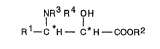Note: Descriptions are shown in the official language in which they were submitted.
o ~
This lnvention relates to a process for the
production of ~-hydroxy-~-aminocarboxylic acids and the
esters thereof.
Some a-hydroxy-~-aminocarboxylic acids are of
interest as structural elements of natural or synthetic
peptides, which are effective as inhibitors of specific
enzymes. They include, in particular, (2R,3S)-3-amino-2-
hydroxy-5-methylhexanoic acid ("Norstatin") and (2R,3S)-3-
amino-2-hydroxy-4-cyclohexyl butyric acid
("Cyclohexylnorstatin") [see, e.g., K. Iizuka et al., J.
Med. Chem., 33, (1990), 2707-2714].
Processes for the production of these compounds
are known. Starting from the corresponding oe-
aminocarboxylic acid having a chain with one less carbon
atom, these processes yield the desired products via the N-
protected a-amino alcohol. The corresponding N-protected
o!-amino aldehyde and the ryanohydrin are obtained by
hydrolysis of the cyano group to the carboxyl group with
hydrocyanic acid [Iizuka et al., loc. cit.; see also D.H.
Rich et al., J. Org. Chem., 45, (1980), 2288-2290]. A
drawback of these processes is the use of an aldehyde as an
intermediate step, which must be produced either by a
reduction oxidation sequence from the carboxylic acid or by
reduction with diisobutyl aluminum hydride from the methyl
ester, which is difficult for synthesising on a large
scale. Other problems follow from the use of hydrocyanic
acid. Moreover, the production of esters of a-hvdroxy-~-
amino acids employing these processes requires an
additional esterification step.
The main ohjective of the invention is to provide
a novel process for the production of a-hydroxy-~-
aminocarboxylic acids and their esters that avoids the
disadvantage of the prior art and is also suitable for
production on an industrial scale.
.~ .
" ~
. ~ -
-. .. . .
'
::
Accordingly, the invention provides a process for
the production of an ~-hydroxy-~-amino carboxylic aoid, an
ester thereof or a salt thereof of the formula:
N~3R4 OH
R~C H - C~H - COOR~ (I)
wherein Rl is a linear or branched alkyl group having 1 to
6 carbon atoms, capable of being optionally substituted
with an aryl group or a cycloalkyl group having 3 to 8 ring
members, R2 is hydrogen or a linear or branched alkyl group
having 1 to 4 carbon atoms, and R3 and R4, independent of
one another, are hydrogen or an amino protective group or
R3 and R4 together form a bifunctional amino protective
group, which process comprises:
(a) reacting an N-protected ~-aminocarboxylic
acid halide of the formuJa:
NR5R6 o
1 1* 11
R -C H - C - X (II)
wherein Rl has the above-mentioned meaning, X is chlorine or
bromine, and R5 and R6 have the same meanings as the above-
mentioned meanings for R3 and R4, provided that Rs and R6 are
not both hydrogen at the same time,
with trimethylsilyl cyanide to form the
corresponding ~-oxonitrile of the formula:
NR5R6 o
~ 1* 11
R - C H - C -CN (III)
wherein Rl, R5 and R6 have the above-mentioned meanings,
(b) converting the cyano group of a compound
(III) into an alkoxy carbonyl group to form the
corresponding ~ oxocarboxylic acid ester of the formula:
NR5R~ O
F~1 _ C * H-- C--CoOR7 ( IV )
` . - ' ~ ' ` ' ' ' ` `
~` ' . , ` .
2 ~
wherein Rl, Rs and R6 have the above-mentioned meanings, and
R7 has the same meaning as the above-mentioned meaning for
R2, provided that R7 is not hydrogen,
(c) selectively reducing the keto group of a
compound (IV) to form the corresponding N-protected a-
hydroxycarboxylic acid ester of the formula:
NR5R6 OH
R1 _C~H-- C~H--CoOR7 (V)
wherein Rl, R5, R6 and R7 have the above-mentioned meanings;
and, optionally,
(d) cleaving the amino protective group and/or
hydrolysing the ester function.
Preferably, the N-protected a-aminocarboxylic
acid halide (II) is employed in the L configuration.
Preferably, an N-protected a-aminocarboxylic acid chloride
is used as the N-protected a-aminocarboxylic acid halide
(II). Preferably, an N-protected L-phenylalanyl chloride
is used as the N-protected a-aminocarboxylic acid chloride.
Preferably, a phthaloyl group is used as the amino
protective group R5R6. Preferably, the cleaving-off of the
phthaloyl group is performed with hydrazine hydrate or
concentrated hydrochloric acid. Preferably, the reaction
with trimethylsilyl cyanide is performed in the presence of
zinc iodide. Preferably, the conversion of the cyano group
in an alkoxy carbonyl group is performed by reaction with
the corresponding alcohol R70H in the presence of a strong
acid and subsequent hydrolysis of the iminoester salt
for~ed as an intermediate product. Preferably, methanol is
used as the alcohol. Preferably, a diastereomer separation
of a-hydroxycarboxylic acid ester (V) is performed between
step (c) and step (d). Preferably, the selective reduction
of the keto group is performed with a borohydride selected
from the group consisting of LiBH4, NaBH4, Ca(B~4)2 and
Zn(BH4)2, as the reducing agent. Also preferably, the
. ., : :. ~
. . . . .
selective reduction of the keto group is performed by
catalytic hydrogenation on a platinum/rhodium mixed
catalyst.
The invention also provides N-protected ~-oxo-~-
aminonitriles of the general formula:
NR5R6 o
11
R - C*H ~ C - CN (III)
wherein R~ is a linear or branched alkyl group having 1 to
6 carbon atoms, capable of being optionally substituted
with an aryl group or a cycloalkyl group having 3 to 8 ring
members, and either R5 is an amino protective group and R6
is hydrogen or an amino protective group or R5 and R6
together form a bifunctional amino protective group.
Preferably, the N-protected ~-oxo-~-aminonitrile
has the (S) configuration. Preferably, R5 and R6 together
form a phthaloyl group. Preferably, Rl is selected from the
group consisting of isobutyl, benzyl, p-hydroxybenzyl and
cyclohexylmethyl.
The invention further provides N-protected ~-oxo-
~-aminocarboxylic acid esters of the general formula:
NR5p,6 o
* 7
R --C H-- C--COOR (IV)
wherein the asymmetric center has the ~S) configuration, Rl
is a linear or branched alkyl group having 1 to 6 carbon
atoms, capable of being optionally substituted with an aryl
group or a cycloalkyl group having 3 to 8 ring members, and
R7 is a linear or branched alkyl group having 1 to 4 carbon
atoms and either R5 is an amino protective group and R6 is
hydrogen or an amino protective group or Rs and R6 together
form a bifunctional amino protective group.
Preferably, R7 is methyl. Preferably, R5 and R6
together are a phthaloyl group. Preferablyr Rl is selected
~3~8
from the group consisting of isobutyl, benzyl, p-
hydroxybenzyl and cyclohexylmethyl.
The process according to the invention is
suitable for the production of ~-hydroxy-~-aminocarboxylic
acids and their esters of the general formula:
IR3R4 I H
F11--C*~l-- C~H--COOR2 (I)
wherein Rl is a linear or branched alkyl group having 1 to
6 carbon atoms, for example, methyl, ethyl, propyl,
isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl
and hexyl. The alkyl group can be substituted with aryl
groups, for example, phenyl, o-, m- and p-tolyl,
chlorophenyl, hydroxyphenyl, methoxyphenyl and cycloalkyl
groups having 3 to 8 ring members, for example,
cyclopropyl, cyclopbutyl, cyclopentyl, cyclohexyl,
cycloheptyl and cyclooctyl. Especially preferred groups
for Rl are isopropyl, isobutyl, sec-butyl, benzyl, p-
hydroxybenzyl and cyclohexylmethyl. Basically Rl may
comprise any substituent which remains non-reactive in the
applied reaction conditions. R2 is hydrogen or a linear or
branched alkyl group having 1 to 4 carbon atoms, i.e.,
methyl,
, :
;', ~ . . ~
,
:"' ' ; ,
- \
ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and tert-
butyl, preferably methyl. R3 and R~, independent from each
other, are hydrogen or amino protective groups, or R3 and R4
together form a bifunctional amino protective group. Herein,
bifunctional amino protective groups are to be understood in
this connection to be those Whi Ch form a heterocyclic ring by
substitution of both hydrogen atoms of an amino group together
with the amino nitrogen atom. Amino protective groups are
especially in use in the peptide synthesis and are known in
the art; a survey of their properties and the methods for
their introduction and cleavage are provided, for example, by
E. Wunsch, Methoden Org. Chem., (Houben-Weyl), 4th Ed.,
(1974), Vol. XV/l, pp. 46-305.
The compounds produced according to the process of the
invention have two chiral centers (wlless other chiral centers
are present in R1 and/or R2), thus each can occur in four
stereoisomeric forms. Since only one of the two chiral
centers is newly formed in the reaction sequence according to
the invention, while the configuration of the other chiral
center is maintained, generally diastereomeric mixtures result
with the use of optically active ~-amino acid derivatives as
the initial material,whjch however, can easily be separated
because of the different physical properties. Therefore, the
process according to the invention is especially suitable for
the production of configurationally homogeneous ~-hydroxy-~-
amlno aclds.
:
.` '
2~3: ~
According to the invention, a chloride or
bromide, preferably chloride, of an ~-amino carboxylic acid
with protected amino function corresponding to general
formula:
NR5R6 o
11
Rl-C*H - C - X (II)
is used as starting material. In formula (II), X is
chlorine or bromine, Rl has the same meaning as in formula
(I), and Rs and R6 have the same meaning as R3 and R4 in
formula (I~, provided that R5 and R6 are not both hydrogen
at the same time.
Basically the process can be per~ormed with any
of the amino protective group which is only slightly
sensitive to acids and bases as well as trimethylsilyl
cyanide and halides and is not cleaved off by the reaction
conditions of steps (a) to (c) of the inv~ntion process.
Suitable protective groups are, for example, p toluene
sulfonyl (tosyl), and the bifunctional protective groups,
the so-called diacyl protective groups, such as maleoyl or
especially phthaloyl. The phthaloyl protective group can
be easily introduced in ~-amino acids according to or
analogously to known processes and again cleaved off with
hydrazine hydrate or strong acids (E. Wunsch, loc. cit.,
pp. 250-263).
,
,
.:
::
.
8 2~3~8
The thus obtained N-phthaloyl-~-aminocarboxylic acids
can be converted also by known methods with halides of
inorganic acids, such as, thionyl chloride, into the amino
acid halides re~lired for the process according to the
invention (see, e.g., P. Stetzel, Methoden Org. Chem.,
(Houben-Weyl), 4th Ed., (1974), Vol. XV/2, pp 355-364).
The N-protected ~-aminocarboxylic acid halides (II)
according to the invention are converted in the first stage
(a) with trimethylsilyl cyanide (cyanotrimethyl silane) into
the corresponding ~-oxonitriles (acyl cyanides) (III), having
the formula:
NR R O
R - 1 H - C - CN III
wherein Rs and R6 are defined above. These compounds (III) are
nov~l and also constitute ~a~t of tliis invention.
No racemization takes place in the reaction with
trimethylsilyl cyanide in contrast to the previously known
process of the reaction of carboxylic acid halides with
copper(I)-cyanide, according to which phthaloylglycyl c~anide
is prodllced, for example, from phthaloylglycyl bromide; but
racemization was observed in the case of optically active -
amino acid derivatives (P. ~tetzel, loc. cit. p. 364).
~.
' : ~
~.
,
The reaction with trimethylsilyl cyanide is
preferably performed in the presence of a catalytic amount
of zinc iodide. A solvent is not necessary, however for
better handling an inert solvent, such as dioxane, diethyl
acetate or tetrahydrofuran, is added. The reaction
temperature is advantageously from about 20 to 100C,
preferably from about 40 to 70C. The trimethylsilyl
chloride or bromide resulting in the reaction can
optionally be distilled off because of its lower boiling
point, and used again for the production of trimethylsilyl
cyanide.
The ~-oxonitriles (III) according to the
invention are converted in step (b) into the ~-
oxocarboxylic acid esters (IV). The ~-oxocarboxylic acid
esters (IV) are novel and also constitute part of the
invention. The ~-oxocarboxylic acid esters (IV) are of the
formula:
NR5R~ O
R1 _C~ H-- C--CoOR7 ( IV)
wherein Rl, R2, R5 and R6 are defined above. It is not
necessary to purify the ~-oxonitrile (III) for the
production o~ the ~-oxocarboxylic acid esters (IV), since
the crude product or even the unworked-up reaction mixture
of the first synthesis step can be used.
Preferably, this reaction is performed in the
manner of the Pinner reaction with an alcohol that
corresponds to the desired ester, i.e., preferably
methanol, and a strong acid. Thus, first a salt of the
corresponding imino ester is formed which readily
hydrolyses with water to the normal ester. The reaction is
preferably performed in an inert polar solvent, such as
diethyl ether or tetrahydrofuran. Because of the
substitution of the cyano group by an alkoxy group as a
possible secondary reaction which leads to an ~-
aminocarboxylic acid ester, it is advantageous not to use
- . . ..:
., ;,
: ::
~ '. .
~3~
an excessive amount of alcohol. By choosing suitable
reaction conditions the portion of the substitution product
can be limited, for example, to 5 to 10 percent. Hydrogen
chloride is preferably used as the acid which can be
introduced in a gaseous state. Because of the high
reactivity of the ~-o~onitriles (III) the Pinner reaction
can be performed at a comparatively low temperature.
The reaction temperature is suitably from about
-~0 to +20C, preferably from about -20 to 0C. It is
also within the scope of this invention to produce ~-
oxocarboxylic acid esters (IV) from ~-oxonitriles (III)
according to another method, for example, by hydrolysis to
~-oxocarboxylic acid and subsequent esterification.
According to the invention in step (c), the keto-
carbonyl group of the ~-oxocarboxylic acid esters (IV) are
selectively reduced to the N-protected ~-hydroxycarbo~ylic
acid esters (V~. Preferably, borohydrides, especially
lithium borohydride, so~ium borohydride, calcium
borohydride, zinc borohydride, or hydrogen in the presence
of a platinum/rhodium mixed catalyst are used as the
reducing agent.
From the reduction process a second asymmetric
center is formed such that depending on whether it was
started from an optically active or from a racemic
carboxylic acid halide (II) generally two diastereomers or
diastereomer pairs result. The proportiGn of the
diastereomers, i.e., the diastereoselectivity of the
reduction, depends on the reduction agent used and the
temperature. Especially high diastereoselectivities were
observed when LiBH4 and Zn(BH4)2 are used as the reducing
agents. Preferably, in each case the diastereomer with
opposite configuration is formed on both asymmetric
centers, i.e., the (2R,3S)- or (2S,3R)-diastereomer.
The reduction with borohydrides, preferably with
Zn(BH4)2, is performed suitably at a temperature of from
about -50 to +25C, preferably from about -30 to 0C.
.
:;
The reduction wlth hydrogen at a platinum/rhodium mixed
catalyst is preferably performed at a hydrogen pressure of
from about 10 to 200 bars, and a temperature of from about
20 to 80C in a non-polar solvent, such as toluene, or a
polar solvent, such as ethyl acetate or tetrahydrofuran.
It was found that the diastereomers resulting
from the reduction can easily be separated at this step,
for example, by column chromatography or recrystallization.
: .
l2
Then, optionally in another step (d), the amino
protective group is cleaved off and/or the ester function is
hydrolyzed. For example, the phthaloyl protective group can
be cleaved off while maintaining the ester function with
hydrazine hydrate or by hydrolysis of the ester with a strong
acid. However, it is possible under less drastic conditions
to hydrolyze only the ester group with acid while maintaining
the phthaloyl protective group.
The following Examples illustrate the performance of the
process according to the invention.
Example 1
N-Phthaloyl-L-phenYlalanyl chloride
((S)-3-~henyl~~p'l~hai.imidopropionyl chloride)
A mixture of 138.2 g (0.47 mol) of ll-phthaloyi~L~
phenylalanine (produced according to A.K. Bose, Org.
Syntheses, Coll., Vol. 5, p. 973) and 167 g (1.4 mol) of
thionyl chloride was refluxed for 3 hours. Then the excess
thionyl chloride was distilled off. The residue was mixed
with 200 ml of toluene and hea~ted to about 80C. After
addition of 380 ml of hexane the solution was cooled for
crystallization to about 4C~ The ~rystallized
product was filtered off and dried. The yield of the
product was 143.9 g (98% of theory). The melting point oE the
product was from 81.9 to 82.4àC. Other data regarding the product
was:
[~]D: -226.7 (c = 2.0; benzene)
.;
' ~:
13 2 ~
H-NMR (CDCl3, 300 MHz) ~: 3.50-3.72 (m, 2H);
5.35 (dd, lH);
7.10-7.24 (m, 5H);
7.65-7.79 (m, 4H).
F.xample 2
(S)-2-Oxo-4-phenyl-3-phthalimidobutane nitrile
A mixture of 25 g (79.7 mmol) of N-phthaloyl-L-
phenylalanyl chloride (produced according to Example 1), 9.5 g
(95.7 mmol) of trimethylsilyl cyanide and 255 mg (0.80 mmol)
of zinc iodide was heated to 60C. After 35 minutes, 30 ml of
tetrahydrofuran was added. The mixture was refluxed for another
80 minutes, then mixed with 200 ml of hexane and cooled to
about 4C. The precipitated product was filtered off, washed
with hexane and dried in a vacuum. The yield of the product
was 21.06 g (87% of theory). Other data regarding the product
was:
[~]D0: -264.2 (c = 1.0; THF)
H-NMR (CDCl3, 300 MHz) ~: 3.38 (dd, lH);
3.65 (dd, lH);
5.20 (dd, lH);
7.08-7.23 (m, 5H);
7.70-7.90 (m, 4H).
- . , , . ~ .
, ~ , , , , .: :
-
. .: ., , ~:
l4 ~31Q8
ExamPle 3
(S)-2-Oxo-4-Phenyl-3-~hthalimidobutYric acid methyl ester
A mixture of 100 ml of diethyl ether and 4 ml of methanol
was saturated with HCl gas at -10C. Then 9.0 g (30 mmol) of
(S)-2-oxo-4-phenyl-3-phthalimidobutane nitrile (produced
according to Example 2) was added. The mixture was stirred
for 45 minutes at -10C and then mixed with 170 ml of water at
-20 to 0C. The aqueous phase was extracted three times with
200 ml of diethyl ether each. The combined ether phases were
washed three times with 150 ml of saturated sodium bicarbonate
solution each and the washing solution was extracted back with
diethyl ether. The combined ether phases were then drled on
magnesium sulfate, concentrated by evaporation on a rotary
evaporator and finally freed of the remaining solvent in a
high vacuum. The yield of the product (viscous oil) was 6.8 9
(68% of theory). Other data regarding the product was:
H-NMR (CDCl3, 300 MHz) ~: 3.40 (dd, lH);
3.60 (dd, lH);
3.84 (s, 3H);
5.57 (dd, lH);
7.10-7.25 (m, 5H);
7.65-7.87 (m, 4H).
: .; ~ . : -
'
. . .
'
~3~
After chromatographic purification the product was able to be
crystallized. The melting point of the product was 75D to
7 6C~ Other data regarding the product was:
[~]D0: -200.4 (c = 1.0; CHCl3)
_ample 4
(2R,3S)- and (2S,3S)-~-HydroxY-4-phenvl _ Phthalimidobutyric
acid methyl ester
4.92 g of (S)-2-oxo-4-phenyl-3-phthalimidobutyric acid
methyl ester (produced according to Example 3) was dissolved
in 50 ml of tetrahydrofuran. The solution was cooled to -25C
and mixed with 95 mg of lithium borohy~rice The reaction
mixture was stirred for 30 minutes at -25 to -20C. Then 200
ml of diethyl ether was added and the mixtllre was washed twice
with 100 ml of 0.1 M hydrochloric acid each. The washing
solution was extracted back twice wlth 100 ml of diethyl ether
each. The combined organic phases were dried on magnesium
sulfate, concentrated by evaporation on a rotary evaporator
and finally freed of the remaining solvent in a high vacuum.
The yield of the produ~t (oil ) was 4.78 9 (96% of theory) .
By integration of the lH-NMR signals the diastereomer ratio of
(2R,3S):(2S,3S) was founc to be about 5:1. (The
configuration a~sign~ent took place after separation,
cleavage of the phthaloyl groups and derivatization with
phosgene to the corresponding oxazolidinone based on the
coupling constants in the 1H-NMR, cf. in this connection K.
. ;; -. .
:.. : , . .
, ,, , . :
.: , .. .
16 ~''3 ~
Iizuka et al., J. Med. Chem., (1990), 33, 2707-2714.) The
diastereomer mixture was able to be pu~ ~iec and se~-dted by cnrc~atography
on silica gel (mobile solvent hexane/dichloromethane/diethyl
ether 2:2:1).
Data for the (2R,3S)-compound was:
Melting point: 91.9-92.9C
[~]D : -107.6 (c = 1.0; CHCl3)
H-NMR (CDCl3, 300 MHz) ~ 3.20-3.40 (m, 2H);
3.63 (s, 3~);
4.48-4.60 (m, 2H);
4.90-5.00 (m, l~I);
7.10-7.30 (m, 5H);
7.67 7.~5 (m, 4H).
Exam~le 5
(2R,3S)-3-Amino-2-hvdroxy-4-phenylbutyric acid h~drochloride
13.25 g (39.1 mmol) of (2R,3S)-/(2S,3S)-2-hydroxy-4-
phenyl-3-phthalimidobutyric acid methyl ester (diastereomeric
ratio = 3:1) was dissolved in 60 ml of acetic acid. The
solution was mixed with 380 ml of concentrated hydrochloric
acid and refluxed for 8 hours. After cooling the mixture was
extracted with diethyl ether continuously for 18 hours in a
liquid-liquid extractor (according to Kutscher-Steudel). The
ether extract and the aqueous phase were separately
concentrated by evaporation. The residue of the ether phase
(7.41 g of brown powder) consisted basically of phthalic acid.
~31~8
The residue of the a~ueous phase (6.38 g) was suspended in 200
ml of acetonitrile and refluxed for about 15 minutes. The
undissolved part (2.6 g) was filtered off, it consisted
basically of (2R,3S)-diastereomer. Data for the (2R,3S)-
diastereomer was:
H-NMR tDMSO-d6, 300 MHz) ~: 2.90 (dd, lH);
3.05 (dd, lH);
3.40 (br.m, lH);
3.90 (d, lH);
7.20-7.43 (m, 5H);
8.20 (br.m, 3H).
Example 6
f2R 3S~-3-Am no-4-c~clohexyl-2-hydr~xybutyric acid
hydrochloride (cyclohexylnorstatin hydrochloride)
500 mg of (2R,3S)-3-amino-2-hydroxy-4-phenylbutyric acid
hydrochloride was dissolved in 5 ml of acetic acid and 5 ml of
water, mixed with 50 mg of rhodium/activated carbon (5~ Rh)
and hydrogenated for 24 hours at room temperature at 20 bars
hydrogen pressure. The catalyst was then filtered off and the
filtrate concentrated by evaporation on a rotary evaporator.
The residue was dried in a high vacuum. The yield of the
.
.. : .,.~ ....
-
.
;. ~ '
13
product was quantitative. Other data regarding the product
was:
H-NMR (DMSO-d6, 300 MHz) ~: 0.75-1.80 (m, 13H);
3.40 (br.m, lH);
4.08 (d, lH).
Example 7
(2R 3S)-2 HYdroxy-4-phenyl-3-phthalimidobut~ric acid
16.8 g of (2R,3S)-2-hydroxy-4-phenyl-3-phthalimidobutyric
acid methyl ester (content according to HPLC 49.6%) was
dissolved in 50 ml of acetic acid and mixed with 100 ml of 32%
hydrochloric acid. The mixture was stirred for 28 hours at
room temperature, and a white solid precipitated. The
precipitate was filtered off and washed with cold water. The
yield of the product was 10.2 g. Other data regarding the
product was:
H-NMR (CDCl3, 300 MHz) ~: 3.15-3.40 (m, 2H);
4.50 (d, lH);
4.95-5.05 (m, lH);
7.08-7.20 ~m, 5H);
7.60-7.80 (m, 4H).
~ o ~
Example 8
(2R.3S)-2-Hvdroxy-~-Phenvl-3-Phthalimidobutyric acid methyl
ester ~reduction with Zn(BH4)2~
2.85 g of sodium borohy~ride and 12.83 g of anhydrous
zinc chloride were stirred for 18 hours at room temperature in
585 ml of anhydrous tetrahydrofuran. Then the resultant
suspension was cooled to -25C and mixed with a solution of
63.5 g of (S)-2-oxo-4-phenyl-3-phthalimidobutyric acid methyl
ester in 50 ml of tetrahydrofuran. The mixture was stirred
for another 1.5 hours at 25C, then diluted with 500 ml of
toluene and extracted three times with 350 ml of o.1 M
hydrochloric acid each. The organic phase was washed three
times with 350 ml of saturated sodium bicarbonate solution
each. The combined aqueous phases were extracted with
toluene. After drying the combined oryanic phases over
magnesium sulfate the solvent was distilled off on a rotary
evaporator and the residue was recrystallized from 160 ml of
toluene. The yield of the product was 33.9 g (53% of theory).
Exam~le 9
(2R,3S)-2-Hydroxy-4-phenYl-3-phthalimidobutyric acid methyl
ester (reduction with Ca(BH4)2)
1.12 g of sodium boronydride and 1.23 g of anhydrous
calcium chloride were stirred for 16 hours at room temperature
in 210 ml of anhydrous tetrahydrofuran. Then the mixture was
,: " ~'~' ,'
, ! ; :
2~83~g
cooled to -25C. A solution of 25 g of (S)-2-oxo-4-phenyl-3-
phthalimidobutyric methyl ester in 40 ml of tetrahydrofuran
was lnstilled in this mixture. The reaction mlxture was
s-tirred for another 30 minutes at -25C, then diluted ~lith 250 ml of
toluene,and washed first with 200 ml of 0.1 M hydrochloric
acid and then three times with 250 ml of saturated sodium
bicarbonate solution each. The further working-up took place
as des~ribed in Example 8 but without recrystallization. The
yield of the product was 22.5 g (89.5% of theory). Other data
for the product was:
~iastereomeric ratio: (2R,3S)/(2S,3S) - 4:1.
Example 10
(2R,3S)-2-Hydroxy-4-phenyl-3-phthalimidobutyric acid methyl
ester (reduction with H2/Pt/Rh)
100 mg of a Pt/Rh mixed catalyst (4% Pt, 1% Rh on
activated carbon) was suspended in a solution of 1 g of (S)-2-
oxo-4-phenyl-3-phthalimidobutyric acid methyl ester in 10 ml
of ethyl acetate. The mixture was hydrogenated for 24 hours
at 50 bars hydrogen pressure and 50C, then filtered off from
the catalyst and concentrated by evaporation on a rotary
evaporator. The yield of the product was 1.0 g, with a
conten~ (HPLC) of 53.5 percent. Other data for the product
was:
Diastereomeric ratio: (2R,3S) ~ 5.