Note: Descriptions are shown in the official language in which they were submitted.
- 1 2083663
FIELD OF THE INVENTION
This invention relates to an improvement in a slurry hydro-
conversion process for heavy hydrocarbonaceous feedstocks wherein a
concentrate of a presulfided catalyst precursor concentrate of
molybdenum, formed in a portion of process feed, is used as a source of
catalyst. The presulfided precursor concentrate can be added directly
to the hydroconversion reactor or it can be premixed with process feed
prior to introduction to the reactor provided that the temperature of
the concentrate, prior to addition or to premixing, is maintained below
about 315C.
BACKGROUND OF THE INVENTION
There is substantial interest in the petroleum industry in
converting heavy hydrocarbonaceous feedstocks to lower boiling liquids.
One type of process suitable for hydroconversion of heavy feedstocks is
a slurry process using a catalyst prepared in a hydrocarbon oil from (a
thermally decomposable, metal compound) catalyst precursor. The cata-
lyst is formed in situ in the hydroconversion zone. See for example,
U.S. Patent Nos. 4,226,742 and 4,244,839.
Further, U.S. Patent Nos. 4,740,295 and 4,740~489, teach a
method wherein a catalyst is prepared from a phosphomolybdic acid
precursor concentrate. The precursor concentrate is sulfided prior to
the final catalyst formation. This presulfiding step is taught to
produce a catalyst having greater control over coke formation. The
presulfiding agent in these two patents requires a hydrogen-sulfide
containing gas or a hydrogen-sulfide precursor and the resulting
catalyst concentrate is used for hydroconversion of heavy hydrocarbo-
naceous materials to lower boiling products.
The term ~hydroconversion~ with reference to a hydro-
carbonaceous oil, is used herein to designate a catalytic process
conducted in the presence of hydrogen in which at least a portion of the
heavy constituents of the oil is converted to lower boiling products.
The simultaneous reduction of the concentration of nitrogenous
2083663
-- 2 -
compounds, sulfur compounds and metallic constituents of the oil may
also result.
All boiling points referred to herein are atmospheric
pressure equivalent boiling points unless otherwise specified.
The process of introducing a catalyst precursor in the form
of a concentrate mixed in a hydrocarbonaceous oil into a hydroconversion
zone containing a heavy hydrocarbonaceous chargestock is disclosed in
U.S. Patent No. 5,053,376, which is incorporated herein by reference.
This procedure has certain advantages when compared with a process
wherein the catalyst precursor is introduced into the hydroconversion
zone without first forming a concentrate; that is, by introducing the
catalyst precursor directly into the feed or directly into the reactor.
The advantages include: (i) ease of mixing the precursor with a small
stream instead of the whole feed; (ii) the ability to store the pre-
cursor concentrate for future use and/or activity certification either
on site or at another site; and (iii) the ability to use a hydrocarbo-
naceous oil, other than the feedstock, as dispersing medium for the
catalyst precursor, when hydrocarbonaceous oil other than the feedstock
can be more optimum for developing catalyst activity. U.S. Patent No.
5,053,376 teaches the use of elemental sulfur as a sulfiding agent.
A catalyst precursor concentrate is usually presulfided and
converted to a catalyst concentrate comprised of solid catalyst
particles dispersed in a hydrocarbonaceous oil. At least a portion of
this catalyst is then introduced into the hydrocarbonaceous chargestock
to be hydroconverted, with or without coal. Typically, a catalyst con-
centrate that contains from about 0.2 wt.$ up to 0.7 wt.% Mo 1s added to
the process feedstock to obtain a blend of feed and concentrate that
contalns from about 0.005 wt.% (50 wppm) to about 0.100 wt.% (1000 wppm)
Mo. This invention is concerned only with introducing a presulfided
catalyst precursor concentrate into a hydroconversion zone and not a
catalyst concentrate.
Attempts to increase the molybdenum content of presulfided,
preformed catalyst concentrates results in loss of catalyst activity
3 2083663
when the concentration is increased above about 0.7 wt.% molybdenum (see
Figure 2). The problem appears to derive from association of small
catalyst particles as the concentration of catalyst particles increases,
thus resulting in loss of catalyst dispersibility.
The presence of higher molybdenum levels in a concentrate is
valued in that such levels minimize the size of the preparation
facilities as well as the volume of the concentrate stream that must be
blended with process feed to maintain a given level of Mo in the
reactor. Reduction of stream volume makes the maintenance of heat
balance simpler and reduces the amount of gas oil entering the reactor
in the case where an atmospheric residuum is used as the vehicle for the
concentrate preparation. Reduction of the volume of gas oil results in
reduction in gas production and in consumption of hydrogen.
It has now been found that this concentration limit of about
0.7 wt.% Mo can be overcome by eliminating the catalyst preforming step
and feeding only the presulfided precursor concentrate into the hydro-
conversion zone. When a presulfided precursor concentrate is used, the
concentration of Mo in the concentrate can be raised to 1.5 wt.%, and
above, without sacrifice in activity of the catalyst that is ultimately
formed when blends of precursor concentrate and process feed (overall Mo
content of 50 to 1000 wppm) are subjected to hydroconversion conditions
(Figure 2).
SUMMARY OF THE INVENTION
In accordance with the present invention, there is provided
a process for hydroconvertlng a hydrocarbonaceous chargestock to lower
boiling products, which comprlses reactlng the hydrocarbonaceous charge-
stock with a presulfided catalyst precursor concentrate in the presence
of hydrogen at hydroconversion conditions, the presulfided catalyst
precursor concentrate having been prepared by the steps which comprise:
(a) forming a mixture of a hydrocarbonaceous oil comprising constituents
boiling above about 565C and an aqueous solution of a precursor
containing a molybdenum polyacid precursor in an amount to provide from
about 0.2 wt.% to 5 weight percent molybdenum, calculated as elemental
~ 4 ~ 2083663
molybdenum, based on the hydrocarbonaceous oil to produce a water-
containing catalyst precursor concentrate, (b) drying the water-
containing catalyst precursor concentrate to remove water and produce a
substantially water-free catalyst precursor concentrate, and (c)
contacting the resultant catalyst precursor concentrate with added
hydrogen sulfide at a temperature from about 10C up to but not
including the temperature of catalyst formation, and a hydrogen sulfide
partial pressure ranging from about 14.7 psia to about 200 psia to
produce a presulfided catalyst precursor concentrate.
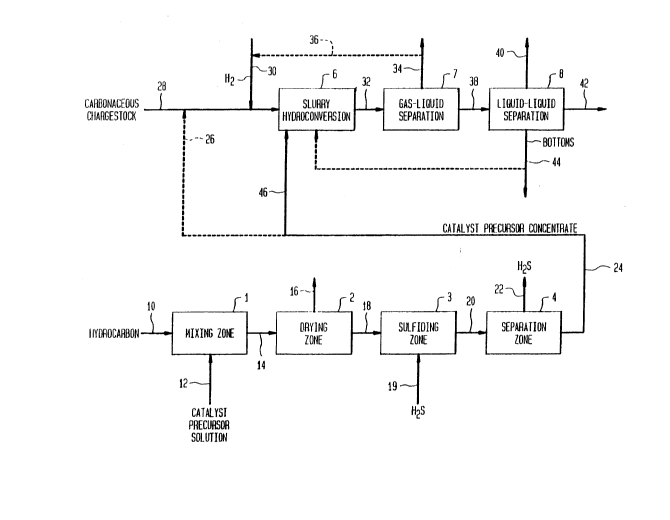
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 is a schematic flow plan of one embodiment of the
invention.
Figure 2 is a graph comparing the catalyst activity of
presulfided precursor concentrate and presulfided-preformed catalyst
concentrate as the wt.% molybdenum in the concentrate increases from 0.2
wt.% to 1.5 wt.%.
Figure 3 is a graph comparing the wt.% of catalyst solids in
the catalyst concentrate stream as a function of catalyst preforming
temperature. The threshold temperature for catalyst solids formation is
about 315C.
DESCRIPTION OF THE PREFERRED EMBODIMENT
Referring now to the Figure 1, a heavy hydrocarbonaceous oil
is introduced by line 10 into mixing zone 1. Suitable hydrocarbonaceous
oils for introduction into mixing zone 1 include hydrocarbonaceous olls
comprising constituents boiling above 565C, preferably having at least
10 wt.% constituents boiling above 565C, such as crude oils, atmo-
spheric residua boiling above 340C, and vacuum residua boiling above
565C. Preferably, the hydrocarbonaceous oil has an initial boiling
point above at least 340C and comprise asphaltenes and/or resins.
Most preferably, the hydrocarbonaceous oils comprise a lighter boiling
oil boiling below 565C and a heavier oil boiling above about 565C in
2083663
a blend comprising at least about 22 wt.% materials boiling above
565C. Preferred concentrations of the 565+C blend include from about
22 to 85 wt.% heavier oil, more preferably from about 30 to 85 wt.%
heavier oil, still more preferably about 40 to 85 wt.% heavier oil, and
most preferably about 45 to 75 wt.% heavier oil, based on the total
weight of the blend (mixture of oils). The light oil may be a gas oil
and heavier oil may be a vacuu0 residuum. Alternatively, an atmospheric
residuum having the appropriate amount of desired constituents may be
used as the oil of line 10.
An aqueous solution of phosphomolybdic acid (or other
suitable water or oil soluble molybdenum compound) is introduced
into mixing zone l via line 12. A sufficient amount of the aqueous
phosphomolybdic acid solution is introduced into mixing zone l to
provide from about 0.2 to 5, preferably from about 0.2 to 3, more
preferably 0.5 to 2 wt.% molybdenum from the phosphomolybdic acid,
calculated as elemental molybdenum based on the hydrocarbonaceous oil.
The resulting mixture is a water-containing catalyst precursor concen-
trate (i.e., wet catalyst precursor concentrate). The wet catalyst
precursor concentrate is removed from mixing zone l via line 14 and
passed to drying zone 2. In drying zone 2, bulk water is removed from
the wet catalyst precursor concentrate by any suitable manner, such as
heating the water-containing catalyst precursor concentrate to a
temperature sufficient to vaporize the water. A temperature ranging
from about 100C to 150C is suitable for this purpose. Water is
removed from drying zone 2 via line 16.
The term "phosphomolybdic acid~ is used herein to designate
aqueous solutions of the reaction product of MoO3 with dilute phosphoric
acid in which the phosphorus to molybdenum atomic ratio ranges from
0.083 to 2.00, preferably from 0.083 to 1.00 and most preferably from
0.083 to 0.50. These solutions can contain one or more phosphomolybdic
acid species such as the 12-molybdophosphoric acid (e.g.
H2PMo12040 x H20) and the dimeric 18-molybdophosphoric acid. Moreover,
the crystalline 12 and 18 acids can be used to prepare the water solu-
tions of phosphomolybdic acid used in the process of the invention. As
- 6 - 2083663
to phosphomolybdic acids refer to Topics in Current Chemistry No. 76,
published by Springer-Verlag of New York, pp. 1-64, 1978.
Although phosphomolybdic acid is most often employed, other
suitable molybdenum compounds convertible (under preparation conditions)
to presulfided catalyst precursor concentrates include: (1) inorganic
metal compounds such as carbonyls, halides, oxyhalides, poly acids such
as isopolyacids and heteropolyacids (e.g., molybdosilicic acid, as well
as phosphomolybdic acid); (2) molybdenum salts of organic acids such as
acyclic and alicyclic aliphatic carboxylic acids and thiocarboxylic
acids containing two or more carbon atoms (e.g., naphthenic acids);
aromatic carboxylic acids (toluic acid); sulfonic acids (e.g., toluene-
sulfonic acid); sulfinic acid; mercaptans, xanthic acids; phenols, di-
and polyhydroxy aromatic compounds: (3) organomolybdenum compounds such
as chelates, e.g., with 1,3-diketones, ethylenediamine, ethylene-
diaminetetraacetic acid, phthalocyanines, etc.; (4) molybdenum salts of
organic amines such as aliphatic amines, aromatic amines and quaternary
ammonium compounds.
The essentially water-free catalyst precursor concentrate is
removed from drying zone 2 and is passed to sulfiding zone 3 via line
18. Of course, if an aqueous solution of catalyst precursor is not
used, then the drying step can be eliminated. In sulfiding zone 3, the
dry catalyst precursor concentrate is contacted with a sulfiding agent
which may be a hydrogen sulfide-containing gas, hydrogen sulfide pre-
cursor, or elemental sulfur. Hydrogen may be present or absent during
the sulfiding step, preferably absent. The sulfiding agent is prefer-
ably a gas comprising from 10 to 100 mole percent hydrogen sulfide. The
sulfiding step is conducted at relatively low total pressures. Suitable
low pressures include a total pressure ranging from about 0 to 450 pslg,
preferably a pressure ranging from about 0 to 100 psig. The hydrogen
sulfide partial pressure may range from about 14.7 psia to 200 psia,
preferably from 20 psia to 100 psia. Suitable temperatures in the
sulfiding zone include a temperature of at least about 10C up to, but
not including the temperature at which the precursor will be converted
to a catalyst, preferably the temperature will be from about 10C to
315C, more preferably from about 40C to 150C. Suitable contact
- 7 - 2083~63
times will range from about S minutes to 120 minutes, depending on the
sulfiding temperature employed. Contact of the essentially dry catalyst
precursor concentrate with the sulfiding agent at the given conditions
produces a presulfided catalyst precursor concentrate.
If elemental sulfur is used, it may be used either as a
sublimed powder, or as a concentrated dispersion of sublimed powder,
such as commercial Flowers of Sulfur, in heavy hydrocarbonaceous oil.
Allotropic forms of elemental sulfur, such as orthorhombic and
monoclinic sulfur are also suitable. The preferred physical form of
sulfur is the sublimed powder (flowers of sulfur), although sulfur may
also be introduced as molten sulfur and as sulfur vapor. The amount of
sulfur added into mixing zone 1 is such that the atomic ratio of sulfur
to molybdenum is from about 1/1 to 8/1, preferably from about 2/1 to 7/1
and more preferably from about 3/1 to 6/1. Preferably, sulfur is
dispersed in the hydrocarbonaceous oil prior to addition of the
molybdenum compound, e.g., the aqueous solution of phosphomolybdic acid.
The sulfur containing precursor mixture is subjected to a brief contact
of from about 5 minutes to 60 minutes at a temperature ranging from
about 300F to 600F to prepare the presulfided precursor concentrate.
The use of elemental sulfur is described in detail in U.S. Patent No.
5,039,392 which is incorporated herein by reference.
The effluent of the presulfiding zone comprising the
presulfided catalyst precursor concentrate and a gaseous phase including
the unreacted sulfiding agent (for purpose of this discussion, H2S) is
passed by line 20 to separation zone 4 in which the gaseous phase
comprising the H2S and any remaining water is separated from the
presulfided catalyst precursor concentrate. The gaseous phase is
removed via line 22. The catalyst precursor concentrate can be fed via
lines 24 and 46 as a separate stream to the reactor. This strea~ ~s
maintained at a temperature up to, but not including the threshold
temperature for catalyst formation. Figure 3 illustrates this. Direct
injection of presulfided concentrate is therefore possible (without loss
of activity) when the inlet line temperature is held below about 315C,
for example in the range of about 100C to 315C, preferably in the
range of about 285-315C. The threshold temperature of catalyst
- 8 - 2083~3
formation is approximately 315C. Insulated nozzles are required.
Furthermore, catalyst precursor injection should be at a point of severe
mixing or turbulence.
Alternately, all or part of the concentrate may be passed
through line 26 to be blended with the reactor chargestock of line 28.
In this alternative, the inlet line temperature at the point where the
concentrate mixes with process feed should not exceed about 315C.
Regardless of the option chosen, it is essential that the temperature of
the presulfided precursor concentrate be maintained below about 315C
until such point that the precursor concentrate can be blended, in more
or less instantaneous fashion (i.e., at a point of high turbulence) with
process feed or with reactor liquid. Maintenance of concentrate temper-
ature below about 315C ensures that formation of catalyst solids does
not occur (Figure 3), thus avoiding loss of activity that occurs when
concentrates that contain above 0.7 wt.% Mo are converted to catalyst
concentrates ~i.e., the presulfided-preformed concentrates of Figure 2).
Line 28 carries a hydrocarbonaceous chargestock which may
have the same boiling point range as the hydrocarbonaceous oil of line
10. It may alternately be a single hydrocarbon or a mixture of hydro-
carbons having the same boiling point range as the hydrocarbonaceous oil
of line 10, or a different boiling point range from the hydrocarbo-
naceous oil of line 10. The hydrocarbonaceous chargestock may be a
hydrocarbonaceous oil or coal in a hydrocarbon diluent. Suitable hydro-
carbonaceous oil chargestocks include crude oils; mixtures of hydro-
carbons boiling above 220C, preferably above 340C, for example, gas
oils, vacuum residua, atmospheric residua, once-through coker bottoms,
and asphalt. The hydrocarbonaceous oil chargestock may be derived from
any source, such as petroleum, shale oil, tar sand oil, oils derlved
from coal liquefaction processes, including coal liquefaction bottoms,
and mixtures thereof. Preferably, the hydrocarbonaceous oils have at
least 10 wt.% materials boiling above 565C. More preferably, the
hydrocarbonaceous oils have a Conradson carbon content ranging from
about 5 to about 50 wt.%. Coal may be added to any of these oils.
Alternatively, slurries of coal in a hydrocarbon diluent may be used as
chargestock to convert the coal (i.e., coal liquefaction). The diluent
.
. . ~.
- 9 - 2~83~3
may be a single type of hydrocarbon or a mixture of hydrocarbons and may
be a light hydrocarbon or a heavy hydrocarbon, as described in U.S.
Patent No. 4,094,765, column 1, lines 54 to column 2, line 43, the
teachings of which are hereby incorporated by reference.
When at least a portion of the presulfided catalyst
precursor concentrate is introduced into the chargestock, the con-
centrate disperses in the chargestock. If the chargestock comprises
coal in a diluent, the presul~ided precursor concentrate is added to the
diluent before, after or simultaneously with the addition of coal to the
diluent. A hydrogen-containing gas is introduced via line 30 into line
28. The mixture of hydrocarbonaceous chargestock and presulfided
catalyst precursor concentrate and hydrogen is passed into slurry
hydroconversion zone 6.
The presulfided catalyst precursor concentrate is added to
the hydrocarbonaceous chargestock in an amount sufficient to provide
from about 10 to about 1000 wppm molybdenum, preferably 50 to 300 wppm
molybdenum, calculated as elemental molybdenum, based on the total
hydroconversion zone chargestock, i.e., presulfided precursor concen-
trate plus hydrocarbonaceous chargestock.
Catalyst effectiveness is reduced when precursor concen-
trates containing more than about 0.7 wt.% Mo are converted to catalyst
concentrates prior to injection into the hydroconversion zone (see
Figure 2). Therefore, it is preferred that ~he direct injection of
presulfided concentrate into the hydroconversion zone, or into charge-
stock prior to the reactor, be so configured that the injection line
and/or nozzle be operated at a temperature below that at which the
precursor concentrate is converted to the corresponding catalyst concen-
trate, i.e., the temperature at which catalyst particles are formed.
As illustrated in Figure 3, there is an apparent threshold
temperature of about 315C at which the presulfided precursor catalyst
concentrate is converted to a catalyst concentrate. Thus, for direct
injection of precursor concentrate into the reactor, the maximum temper-
ature for the injection line and/or nozzle will be preferably held to a
- lo- 2~83663
maximum of about 315C, more preferably below 315C and most preferably
between 285C-315C. Once the presulfided precursor concentrate is
injected into the reactor liquid in the reaction zone, there is con-
current formation of catalyst particles and dilution of catalyst
particles in the reactor liquid, which condition results in retention of
the maximum catalyst effectiveness.
Alternatively, presulfided precursor concentrates that
contain more than about 0.7 wt.% Mo can be preblended with part or all
of the chargestock prior to injection into the hydroconversion zone.
Preferably, in this mode of operation, the mixing of presulfided
precursor concentrate with chargestock results in a stream in which the
presulfided precursor concentrate, expressed as concentration of Mo, is
less than about 0.7 wt.% and more preferably below about 0.5 wt.%.
Further, in this mode of operation, the temperature of the presulfided
precursor concentrate will also be held below about 315C.
Suitable hydroconversion operating conditions are summarized
in Table 3.
TABLE I
ConditionsBroad Range Preferred Range
Temperature, C 425 to 510435 to 470
H2 Partial Pressure, psig 50 to 5000100 to 2500
The hydroconversion zone effluent is removed by line 32 and
passed to a gas-liquid separation zone 7 wherein the normally gaseous
phase is separated from a normally liquid phase. The gaseous phase is
removed from separation zone 7 via line 34. Alternatively, the gaseous
phase,which comprises hydrogen, may be recycled via line 36, preferably
after removal of undesired constituents to hydroconverslon zone 6 via
line 30. The normally liquid phase, which comprises the molybdenum-
containing catalytic solids and a hydroconverted hydrocarbonaceous oil
product is passed via line 38 to liquid-liquid separation zone B for
fractionation by conventional means, including distillation into various
fractions, such as light, medium, and heavy boiling heavy bottoms
' : ' .,
,
'
.
,
- 11- 2083~3
fractions. The light fraction is removed via line 40. The medium
boiling fraction is removed via line 42 and collected for further use or
processing. The heavy bottoms fraction is removed via line 44, and if
desired, at least a portion of this bottoms fraction may be recycled to
hydroconversion zone 6.
Furthermore, if desired, the catalytic solids may be
separated from the hydroconverted oil product and the separated solids
may be recycled to hydroconversion zone 6.
The following examples are presented to illus~rate the
invention.
EXAMPLES
Example 1 - Preparation of presulfided catalyst precursor
concentrate that contains 0.44 wt.% Mo (preparation R-1839-CP)
This preparation was carried out in a standard Autoclave
Engineer's 300 cc autoclave with Magnedrive stirrer. The concentrate was
prepared in whole Cold Lake bitumen, which bitumen had an initial
boiling point of about 232C, 50 wt % of bottoms that had a boiling
point greater than 524C, and a Conradson Carbon content of 13.4 wt.%.
Phosphomolybdic acid solutions were prepared from crystalline phospho-
molybdic acid (20 MoO3 . 2 H3P04 . 48 H20) supplied by Fisher Scientific.
The autoclave was charged with 90 9. of bitumen, flushed
with nitrogen and heated to 80C with stirring. At this point, 10 9. of
a solution of phosphomolybdic acid in deionized water (4 wt.~ Mo in
solution) was injected into the autoclave and stirring was continued at
80C for lO minutes. At the end of this period a flow of nitrogen was
started through the autoclave and the temperature was raised to 149C
and maintained at 149C for 10 minutes to remove water. Sulfiding was
then carried out by adding 50 psig hydrogen sulfide for a 30 minute
stirred treatment at 149C. The autoclave was subsequently vented,
flushed with hydrogen and cooled to room temperature. There was
- . , - ~ . . . .
2083663
- 12 -
recovered about 90 g. of presulfided catalyst precursor concentrate that
contained 0.44 wt.% Mo.
Example 2 - Preparation of presulfided catalyst precursor
concentrate that contains 0.5 Wt.% Mo (preparation R-2589-CP)
The procedure of Example 1 was repeated with the exceptions
that whole Athabasca Bitumen was substituted for whole Cold Lake
Bitumen and that 9 g. of a 5 wt % Mo solution was injected. This
Athabasca Bitumen, comparable to the Cold Lake Bitumen, had an initial
boiling point of about 260C, contained 47 wt.% of components boiling
above 525C and 12.3 wt.% Conradson Carbon. Upon completion of the
sulfiding step there was recovered 91 g. of presulfided catalyst
precursor concentrate that contained O.S5 wt.% Mo.
Example 3 - Preparation of presulfided catalyst precursor
concentrate that contains 1.0 Wt.% Mo (preparation R-2590-CP)
The procedure of Example 2 was followed, except that the
concentration of phosphomolybdic acid in deionized water was increased
such that the concentration of Mo in the solution was 6.0 wt.%, and the
amount of solution injected into 90 g. of Athabasca Bitumen was
increased to 15.0 9. There was recovered 92 9. of presulfided catalyst
precursor concentrate that contained 0.98 wt.% Mo.
Example 4 - Preparation of presulfided catalyst precursor
concentrate that contains 1.5 Wt.% Mo (preparation R-2596-CP)
The procedure of Example 2 was followed except that the
concentration of phosphomolybdic acid in deion~zed water was increased
such that the concentration of Mo in the solution was 7 wt.%, and the
amount of solution injected into the Athabasca Bitumen was increased to
20.0 g. Also, H2S pressure in the sulfiding step was increased to 100
psig. There was recovered 103 9. of presulfided catalyst precursor
concentrate that contained 1.5 wt.% Mo.
.i
- 13 - 2083663
Example 5 - Preparation of presulfided and preformed catalyst
concentrate that contains 0.44 Wt.% Mo (preparation R-1851-CP)
The procedure of Example 1 was followed through completion
of the sulfiding step and removal of excess H2S. At this point the
reactor was sealed and the temperature was increased to 371C and held
for a period of 20 minutes, which step resulted in conversion of the
presulfided preformed catalyst concentrate to a sulfided catalyst
concentrate. Upon cooling and venting the reactor there was obtained 90
9. of catalyst concentrate that contained 0.44 wt.% Mo.
When 30 g. of catalyst concentrate was diluted with 90 g. of
toluene and filtered, there was recovered 0.84 g. (2.8 wt.% ) of
preformed catalyst solids.
ExamPle 6 - Preparation of presulfided and preformed catalyst
concentrate that contains 0.86 Wt.% Mo (preparation R-2511-CP)
The procedure of Example 5 was followed with the following
exceptions; whole Athabasca Bitumen (see Example 2) was substituted for
whole Cold Lake Bitumen, the concentration of phosphomolybdic acid in
deionized water was increased such that the concentration of Mo in
solution was 6 wt.%, the amount of solution injected was increased to
11.7 9. and H2S pressure in the sulfiding step was increased from 50 to
100 psig.
Upon completion of the preforming step the reactor was
vented while still at 371C and then cooled to room temperature. Some
light hydrocarbon liquid was removed from the reactor upon venting.
There was recovered 88.2 9. of presulfided and preformed catalyst
concentrate that contained 0.86 wt.% Mo. The concentration of toluene-
insoluble, filterable catalyst solids was 6.1 wt.~ in the concentrate.
Example 7 - Preparation of presulfided and preformed catalyst
concentrate that contains 1.05 Wt.% Mo (preparation R-2510-CP)
- 14 - 20836~3
The procedure of Example 6 was followed except that the
amount of phosphomolybdic acid solution added to the 9o 9. of bitumen
was increased to 15 9. Upon completion of the preforming step there was
recovered 86 9. of presulfided and preformed concentrate that contained
1.05 wt.% Mo. The concentration of toluene-insoluble, filterable
catalyst solids in the concentrate was 6.9 wt.%.
Example 8 - Preparation of presulfided and preformed catalyst
concentrate that contains 1.52 wt.% Mo (preparation R-2508-CP)
The procedure of Example 6 was followed except that the
concentration of phosphomolybdic acid in deionized water was increased
such that the concentration of Mo in solution was 7 wt.%, and the amount
of solution injected was increased to 20.0 g. Upon completion of the
preforming step there was recovered 88 9. of presulfided and preformed
concentrate that contained 1.60 wt.% Mo. The concentration of toluene
insoluble, filterable catalyst solids in the concentrate was not
determined.
ExamDle 9 - Test of presulfided precursor concentrate of Example 1
under hydroconversion conditions (Test no. R-1842)
A hydroconversion run was carried out with a Cold Lake Crude
vacuum bottoms feed, which had an initial boiling point of about 490C
and which contained 94.8 wt.% of components boiling above 524C and
23.4 wt.% Conradson Carbon.
To a 300 cc Autoclave Engineer's autoclave, equipped with
Magnedrive stirrer, was charged 109.5 9. of Cold Lake vacuum bottoms,
5.00 9. of whole Cold Lake crude (see Example 1 for feed properties) and
5.50 9 of the presulfided catalyst concentrate of Example 1. The amount
of concentrate charged was sufficient to give a Mo concentration of 201
wppm in the total reactor charge, i.e., the combined weight of vacuum
bottoms, whole Cold Lake crude and catalyst preparation. The autoclave
was next flushed with hydrogen, sealed and stirred for 10 minutes at
93C to mix the reactor contents.
-
~
- 15 - ~083663
Upon cooling to room temperature, the autoclave was charged
with 13~0 psig hydrogen. With stirring the autoclave was heated to
385C and held at 385C for 20 minutes. At this point, autoclave
pressure was adjusted to 2100 psig, a flow of hydrogen was started
through the autoclave to maintain an outlet gas rate of 0.36 liter/min.
(as measured at room temperature and atmospheric pressure) and the
temperature was increased to 443C.
Upon completion of a three hour stirred contact at 443C
under 2~ psig w'th hydrogen flow gf 0.36 llmin., the ~low of hydrogen
was stopped and the autD~lave w~s c~led qui~kIy t~ 93aC.
Gaseous products were vented form the autoclave at 93C,
scrubbed with dilute caustiC solution to remove hydrogen sulfide, passed
through a wet test meter to determine volume and then collected for
analysis by mass spectrometry. Gaseous products that were collected
during the flow-through portion of the run were d1SO col1ected and
analyzed. Some light liquid product, b.p. 16C-316C, swept from the
autoclave during the flow-through period was collected in a cold,
high-pressure knockout vessel that was attached to the autoclave gas
exit line.
- Liquid and solid products that remained in the autoclave
were removed by washing with toluene, and the toluene wash was then
filtered to recover solids, which solids comprise catalyst residues,
demetallation products and toluene insoluble coke. After washing with
toluene and drying under vacuum, the solids were weighed and analyzed
for carbon content.
The yield of toluene insoluble coke was calculated as
follows:
9. dry solids X wt frac carbon
Tol. Insol. Coke, = 0.85* X 100
Wt.% on 524+C Feed
9. 524+C Feed
(* Empirical factor for converting 9. carbon to 9. coke)
20836~3
- 16 -
Unconverted feed boiling at a temperature greater than
524C (vacuum bottoms) was recovered from the toluene wash by
distillation and the product was analyzed for Conradson carbon content.
The yield of toluene insoluble coke was 1.73 wt.% on feed
boiling at a temperature greater than 524C (i.e., 524+C bottoms) and
conversion of Conradson Carbon to non-coke products was 61%.
Example 10 - Test of presulfided precursor concentrate of Example
2 under hydroconversion conditions (test no. R-2592)
The hydroconversion test described in Example 9 was repeated
with a reactor char~e consisting of 109.5 9. of vacuum Cold Lake, 4.10
9. of whole Cold Lake Crude and 6.40 9. of the presulfided catalyst
concentrate of Example 2. The concentration of Mo in the reactor charge
was 266 wppm.
The yield of toluene insoluble solids from this test was
1.91 wt.% on 524+C feed and conversion of Conradson Carbon to non-coke
products was 67%.
Example 11 - Test of presulfided precursor concentrate of Example
3 under hydroconversion conditions (test no. R-2593)
The hydroconversion test described in Example 9 was repeated
with a reactor charge consisting of 109.5 9. of vacuum Cold Lake
bottoms, 7.23 9. of whole Cold Lake crude and 3.27 9. of the presulfided
catalyst precursor concentrate of Example 3. The concentration of Mo in
the reactor charge was 270 wppm.
The yield of toluene insoluble coke was 1.82 wt.% on 524+C
feed and conversion of Conradson Carbon to non-coke products was 66 %.
ExamDle 12 - Test of presulfided precursor concentrate of Example
4 under hydroconversion conditions (test number R-2601)
20836~3
- 17 -
The hydroconversion test described in Example 9 was repeated
with a reactor charge consisting of 109.5 g. of vacuum Cold Lake
bottoms, 8.36 g. of whole Cold Lake crude and 2.14 9. of the presulfided
precursor concentrate of Example 4. The Mo concentration in the reactor
charge was 267 wppm.
The yield of toluene insoluble coke was 1.81 wt.% on 524+C
feed, and Conradson carbon conversion to non-coke products was 65%.
Example 13 - Test of presulfided/preformed catalyst concentrate of
Example 5 under hydroconversion conditions (test number R-1853)
The hydroconversion test described in Example 9 was repeated
with a reactor charge that consisted of 109.5 g. of vacuum Cold Lake
bottoms, 5.00 g. of whole Cold Lake crude and 5.50 g. of
presulfided/preformed catalyst concentrate of Comparative Example 5. The
Mo concentration in the reactor charge was 202 wppm.
The yield of toluene insoluble coke was 1.54 wt.~ on 524+C
feed, and Conradson Carbon conversion to non-coke products was 67%.
Example 14 - Test of presulfided/preformed catalyst concentrate of
Comparative Example 6 under hydroconversion conditions (test number
R-2514)
The hydroconversion test described in Example 9 was repeated
with a reactor charge that consisted of 109.5 g. of vacuum Cold Lake
bottoms, 7.01 9. of whole Cold Lake crude and 3.49 g. of the
presulfided/preformed catalyst concentrate of Comparative Example 6. The
Mo concentration in the reactor charge was 250 wppm.
The yield of toluene insoluble coke was 2.11 wt.% on 524+C
feed and Conradson Carbon conversion to non-coke products was 65%.
Example 15 - Test of presulfided/preformed catalyst concentrate of
Comparative Example 7 under hydroconversion conditions (test number
R-2521)
2083663
- 18 -
The hydroconversion test described in Example 9 was repeated
with a reactor charge that consisted of 109.5 9. of vacuum Cold Lake
bottoms, 7.77 9. of whole Cold Lake crude and 2.73 9. of the
presulfided/preformed catalyst concentrate of Comparative Example 7. The
Mo concentration in the reactor charge was 238 wppm.
The yield of toluene insoluble coke was 2.43 wt.% on 524+C
feed and conversion of Conradson Carbon to non-coke products was 73%.
Example 16 - Test of presulfided/preformed catalyst concentrate of
Comparative Example ~3 under hydroconversion conditions ttest number
R-2517)
The hydroconversion test described in Example 9 was repeated
with a reactor charge that consisted of 109.5 9. of vacuum Cold Lake
bottoms, 8.62 9. of whole Cold Lake crude and 1.88 9. of the
presulfided/preformed catalyst concentrate of comparative Example 8. The
Mo concentration in the reactor charge was 251 wppm.
The yield of toluene insoluble coke was 4.92 wt.% on 524~C
feed and conversion of Conradson Carbon to non-coke products was 46%.
Example 17 - Preparation of presulfided/preformed catalyst
concentrate using H2S as sulfiding agent and preforming at 360C
(preparation R-2535-CP~
A presulfided/preformed catalyst concentrate was prepared
according to the following procedure:
Step A - To an aqueous solution of 20 g. of phosphomolybdic
acid (Climax Molybdenum Lot Number 1768-37) which contained 5.18 wt.$ Mo
and 0.17% P there was added 0.19 9. of phosphoric acid (85 wt.% H3P04).
A sample of 8.82 9. of the resultant modified solution was then injected
into 90 9. of whole Athabasca Bitumen while stirring at 80C in a
standard 300 cc autoclave from Autoclave Engineers. Stirring was
continued for 10 minutes after injection of the solution.
- 19- 2~83663
Step B - The autoclave was then heated with stirring to a
temperature of 149C and held for 10 minutes with nitrogen flow-through
to remove water.
Step C - At this point the autoclave was charged with 100
psig H2S and stirring was continued at 150C for an additional period of
30 minutes.
Step D - Excess hydrogen sulfide was removed from the
autoclave by venting he autoclave while still at 150C and then
sweeping with nitrogen.
SteD E - Wlth the autoclave free of excess H2S and sealed,
the temperature was increased to 360C for a 30-minute stirred contact
to form the catalyst, i.e., the catalyst concentrate.
Upon dilution of a 30 9. sample of the resultant concentrate
with 150 g. of toluene and then filtering over a #2 Whatman paper, there
was recovered 1.19 g of catalyst solids that contained 11.6 wt.% Mo.
Thus, the concentration of catalyst solids in this catalyst concentrate,
which was preformed at 360C, was 3.96 wt.%.
Example 18 - Preparation of presulfided/preformed catalyst
concentrate using H2S as sulfiding agent and preforming at 331C
(preparation R-2567-CP)
The procedure of Example 17 was repeated except that the
temperature used to preform the catalyst in Step E was 331C.
The resultant catalyst concentrate contained 4.03 wt.%
catalyst sollds, which sollds contained 11.3 wt.% Mo.
Examole 19 - Preparation of presulfided/preformed catalyst
concentrate using H2S as sulfiding agent and preforming at 318C
(Preparation R-2744-CP)
2083~3
- 20 -
The procedure of Example 17 was repeated except that the
temperature used to preform the catalyst in Step E was 318C.
The resultant catalyst concentrate was not completely formed
and contained only 1.77 wt.% solids.
Example 20 - Preparation of presulfided/preformed catalyst
concentrate using sulfur as sulfiding agent and preforming at 360C
(preparation R-2534-CP).
Preparation of presulfided/preformed catalyst concentrate
using sulfur as sulfiding agent was carried out in a manner similar to
that described in Example 17, except that Steps C and D (associated with
use of H2S were omitted.
~ The procedure of Example 17 was repeated except
that a blend of 89.14 g. of whole Athabasca Bitumen with 0.86 g. of
Flowers of sulfur was substituted for the 90 9. charge of whole
Athabasca Bitumen.
Step B - The procedure described in Step B of Example 17 was
repeated.
Ste~ C - Upon completion of the drying step of Step B, the
autoclave was sealed and then heated with stirring to a temperature of
360C to sulfide and to form the catalyst. The time required for
heating the autoclave from 149C to 360C was 20 minutes, which
interval provided time for formation of the presulfided precursor.
The resultant presulfided/preformed catalyst concentrate
contained 4.87 wt.% catalyst solids, whlch solids contained 9.34 wt.%
Mo.
~ X3DQ~ 21 - Preparation of presulfided/preformed catalyst
concentrate using sulfur as sulfiding agent and preforming at 343C
(preparation R-2552-CP)
2083663
- 21 -
The procedure of Example 20 was repeated except that the
preforming temperature used in Step C was 343C.
The resultant presulfided/preformed catalyst concentrate
contained 4.56 wt.% catalyst solids, which solids contained 10.0 wt.%
Mo.
Example 22 - Preparation of presulfided/preformed catalyst
concentrate using sulfur as sulfiding agent and preforming at 327C
(preparation R-2566-CP)
The procedure of Example 20 was repeated except that the
preforming temperature used in Step C was 327C.
The resultant presulfided/preformed catalyst concentrate
contained 4.60 wt.% catalyst solids, which solids contained 9.9 wt.% Mo.
Example 23 - Preparation of presulfided/preformed catalyst
concentrate using sulfur as sulfiding agent and preforming at 327C
(preparation R-2575-CP)
The procedure of Example 20 was repeated except that the
preforming temperature of Step C was 315C.
The resultant concentrate did not contain filterable solids.
Example 24 - Preparation of presulfided/preformed catalyst
concentrate using sulfur as sulfiding agent and preforming at 300C
(preparation R-2557-CP)
The procedure of Example 20 was repeated except that the
preforming step of Step C was 300C.
The resultant concentrate did not contain filterable solids.
; '
.:, ~:
.,
~ .
:
...