Note: Descriptions are shown in the official language in which they were submitted.
~ ~ J 7-~L~
The specific regulation o unwanted gene expression by
complementary nucleic acids, so-called an~isense
oligonuceotides, would represent a great advance in
chemotherapy. It could be of Lmportance for the treatment
of genetically related disorders and for defence against
exogenous nucleic acids, fox example after infection by
bacteria, fungi or viruses.
The antisense principle is based on the ability of the
so-called antisense oligonucleotides to hybridise with
the mRNA to be inhibited, and in this way to prevent
protein biosynthesis.
There is evidence in the literature for the ac~ion of
antisense oiigonucleotides. Thus, inter alia, their
antiviral action, for example ~gain~t retroviruses such
lS as HIV, is discussed ~P. C. Hoyle, E. C. Cooper, Adv.
Appl. Biotechnol. Ser. 2, 1989, 35).
The use of nature-identical phosphodiester
oligonucleotides is prevented by their great sensitivity
to enzymatic degradation by endogenous endo- and
exonucleases.
The present invention relates to antisense
oligonucleotide analogues with achiral phosphoramidate
internucleotide linkage which are derived from 5'-amino-
2',5'-dideoxyfur~noses and may be modified at the 5~ end
Le A 28 522 - 1 -
23189-7387
by lipid residues. These oligonucleotide analogues have an
increased resistance to endo- and exonucleases and display anti-
viral actions.
Oligonucleotide analogues which contain one or more
phosphoramidate internucleotide linkages have been described
previously (W. sannwarth~ Helv. Chim. Acta 71 (1988), 1517; M. Mag,
J.W. Engels, Nucleic Acids Res. 17 (1989), 5973; S. M. Gryaznov,
N. I. Sokolova, Tetrahedron Lett. 31 (1990), 3205).
These compounds do not have improved properties compared
with unmodified phosphodiester oligonueleotides. Antiviral
activities have not been disclosed for these compounds.
Syntheses of oligonucleotide sequences which eontain
phosphoramidate internueleotide linkages throughout have not been
described.
However, introduetion of phosphoramidate internucleo-
tide linkages throughout, or almost throughout, results in
adequately nuelease-stable molecules with antiviral aetion. It
is also possible to use these structures as gene probes (Review:
J. A. Matthews, L. J. Xrieka, Analytical Bioehemistry 169
~0 (1988), 1).
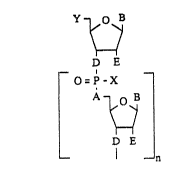
The invention relates to oligonueleotides of the
general strueture I:
B
D E
O=P~X
A ~ B
D E
_ I ~ n
where
A can be NX or NR2 throughout,
~ can be, independently of one another, a na~urally
occurring or modified pyrimidine or purine
nucleobase or else OH, oR2 or H,
D can be O, S, NH or NR2,
E can be, independently of one another, H, OH, oR2,
azide or halogen,
10 X can be -O~, S~, -oR2, -SR2 or alkyl,
R2 can be alkyl, aryl, aralkyl or cycloalkyl.
By alkyl iB meant here a branched or unbranched
alkyl radical with 1 to 20 C atom~. Alkyl radicals
with 1 to 10 C atoms are preferred, particularly
preferably methyl, ethyl, propyl, allyl, 2,2-
dimethylallyl, benzyl.
Le A 28 522 - 3 _
a~L~
By aryl i~ meant here phenyl, naphthyl, optionally
~ub~titutad, for example by halog0n or nitro.
By aralkyl i5 meant here an aralkyl radical with 1
to 18 carbon atoms in the straight-chain or branched
alkyl moiety and 6 to 12 C atom in the aryl moiety,
where aryl represents phenyl or n~phthyl.
By cycloalkyl is meant here a cyclic alkyl radical
with 3 to 8 carbon atoms, preferably with 3 to 6
carbon atoms, par~icularly preferably cyclopentyl
and cyclohexyl.
Y can be -NH2, -NHR2, -NR2 or N ? --OR
H o~
R~ can be alkyl, aryl, hetaryl, aralkyl, cycloalkyl,
lipid.
By alkyl is meant here a branched or unbranched
alkyl radical with 1 to 30 C atoms. Alkyl radicals
with 1 to 18 C atoms are preferred, and alkyl
radicals with 6 to 18 C atoms are very particularly
preferred.
2~
By aryl is meant here, for example, phenyl or
naphthyl, optionally substituted, for example by
halogen or nitro.
By hetaryl is meant, for example, pyridyl, acridinyl
or carbazolyl.
Le A 28 522 - 4 -
By aralkyl is meant an aralkyl radical with l to 18
C atoms in the traight-chain or branched alkyl
moiety and 6 to 12 C atoms in the aryl moiety, where
aryl represen~s phenyl or naphthyl.
By cycloalkyl is meant cyclic alkyl radical with
3 to 8 carbon atoms, preferably with 3 to 6 carbon
atoms, particularly preferably cyclopentyl and
cyclohexyl.
n can be 3 to 50.
n i~ preferably 10 to 30 and particularly preferably 18
to 28.
Le A 28 522 - 5 -
~s~
2318g-7387
The activated monomers are synthesised by known processes
(~. Mag., J. W. Engels, Nucleic Acids Res. 17 (1989),
5979).
HO ~ T N3 ~ T ~N ~ T
OH OH OH
1a 3a 4a
M~rrHN ~ T Mn~THN ~ T
OH ~
N~P~o ~ CN
~a 6a
where T indicates thymine
Le A 28 522 - 6 -
2~i_t~
M~rr= - C ~ O~H3
_~ _
Thymidine is reacted with lithium azide, tetra-
bromomethane and triphenylphosphine in
dimethylformamide. The azido group is reduced with
triphenylphosphine in pyridine to the amino groups,
and the amino group is subsequently protected with
the monomethoxytrityl group. The activated
derivative 6a can be obtained from 5a using
cyanoethyl N,N,N',N'-tetraisopropyl-phosphoro-
diamidite.
HO ~ B Ts ~ B N3 ~ B
OH OH OH
1~-e) ~(~e) 3(~e~
Le A 28 522 - 7 -
~7~
H N~B MM~ ~B MM HN~B
OH OH ~
_,~N~P~o~
4(b-e) 5(b-e) 6(b~)
b: B = N~lo
o N
NH- C ~
c: B = N 3~N
Ts -- ~SO2 ~ CH3
Le A 28 522 - 8 -
o
d: B~ ~ ~ NH o
- N N~N~-C
e. B= ~ ~ NH O
N N ~H-C - CH2 ~
N~-benzoyl-2'-deoxycytidine lb, N4-benzoyl-2'-deoxy-
adenosine lc, N2-isobutyryl-2'-deoxyguanosin~ ld and
N2-phenylacetyl-2'-deoxyguanosine le are prepared by the
transient protection method (G.S.Ti., B. L. Gaffney,
R. A. Jones, J. Am. Chem. Soc. 104 (1982), 1316;
F. Benseler, L. W. McLaughlin, Synthesis 1986, 45).
The monotosylates 2 (b-e) are obtained from the building
blocks 1 (b-e) and are converted with lithium a~ide into
the 5'-azido compounds 3 (b e).
The synthetic route described for the thymidine
derivative is utilised for sub equent reaction step~.
The lipid residues, for example undecanol, are activated
analogously by reacting with cyanoethyl N,N,N',N'-tetra-
Le A 28 522 - g -
~ 23189-7387
isop~opyl-phosphorodiamidite to yield, for example to 7
,OCH2CH2CN
CH3-(CH2)l0 ~ `N
7 ) \
Linkage of the 5~-deoxyaminonucleotide building blocks to
give oligonucleotides
The linkage of the monomer building blocks to give
oligonucleotides is preferably carried out under ~he
conditions of solid-phase synthesis based on a described
process (M. Mag, J. W. Engels, Nucl. Acids Res, 17, 5973
(1989); W. Bannwar~h, Helv. Chim. Acta 71, 1517 (1988)).
The oligonucleotide analogues according to the invention
are linked in the final reaction step, for example, with
the phosphoramidite rea~ent 7. Af~er ths protec~ive
groups have been removed and the sequenco ha3 been
cleaved off the ~olid phass by base treatment, the
reaction product is preferably isolated ~y preparative
HPLC, the chromatographic behaviour being determined in
each case by the lipid residue which permits easy removal
from by-products.
Oligonucleotide sequences such as the following have
been synthesized in this way.
RNHGNHCNHTNHCNHCNHGNHANHGNHGNHCNHTNHINHANIiaNHANHTG
Le A 28 522 - 10 -
R = CH3-(cH2)lo - O -P - 8
OH
The corresponding pharmaceutical formulations contain,
besides the pho~phoramidate oligonucleotide~, the
auxiliaries customary for parenteral formulations such
S as, for example, buffers and/or stabilisers or liposome
formulations. Topical application is also conceivable.
Examples of formulations which can be employed for this
purpose are ointments, creams, solutions or plasters
which, besides the acti~e compound, contain the
pharmaceutical auxiliaries which are suitable for this
application.
Svnthesis examples
Example 1:
5'-O-(4-Toluenesulphonyl)-N~-phenylacetyl-2'-
deoxyguanosine
N2-Phenylacetyl-2'-deoxyguanosine (13.9 g; 36 mmol) and
4-toluenesulphonyl chloride (21.0 g; 108 mmol) are
stirred in anhydrous pyridine (140 ml) at room
temperature for 70 min. After this, water (5 ml) is added
at 0C, the mixture is stirred at room temperature for 20
min, concentrated under high vacuum and codistilled
several times with toluene, the residue i8 taken up in
ethyl acetate (150 ml) and washed once each with 5%
Le A 28 522
NaHC0~ solution and NaCl solution (50 ml each)~ and the
organic phase is dried (MgS04) and concen~rated. The
crude product is purified by chromatography (dichloro-
methane/methanol 15:1).
Yield: 11.3 g (58~).
Example 2:
5'-Azido-N2-phenylacetyl-2',5'-dideoxyguanosine
Lithium azide (5.0 g, 100 mmol) is added to a solu~ion of
5'-0-(4-toluenesulphonyl)-N2-phenylacetyl-2'-deoxy-
guanosine (11.0 g; 20 mmol) in N,N-dimethylformamide
(120 ml) and the solution is stirred at 50C for 7.5 h.
It is cooled, concentrated under high vacuum and
codistilled several times with toluene, and the residue
is taken up in ethyl acetat2 (500 ml) and extracted by
shaking twice each with NaHC03 solution (60 ml each time)
and NaCl solution (100 ml each time). The combined
aqueous phase is back-extracted several times with ethyl
acetate. The combined organic phases are dried (MgS0~)
and concentrated. The crude product is purified by
chromatography (dichloromethane/methanol 13:1 ~ 10:1).
Yield: 8.0 g (95~).
Example 3:
5'-Amino-N2-phenylacetyl-2',5'-dideoxyguanosine
5'-Azido-N2-phenylacetyl-2',5'~dideoxyguanosine (8.2 g;
29 mmol) and triphenylphosphine (7.9 g; 30 mmol) are
dissolved in pyridine (150 ml) and left at room
Le A 28 522 - 12 -
temperature for 16 h. Water (25 ml) is then added and the
mixture is stirred for 2 h. The reaction solution is
poured into water (200 ml), filtered and the aqueous
phase is extracted twica with ethyl acetate (100 ml each
time). ~he combined organic phase is then extracted five
times with water (100 ml each time). The combined aqueous
phase is freeze-dried and provid0s the crude product as
a yellowish powder which is immediately reacted further.
Yield: 7.1 g (92%)
Example 4:
5~-(4-Methoxytriphenylmethyl)amino-N2-phenylacetyl-2~,5'-
dideoxyguanosine
5'-Amino-NZ-phenylacetyl-2~,5~-dideoxyguanosine (7.1 g;
18.5 mmol) is dissolved in pyridine (200 ml), chloro(4-
methoxy)triphenylmethane (17.0 g; 55.5 mmol), 4-(N,N-
dimethylamino)pyridine (490 mg; 3.8 mmol) and
triethylamine (2.7 ml; 19 mmol) are added, and the
mixture is stirred at room temperature for 2.5 h. It is
then concentrated under high vacuum and codistilled
several times with toluene, the residue is taken up in
dichloromethane (500 ml) and extracted by shaking twice
each with NaHCO3 solution and NaCl solution, and the
organic phase is dried (MgSO4) and concentrated.
Purification by chromatography follows
(dichloromethane/methanol 20:1 10:1).
Yield: 2.4 g (20%).
Le A 28 522 - 13
Example 5:
5~-(4-methoxytriphenylmethyl)amino-N2-phenylacetyl-2',5'-
dideoxyguanosine 3'-0-(2-cyanoethyl-N,N-diisopropyl-
amino)phosphite
5'-(4-Methoxy~riphenylmethyl)amino-N2-phenylacetyl-2',5'-
dideoxyguanosine (2.2 g; 3.3 mmol) and tetrazole (176 mg;
2.5 mmol) are dissolved, s~ringently excluding air and
moisture, in absolute dichloromethane/acetonitrile (1:1;
70 ml), 2-cyanoethyl N,N,N~,N'-tetraisopropyl-phosphoro-
diamidite (1.65 ml; 5.3 mmol) is added, and the mixture
is stirred at room temperature for 1.5 h. After the
reaction is complete, butanol ~3 ml) is added, and the
mixture is then stirred for 10 min, diluted with
dichloromethane (500 ml), rapidly extracted once each
with 5~ NaHCO3 solution (150 ml) and NaCl solution
(250 ml), dried (MgSO4) and concentrated. The crude
product is dissolved in dichloromethane/ether (1:1,
14 ml) and precipita~ed in n-pentane (450 ml) cooled in
dry ice.
Yield: 1.67 g (59%).
Exam~le 6:
Undecane l-O-(2-cyanoethyl-N,N-diisopropylamino)phosphite
Bis-(diisopropylamino)-(2-cyanoethoxy)-pho~phine (3.3 g,
11 mmol) is added to a stirred solution of undecanol
(1.72 g, 10 mmol), diisopropylamine (0.5 g, 5 mmol) and
tetrazole (350 mg, 5 mmol) in 30 ml of dichloromethane
under argon over the course of 20 min.
Le A 28 522 - 14 -
2318g-7387
After the reaction is complete, the mixture is ex~racted
by shaking with cold 10% strenqth sodium carbonate
solution. The organic phase is evaporated, and ~he
product is chromatographed on silica gel.
Yield: 3.3 g (73%).
Example 7:
Synthesis of the exemplary sequence detailed below:
R~GN}~CN~T~ HCNHC~C,"~ANXGNHG~CNHTNHCNHA~`.~GNHANHTC
R = CH3-(CH2)l0-- P
OH
The synthesis wa~ carried out in an Applied Biosystems
380B automatic DNA synthesiser. Used as solid phase was
a commercially available (Applied Biosystems) 10 ~mol (or
1 ~mol) controlled pore glass support to which 2-deoxy-
cytidine that is protected at the 5'-position by a 4,4'-
dimethoxytrityl group is bonded via the 3'-hydroxyl group.
The 4,4'-dimethoxytrityl protective group is eliminated
by treatment with 2.5% strength dichloroacetic acid in
methylene chloride, and the excess acid and the protective
group are subsequently washed out with acetonitrile. The
chain extension is carried out by adding a 0.1 M solution
of 5'-N~-mono-methoxytrityl-protected nucleotide building
block 6a in acetonitrile in the presence of tetrazole.
Subsequent
Le A 28 522 - 15 -
231~9-7387
oxidation to pentavalent phosphorus is carried out by treatment
with a 0.2 M iodine solution in pyridine/H2O/THF/1:1:8O To
avoid wrong sequences, subsequently unreacted 5'-hydroxyl or 5'-
amino groups (in the subsequent cycles) are blocked by treatment
with acetic anhydride/dimethylaminopyridine in pyridine/
acetonitrile.
The next reaction cycle starts with elimination of the
monomethoxytrityl protective group by treatment with 2.5% strength
dichloroacetic acid. The reaction with subsequent 5'-amino-
nucleotide building blocks 6 is carried out in analogy to the
description given above, using the compound of formula 6a to
introduce thymine and the corresponding adenine-, cytosine- or
guanine-containing compound to introduce the respective other
bases (A), (C) or (G).
In the last reaction step, a 0.1 M solution of unde-
canol ~-cyanoethyl phosphoramidite reagent 7 in acetonitrile is
coupled on in the presence of tetrazole after acid elimination of
the last monomethoxytrityl protective group. Iodine oxidation is
followed by cleavage off the polymeric support and removal of the
protective groups by treatment with concentrated ammonia at 55C
~or 16 h.
The reaction product is isolated by preparative HPLC on
an RP 18 column with a linear rising gradient of acetonitrile in
0.1 M triethylammonium acetate; yield: 2.8 mg (4.8%).
Nuclease stability of olignonucleotide amidates
Besides the chain length, sequence and cell perme-
ability,
- 16 -
~r~t7~
Lmportant for the biological action of antisense
oligonucleotides is the nuclease resistance. The
synthesised oligonucleotides with 5'-amidate inter-
nucleotide linkage were tnerefore compared with natural
S oligonucleotide diesters in respect of their nuclease
stability.
The antisense oligonucleotide diesters and amidates were
for this purpose radioactively labelled at the S' end
with 32P-ATP and their degradation with various defined
nucleases and cell extracts was examined by polyacryl-
amide gel electrophoresis and autoradiography and
quantitatively determined in a scintillation counter by
isola~ion of the degradation products from the gel.
Natural oligonucleotide diesters have only low nuclease
stability. They are completely degraded within 30 minutes
to 1 hour. Oligonucleotide amidates, by contrast, are 10
to 100 times more resistant to nucleases and are
therefore particularly well suited for use as antiviral
antisense oligonucleotide inhibitors.
Antiviral actions:
In vitro translation assay
The antiviral, translation-inhibiting activity of the
amidates was shown in a cell-free in vitro translation
assay. For this, in vitro synthesised TAR-tat mRNA was
placed in a rabbit reticulocyte translation system and
Le A 28 522 - 17 -
the ta~ protein synthesis was measurad quantitatively
with and without oligonucleotide.
In detail: 1 ~g of in vitro synthesised TAR-tat mRNA is
mixed with a required amount of oligonucleotide
(0.2-2 ~9) in a volume of 2,5 .~1. For the translation, 10 ~1 of
preincubated translation mix (from Promegs) with
radioactive 35S-cysteine are pipetted into the 2.5 ~l and
incubated at 30C for 90 min. An aliquot of the mixture
is removed and heated with SDS loading bUf fer and
fractionated on a 6-20~ discontinuous SDS PAGE. After
fixing and drying, the gel is autoradiographed and the
resulting tat protein bands are evaluated by
densitometry.
Result: the inhibition of the translation of the tat
protein by the assayed amidates is 3 times greater than
that of diester oligonucleotides of identical sequence in
various concentrations.
Antiviral action in a cell culture assay
The phosphoramidate oligonucleotides were tested on the
virus isolates detailed below:
HI~-lD3~ Isolation by Dr. von Briesen and Prof.
Dr. H. Rubsamen-Wai~mann, Georg-Speyer-
Haus, Frankfurt am Main, 1985.
Le A 28 522 - 18 -
HIV-2RoD Isolation by Pasteur Institute, Paris,
1986.
Used as assay system were peripheral blood lymphocytes
( PBL ) from HIV-seronegative donors which had been
preactivated with phytohaemagglutinin. The substances
were added to the cell culture immediately after the
infection. The cell suspensions were cultivated with
various concentrations o-f the compounds in an incubator
for 4-6 days. Virus replication was detected by
evaluation under the light microscope of the HIV-induced
syncytia formation.
Table 1:
Inhibition of virus-induced syncytia formation in the
presence of phosphoramidate oligonucleotide 8
Test HIV-l HIV-2
~g/ml ~mol/l ~g/ml ~mol/l
1 5-10 0.9-1.9 _
2 2-5 0.4-0.9 15-25 2.8-4.7
3 2-5 0.4-0.9 15-25 2.8-4.7
4 5-lO 0.9-1.9 ~ ~ l
The test substance distinctly inhibits HIV-1-induced
syncytia formation in concentrations above 2-5 ~g/ml,
that is to say 0.4-O.g ~mol/l.
Le A 28 522 - 19 -