocycles
described in Part I i8 illustrated in the following
schemes and descriptions.
The synthesis of Angiotensin II Antagonists
incorporating a substituted benzyl group as shown in
Formula I may be accomplished by reactions in the
presence of a base of a heterocyclic compound (as
described in Part I) with a benzylic compound bearing
a good leaving group, and the appropriate
substituents R9, R10, Rll, R12, g, Y and Z as shown
in Formula I. Alternatively, compounds with
structures according to Formula I may also be
synthesized in stages from a benzyl-substituted
heterocycle which contains the substituents R9, R10
and X, followed by reaction with an intermediate
20 - (such as a substituted alpha-bromophenylacetic ester)
which introduces the substituents at Rll, R12 and Z.
Examples of this latter methodology in which a
benzyl-substituted heterocyclic intermediate is
prepared first, and then elaborated to afford
compounds with structures described by Formula I, are
shown in the Schemes II-l, II-2 and II-3. The
preparation of compound 5 of Formula I wherein:
WO91/11~9 PCT/US91/~ss7
,.
207~627
_ 143 -
-Al-A2-A3-A4- = -C~=C~-C~=C~-, B= a single bond, Rl=
butyl, R9, R10 and Rll are ~ 0, Y- a ~ingle bond~
Z= C02~ and R12= phenyl appear~ in Scheme II-l and in
Example 1 of the experimental section. Deprotonation
of 2-butylbenzimidazole with strong bases such as
sodium hydride or potassium tert-buto~ide in DMF for
a period of 1-24 hours at temperatures of 20-lOO-C,
followed by al~ylation with 4-benzylo~ybenzyl
chloride affords the protected ether 2. The benzyl
lo ether is ne~t removed by hydrogenolysis using
hydrogen and an appropriate catalyst ~uch a~ Pd/C,
Pd(0~)2/C or Pt/C which affords the intermediate
phenol 3. The phenolic proton is then abstracted,
and the phenolate is al~ylated with methyl
2-bromophenylacetate to furnish ester 4. Finally,
the ester is hydrolyzed and the free acid 5 is
obtained.
WO 91/11999 Pcr/us9l/oo9~7
:
,~ 2Q7S6~7
- 144 -
SC~MF II-l
~ ~ ~ D~ ~ ~'
~nO Cl r~nO'13J n-
\~"~t~H. DM~ \~3
J3~ 3 ~CO~ oJ3J
OH, M~ OH \~
2) }~
~CO~ H
The synthesis of compound 10 of Formula I
wherein: -Al-A2-A3-A4- = -C(CH3)=C~-C~=N-, B= a
single bond Rl= n-propyl, R9, R10 and Rll are E, X=
O, Y= a single bond, Z= C02~ and R12= phenyl is
presented in Scheme II-2 and in Example 2 of the
experimental section. Deprotonation of 7-methyl-2-
WO91/11~ pcT/us9l/oo9s7
20756~7
- 145 -
propylimidazo[4,5-~]pyridine (6) with sodium hydride
in DMF, followed by treatment with 4-benzyloxybenzyl
chloride gives compound 7. The benzyl ether is
removed by hydrogenolysis to give the phenol 8, which
is then deprotonated with potassium hydride and
18-crown-6 in DMF and al~ylated with methyl
2-bromophenylacetate to give the ester 9. Basic
hydrolysis of 9 gives the free acid 10. Al~ylation
of the phenol 8 with substituted 2-bromophenylacetic
lo esters, followed by ester hydrolysis leads to
compounds of Formula I where R12 is a substituted
phenyl ~roup such as those shown in Examples 6 (R12=
2,6-dichlorophenyl), 7 (Rl2= 2-nitrophenyl), 8 (R12=
cyclohexyl), 9 (R12= n-propyl), and lO (R12=
o-carboxyphenyl) in the experimental section.
20-
WO91/11999 PCT/US91/~9~7
.
. " ; 207562~
- 146 -
S t~ MF`. T T--2
~nO ~~
~' ~ '
15 ~,J~ ¢~co,l~ ,0~ 9
~CO,~
1~ ~O~ or~
2) Kl
~C0,31
The synthesi 6 of compound 15 of Formula I
wherein: -Al-A2-A3-A4- = -C(CH3)=C~-C(C~3)=N-, B= a
single bond, Rl= ethyl, R9, R10 and Rll are ~, X= 0,
Y= a sin~le bond, Z= C02~ and R12= 2-methylphenyl is
shown in Scheme II-3 and in Eæample 24 of the
WO91/11~ PCT/US91/~s~7
2075627
- 147 -
experimental section. Deprotonation of
5,7-dimethyl-2-ethylimidazot4,5-~]pyridine (11) with
a strong base such as sodium hydride in DMF, followed
by treatment with 4-benzyloxybenzyl chlorite produces
the ether 12. The benzyl ether is removed by
hydrogenolysis to give the phenol 13, which is then
deprotonated with potassium hydride and 18-crown-6 in
DME and alkylated with methyl 2-bromo-2'-methylphenyl-
acetate to give the ester 14. Alkaline hydrolysis of
lo 14 gives the free acid 15. Reaction of the phenol 13
with other substituted alpha-bromophenylacetic esters
followed by alkaline hydrolysis leads to additional
derivatives in this heterocycle series, such as
Examples 25 (R12= 2-chlorophenyl), 26 (R12=
2-bromophenyl) and 40 (R12= 2-phenylethyl) in the
experimental section.
WO 91/11999 PCI/US91/00957 ~;
....
- 148 - 2073~7
SCHEME I I -3
~1BnO ~ o~C~3
CH3
~CH3
H~, Pd~C ~J
~OH J~J 13
HO
2 0 ~H DME ~[3J 1 4
cro~n-6 0
~CO
CH3
N~CH3
1 ) NaO~ ~30H ,¢~J 1 5
2) HCl O
~CO2H
~C~
&lJB~ 11~ lJTE S~EEr
WO91/11~9 PCT/US91/~9~7
2075627
- 149 -
benzaldehydes (18) with trimethylsilyl cyanide
affords the trimethylsilyl-cyanohydrins 19.
Treatment of 19 with acidic ethanol produces the
hydroxy esters ~0, and subsequent reaction with
carbon tetrabromide and triphenylphosphine provides
the substituted 2-bromophenylacetic esters 17.
SC~F.MF. TI-4
R R ~r
~02H 1 ) SOCl~ r2 ~
16 17
1 5 SC~F.~E II-5
o~s
~fHD ~SlCN. ~CN ~N }~Cl. EtOH
2 0 1 ~-cro~-6
1~ 19
OH R
2 ~ ~02Et PPh3. C~r~ Et
17
WO91/11~ PCT/US91/00957
2075~27
- 150 -
The synthesis of Angiotensin II Antagonists
incorporating a substituted benzyl element defined by
Formula I may also be accomplished by the alkylation
reaction of a heterocycle (as described in Part I)
with a benzylic intermediate bearing a good lea~ing
group, and with all of the appropriate substituents
~9 R10 Rll ~12, ~, y and Z in place. Thi~
approach which i6 generally preferred when either R9
or R10 are non-hydrogen, is illustrated in Scheme
II-6 and in Examples 3, 4, 27-31, 33, and 46 of the
experimental section. Deprotonation of p-cresol (21)
with strong bases such as potassium hydride or
potassium tert-butoxide in DM and alkylation with
methyl 2-bromo-2-phenylacetate gives the ether 22.
Bromination of 22 at the benzylic methyl group with
N-bromosuccinimide gi~es the alkylating agent 23.
Deprotonation of 5,7-dimethyl-2-ethylimidazo[4,5-b]-
pyridine (11) with sodium hydride in DMF, followed by
reaction with bromide 23, and subsequent ester
hydrolysis provides the acid 24.
WO 91/11999 PCI/US91/00957
- 151- 207~627
S C~IEME I I - 6
~3~CH3 1 8 - cr o~- 6 ~l~cH3
HO ~CO2~ ~CO2M~
J~r
NBS, AI BN o Na H,DMF
CC14 reflux ~COzMe C~l
CH3 1 1
1 ) NaOH, ~OH, \ <,N~
2 ) HC1 N~CH3
0~
~CO2H
24
SUE~ 111 ~JTE SHEEr
WO 91/11~ 2 0 7 ~ 6 2 7 PCT/US91/~57
-15lA-
A strategy similar to that of Scheme II-6
is applied when substitution at Rll is desired as
shown in Scheme II-7. Intermediate ethers such as 22
in Scheme II-6 are deprotonated with strong bases
such as lithium bis(trimethylsilyl)amide in THF and
can then be reacted with an alkylating agent such as
an alkyl halide or mesylate. In this case, reaction
of the anion derived from ether 22 with methyl iodide
affords the alkylated product 25. Reaction of 25
lo with N-bromosuccinimide gives bromide 26, which is in
turn used for alkylation of a heterocyclic compound
from Part I. Scheme II-7 illustrates the alkylation
of heterocycle 6 with bromide 26 which after ester
hydrolysis affords acid 27.
2s
SUBSTITUTE SHEET
WO 91/11999 PCI/US91/00957
-152- 207~27
SCHEME II-7
~,CH3 LiN( S1~33) 2~ f H3
CH3I H3C ~
~COzl~ ~CO2~
22
H3C NaH, D~
CCl~. r~Flux [3~Co~ ~3
CH3
2 0 N~
1~ NaOH, ~OH ~ J
2 ) HCl H3C
2 5 2C7O2 H
SaJBSTITUTE SHEET
WO91/11~ PCT/US91/O~gs7
20756~7 ~
-152A-
The synthesis of compound 32 of Formula I
wherein: -Al-A2-A3-A4- = -C(CH3)=C~-C(CH3)=N-, B= a
single bond, Rl= ethyl, R9, R10 and Rll are ~, X= 0,
Y= CH2, Z= C02H and R12= phenyl is shown in Scheme
II-8. In this example, p-hydroxybenzyl alcohol (28)
is selectively alkylated at the phenolic hydroxyl
group with methyl bromoacetate when they are refluxed
with potassium carbonate in acetone. After the
remaining hydroxyl group is protected as a
lo tert-butyldimethylsilylether, this ether (29) may
then be deprotonated with a strong base such as
potassium bis(trimethylsilyl)amide and reacted with
an alkylating agent in a manner similar to that shown
for intermediate 22 in Scheme II-7. Alkylation of
ether 29 with benzyl bromide provides 30. Silylether
hydrolysis of 30 and bromination of the resulting
alcohol affords an alkylating agent (31) which is
then used to alkylate a heterocyclic compound from
Part I. Alkylation of the anion derived from
heterocycle 11, followed by ester hydrolysis affords
the acid 32 shown in Scheme II-8 and described in
Example 41 of the experimental section.
SlJBSTITUT~ SHEET
WO 91/119g9 PCI/US91/00957
-153- 207362~
SCHEME II-8
.,
~H 1 ) Br CH2co2cH3 ~OT8D~
,~J K2CO3, acet one ~
HO 2) t-~u~2SlCl I 29
28 DM~P, CH2Cl2 CO2~
~--OT~D~S
1 ) KN(Si~3)z 13~,~J 1 ) n-~u~NF, T~
2) PhCH2~3r 2~ 2) C~3r4, PPh3
CH2Cl2
~r CH3
N$~ 1 ) NaH, DMF
81 H
CH3
1 ) NaOH, ~OH ~3~CH3
2 5 [3~`co2 H
SUBSTITUTE SHE~Er
WO 91/11~ 2 0 7 5 ~ 2 7 PCT/US91/ ~ 7
--153A--
Scheme II-9 illustrates the preparation of
an antagonist of Formula I wherein: -Al-A2-A3-A4-=
-C(CH3)=CE-CH=N-, B= a single bond, Rl= propyl, R9,
R10 and Rll are ~, g is a single bond, Y= 0, Z= C02~
and, R12= phenyl. In this example, the Hell-Volhard-
Zelinsky reaction converts 4'-methylphenylacetic acid
(33) to the alpha-bromoester 34, which is in turn
reacted with the potassium salt of phenol to yield
35. Benzylic bromination of 35 provides alkylating
agent 36 which is then reacted with a heterocyclic
species described in Part I. When the sodium salt of
heterocycle 6 is alkylated with the bromide 36 in
DME, ester 37 is obtained. Alkaline hydrolysis of
ester 37 then provides acid 38, which is also the
product of Example 17 in the experimental section.
8UBS ~ JTE SHEEl-
WO 91/11999 PCI/US91/00957
. .
... .
-154- 2a75~2~
S (~F.M~. I I - 9
.
HO~C~ 1 ) SOCla, Br~ ~eozc~fH3
33 2~ ~OH Br 34
Phe no 1 f ~CH3
KH, DMF ~ozc~l NE~ IBN
1 8-crown-6 ~O CCl~ rcflux
~1 35
~3r NaX DMF
~02C~ CH3
~ 3 5 ~NJ~;3 6
H
CH3 CH3
25N~ 1 ) NaOX ~5eOH N~
~o c~3J 2) HCl Ho2C~3J38
~ 37 [~f
SUBS~ITUTE SHEET
WO 91/11~ 2 0 ~ 5 6 2 7 PCT/US91/~957
- 155 -
Schemes II-10 and II-ll illustrate the
preparation of analogs where -Al-A2-A3-A4- =
-C(C~3)=C~-C~ , B= a single bond, Rl= n-propyl, R9
and R10 are ~, Y= a single bond, R12 is phenyl, Z=
C02~ and X is either methyne or methylene. A
Reformatsky reaction is first employed to prepare
methyl 3-hydro~y-3-(4-methylphenyl)-2-phenyl-
propanoate (39) from the starting materials shown in
Scheme II-10. When heated in the presence of
p-toluenesulfonic acid in benzene 39 i8 dehydrated to
the trans-stilbene derivative 40, and then benzylic
bromination of 40 gives the alkylating agent 41.
Deprotonation of heterocycle 6 with sodium hydride in
DMF and treatment with 41 gives ester 42. Alkaline
lS hydrolysis of 42 affords the product 43, in which X
is a methyne group (Rll is absent) doubly bonded to
the carbon atom bearing substituents R12 and Z as
shown in Scheme II-ll and in Example 11. Catalytic
hydrogenation of 43 gives the derivati~e 44 where X
is a methylene group and Rll is a hydrogen atom
(Scheme II-ll; Example 12 in the e~perimental
section).
WO 91/11999 PCI~US91/00957
2075627
-- 156 --
S t~F.MF. T I--~ O
S E~n' H,cJ~f ~ 39
p- n OH ~~ N ~r
~ t 1
~ CCl4. r~ x
SC~MF II-ll
c~3 CH3
H ~
6 0~ Z
CH3 CH3
N~O~ ~aOH ~1 H2, Pd- C
2 5 ~3J Et OAc ,13J
~CO~H 43 ~H
WO91/11999 `. 20~ 5 6~ PCT/USsl/OQ9~7
- 157 -
The synthesi~ of compound 47 of Formula I
which has the same substituents a~ compound 10
(Scheme II-2) with the exception that Z is a
tetrazol-5-yl group, is illustrated in Scheme II-12.
Exposure of ester 9 to excess ammonia in methanol
produces the corresponding amide which is then
dehydrated with phosphorous oxychloride and
triethylamine to give the nitrile 45. Reaction of
the nitrile 45 with trimethylstannyl azide in
lo refluxing toluene provides the tetrazole derivative
46.
S~MF II-12
CH, CH3
1 ) N~33, ~OH ~3
2 0 J3J 2) POCl~, Et ,N J3J
0~0~a 9 E~N 45
1) ~,SnN3 ~
tolu n- r-rlu~
2 ) ~Ac ,13J
O 46
~ `r
WO 91/11~99 2 0 7 5 6 2 7 Pc~/us9l/00957
- 158 -
Scheme II-13 illustrates the preparation of
a tetrazole analog (52) similar to structure 46
wherein Rl2 is a 2-chlorophenyl group. In this
synthesis, the ester group of intermediate 47 is
converted to a nitrile prior to alkylating a
heterocycle (Part I) with this substituted benzyl
element. Thus, reaction of ester 47 with ammonia in
methanol, followed by dehydration of amide 48
produces nitrile 49. Benzylic bromination affords
50, which is then reacted with the sodium salt of
heterocycle 6 in DMF to give intermediate 51.
Finally, reaction of nitrile 51 with trimethylstannyl
azide in refluxing toluene gives the tetrazole 52
shown in Scheme II-13 (Example 15).
lS
SUB~ 111 ~JTE SHEET'
wo 91~11999 ` 2 0 7 ~ 6 2 ~ PCI`/US91/0(~957
-158A-
SC~FME II-13
~f NH3, I~OH ,~f H~
~CO2~3 H~~248
POCl~. Et~N oJ~f NE3S, AIBN,
~N CCl4, re~lux
o~f ~ 6
~ ~33
o~lJ t oluene re~ lux J3J 52
~ 2) ~P.c Cl
SUBSTITUTE ~HEE r
WO 91/11~9 2 0 7 ~ ~ 2 7 PCT/US91/00957
- 159 -
The preparation of a deriYati~e of ~ormula
I analogous to tetrazole 47 (Scheme II-12) which haæ
a methylene group for the ~ substituent, is shown in
Scheme II-15 and in E~ample 16 of the e~perimental
section. In this synthesis, phenylacetonitrile is
deprotonated with lithium bis(trimethyl~ilyl)amide
and then al~ylated with the tert-butyldimethylsilyl-
ether of p-hydroxymethylbenzyl bromide (preparation
of bromide S3 i8 shown in Scheme II-14 and Example 16
lo f the e~perimental section) to yield nitrile 54.
The silylether group in compound 54 is directly
converted to the bromide 55 with carbon tetra~romide,
triphenylphosphine and acetone in dichloromethane
(Mattes, ~.; Benezra, C. Tetrahedron Lett., 1987,
1697). Alkylation of the sodium salt of heterocycle
6 with bromide 5~, followed by reaction of 56 with
trimethylstannyl azide in refluxing toluene yields
the tetrazole ~7.
S~F~E II-14
Br~[~ 2H E3H3, T~ }~r [~OH
t - 9uMe2S iCI. DM~P 9r ~BD~
i-PrEt2N. CH2C12
WO 91/11~ 2 0 7 5 6 2 7 PCT/US91/~957
- 160 -
.
S ~ T - ~ 5
1 ) LIN~Sl~)~ THF ~BD~;
r~ . CEir,
E~ S3 ~CN C~Cl~. el~tone
5q
10 ~ ~36
DMF ,13J
56
~ ~N
1 ) ~SnN3CH~
toul~ne r~flux, ~J~
2) ~Ac ~
,~J 57
2 0 ~
Scheme II-16 illustrates the preparation of
a derivative of ~ormul-a I where -Al-A2-A3-A4- =
2~ -C(CH3)=CH-C(CH3)=N-. Rl is ethyl, B i8 a sin~le
bond, R9, R10 and Rll are ~, X= 0, I= a ~inEle bond,
R12 is 2-methylphenyl. and Z is a phosphonic acid
group. Reaction of o-tolualdehyde (58) with
dimethylphosphite in the presence of triethylamine
affords the phosphonate ester 59. ~romination of the
hydro~yl group of 59 with carbon tetrabromide and
triphenylphoSphine in dichloromethane gi~es bromide
60. Deprotonation of p-hytroxybenzyl alcohol with
~ WO91/11~9 PCT/US91/~9~7
2a75s27
- - 161 -
sodium hydride in DME followed by addition of bromide
60 affords intermediate 61. A second bromination
reaction (CBr4, PPh3, C~2C12) converts alcohol 61 to
the bromide 62 which is then used to alkylate a
heterocyclic compound described in Part I. Scheme
II-16 illustrates the case where the anion of
heterocycle 11 is reacted with bromide 62 to give
upon workup, the phosphonate mono-ester 63 (Example
67). Phosphonic acid 64 may be obtained by treatment
lo Of ester 63 with trimethylsilyl bromide.
SC~MF. II-16
OH ~r
~ (~0)2POH ~OMa)2 PPH3. C~r~ ~O~le)2
CH3 H3 C}32Cl, H3
5~ 59 60
2 0HO~OH ,~f PPH3 . C~r ~ o~
(~PO( ~) 2 CH2Cl2 [3~PCI( O~k) 2
CH3 CH3
25~ ~cH~ O ~slrr O
3 ~POC O~S ) OH ~PO~ OH) 2
WO 9l/llg99 2 0 7 5 S 2 7 pcr/us9l/on4~7
- 162 -
The ~ynthesis of a derivative of Formula I
where Z is an acyl-~ulfonamide group is illu~trated
in Scheme II-17. Alkylation of the anion deri~ed
from heterocycle 11 with bromide 6~ (synthesis
described in Example 28 of the experimental ~ection)
and alkaline hydrolysis of the resulting e~ter (66)
affords the acid 67 (Example 29). Reaction of acid
67 with 1,1'-carbonyldiimidazole in T~F at elevated
temperatures gives an acylimidazolide which may be
reacted with a ~ulfonamide (benzenesulfonamide in
this example) and DBU in THF to provide the target
compound (68) where Z is the acyl-sulfonamide group.
SC~E~F II-17
- 05 ~CO,~
c~ C~33
2s ~ ~ ~ ~
N~OH Cl~J 1~ CD~. ~XF Cl_~J N~CH3
o~J o7 2~ P~-90,N~,. DE~o,~J o~
~HN` -'[3
~ WO91/11~9 PCT/USgl/~9~7
2a75~27
- 163 -
Precursors for the synthesis of AII
Antagonists incorporating a 8Ubstituted benzyl
element wherein either substituents R9 or R10 are
non-hydrogen include substituted p-cresols (Scheme
II-6), 4-hydroxybenzyl alcohols, 4-hydroxybenz-
aldehydes, 4-hydroxybenzoic acids and their esters as
shown in Schemes II-18-20.
Commercially available benzyl alcohols such
as 3-chloro-4-hydroxy-5-methoxybenzyl alcohol may be
lo selectively alkylated by alpha-bromophenylacetic
esters when they are refluxed together in the
presence of bases such as anhydrous potassium
carbonate, giving 2-phenoxyesters like 69 shown in
Scheme II-18. Conversion of the benzyl alcohol group
in 69 to a bromide (CBr4, PPh3, CH2C12) affords an
alkylating agent (70). A heterocyclic compound from
Part I (11) is then alkylated with bromide 70;
hydrolysis of the intermediate ester affords 71, the
product of Example 37 in the experimental section.
Alternati~ely, a heterocyclic compound from Part I
may be directly coupled with benzyl alcohols like 69
using Mitsunobu reaction conditions (diethyl
azodicarboxylate, PPh3, THF). Again, hydrolysis of
the resulting ester completes the synthesis.
WO 9 1 / 1 1 999 2 0 7 5 6 2 7 Pcr/ us 9 1 /0~95 7
'~
-- 164 --
S ~F~ 18
Cl~H acet ~ne, he-~t
~ [3~o2Me ~CO
Cl~r
PPh ~, CBr ~ O
CH2Cl2 ~co2~
\~ 1 )N~ DMF N OH ~H3
11 Cl`~fb ~J
~CO, ~b ~ ~
Scheme II-19 illustrates the use of
commercially available 3-ethoxy-4-hydro~ybenzaldehyde
(72~ to prepare an AII Antagonist of ~ormula I
bearing a 3-ethoxy group (R9) on the substituted
benzyl element- Alkylation of the phenolic group of
72 with methyl 2-bromophenylacetate gives the
WO91/119~ pcT/us9l/~9s7
2075~2~
- 165 -
aldehyde 73 which is then reduced to a benzyl alcohol
with sodium borohydride in methanol or ethanol. The
alcohol i5 con~erted to the bromide 74, and the
~ynthesis of product 75 (Example 34) is completed as
pre~iously described.
S~ E II-l9
c,~o~f ~c~tone, raf lux C~f
72 ~02~'b [~C02~
C2H~~ r
H~. EtOH, J~l
2 ~ PPh~, C~r ,~
CH2Cl2 ~C27 q
2 5 \~N~ 1 ? N~ DMF NrOH \~H~
H CE~3 2)c H ~ I~OH C~3J
(3~co, Me ~C2 H
W091/11~9
; - 2 0 7 S 6 ~ 7 PCT/US91/~957
. ',
- 166 -
Substituted 4-hydroxybenzoic esters are
also convenient precursor6 for the synthesis of the
~ubstituted benzyl element defined in AII Antagonists
of Formula I. In this approach, the phenolic
hydroxyl group is usually first protected with a
suitable protecting group, the ester is then reduced
to a hydro~ymethyl group, and deprotection affords a
4-hydroxybenzyl alcohol deri~ative. Scheme II-20
illustrates the preparation of derivati~e 80 usin~
this sequence starting from methyl 3,5-dichloro-
4-hydroxybenzoate (76). Silylation of phenol 76
followed in turn by lithium aluminum hydride
reduction of the ester and silylether deprotection
affords 3,5-dichloro-4-hydroxybenzyl alcohol (77).
Phenol 77 was then selectively alkylated with methyl
2-bromophenylacetate, and the synthesis of derivative
80 (Example 38) was completed using the previously
described methods.
WO91/11~9 PCT/USgl/~ss7
~ .
207SS27
- 167 -
S C~MF. T I--2 0
1 ) t-~,Bl~l Cl ~co,Cl~f~
Cl ~fO~DM~P, CH~Cl~ ~f c-tor~ . r-l-ux 0~
~J 2) L~
Tl ~) ~4N~. ~ 77 ~ 7e
PP~ ~-$~ \~H~ Cl
~)~ 1~ N~' D~F
79 2) N~O}i. 1~0~ 0~
~O~H 00
A variety of 2-substituted phenols are
selectively carbo~ylated when refluxed with carbon
tetrachloride, 50Z agueous sodium hydroxide and
powdered copper (European Patent Application
2 #193,8~3, 10-Sept-86) to afford the corresponding
substituted 4-hydroxybenzoic acids. This reaction
may be added to the synthetic sequence when it is
convenient to derive the desired substituent on the
benzyl portion of the target AII Antagonist from a
readily available 2-substituted phenol. This
strategy is illustrated for the preparation of
derivative 84 shown in Scheme II-21. Carboxylation
of 2-ethylphenol provides 3-ethyl-4-hydro~ybenzoic
acid (81). Acid 81 is then esterified, silylated,
3 reduced and desilylated to give the 3-ethyl-4-
WO 91/11999 2 0 7 ~ 6 2 7 PCT/US91/~9~7
- 168 -
hydroxybenzyl alcohol 82. Alcohol 82 may then be
used to complete the synthesis of AII Antagonist 84
shown in Scheme II-21 using the previously discussed
methodology.
SCHEME II-21
C~ 3 CCl, Cu 2Hs~CO~H
heat 81
1 ) 2~0~ H2SO"
2) t-~3uMQ2SlCl C2H~ ~bH
DM~P. CH2Cl2,
3) LlAlH4 THF HD
1 S 4) n- E~u"NF. THF
82
1 ) ~3r
~O~Q
~ K2CO3, ~r
acetone, reflu,x
2) PPh3. CBr"
CH2Cl~ ~ 2MQ
~ 83
CH3 CH~
\ <~
2) NaOH, M~OH O
[3~Co2H 84
~;UB~ ~ JTE SHEEr
WO91/11~9 ~ - PCT/US9lt~9~7
2 ~ 73 6 2 7
- 169 -
The Claisen rearrangement of
phenyl-allylethers offer~ another useful technigue
for the introduction of alkyl ~ubstitutents (R9 or
RlO) at the meta position of the substituted benzyl
element. In Scheme II-22, Claisen rearrangement at
185-C of allyl ether 85 provides the allylphenol 86.
Silylation of this phenol (86), followed by reduction
of the ester group and bromination leads to the
benzyl bromide 87. Alkylation of a heterocyclic
species from Part I, such as imidazopyridine 11,
followed by silylether removal gives intermediates
related to 88. Alkylation of 88 with methyl
2-bromophenylacetate followed by alkaline hydrolysis
gives a derivative of ~ormula I (89) wherein R9 is a
meta-allyl group (Example 42). ~ydrogenation of
intermediate 88 followed by the same sequence
provides derivative 90 where R9 is the meta-propyl
group as shown in Scheme II-22 and described in
Example 43 of the experimental section.
~ WO 9~ 2 0 7 5 6 2 7 PCT/US91/00957
- 170 -
St~.~F. T I -~ 2
q ~ Cl
5,~J~ 18~C, ~DM1~P. CB~C1
O ~Cl D2) L~Al~, ~F 5~8
es W~ c}~ci, 87
C~, CH,
) N~ Dt~ ~3J ~B
Z) n-E~u~NF, THF ~X~
C~ C~
--~3J~ 9 or ~3J
0~CO~H ~0~ H
The Claisen rearrangement strategy for the
introduction of a meta-alkyl ~ubstituent onto the
substituted benzyl element of an AII Antagonist of
~ormula I may be exercised twice when it is desired
that both R9 and R10 be meta-alkyl substituents.
Thus, allyl phenol 86 may be con~erted to its
O-allylether and subjected to a second Claisen
rearrangement to provide the phenol (91) shown in
Scheme II-23. Silylation of phenol 91, followed by
catalytic hydrogenation and reduction of the ester
group with lithium aluminum hydride gives the benzyl
alcohol 92. A Mitsunobu reaction of the benzyl
alcohol 92 with a heterocyle (11) described in Part
WO91/11999 - PCT/US91/~gs7
2~75G27
- 171 -
I, followed by silylether deprotection gives an
intermediate related to 93. The phenolic hydroxyl
group of 93 may then be alkylated with a substituted
alpha-bromoester and the ester hydrolyzed to yield
the acid 94 in which R9 and R10 are meta-propyl
groups as shown in Scheme II-23 and Example 52.
SUBSTITUTE SHEE~
WO 91/11999
` `- 2 0 7 5 6 2 7 PCr/US91/009~7
-1 71A-
SC~EME II-23
~CO2~ ~ Br, K2CO3
acetone, reflux~
.~ ~ Cl
86 2) 1 85C, ~l
) t - Bu~2sicl
~ DM~P, CH2Cl2
HO~' 2 ) H2, Rh/C, Et OH
91 3) LiAlH4, THF
3~H
TBD~O l'
92
2s
SUB~;TITUTE SHEEr
WO 91/11999 PCI/US91/009~7
-- 20~ a 627
-171B-
SCHEM~ 23 cont ' d
~ CH3
2 ) n- Bu4NF, T~ 9 3
H~ ~
~o Br CH3
~C2 ~ N~CH3
acetone, reflux ~
2) NaO~ ~OH O~J g4
2 0 ~ o~2 H
SUBSTITUTE SHEEl
WO9l/llW9 2 0 7 ~ 6 2 7 PCTIUS91/00957
- 172 -
The synthesis of compounds of Formula I
wherein: -Al-A2-A3-A4- = -C(CH3)=CH-C(CH3)=N-, B= a
single bond Rl= ethyl, R~, R10 and Rll are H, Y= a
single bond, Z= C02H R12= phenyl, and X= NR, are
presented in the following two Schemes. To access
these analogs, a heterocycle (ie. ~1) defined in Part
I is alkylated with p-nitrobenzyl bromide to yield
nitro compounds such as 95 in Scheme II-24.
Catalytic hydrogenation of the nitro group provides
an aniline derivative ~96) which is then alkylated by
an alpha-bromoester. The ester 97 is subsequently
hydrolyzed to afford a derivative of Formula I (98)
where X= NH (Example 57).
SUB~ I 11 IJTE SHEEr
PCI/US91/00957
WO 91/11999
-172A- 2075627
SCHE~ 4
CH3
CH3 1 ) N~ ~. Dl~ r \~H3
H 2) ~3 ~2N
H2~ Pd/C \~q 1 ) Na~, DMF
~:H3 2 ) 9r
,~ 96 ~CO~Me
}~N
\~N~ ~H3
,13J 2*10H J3J 9 9
[3~co,~ [3~o,H
SUB~ ~ JTE SHE~
wo 9~ ~g 2 0 7 ~ 6 2 7 PCT/US91/~957
- 173 -
The preparation of AII Antagonists of
Formula I similar to 98 in Scheme II-24 but having X=
NR may be accomplished by methodology shown in Scheme
II-25. The substituted aniline (96) presented abo~e,
is readily con~erted to the N-tert-butylcarbamate
(~OC) 99. Carbamates such as 99 may be deprotonated
at the amide nitrogen atom when reacted with bases
such as sodium hydride in DMF, and then reacted with
an alkyl halide. Subsequent treatment of the
lo intermediate with trifluoroacetic acid remove~ the
~OC group providing the mono-al~ylated aniline
derivative 100. The aniline nitrogen in 100 may be
deprotonated again with sodium hydride in DMF and
alkylated a second time with a substituted
lS alpha-bromoester to provide esters such as 101.
Alternatively~ the order of introduction of the
substituents on the nitrogen atom may be re~er~ed.
Intermediate 97 (Scheme II-24) may also be
deprotonated by strong base~ such as lithium
bis(trimethylsilyl)amide in THF and then reacted with
an alkyl halide to yield similar products (101).
~ster 101 prepared by either synthetic route, is then
hydrolyzed to afford the targeted AII Antagonists
(102) of Formula I where ~= NR.
WO 91/11999 PCI~/US91/009s7
- 2075~27
-- 174 --
SCHEME II-25
CH3
N~CH3BOC20,
~[3J Et 3N, CH2Cl2,
H2N
CH3
\ <~N~ 1 ) Na~ D~
N~CH3 ~3r
¦~IJ 99 2) CF3C02H.
BOC~N~ CH2Clz
SUBSTITUTE SHEET
WO 9~ 9g9 2 0 7 5 6 2 7 PCI/US91/~00957
-174A-
SC~MF II-25 cont ' d
CH3 13r
~H3 ~CO2~$ CH3
100 NaH, DMF \ \~N~H3
H ~J 101
N~H3 ~~
J3' 2 ) ~ E3r
H~ 97
~~ NaOH, MaOH
CH3
N~H3
102
~O~ H
SUBSTITUTE SHEEl
W091/11~9 ~ PCT/US91/~957
2~75~27
-175-
The compounds of this invention form salts
with various inorganic and organic acids and bases
which are also within the scope of the invention.
Such salts include ammonium salts, alkali metal salts
li~e sodium and potassium salts, al~aline earth metal
salts li~e the calcium and magnesium salts, salts
with organic bases; e.g., dicyclohexylamine salt6,
N-methyl-D-glucamine, salt6 with amino acids like
arginine, lysine, and the like. Also, ~alts with
lo organic and inorganic acids may be prepared; e.g.,
~Cl, HBr, ~2S04, H3P04, methane-sulfonic,
toluenesulfonic, maleic, fumaric, camphorsulfonic.
The non-toxic, physiologically, acceptable salts are
preferred, although other salts are also useful;
e.g., in isolating or purifying the product.
The salts can be formed by conventional
means such as by reacting the free acid or free base
forms of the product with one or more equivalents of
the appropriate base or acid in a solvent or medium
in which the salt is insoluble, or in a sol~ent such
as water which is then removed in ~acuo or by
freeze-drying or by exchanging the cations of
anexisting salt for another cation on a suitable ion
exchange resin.
Angiotensin II (AII) is a powerful arterial
vasoconstrictor, and it exerts its action by
interacting with specific receptors present on cell
membranes. The compounds described in the present
invention act as competitive antagonists of AII at
the receptors. In order to identify AII antagoni~ts
and determine their efficacy in vitro, the following
~wo ligand-receptor binding assays were established.
WO 91/11~ 2 0 7 5 6 2 7 PCT/US91/00957
-176-
~eceptor binding assay using rabbit aortae membrane
pre~ration
Three frozen rabbit aortae (obtained from
Pel-Freeze Biologicals) were su6pended in 5 mM
Tris-0.25M Sucrose, p~ 7.4 buffer (50 mL) homogenized,
and then centrifuged. The mi~ture was filtered
through
a cheesecloth and the supernatant was centrifuged for
30 minutes at 20,000 rpm at 4-C. The pellet thus
obtained was resuspended in 30 mL of 50 mM Tris-5 mM
MgC12 ~uffer containing 0.2% Bovine Serum Albumin and
0.2 mg/mL ~acitracin and the suspension was used for
100 assay tubes. Samples tested for screening were
done in duplicate. To the membrane preparation (0.25
mL) there was added 125I-SarlIle8-
angiotensin II [obtained from New England Nuclear]
(10 mL; 20,000 cpm) with or without the test sample
and the mi~ture was incubated at 37C for 90
minutes. The mixture was then diluted with ice-cold
50 mM Tris-0.9% NaCl, pH 7.4 (4 mL) and filtered
through a glass fiber filter (GF/B Whatman 2.4~
diameter). The filter was soaked in scintillation
cocktail (10 mL) and counted for radioactivity using
Packard 2660 Tricarb liquid scintillation counter.
The inhibitory concentration (ICsO) of potential AII
antagonist which gives 50% displacement of the total
specifically bound 125I-SarlIle8-angiotensin
II was presented as a measure of the efficacy of such
compounds as AII antagonists
WO91/11~ PCT/US91/00957
2075627
-177-
Receptor assay using Bovine adrenal cortex
preparAtion
Bovine adrenal cortex was selected as the
source of AII receptor. Weighed tissue (0.1 g is
needed for 100 assay tubes) was suspended in Tris ECl
(50 mM), pH 7.7 buffer and homogenized. The
homogenate was centrifuged at 20,000 rpm for 15
minutes. Supernatant was discarded and pellets
resuspended in buffer tNa2~P04 (10 mM)-NaCl (120
mM)-disodium EDTA (5 mM) containing phenylmethane
sulfonyl fluoride (PMSF)(0.1 mM)]. (For screening of
compounds, generally duplicates of tubes are used).
To the membrane preparation (O.5 mL) there was added
3H-angiotensin II (50 mM) (10 mL) with or without the
test sample and the mi~ture was incubated at 37-C for
l hour. The mixture was then diluted with Tris
buffer (4 mL) and filtered through a glass fiber
filter (GF/B Whatman 2.4" diameter). The filter was
soaked in scintillation cocktail (lO mL) and counted
for radioactivity using Packard 2660 Tricarb liquid
scintillation counter. The inhibitory concentration
(IC50) of potential AII antagonist which gives 50%
displacement of the total specifically bound
3H-angiotensin II was presented as a measure of the
efficacy of such compounds as AII antagonists.
The potential antihypertensive effects of
the compounds described in the present invention may
be evaluated using the methodology described below:
Male Charles River Sprague-Dawley rats (300-375 gm)
were anesthetized with methohexital (Brevital; 50
mg/~g i.p.) and the trachea was cannulated with PE
wo 9l/ll~g 2 ~ ~ 5 6 ~ ~ PCT/US91/00957
-178-
205 tubing. A stainles6 ~teel pithing rod (1.5 mm
thick, 150 mm long) was inserted into the orbit of
the right eye and down the spinal column. The rats
were immediately placed on a ~arvard Rodent
Ventilator (rate - 60 ~trokes per minute, ~olume -
1.1 cc per 100 grams body weight). The right carotid
artery was ligated, both left and right ~agal ner~es
were cut, and the left carotid artery was cannulated
with PE 50 tubing for drug administration, and body
temperature was maintained at 37C by a thermostati-
cally controlled heatin~ pad which received input
from a rectal temperature probe. Atropine (1 mg/kg
i.v.) was then administered, and 15 minutes later
propranolol (1 mg/kg i.v.). Thirty minutes later
angiotensin II or other agonists were administered
intravenously at 30 minute intervals and the increase
in the diastolic blood pressure was recorded before
and after drug or vehicle administration.
Using the methodology described above,
representative compounds of the invention were
evaluated and found to exhibit an activity of at
least IC50 ~ ~0 mM thereby demonstrating and
confirming the utility of the compounds of the
invention as effective AII antagonists.
Thus, the compounds of the invention are
useful in treating hypertension. They are also of
value in the management of acute and chronic
congestive heart failure, in the treatment of
secondary hyperaldosteronism, primary and secondary
pulmonary hyperaldosteronism, primary and ~econdary
pulmonary hypertension, renal failure and renal
WO91/11~ PCT/US91/~gs7
2075S27
-179-
vascular hypertension, and in the management of
vascular disorders such as migraine or Raynaud 1 8
disease. The application of the compounds of this
invention for these and similar disorders will be
apparent to those skilled in the art.
The compounds of this invention are also
useful to treat elevated intraocular pressure and can
be administered to patients in need of such treatment
with typical pharmaceutical formulations ~uch as
lo tablets, capsules, injectables, as well as topical
ocular formulations in the form of solutions,
ointments, inserts, gels and the like.
Pharmaceutical formulations prepared to
treat intraocular pressure would typically contain
about 0.1% to 1~% by weight, and preferably 0.5% to
2.0% by weight of a compound of this invention.
In the management of hypertension and the
clinical conditions noted above, the compounds of
this invention may be utilized in compositions such
as tablets, capsules or elixirs for oral administra-
tion, suppositories for rectal administration,
sterile solutions or suspensions for parenteral or
intramuscular administration, and the like. The
compounds of this invention can be administered to
2s patients (animals and human) in need of such
treatment in dosages that will provide optimal
pharmaceutical efficacy. Although the dose will vary
from patient to patient depending upon the nature and
severity of disease, the patient's weight, special
diets then being followed by a patient, concurrent
medication, and other factors which those s~illed in
~ WO 91/11~ 2 0 7 5 S ~ 7 PCT/US91/00957
-180-
the art will recognize, the do~age range will
generally be about 1 to 1000 mg per patient per day
which can be administered in single or multiple
doses. Perferably, the dosage range will be about
2.5 to 250 mg per patient per day; more preferably
about 2.5 to 75 mg per patient per day.
The compounds of thi~ invention can also be
administered in combination with other antihyperten-
sives and/or diuretics and/or angiotensin converting
enzyme inhibitors and/or calcium channel blockers.
For example, the compounds of this invention can be
given in combination with such compounds as amiloride,
atenolol, bendroflumethiazide, chlorothalidone,
chlorothiazide, clonidine, cryptenamine acetates and
cryptenamine tannates, deserpidine, diazoxide,
~uanethidene sulfate, hydralazine hydrochloride,
hydrochlorothiazide, metolazone, metoprolol tartate,
methyclothiazide, methyldopa, methyldopate hydro-
chloride, minoxidil, pargyline hydrochloride,
polythiazide, prazosin, propranolol, rauwolfia
serpentina, rescinnamine, reserpine, sodium
nitroprusside, spironolactone, timolol maleate,
trichlormethiazide, trimethophan camsylate,
benzthiazide, guinethazone, ticrynafan, triamterene,
acetazolamide, aminophylline, cyclothiazide,
ethacrynic acid, furosemide, merethoxylline procaine,
sodium ethacrynate, captopril, delapril hydrochloride,
enalapril, enalaprilat, fosinopril sodium, lisinopril,
pentopril, quinapril hydrochloride, ramapril,
teprotide, zofenopril calcium, diflunisal, diltiazem,
felodipine, nicardipine, nifedipine, niludipine,
nimodipine, nisoldipine. nitrendipine, and the like,
as well as admixtureS and combinations thereof.
WO91/11~9 PCT/US91/~s~7
2075627
-181-
Typically, the individual daily dosages for
the~e combination~ can range from about one-fifth of
the minimally recommended clinical dosages to the
ma~imum recommended levels for the entities when they
are given singly.
To illustrate these combinations, one of the
angiotensin II antagonists of this invention
effective clinically in the 2.5-250 milligrams per
day range can be effectively combined at levels at
the 0.5-250 milligrams per day range with the
following compounds at the indicated per day dose
range: hydrochlorothiazide (15-200 mg), chlorothiazide
(125-2000 mg), ethacrynic acid (15-200 mg), amiloride
(5-20 mg), furosemide (5-80 mg), propranolol (20-480
mg), timolol maleate (5-60 mg), methyldopa (6~-2000
mg), felodipine (5-60 mg), nifedipine (5-60 mg), and
nitrendipine (5-60 mg). In addition, triple drug
combinations of hydrochlorothiazide (15-200 mg) plus
miloride (5-20 mg) plus angiotensin II antagonist of
this invention (3-200 mg) or hydrochlorothiazide
(15-200 mg) plus timolol maleate (5-60) plus an
angiotensin II antagonist of thiæ invention (0.5-250
mg) or hydrochlorothiazide (15-200 mg) and nifedipine
(5-60 mg) plus an angiotensin II antagonist of this
invention (0.5-250 mg) are effective combinations to
control blood pressure in hypertensive patients.
Naturally, these dose ranges can be adjusted on a
unit basis as necessary to permit divided daily
dosage and, as noted above, the dose will vary
depending on the nature and severity of the disease,
weight of patient, ~pecial diets and other factors.
wo 9~ 9 `2 0~ ~ G ~ PCT/US91/~957
-182-
Typically, these combinations can be
formulated into pharmaceutical compositions as
discussed below.
About 1 to 100 mg of compound or mixture of
compounds of Formula I or a physiologically
acceptable salt is compounded with a physiologically
acceptable ~ehicle, carrier, excipient, binder,
preservative, stabilizer, flavor, etc., in a unit
dosage form as called for by accepted pharmaceutical
o practice The amount of active substance in these
composltlons or preparations is such that a suitable
dosage in the range indicated is obtained.
Illustrative of the adjuvants which can be
incorporated in tablets, capsules and the like are
the following: a binder such as gum tragacanth,
acacia, corn starch or gelatin; an excipient such as
microcrystalline cellulose; a disintegrating agent
such as corn starch, pregelatinized starch, alginic
acid and the like; a lubricant such as magnesium
stearate; a sweetening agent such as sucrose, lactose
or saccharin; a flavoring agent such as peppermint,
oil of wintergreen or cherry. When the dosage
unitform is a capsule, it may contain, in addition to
materials of the above type, a liquid carrier such as
fatty oil. Various other materials may be present as
coatings or to otherwise modify the physical form of
the tosage unit. For instance, tablets may be coated
with shellac, sugar or both. A syrup or eli~ir may
contain the active compound, sucrose as a sweetening
agent, methyl and propyl parabens as preservatives, a
dye and a flavoring such as cherry or orange flavor.
WO91/11~ PCT/US91/~9~7
~7~i627
-183-
Sterile compositions for injection can be
formulated according to conventional pharmaceutical
practice by dissolving or su6pending the active
substance in a vehicle such a6 water for injection, a
naturally occuring vegetable oil li~e se6ame oil,
coconut oil, peanut oil, cottonseed oil, etc., or a
synthetic fatty vehicle like ethyl oleate or the
like. Buffers, preserYatives, antioxidants and the
like can be incorporated as required.
The compounds of this invention are also
useful to treat elevated intraocular pressure and can
be administered to patients in need of such treatment
with typical pharmaceutical formulations such as
tablets, capsules, injectables, as well as topical
ocular formulations in the form of solutions,
ointments, inserts, gels and the like. Pharmaceutical
formulations prepared to treat intraocular pressure
would typically contain about 0.1% to 15% by weight,
and preferably 0.5% to 2.0% by weight of a compound
f this invention.
Thus, the compounds of the invention are
useful in treating hypertension. They are also of
value in the management of acute and chronic
congestive heart failure, in the treatment of
~econdary hyperaldosteronism, primary and secondary
pulmonary hypertension. renal failure such as
diabetic nephropathy. glomerulonephritis, scleroderma,
and the like, renal vascu~ar hypertension, left
ventricular dysfunction, diabetic retinopathy, and in
the management of ~ascular disorders 6uch as migraine
or Raynaud's di8ease- The application of the
WO91/11~ PCT/US91/OOs~7
.' 2~7562~
-184_
compounds of this invention for these and similar
disorders will be apparent to those skilled in the
art.
The useful central nervous system (CNS)
activities of the compounds of this invention are
demonstrated and exemplified by the ensuing assays.
COGNIllv~; F~NCTTON A~SAY
The efficacy of these compounds to enhance
cognitive function can be demonstrated in a rat
passive a~oidance assay in which cholinomimetics such
as physostigmine and nootropic agents are ~nown to be
active. In this assay, rats are trained to inhibit
their natural tendency to enter dark areas. The test
apparatus used consists of two chambers, one of which
is brightly illuminated and the other i5 dark. Rats
are placed in the illuminated chamber and the elapsed
time it takes for them to enter the darkened chamber
is recorded. On entering the dark chamber, they
receive a brief electric shoc~ to the feet. The test
animals are pretreated with 0.2 mg/~g of the
muscarinic antagonist scopolamine which disrupts
learning or are treated with scopolamine and the
compound which is to be tested for possible reversal
of the scopolamine effect. Twenty-four hours later,
the rats are returned to the illuminated chamber.
Upon return to the illuminated chamber, normal young
rats who have been subjected to this training and who
have been treated only with control vehicle take
longer to re-enter the dark chamber than test animals
who have been exposed to the apparatus but who ~Aave
WO91/11~ PCT/USgl/~ss7
- 2~75~27
-185-
not received a shoc~. Rats treated with scopolamine
before training do not ~how this hesitation when
tested 24 hours later. ~fficacious test compounds can
overcome the disruptive effect on learning which
scopolamine produces. Typically, compounds of this
invention should be efficacious in this passive
avoidance assay in the dose range of from about 0.1
mg/kg to about 100 mg/kg.
~N~TOI.YTIC ASSAY
The anxiolytic activity of the invention
compounds can be demonstrated in a conditioned
emotional response (CER) assay. Diazepam is a
clinically useful anxiolytic which is active in this
assay. In the CER protocol, male Sprsgue-Dawley rats
(250-350 g)
are trained to press a lever on a variable interval
(VI) 60 second schedule for food reinforcement in a
standard operant chamber over weekly (five days per
week) training sessions. All animals then receive
daily 20 minute conditioning sessions, each session
partitioned into alternating 5 minute light (L) and 2
minute dark (D) periods in a fixed LlDlL2D2L3
sequence. During both periods (L or D), pressing a
lever delivers food pellets on a VI 60 second
schedule: in the dark (D), lever presses also elicit
mild footshock (0.8 mA, 0.5 sec) on an independent
shock presentation schedule of VI 20 seconds. Lever
pressing is suppressed during the dark periods
reflecting the formation of a conditioned emotional
response (C~R).
~------~~~~ WO91/11~9 2 07 s~7 PCT/US91/0095
-186-
Drug te~ting in this paradigm i6 carried out
under e~tinction conditions. During e~tinction,
animals learn that responding for food in the dar~ iæ
no longer punished by shock. Therefore, response
rates gradually increase in the dar~ periods and
animals treated with an anxiolytic drug ~how a more
rapid increase in response rate than vehicle treated
animals. Compounds of this in~ention should be
efficacious in this test procedure in the range of
from about 0.1 mgtkg to about 100 mgt~g.
D~P~SSION ASSAY
The antidepressant activity of the compounds
f this invention can be demonstrated in a tail
suspension test using mice. A clinically useful
antidepressant which serves as a positive control in
this assay is desipramine. The method is based on
the observations that a mouse 5uspended by the tail
shows alternate periods of agitation and immobility
and that antidepressants modify the balance between
these two forms of behavior in favor of agitation.
Periods of immobility in a 5 minute test period are
recorded using a keypad linked to a microcomputer
which allows the experimenter to assign to each
animal an identity code and to measure latency,
duration and frequency of immobile periods.
Compounds of this invention should be efficacious in
this test procedure in the ran~e of from about 0.1
mglkg to about 100 mg/kg.
WO91/11~9 PCT/US91/~ss7
- 2075627
-187-
S~T~OP~RF~IA ASSAY
The antidopaminergic activity of the
compounds of this invention can be demonstrated in an
apomorphine-induced stereotypy model. A cli~ically
useful antipsychotic drug that i8 used as a positive
control in this assay is haloperidol. The assay
method is based upon the observation that stimulation
of the dopaminergic system in rats produces stereo-
typed motor behavior. There is a strong correlationbetween the effectiveness of clas~ical neuroleptic
drugs to block apomorphine-induced stereotypy and to
prevent schizophrenic symptoms. Stereotyped behavior
induced by apomorphine, with and without pretreatment
with test compounds, is recorded using a keypad
linked to a microcomputer. Compounds of the inven-
tion should be efficacious in this assay in the range
of from about 0.1 mg/kg to about 100 mg/kg.
In the treatment of the clinical conditions
noted above, the compounds of this invention may be
utilized in compositions such as tablets, capsules or
elixirs for oral administration, suppositories for
rectal administration, sterile solutions or suspen-
~ions for parenteral or intramuscular administration,
and the like. The compounds of thi5 invention can be
administered to patients (animals and human) in need
of such treatment in dosages that will provide
- optimal pharmaceutical efficacy. Although the dose
will vary from patient to patient depending upon the
nature and ~everity of disease, the patient~s
weight, special diets then being followed by a
patient, concurrent medication. and other factors
WO91/11~9 PCT/US91/~957
20~562~ ~ ~
-188-
which those skilled in the art will recognize, the
dosage range will generally be about 5 to 6000 mg.
per patient per day which can be administered in
single or multiple doses. Perferably, the dosage
range will be about 10 to 4000 mg. per patient per
day; more preferably about 20 to 2000 mg. per patient
per day.
In order to obtain maximal enhancement of
cognitive function, the compounds of this invention
may be combined with other cognition-enhancing
agents. These include acetylcholinesterase inhibitors
such as heptylphysostigmine and tetrahydroacridine
(THA; tacrine), muscarinic agonists such as
oxotremorine, inhibitors of angiotensin-converting
enzyme such as octylramipril, captopril, ceranapril,
enalapril, lisinopril, fosinopril and zofenopril,
centrally-acting calcium channel blockers such as
nimodipine, and nootropic agents such as piracetam.
In order to achieve optimal anxiolytic
activity, the compounds of this invention may be
combined with other anxiolytic agents such as
alprazolam, lorazepam, diazepam, and busipirone.
In order, to achieve optimal antidepressant
activity, combinations of the compounds of this
invention with other antidepressants are of use.
These include tricyclic antidepressants such as
nortriptyline, amitryptyline and trazodone, and
monoamine oxidase inhibitors such as tranylcypromine.
In order to obtain maximal antipsychotic
activity, the compounds of this invention may be
combined with other antipsychotic agents ~uch as
promethazine, fluphenazine and haIoperidol.
WO91/11~9 PCT/US91/oogs7
: 2075627
-189-
The following examples illustrate the
preparation of the compounds of ~ormula I and their
incorporation into pharmaceutical compositions and as
such are not to be considered as limiting the
invention set forth in the claims appended hereto.
F.~A~ple 1
2-Butyl-1-[4-(1-carboxy-1-phenyl)methoxyphenyl]methyl-
10 benzimidazole
Step A: Preparation of 1-(4-benzyloxyphenyl)-methyl-
2-~utylbenzimidazole
A suspension of 1.50 g (8.62 mmol) of
2-butylbenzimidazole (described in European Patent
Application #400,835, 12-May-90) and NaH (272 mg,
1.05 eq) in DMF (20 mL) was stirred 25 minutes.
Next, 4-benzyloxybenzyl chloride (2.10 g, 1.05 eg)
was added to the reaction mixture. After stirring
overnight, the reaction mixture was concentrated
in vacuo and the residue was chromatographed on a
medium pressure liquid chromatograph eluted with 30%
ethyl acetate/hexane to yield 3.08 g (96%) of the
title compound.
lH NMR (300 MHz, CDC13, ppm): ~ O.9-1.0 (t, 3H), 1.35
(m, 2H), 1.75-1-9 (m, 2H), 2-8-2.9 (t, 2~), 5.0 (s,
2H), 6.85-6.95 (d, 2H), 6.95-7.05 (d, 2H), 7.15-7.45
(m, 8~),7.75-7.80 (d, lH).
FAB-MS: m/e 371 (M+H).
WO 91/11999 PCr/US91/OOg57
-; 207562~
--190--
Step B: Preparation of 2-butyl-1-(4-hydroxyphenyl~-
~ethylbenzi~idazole
A solution of the pr~duct of Step A (1.00 g,
2.70 mmol) dissolved in 20 mL of MeO~, wa~ added
0.100 g of a lOZ PdlC catalyst and the reaction
mixture was ~tirred under an ~2 atmosphere (1 atm)
for 6.5 hours. The reaction mixture was filtered
through MgS04 and the filtrate concentrated ~n v~cuo
to yield 0.628 g (83%) of the title compound.
1~ NMR (300 MHz, CD30D, ppm): ~ 0.9-1.0 (t, 3~),
1.35-1.50 (m, 2~), 1.65-1.8 (m, 2~), 2.9-3.0 (t, 2~),
5.4 (s,2H),6.7-6.8 (d, 2H), 6.9-7.0 (d, 2~),
7.25-7.35 (m, 2H), 7.4-7.5 (m, lH), 7.6-7.7 (m, lH;
FAB-MS: m/e 281 (M+H).
Step C: Preparation of 2-butyl-l-t4-(l-carbometh
l-phenyl)methoxyphenyll~ethylbe~zimidazole
To a solution of the product of Step B (100
mg, O.357 mmol) in DMF (1 mL) was added Na~ (11 mg,
1.0 eq). After stirring the reaction mixture for 15
minutes, a solution of methyl 2-bromophenylacetate
(82 mg, 1.0 eq) in DMF (500 mL) was added and the
reaction mixture was stirred for 60 hours. The
reaction mixture was quenched with saturated ammonium
chloride solution and the organic layer was
concentrated in vacuo. The residue was dissolved in
ethyl acetate and washed with water and then brine.
The organic layer was dried (MgS04), filtered and
concentrated in ~acuo and the residue was purified on
a silica gel flash chromatography column (120 x 40
mm) eluted with 50% ethyl acetatelhexane to yield 26
mg (17%) of the title compound.
- - - - - - - - - -
WO91/11~ PCT/US91/00957
` .: ~. ;
20~627
-191-
lH NMR (300 MHz, CDC13, ppm): ~ O.9-1.0 (t, 3E),
1.35-1.50 (m, 2H), 1.65-1.75 (m, 2H), 2.75-2.85 (t,
2H), 3.7 (s, 3H), 5.25 (s, 2H), 5.6 (~, lE), 6.8-6.9
(d, 28), 6.9-7.0 (d, 2H), 7.15-7.25 (m, 3H), 7.35-7.45
(m, 3H), 7.5-7.6 (m, 2H), 7.7-7.8 (d, 2H).
FAB-MS: m/e 429 (M+H).
Step D: Preparation of 2-butyl-1-~4-(1-carboxy-1-
~henyl)methoxyphenyll~et~ylbenzimidazole
To a solution of the product of Step C (25
mg, 0.058 mmol) in 2.0 mL of MeOH was added 1 N ~OH
(0.5 mL). The reaction mixture was stirred for 1
hour and then concentrated ~n vacuo. The residue was
dissolved in water, acidified to pH 4 with 1 N HCl,
and the resulting precipitate was extracted into
chloroform. The organic extracts were dried (MgS04),
filtered and evaporated in vacuo, and then
precipitated from HCl/EtOAc to yield 7.1 mg (30~/~) of
the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ O.55-0.65. (t, 3H),
1.05-1.2 (m, 2H), 1.40-1.50 (m, 2H), 2.75-2.85 (t,
2H), 5.15-5.30 (q, 2H), 5.6 (s, lH), 6.85-7.00 (m,
4~), 7.2-7.4 (m, 6H), 7.65-7.75 (m, 2H), 7.75-7.85
(m, lH).
FAB-MS: m/e 415 (M+H).
WO91/11~ PCT/US91/009~7
., . , ~ .
207~627
-192-
F~A~Dle ~
3-~4-(1-carboxy-l-phenyl)methoxyphenyl]methyl-7-
methy~ ropyl-3~-imid~zor4~5-blpyridi~e
S
Ste~ A: Preparation of 2,3-diamino-4-picoline (cf.
Lappin, G. R.; Slezak, F. B. J. Am. Chem.
Soc.. l950. 7Z. 2806-7~
To a ~lurry of 2-amino-4-methyl-3-nitropyri-
dine (10.0 g, 65.3 mmol) in 350 mL of 95% EtOH was
added 500 mg of a 10% Pd/C catalyst. The mixture was
stirred under a H2 atmosphere (1 atm) for 36 hours.
~iltration and evaporation gave 8.05 g of a black
solid which was used directly in the next step.
Step B: Preparation of 7-methyl-2-propylimidazo
t4,5-b]pyridine (cf. Lappin, G. R.; Slezak,
F. ~. J. Am. Chem. Soc.. ~950. 72. 2806-7)
A mixture of butyric acid (6.57 mL, 71.9
mmol), 2,3-diamino-4-picoline (8.05 g, 65.4 mmol),
and polyphosphoric acid (50 g) was heated to 100C
with stirring for 3 hours, and monitored by tlc of
N~40H neutralized aliquots. Basification (NH40H),
extraction (CH2C12, 4 ~ 50 mL), drying (K2C03),
purification (by filtering through 100 g silica gel,
EtOAc elution), and concentration gave 10.0 g (87%)
of the title compound as an amorphous tan solid which
was judged pure by lH NMR and tlc: mp 110-112C
(without recrystallization).
l~ NMR (300 M~z, CDC13, ppm): ~ 8.13 (d, lH, J=5 Hz),
7.01 (d, lH, J=5 Hz), 3.01 (t, 2H, J=7.8 Hz), 2.67
(s, 3~), 2.07-1.93 (m, 2H), 1.06 (t, 3~, J=7.5 ~z).
WO91/11~9 - pcT/ussl/~ss7
207~627
-193-
Step C: Preparation of 3-(4-benzyloxyphenyl)methyl-
7-methyl-2-~ropYl-3R-imidazor4~5-blpyridine
A suspension of 7-methyl-2-propylimidazo-
t4,5-b~pyridine (1.00 g, 5.71 mmol) and NaH (189 mg,
1.1 eq) in DME (25 mL) was stirred for 1 hour and
then cooled to 0-C. 4-Benzylo~ybenzyl chloride (1.46
g; 1.1 eg) was then added and the ice bath removed.
The reaction mixture was stirred for 2.5 hours and
was then concentrated LB vacuo. The re~idue was
chromatographed on a silica gel flash chromatography
column (30 x 100 mm) eluted with 30'~ ethyl
acetate/hexane to yield 1.07 g (50%) of product.
H NMR (300 MHz, CDC13, ppm): ~ O.9-105 (m, 3H),
1.7-1.85 (m, 2H), 2.65-2.85 (m, 5H), S.0-5.1 (m, 2H),
5-4-5-5 (m, 2H), 6.8-6.95 (m, 2H), 6.95-7.15 (m, 3H),
7.2-7.5 (m, 5H), 8.15-8.25 (m, lH).
Step D: Preparation of 3-(4-hydroæyphenyl)methyl-7-
methyl-2-pro~yl-3H-imid~zor4.5-blpyridine
To a solution of the product of Step C (0.60
g, 1.62 mmol) in 10 mL of MeOH was added 60 mg of a
10% Pd/C catalyst and was stirred under a H2
atmosphere (1 atm) for 7 hours. The reaction miæture
was then filtered through magnesium sulfate and the
filtrate was concentrated ~n ~acuo to yield 0.372 g
(82%) of the title compound.
lH NMR (300 M~z, CD30D, ppm): ~ O.95-1.05 (t, 3H),
2.65-2.8 (m, 2H), 2.7 (s, 3H), 2.9-3.0 (t, 2H), 5.5
(s, 2H), 6.7-6.8 (d, 2~), 7.0-7.1 (d, 2H), 7.2-7.5
(d, lH), 8.25-8.3 (d, lH).
WO 91/11~9 ~ Q 7 ~ 6 27 PCT/US91/00957
-194-
Step F: Preparation of 3-t4-(1-carbomethoxY-l-
phenyl)-metho~yphenyl]methyl-7-methyl-2-
propyl-3~ idAzor4.5-blpyridine
To a 6uspension of K~ (45 mg, 1.1 eg) in DME
(0.5 mL) was added the product of Step D (100 mg,
0.365 mmol) followed by 18-crown-6 (20 mg, 0.2 eg).
After stirring the reaction mixture for 0.5 hour
until the foaming subsided, a solution of methyl
2-bromophenylacetate (81 mg, 1.O eg) in DME (0.5 mL)
was added and the reaction mixture was stirred 2
hours and was then concentrated in vacuo. The
residue was purified on a silica gel flash chromato-
graphy column (120 x 20 mm) eluted with 50% ethyl
acetate/hexane to yield 67 mg (44%) of the title
COmpound
1~ NMR (300 M~z, CDC13, ppm): ~ O.9-1.0 (t, 3H)
1.65-1.80 (m, 2~), 2.65 (s, 3H), 2.7-2.8 (t, 2~), 3.7
(s, 3H), 5.4 (s, 2~), 5.6 (s, lH), 6.8-6.9 (d, 2~),
6.95-7.1 (m, 3H), 7.35-7.45 (m, 3H), 7.5-7.6 (m, 2H),
8-2 (d, lH)
Ste~ F: Preparation of 3-~4-(1-carbomethoxy-1-
phenyl)methoxyphenyl]methyl-7-methyl-2-
propyl-3~-imidazor4.5-blpyridine
To a solution of the product of Step E (21
mg, 0.0490 mmol) in MeO~ (1 mL) was added lN NaO~ (1
mL). The reaction mixture was stirred for 1.5 hours,
and was then concentrated in ~acuo. The residue was
partitioned between brine and T~F. The organic layer
was separated from the aqueous layer, dried (MgS04),
filtered, and evaporated in vacuo. The residue was
purified on a silica gel flash chromatography column.
WO91/11~ ~ PCT/US91/009~7
2075627
-195-
(15 x 130 mm) eluted first with 20% methanol/ethyl
acetate, and later with 50% methanoltethyl acetate.
The product fractions were concentrated Ln ~cuo,
redissolved in ethyl acetate and filtered to yield 16
mg (76~Z) of the title compound.
1~ NMR (300 M~z, CDC13, ppm): ~ O.8-0.9 (t, 3~),
1.5-1.7 (m, 2~), 2.2-2.3 (m, 5H), 5.1 (s, 1~), 5.25
(s, 2H), 6.6-6.6 (d, 2~), 6.7-6.8 (d, 2~), 6.9-7.0
(m, 4H), 7.15-7.25 (m, 2H), 8.1 (d, 1~).
FAB-MS: m/e 438 (M+H).
F~A~le 3
3-t4-(1-Carboxy-1-(4-chlorophenyl)methoxyphenyl]-
methyl-7-methyl-2-~ropyl-3~-imidazor4.5-bl~yridine
Step A: Preparation of methyl 2-bromo-2-(4-chloro-
phenyl)acetate
A mixture of 4-chlorophenylacetic acid (5.00
g. 29.3 mmol) and thionyl chloride (2.67 mL, 1.25 eq)
were heated at reflux while bromine (1.51 mL, 1.0 eq)
was added from a dropping funnel o~er 15 minutes.
The reaction mixture was heated at reflux 19.5 hours,
and then cooled to room temperature. Methanol (30
mL, 25 eq) was then added slowly, as an exotherm and
violent bubbling resulted. The reaction mixture was
then concentrated ~n vacuo. The residue was
partitioned between water and ether and the aqueous
phase was then extracted twice with ether. The
combined ether portions were washed with 5Z Na~S03,
dried (MgS04), filtered, and concentrated ~n ~acuo.
The residue was purified on a silica gel flash
WO 91/11~ 2 0 7 5 6 2 7 PCT/US91/~957
-196-
chromatography column (170 x 45 mm) eluted with 15%
ethyl acetate/hexane to yield 2.89 g (37%) of the
title compound.
1~ NMR (300 MHz, CDC13, ppm): ~ 3.8 (s, 3E), 5.35 (s,
lH), 7.2-7.3 (d, 2H), 7.45-7.55 (d, 2H).
EI-MS: m/e 262, 264, 266 (M+, 10:13:3 ratio).
Step ~: Preparation of methyl 2-(4-methylphenoxy)-
4-chlorophenylacetate
To a O~C suspension of ~ (530 mg, 1.0 eq)
in DME (10 mL) was quickly added p-cresol (500 mg,
4.63 mmol), and the reaction mixture was stirred at
room temperature. After stirring 10 minutes hydrogen
evolution had subsided, and 50 mg of 18-crown-6 was
added followed by the product of Step A (1.22 g, 1.0
eq). The reaction mixture was stirred 2.5 hours and
was then concentrated in v~cuo. The residue was
purified on a silica gel flash chromatography column
(140 x 30 mm) eluted with 5% ethyl acetate/hexane to
yield 0.744 g (55%) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 2.3 (5, 3H), 3.75 (s,
3H), 5.6 (s, lH), 6.8-6.9 (d, 2H), 6.9-7.1 (d, 2~),
7.3-7.4 (d, 2~), 7.5-7.6 (d, 2H).
~I-MS: m/e 290, 292 (Ml, 3:1 ratio).
2s
Step C: Preparation of methyl 2-(4-bromomethyl-
~henoxy)-4-chlorophenylacetate
A solution of the product of Step B (200 mg,
0.690 mmol), NB5 (117 mg, 0.95 eq) and AIBN (10 mg,
catalytic amount) in CC14 (5 mL) was heated at reflux
for 2 hours and then cooled and concentrated in vacuo.
The residue was purified on a silica gel flash
WO91/11~9 PCT/US91/~957
2~75~27
-197-
chromatography column (30 x 130 mm) eluted with 5%
ethyl acetate/hexane to yield 128 mg (50%) of the
title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 3.75 (s, 3H), 4.5 (s,
2~), 5.6 (s, 1~), 6.8-6.9 (d, 2H), 7.25-7.35 (d, 2~),
7.35-7.45 (d, 2H), 7.5-7.6 (d, 2~).
Ste~ D: Preparation of l-t4-(1-carbomethoxy-1-(4-
chlorophenyl))methoxyphenyl]methyl-7-methyl-
2-propyl-3H-imidazor4.5-blpyridine
To a suspension of NaH (4.3 mg, 1.0 eq) in
DMF (1 mL) was added 7-methyl-2-propylimidazot4~5-b]
pyridine (25 mg, 0.143 mmol). Next a solution of
product from Step C (53 mg, 1.0 eq) in DMF (1 mL) was
added. The reaction mixture was stirred for 2 hours
and then concentrated L~ vacuo. The residue was
purified on a silica gel flash chromatography column
(20 x 230 mm) eluted with 50% ethyl acetate/hexane to
yield 17 mg (26%) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ O.9-1.0 (t, 3H),
1.65-1.8 (m, 2H), 2.65 (s, 3~), 2.7-2.8 (t, 2H), 3.7
(s, 3H), 5.4 (s, 2H), 5.55 (s, lH), 6.8-6.9 (d, 2~),
7.0-7.1 (m, 3~), 7.3-7.4 (d, 2H), 7.4~-7.5 (d, 2H),
8.2 (d, lH).
2~ FAB-MS: m/e 464, 466 (M~l, 3:1 ratio).
Step E: Preparation of 1-[4-(1-carbo~y-1_(4_
chlorophenyl))metho~yphenyl]methyl-7-methyl-
2-~ropyl-3H-imidazor4.5-blpyridine
To a solution of product from Step D (16 mg,
0.035 mmol) in MeOH (1 mL) was added lN NaOH (1 mL).
The reaction mixture was stirred 10 minutes, and then
WO 91/11~ 2 0 ~ ~ 6 2 7 PCT/US91/00957
"
-198-
concentrated Ln vAcl~o. Water was added to the
residue and the mixture was acidified to pH 2 with 1
N ~Cl. The aqueous layer was then extracted 3 times
with chloroform, the combined organic layers were
dried (MgS04), filtered and concentrated ~n ~cuo to
yield 12 mg (70Z) of the titled product.
H NMR (300 M~z, CDC13, ppm): ~ O.75-0.85 (t, 3H),
1.55-1.7 (m, 2H), 2.65 (s, 3H), 2.9-3.0 (t, 2~), 5.4
(s, 2H), 5.55 (s, lH), 6.85-6.95 (d, 2H), 7.05-7.1
lo (d, 2H), 7.15 (d, lH) 7.3-7.4 (d, 2H), 7.5-7.6 (d,
2H), 8.3 (d, lH).
FAB-MS: m/e 450, 452 (M+l, 3:1 ratio).
~ le 4
3-r4-(1-Carboxy-1-(2-chlorophenyl))methoxyphenyl]-
methyl-7-methyl-2-~ropyl-3H-imidazor4.5-b~pyridine
Step A: Preparation of methyl 2-bromo-(2'-chloro)-
phenylacetate
Commercially available 2-chlorophenylacetic
acid (5.00 g, 29.3 mmol) was converted to 2.13 g
(28%) of the title compound in a procedure ~imilar to
that described in Step A of Example 3.
lH NMR (300 M~z, CDC13, ppm): ~ 3.8 (s, 3~), 5.95 (s,
lH), 7.25-7.45 (m, 3H), 7.7-7.8 (m, 1~).
Step B: Preparation of methyl 2-(2-chlorophenyl)-2-
(4-methylpheno~y)acetate
The product of Step A (1.22 g, 4.63 mmol)
was used to alkylate p-cresol (0.5 g, 4.63 mmol)
using the procedure described in Step ~ of Example 3,
and afforded 1.03 g (77%) of the title compound.
WO91/11~9 PCT/US91/~957
- 207~627
-199-
H NMR: (300 MHz, CDC13, ppm): ~ 2.25 (~, 3H), 3.8
(~, 3H), 6.12 (~, lE), 6.85 (d, 2H), 7.05 (d, 2H),
7.28-7.35 (m, 2H), 7.40-7.45 (m, lH), 7.63-7.70 (m,
lH).
EI-MS: m/e 290, 292 (M+).
Ste~ C: Preparation of methyl 2-(4-bromomethylphen-
oxy)-2-(2-chloro~he~yl)acetate
The product of Step B (0.200 g, 0.69 mmol)
was brominated with NBS (117 mg, 0.66 mmol) and
purified using the procedure described in Step C of
Example 3, and afforded 0.186 g (73Z) of the title
compound.
lH NMR (300 MHz, CDC13, ppm): ~ 3.8 (s, 3H), 4.5 (s,
2H), 6.15 (s, lH), 6.85-6.95 (d, 2H), 7.25-7.35 (m,
4H); 7.4-7.5 (m, lH), 7.6-7.7 (m, lR).
~I-MS: m/e 368, 370, 372 (M+l, 10:13:3 ratio).
Step D: Preparation Of 3-t4-(1-carbomethoxY-l-
(2-chlorophenyl))methoxyphenyl]methyl-7-
methyl-2-propyl-3~-imidazor4.5-bl~yridine
The product of Step C (0.100 g, 0.27 mmol)
was used to alkylate 0.047 g of 7-methyl-2-propyl-
imidazo[4,5-b]pyridine (E~ample 2, Step B) according
to the procedure described for Step D of Example 3,
which after purification afforded 0.040 g (32%) of
the title compound.
H NMR (300 M~z, CDC13, ppm): ~ O.9-1.0 (t, 3H),
1.65-1.8 (m, 2H), 2.65 (s, 3H), 2.7-2.8 (t 2H), 3.75
(s, 3H), 5.4 (s, 2H), 6.1 (s, lH), 6.8-6.9 (d, 2H),
7.0-7.1 (m, 3H), 7.25-7.35 (m, 2H), 7.35-7.45 (m,
lH), 7.55-7.65 (m, lH), 8.2 (d, lH).
~AB-MS: m/e 464, 466 (M+l, 3:1 r~tio).
WO9l/ll~9 æo~ ~ 627 PCT/US9l/~957
-200-
Step F: PreparatiOn of 3-t4-(l-carboxy-l-(2-chlor
phenyl))metho~yphenyl]methyl-7-methyl-2-
~ropyl-3H-imidazor4.5-bl~yridine
The product of Step D (O.040 g, O.086 mmol)
was dissolved in 1.0 mL of methanol and 1.0 mL of 1 N
NaOH was added. The hydrolysis was complete in 5
minutes, and the solution was then concentrated
in VACUO. The residue was chromatographed on silica
gel (130 ~ 20 mm) eluted with ethyl acetate/hexane/
acetic acid (19:5:1) to afford 33 mg (85Z) of the
title compound.
1~ NMR (300 MHz, CD30D, ppm): ~ 0.85-0.95 (t, 3H),
1.55-1.75 (m, 2H), 2.65 (s, 3H), 2.8-2.9 (t, 2H), 5.5
(s, 2H), 6.05 (s, 1~, 6.85-6.95 (d, 2~), 7.05-7.20
(m, 3H), 7.25-7.35 (m, 2H), 7.35-7.45 (m, 1~),
7.55-7.65 (m lH). 8.2 (d, 1~).
FAB-MS: m/e 450,452 (M~l).
E~ample 5
3-[4-(1-Carboxy-1-(3-chlorophenyl))methoxyphenyl]-
methyl-7-methyl-2-propyl-3~-imidazor4.5-blpyridine
Step A: Preparation of methyl 2-bromo-2-(3-chloro-
phenyl)acetate
Commercially available 3-chlorophenylacetic
acid (5.00 g, 29.3 mmol) was converted to 1.70 g
(22%) of the title compound in a procedure similar to
that described in Step A of Example 3.
1~ NMR (300 MHz, CDC13, ppm): ~ 3.80 (s, 3~) 5.30 (s,
lH) 7.25-7.60 (m, 4H).
WO91/11~9 PCT/US91/~957
2~7~;627
-201-
Step B: Preparation of 3-t4-(1-carbomethoxy-1-(3-
chlorophenyl))methoxyphenyl~methyl-7-methyl-
2-propy]-3~-imid~zor4.~-b~pyridine
To a solution of 0.150 g (0.54 mmol) of the
product of Step D of Example 2 dis~olved in 1.0 mL of
DMF was added 61 mg of RH, and 0.141 g of 18-crown-6.
The reaction was stirred under an N2 atmosphere for
20 minutes, and then 0.155 g of the product of Step A
dissolved in 0.5 mL of DMF was added. The reaction
was stirred an additional 30 minutes, then
partitioned between water and ethyl acetate. The
organic layer was washed with water, dried (MgS04),
filtered and evaporated in vacuo. The residue was
purified on a silica gel flash chromatography column
eluted first with 30Z ethyl acetate/hexane then with
50% ethyl acetate/hexane to afford 0.087 g (35%) of
the title compound.
H NMR (300 MHz, CDC13, ppm): ~ O.90-1.00 (t, 3H),
1.65-1.85 (m, 2H), 2.65 (s, 3H), 2.70-2.85 (t, 2H),
3.75 (s, 3H), 5.45 (s, 2H), 5.55 (s, lH), 6.80-6.90
(d, 2H), 7.00-7.10 (m, 3H), 7.25-7.35 (m, 2H),
7.30-7.35 (m, lH), 7.55 (br s, 1~), 8.15-8.25 (d, lH).
Step C: Preparation of 3-t4-(1-carboxy-1-(3-chloro-
phenyl))methoxyphenyl~methyl-7-methyl-2-
propyl-3~-imidazot4.5-bl~yridine
To a ~olution of 0.087 g (0.19 mmol) of the
product of Step B dissolved in 2 mL of methanol was
added 1 mL of 2 N NaOH and the reaction was stirred
for 2 hours at room temperature. The reaction
mixture was concentrated in vacuo and applied to a
.:, WO 91/11999
PCT/US91/009S7
2075627
-202-
silica gel flash chromatography column eluted with
CHC13/MeO~/~OAc (100:3:1) to afford 0.049 g (58%) of
the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ 0.90-1.00 (t, 3H),
1.60-1.75 (m, 2~), 2.65 (s, 3~), 2.80-2.90 (t, 2~),
5.50 (s, 2~), 5.70 (s, l~), 6.90-6.95 (d, 2H),
7.05-7.20 (m, 3H), 7.30-7.35 (m,2H), 7.45-6.50 (m,
2H), 7.45-7.50 (m, lH), 7.55 (br s, 1~), 8.15-8.25
(d, lH).
~AB-MS: m/e 450,452 (M+l).
~mple 6
3-[4-(1-carboxy-1-(2,6-dichlorophenyl))methoxyphenyl3-
methyl-7-methyl-2-~ro~yl-3E-imidazor4.5-blpyridine
Step A: Preparation of methyl 2-bromo-2',6'-dichloro-
phenylacetate
Commercially available 2,6-dichlorophenyl-
acetic acid (5.00 g, 24.4 mmol) was converted to 2.60
g (35%) of the title compound in a procedure similar
to that described in Step A of Example 3.
H NMR (300 MHz, CDC13, ppm): ~ 3.80 (6, 3H), 6.70
(s, 1~), 7.20-7.30 (m, lH), 7.35-7.40 (d, 2H).
Step B: Preparation of 3-t4-(l-carbomethoxy-l-(2~6
dichlorophenyl))methoxyphenyl]methyl-7-
methyl-2-propyl-3E-imidazor4.5-blpyridine
The product of Step D of Example 2 (0.100 g,
0.36 mmol) was deprotonated (41 mg KH, 94 mg
18-crown-6. 1.0 mL DMF) and alkylated with 0.117 g
(0.39 mmol) of the product of Step A according to the
procedure described in Step B of Example 5. Purifica-
WO91/11~9 PCT/US91/~957
~ ` 207~627
-203-
tion on a silica gel flash chromatography column
eluted with 40% ethyl acetate/hexane afforded 0.085 g
(48X) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ O.90-1.00 (t, 3H),
1.60-1.80 (m, 2E), 2.65 (s, 3~), 2.70-2.80 (t, 2~),
3.80 (s, 3~), 5.40 (s, 2~), 6.40 (s, 1~), 6.60-6.65
(d, lH), 6.85-6.90 (d, lH), 6.90-7.05 (m, 5~),
7.15-7.35 (m, 2~), 8.15-8.35 (d, lH).
Step C: Preparation Of 3-t4-(1-carboxy-1-(2,6-
dichlorophenyl))methoxyphenyl]methyl-7-
methyl-2-propyl-3~-imidazor4.5-b~pyridine
To a solution of 0.085 g (0.17 mmol) of the
product of Step ~ dissolved in 1.0 mL of methanol was
added 1.0 mL of 1 N NaOH and the reaction was stirred
for 3 hours at room temperature. The reaction
mixture was concentrated in vacuo and applied to a
silica gel flash chromatography column eluted with
CHC13/MeOH/NH40H (80:15:1) to afford 0.070 g (84%) of
the title compound.
H NMR (300 M~z, CD30D, ppm): ~ O.90-1.00 (t, 3H),
1.55-1.75 (m, 2H), 2.65 (s, 3H), 2.80-2.90 (t, 2H),
5.50 (s, 2H), 6.30 (s, lH), 6.95-7.10 (m, 2H),
7.30-7.40 (d, 2H), 8.20 (d, lH).
FAB-MS: m/e 484 (M+l).
Example 7
3-t4-(1-carboxy-1-(2-nitrophenyl))methoxyphenyl3-
methyl-7-methYl-2-~ropyl-3~-imidazor4.5-bl~yridine
Step A: PreparatiOn of methyl 2'-nitrophenylacetate
To a flask charged with 5 mL of methanol was
introduced a fine stream of hydrogen chloride gas
WO 91/11~ 2 0 7 5 6 2 7 PCT/US91/~957
-204-
until the solution was saturated. The hydrogen
chloride was 6topped, and 0.50 g (2.7 mmol) of
2'-nitrophenylacetic acid was added and the reaction
mixture was stirred for 1.5 hours. The reaction
mixture was then partitioned between ethyl acetate
and water, the organic layer was washed with
saturated NaHC03, brine, dried (MgS04), filtered and
evaporated to afford 0.529 g (98%) of the title
compound.
lH NMR (300 M~z, CDC13, ppm): ~ 3.75 (~, 3H), 4.05
(s, 2H), 7.25-7.30 (d, lH), 7.45-7.55 (m, lH),
7.55-7.65 (m, lH), 8.20-8.25 (d, lH).
FAB-MS: m/e 196 (M+l).
5 Step B: Preparation of methyl 2-bromo-2'-nitrophenyl-
acetate
To a solution of the product of Step A
dissolved in 10 mL of carbon tetrachloride was added
0.441 g of N-bromosuccinimide and 25 mg of AIBN and
the reaction was heated at reflux for 14 hours. The
mixture was then cooled and evaporated and the
residual oil was purified on a silica gel flash
chromatography column eluted with 5% ethyl acetate.
Evaporation of the purified fractions afforded 0.335
2s g (50~/) of the title compound.
1~ NMR (300 MHz, CDC13, ppm): ~ 3.80 (s, 3H), 6.10
(s, lH), 7.50-7.60 (m, lH), 7.70-7.80 (m, lH),
8.00-8.10 (m, 2H).
WO91/11~ PCT/US91/00957
207~627
-205-
Step C: Preparation Of 3-t4-(1-carbomethoxy-1-(2-
nitrophenyl))methoxyphenyl]methyl-7-methyl-
2-propylimidazo r 4.5-blpyridine
A solution of 0.364 g (1.30 mmol) of 3-(4-
hydroxyphenyl)methyl-7-methyl-2-propyl-3~-imidazo
~4,5-b]pyridine (Step D, Example 2) dissolved i~ 5 mL
of DMF was deprotonated (O.163 g ~H, 0.376 g
18-crown-6) and alkylated with 0.355 g (1.30 mmol) of
the product of the previous step similarly to the
procedure described in Example 2, Step E. Purifi-
cation on a silica gel flash chromatography column
eluted with 40% ethyl acetate/hexane afforded 0.296 g
(48Z) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ O.95-1.05 (t, 3H),
1.70-1.80 (m, 3H), 2.65 (s, 3H), 2.70-2.80 (t, 2~),
3.75 (s, 3H), 5.40 (s, 2H), 6.65 (s, 1~), 6.85-6.95
(d, 2H), 7.00-7.10 (m,3H), 7.50-7.55 (t, 1~),
7.60-7.65 (t, lH), 7.80-7.85 (d, lH), 8.05-8.10 (d,
lH), 8.20-8.25 (d, lH).
20-
Step D: Preparation of 3-[4-(1-carboxy-1-(2-nitro-
phenyl))methoxyphenyl]methyl-7-methyl-2-
propyl-3H-imidazor4.5-blpyridine
Hydrolysis of 0.030 g (0.063 mmol) of the
ester prepared in the previous ~tep was performed in
a manner similar to that described in Example 2, Step
F. Purification on a silica gel flash chromatography
column eluted with CHC13/MeOH/~OAc (100:3:1) afforded
0.023 g (74%) of the title compound.
lH NMR (300 M~z, CD30D, ppm): ~ O.90-1.00 (t, 3H),
1.60-1.75 (m, 2H), 2.65 (s, 3H), 2.80-2.90 (t, 2H),
WO91/11~9 ~7S~ pCT/US9l/009~7
-206-
~.50 (~, 2H), 6.45-6.60 (~r s, lH), 6.90-7.00 (d,
2H), 7.05-7.15 (d, 2H), 7.15-7.20 (d, 2H), 7.50-7.60
(t, lH), 7.60-7.70 (t, lH), 7.75-7.80 (t, lH),
8.00-8.05 (d, lH), 8.20-8.25 (d, lH).
FAB-MS: m/e 461 (M+l).
F.~n~le 8
3-[4-(1-Carboxy-l-cyclohexyl)methoxyphenyl~methyl-7-
~ethyl-2-propyl-3~-imidAzor4.5-blpyridine
Step A: Preparation of 3-[4-(1-carbomethoxy-1-cyclo-
hexyl)methoxyphenyl]methyl-7-methyl-2-propyl-
3~-imidazor4.5-blpyridine
A solution of 0.200 g (0.71 mmol) of 3-(4-
hydroxyphenyl)methyl-7-methyl-2-propyl-3H-imidazo
t4,5-b3pyridine (Step D, Example 2) dissolved in 0.5
mL of DMF was deprotonated (82 mg ~H, 0.188 ~
18-crown-6) and al~ylated with 0.184 g (0.78 mmol) of
2~ commercially available methyl 2-bromo-2-cyclohexyl-
acetate similarly to the procedure described in
Example 2, Step E. Purification on a silica gel
flash chromatography column eluted with 30% ethyl
acetate/hexane afforded 0.029 g (10%) of the title
2s Cmpound.
lH NMR (300 M~z, CDC13, ppm): ~ 0.97 (t, J=8 Hz, 3H),
1.10-1.90 (m, 13H), 2.66 (s, 3H), 2.78 (t, J=8 ~z,
2~), 3.70 (F, 3H), 4.31 (d, J=6 ~z, lH), 5.40 (s,
2H), 6.77 (d, J=10 Hz, 2H), 7.02-7.10 (m, 3H), 8.10
(d, J=6 ~z, 1~).
WO91/11~ PCT/US91/~s~7
207~i~2~
-207-
Step B: Preparation of 3-t4-(l-carbo2y-l-cyclohexyl)
methoxyphenyl~methyl-7-methyl-2-propyl-3~-
imidazor4.5-blpyridine
~ydrolysis of 0.029 g (0.066 mmol) of the
ester prepared in Step A was performed in a manner
similar to that described in Example 2, Step F.
Purification on a silica gel flash chromatography
column eluted with CHC13/MeOH/~OAc (100:3:1) afforded
0.017 g (61%) of the title compound.
1~ NMR (300 M~z, CD30D, ppm): ~ 0.95 (t, J=8 ~z, 3H),
1.10-1.90 (m, 13~), 2.65 (s, 3~), 2.85 (t, J=8 Hz,
2H), 4.38 (d, J=6 ~z, 1~), 5.48 (s, 2H), 6.84 (d,
J=10 Hz, 2H), 7.06 (d, J=10 Hz, 2H), 7.15 (d, J-6 Hz,
lH), 8.20 (d, J=6 Hz, lH).
FAB-MS: m/e 422 (M+l).
F~ple 9
3-t4-(l-carboxy-l-propyl)methoxyphenyl]methyl-7
methyl-2-~ropyl-3~-imidazor4.5-blpyridine
Step A: Preparation of 3-t4-(1-carbomethoxy-1-
propy})methoxyphenyl]methyl-7-methyl-2-
p~opyl-3~-imidazor4.5-blpyridine
To a suspension of ~ (22 mg, 1.1 eq) in
0.25 mL of DMF under N2 was added 0.050 g (0.18 mmol)
of the product of Step D in Example 2, and the
reaction mixture was stirred for 15 minutes. When
the K~ had dissolved, 18-crown-6 (10 mg, 0.2 eq) was
added, followed by ethyl 2-~romopentanoate (30 uL,
l.0 eq). After 5 minutes, tlc showed that the ~romide
had disappeared~ ~ut that some of the starting
`- WO 91/11999
PCT/US91/~957
207~i 62~ ~
-208-
phenolic intermediate remained. The reaction mixture
was quenched with saturated ~mmonium chlor~de and
concentrated in vacuo. The residue was chromato-
graphed on silica gel (120 x 20 mm) eluted with 30%
ethyl acetate/hexane. The product was isolated in a
19Z yield (14 mg).
1~ NMR (300 MHz, CDC13 ppm): ~ O.9-1.0 (m, 6~),
1.15-1.25 (t, 3H), 1.4-1.6 (m, 2H), 1.7-2.0 (m, 4H),
2.65 (s, 3H), 2.7-2.8 (t, 2H), 4.1-4.2 (m, 2H),
4.5-4.6 (m, lH), 5.4 (s, 2~), 6.7-6.8 (d, 2H),
7.0-7.1 (m, 3H), 8.2 (d, lH).
FAB-MS: m/e 410 (M+l).
Ste~ B: Preparation of 3-~4-(1-carboxy-1-propyl)-
methoxyphenyl~methyl-7-methyl-2-propyl-3~-
i~idazor4.5-blpyridine
To a solution of 21 mg (0.049 mmol) of the
product of Step ~ dissolved in 1 mL of ethanol was
added 0.25 m~ of 1 N NaOH. The reaction mixture was
stirred overnight, after which time tlc indicated the
consumption of the starting material. The reaction
mixture was neutralized with 1 N HCl and concentrated
La vacuo. The residue was purified on a silica gel
flash chromatography column (90 x 10) eluted with
ethyl acetate/hexane/acetic acid (76:20:4). The
product fractions were combined and evaporated,
redissolved in ethyl acetate and reconcentrated in
vacuo several times to remove residual acetic acid.
Drying in ~acuo afforded 13 mg (93Z) of the title
CmpOund~
WO91/11~ PCT/US91/~9S7
2075627
-209-
H NMR (300 MHz, CD30D, ppm): ~ 0.9-1.0 (m, 6H),
1.4-1.6 (m, 2H), 1.6-1.8 (m, 2~), 1.8-1.9 (m, 2H),
2.65 (s, 3H), 2.8-2.9 (t, 2H), 4.5-4.6 (t, lH), 5.5
(s, 2H), 6.8-6.9 (d, 2H), 7.0-7.1 (d, 2H), 7.15 (d,
lH), 8-2 (d, lH).
FAB-MS: m/e 382 (M+l).
~ 1e 10
1o 3-[4-(1-Carboxy)-1-(2-carboxyphenyl)methoxyphenyl]-
methyl-7-methyl-2-propyl-3H-imidazor4~5-blpyridine
Step A: Pre~aration of dimethyl homo~hthal~te
To homophthalic acid (500 mg, 2.78 mmol)
cooled to O-C was added a saturated solution of
HC1/CH30H (20 mL). The solution was gradually warmed
to room temperature and stirred ~or 3 days. The
reaction was concentrated in vacuo, the residue was
partitioned between ethyl acetate and water, and the
organic layer washed with saturated NaHC03 and
brine. The organic layer was concentrated n vacuo,
then redissol~ed in ethyl acetate, dried (MgS04),
filtered and evaporated to afford 0.470 g (81%) of
the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 3.7 (s, 3H), 3.9 (s,
3H), 4.0 (s, 2H), 7.2-7.2 (m, lH), 7.25-7.35 (m, lH),
7 45-7.55 (m, lH), 8.0-8.1 (m, lH).
EI-MS: m/e 208 (M+).
Step B: Preparation of methyl 2-bromo-(2'-carbo-
metho~y)phenyl~cetate
To a solution of the product of Step A (464mg, 223 mmol) in 10 mL CC14 were added NBS (377 mg,
W091/ll ffl PCT/US91/~gs7
2075627
-210-
0.95 eq) and a catalytic amount of AIBN. The
reaction mixture was refluxed overnight, and then
concentrated in vac~o. The residue was chromatogra-
phed on silica gel (140 x 40 mm) eluted with 8% ethyl
acetate/hexane. The product was isolated in a 65%
yield (398 mg).
1~ NMR (300 MHz, CDC13, ppm): ~ 3.8 (s, 3~), 3.9 (s,
3~), 6.6 (s, 1~), 7.35-7.45 (t, lH), 7.55-7.65 (t,
1~), 7.8-7.9 (d, lH), 7.95-8.0 (d, 1~).
Step C: Preparation of 3-t4-(1-carbomethoxy-1-(2-
carbomethoxyphenyl))methoxyphenyl]methyl-7-
~ethyl-2-propyl-3~-imidazor4.5-bl~yridine
To a suspension of KH (22 mg, 1.1 eq) in 250
uL DMF under N2 was added 50 mg (0.178 mmol) of
3-(4-hydroxyphenyl)methyl-7-methyl-2-propyl-3~-
imidazot4,5-b]pyridine (Example 2, Step D), and the
reaction mixture was stirred for 1 hour. To this
solution were added 18-crown-6 (47 mg, 1.1 eq) and
20 - the product of Step ~ (56 mg, 1.1 eq). The reaction
mixture was stirred at room temperature overnight.
The reaction mixture was then guenched with saturated
ammonium chloride and concentrated ~n vacuo. The
residue was partitioned between ethyl acetate and
water, the organic layer extracted with saturated
NaHC03 and concentrated in ~acuo. The product was
purified on a silica gel flash chromatography column
(130 x 20 mm) eluted with 40Z ethyl acetate/hexane to
afford 33 mg (38%) of the title compound.
WO91/11~ PCT/USgl/~ss7
- ~''"' 207~627
-211-
lH NMR (300 M~z, CDC13, ppm): ~ O.9-1.0 (t, 3~),
1.65-1.8 (m, 2H), 2.65 (s, 3H), 2.7-2.8 (t, 2H), 3.7
(s, 3H), 3.9 (s, 3~), 5.2 (s, 2H), 6.8-6.95 (m, 3H),
7.0-7.1 (m, 3H), 7.35-7.45 (m, lH), 7.45-7.55 (m,
lH), 7.7 (m, lH), 7.9-8.0 (m, lH), 8.2 (d, lH).
~AB-MS: m/e 488 (M+l).
Step D: Preparation of 3-t4-(1-carboxY)-1-(2-
carbo~yphenyl)methoxyphenyl]methyl-7-methyl-
2-~ropyl-3~-imidazor4.5-b~pyridine
To a methanol solution (1.5 mL) of the
product of Step C (33 mg, 0.068 mmol) wa~ added 1.5
mL of 1 N NaOH. The reaction mixture was ~tirred at
room temperature for 4.5 hours, and was then
neutralized with 1 N ~Cl and concentrated in vacuo.
The residue was chromatographed on silica gel (120 x
20 mm) eluted with CHC13/MeOH/HOAc (100:10:2) to
afford 32 mg (94%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ O.85-0.95 (t, 3H),
1.5-1.7 (m, 2H), 2.6 (s, 3H), 2.7-2.8 (t, 2~), 5.35
(s, 2H), 6.3 (s, lH), 6.8-7.0 (m, 4~), 7.05-7.30 (m,
3H), 7.55-7.7 (m, 2H), 8.15 (d, 2H).
~rA~ le 11
(z)-3-t(4-((2-Carboxy-2-phenyl)ethenyl)phenyl)-
methyll-7-methyl-2-pro~Yl-3H-imidazor4~5-bl~yridine
Step A: Preparation of methyl 3-hydroxy-3-(4-meth
phenyl)-2-phenylpropionate
To a solution of methyl 2-bromophenylacetate
(4?3 mg; 2.065 mmol) and 4-methylbenzaldehyde (188
WO91/11~9 PCT/USgl/~ss7
20~5~27 ~
-212-
mg, 1.57 mmol) in dry THF (5 mL) under N2 was added
powdered zinc (201 mg, 3.09 mmol). After ~tirring at
reflux for 10 min, a few crystals of iodine were
added. The mixture was refluxed under N2 for 4
hours, then allowed to stand at room temperature
overnight. The next day the mixture was diluted in
Et20 and 1 N HCl was added. The biphasic mixture was
stirred until the zinc had completely dissolved in
the aqueous layer. The organic layer was separated,
lo washed with brine, dried (MgS04), filtered, and
concentrated in VACUO. The residue which contained
two diastereomers (Rf=0.22, 0.17 in 15%
EtOAc/hexane), was purified on a ~ilica gel flash
chromatography column eluted with 15X ethyl
acetate/hexane. A total of 283 mg (67%) of both
diastereomers was isolated. The 300 MHz lH NMR
spectra of each diastereomer was consistent with its
structure.
Step B: Preparation of methyl (Z)-3-(4-methylphenyl)-
- 2-ghenyl~ropenoate
To a solution of a mixture of the diastereo-
isomers from Step A (34 mg, 0.126 mmol) in dry benzene
(3 mL) was added a few crystals of p-TsO~ and the
mixture was heated to reflux. After several minutes
tlc analysis indicated complete reaction. The mi~ture
was diluted with Et20, washed with saturated NaHC03,
brine, dried (MgS04), filtered and concentrated
in vacuo to provide 31 mg (98/c) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ Z.27 (s, 3H), 3.79
(s, 3H), 6.92 (g, 4~), 7.25 (m, 2H), 7.40 (m, 3H),
7.82 (s, 1~).
WO91/11~9 PCT/US91/~957
2075627
-213-
Ste~ C: Preparation of methyl (Z)-3-(4-bromomethyl-
phenyl)-2-~heny~propeno~te
To a solution of the product of Step B (31
mg, 0.123 mmol) in dry CC14 (1 mL~ under N2 were
added NBS (20 mg, 0.9 eq) and a catalytic amount of
AIBN. The mixture was stirred at reflux under N2 for
1.5 hours. The mixture was cooled to room tempera-
ture, diluted with Et2O and filtered to remove the
precipitated succinimide. The filtrate was concen-
trated in vacuo and the residue was purified on asilica gel flash chromatography column eluted with
15:1 hexane/EtOAc to afford 27.4 mg (67%) of the
title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 3.80 (s, 3H), 4.49
(s, 2H), 7.02 (d, 2H), 7.18 (d, 2~), 7.24 (d, 1~),
7.39 (m, 4~), 7.82 (s, 1~).
Step D: Preparation of (Z)-3-~(4-(2-carbomethoxy-2-
phenylethen-l-yl)phenyl)methyl]-7-methyl-2-
propyl-3~-imidazor4.5-blpyridine
To a solution of 30 mg (0.171 mmol) of
2-propyl-7-methylimidazO[4,5-b]pyridine (Example 2,
Step D) dissolved in 1 mL of dry DME was added 11 mg
of a 60% oil dispersion of Na~ (1.5 eq) and the
reaction mixture was stirred under an N2 atmosphere.
After stirring at room temperature for 30 minutes the
product from Step C (27.4 mg, 0.082 mmol) dissolved
in 0.5 mL DMF was added ~ia syringe. The reaction
mixture was quenched with saturated N~4Cl solution,
and the solvent was removed under in vacuo. The
residue was dissolved in EtOAc and washed with E2O
and brine. The organic layer was dried (MgSO4),
WO 91/11999 Pcr/us9l/oo9s7
- 2~7~627
-214-
filtered, and concentrated ~n ~cuo. The residue was
purified on a silica gel flash chromatography column
eluted with 50% ethyl acetate/hexane to provide 13.2
mg (38%) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ O.9S (t, 3H), 1.72
(q, 2H), 2.68 (s, 3H), 2.71 (q, 2H), 3.78 (s, 3H),
5.40 (s, 2H), 6.91 (q, 4H), 7.02 (d, lH), 7.19 (dd,
lH), 7.32 (m, 3H), 7.79 (s, lH), 8.16 (d, lH).
Step ~: Preparation of (Z)-3-t(4-((2-carboxy-2-
phenyl)ethen-l-yl)phenyl)methyl]-7-methyl-2-
propyl-3H-imidazor4.5-blpyridine
To a solution of the product of Step D (13.2
mg, 0.031 mmol) in MeOH (1 mL) was added 1 N NaOH
1~ (0.138 mL, 4.5 eq). After stirring overnight the
mixture was quenched with 0.5 mL of acetic acid and
concentrated in ~acuo. The product was purified on a
silica gel flash chromatography column eluted with
he~ane/EtOAc/HOAc (75:50:1) to afford 10.8 m~ (85-/.)
of the title compound.
H NMR (300 M~z, CD30D, ppm): ~ O.91 (t, 3H), 1.63
(q, 2H), 2.62 (s, 3H), 2.79 (t, 2H), 5.47 (s, 2H),
6.87 (d, 2H), 6.96 (d, 2H), 7.12 (m, 3H), 7.31 (m,
3H), 7.79 (s, lH), 8.12 (d, lH).
F.~ple 12
3-[(4-((2-Carboxy-2-phenyl)ethyl)phenyl)methyl]-7-
methyl-2-propyl-3H-imidazor4.~-blpyridine
WO91/ll ffl PCT/US91/00957
2075(i27
-215-
Ste~ A: Preparation of 3-[(4-((2-carbo~y-2-phenyl)-
ethylphenyl)methyl]-7-methyl-2-propyl-3~-
imidazor4.5-blpyridine
To a ~olution of 2.2 mg (0.0054 ~ol) of the
product of Step E of Example ll in 1 mL of EtOAc was
added a catalytic amount of 10% Pd on carbon. A
hydrogen atmosphere was secured with a balloon and
the mixture was stirred for 1 hour. The catalyst was
removed by filtering the mixture through a pad of
celite, and the filtrate was concentrated Ln ~acuo to
provide 1.3 mg (59%) of the title compound.
H NMR (300 MHz, CDC13, ppm): ~ O.87 (t, 3H), 1.63
(q, 2~), 2.62 (s, 3~), 2.67 (t, 2H), 5.30 (5, 2H),
6.80 (s, 4~), 6.92 (d, lH), 7.01 (m, SH), 8.12 (d,
lH).
F~ple 13
3-[4-(1-carboxy-1-methyl-1-phenyl)methoxyphenyl3-
methyl-7-methyl-2-~ro~yl-3~-imidazor4~5-bl~yridine
Step A: Preparation of methyl 2-(4-methylphenoxy)-
~henylacetate
To a cooled (0C) suspension of ~ (2.12 g,
1.0 eq) in DMF (30 mL) was added a solution of
p-cresol (2.00 g, 18.5 mmol) in 20 mL of DMF. The
reaction mixture was stirred 15 minutes, then
18-crown-6 (200 mg) was added followed by a solution
of 4.24 g (18-5 mmol) of methyl 2-bromophenylacetate
dissolved in 10 mL of DME. The reaction mixture was
stirred 45 minutes, then partitioned between ethyl
acetate and water. The organic layer was washed with
WO91/11~ PCT/US91/009~7
207562~
-216-
water, brine, dried (MgS04~, filtered and evaporated
~n vacuo. The re~idue was purified on a silica gel
flash chromatography column (150 x 40 mm) eluted with
5% ethyl acetate/hexane to yield 2.63 g (58%) of the
title compound.
lH NMR (300 M~z. CDC13): ~ 2.3 (s, 3H), 3.75 (s, 3H),
5.6 (s, lH), 6.8-6.9 (d, 2H~, 7.0-7.1 (d, 2E),
7.3-7.45 ~m, 3H), 7.5-7.6 (d, 2H).
FAB-MS: m/e 257 (M+l).
Step B: Preparation of methyl 2-(4-methylphenoxy)-
2-phenylpropanoate
A solution of the product of Step A (50 mg,
O.195 mmol) in 500 uL of T~F was cooled to -78OC. A
solution of lithium bis(trimethylsilyl)amide (195 uL,
1.O M in THF) was added and the reaction was stirred
for 15 minutes. Methyl iodide (12 uL, 1.0 eg) was
added and the cooling bath remo~ed. After 8 minutes,
the reaction mixture was quenched with saturated
N~4Cl and concentrated in vacuo. The residue was
purified on a on a silica gel flash chromatography
column (140 x 20 mm) eluted with 2.5% ethyl
acetate/hexane to yield 26 mg (49%) of the title
compound.
2~ lH NMR ~300 MHz, CDC13, ppm): ~ 1.9 (s, 3H), 2.3 (s,
3H), 3.75 (s, 3H), 6.7-6-8 (d, 2H), 7.0-7.1 (d, 2H),
7.3-7.45 (m, 3H), 7.6-7.7 (d, 2H).
~AB-MS: m/e 271 (M+l).
0 Step C: Preparation of methyl 2-(4-bromomethyl-
phenoxy~-2-phenylpropanoate
A solution of the product of Step B (26 mg.
~ 0.096 mmol) NBS (16 mg, 0-95 eq) and AIBN (2 mg,
WO91/11~ PCT/US91/~9~7
207~1~2~
-217-
catalytic amount) in CC14 (2 mL) was heated to reflux
for 1 hour and then concentrated Ln v~cllo. The
residue was purified on a silica gel fla~h chromato-
graphy column (125 x 20 mm) eluted with 5% ethyl
acetate/hexane to yield 21 mg (62%) of the title
compound.
lH NMR (300 MHz, CDC13, ppm): ~ 1.9 (s, 3~), 3.75 (s,
3H), 4.5 (s, 2H), 6.9-6.9 (d, 2H), 7.2-7.3 (d, 2H),
7.3-7.5 (m, 3H), 7.6-7.7 (d, 2H).
Step D: Preparation of 1-[4-(1-carbomethoxy-1-
methyl-l-phenyl)methoxyphenyl]methyl-7-
methyl-2-~ro~yl-3~-imidazor4.5-bl~yridine
To a suspension of NaH (2.0 mg, 1 eq) in DM~
(0.25 mL) was added lO mg (l.0 eq) of 7-methyl-2-
propylimidazot4,5-b]pyridine (Example 2, Step B), and
the reaction mixture was stirred 15 minutes. Next, a
solution of the product of Step C (20 mg, 0.057 mmol)
in DMF (0.5 mL) was added. After stirring 1.5 hours,
the reaction mixture was concentrated ~n ~acuo. The
residue was purified on a silica gel flash chromato-
graphy column (140 x 15 mm) eluted with 50% ethyl
acetate/hexane to yield 12 mg (48/o) of the titled
product.
lH NMR (300 M~z, CDC13, ppm): ~ O.9-1.0 (t, 3H),
1.65-1.8 (m, 2H), 1.85 (s, 3H), 2.7 (s, 3H), 2.75-2.85
(t, 2H), 3.7 (s, 3H), 7.55-7.65 (m, 2H),8.2 (d lH).
EAB-MS: m/e 444 (M+l).
WO 91/11~ 2 0 ~ 5 6 2 7 PCT/US91/~957
-218-
Step F: Preparation Of 3-t4-(1-carbo~Y-l-methyl-l-
phenyl)methoxyphenyl]methyl-7-methyl-2-
propyl-3~-imidazor4.5-blpyridine
To a solution of the product of Step D (12
mg, O.027 mmol) in MeOH (2 mL) was added 2 mL of a 1
N NaOH solution. The reaction mi~ture was stirred
for 45 minutes, and was then concentrated Ln ~Acuo.
The residue was taken up in water and acidified to pH
2 with 1 N HCl. Next, the agueous layer was diluted
with water and extracted 3 times with chloroform.
The combined organic layers were dried (MgS04),
filtered, and concentrated ~n vacuo to yield 8.3 mg
(70%) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 0.9-1.0 (t, 3H),
1.7-1.85 (m, 2H), 1.8 (s, 3H), 2.8 (s, 3~), 3.1-3.2
(t, 2H), 5.5 (s, 2~), 6.75-6.85 (d, 2H), 7.0-7.1 (d,
2H), 7.25-7.4 (m, 4H), 7.5-7.6 (m, 2H), 8.45 (d, 1~).
FAB-MS: m/e 450, 452 (M+l, 3:1 ratio).
~A~yle 14
7-Methyl-2-propyl-3-[4-(1-(tetrazol-5-yl)-1-phenyl)-
methoxy~henyllmethyl-3H-imidazor4.5-blpyridine
~ Step A: Preparation of 3-[4-(1-carboxamido-1-phenyl)-
methoxyphenyl3methyl-7-methyl-2-propyl-3H-
imidazor4.5-blpvridine
A solution of the product of Example 2, Step
E (125 mg, 0.291 mmol) in MeOH (10 mL) was cooled to
0C and ammonia was bubbled through the mixture for
1.5 hours. The flask was stoppered and stirred for 6
hours, during which time the product precipitated.
WO91/11~ PCT/US91/~gs7
207562~
-219-
The reaction mixture was concentrated Ln ~acuo to
yield 105 mg (88Z) of product (Rf=0.50 5% methanol/
ethyl acetate) which was used in the next step without
further purification.
lH NMR (300 MHz, CDC13, ppm): ~ O.9-1.0 (t, 3~),
1.7-1.8 (m, 2H), 2.65 (5, 3H), 2.7-2.8 (t, 2~), 5.4
(s, 2H), 5.45 (s, lH), 5.5-5.6 (s, lH), 6.55-6.65 (s,
lH), 6.8-6.9 (d, 2H~, 7.0-7.1 (m, 3H), 7.3-7.4 (m,
3~), 7.45-7.55 (m, 2H), 8.2 (d, lH).
Step B: Preparation of 3-[4-(1-cyano-1-phenyl)-
methoxyphenyl]methyl-7-methyl-2-propyl-3H-
imidazor4.5-blpyridine
To a 0C suspension of the product of Step A
(83 mg, 0.20 mmol) in phosphorous oxychloride (0.51
mL, 27 eq) was added Et3N (61 uL, 2.2 eg) over 55
minutes. After the addition was complete, the
reaction mixture was warmed to room temperature over
2 hours and then heated to reflux for 45 minutes.
Next, the reaction mixture was concentrated in vacuo,
and the residue was partltioned between ice water and
toluene. The aqueous layer was extracted three times
with toluene. The combined extracts were washed with
0.5 N NaOH and then with water. The organic layer
was separated and concentrated in ~acuo to yield 59
mg (74%) of crude product (Rf=0.35 in 50Z ethyl
acetate/hexane), which was used in the next step
without purification.
WO 91/11999
PCT/US91/~957
2075~7 `-`
-220-
Step C: Preparation of 7-methyl-2-propyl-3-~4_(1_
(tetrazol-5-yl)methoxyphenyl]methyl-3~-
imidazo-r4.5-b~pyridine
To a solution of the product of Step B (29
mg, 0.073 mmol) di~sloved in 0.5 mL of toluene was
added 15 mg (1.2 eq) of Me3SnN3 and the reaction
mixture was refluxed for 22 hours. An additional 15
mg (1.2 eq) of ~e3SnN3 was then added, and the
refluxing continued for another hour. The reaction
mixture was then poured into ethyl acetate/ether and
washed with saturated N~4Cl and brine. The organic
layer was separated and concentrated Ln ~cuo. The
residue was purified on a silica gel flash chromato-
graphy column (120 x 30 mm) eluted with chloroform/
methanol/acetic acid (100:5:1) to yield 5 mg (16%) of
the title compound.
lH NMR (300 MHz, CD30D, ppm~: ~ 0.9-1.0 (t, 3~),
1.6-1.7 ~m, 2H), 2.65 (s, 3H), 2.8-2.9 (t, 2H), 5.5
(s, 2H), 6.8 (s, 1~), 6.95-7.1 (m, 5H), 7.1-7.2 (d,
lH), 7.3-7.4 (m, 2~), 7.45-7.55 (m, 2H), 8.2 (d, lH).
~AB-MS: m/e 440 (M+l).
F~Ample 15
7-Methyl-2-propyl-3-t4-(1-(tetrazol-5-yl)-1-(2-chloro-
phenyl~metho~yphenyllmethyl-3H-imidazor4 5-blpyridine
Step A: Preparation of 2-(4-methylpheno~y)-2-(2-
chlorophenyl)acetamide
To a solution of 0.700 g (2.41 mmol) of
methyl 2-(2-chlorophenyl)-2-(4-methylpheno~y)acetate
(Example 4, Step ~) in 10 mL of methanol, ~tirred at
WO91/11~ PCT/US91/~957
20~5627
-221-
0C (ice-bath) was added anhydrous NH3 for 45 minutes.
The reaction mixture wa~ warmed to room temperature
and stirred o~ernight. The reaction mixture was then
concentrated Ln v~c--o and afforded O.658 g (99%) of
title compound.
lH NMR (300 M~z, CD30D, ppm): ~ 2.25 (s, 3H), 6.0 (s,
lH), 6.8-6.9(d, 2H), 7.0-7.1 (d, 2H), 7.25-7.35 (m,
2H), 7.4-7.5 (m, lH), 7.55-7.6 (m, lH)
FAB-MS: m/e 276, 278 (M+l, 3:1 ratio).
Step ~: Preparation of 2-(4-methylphenoxy)-2-(2~-
chlorophenyl)aceto~itrile
The product of Step A (0.650 g, 2.36 mmol)
was added to phosphorous oxychloride (5.95 mL, 27 eq)
at 0C followed ~y slow addition of 0.73 mL (2.2 eq)
of triethylamine. The reaction mixture was warmed to
room temperature for 10 minutes, refluxed for 50
minutes and then cooled to room temperature. The
reaction mixture was concentrated in vacuo, and
partitioned between ice water and toluene. The
aqueous layer was extracted 3 times with toluene, and
the combined toluene extracts were washed with 0.5 N
NaOH and H20. The crude product was concentrated
~a vacuo and purified on a silica gel flash chromato-
graphy column (140 x 30 mm) eluted with 2% ethylacetate/hexane to afford 0.555 g (9lZ) of the title
compound.
H NMR (300 M~z, CD30D, ppm): ~ 2.35 (s, 3H), 6.15
(s, lH), 6.95-7.05 (d, 2H), 7.1-7.2 (d, 2H), 7.4-7.6
(m, 3H), 7.8-7.9 (m, lH).
~I-MS: m/e 257,259 (M+, 3:1 ratio).
WO 91/11~ ~ 07 ~ 6~ PCT/US91/00957
-222-
Step C: Preparation of 2-(4-bromomethylphenoxy)-2_
(2-chlorophe~yl)~cetonitrile
To a CC14 solution (5 mL) of the product
(0.200 g) of Step B was added NBS (0.132 g, 0.95 eq)
and a catalytic amount of AIBN. The reaction mi~ture
was refluxed overnight. TLC indicated clean conver-
sion to product, and the reaction mixture was cooled
to room temperature and concentrated ~n VACUO. The
crude product was purified on a ~ilica gel flash
chromatography column (120 x 30 mm) eluted with 5%
ethyl acetate in hexane to afford 0.178 g (72%) of
the title compound.
lH NMR (300 M~z, CDCl3 ppm): ~ 4.5 (s, 2H), 6.2 (s,
1~), 7.0S-7.15 (d, 2H), 7.35-7.55 (m, 5H), 7.8-7.9
(m, lH).
Step D: Preparation of 3-[4-(l-cyano-1-(2-chloro-
phenyl)methoxyphenyl~methyl-7-methyl-2-
~ropyl-3~-imidazor4.5-blpyridine
To a DMF suspension (0.5 mL) of NaH (7.5 mg,
0.252 mmol) under N2 was added 0.040 g (0.23 mmol) of
7-methyl-2-propylimidazO~4,5-b]pyridine (Example 2,
Step B) and the reaction mixture was ~tirred at room
temperature for 1.5 hours. To the resulting sodium
salt was added the product of Step C (84 mg, 1.1 eq)
and the reaction mixture was stirred for 5 hours.
The reaction mixture was then guenched with saturated
NH4Cl and concentrated in vacuo The crude product
was purified on a silica gel flash chromatography
column (130 x 20 mm) eluted with 30% ethyl
acetate/hexane to afford 24 mg (24%) of the title
compound.
WO91/11~ PCT/US91/~957
2 0 1~ 6 2 7
-223-
H NMR (300 MHz, CDC13, ppm): d 0.9-1.0 (t, 3H),
1.7-1.85 (m, 2H), 2.7 (s, 3H), 2.75-2.85 (t, 2H),
5.45 (8, 2H), 6.15 (6, lH), 6.95-7.05 (m, 3H),
7.1-7.2 (d, 2H), 7.35-7.5 (m, 3H), 7.75-7.85 (m, lH),
8-2 (d, lH).
FAB-MS: m/e 431 (M+l).
Step E: Preparation of 7-methyl-2-propyl-3-t4-(1-
(tetrazol-5-yl)-1-(2-chlorophenyl)methoxy-
phenyllmethyl-3~-imidazor4.5-blpyridine
To a toluene solution (1 mL) of the product
of Step D (24 mg, 0.056 mmol) was added trimethyl-
stannyl azide (14 mg, 1.2 eq) and the mixture was
refluxed for 48 hours. The reaction mi~ture was then
concentrated in vacuo and purified on a silica gel
flash chromatography column (130 x 15 mm) eluted with
CHC13/MeOH/HOAc (100:3:1) to afford 13 mg (50%) of
the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ 0.85-0.95 (t, 3H),
2.55-2.7 (m, 2H), 2.65 (s, 3H), 2.8-2.9 (t, 2H), 5.5
(s, 2~), 6.9-7.0 (d, 2H), 7.05-7.2 (m, 4H), 7.3-7.5
(m, 3H), 7.55-7.65 (m, lH), 8.2 (d, lH).
~AB-MS: m/e 474, 476 (M~l).
2~ le 16
7-Methyl-2-propyl-3-t4-(2-phenyl-2-(tetrazol-5-yl)-
ethyl~phenyllmethyl-3H-imidazor4.5-blpyridine
WO91/11~9 PCT/US91/~ss7
2~)75627 - -
-224-
Step A: Prepar~tio~ of 4-(bromo~ethyl)benzyl Alcoh
A suspension of 4-bromomethylbenzoic acid
(5.04 g, 23.3 mmol) in TEF (30 mL) was cooled to 0-C
and treated with borane/TEF (35 mmol). The ice bath
was removed and the mixture was allowed to warm to
room temperature and stirred for 1.5 hours. The
excess borane was quenched first with MeOH, then with
water. The reaction mixture was then concentrated
in vacuo and redis~olved in ethyl acetate. The ethyl
acetate layer was washed with 5% HCl, water, NaHCO3,
brine, dried (MgSO4), filtered, and evaporated
in vacuo to afford 4.44 g (94~Z) of the titled product.
H NMR: (300 MHz, CDC13,ppm): d 7.38 (q, 4H), 4.70
(s, 2H), 4.51 (s, 2H).
FAB MS: m/e 202 (M+l).
Step B: Preparation of 4-(bromomethyl)-1-(tert-butyl-
dimethylsilylo~ymet~yl)benzene
To a solution of the product of Step A (4.44
g. 22.1 mmol) in CH2C12 were added N,N-diisopropyl-
ethylamine (1.2 eq.), 4-dimethylaminopyridine (0.1
eq), and tert-butyldimethylsilyl chloride (1.2 eq).
The reaction mixture was stirred for 1.5 hours at
room temperature, then concentrated Ln v~cuo. The
2~ residue was dissolved in ethyl acetate and washed
with water, brine, dried (MgSO4), filtered, and
concentrated La vacuo. The residue was chromatogra-
phed on silica gel eluted with 2.5Cb ethyl acetate/
hexane to afford 5.0 g (71 %) of the titled product.
lH NMR (300 MHz, CDC13, ppm): ~ 7.34 (q, 4H), 4.74
(s, 2H), 4.59 (s, 2H), 0.95 (s, 9H), 0.11 (s, 6H).
-
WO91/11~ PCT/US91/~9~7
2075~27
-225-
Ste~ C: Preparation of 3-~4-(tert-butyldimethyl-
silyloxymethy~henyll-2-~henyl~ropanonitrile
A solution of phenylacetonitrile (1.5 mL,
12.7 mmol) in T~F (40 mL) containing EMPA (11 mL,
63.4 mmol) was cooled to -78-C and treated with 16 mL
(16 mmol) of a 1.0 M TEF solution of lithium
bis(trimeth-
ylsilyl)amide dropwise while the temperature was
maintained at -78C. The reaction was stirred at
-780C for 1.5 hours, and a solution of the product of
Step B (2.00 g, 6.34 mmol) in THF (8 mL) was added
dropwise with the temperature maintained below -70cC.
The reaction temperature uas maintained below -68C
for 3 hours. The reaction was quenched ~t this
temperature by addition of 1 N NaHS04. After warming
to room temperature, the mixture was partitioned
between EtOAc and water, and the combined organic
layers were washed with water, ~aturated NaRC03,
brine, dried (MgS04), filtered, then concentrated
in vacuo. The residue was chromatographed on a
silica gel flash chromatography column eluted with 5%
ethyl acetate/he~ane to afford 1.5 g (67%) of the
title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 7.40-7.30 (m, 3H),
7.30-7.22 (m, 4H), 7.10 (d, 2~), 4.73 (s, 2~), 3.98
(t, lH), 3,23-3.08 (m, 2H), 0.94 (s, 9H), 0.10 (s,
6~).
~AB MS: m/e 294 (M+ less tert-Bu).
WO91/11~ PCT/US91/~957
207~627
-226-
Step D: Preparation of 3-t4-(bromomethYl)phenyl]-
?-phenylpro~no~itrile
To a cooled ~0C) colution of 1.5 g ( 4.27
mmol) of the product of Step C dissolved in 20 mL of
acetonitrile, was added 2.12 g (6.4 mmol) of carbon
tetrabromide, and 1.68 ~ (6.40 mmol) of triphenyl-
phosphine. The reaction mixture was ~tirred 30
minutes at 0C, then allowed to warm to room tempera-
ture and 0.5 mL (6.4 mmol) of acetone was added. The
reaction mixture was stirred an additional 16 hours
at room temperature, then filtered and e~aporated
in ~acuo. The residue was purified on a silica gel
flash chromatography column eluted with 5% ethyl
acetate/hexane to afford 0.~75 g (4~%) of the title
Compound.
lH NMR (300 M~z, CDC13, ppm): ~ 7.48-7.10 (m, ~H),
4.50 (s, 2H), 4.00 (t, lH), 3.26-3.10 (m, 2H).
FAB MS: m/e 299, 301 (M+l).
Step F: Preparation of 3-~4-(2-cyano-2-phenylethyl)-
phenyl~methyl-7-methyl-2-propyl-3~-imida~o
r4.5-blpyridine
To a solution of 7-methyl-2-propylimidazo
t4,5-b]pyridine (106 mg, 0.61 mmol) in DMF (3 mL) was
added NaH (0.91 mmol). The suspension was stirred at
room temperature for 30 minutes, at which time a
solution of the product of Step D (200 mg, 0.667
mmol) in DMF (2 mL) was added. The mi~ture was
stirred for 2 hours at room temperature, quenched
with water, and then concentrated in vacuo. The
residue was partitioned ~etween water and EtOAc, and
WO91/11~ PCT/US91/009~7
.
20756~7
-227-
the combined organic layer~ were washed with brine,
dried (R2C03), filtered, and evaporated Ln vAcuo.
The residue was chromatographed on a silica gel flash
chromatography column eluted with 50% ethyl acetate/
hexane to afford 46 mg (19%) of the title compound.
1~ NMR (300 M~z, CD30D, ppm): ~ con~istent with
structure.
FAB MS: m/e 395 (M+l).
Step F: Preparation of 7-Methyl-2-propyl-3-[4-(2-
phenyl-2-(tetrazol-5-yl)ethyl)phenyl]methyl-
3~-imidazor4.5-blpyridine
To a solution of the product of the product
of Step E (46 mg, 0.12 mmol) in toluene (2 mL) was
added 29 mg (0.14 mmol) of trimethylstannyl azide and
the reaction mixture was refluxed for 24 hours. The
reaction mixture was concentrated ~B vacuo. The
residue was dissolved in T~F and treated with 12 N
~Cl (5 drops) for 5 minutes at room temperature. The
miæture was concentrated ~n vacuo and purified on a
silica gel flash chromato~raphy column eluted with
C~C13/MeO~/NH40H (80:20:2) to afford 19.6 mg (38%) of
the title compound.
1~ NMR (300 M~z, CD30D, ppm): ~ 8.18 (d, lH),
7.28-7.12 (m, 6~), 7.05 (d, 2H), 6.93 (d, 2H), 5.48
(s, 2H), 4.58 (t, lH), 3.59-3.49 (m, 1~), 3.38-3.30
(m, 1~), 2.78 (t, 2~), 2.65 (s, 3~), 1.68-1.52 (m,
2H), 0.89 (t, 3~).
FAB MS: m/e 438 (M+l).
WO91~ PCT/US91/~gs7
20756~
-228-
F.~A~ple 17
3-~4-(l-carbo2y-l-phenoxy)methylphenyl]methyl-7-
~ethyl-~-propyl-3~-imidazor4.5-blpyridine
Step A: Preparation of methyl 2-bromo-4'-methyl-
phenylacetate
A mixture of 4-methylphenylacetic acid (5.00
g, 33.3 mmol) and thionyl chloride (2.67 mL, 36.6
mmol) were heated to reflux. Bromine (1.51 mL, 29.3
mmol) was added dropwise to the reaction mixture over
10 minutes and then the mixture was refluxed overni~ht
(17 hours). The reaction mixture was cooled to room
temperature and 34 mL of methanol was added slowly.
The reaction mixture was concentrated ~n ~acuo and
chromatographed on silica gel (45 x 120 mm) eluted
with 2% ethyl acetate/hexane to afford 3.18 g (37%)
of the titled compound.
lH NMR (300 M~z, CDC13, ppm): d 2.35 (s, 3~), 3.8 (s,
3H), 5.35 (s, 1~), 7.1-7.2 (d, 2E), 7.4-7.5 (d, 2H).
FAB-MS: m/e 243, 241 (M+l, 1:1 ratio).
Step B: Preparation of methyl 2-pheno2y-2-(4-methyl-
phenyl)acetate
To a suspension of ~H (244 mg, 2.13 mmol) in
2 mL of DM~ at room temperature under N2 was added a
DMF solution (1 mL) of phenol (200 mg, 2.13 mmol).
When the KH had completely dissolved, the reaction
mi2ture was cooled to 0C. A DMF solution (1 mL) of
the product of Step A (517 mg, 2.13 mmol) was then
added by syringe- After the addition was complete,
- - - - - - - - - - -
WO91/11~9 PCT/US91/~957
207~627
. .
-229-
the reaction mixture was allowed to warm to room
temperature, and was then stirred for 2 hours. The
reaction mixture was quenched with saturated N~4Cl
and concentrated ~n ~cuo. The residue was purified
on a silica gel flash chromatography column (130 x 40
mm) eluted with 5X ethyl acetate/hexane to afford
0.126 g (23%) of the title compound.
H NMR (300 MHz, CDC13, ppm): ~ 2.35 (s, 3H), 3.75
(s, 3H), 5.65 (s, lH), 6.9-7.0 (m, 3H), 7.1-7.3 (m,
lo 4H), 7.4-7.5 (d, 2H).
EI-MS: m/e 256 (M+).
Step C. Preparation of methyl 2-phenoxy-2-(4-bromo-
methylphenyl)acetate
To a CC14 solution (1 mL) of the product of
Step B (126 mg, 0.495 mmol) was added N-bromosuccin-
imide (44 mg, 246 mmol) and a catalytic amount of
AIBN. The solution was refluxed for 30 minutes and
then concentrated Ln vacuo. The residue was purified
on a silica gel flash chromatography column (150 x 30
mm) eluted with 5% ethyl acetate/hexane to afford 27
mg (33%) of the title compound.
lH NMR (300 M~z, CDC13 ppm): ~ 3.75 (s, 3~), 4.5 (s,
2H), 5.65 (s, lH), 6.9-7.0 (m, 3H), 7.25-7.35 (m,
2~), 7.4-7.5 (d, 2H), 7.55-7.6 (d, 2H).
EI-MS: m/e 334, 336 (M~, 1:1 ratio).
Step D. Preparation of 3-[4-(1-carbomethoxy-1-
phenoxy)methylphenyl]methyl-7-methyl-2-
propyl-3H-imidazor4.5-blpyridine
To a DMF suspension (0.1 mL) of NaH (2.4 mg;
0.081 mmol) under N2 was added 1.0 eq. of 7-methyl-2-
WO91/11~ PCT/US91/~gs7
- 207~627
-230-
propylimidazo~4,5-b]pyridine (14 mg). After 15
minutes, the Na~ had completely reacted. The reaction
mixture was then treated with a DMF solution (0.4 mL)
of the product of Example 9, Step C (27 mg, 0.081
mmol). The reaction mixture was stirred for 5 hours,
and then concentrated in vacuo. Th~ crude product
was chromatographed on silica gel (140 x 20 mm)
eluted first with 300 mL of 30% ethyl acetate/hexane,
then with 50% ethyl acetate/hexane to afford 5.2 mg
(15%) of the titled compound.
lH NMR (300 MHz, CDC13, ppm): ~ O.9-1.0 (t, 3H),
1.65-1.85 (m, 2~), 2.7 (5, 3H), 2.75-2.85 (t, 2~),
3.7 (s, 3~), 5.5 (s, 2H), 5.65 (s, 1~), 6.9-7.1 (m,
4~), 7.1-7.2 (m, 2H), 7.2-7.3 (m, 2~), 7.5 (d, 2~),
8-2 (d, lH).
Step E: Preparation of 3-t4-(1-carboxy-1-phenoxy)-
methylphenyl3methyl-7-methyl-2-propyl-3~-
imidazo-r4.5-blpyridine
To a methanol solution (50 uL) of the product
of F.xample 9, Step D (5.2 mg, O.012 mmol) was added 1
N NaO~ (12.1 uL, 1.0 eq). The hydrolysis was complete
after stirring for 4 days. The reaction mixture was
chromatographed on silica gel (140 x 10 mm) eluted
with ethyl acetate/hexane/acetic acid (30:20:1) to
afford 3.0 mg (57%) of the title compound.
H NMR (300 MHz, CD30D, ppm): ~ O.9-1.0 (t, 3E),
1.6-1.8 (m, 2~), 2.65 (s, 3H), 2.8-2.9 (t, 2H), 5.4
(s, 1~), 5.6 (s, 2~), 6.8-7.0 (m, 3~), 7.1-7.3 (m,
5H), 7.55-7.65 (d, 2H), 8.2 (d, 1~).
FAB-MS: m/e 416 (M+l).
WO91/11~9 PCT/US91/~9~7
207~627
-231-
F~ le 18
3-[4-(1-Carboxy-1-(2-methylphenyl))metho~yphenyl]-
methyl-7-methyl-2-propyl-3~-imidazor4.5-bl~yridine
General procedure for the synthesis of 2-bromophenyl-
acetic esters from benzaldehydes (Steps A-C):
Step A: Preparation of 2-trimethylsilylo~y-2-(2-
methyl~henyl~acetonitrile
To a solution of 1.00 g (8.33 mmol) of
2-methylbenzaldehyde dissolved in 20 mL of dichloro-
methane was added 1.33 mL (10.0 mmol) trimethylsilyl-
cyanide, 1-2 mg of potassium cyanide, 1-2 mg of
18-crown-6, and the reaction mixture was stirred at
room temperature for 3 hours. The reaction mixture
was then diluted into diethyl ether, washed with 5%
NaHC03, brine, dried (MgS04), filtered and evaporated.
The residual oil was used directly in the next step.
Step B: Preparation of ethyl 2-hydroxy-2-(2-methyl-
~henyl)acetate
To a stirred 0C (ice-water bath) solution
of 1.83 g (8.35 mmol) of the product of Step A
dissolved in 10 mL of ethanol was introduced a slow
stream of anhydrous hydrogen chloride gas. After 5
minutes the hydrogen chloride was turned off and the
reaction mixture was stoppered and stirred at room
temperature 14 hours. The reaction was then poured
into ice-water and extracted into chloroform. The
chloroform solution was filtered through a 60 mL
WO91/11~ PCT/USgl/~9~7
,,. ~ ..
20~5~æ7
-232-
sintered funnel filled with silica gel and the silica
gel was wa~hed with additional chloroform. The
combined filtrate was evaporated ~n VACUO to afford
0.437 g (27%) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 1.15-1.25 (t, 3~),
2.40 (s, 3~), 3.35-3.45 (d, lH), 4.05-4.30 (m, 2H),
5.30-5.35 (d, 1~), 7.05-7.20 (m, 4~).
Step C: Preparation of ethyl 2-bromo-2-(2-methyl-
~henyl)acetate
To a cooled (0C) solution of 0.425 g (2.19
mmol) of the product of Step B dissolved in 10 mL of
dichloromethane was added 0.717 g (2.74 mmol) of
triphenylphosphine followed by 0.908 g ( 2.74 mmol)
of carbon tetrabromide. After 30 minutes the
reaction was allowed to warm to room temperature and
stirring was continued for 2 hours. The raction
mixture was evaporated Ln V~CUQ and the residue was
purified on a silica gel flash chromatography column
eluted with 5% ethyl acetatelhexane to afford 0.373 g
(66~/c) of the title compound.
1~ NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3~),
2.40 (s, 3~), 4.15-4.30 (m, 2H), 5.60 (s, 1~),
7.10-7.25 (m, 3~), 7.55-7.65 (m, 1~).
WO91/11~ PCT/US91/~957
2~75~27
-233-
General procedure for the al~ylation of i~idazo~4,5-b]
pyridi~es with 2-~romophenylacetic esters:
Step D: Preparation of 3-[4-(1-carbomethoy )-1-(2-
methylphenyl)methoxyphenyl~methyl-7-methyl-
2-progyl-3~-imid~zor4.5-bl~yridi~e
To a suspension of 37 mg (0.32 mmol) of a
35% oil dispersion of potassium hydride in 0.5 mL of
DMF was added 0.090 g (0.32 mmol) of 3-(4-hydroxy-
phenyl)methyl-7-methyl-2-propyl-3H-imidazot4,5-b]
pyridine (Example 2, Step D) and the reaction was
stirred under an N2 atmosphere. After stirring for
15 minutes, 0.085 g of 18-crown-6 was added followed
by addition of a solution of 0.090 g (0.35 mmol) of
the product of Step C dissolved in 0.75 mL of DMF.
The reaction mi~ture was stirred for 4 hours, then
concentrated in ~acuo. The residue was purified on a
silica gel flash chromatography column eluted with
40% ethyl acetate/hexane to afford 0.099 g (68%) of
the title compound.
1~ NMR (300 M~z, CDC13, ppm): ~ O.90-1.00 (t, 3E),
1.15-1.25 (t, 3H), 1.65-1.80 (m, 2E), 2.45 (s, 3~),
2.65 (s, 3H) 2.70-2.80 (t, 2H), 4.05-4.25 (m, 2H),
5.35 (s, 2~), 5.75 (s, 1~), 6.75-6.85 (d, 2~),
2s 6.95-7.05 (m, 3~), 7.10-7.25 (m, 3~), 7.45-7.55 (m,
1~), 8.15-8.20 (d, 1~).
~AB-MS: m/e 458 (M+l).
WO9l~ll ffl 2 0 ~ 5 1~ 2 7 Pcr/usgl/oo9s7
-234-
General procedure for ester hydrolysis:
Step F: Preparation of 3-t4-(1-carbo~Y)-1-(2-methyl-
phenyl)methoxyphenyl~methyl-7-methyl-2-
propyl-3~-imidAzor4.5-blpyridi~e
To a solution of 0.097 g (0.21 mmol) of the
product of Step D dissolved in 3 mL of ethanol was
added 1 mL of a 1 N NaOH solution. The reaction
mixture was stirred at room temperature for 1.5
hours, neutralized to pH 7 with 1 N ECl and then
concentrated Ln ~acuo. The residue was purified on a
silica gel flash chromatography column eluted with
C~C13tMeOH/NH40R (80:15:1) to afford 0.076 g (84Z) of
the title compound.
lH MMR (300 M~z, CDC13, ppm): ~ O.90-1.00 (t, 3~),
1.65-1.75 (m, 2H), 2.50 (s, 3H), 2.70 (s, 3H),
2.85-2.95 (t, 2~), 5.50 (s, 2~), 5.85 (s, 1~),
6.85-6.95 (d, 2H), 7.05-7:15 (d, 2H), 7.15-7.25 (m,
4H), 7.45-7.55 (d, lR), 8.20-8.25 (d, 1~).
FAB-MS: m/e 430 (M~l).
Fx~le 19
3-[4-(1-Carboxy-1-(2-ethoxyphenyl))methoxyphenyl]-
methyl-7-methyl-2-~ropyl-3R-i~idazor4.5-blpyridine
Step A: Preparation of ethyl 2-bromo-2-(2-ethoxy-
phenyl~acetate
Using the general procedure for the synthesis
of 2-bromophenylaCetic esters from benzaldehydes
(Steps A-C, Example 18), 1.00 g (6.67 mmol) of
WO 91/119g9 PCI/US91/009s7
207562~
-235-
2-etho~ybenzaldehyde was converted to 0.291 g (1.01
mmol) of the title compound in 15% overall yield.
H MMR (300 MHz, CDC13, ppm): d 1.30-1.40 (t, 3H),
1.45-1.55 (t, 3H), 4.00-4.10 (m, 2H), 4.15-4.30 (m,
2H), 5.85 (s, lH), 6.80-6.85 (d, lE), 6.90-7.00 (t,
lH), 7.00-7.30 (t, lH), 7.55-7.65 (d, lH).
Step B: Preparation of 3-t4-(l-carbomethoxy)-1-(2-
ethoxyphenyl)metho~yphenyl]methyl-7-methyl-
2-~ropyl-3~-imidazor4.5-blpyridine
Using the general procedure for the
alkylation reaction described in Step D of Example
18, 0.089 g (0.32 mmol) of 3-(4-hydroxyphenyl)methyl-
7-methyl-2-propyl-3H-imidazot4,5-b~pyridine (Example
2, Step D) was alkylated with 0.100 g (0.35 mmol) of
the product of Step A, to afford 0.107 g (69%) of the
title compound.
H NMR (300 M~z, CDC13, ppm): ~ O.85-0.95 (t, 3H),
1.10-1.20 (t, 3H), 1.30-1.40 (t, 3H), 1.60-1.76 (m,
2H), 2.65 ts, 3H), 2.70-2.80 (t, 2H), 4.00-4.25 (m,
4H), 5.35 (s, 2H), 6.05 (s, lH), 6.80-7.05 (m, 6H),
7.20-7.30 (m, 2H), 7.40-7.50 (d, lH), 8.15-8.20 (d,
lH).
2s Step C: Preparation of 3-t4-(1-carbo~Y)-1-(2-ethoxy-
phenyl)methoxyphenyl]methyl-7-methyl-2-
propyl-3H-imidazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.107 g
(0.22 mmol) of the product of Step ~ was converted to
0.087 g (86%) of the title compound.
WO91/11~9 PCT/US91/009~7
20756~
-236-
lH NMR (300 M~z, CD30D, ppm): ~ O.90-1.00 (t, 3H),
1.35-1.45 (t, 3H), 1.50-1.65 (m, 2~), 2.70 (s, 3H),
2.80-2.90 (t, 2H), 4.05-4.20 (m, 2H), 5.50 (~, 2H),
6.05 (~, lH), 6.90-7.05 (m, 3H), 7.05-7.10 (m, 3H),
7.15-7.20 (d, lH), 7.25-7.35 (t, lH), 7.45-7.50 (d,
lH), 8.20-8.25 (d, 1~).
FAB-MS: m/e 460 (M+l).
F.~ ple 20
3-[4-(1-Carboxy-1-(2-(1-hexyloxy)phenyl))methoxy-
phenyl]methyl-7-methyl-2-propyl-3H-imidazot4,5-b]
pyr~dlne
Step A: Preparation of ethyl 2-bromo-2-(2-(1-
hexyloxy)phenyl)acetate
Using the general procedure for the synthesis
of 2-bromophenylacetic esters from benzaldehydes
(Steps A-C, Example 18), 0.50 g (2.43 mmol) of
2-hexyloxybenzaldehyde was converted to 0.088 g (0.26
mmol) of the title compound in llZ overall yield.
lH NMR (300 MHz, CDC13, ppm): ~ O.85-0.95 (t, 3~),
1.20-1.55 (m, 9H), 1.75-1.85 (m, 2H), 3.95-4.05 (t,
2H), 4.15-4.30 (m, 2H), 5.85 (s, 1~), 6.80-6.85 (d,
lH), 6.90-7.00 (t, 3H), 7.20-7.30 (t, lH), 7.55-7.65
(d, lH).
Step B: Preparation of 3-t4-(1-carbomethoxy)-l-(2-(1-
hexyloxy)phenyl)methoxyphenyl]methyl-7-
methYl-2-propyl-3~-imidazor4~5-blpyridine
WO91~ PCT/USgl/~957
2075~7
-237-
Using the general procedure for the
alkylation reaction described in Step D of E~ample
18, 0.065 g (0.23 mmol) of 3-(4-hydroxyphenyl>methyl-
7-methyl-2-propyl-3H-imidazo[4,5-b]pyridine (Example
2, Step D) was alkylated with 0.088 g (0.25 mmol) of
the product of Step A, to afford 0.090 g (71Z) of the
title compound.
lH NMR (300 MHz, CDC13, ppm): d 0.80-0.90 (t, 3~),
0.90-1.00 (t, 3H), 1.15-1.50 (m, 9H), 1.65-1.80 (m,
lo 4H), 2.65 (s, 3~), 2.70-2.80 (t, 2H), 3.95-4.05 (t,
2E), 4.05-4.25 (m, 2H), 5.35 (s, 2~), 6.05 (s, 1~),
6.80-7.05 (m, 6H), 7.20-7.30 (m, 2~), 7.40-7.45 (d,
lH), 8.15-8.20 (d, lH).
Step C: Preparation of 3-[4-(1-carboxy)-1-(2-(1-
hexyloxy)phenyl)methoxyphenyl3methyl-7-
methyl-2-~ropyl-~-imidazor4.5-bl~yridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.090 g
(0.17 mmolj of the product of Step B was converted to
0.072 g (85%) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 0.80-0.90 (t, 3H),
0.90-1.00 (t, 3H), 1.20-1.35 (m, 4~, 1.50-1.60 (m,
2H), 1.60-1.85 (m, 2H), 2.65 (s, 3~), 2.80-2.90 (t,
2s 2H), 3.95-4.05 (m, 2~), 5.50 (s, 2H), 6.05 (s, 1~),
6.90-7.05 (m, 4~), 7.05-7.15 (d, 2H), 7.15-7.20 (d,
1~), 7.25-7.35 (t, lH), 7.40-7.45 (d, lH), 8.20-8.25
(d, 1~).
FAB-MS: m/e 516 (M+l).
wo 9~ ~g ~ ~ 7 ~ 6 2 ~ PCT/US91/00957
-238-
F.~A~le 21
3-t4-(1-Carboxy-1-(2-methoxyphenyl))methoxyphenyl3-
methyl-7-methyl-2-pro~yl-3~-imidazor4~5-b~pyridine
S
Ste~ A: Preparation of ethyl 2-bromo-2-(2-methoxy-
~henyl)acetate
Using the general procedure for the synthesisof 2-bromophenylacetic esters from benzaldehydes
(Steps A-C, Example 18), 1.00 g (7.35 mmol) of
2-methoxybenzaldehyde was converted to 0.736 g (2.69 -
mmol) of the title compound in 37% overall yield.
lH MMR (300 MHz, CDCl3, ppm): ~ 1.20-1.30 (t, 3H),
3.85 (s, 3H), 4.15-4.30 (m, 28), 5.85 (c, lH),
6.80-6.90 (d, lH), 6.90-7.00 (t, lH), 7.25-7.35 (t,
lH), 7.55-7.65 (d, lH).
Step B: Preparation of 3-[4-(1-carbomethoxy)-1-(2-
methoxyphenyl)methoxyphenyl3methyl-7-methyl-
2-propyl-3~ idazor4.5-blpyridine
Using the general procedure for the
alkylation reaction described in Step D of E~ample
18, 0.090 g (0.32 mmol) of 3-(4-hydroxyphenyl)methyl-
7-methyl-2-propyl-3H-imidazot4,5-b]pyridine (Example
2, Step D) was alkylated with 0.096 g (0.35 mmol) of
the product of Step A, to afford 0.126 g (83Z) of the
title compound.
lH NMR (300 MHz, CDC13, ppm): ~ O.90-1.00 (t, 3H),
1.15-1.25 (m, 3H), 1.65-1.80 (m, 2H), 2.65 (s, 3H),
2.70-2.80 (t, 2H), 4.05-4.25 (m, 2H), 5.35 (s, 2H),
6.05 (s, lH), 6.80-7.05 (m, 7H), 7.25-7.35 (m, lH),
7.45-7.50 (d, lH), 8.15-8.20 (d, lH~.
FAB-MS: m/e 474 (M+l).
WO91/11~ PCT/US91/~ss7
2075S27
-239-
Step C: Preparation of 3-~4-(1-carboxy)-1-(2-methoxy_
phenyl)methoxyphenyl]methyl-7-methyl-2-
pro~yl-3~-imid~zor4.5-bl~yridi~e
Using the general procedure for ester
hydrolysis descri~ed in Step F of Example 19, 0.123 g
(0.26 mmol) of the product of Step B was converted to
0.095 g (82%) of the title compound.
lH NMR (300 M~z, CD30D, ppm): ~ 0.90-1.00 (t, 3H),
1.60-1.80 (m, 2H), 2.60 (s, 3~), 2.80-2.90 (t, 2~),
lo 3-90 (s, 3H), 5.50 (s, 2H), 6.00 (s, lH), 5.90-7.15
(m, 6H), 7.15-7.20 (d, lH), 7.20-7.25 (t, lH),
7.45-7.55 (d, lH), 8.20-8.25 (d, lH).
FAB-MS: m/e 446 (M+l).
~ ple 22
3-~4-(1-Carboxy-l-(naphth-l-yl))methoxyphenyl~methyl-
7-methyl-2-propyl-3~-imidazor4.5-blpyridine
Step A: Preparation of ethyl 2-bromo-2-(naphth-1-
yl)aceta~e
Using the ~eneral procedure for the synthesis
of 2-bromophenylacetic esters from benzaldehydes
(Steps A-C, Example 18), 1.00 g (6.40 mmol) of l-naph-
thaldehyde was converted to 0.694 ~ (2.69 mmol) ofthe title compound in 37% overall yield.
lH MMR (300 M~z, CDC13, ppm) ~ 1.20-1.30 (t, 3H),
4.15-4.3S ~m, 2~), 6-15 (~, lH), 7.40-7.65 (m, 3H),
7.75-7.9S (m, 3~), 8.05-8.15 (d, 1~).
EI-MS: m/e 292, 294 (M+, 1:1 ratio).
` WO91/11~9 PCT/USgl/00957
~0~2~ -~
-240-
Step B: Preparation of 3-t4-(1-carbometho~y-1-
(naphth-l-yl)~methoxyphenyl]methyl-7-methyl-
2-~ropyl-3~-imidazot4.5-blpyridine
Using the general procedure for the al~yla-
tion reaction described in Step D of Egample 18, 0.090
g (0.32 mmol) of 3-(4-hydroxyphenyl)methyl-7-methyl-2-
propyl-3H-imidazot4,5-b~pyridine (Example 2, Step D)
was alkylated with 0.103 g (0.35 mmol) of the product
of Step A, to afford 0.132 g (84%) of the title
CmPound.
lH NMR (300 MHz, CDC13, ppm) ~ O.85-0.95 (t, 3H),
1.05-1.15 (t, 3H), 1.60-1.80 (m, 2H), 3.65 (s, 3H),
3.70-3.80 (t, 2~), 4.05-4.25 (m, 2H), 5.35 (s, 2H),
6.20 (s, lH), 6.80-6.90 (d, 2H), 6.95-7.05 (m, 3H),
7.40-7.60 (m, 3H), 7.65-7.75 (d, lH), 7.80-7.90 (m,
2H), 8.15-8.20 (d, 2H), 8.20-8.30 (d, lH).
FAB-MS: m/e 494 (M+l).
Step C: Preparation of 3-[4-(1-carboxy-l-(naphth-1-
yl))methoxyphenyl]methyl-7-methyl-2-propyl-
3H-imidazor4.5-bl~yridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.125 g
(0.25 mmol) of the product of Step B was cOnverted to
0.108 g (91%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ 0.90-1.00 (t, 3H),
1.50-1.75 (m, 2H), 2.70 (~, 3H), 2.80-2.90 (t, 2~),
6.50 (s, 2H), 6.20 (s, lH), 6.95-7.05 (d, 2H),
7.05-7.15 (d, 2H), 7.15-7.20 (d, lH), 7.45-7.55 (m,
3H), 7.80-7.95 (m, 2H), 8.15-8.25 (d, lH), 8.35-8.45
(d, lH).
FAB-MS: m/e 466 (M~l).
WO91/11~ - PCT/US91/~9S7
207~627
-241-
T~ple ~3
3-[4-(1-Carboxy-1-(3-methylnaphth-2-yl))methoxy-
phenyl]methyl-7-methyl-2-propyl-3~-imidazot4,5-b]
pyridi~e
Step A: Preparation of ethyl 2-bromo-2-(3-methyl-
naphth-2-yl)acetate
Using the general procedure for the synthesis
Of 2-bromophenylacetic esters from benzaldehydes
(Steps A-C, Example 18), 1.00 g (5.88 mmol) of
3-methyl-2-naphthaldehyde was converted to 0.953 g
(3.10 mmol) of the title compound in 53% overall
yield.
1~ NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3~),
2.55 (s, 3H), 4.20-4.40 (m, 2~), 5.60 (s, lH),
7.40-7.50 (m, 2H), 7.65 (s, 1~), 7.70-7.75 (d, 2H),
7.75-7.85 (d, 1~), 8.10 (s, 1~).
EI-MS: m/e 306, 308 (M+, 1:1 ratio).
Step B: Preparation of 3-t4-(1-carbomethoxy-1-(3-
methylnaphth-2-yl))methoxyphenyl]methyl-7-
methyl-2-propyl-3H-imidazor4.5-bl~yridine
Using the general procedure for the
alkylation reaction described in Step D of Example
18, 0.090 g (0.32 mmol) of 3-(4-hydroxyphenyl)methyl_
7-methyl-2-propyl-3H-imidazo[4,5-b]pyridine (Example
2, Step D) was alkylated with 0.108 g (0.35 mmol) of
the product of Step A, to afford 0.121 g (75/O) of the
title compound.
WO91/11~9 Z O ~ 5 6 2 7 PCT/US91/~9s7
-242-
1~ NMR (300 M~z, CDC13, ppm): ~ O.85-0.95 (t, 3E),
1.15-1.25 (m, 2H), 1.65-1.75 (m, 2H), 2.60 (~, 3H),
2.65 (s, 3H), 2.70-2.80 (t, 2H), 4.05-4.30 (m, 2H),
5.40 (s, 2H), 5.85 (s, lH), 6.80-6.90 (d, 2H),
6.95-7.05 (m, 3H), 7.35-7.45 (m, 2H), 7.65 (8, lH),
7.70-7.80 (m, 2H), 8.00 (s, lH), 8.15-8.20 (d, lH).
FAB-MS: m/e 508 (M+l).
Step C: Preparation of 3-[4-(1-carboxy-1-(3-
1o methylnaphth-2-yl))methoxyphenyl]methyl-7-
methyl-2-propyl-3H-imidazor4 5-bl~yridine
Using the general procedure for ester
hydrolysis described in Step F of Example 19, 0.090 g
~0.32 mmol) of the product of Step B was converted to
0.121 g (75/O) of the title compound.
H NMR (300 MHz, CD30D, ppm): ~ 0.85-0.95 (t, 3H),
1.60-1.75 (m, 2H), 2.60 (s, 3H), 2.54 (s, 3H),
2.80-2.90 (t, 2H), 5.50 (s, 2H), 5.85 (s, lH),
6.95-7.05 (d, 2H), 7.05-7.15 (d, 2H), 7.35-7.50 (m,
2H), 7.65 (s, lH), 7.75-7.85 (t, 2H), 8.00 (s, lH),
8.15-8.25 (d, lH).
~AB-MS: m/e
F.~A~P1 e 24
3-[4-(1-Carboxy-1-(2-methylphenyl))methoxyphenyl]-
methyl-5.7-dimethyl-2-ethYl-3~-imid_ZOr4.5-blpyridine
WO91/11~ PCT/US91/00957
207~27
-243-
Step A: Preparation of 2-nitramino-4,6-dimethyl-
~yridine
2-Amino-4,6-dimethylpyridine (10.0 g, 81.8
mmol) was added portion-wise to 65 mL of ~2S04 (conc.
d=1.84) which was stirred (mechanical) at 0C. After
complete addition, the mixture was warmed to room
temperature until the mixture became homogeneous.
The solution was then cooled to -10C and a pre-cooled
(0C) mixture of conc EN03 (11.5 mL, d = 1.40) and
H2S04 (8.2 mL, d = 1.84) was added at such a rate as
not to raise the internal reaction temperature above
-9C. Ten minutes after the addition was complete
this cooled (-10C) mixture was poured onto 400 g of
crushed ice. The resulting slurry was neutralized by
the addition of conc N~40H (to p~ 5.5) while cooling
(ice bath). The solid was isolated by filtration,
and dried at room temperature to give 13.3 g of the
title compound as a white solid.
Step B: Preparation of 2-amino-3-nitro-4,6-dimethyl-
pyridine
To 75 mL of stirred conc H2S04 cooled to
-5C (ice-salt bath) was added 4,6-dimethyl-2-
nitraminopyridine (13.2 g, 79 mmol) portion-wise at
such a rate as to maintain the internal temperature
below -3C. The mixture was warmed to O-C until
homogeneous (30 minutes) at which time tlc (SiO2, 1:1
EtOAc/hexanes on a N~40H neutralized aliquot)
indicated that the rearrangement was complete. The
mixture was poured onto 400 g of crushed ice and the
pH was adjusted to 5.5 by the addition of conc
WO91/11~ PCT/USgl/00957
20~562~
-244-
NH40H. The resulting yellow slurry was cooled to
0C, filtered, washed with cold water (50 mL), and
dried at room temperature to gi~e 10.32 g of a
mixture of the title compound and the 5-nitro isomer
in a 55:45 ratio (determined by 1~ N~R). Thi8 mixture
was used directly in the next step.
Ste~ C: Preparation of 5,7-dimethyl-2-ethylimidazo
r4~5-bl~yridine
To a mixture of 8.44 g of a 55:45 mixture of
2-amino-3-nitro-4,6-dimethylpyridine and 2-amino-5-
nitro-4,6-dimethylpyridine in MeOH (1.2 L) was added
10 % Pd/C (2.4 g). The reaction vessel was evacuated
then purged with ~2 at 1 atm. and stirred vigorously
for 18 h. Filtration through a celite pad, and
concentration gave 6.65 g of a mixture of 2,3-diamino-
4,6-dimethylpyridine and 2,5-diamino-4,6-dimethyl-
pyridine as a dark solid. To 5.40 g (39.4 mmol) of
this mixture was added propionic acid (B.80 mL, 118
mmol) followed by polyphosphoric acid (100 mL). This
stirred mixture was heated to 90C for 3 h then to
100C for 1 hour. The inside walls of the flask were
scraped with a spatula to assist dissolution of the
solids. After the reaction was complete, the warm
mixture was poured onto 300 g of ice and the mixture
was made basic with N~40H. The migture was egtracted
(4 x 50 mL CH2C12), dried (K2C03) and concentrated to
give a mixture of the title compound and 4,6-dimethyl-
2,5-bis(propionamido)pyridine. Purification (SiO2,
5% MeO~/ EtOAc) gave 1.66 g of the title compound as
the ~lower eluting component.
WO91/11~ PCT/US91/009s7
207~627
-245-
H NMR (CD30D, 300 MHz, ppm): ~ 6.95 (~, lH), 2.92
(q, J=7.8 Hz, 2H), 2.54 (apparent s, 6H), 1.40 (t,
J=7.8 Hz, 3H).
steP D: Preparation of 3-(4-(benzyloxy)phenyl)-
methyl-5,7-dimethyl-2-ethyl-3~-imidazo
r4.5-blpyridine
To a suspension of 0.503 g (12.5 mmol) of a
60% oil dispersion of sodium hydride in 20 mL of DM~
was added 2.0 g (11.4 mmol) of the product of Step C
and the mixture was stirred at room temperature.
After 25 minutes, 2.92 g (12.5 mmol) of 4-benzyloxy-
benzyl chloride and a catalytic amount of sodium
iodide were added and the reaction was stirred for an
additional 4 hours. The reaction mixture was then
partitioned between ethyl acetate and water. The
organic layer was separated, washed with water,
brine, dried (MgS04), filtered and evaporated. The
residual oil was purified on a silica gel flash
20- chromatography column eluted with 30% ethyl
acetate/hexane to afford 3.33 g (79%) of the title
compound.
lH NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3H),
2.55 (s, 3H), 2.60 (s, 3H), 2.70-2.80 (m, 2~), 5.00
(s, 2H), 5.35 (s, 2H), 6.80-6.90 (m, 3H), 7.00-7.10
(d, 2H), 7.25-7.45 (m, 5H).
FAB-MS: m/e 372 (M+l).
WO91/11~ PCT/US91/00957
~ .~
~o756~
-246-
Step ~.: Preparation of 5,7-dimethyl-2-ethyl-3-(4
hydro~yphenyl)methyl-3H-imidazo~4,~-b]
~yridine
A ~olution of 1.40 g (3.77 mmol) of the
product of Step D di~solved in 38 mL of methanol was
placed in a Parr hydrogenation flas~ and 0.140 g of
10% Pd/C catalyst was added. The reaction mixture
was placed in a Parr apparatus and roc~ed under a
hydrogen atmosphere (32 psig) for 2 hours. The
reaction mixture was then filtered through a plug of
~ilica gel eluted with 20Z methanol/chloroform to
remo~e the catalyst. Evaporation of the filtrate and
drying in vacuo afforded 0.650 g (61%) of the title
compound.
1~ NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3~),
3.64 (s, 3H), 3.66 (s, 3H), 2.85-2.95 (m, 2~), 5.45
(s, 2H), 6.70-6.80 (d, 2H), 6.95-7.05 (m, 3H).
FAB-MS: m/e 282 (M+l).
Step F: Preparation of 3-[4-(1-carboethoxy-1-(2-
methylphenyl)metho~yphenyl]methyl-5,7-
dimetbyl-2-ethyl-3H-imidazor4.5-blpyridine
To a ~uspension of 0.108 g (0.94 mmol) of
potassium hydride in 2 mL of DMF was added 0.264 g
(0 94 mmol) of the product of Step E and the reaction
mixture was stirred under an N2 atmosphere. After 10
minutes, 0.248 g (0.94 mmol) of 18-crown-6 and a
solution of 0.266 g (1.03 mmol) of ethyl 2-bromo-2-(2-
methylphenyl)acetate (Example 18, Step C) dissolved
in 1 mL DME were added and the reaction was then
stirred an additional 15 minutes. The reaction
WO91/11~ PCT/US91/009~7
207~627
-247-
mi~ture was then partitioned between ethyl acetate
and water. The organic layer was separated, washed
with water, brine, dried (MgS04), filtered and
evaporated. The residual oil was purified on a
silica gel flash chromatography column eluted with
40% ethyl acetate/hexane to afford 0.338 g (79%) of
the title compound.
H NMR (300 MHz, CDC13, ppm): ~ 1.10-1.20 (t, 3H),
1.20-1.30 (t, 3H), 2.45 (s, 3H), 2.55 (s, 3H), 2.60
lo (s, 3~), 2.70-2.80 (m, 2H), 4.05-4.25 (m, 2H), 5.35
(s, 2H), 5.70 (s, lH), 6.75-6.85 (d, 2H), 6.88 (s,
lH), 6.95-7.05 (d, 2H) 7.15-7.25 (m, 3H), 7.45-7.55
(d, lH).
FAB-MS: m/e 458 (M+l).
Step G: Preparation of 3-t4-(1-carbo~Y-1-(2-methyl-
phenyl)methoxyphenyl]methyl-5,7-dimethyl-2-
ethyl-3~-imidazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.060 g
(0.13 mmol) of the product of Step ~ was converted to
0.054 g (96%) of the title compound.
lH NMR (300 M~z, CD30D, ppm): ~ 1.20-1.30 (t, 3H),
2.50 (s, 3H), 2.60 (s, 3H), 2.65 (s, 3H), 2.85-2.95
2s (m, 2H), 5.50 (s, 2H), 5.85 (s, lH), 6.90-7.00 (d,
2H), 7.05-7.15 (m, 3H), 7.15-7.25 (m, 3H), 7.50-7.55
(d, lH).
~AB-~S: m/e 430 (M+l).
WO91/11~9 PCT~US91/~ss7
-.
2~7~2
-24~-
~ ple ~5
3-[4-(1-Carboxy-1-(2-chlorophenyl))metho~yphenyl]-
~ethyl-5.7-dimethy~-2-ethyl-3~ idazor4.5-bl~yridi~e
Step A: Preparation of 3-r4-(1-carboetho~y-1-(2-
chlorophenyl)methoxyphenyl]methyl-5,7-
dimet~yl-2-ethyl-3R-imidazor4.5-blpyridine
Using the procedure described in Step F of
Example 24, 0.050 g (0.18 mmol) of 5,7-dimethyl-2-
ethyl-3-(4-hydroxyphenyl)methyl-3~-imidazot4,5-b]
pyridine (Example 24, Step E) was al~ylated with
0.052 g (0.20 mmol) of methyl 2-bromo-(21-chloro)-
phenylacetate (Example 4, Step A) to afford 0.053 g
(64%) of the title compound.
Step B: Preparation of 3-~4-(1-carboxy-1-(2-chloro-
phenyl)methoxyphenyl~methyl-5,7-dimethyl-2-
ethyl-3~-imidazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.048 g
(0.10 mmol) of the product of Step A was converted to
0.036 g (78Z) of the title compound.
lH NMR (300 M~z, CD30D, ppm): ~ 1.20-1.30 (t, 3H),
2.60 (s, 3E), 2.63 (s, 3H), 2.85-2.95 (m, 2~), 5.50
(s, 2H), 6.05 (s, lH), 6.90-7.00 (d, 2H), 7.05-7.15
(m, 3H), 7.30-7.40 (m, 2R), 7.40-7.50 (m, lR),
7.60-7.65 (m, 1~).
~AB-MS: m/e 450, 452 (M+l, 3:1 ratio).
WO91/11~ PCT/US91/~9~7
- 2075627
-249-
~ ple 26
3-~4-(1-Carboxy-1-(2-bromophenyl))methoxyphenyl]-
methyl-5,7-dimet ~1-2-etl~yl-3~-imid~zor4.5-blpyridirle
s
Ste~ A: Preparation of methyl 2-bromo-2-(2-~romo-
~henyl~cetate
Commercially available 2-bromophenylacetic
acid (5.00 g, 23.3 mmol) was converted to 4.89 g
10 (68%) of the title compound in a procedure similar to
that described in Step A of Example 3.
1~ NMR (300 MHz, CDC13, ppm): ~ 3.80 (s, 3H), 5.90
(s, lH), 7.15-7.20 (t, lH), 7.20-7.25 (t, lH),
7.50-7.55 (d, lH), 7.70-7.75 (d, lH).
FAB-MS: m/e 306 ~ 308 ~ 310 (M+l, 1:2:1 ratio).
Step B: Preparation of 3-[4-(1-carbomethoxy-1-(2-
bromophenyl)methoxyphenyl]methyl-5,7-
dimethyl-2-ethyl-3~-imidazor4.5-blpyridine
Using the procedure described in Step F of
Example 24, 0.050 g (0.18 mmol) of 5,7-dimethyl-2-
ethyl-3-(4-hydroxyphenyl)methyl-3H-imidazot4,5-b]
pyridine (Example 24, Step E) was alkylated with
0.060 g (0.20 mmol) of the product of Step A to
afford 0.077 g (85Z) of the title compound.
lH NMR (300 MHz ~ CDC13, ppm): ~ 1.20-1. 30 (t, 3~) ~
2.55 (s, 3H), 2.60 (s, 3H), 2.65-2.75 (m, 2H), 3.75
(s, 3H), 5.35 (s, 2H), 6.05 (s, lH), 6.80-6.90 (m,
3H), 6.95-7.05 (d, 2H), 7.15-7.35 (m, 2H), 7.50-7.60
(d, 2H)
FAB-MS: m/e 508, 510 (M~ 1 ratio).
WO 91/11~9 2 ~ 7 5 6 2 7 PCT/US91/~957
-250-
Ste~ C: Preparation of 3-t4-(1-carboxY-1-(2-
bromophenyl)methoxyphenyl]methyl-5,7-
dimethyl-2-ethyl-~-imidazor4.5-bl~yridine
Using the general procedure for e~ter
hydrolysis described in Step E of E~ample 19, 0.077 g
(0.15 mmol) of the product of Step B was converted to
0.065 g (86%) of the title compound.
1~ NMR (300 MHz, CD30D, ppm): ~ 1.20-1.30 (t, 3~),
2.64 (s, 3H), 2.66 (s, 3H), 2.85-2.95 (m, 2H), 5.50
lo (s, 2~), 5.95 (s, 1~), 6.90-7.00 (d, 2~), 7.05-7.15
(m, 3H), 7.20-7.30 (t, lH), 7.30-7.40 (t, lH),
7.60-7.70 (m, 2H).
~AB-MS: m/e 494, 496 (M+l, 1:1 ratio).
F~ple 27
3-t4-(1-Carboxy-l-phenyl)methoxyphenyl]methyl-5,7-
dimethyl-2-ethyl-3~I-imidazor4.S-blpyridine
Step A: Preparation of methyl 2-(4-bromomethyl-
phenoxy)-2-~henylacetate
To a solution of 0.710 g (2.77 mmol) of
methyl 2-(4-methylphenogy)phenylacetate (Step A of
Example 13) dissolved in 10 mL of CC14, was added
0-494 g (2.77 mmol) of N-bromosuccinimide, and 25 mg
of AIBN (catalytic amount). The mi~ture wa~ stirred
and heated to reflux for 4 hours, then cooled to room
temperature and concentrated in vacuo. The residue
was purified on a silica gel flash chromatography
column (25 ~ 170 mm) eluted with 5% ethyl acetate/
hexane to afford 0.509 g (5~/.) of the title compound.
WO91/11~9 PCT/US91/009~7
20~5627
-251-
lH NMR (300 M~z, CDC13, ppm): ~ 3.75 (s, 3H), 4.5 (s,
2H), 5.65 (s, lH), 6.9-7.0 (d, 2H), 7.3-7.35 (d, 2~),
7.35-7.5 (m, 3H), 7.5-7.6 (d, 2~).
EI-MS: mte 334, 336 (M+, 1:1 ratio).
Step ~: Preparation of 3-~4-(1-carbomethoxy-1-
phenyl)methoxyphenyl]methyl-5,7-dimethyl-2-
ethyl-3R-i~idazor4.5-bl~yridine
To a suspen~ion of 12 mg of a 60% oil
dispersion of sodium hydride in 1 mL of DMF was added
0.050 g (0.29 mmol) of 5,7-dimethyl-2-ethylimidazo
t4,5-b]pyridine (Example 24, Step C) and the mixture
was stirred at room temperature under an N2
atmosphere. After 15 minutes, a solution of 0.105 g
(0.32 mmol) of the product of Step A dissolved in 1
mL of DMF was added and the reaction was stirred an
additional 75 minutes. The mixture was then
concentrated in vacuo and purified on a silica gel
flash chromatography column eluted with 50% ethyl
acetate/hexane to afford 0.068 g (55%) of the title
compound.
lH NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3H),
2.55 (s, 3H), 2.60 (s, 3H), 2.70-2.80 (m, 2H), 3.70
(s, 3H), 5.35 (s, 2H), 5.55 (s, lH), 6.80-6.90 (m,
3H), 7.00-7.05 (d, 2H), 7.25-7.45 (m, 3H), 7.45-7.55
(m, 2H).
FAB-MS: m/e 430 (M+l).
Step C: Preparation of 3-t4-(1-carboxY-l-Phenyl)-
methoxyphenyl]methyl-5,7-dimethyl-2-ethyl-
3~-imidazor4.5-blpyridine
WO 91/11999 PCI'/US91/009s7
.. -. 2o~62~ ,.
-252-
Using the general procedure for ester
hydrolysis described in Step E of Egample l9, 0.065 g
(0.15 mmol) of the product of Step B was converted to
O . 044 g (70%) of the title compound~
lH NMR (300 MHz, CD30D, ppm): ~ 1.20-1.30 (t, 3~,
3.60 (s, 3H), 3.65 (s, 3H), 2.85-2.95 (m, 2~), 5.50
(s, 2H), 5.65 (8, lH), 6.90-7.00 (d, 2~), 7.05-7.15
(m, 3~), 7.35-7.45 (m, 3~), 7.55-7.65 (m, ZH).
FAB-MS: m/e 416 (M+l).
~ le 28
3-t3-Chloro-4-((1-carboxy-1-phenyl)methoxy)phenyl]-
~ethyl-7-methyl-2-~ropyl-3H-imidazor4.5-bl~yridi~e
Step A: Preparation of methyl 2-(2-chloro-4-methyl-
pheno~y~-2-phenylacetate
To a suspension of 0.282 g (7.04 mmol) of a
60% oil dispersion of sodium hydride in DME was added
1.00 g (7.04 mmol) of 2-chloro-4-methylphenol and the
mixture was stirred under an N2 atmosphere at room
temperature. After 10 minutes, a solution of 1.94 g
(8.45 mmol) of methyl 2-bromophenylacetate dissolved
in 10 mL of DM~ was added and the reaction was
stirred an additional 1.5 hours. The reaction was
then diluted into ethyl acetate, uashed with water,
dried (MgS04), filtered and evaporated. The residue
was purified on a silica gel flash chromatography
column eluted with 4% ethyl acetate/hexane to afford
1.70 g (83%) of the title compound.
WO 91/11999 PCI~/USsl/ooss7
2075627
-253-
1~ NMR (300 MEz, CDC13, ppm): ~ 2.20 (s; 3E), 3.70
(~, 3~), 5.60 (~ ), 6.70-6.80 (d, 1~), 6.85-6.95
(d, 1~), 7.20 (br s, lH), 7.20-7.30 (m, 3H),
7.~5-7.65 (m, 2~).
EI-MS: m/e 290 (M+).
Step ~: Preparation of methyl 2-(2-chloro-4-bromo-
methylpheno~y)-2-~henylacetate
To a solution of 1.70 g (5.86 mmol) of the
product from Step A dissolved in 20 mL of CC14 was
added 1.04 g (5.86 mmol) of N-bromosuccinimide and 50
mg (catalytic amount) of AIBN. The reaction mixture
was stirred and heated at reflux for 7 hours, then an
additional 0.20 g of NBS was added. The reaction was
refluxed for 48 hours, then cooled and concentrated
in vacuo. The residue was purified on a silica gel
flash chromatography column eluted with 10Z ethyl
acetate/hexane to afford 0.730 g (34%) of the title
compound.
lH NMR (300 M~z, CDC13, ppm): ~ 3.70 (s, 3~), 4.40
(s, 2H), 5.65 (s, lH), 6.75-6.85 (d, 1~), 7.10-7.20
(d, lH), 7.30-7.45 (m, 4~), 7.55-7.65 (m, 2~).
FAB-MS: m/e 369 (M+l).
Step C: Preparation of 3-~3-chloro-4-((1-carbo-
methoxy-l-phenyl)methoxy)phenyl]methyl-7-
methyl-2-~ro~yl-3H-imidazor4.5-bl~yridine
The product of Step B (0.127 g, 0.34 mmol)
was used to alkylate 0.050 g (0.29 mmol) of 7-methyl-
2-propylimidazO[4,5-b]pyridine (Example 2, Step B)
according to the procedure described for Step D of
Example 3, which after purification afforded 0.059 g
(45%) of the title compound.
WO 91/11999 2 0 7 5 6 2 ~ Pcr/usgl/oog57
-254-
lH NMR (300 M~z, CDC13, ppm): ~ 0.90-1.00 (t, 3H),
1.65-1.80 (m, 2H), 2.65 (s, 3H), 2.65-2.80 (t, 2H),
3.70 (s, 3H), 5.35 (s, 2H), 5.60 (8, lH), 6.70-6.75
(d, lH), 6.85-6.95 (d, lH), 7.00-7.05 (d, 1~), 7.20
(br s, lH), 7.30-7.45 (m, 3H), 7.50-7.60 (m, 2R),
8.15-8.20 (d, lH).
FAB-MS: m/e 464 (M+l).
Step D: Preparation of 3-t3-chloro-4-((1-carboxy-1-
phenyl)methoxy)phenyl]methyl-7-methyl-2-
~ro~yl-3~ idazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.059 g
(0.13 mmol) of the product of Step C was converted to
0.040 g (70%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ 0.90-1.00 (t, 3H),
1.65-1.80 (m, 2H), 2.70 (s, 3H), 2.85-2.95 (t, 2H),
5.50 (s, 2H), 5.75 (s, lH), 6.95-7.10 (m, 2H),
7.15-7.20 (d, lH), 7.25 (br s, 1~), 7.35-7.45 (m,
3H), 7.60-7.70 (m, 2H), 8.20-8.25 (d, lH).
FAB-MS: m/e 450 (M+l).
T.~A~le 29
2s 3-[3-Chloro-4-((1-carboxy-1-phenyl)methoxy)phenyl]-
methyl-5.7-dimethYl-2-ethYl-3~-imidazor4.5-blpyridi~e
Step A: Preparation of 3-~3-chloro-4-((1-carbo-
metho~y-l-phenyl)methoxy)phenyl]methyl-5,7-
dimethyl-2-ethyl-3~-imidazor4.5-bl~yridine
WO 91/11999 PCI~/US91/009s7
2075627
-255-
The product of Example 28, Step B (0.127 g,
0.34 mmol) was used to al~ylate 0.050 g (0.29 mmol)
of 5,7-dimethyl-2-ethylimidazo[4,5-b]pyridine
(Example 24, Step C) according to the procedure
described for Step B of Example 27, which after
purification afforded 0.080 g (61%) of the title
compound.
lH NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3~),
2.55 (s, 3H), 2.60 (s, 3H), 2.65-2.80 (m, 2H), 3.70
(s, 3H), 5.35 (s, 2~), 5.60 (s, 1~), 6.80-6.85 (d,
lH), 6.85-6.95 (m, 2H), 7.20-7.25 (d, lH), 7.30-7.45
(m, 3~), 7.50-7.60 (m, 2H).
FAB-MS: m/e 464 (M+l).
Step B: Preparation of 3-t3-chloro-4-((1-carbo~y-1-
phenyl)metho~y)phenyl]methyl-5,7-dimethyl-2-
ethyl-3H-imidazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step ~ of Example 19, 0.080 g
(0.17 mmol) of the product of Step A was converted to
0.047 g (60%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ 1.25-1.35 (t, 3H),
2.60 (s, 3H), 2.65 (s, 3H), 2.85-2.95 (m, 2H), 5.50
(s, 2~), 5.75 (s, lH), 6.95-7.10 (m, 3H), 7.25 (s,
lH), 7.35-7.45 (m, 3H), 7.60-7.70 (m, 2H).
FAB-MS: m/e 450 (M+l).
WO 91/11~9 ~ 07 5 6 ~7 PCT/US91/~957
,
-256-
~ ple 30
3-t3-Benzoyl-4-((l-carboxy-l-phenyl)methoxy)phenyl]-
metl~,yl-5 . 7-dimet~url-2-etl~yl-3~ nidAzor4 . 5-bl~yridi~e
Step A: Preparation of methyl 2-(2-benzoyl-4-methyl-
~heno~y)-2-phe~ylAcet~te
To a solution of 1.00 g ~4.72 mmol) of
2-hydroxy-5-methylbenzophenone and 1.19 g (5.19 mmol)
f methyl 2-bromophenylacetate in 10 mL of acetone
was added 1.30 g (9.44 mmol) of ~2C03 and the mixture
was stirred and refluxed for 14 hours. The mi~ture
was cooled, filtered and evaporated Ln v~cuo and the
residue was purified on a silica gel flash chromato-
graphy column eluted with 5/. ehtyl acetate/hexane toafford 0.320 g (19%) of the title compound.
1~ NMR (300 MHz, CDC13, ppm): ~ 2.30 (s, 3H), 3.60
(s, 3~), 5.50 (s, 1~), 6.65-6.75 (d, lE), 6.90-7.00
(d, 2H), 7.10-7.25 (m, 4~), 7.30 (s, 1~), 7.40-7 50
(m, 2H), 7.50-7.55 (m, lH), 7.80-7.90 (m, 2H).
FAB-MS: m/e 361 (M+l).
Step B: Preparation of methyl 2-(2-benzoyl-4-bromo-
methylphenoxy)-2-phenylacetate
To a solution of 0.314 g (0.87 mmol) of the
product of Step A dissolved in 10 mL of CC14 was
added 0.155 g (0.87 mmol) of N-bromosuccinimide and
15 mg (catalytic amount) of AIBN. The mixture was
stirred at reflux for 7 hours, then cooled filtered
and evaporated ~s vacuo. The residue was purified on
a silica gel flash chromatography column eluted with
WOgl/ll~9 PCT/US91/009~7
207562~
-257-
15% ethyl acetate/hexane to afford 0.136 g (36%) of
the title compound.
H NMR (300 MHz, CDC13, ppm): ~ 3.65 (s, 3H), 4.50
(s, 2E), 5.55 (s, lE), 6.75-6.85 (d, 1~) 6.90-7.00
(d, 2H), 7.10-7.25 (m, 4~), 7.40-7.60 (m, 4H),
7.80-7.90 (d, 2H).
FAB-MS: m/e 440 (M+l).
Step C: Preparation of 3-~3-benzoyl-4-((1-carbo-
methoxy-1-phenyl)methoxy)phenyl]methyl-5,7-
dimethyl-2-ethyl-3H-imidazor4.5-blpyridine
The product of Step B (0.136 g, 0.31 mmol)
was used to alkylate 0.049 g (0.28 mmol) of
5,7-dimethyl-2-ethylimidazot4,5-b]pyridine (Example
24, Step C) according to the procedure described for
Step B of Example 27, which after purification
afforded 0.066 g (44%) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 1.20-1.30 (t, 3~),
2.50 (s, 3H), 2.55 (s, 3H), 2.65-2.80 (m, 2H), 3.80
(s, 3~), 5.30 (s, 3H), 6.80-7.30 (m, 14~).
FAB-MS: m/e 534 (M+l).
Step D: Preparation of 3-[3-benzoyl-4-((1-carboxy-
l-phenyl)methoxy)phenyl]methyl-5,7-dimethyl-
2-ethyl-3R-i~idazor4.5-bl~yridine
Using the generzl procedure for ester
hydrolysis described in Step E of Example 19, 0.060 g
(0.11 mmol) of the product of Step C was converted to
0.031 g (53%) of the title compound.
lH NMR (300 M~z, CD30D, ppm): ~ 1.20-1.30 (t, 3H),
2.55 (s, 3~), 2.60 (s, 3H), 2.70-2.80 (m, 2H), 5.50
(s, 3H), 6.85-7.35 (m, 14H).
FAB-MS: m/e 520 (M+l).
WO 91/11~ 2 0 ~ 5 ~ 2 7 PCT/US91/~957
-258-
~ le 31
3-t3-Acetyl-4-((1-carboxy-1-phenyl)methoxy)phenyl]-
methyl-5~7-di~ethyl-2-et~yl-3~-imid~zsr4~5-blpyridine
Step A: Preparation of methyl 2-(2-acetyl-4-methyl-
~heno1~y~-2-~henylAcetAte
Using the ~2CO3/acetone conditions for
phenol alkylation described in`Step A of Example 30,
1.00 g (6.67 mmol) of 2'-hydroxy-5l-methylaceto-
phenone was alkylated with 1.68 g (7.34 mmol) of
methyl 2-bromophenylacetate to afford 1.25 g (63%) of
the title compound.
1~ NMR (400 MHz, CDC13, ppm): ~ 2.25 (s, 3H), 2.70
(s, 3~), 3.70 (s, 3H), 5.70 (d, 1~), 6.65-6.75 (d,
lH), 7.10-7.20 (d, lH), 7.30-7.45 (m, 4H), 7.50-7.60
(m, 2H).
FAB-MS: m/e 299 (M+l).
Step B: Preparation of methyl 2-(2-acetyl-4-bromo-
methylphenoxy)-2-phenylacetate
To a solution of 1.25 g (4.19 mmol) of the
product of Step A dissolved in 15 mL of CC14 was
added 0.821 g (4.61 mmol) of N-bromosuccinimide and
20 mg (catalytic amount) of AIBN. The mixture was
stirred at reflux for 3.5 hours, then cooled,
filtered and evaporated in vacuo. The residue was
purified on a silica gel flash chromatography column
eluted with 15% ethyl acetate/hexane to afford 0.431
g (27%) yield of the title compound.
-
WO91/11~9 ~ PCT/US91/~957
207~S27
-259-
H NMR (400 M~z, CDCl3, ppm): ~ 2.75 (s, 3H), 3.75
(s, 3H), 4.45 (s, 2H), 5.70 (~, lH), 6.75-6.80 (d,
2H), 7.35-7.45 (m, 4H), 7.50-7.60 (m, 2H), 7.75 (s,
lH).
Step C: Preparation of 3-t3-acetYl-4-((1-carbo-
methoxy-l-phenyl)methoxy)phenyl]methyl-5,7-
di~et~yl-2-et~yl-3H-imidazor4.5-bl~yridine
The product of Step B (0.119 g, 0.31 mmol)
was used to al~ylate 0.050 g (0.28 mmol) of 5,7-
dimethyl-2-ethylimidazo[4,5-b]pyridine (E~ample 24,
Step C) according to the procedure described for Step
B of Example 27, which after purification afforded
0.040 g (30%) of the title compound.
lH NMR (400 M~z, CDC13, ppm): ~ 1.20 (s, 3H),
1.20-1.30 (t, 3H), 2.55 (s, 3~), 2.60 (s, 3H),
2.70-2.80 (m, 2H), 3.75 (s, 3~), 5.35 (s, 3H), 6.85
(s, lH), 6.95-7.00 (d, lH), 7.05-7.10 (s, lH), 7.15
(s, lH), 7.20-7.30 (m, 3H), 7.50-7.55 (m, 2H).
FAB-MS: m/e 472 (M+l).
Step D- Preparation of 3-~3-acetyl-4-((1-carbo~y-1-
phenyl)methoxy)phenyl]methyl-5,7-dimethyl-2-
ethyl-3H-imidazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.038 g
(0.08 mmol) of the product of Step C was converted to
0.016 g (43%) of the title compound.
lH NMR (400 MHz, CD30D, ppm): ~ 1.15 (s, 3~),
1.20-1.30 (t, 3H), 2.55 (s, 3H), 2.60 (s, 3H),
2.85-2.95 (m, 2H), 5.50 (s, 3H), 6.95-7.05 (m, 2H),
7.05 (s, lH), 7.10 (s, lH), 7.25-7.35 (m, 3H),
7.60-7.70 (m, 2H).
FAB-MS: m/e 458 (M+l).
WO 91/11999 2 ~ ~ ~ 5 2 7 Pcr/usgl/009s7
-260-
~Am~le 32
3-~4-(tl-carboxy-l-phenyl)methoxy)-3-metho~yphenyl]-
~ethyl-5.7-dimethyl-2-ethyl-3~ id~zor4.5-bl~yridine
Step A: Preparation of methyl 2-(4-hydroxymethyl-2-
~etho~ypheno~y)-2-~henylAcet~te
Using the ~2C03/acetone conditions for
phenol alkylation described in Step A of Example 30,
1.00 g (6.49 mmol) of 4-hydroxy-3-methoxybenzyl
alcohol was alkylated with 1.64 g (7.14 mmol) of
methyl 2-bromophenylacetate to afford 0.495 g (25%)
of the title compound.
1~ NMR (300 MHz, CDC13, ppm): ~ 1.20-1.30 (t, 1~),
3-70 (s, 3~), 3.85 (s, 3~), 4.60 (d, 2E), 5.65 (s,
1~), 6.75-6.85 (m, 2H), 6.95 (s, lH), 7.30-7.40 (m,
3H), 7.50-7.60 (m, 2H).
FAB-MS: m/e 303 (M+l).
Step B: Preparation of methyl 2-(4-bromomethyl-2-
methoxyphenoxy)-2-phenylacetate
To a cooled (0C) solution of 0.490 g (1.62
mmol) of the product of Step A dissol~ed in 8 mL of
CH2C12 was added 0.673 g (2.03 mmol) of carbon
tetrabromide and 0.531 g (2.03 mmol) of triphenyl-
phosphine. The reaction mixture was ~tirred for 1.5
hours and was allowed to slowly warm to room temper-
ature. The reaction mixture was then concentrated
in vacuo, and purified on a silica gel flash chromato-
graphy column eluted with 15% ethyl acetate/hexane toafford 0.478 g (81%) of the title compound.
WO91/11~9 PCT/US91/~957
,
2075627
-261-
H MMR (300 MHz, CDC13, ppm): ~ 3.75 (s, 3R), 3.85
(s, 3H), 4.40 (8, 2H), 5.60 (s, lH), 6.75-6.85 (m,
2H), 6.90 (s, lH), 7.30-7.40 (m, 3H), 7.50-7.60 (m,
2H).
Ste~ C: Preparation of 3-~4-((1-carbometho~y-1-
phenyl)methoxy)-3-metho~yphenyl~methyl-5,7-
di~ethyl-2-ethyl-3H-imidAzor4.5-b~pyridine
The product of Step B (0.115 g, 0.31 mmol)
was used to alkylate 0.050 g (0.28 mmol) of 5,7-
dimethyl-2-ethylimidazot4,5-b]pyridine (Example 24,
Step C) according to the procedure described for Step
of Example 27, which after purification afforded
0.084 g (64%) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 1.20-1.30 (t, 3H),
2.55 (s, 3H), 2.60 (s, 3E), 2.70-2.80 (m, 2H), 3.70
(s, 3H), 3.75 (s, 3H), 5.35 (s, 2H), 5.55 (s, lH),
6.50-6.60 (d, lH), 6.70-6.75 (d, lH), 6.80 (s, lH),
6.85 (s, lH), 7.30-7.40 (m, 3E), 7.45-7.55 (m, 2H).
FAB-MS: m/e 460 (M+l).
Step D: Preparation of 3-t4-((1-carbo~Y-l-phenyl)-
methoxy)-3-methoxyphenyl]methyl-5,7-dimethyl-
2-ethyl-3~-imidazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.080 g
(O.17 mmol) of the product of Step C was converted to
0.064 g (82%) of the title compound.
1~ NMR (300 MHz, CD30D, ppm): ~ 1.20-1.30 (t, 3H),
2.60 (s, 3~), 2.62 (s, 3H), 2.85-2.95 (m, 2~), 3.80
(s, 3H), 5.50 (s, 3H), 6.50-6.60 (d, lH), 6.80-6.90
(d, lH), 6-95 (s, lH), 7.05 (s, lH), 7.30-7.40 (m,
3H), 7.55-7.65 (m, 2H).
FAB-MS: m/e 446 (M+l).
-
WO 91/11~9 2 07 ~ 62~ PCT/US91/~9S7
-262-
F.~ le 33
3-r3-tert-Butyl-4-((1-carboxy-1-phenyl)methoxy)-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazo[4,5-b]
~yridine
Step A: Preparation of methyl 2-(2-tert-butyl-4-
methylpheno~y)-2-~henylacetate
To a suspension of 1.05 g (9.15 mmol) of a
35% oil dispersion of RH in 15 mL of DMF was added
1.50 g (9.15 mmol) of 2-tert-butyl-4-methylphenol and
the mixture was stirred under N2 at room temperature.
After 10 minutes, 2.41 g (91.5 mmol) of 18-crown-6
and then a solution of 2.30 g (10.1 mmol) of methyl
2-bromophenylacetate dissolved in 10 mL of DMF were
added. THe reaction mi~ture was stirred 17 hours,
then partitioned between ethyl acetate and water.
The organic layer was separated, washed with water,
dried (MES04), filtered and evaporated. The residue
was purified on a silica gel flash chromatography
column eluted with 5% ethyl acetate/hexane to afford
0.750 g (26%) of the title compound.
H NMR (300 MHz, CDC13, ppm): ~ 1.43 (s, 9~), 2.24
(s, 3~), 3.68 (s, 3H), 5.64 (s, lH), 6.56 (d, J=10
Hz, lH), 6.86 (dd, J=2, 10 Hz, lH), 7.12 (d, J=2 ~z,
lH), 7.30-7.44 (m, 3H), 7.54-7.62 (m, 2H).
Step B: Preparation of methyl 2-(2-tert-butyl-4-
bromomethylphenoxy)-2-~henylacetate
30To a solution of 0.494 g (1.58 mmol) of the
product of Step A dissolved in 10 mL of CC14 was
WO91/11~ PCT/US91/~9~7
,
207562~
-263-
added 0.310 g (1.74 mmol) of N-bromosuccinimide and
15 mg (catalytic amount) of AIBN and the mixture was
heated at reflux for 3.5 hours. The reaction was
cooled, filtered and evaporated ~n vacuo. The
residue was purified on a ~ilica gel flash chromato-
graphy column eluted with 3% ethyl acetate/hegane to
afford 0.134 g (22Z) of the title compound.
1~ NMR (300 M~z, CDC13, ppm): ~ 1.44 (æ, 3~), 3.70
(s, 3~), 4.45 (s, 2~), 5.64 (s, lH), 6.70 (d, J=10
Hz, lH), 7.12 (dd, J=2, 10 Hz, lH), 7.22 (d, J=2 Hz,
1~), 7.32-7.42 (m, 3~), 7.53-7.60 (m, 2H).
Step C: Preparation of 3-~3-tert-butyl-4-((1-carbo-
methoxy-l-phenyl)methoxy)phenyl]methyl-5,7-
dimethyl-2-ethyl-3~-imidazor4.5-bl~yridine
The product of Step B (0.128 g, 0.33 mmol)
was used to alkylate 0.052 g (0.30 mmol) of 5,7-
dimethyl-2-ethylimidazo[4,5-b3pyridine (Example 24,
Step C) according to the procedure described for Step
~ of Example 27, which after purification afforded
0.088 g (62~/o) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 1.20-1.3~ (t, 3~),
1.40 (s, 9~), 2.55 (s, 3H), 2.60 (s, 3E), 2.75-2.85
(m, 2~), 3.65 (s, 3~), 5.30 (s, 2H), 5.60 (s, 1~),
6.45-6.55 (d, lH), 6.70-6.80 (d, 1~), 6~85 (br s,
lH), 7.25-7.40 ~m, 4H), 7.50-7.55 (m, 2H).
~AB-MS: m/e 486 (M+l).
Step D~ Preparation of 3-[3-tert-butyl-4-((1-carboxy-
1-phenyl)methoxy)phenyl3methyl-5,7-dimethyl-
2-ethy~-3H-imidazor4.~-blpyridine
WO91/11~ PCT/US91/Oo9s7
20756~7 ~ :`
-264-
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.080 g
(O.16 mmol) of the product of Step C was converted to
0.056 g (73%) of the title compound.
lH NMR (300 M~z, CD30D, ppm): ~ 1.25-1.35 (t, 3~),
1.40 (s, 9H), 2.63 (s, 3H), 2.65 (8, 3H), 2.85-2.95
(m, 2H), 5.50 (s, 2H), 5.65 (s, lH), 6.75-6.85 (d,
lH), 6.90-6.95 (d, lH), 7.05 (s, 1~), 7.30 (s, lH),
7.35-7.45 (m, 3H), 7.60-7.70 (m, 2H).
EAB-MS: m/e 472 (M+l).
F~ple 34
3-t4-((1-carboxy-1-phenyl)metho~y)-3-ethoxyphenyl]-
methyl-5.7-dimethyl-2-ethyl-3H-imidazor4.5-blpyridine
Step A- Preparation of methyl 2-(2-ethoxy-4-formyl-
phenoxy)phenylacetate
Using the K2C03/acetone conditions for
phenol alkylation described in Step A of Example 30,
1.00 g (6.02 mmol) of 3-ethoxy-4-hydroxybenzaldehyde
was alkylated with 1.52 g (6.62 mmol) of methyl
2-bromophenylacetate to afford 1.74 g (92%) of the
title compound.
2~ lH NMR (300 ~z, CDC13, ppm): ~ 1.40-1.50 (t, 3H),
3.85 (s, 3~), 4.10-4.20 (m, 2H), 5.75 (s, lH), 6.95
(s, lH), 7.20-7.35 (m, 5H), 7.50-7.60 (m, 2H), 9.85
(s, lH).
FAB-MS: m/e 315 (M+l).
-
WO91/11~9 PCT/US91/00957
- - 2075~27
-265-
Step B: Preparation of methyl 2-(2-ethoxy-4-hydroxy-
met}~ heno~ h enyl ~ c et at e
A stirred solution of 1.74 g (5.54 mmol) of
the product of Step A dissolved in 22 mL of methanol
was treated with 0.105 g (2.8 mmol) of sodium
borohydride at room temperature. After 5 minutes the
reaction mixture was partitioned between ethyl
acetate and 1 N hydrochloric acid, and the organic
layer was separated. The product was washed with
water, brine, dried (M~S04), filtered and evaporated
n vacuo. The residue was purified on a silica gel
flash chromatography column eluted with 30/O ethyl
acetate/hexane to afford 1.15 g (66%) of the title
compound.
1~ NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 1~),
1.35-1.45 (t, 2H), 3.70 (s, 3H), 4.05-4.15 (m, 2H),
4.60 (s, 2H), 5.65 (s, 1~), 6.75-6.80 (d, lH),
6.85-6.95 (m, 2H), 7.30-7.40 (m, 3H), 7.50-7.60 (m,
2~).
~AB-MS: m/e 317 (M+l).
Step C: Preparation of methyl 2-(4-bromomethyl-2-
ethoxy~heno2y)phenylacetate
To a stirred and cooled (~C) solution of
1.15 g (3.64 mmol) of the product of Step B dissolved
in 18 mL of CH2C12 was added 1.51 g (4.55 mmol) of
carbon tetrabromide and 1.19 g (4.55 mmol) of
triphenylphOSphine. After the addition the reaction
mixture was stirred 30 minutes and allowed to warm to
room temperature. The mi~ture was then evaporated
in ~acuo and purified on a silica gel flash chromato-
graphy column eluted with 15% ethyl acetate/hexane to
afford 1.21 g (88%) of the title compound.
WO91/11~9 PCT/USgl/oogs7
.,
207~627
-266-
lH NM~ (300 M~z, CDC13, ppm): ~ 1.3S-1.45 (t, 3~),
3.75 (8, 3~), 4.05-4.15 ~m, 2~), 4.40 (~, 2~), 5.65
(s, 1~), 6.80-6.90 (m, 2~), 6.9S (s, 1~), 7.35-7.4S
(m, 3H), 7.50-7.60 (m, 2H).
FAB-MS: m/e 378, 380 (M~l).
Ste~ D: Preparation of 3-t4-((1-carbomethoxy-1-
phenyl)methoxy~-3-ethoxyphenyl]methyl-5,7-
~imethyl-2-ethyl-3H-imidazor4.5-blpyridine
The product of Step C (0.119 g, 0.31 mmol)
was used to alkylate 0.055 g (0.28 mmol) of 5,7-
dimethyl-2-ethylimidazo[4~5-b]pyridine (Example 24,
Step C) according to the procedure described for Step
~ of Example 27, which after purification afforded
lS 0.084 g ~62Z) of the title compound.
lH NMR (400 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3~),
1.30-1.40 (t, 3H), 2.55 (s, 3E), 2.60 (s, 3H),
2.70-2.80 (m, 2H), 3.70 (s, 3H), 3.90-4.00 (m, 2~),
5.30 (s, 2H), 5.60 (s, 1~), 6.50-6.55 (d, 1~),
6.75-6.80 (m, 2H), 6.85 (s, 1~), 7.30-7.40 (m, 3~),
7.50-7.55 (m, 2H).
FAB-MS: m/e 474 (M+l).
Step E: Preparation of 3-t4-((l-carboxy-l-phenyl)-
methoxy)-3-ethoxyphenyl]methyl-5,7-dimethyl-
2-ethyl-3H-imidazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step E of E~ample 19, 0.080 g
(0.17 mmol) of the product of Step D was con~erted to
0.069 g (88Z) of the title compound.
WO91/11~9 PCT/US91/oogs7
- 2075627
-267-
1~ NMR (400 M~z, CD30D, ppm): ~ 1.20-1.30 (t, 3~),
1.30-1.40 (t, 3~), 2.55 (s, 3H), 2.60 (8, 3H),
2.80-2.90 (m, 2H), 3.95-4.05 (m, 2~), 5.45 (s, 2H),
5.50 (s, lE), 6.50-6.55 (d, lH), 6.80-6.85 (m, 2~,
S 7.25-7.35 (m, 3H), 7.50-7.55 (m, 2~).
F~ e 35
3-[4-((l-Carboxy-l-phenyl)metho~y)-3-methylphenyl~-
methyl-5.7-dimethyl-2-ethyl-3~-imidazor4.5-blpyridine
Step A: Preparation of methyl 2-(4-formyl-2-methyl-
phenoxy)phenylacetate
Using the ~2C03tacetone conditions for
phenol alkylation described in Step A of Example 30,
O.50 g (3.68 mmol) of 4-hydroxy-3-methylbenzaldehyde
was alkylated with 0.926 g (4.05 mmol) of methyl
2-bromophenylacetate to afford 1.00 g (96%) of the
title compound.
1~ NMR (400 M~z, CDC13, ppm): ~ 2.40 (s, 3~), 3.70
(s, 3H), 5.70 (s, lH), 6.80 (d, 1~), 7.35-7.45 (m,
3~), 7.55-7.60 (m, 2~), 7.65 (d, lH), 7.70 (s, 1~),
9.85 (s, 1~).
FAB-MS: m/e 285 (M+l).
Step B: Preparation of methyl 2-(4-hydroxymethyl-2-
methylphenoxy~phe~ylacetate
A stirred solution of 1.00 g (3.52 mmol) of
the product of Step A dissolved in 14 mL of methanol
was treated with 0.067 g (1.8 mmol) of sodium
borohydride at room temperature. After 15 minutes
WO91/11~ PCT/US91/00957
- 2o~627
-268-
the reaction mixture was partitioned between ethyl
acetate and 1 N hydrochloric acid, and the organic
layer was separated. The product was washed with
water, brine, dried (MgS04), filtered and evaporated
in vacuo. The residue was purified on a ~ilica gel
flash chromatography column eluted with 30% ethyl
acetate/hexane to afford 0.660 g (66%) of the title
compound.
lH NMR (400 M~z, CDC13, ppm): ~ 2.10 (s, lH), 2.35
lo ~s, 3H), 3.70 (s, 3H), 4.55 (s, 2H), 5.65 (s, lH),
6.70 (d, lH), 7.05 (d, lH), 7.15 (s, lH), 7.35-7.45
(m, 3H), 7.55-7.65 (m, 2H).
FAB-MS: m/e 287 (M~l).
Step C: Preparation of methyl 2-(4-hydroxymethyl-2-
methylphenoxy)phenylacetate
To a stirred and cooled (0CC) solution of
0.660 g (2.31 mmol) of the product of Step B dissolved
in 12 mL of CH2C12 was added 0.957 g (2.89 mmol) of
carbon tetrabromide and 0.756 g (2.89 mmol) of
triphenylphosphine. After the addition the reaction
mixture was allowed to warm to room temperature and
was stirred overnight. The mixture was then
evaporated in vacuo and purified on a silica gel
flash chromatography column eluted with 15% ethyl
acetate/hexane to afford 0.704 g (87%) of the title
compou~d.
1~ NMR (400 MHz, CDC13, ppm): ~ 2.30 (s, 3H), 3.70
(s, 3H), 4.45 (s, 2H), 5.60 (s, lH), 6.65 (d, lH),
7.10 (d, lH), 7.20 (s, lH), 7.35-7.45 (m, 3H),
7.55-7.60 (m, 2H).
FAB-MS: m/e 269 (M~l).
WO91/11~ PCT/USgl/oogs7
2075~27
-269-
Step D: Preparation of 3-[4-((1-carbomethoxy_l_
phenyl)methoxy)-3-methylphenyl]methyl-5,7-
dimethyl-2-ethyl-3~-imidazor4.5-bl~yridine
The product of Step C (0.110 g, 0.31 mmol)
was used to al~ylate 0.050 g (0.28 mmol) of 5,7-
dimethyl-2-ethylimidazot4,5-b]pyridine (Example 24,
Step C) according to the procedure described for Step
B of Example 27, which after purification afforded
0.041 g (33%) of the title compound.
lH NMR (400 MHz, CDC13, ppm): ~ 1.20-1.30 (t, 3~),
2.25 (s, 3H), 2.55 (s, 3H), 2.60 (s, 3H), 2.70-2.80
(m, 2H), 3.65 (s, 3H), 5.35 (s, 2H), 5.55 (s, lH),
6.60 (d, lH), 6.80 (d, lH), 6.85 (s, 1~), 6.95 (s,
lH), 7.30-7.40 (m, 3H), 7.50-7.55 (m, 2H).
FAB-MS: m/e 444 (M+l).
Step ~: Preparation of 3-[4-((1-carbo~y-1-phenyl)-
methoxy)-3-methylphenyl]methyl-5,7-dimethyl-
2-ethyl-3H-imidazor4.5-~lpyridine
Using the general procedure for ester
hydrolysis described in Step E of E~ample 19, 0.040 g
(0.09 mmol) of the product of Step D was converted to
0.023 g (60%) of the title compound.
lH NMR (400 MHz, CD30D, ppm): ~ 1.15-1.25 (t, 3H),
2,25 (s, 3H), 2.58 (s, 3H), 2.60 (s, 3H), 2.80-2.90
(m, 2H), 5.45 (s, 2H), 5.60 (s, lH), 6.75 (d, lH),
6.85 (d, lH), 6.95 (s, lH), 7.05 (s, lH), 7.30-7.40
(m, 3H~, 7.55-7.65 (m, 2H).
FAB-MS: m/e 430 (M+l).
WO91/11~9 PCT/US91/~s~7
` 20~562~
- 270 -
F.~AnU 1e 36
3-[4-((1-Carbo~y-1-(2-methylphenyl))methoxy)-3-chloro-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazo[4,5-b]-
pyridine
Step A: Preparation of methyl 4-tert-butyldimethyl-
silylo~y-3-chlorobenzoate
To a solution of 5.00 g (26.8 mmol) of methyl
3-chloro-4-hydro~ybenzoate dissolved in 40 mL of
CH2C12 was added 6.55 g (53.6 mmol) of 4-dimethyl-
aminopyridine, 4.85 g (32.2 mmol) of tert-butyl-
dimethylchlorosilane and the mi~ture was stirred at
room temperature for 3.5 hours. The reaction mi~ture
was filtered, diluted with ethyl acetate, washed with
0.1 N ~Cl, saturated NaHC03, and brine. The product
layer was then dried (MgS04), filtered and evaporated
in vacuo to afford 8.05 g (100Z) of the title
compound.
lH NMR (300 M~z, CDC13, ppm): ~ 0.25 (s, 6H), 1.00
(s, 9H), 3.85 (s, 3H), 6.85 (d, lH), 7.80 (d, lH),
8.05 (s, lH).
FAB-MS: m/e 301, 303 (M+l).
Step B: PreparatiOn of 4-tert-butyldimethylsilyloxy-
3-chlorobenzyl alcohol
To a stirred and cooled (0C) solution of
8.00 g (26.7 mmol) of the product of Step A dissolved
in 50 mL of anhydrous THF was added 53.3 mL (53.3
mmol) of a 1 M solution of lithium aluminum hydride
in THF. After the addition was complete the reaction
WO91/11~9 PCT/US91/oogs7
.; :
2075627
- 271 -
mixture was allowed to warm to room temperature and
stirred 2 hours. The stirred reaction was then
quenched by dropwise addition of 2.5 mL water, then
2.5 mL of 15% NaOH, and finally 7.5 mL water. The
reaction mixture was then filtered and concentrated
~n vacuo. The residue was dissolved in ethyl
acetate, washed with 1 N HCl, sturated NaHC03, dried
(MgS04), and evaporated ~n vacuo to afford 4.0 g
(53~/c) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ O.20 (s, 6H), 1.00
(s, 9H), 1.80 (br s, lH), 4.55 (s, 2H), 6.85 (d, lH),
7.10 (d, lH), 7.35 (s, lH).
FAB-MS: m/e-255, 257 (M+l).
Step C: Preparation of 4-tert-butyldimethylsilyl-
o~y-3-chlorobe~zyl bromide
To a stirred and cooled (0C) solution of
4.00 g (14.1 mmol) of the product of Step ~ dissolved
in 70 mL of CH2C12 was added 5.84 g (17.6 mmol) of
carbon tetrabromide and 4.61 g (17.6 mmol) of
triphenylphosphine. After the addition the reaction
mixture was allowed to warm to room temperature and
was stirred 3 hours. The mi~ture was then evaporated
in vacuo and purified on a silica gel flash chromato-
2s graphy column eluted with 2Z ethyl acetate/hexane toafford 4.50 g (92%) of the title compound.
H NMR (300 M~z. CDC13, ppm): ~ O.20 (s, 6H), 1.00
(s, 9H), 4.40 (s, 2H), 6.80 (d, lH), 7.15 (d, lH),
7.35 (s, lH).
WO91/11~ 20~ ~æ~ PCT/US91/~957
- 272 -
Step D: Preparation of 3-(4-tert-butyltimethyl-
~ilyloxy-3-chlorophenyl)methyl-5,7-dimethyl-
?-et~yl-3~-imid~zor4.5-bl~yridine
The product of Step C (1.79 g, 5.15 mmol)
was used to al~ylate 0.750 g (4.29 mmol) of 5,7-
dimethyl-2-ethylimidazo[4,5-b~pyridine (Example 24,
Step C) according to the procedure described for Step
of Example 27, which after purification afforded
0.294 g (15%) of the title compound.
lH NMR (300 MXz, CDC13, ppm): ~ O.20 (s, 6H), 1.00
(s, 9H), 1.20-1.30 (t, 3H), 2.58 (s, 3H), 2.60 (s,
3H), 2.70-2.80 (m, 2H), 5.35 (s, 2H), 7.75 (d, lH),
7.85-7.90 (m, 2H), 7.15 (s, lH).
FAB-MS: m/e 430, 432 (M+l).
Step E: Preparation of 3-(3-chloro-4-hydroxyphenyl)-
methyl-5,7-dimethyl-2-ethyl-3H-imidazo
r4.5-blpyridine
To a solution of 0.294 g (0.661 mmol) of the
product of Step D dissolved in 4 mL of THF was added
0.69 mL (0.69 mmol) of a 1.0 M solution of tetra-n-
butylammonium fluoride in l~, and the reaction
mixture was stirred 2 hours at room temperature. The
reaction was then concentrated in vacuo and purified
by filtration through a silica gel pad eluted with
chloroform. Evaporation of the filtrate and drying
L~ vacuo afforded 0.188 g (87%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ 1.25-1.35 (t, 3H),
2.65 (s, 3H), 2.67 (s, 3H), 2.85-2.95 (m, 2H), 5.45
(s, 2H), 6.90 (d, lH), 6.95 (d, lH), 7.05 (~, lE),
7.15 (~, lH).
FAB-MS: m/e 316, 318 (M+l).
WO91/11~ PCT/US91/~9~7
2075~27
- 273 -
Ste~ F: Preparation of 3-[4-((1-carbomethoxy-1-(2-
methylphenyl))metho~y)-3-chlorophenyl~methyl-
5,7-dimethyl-2-ethyl-3~-imidazo~4,5-b~
~yridi~e
To a suspension of 7 mg of a 60% oil disper-
sion of sodium hydride in 0.75 mL of DMF was added
O.050 g (0.16 mmol) of the product of Step E and the
reaction mi~ture was stirred 10 minutes under an N2
atmosphere. A solution of methyl 2-bromo-2'-methyl-
lo phenylacetate (prepared from 2'-methylphenylacetic
acid via a Hell-Volhard-Zelins~y reaction similar to
Step A of Example 17) dissolved in O.75 mL of DMF was
then added and the reaction was stirred for 2 hour~
at room temperature. The reaction mixture was then
partitioned between ethyl acetate and water, the
organic layer was separated, dried (MgS04), filtered
and e~aporated. The residue was purified on a silica
gel flash chromatography column eluted with EtOAc/
hexane/CHC13 (50:40:10) to afford 0.070 g (92%) of
the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 1.25-1.35 (t, 3H),
2.45 (s, 3H), 2.60 (s, 3H), 3.65 (s, 3H), 2.75-2.85
(m, 2H), 3.70 (s, 3H), 5.35 (s, 2H), 5.60 (s, lH),
6.75 (d, lH), 6.85-6.95 (m, 2H), 7.15-7.40 (m, 4H),
7.55-7.65 (m, lH).
FAB-MS: m/e 478, 480 (M~l, 3:1 ratio).
WO91/11~ PCT/USgl/~9~7
297~i27
- 274 -
Ste~ G: Preparation of 3-t4~ -carboxy-l-(2-meth
phenyl))methoxy)-3-chlorophenyl]methyl-5,7-
di~ethyl-2-et~yl-3~-imidAzor4.5-bl~yridi~e
Using the general procedure for ester
hydrolysis described in Step E of E~ample 19, 0.040 g
(0.15 mmol) of the product of Step F was con~erted to
0.066 g (97%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): d 1.25-1.35 (t, 3H),
2.50 (s, 3H), 2.63 (s, 3H), 2.65 (5, 3H), 2.85-2.95
lo (m, 2H), 5.50 (s, ZH), 5.80 (s, lH), 6.95-7.10 (m,
3H), 7.20-7.30 (m, 4H), 7.60 (d, lH).
FAB-MS: m/e 464, 466 (M+l, 3:1 ratio).
F~m~le 37
3-t4-((1-Carboxy-l-phenyl)methoxy)-3-chloro-5-
methoxyphenyl~methyl-5,7-dimethyl-2-ethyl-3H-imidazo
r4.5-blpyridine
Step A- Preparation of methyl 2-(2-chloro-4-hydroxy-
~ethyl-6-methoxy~henoxy~-2-phenylacetate
Using the K2C03/acetone conditions for
phenol alkylation described in Step A of Example 30,
0.50 g (2.65 mmol) of 3-chloro-4-hydroxy-5-methoxy-
benzaldehyde was alkylated with 0.668 g (2.92 mmol)
of methyl 2-bromophenylacetate to afford 0.570 g
(64Z) of the title compound.
1~ NMR (300 MHz, CDC13, ppm): ~ 1.65-1.75 (t, lE),
3.70 (s, 3E), 3.80 (s, 3H), 4.55 (d, 2H), 5.75 (s,
lH), 6.80 (s, lH), 6.90 (s, lH), 7.30-7.40 (m, 3H),
7.50-7.60 (m, 2H).
FAB-MS: m/e 337, 339 (M+l, 3:1 ratio).
WO91/11~ PCT/US91/00957
2075627
- 275 -
Ste~ B: Preparation of 2-(4-bromomethyl-2-chloro-6_
metho~y~h~noxy)-2-~henylacet~te
To a stirred and cooled (0C) solution of
0.570 g (1.69 mmol) of the product of Step A dissolved
in 6 mL of CH2C12 was added 0.702 g (2.11 mmol) of
carbon tetrabromide and 0.555 g (2.11 mmol) of
triphenylphosphine. After the addition the reaction
mixture was allowed to warm to room temperature and
was stirred 4 hours. The mixture was then e~aporated
in vacuo and purified on a silica gel flash chromato-
graphy column eluted with 20% ethyl acetate/hexane to
afford 0.580 g (86%) of the title compound.
H NMR (300 MHz, CDC13, ppm): ~ 3.75 (s, 3H), 3.80
(s, 3H), 4.35 (s, 2H), 5.65 (s, lH), 6.80 (s, lH),
6.95 (s, lH), 7.30-7.40 (m, 3H), 7.50-7.60 (m, 2H).
FAB-MS: m/e 398, 400, 402 (M+l).
Step C: Preparation of 3-t4-((1-carbometho~y-1-
phenyl)methoxy)-3-chloro-5-methoxyphenyl]-
methyl-5,7-dimethyl-2-ethyl-3H-imidazot4,5-b]
pyridine
The product of Step B (0.126 g, 0.31 mmol)
was used to alkylate 0.050 g (0.29 mmol) of
5,7-dimethyl-2-ethylimidazot4,5-b]pyridine (Example
24, Step C) according to the procedure described for
Step B of Example 27, which after purification
afforded 0.092 g (65%) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3H),
2.55 (s, 3H), 2-60 (s, 3H), 2.70-2.80 (m, 2H), 3.65
(s, 3H), 3.70 (s, 3H), 5.30 (s" 2H), 5.70 (s, lH)
6.63 (s, 1~), 6-68 (s, lH), 6.90 (s, lH), 7.25-7.35
(m, 3H), 7.45-7.55 (m, 2H).
FAB-MS: m/e 494, 496 (M+l).
wo 9l/ll~g 2 0 ~ 5 6 ~ ~ PCT/US91/00957
- 276 -
Step D: Preparation of 3-t4-((1-carboxY-l-phenyl)-
metho~y)-3-chloro-5-methoyphenyl3methyl-5,7-
dimethyl-2-et~ -3~ dAzOr4.S-blpyridi~e
Using the general procedure for e~ter
hydrolysis described in Step E of Example 19, 0.090 g
(0.18 mmol) of the product of Step C was converted to
0.070 g (80%) of the title compound.
lH NMR (300 M~z, CD30D, ppm): ~ 1.20-1.30 (t, 3E),
2.60 (s, 3H), 2.65 (s, 3H), 2.80-2.90 (m, 2H), 3.75
lo (s, 3H), 5.45 (s, 2H), 5.70 (s, lE), 6.60 (s, 1~),
6.85 (s, lH), 7.05 (s, lH), 7.35-7.45 (m, 3E),
7.45-7.55 (m, 2H).
FAB-MS: m/e 480, 482 (M+l, 3:1 ratio).
F~am~le 38
3-t4-((1-Carboxy-l-phenyl)methoxy)-3,5-dichloro-
phenyl]methyl-5,7-dimethyl-2-ethyl-3H-imidazo[4,5-b]
pyr~dlne
Step A: Preparation of methyl 4-tert-butyldimethyl-
silylo~y-3.5-dichlorobenzoate
To a solution of 10.00 g (45.2 mmol) of
methyl 3,5-dichloro-4-hydroxybenzoate dissolved in
100 mL of CH2C12 was added 11.06 g (90.2 mmol) of
4-dimethylaminopyridine and 8.18 g (54.2 mmol) of
tert-butyldimethylChlorOSilane and the mixture was
stirred under N2 for 5 hours. The reaction mixture
was then filtered and the filtrate was diluted with
ethyl acetate. The solution was washed with water, 1
N HCl, saturated NaHC03, dried (MgS04), filtered and
WO91/11~9 PCT/US91/~gs7
i 2075627
- 277 -
evaporated. The residue was purified on a silica gel
flash chromatography column eluted with 5% ethyl
acetate/hexane to afford 7.70 g (51%) of the title
compound.
lH NMR (300 MHz, CDC13, ppm): ~ O.30 (s, 6~), 1.00
(s, 9H), 3.90 (s, 3H), 7.95 (s, 2~).
FAB-MS: m/e 335, 337, 339 (M+l).
Step B: Preparation of 4-tert-butyldimethylsilyloxy-
lo 3~5-dichlorobenzyl alcohol
To a stirred and cooled (0C) solution of
7.70 g (23.0 mmol) of the product of Step A dissolved
in 50 mL of anhydrous T~F was added 23.0 mL (23.0
mmol) of a 1 M solution of lithium aluminum hydride
in T~F. After the addition was complete the reaction
mixture was allowed to warm to room temperature and
stirred 3.5 hours. The stirred reaction was then
quenched by dropwise addition of 0.88 mL water, then
0.88 mL of 15% NaOH, and finally 2.62 mL water. The
reaction mixture was then filtered and concentrated
in vacuo. The residue was dissolved in ethyl
acetate, washed with 1 N ~Cl, ~turated Na~C03, dried
(MgS04), and e~aporated La vacuo to afford 1.83 g
(26%) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 0.30 (s, 6H), 1.05
(s, 9H), 1.80 (br s, lH), 4.55 (s, 2H), 7.22 (s, 2H).
FAB-MS: m/e 306 (M+l).
WO91/11~9 PCT/USgl/00957
2o7~627
- 278 -
Ste~ C: Preparation of 3,5-dichloro-4-hydro~ybenzyl
~lcohol
To a solution of 1.83 g (5.96 mmol) of the
product of Step B dissolved in 6 mL of '1~ was added
5.96 mL (5.96 mmol) of a 1 M solution of tetra-n-
butylammonium fluoride in THF and the reaction
mi~ture was stirred at room temperature 30 minutes.
The solution was then e~aporated Ln y~cuo a~d the
residue was purified on a silica gel flash chromato-
lo graphy column eluted with 4% methanol/chloroform to
afford 0.733 g (64%) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 1.90-4.10 (br, 2H)
4.60 (s, 2H), 7.27 (s, 2H).
FAB-MS: m/e 192 (M+l).
Step D: Preparation of methyl 2-(2,6-dichloro-4-
hydroxymethylpheno~y)-2-~henylacetate
Using the K2C03/acetone conditions for
phenol alkylation described in Step A of E~ample 30,
0-400 g (2.07 mmol) of the product of Step C was
alkylated with 0.522 g (2.28 mmol) of methyl
2-bromophenylacetate to afford 0.144 g (20Z) of the
title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 1.70-1.80 (t, 3H),
2s 3.75 (s, 3H), 4.55 (d, 2H), 5.75 (s, 1~), 7.22 (s,
2H), 7.30-7.40 (m, 3H), 7.45-7.55 (m, 2H).
FAB-MS: m/e 341, 343, 345 (M+l, 10:6:1 ratio).
WO91/11~9 PCT/USgl/~957
- 20~5627
- 279 -
Step ~: Preparation of methyl 2-(4-bromomethyl-2,6-
dichloropheno~y)-2-phenyl~cet~te
To a stirred and cooled (0C) ~olution of
O.140 g (O.41 mmol) of the product of Step D
dissolved in 2 mL of CH2C12 was added 0.170 g (0.51
mmol) of carbon tetrabromide and 0.135 ~ (0.51 mmol)
of triphenylphosphine. After the addition the
reaction mixture was allowed to warm to room temper-
ature and was stirred overnight. The mixture was
lo then evaporated in vacuo and purified on a silica gel
flash chromatography column eluted with 15/. ethyl
acetate/hexane to afford 0.130 g (78Z) of the title
compound.
lH NMR (300 MHz, CDC13, ppm): ~ 3.75 (s, 3H), 4.30
(s, 2H), 5.75 (s, lH), 7.27 (s, 2H), 7.30-7.40 (m,
3H), 7.45-7.55 (m, 2H).
FAB-MS: m/e 405 (M+l).
Ste~ F: Preparation of 3-[4-((1-carbomethoxy-1-
phenyl)methoxy)-3,5-dichlorophenyl~methyl-
5,7-dimethyl-2-ethyl-3H-imidazo~4,5-b]
pyridine
The product of Step E (0.126 g, 0.31 mmol)
was used to al~ylate 0.050 g (0.29 mmol) of 5,7-
dimethyl-2-ethylimidazo~4,5-b]pyridine (Example 24,
Step C) according to the procedure described for Step
B of Example 27, which after purification afforded
0.096 g (68%) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 1.20-1.30 (t, 3~),
2.58 (s, 3H), 2.62 (s, 3H), 2.65-2.75 (m, 2H), 3.75
(s, 3H), 5.30 (s, 2~), 5-75 (s, 1~), 6.90 (s, lH),
7.00 (s, 2H), 7.25-7.35 (m, 3H), 7.45-7.55 (m, 2E).
FAB-MS: m/e 498, 500, 502 ~M+l).
WO91/11~ PCT/USgl/o~s~7
20~62~ -`
- 280 -
Step G: Preparation of 3-t4-((1-carboxy-1-phenyl)-
methoxy)-3,5-dichlorophenyl]methyl-5,7-
dimethyl-2-et~yl-3~ idazor4.5-bl~yridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.092 g
(0.18 mmol) of the product of Step F was con~erted to
0.080 g (90%) of the title compound.
lH MMR (300 M~z, CD30D, ppm): ~ 1.20-1.30 (t, 3H),
2.61 (s, 3H), 2.65 (s, 3H), 2.80-2.95 (m, 2H), 5.45
1~ (s, 2H), 5.65 (s, lH), 7.05 (s, 2H), 7.25-7.35 (m,
3H), 7.45-7.55 (m, 2H).
FAB-MS: m/e 484, 486, 488 (M+l, 10:6:1 ratio).
F~ le 39
1~
3-[4-((1-Carboxy-l-phenyl)methoxy)-2-chlorophenyl]-
methyl-5 7-dimethyl-2-ethyl-3H-imidazor4.5-blpyridine
Step A: Preparation of methyl 2-(3-chloro-4-formyl-
phenoxy)-2-phenylacetate
Using the K2C03/acetone conditions for
phenol alkylation described in Step A of Example 30,
1.00 g (6.41 mmol) of 2-chloro-4-hydroxybenzaldehyde
was alkylated with 1.61 g (7.05 mmol) of methyl
2-bromophenylacetate to afford 1.49 g (76%) of the
title compound.
H NMR (300 MHz, CDC13, ppm): ~ 3.75 (s, 3H), 5.68
(s, lH), 6.90 (d, 1~), 7.00 (s, lH), 7.35-7.45 (m,
3H), 7.50-7.60 (m, 2H), 7.85 (d, lH), 10.30 (s, lH).
FAB-MS: m/e 305, 307 (M+l).
WO91/11~ PCT/US91/~ss7
207~627
- 281 -
Step B: Preparation of methyl 2-(3-chloro-4-hydr
met~ylphe~oxy)-2-~henylAcetate
A stirred solution of 1.49 g (4.90 mmol) of
the product of Step A dissolved in 20 mL of methanol
was treated with 0.093 g (2.46 mmol) of sodium
borohydride at room temperature. After S minutes the
reaction mixture was partitioned between ethyl
acetate and 1 N hydrochloric acid, and the organic
layer was ~eparated. The product was washed with
water, brine, dried (MES04), filtered and evaporated
~n ~acuo. The residue was purified on a ~ilica gel
flash chromatography column eluted with 25% ethyl
acetate/hexane to afford 1.380 g (92%) of the title
compound.
1~ NMR (400 MHz, CDC13, ppm): ~ 1.80-1.85 (t, 1~),
3.75 (s, 3~), 4.70 (d, 2~), 5.60 (s, lH), 6.85 (d,
lH~, 7.00 (s, 1~), 7.30-7.45 (m, 4~), 7.50-7.60 (m,
2H).
Step C: Preparation of methyl 2-(4-bromomethyl-3-
chloro~henoxy)-2-phenylacetate
To a stirred and cooled (O-C) solution of
1.38 g (4.51 mmol) of the product of Step D dissolved
in 18 mL of CH2C12 was added 1.87 g (5.64 mmol) of
carbon tetrabromide and 1.48 g (5.64 mmol) of
triphenylphosphine. After the addition the reaction
mixture was allowed to warm to room temperature and
was stirred 3 hours. The miæture was then evaporated
in vacuo and purified on a silica gel flash chromato-
graphy column eluted with 10/. ethyl acetate/he~ane toafford 1 60 g (96%) of the title compound.
WO91/11~9 PCT/US91/~ss7
20~6~
- 282 -
H NMR (400 MHz, CDC13, ppm): ~ 3.75 (s, 3H), 4.55
(s, 2H), 5.60 (s, lH), 6.80 (d, lH), 7.00 (s, lH),
7.30 (d, lH), 7.35-7.45 (m, 3H), 7.50-7.60 (m, 2H).
FAB-MS: m/e 369, 371, 373 (M+l).
Step D: Preparation of 3-t4-((1-carbometho~y-1-
phenyl)methoxy)-2-chlorophenyl~methyl-5,7-
dimethyl-2-ethyl-3~-imidAzor4.5-blpyridine
The product of Step C (0.116 g, 0.31 mmol)
was used to alkylate 0.050 g (0.29 mmol) of 5,7-
dimethyl-2-ethylimidazo~4,5-b]pyridine (E~ample 24,
Step C) according to the proc~dure described for Step
B of Example 27, which after purification afforded
0.094 g ~69%) of the title compound.
lH NMR (400 MHz, CDC13, ppm): ~ 1.20-1.30 (t, 3H),
2.55 (s, 3H), 2.62 (s, 3H), 2.65-2.75 (m, 2H), 3.70
(s, 3H), 5.45 (s, 2H), 5.55 (s, lH), 6.45 (d, lH),
6.65 (d, lH), 6.90 (s, lH), 7.05 (s, lH), 7.35-7.40
(m, 3H), 7.45-7.55 (m, 2H).
FAB-MS: m/e 464, 466 (M+l, 3:1 ratio).
Step ~: Preparation of 3-[4-((1-carboxy-1-phenyl)-
methoxy)-2-chlorophenyl~methyl-5,7-dimethyl-
2-ethyl-3H-imidazor4.5-blpyridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.090 g
(0.18 mmol) of the product of Step D was converted to
0.030 g (34%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ 1.25-1.35 (t, 3H),
2.60 (s, 3H), 2.65 (s, 3H), 2.80-2.90 (m, 2H),
5.55-5.65 (m, 3H), 6.50 (d, lH), 6.85 (d, lH), 7.05
(s, lH), 7.15 (s, lH), 7.35-7.45 (m, 3H), 7.55-7.65
(m, 2H).
FAB-MS: m/e 450, 452 (M+l, 3:1 ratio).
WO 91/11999
PCT/US91/00957
20756~7
- 283 -
F.Y~ple 40
3-[4-((1-Carboxy-1-(3-phenyl)propyloxy)phenyl3methyl-
5.7-dimet~yl-2-etbyl-3~ idAzor4.5-b~pyri~i~e
Step A: Preparation of 3-t4-((1-carboetho~Y-1-(3-
phenyl)propo~y)phenyl~methyl-5,7-d~methyl-2-
ethyl-3~I-imid~zor4~5-blpyridine
To a suspension of 20 mg (0.18 mmol) of a
35~/ oil dispersion of ~H in 0.4 mL of anhydrous DMF
was added 0.050 g (0.18 mmol) of 5,7-dimethyl-2-ethyl-
3-(4-hydroxyphenyl)methyl-3~-imidazot4,5-b]pyridine
(Example 24, Step E) and the reaction mi~ture was
stirred under N2 at room temperature. After 15
minutes, 0.050 g (0.18 mmol) of 18-crown-6 and a
solution of 0.053 g (0.20 mmol) of ethyl 2-bromo-4-
phenylbutanoate dissolved in 0.4 mL of DMF were
added. The reaction mixture was stirred at room
temperature 4 hours, then partitioned between ethyl
acetate and water. The organic layer was ~eparated,
washed with water, dried (MgS04), filtered and
evaporated. The residue was purified on a silica gel
flash chromatography column eluted with 50% ethyl
acetate/hexane to afford 0.063 g (75%) of the title
COmpound.
1~ NMR (300 M~z, CDC13, ppm): ~ 1.17 (t, J=8 Hz, 3~),
1.27 (t, J=8 Hz, 3~), 2.11-2.30 (m, 2E), 2.56 (s,
3~), 2.60 (s, 3H), 2.70-2.90 (m, 4H), 4.15 (q, J~8
Hz, 2H), 4.50 (td, J=6, 7 Hz, lH), 5.35 (s, 2H), 6.74
(d, J=10 Hz, 2H), 6.86 (s, lE), 7.04 (d, J=10 Ez,
2H), 7.10-7.28 (m, 5~).
FAB-MS: m/e 472 (M+l)
WO91/11~9 PCT/USgl/~ss7
207 56~7 `-``
- 2B4 -
Step B: Preparation of 3-t4-((1-carboxY-1-(3-phenyl)-
propoxy)phenyl]methyl-5,7-dimethyl-2-ethyl-
3~-imid~zor4.5-bl~yridine
Using the general procedure for ester
hydrolysis described in Step E of E~ample 19, 0.063 g
(O.13 mmol) of the product of Step A was converted to
0.047 g (80%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ 1.27 (t, J~8 Hz, 3H),
2.16 (m, 2H), 2.63 (s, 3~), 2.66 (s, 3H), 2.80-2.95
lo (m, 4H), 4.55 (t, J=6 Hz, lH), 5.52 (s, 2H), 6.86 (d,
J=10 Hz, 2H), 7.07 (s, lR), 7.12 (d, J=10 ~z, lH),
7.15-7.32 (m, 5H).
FAB-MS: m/e 444 (M+l).
F.~rA~1e 41
3-[4-((1-Carboxy-1-(2-phenyl)ethoxy)phenyl]methyl-5,7-
dimethyl-2-ethyl-3R-imidazor4 5-bl~yridine
Step A: Preparation of methyl 2-(4-hydroxymethyl-
~henoxy)acetate
Using the K2C03/acetone conditions for
phenol alkylation described in Step A of Example 30,
8.00 g (64.5 mmol) of 4-hydroxybenzyl alcohol was
25 alkylated with 11.84 g (77.4 mmol) of methyl bromo-
acetate to afford 6.00 g (48%) of the title compound.
H NMR (300 MHz, CDC13, ppm): ~ 1.74 (br s, lH), 3.76
(s, 3H), 4.61 (br s, 4H), 6.86 (d, J=10 Hz, 2H), 7.26
(d, J=10 Hz, 2E).
FAB-MS: m/e 305 (M+1).
WO91/11~ PCT/US91/~957
2075627
- 285 _
Step B: Preparation of methyl 2-(4-tert-butyl-
dimet~ylsilyl~y~ethy~phenoyy)~cet~te
To a solution of 4.00 g (20.4 mmol) of the
product of Step A dissolved in 30 mL of C~2C12 was
added 5.00 g (40.8 mmol) of 4-dimethylaminopyridine
and 3.69 g (24.5 mmol) of tert-butyldimethylchloro-
silane and the mi~ture was stirred under N2 for 5
hours. The reaction mi~ture was then filtered and
the filtrate was diluted with ethyl acetate. The
solution was washed with water, 1 N HCl, saturated
NaHC03, dried (MgS04), filtered and evaporated.
Drying in vacuo afforded 6.30 g (99%) of the title
compound.
1~ NMR (300 M~z, CDC13, ppm): ~ O.09 (s, 6~), 0.94
(s, 9~), 3.80 (s, 3H), 4.63 (s, 2~), 4.68 (s, 2~),
6.86 (d, J=10 ~z, 2H), 7.26 (d, J=10 ~z, 2H).
FAB-MS: m/e 311 (M+l).
Step C: Preparation of methyl 2-(4-tert-butyl-
dimethylsilyloxymethylphenoxy)-3-phenyl-
propanoate
A solution of 1.00 g (3.23 mmol) of the
product of Step B dissolved in 4 mL of toluene was
stirred under a N2 atmosphere and cooled to 0CC with
an ice-water bath. To this solution was added 9.68
mL (4.85 mmol) of a 0.5 M solution of potassium
bis(trimethylSilyl)amide in toluene. After a 15
minute interval, 0.46 mL (3.88 mmol) of benzyl
bromide was added and the reaction mixture was
allowed to warm to room temperature and stirred 1
hour. Several milliliters of methanol were added to
consume excess base, and the reaction was partitioned
wo 9~ 2 ~ 7 ~ 6 2 ~ PCT/US91/~9~7
- 286 -
between ethyl acetate and 1 N ECl. The organic layer
was separated, washed with ~aturated NaHC03, brine,
dried (MgS04), filtered and evaporated. The residue
was purified on a ~ilica gel flash chromatography
column eluted with 15% ethyl acetate/he~ane to afford
0.540 g (42%) of the title compound.
H NMR (300 MHz, CDC13, ppm): ~ O.09 (s, 6H), 0.94
(s, 9H), 3.24 (m, 2H), 3.70 (s, 3H), 4.64 (s, 2H),
4.80 (dd, J=6,7 Hz, lH), 6.B0 (d, J=10 Hz, 2H),
lo 7-09-7-35 (m, 7H).
FAB-MS: m/e 401 (M+l).
Step D: Preparation of methyl 2-(4-hydroxymethyl-
~henoxy)-3-~henyl~ro~anoate
To a solution of 0.520 g (1.30 mmol) of the
product of Step C dissolved in 3 mL of anhydrous T~F
was added 1.3 mL (1.3 mmol) of a 1 N solution of
tetra-n-butylammonium fluoride in THF and the mixture
was stirred at room temperature 1.5 hours. The
mixture was then concentrated Ln vacuo and the
residue was purified on a silica gel flash chromato-
graphy column eluted with 70% ethyl acetate/hexane to
afford 0.230 g (62%) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 1.55 (br s, lH), 3.23
(m, 2~), 3.66 (s, 3H), 4.56 (d, J=9 Hz, 2H), 4.74
(dd, J=6,7 ~z, lH), 6.83 (d, J=10 Hz, 2H), 7.10-7.30
(m, 7H).
FAB-MS: m/e 285 (M+l).
WO91/11~ PCT/US91/~ss7
2D7~627
- 287 -
Ste~ F: Preparation of methyl 2-(4-bromomethyl_
pheno~y~-3-~henyl~rop~noAte
To a cooled (O-C) ~olution of 0.230 g (0.80
mmol) of the product of Step D dissolved in 4 mL of
CH2C12 was added 0.263 g (1.00 mmol) of carbon
tetrabromide and 0.333 g (1.00 mmol) of triphenyl-
phosphine. After 15 minutes at O-C, the reaction
mixture was allowed to warm to room temperature and
was stirred for an additional 6 hours. The reaction
was then concentrated ~ vacuo and purified on a
silica gel flash chromatography column eluted with 5%
ethyl acetate/hexane to afford 097 g (35%) of the
title compound.
lH NMR (300 M~z, CDC13, ppm): ~ 3.20 (m, 2H), 3.80
(s, 3H), 4.44 (s, 2H), 4.77 (dd, J=6,7 Hz, lH), 6.76
(d, J=10 Hz, 2H), 7.17-7.35 (m, 7H).
Step F: Preparation of 3-[4-((1-carboethoxy-1-(2-
phenyl)ethoxy)phenyl]methyl-5,7-dimethyl-2-
ethyl-3H-imidazor4.5-blpyridine
The product of Step E (0.094 g, 0.27 mmol)
was used to alkylate 0.040 g (0.23 mmol) of 5,7-
dimethyl-2-ethylimidazo~4,5-b]pyridine (Example 24,
Step C) according to the procedure described for Step
B of Example 27, which after purification afforded
0.054 g (53%) of the title compound.
lH N~R (300 M~z, CDC13, ppm): ~ 1.26 (t, J=8 Hz, 3H),
2.55 (s, 3H), 2.60 (s, 3H), 2.74 (q, J=8 Hz, 2H),
3.20 (m, 2H), 3.65 (s, 3H), 4.72 (dd, J=6,7 ~z, lH),
5.34 (s, 2H), 6.70 (d, J=10 ~z, 2H), 6.83 (s, lH),
7.02 (d, J=10 Hz, 2H), 7.16-7.30 (m, 5H).
FAB-MS: m/e 444 (M+l).
WO91/11~ PCT/US91/~ss7
207~627
- 288 -
Step G: Preparation of 3-t4~ -carboxy-l-(2-phenyl)-
etho~y)phenyl]methyl-5.7-dimethyl-2-ethyl-3~-
imid~zor4.5-blpyridi~e
Using the ~eneral procedure for ester
hydrolysis described in Step E of Example 19, 0.054 g
(0.12 mmol) of the product of Step A was converted to
0.042 g (81%) of the title compound.
1~ MMR (300 M~z, CD30D, ppm): ~ 1.25 (t, J=8 ~z, 3H),
2.60 (s, 3~), 4.63 (s, 3~), 4.85 (g, J=8 ~z, 2~),
lo 3.18-3.25 (m, 2~), 4.78 (dd, J=6,7 Hz, 1~), 5.45 (s,
2~), 6.80 (d, J=10 ~z, 2~), 7.02-7.10 (m, 3~),
7.15-7.35 (m, 5H).
FAB-MS: m/e 430 (M+l).
T~ le 42
3-[4-((1-Carboxy-l-phenyl)methoxy)-3-(2-propen-1-yl)-
phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazot4,5-b]
pyridine
Ste~ A: Preparation of methyl 4-(2-propen-1-yl)oxy-
benzoate
A 2 L flask was equipped with a mechanical
stirrer, a reflux condenser and a stopper, then
charged with 50.05 g (0.329 mol) of methyl 4-hydroxy-
benzoate, 960 mL of acetone, 22.50 g (1.625 mol) of
anhydrous potassium carbonate, 80.5 mL (112.6 g,
0.932 mol) of allyl bromide and the mixture was
stirred and refluxed for 14 hours. The mixture was
cooled to room temperature, filtered and concentrated
to an oil. The residual oil was purified by
distillation (97-C @ 0.03 mm Eg) to afford 53.52 g
(86~/~) of the title compound.
WO91/11~ PCT/US91/0095~
: 20756~7
- 289 -
1~ NMR (300 M~z, CDC13, ppm): ~ 3.84 (s, 3~), 4 56
(d, J=7 ~z, 2~ .Z8 (dd, J=3,12 Hz, 1~), 5.40 (dd,
J=3,19~z, 1~), 5.96-6.10 (m, 1~), 6.90 (d, J=10 ~z,
2H), 7.96 (d, J=10 Hz, 2~).
FAB-MS: m/e 193 (M~l).
Ste~ ~: Preparation of methyl 4-hydro~y-3-(prop-2-
en-l-yl)benzoate
A solution of 15.05 g (78.3 mmol) of the
lo product of Step A in 25 mL of 1,2-dichlorobenzene was
magnetically stirred and refluxed (183-C~ under an
argon atmosphere for 18 hours. At this point, the
reaction mi~ture was cooled to room temperature and
applied to a 6 cm diameter by 18 cm silica gel flash
chromatography column and eluted with 25Z ethyl
acetate-hexane to separate the 1,2-dichlorobenzene,
then with 40% ethyl acetate-hexane to elute the
product. The product fractions were concentrated
in Y~CUO and the residual oil was crystallized from
2~ hexane to afford 13.70 g (91%) of the title compound
1~ NM~ (300 M~z, CDC13, ppm): ~ 3.42 (d, J=8 Hz, 2H),
3.88 (s, 3~), 5.14-5.20 (m, 2~), 5.48 (s, lH),
5.94-6.06 (m, lE), 6.82 (d, J=12 H~ ), 7.80-7.85
(m, 2~).
~AB-MS: m/e 193 (~+1).
Step C: Preparation of methyl 4-(tert-butyldimethyl-
silylD~y)-3-~2-~ro~en-1-yl)~e~zo~t~
To a ~olution of 5.168 g (26.9 mmol) of the
product of Step B in 50 mL of dichloromethane was
added 4.40 mL (2.95 mmol) of triethylamine, 4.46 g
WO91/11~ ~ 0 7 5 ~ 2 7 PCT/US91/00957
- 290 -
(2.95 mmol) of tert-butyldimethylchlorosilane,0.100 g
of 4-dimethylaminopyridine, and the reaction mi~ture
was ~tirred at room temperature for 2 hours. The
mixture was then diluted with S0 mL dichloromethane,
washed with lO0 mL 1 N hydrochloric acid, dried
(MgS04), filtered and evaporated. The residual oil
(7.993 g, 97%) was used in the next ~tep without
further purification.
lH NMR (300 M~z, CDC13, ppm): ~ 0.24 (s, 6E), 1.02
(s, 9H), 3.36 (d, J=8 Hz, 2~), 3.84 (s, 3H),
4.98-5.08 (m, 2H), 5.88-6.03 (m, lH), 6.78 (d, J=ll
Hz, lH), 7.76-8.40 (m, 2H).
FAB-MS: m/e 307 (M+l).
Step D Preparation of 4-(tert-butyldimethyl-
silyloxy)3-(2-propen-1-yl)benzyl alcohol
To a magnetically stirred solution of 8.523
g (28.0 mmol) of the product from Step C in 35 mL of
anhydrous T~F was added 15.0 mL of a 1.0 M ~olution
f lithium aluminum hydride in TJF, and the reaction
- mi~ture was stirred under a nitrogen atmosphere for 2
hours. At this point, the reaction was quenched by
cautious addition of 10 mL water, the re~ulting
precipitate was di~solved by addition of 1.0 N
hydrochloric acid and the product was extracted into
ethyl acetate. The organic layer was separated,
dried (MgS04), filtered and e~aporated in ~acuo to
afford 7.258 g (93%) of the title compound.
lH NMR (300 M~z, CDC13, ppm): ~ O.20 (s, 6H), 1.00
(s, 9H), 3.34 (d, J=8 Hz, 2H), 3.84 (s, lH), 4.57 (s,
2H), 4.97-5.07 (m, 2H), 5.88-6.03 (m, lH), 6.86 (d,
J=10 Hz, lH), 7.05-7.14 (m, 2H).
FAB-MS: m/e 279, 261 (M+l).
~ WO91/11~9 PCT/US91/oogs7
2075627
- 291 -
Step F: Preparation of 4-(tert-butyldimethyl_
silyl)3-(7-prope~-1-y~benzy~ bromide
To a magnetically stirred solution of 7.258
g (26 mmol) of the product from Step D and 10.281 g
(31 mmol) of carbon tetrabromide in 50 mL of dry
dichloromethane was slowly added 8.131 g (31 mmol) of
triphenylphosphine at 0C under a nitrogen atmosphere.
The reaction mixture was stirred 45 minutes and
allowed to warm to room temperature. At this point,
lo the reaction mixture was applied to a silica gel
flash chromatography column and was eluted with
dichloromethane. Evaporation of the product
fractions and drying in vacuo afforded 7.651 g (86%)
of the title compound as a ~iscous oil.
lH NMR (300 M~z, CDC13, ppm): ~ 0.23 (s, 6H), 1.00
(s, 9H), 3.34 (d, J=8 Hz, 2H), 4.45 (s, 2H),
4.98-5.09 (m, 2H), 5.86-6.02 (m, lE), 6 . 74 (d, J=10
Hz, lH), 7.08-7.16 (m, 2H).
~AB-MS: m/e 343, 341 (M+l).
Ste~ F: Preparation of 5,7-dimethyl-2-ethyl-3-~4-
tert-butyldimethylsilyloxy-3-(2-propen-1-yl)-
~henyllmethyl-3H-imidazo r 4.5-blpyridine
To a solution of 1.029 g (5.9 mmol) of
5,7-dimethyl-2-ethylimidazO[4,5-b]pyridine (Example
24, Step C) dissolved in 10 mL of dry DMF was added
0.258 g (6.5 mmol) of a 60% oil dispersion of sodium
hydride and the reaction mixture was stirred under a
nitrogen atmosphere for 1 hour. At this point,
hydrogen evolution had ceased, and a solution of
2.210 g of the product of Step E dissol~ed in 2.0 mL
WO91/11~9 PCT/USgl/00957
2 0 7 5 6 2 ~ --
- 292 -
of dry DMF was added via ~yringe. The reaction was
~tirred an additional 2 hours at room temperature and
then partitioned between ethyl acetate and water.
The organic layer was extracted, washed with brine,
dried (MgS04), filtered and evaporated. The residual
oil was purified on a silica gel flash chromatography
column eluted with 50'~ ethyl acetate-hexane.
F.vaporation of the purified fraction and drying
in vacuo afforded 1.519 g (59%) of the title compound.
1~ NMR (300 M~z, CDC13, ppm): ~ O.16 (s, 6H), 0.96
(s, 9~), 1.27 (t, J=9 ~z, 3H), 2.57 (s, 3H), 2.60 (8,
3H), 2.76 (q, J=9 Hz, 2H), 3.28 (d, J=8 ~z, 2H),
4 93-5.03 (m, 2~), 5.33 (s, 2~), 5.81-5.95 (m, lH),
6.64 (d, J=10 Hz, lH), 6.76 (dd, J=3,10 Hz, lH), 6.86
(s, lH), 7.00 (d, J=3 ~z, lH).
FAB-MS: m/e 436 (M+l).
Step G: Preparation of 5,7-dimethyl-2-ethyl-3_[4_
hydroxy-3-(2-propen-1-yl)phenyl]methyl-3~-
imidazor4.5-blpyridine
To a solution of 1.519 g (3.48 mmol) of the
product of Step F in 8.0 mL of anhydrous THF was
added 3.6 mL of a 1.0 M solution of tetra-n-butyl-
ammonium fluoride in T~F and the reaction mixture was
stirred for 2.5 hours at room temperature. The
reaction mixture was then evaporated ~n vacuo and the
residual oil was chromatographed on a silica gel
flash chromatography column eluted with ethyl
acetate. The purified fractions were combined,
evaporated and dried ~n vacuo to afford the title
compound.
WO91/11~9 PCT/US91/ooss7
'' . 2075627
- 293 -
lH NMR (300 M~z, CDC13, ppm): ~ 1.24 (t, J=9 ~z, 3H),
2.57 (s, 3H), 2.60 (s, 3H), 2.73 (q, Jc9 Hz, 2E),
3.31 (d, J=8 Hz, 2H), 5.03-S.10 (m, 2E), 5.33 (~,
2H), 5.88-6.02 (m, lH), 6.36 (d, J=10 Ez, 1~), 6.48
(dd, J=3,10 Hz, lH), 6.84-6.89 (m, 2H), 7.37 (br 8,
lH).
FAB-MS: m/e 322 (M+l).
Ste~ ~: Preparation of 5,7-dimethyl-2-ethyl-3-t4-
((1-carbomethoxy-1-phenyl)metho~y)-3-
(2-propen-1-yl)phenyl]-methyl-3H-imidazo~4,5-
bl pyridine
To a solution of 0.171 g (0.53 mmol) of the
product of Step G dissolved in 2.5 mL of anhydrous
DMF was added 0.023 g (0.58 mmol) of a 60% oil
dispersion of sodium hydride and the reaction mixture
was stirred under a nitrogen atmosphere for 30
minutes at room temperature. A solution of 0.134 g
of methyl a-bromophenylacetate in 1.0 mL of DMF was
then added ~ia syringe and the reaction mixture was
stirred an additional 1.5 hours. The reaction mi~ture
was then partitioned between ethyl acetate and water,
the organic layer was separated, dried (MgS04),
filtered and evaporated. The residual oil was
purified on a silica gel flash chromatography column
eluted with 75% ethyl acetate-hexane to afford after
evaporation of the purified fractions and drying
in vacuo 0.212 g (8570) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ 1.26 (t, J=9 Hz, 3H),
2.57 (s, 3H), 2.60 (s, 3H), 2.76 (q, J=9 Hz, 2H),
3.45 (d, J=8 Hz, 2~), 3.66 (s, 3H), 4.99-5.07 (m,
WO91/11~9 PCT/USgl/ooss7
207~27
- 294 -
2H), 5.34 (s, 2~), 5.57 (s, lH), 5.88-6.04 (m, lE),
6.59 (d, J=10 ~z, lH), 6.79 (dd, J=3,10 ~z, l~), 6.86
(s, 1~), 7.05 (d, J=3 Hz, lE), 7.30-7.40 (m, 3H),
7.48-7.56 (m, 2H).
FAB-MS: m/e 470 (M+l).
Ste~ I: Preparation of 3-~4-((1-carboxy-1-phenyl)-
methoxy)-3-(2-propen-1-yl)phenyl]methyl-5,7-
dimethyl-2-ethyl-3~-imidazor4.5-bl~ysidine
To a solution of 0.141 g (0.30 mmol) of the
product of Step H dissolved in 2.0 mL of methanol was
added 0.25 mL of a 1.0 N solution of sodium hydroxide
and the reaction was stirred at room temperature for
2 hours. The reaction mi~ture was then adjusted to
pH 7 with 1.0 N hydrochloric acid and then partitioned
between ethyl acetate and water. The organic layer
was separated, dried (MgS04), filtered, evaporated
and then purified on a silica gel flash chromatography
column eluted with chloroform-methanol-conc. ammonium
20- hydroxide (80:15:1). Evaporation of the purified
fractions and drying ~a vacuo afforded 0.092 g (69%)
of the title compound as a white amorphous solid.
lH NM~ (300 M~z, CD30D, ppm): ~ 1.27 (t, J=9 ~z, 3~),
2.61 (s, 3H), 2.64 (s, 3H), 2.89 (q, J=9 ~z, 2H),
3.40-3.52 (m, 2~), 4.95-5.06 (m, 2H), 5.53 (s, 2H),
S.73 (s, 1~), 5.94-6.13 (m, 1~), 6.84 (d, J=10 ~z,
1~), 6.95 (dd, J=3,10 Ez, 1~), 7.06 (br s, 2H),
7.36-7.44 (m, 3H), 7.57-7.64 (m, 2~).
FAB-MS: m/e 456 (M+l).
WO 91/11999 PCI'/US91/009s7
2075627
- 295 -
~ le 43
3-t4-((1-Carboxy-l-phenyl)methoxy)-3-propylphenyl]-
methyl-5.7-dimet~yl-2-ethyl-3~-imidazor4.5-bl~yridine
Step A: Preparation of 5,7-dimethyl-2-ethyl-3-[4-
hydroxy-3-propylphenyl]methyl-3H-imidazo
r4.5-blpyridine
A solution of 0.255 (0.79 mmol) of the
product of Example 1, Step G in 10 mL ethanol was
placed in a small Parr hydrogenation flask and 50 mg
of a 10% palladium on carbon catalyst was added. The
reaction mixture was then shaken in a Parr apparatus
under a 45 psig hydrogen atmosphere for 1 hour at
room temperature. The reaction mixture was then
remo~ed from the flask, filtered, evaporated and
dried ~a vacuo to afford 0.239g (93%) of the title
compound which was used in the next step without
further purification.
lH NMR (300 MHz, CDC13, ppm): ~ O.90 (t, J=9 Ez, 3~),
1.24 (t, J=10 Hz, 3H), 1.50-1.62 (m, 2H), 2.48 (t,
J=8 Hz, 2H), 2.56 (s, 3H), 2.59 (s, 3H), 2.72 (q, J=9
Hz, 2H), 5.32 (s, 3H), 6.23 (d, J=10 Hz, lH), 6.38
(dd, J=3,10 Hz, lH), 6.79 (d, J=3 Hz, lH), 6.87 (s,
lH), 7.68 (br s, lH).
FAB-MS: m/e 324 (M+l).
wo gl,ll~g 2 0 7 ~ 6`2 ~ PCT/US91/~957
- 296 -
Step B: Preparation of 5,7-dimethyl-2--ethyl-3-t4-
((l-carbomethoxy-l-phenyl)methoxy)-3-propyl-
phenyllmethyl-3~-imidazor4.5-blpyridine
To a solution of 0.062 g (0.19 mmol) of the
product of Step A in 1.5 mL of anhydrous DM~ was
added 8.4 mg of a 60% oil dispersion of sodium
hydride and the reaction mixture was stirred under a
nitrogen atmosphere. After the reaction mi~ture had
stirred 30 minutes at room temperature, hydrogen
evolution had ceased, and a solution of 0.048 g of
methyl 2-bromophenylacetate in 0.5 mL of dry DMF was
added via syringe. The reaction mixture was stirred
an additional 1.5 hours and then partitioned between
ethyl acetate and water. The organic layer was
separated, washed with brine, dried (MgS04),
filtered, evaporated and then chromatographed on a
silica gel flash chromatography column eluted with
50% ethyl acetate-hexane. The purified fractions
were combined, evaporated and dried in vacuo to
afford 59 mg (66%) of the title compound as a ~iscous
oil.
lH NMR (300 MHz, CDC13, ppm): ~ O.90 (t, J=9 Hz, 3H),
1.25 (t, J=9 Hz, 3H), 1.54-1.68 (m, 2H), 2.56-2.66
(m, 2H), 2.58 (s, 3H), 2.62 (s, 3H), 2.76 (q, J=9 Hz,
2H), 3.66 (s, 3H), 5.35 (s, 2H), 5.57 (s, lH), 6.59
(d, J=10 Hz, lH), 6.81 (dd, J=3,10 Hz, lH), 6.87 (s,
lH), 6.99 (d, J=3 Hz, lH), 7.28-7.40 (m, 3H),
7.49-7.56 (m, 2H).
FAB-MS: m/e 472 (M+l).
WO91/11~ PCT/US91/~957
~ 2075627
- 297 -
Step C: Preparation of 3-[4-((1-carboxy-1-phenyl)-
methoxy)-3-propylphenyl]methyl-5,7-dimethyl-
2-et~yl-3~-imidazor4.5-blpyridine
To a ~olution of 0.042 g (0.09 mmol) of the
product of Step B tissol~ed in 1.0 mL of methanol was
added 0.1 mL of a 1.0 N solution of sodium hydroxide
and the reaction mixture was stirred for 3 hours at
room temperature. The reaction mixture was then
adjusted to pH 7 with 1.0 N hydrochloric acid and
then partitioned between ethyl acetate and water.
The organic layer was separated, dried (MgSO4),
filtered, evaporated and then purified on a ~ilica
gel flash chromatography column eluted with
chloroform-methanol-conc. ammonium hydroxide
(80:15:1). E~aporation of the purified fractions and
drying L~ vacuo afforded 0.021 g (51%) of the title
compound as a white amorphous solid.
lH NMR (300 MHz, CD30D, ppm): ~ O.90 (t, J=8 Hz, 3H),
1.26 (t, J=9 Hz, 3H), 1.53-1.67 (m, 2H), 2.61 (s,
3H), 2.65 (s, 3H), 2.66-2.80 (m, 2H), 2.86 (q, J=8
Hz, 2H), 5.49 (s, 2H), 5.54 (s, lH), 6.80 (d, J=10
Hz, lH), 6.90 (dd, J=2, 10 Hz, lH), 6.98 (d, J=2 Hz,
lH), 7.06 (s, lH), 7.30-7.44 (m, 3H), 7.60-7.66 (m,
2H).
FAB-MS: m/e 458 (M+l).
T.~rA~1e 44
3-[4-((1-Carboxy-1-(2-methylphenYl))methoXy)-3-propyl-
phenyl~methyl-5~7-dimethyl-2-ethyl-3H-imidazo~4~5-b]
~yridine
WO 9~ S 6~ PCT/US91/00957
- 298 -
Step A: Preparation of 5,7-dimethyl-2-ethyl-3_t4-((1-
carbometho~y-l-(2-methylphenyl))methoxy)-
3-propylphenyl]-methyl-3H-imidazot4,5-b]
pyridi~e
To a suspension of 5.9 mg (2.45 mmol) of a
60% oil dispersion of NaH in 0.8 mL of DMF was added
0.066 g (0.20 mmol) of 5,7-dimethyl-2-ethyl-3-~4-
hydroxy-3-propylphenyl3methyl-3H-imidazot4,5-b]
pyridine (Example 43, Step A) and the mixture was
lo stirred at room temperature. After 20 minutes, a
solution of 0.060 g (2.45 mmol) of methyl 2-bromo-2'-
methylphenylacetate (prepared from 2'-methylphe`nyl-
acetic acid via a Hell-Volhard-Zelinsky reaction
similar to Step A of Example 17) dissol~ed in 0.5 mL
of DMF was added and the reaction mixture was stirred
an additional 1.5 hours. The reaction was then
partitioned between ethyl acetate and water, the
organic layer was separated, washed with water, dried
(MgS04), filtered and evaporated Ln ~acuo. The
residue was purified on a silica gel flash chromato-
graphy column eluted with 50% ethyl acetate/hexane to
afford 0.082 g (83%) of the title compound.
H NMR (300 M~z, CDC13, ppm): ~ O.90 (t, J=9 Hz, 3H),
1.25 (t, J=8 Hz, 3H), 1.50-1.65 (m, 2H), 2.44 (s,
3H), 2.56 (s, 3H), 2.60 (~, 3H), 2.52-2.65 (m, 2H),
2.75 (q, J=8 Hz, 2H), 3.67 (s, 3H), 5.34 (s, 2H),
5.74 (s, lH), 6.55 (d, J=10 Hz, lH), 6.78 (dd, J=2,
10 Hz, lH), 6.86 (s, lH), 6.98 (d, J=2 Hz, lH),
7.15-7.28 (m, 3H), 7.50-7.56 (m, lH).
FAB-MS: m/e 486 (M+l).
WO91/11~9 PCT/US91/00957
2075627
- 299 -
Ste~ B: Preparation Of 3-t4-((l-carboxy-1-(2-methyl-
phenyl~)metho~y)-3-propylphenyl~methyl-5,7-
~imet~yl-2-et~yl-3~-imidazor4.5-bl~yridine
Using the general procedure for e~ter
hydrolysis described in Step E of Example 19, 0.076 g
(0.16 mmol) of the product of Step A was con~erted to
0.040 g (~4%) of the title compound.
H NMR (300 MHz, CD30D, ppm): ~ 0.88 (t, J=9 Hz, 3H),
1.27 (t, J=8 Hz, 3H), 1.50-1.65 (m, 2~), 2.49 (s,
3H), 2.62 (s, 3~), 2.65 (s, 3H), 2.~-2.70 (m, 2H~,
2.88 (q, J=8 ~-, 2H), 5.48 (s, 2~), 5.83 (æ, lH),
6.78 (d, J=10 Hz, lH), 6.90 (dd, J=2,10 ~z, lH), 6.99
(d, J=2 Hz, 1~), 7.06 (s, 1~), 7.20-7.27 (m, 3~),
7.54-7.60 (m, lH).
lS FAB-MS: m/e 472 (M+l).
~m~le 45
3-[4-((1-Carboxy-1-(2-chlorophenyl))methoxy)-3-propyl-
phenyl]methyl-5,7-dimethyl-2-ethyl-3~-imidazot4,5-b]
pyridlne
Step A: Preparation of 5,7-dimethyl-2-ethyl-3-[4-((1-
carbomethoxy-1-(2-chlorophenyl))methoxy)-
3-propylphenyl]-methyl-3H-imidazot4,5-b]
~yridine
To a suspension of 6.3 mg (2.63 mmol) of a
60% oil dispersion of Na~ in 1.O mL of DMF was added
0.071 g (0.22 mmol) of 5,7-dimethyl-2-ethyl-3_~4_
hydroxy-3-prOpylphenyl]methyl-3~-imidaZO[4,5-b]
pyridine (Example 43, Step A) and the mi~ture was
wo 9~ 2 0 7 ~ ~ 2 ~ PCT/US91/~9~7
- 300 -
stirred at room temperature. After 20 minutes, a
solution of 0.069 g (0.26 mmol) of methyl 2-bromo-2'-
chlorophenylacetate (Example 4, Step A) dis~olved in
O.75 mL of DMF was added and the reaction mi~ture was
~tirred an additional 2 hours. The reaction was then
partitioned between ethyl acetate and water, the
organic layer was ~eparated, washed with water, dried
(MgS04), filtered and evaporated Ln vacuo. The
residue was purified on a silica gel flash chromato-
graphy column eluted with 50% ethyl acetate/hexane toafford 0.098 g (88%) of the title compound.
lH NMR (300 MHz, CDC13, ppm): ~ O.89 (t, J=9 Hz, 3H),
1.26 (t, J=8 Hz, 3H), 2.55 (s, 3H), 2.59 (s, 3H),
2.52-2.63 (m, 2H), 2.74 (q, J=8 Hz, 2H), 3.70 (s,
3~), 5.34 (s, 2H), 6.03 (s, lH), 6.62 (d, J=10 ~z,
lH), 6.78 (dd, J=2,10 Hz, lH), 6.86 (s, lH), 6.97 (d,
J=2 Hz, lH), 7.24-7.32 (m, 2H), 7.34-7.42 (m, 1~),
7.57-7.64 (m, lH).
FAB-MS: m/e 506 (M+l).
Step B: Preparation Of 3-t4-((1-carboxy-1-(2-
chlorophenyl))metho~y)-3-propylphenyl]methyl-
5,7-dimethyl-2-ethyl-3~-imidazo~4,5-b~
pyridine
Using the general procedure for ester
hydrolysis described in Step E of Example 19, 0.098 g
(0.19 mmol) of the product of Step A was converted to
0.072 g (76%) of the title compound.
lH NMR (300 MHz, CD30D, ppm): ~ O.85 (t, J=9 Hz, 3H~,
1.25 (t, J=8 Hz, 3~), 1.60-1.74 (m, 2H), 2.61 (s,
3H), 2.64 (s, 3H), 2.52-2.60 (m, 2H), 2.88 (q, J=8
DEMANDES OU BREVETS VOLUMINEIJX
LA PRESENTE PARTIE DE ~; I I t DEMANDE OU CE BREVET
COMPP~END PLUS D'UN TOME.
CECI EST LE TOME ~ DE 2
NOTE: Pour les tomes additionels, veuillez c~ntacter le Bureau canadien des
brevets
~ ~7~7
J U IVI BO APPLI CATIO NS /PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE
THAN ONE VO~UME
THIS IS VOLUME 1 -_ OF
NOTE: Ear additicnat v~lumes please c~ntacl the Canadian Patent Office