Note: Descriptions are shown in the official language in which they were submitted.
WO91/18g86 PCT/AU91/00229
MAR~ER FOR YEAST STRAIN IDENTIFICATION
Abbreviations: GUS ~-glucuronidase; MUG 4--methyl umbelll~eryl
glucuronide; SSPE 0.18M NaCl,0.015M sodium citrate
(p~7.0),0.001M EDTA; x-gluc 5-bromo-4-chloro-3-indolyl
glucuronide.
BACKGROUND TO THE INVENTION
Adva~ces in wine fermentations have followed from the
application of pure yeast culture lnoculation. This practice
enables strain differences to be utilized in the production
of a wide range of wines, and also provides greater
reliability and control of fermentation resulting in wines
with fewer flavour defects (1). More recently, the
availability of active dried yeasts has given the winernaker
even greater scope for exploiting oenological properties of
different strains. Raw materials of wine making, however,
contain an unknown load of microorganisms, some of which are
capable of producing off-flavours and spoiling wine. A
suitable method, therefore, to identify and monitor specific
wine yeast strains throughout fermentation is necessary for
the optimization of inoculation and sterilisation processes.
The potential for genetic markers in yeast strain
identification has been recognized and deliberately marked
oenological strains were developed by Vezinhet and Lacroix in
198~ (2). An oenological strain, commercialized as 'marked
K1', was developed by selecting for natural mutants in a
population of the Lalvin V yeast - it is double marked with
diuron and erythromycin resistance genes. While studies
involving marked strain K1 have provided an insight into the
kinetics o~ yeast populations during fermentation (3,4), a
limitation still exists in that wine yeast strains oE choice
cannot be eAsily marked.
A procedure is described here which utilizes recombinant
DNA technology to introduce the E.coli ~-glucuronidase (GUS)
gene as a marker into any desired yeast strains. The GUS
gene was developed as a reporter gene system for use in
nematodes and, more recen~ly, in the study of plant gene
expression (5,6). The advantages of the GUS sys~em as a
marker in yeast strains include its low background levels in
. SUBSTITUTE SII-IEET
_~______
WO91/18986 PCT/AU91/00229
~rt~ ; 2
SaccA~romyces cerevisiae and its ease of assay by
fluorimetry, spectrophotometry and agar plate tests.
SUMMARY OF THE INVENTION
The present invention provides a novel plasmid
comprising a ~-glucuronidase (GUS) coding region operably
linked to yeast promoter and texminatur sequences, whexeby
said coding region is expressible in yeast. Suitable
promoter and terminator sequences are any which allow
expression in yeast and are, in particular, Saccharomyces
cerevisiae alcohol dehydrogenase promoter and terminator
sequences.
The plasmid of the present invention is produced by
ligating the promoter and terminator sequences to the GUS
gene, and inserting the resultant novel construct into an
appropriate carrier plasmid. Preferably, the carrier plasmid
comprises a DNA sequence which is substantially identical to
a DNA sequence on the host yeast chromosome, thereby
assisting integration of the novel construct into that
chromosome. ~ ~uitable carrier plasmid is pWX509, comprising
the SMRl-410 gene. ~he SMRl-410 gene has two functions: (a)
it serves as a selection marker, and (b) being almost
identical in sequence to the yeast ilv~2 gene, it enhances
integra~ion of the novel co~struct into the host chromosQme.
The carrier plasmid is used to introduce the novel
construct into a yeast strain (e.g. an industrial or
polyploid yeast strain, in particular a wine yeast strain) by
integration into a host chromosome. Assay for GUS activity
can then be used to determine the proportion of the mar~d
strain in the total yeast population.
Most substrates for the GUS enzyme are incapable of
crossing the yeast cell membrane, by natural transport.
Therefore, the present assay system comprises : (a) adding a
substrate for the GUS enzyme te.g. x-gluc or MUG) to a sample
of a yeast strain; (~) inducing permeation in the cell 3
membrane of the yeast strain; (c) incubating the yeast
s~rain for sufficient time to ~orm detectable quantities of a
product of the GUS enzyme reaction; and (d) testing for said
WO91/18986 PCF/AU91/00229
3 ~jr~ 3~
product.
The GUS gene is used as a marker gene to monitor yeast
strains, particularly in the field of industrial alcoholic
fermentation processes.
In one embodiment o~ the invention, the GUS gene is used
to monitor killer yeast strains in mixed culture ferments.
It has been found that a marked yeast killer strain c~n
be used in a mixPd culture inoculum to quantify directly the
effect of killer toxin on a sensitive yeast strain undex
fermentation conditions. As a wide range of yeast strain~s can
be readily marked, this system of analysis is unlimited in
application and furthermore provides a simple and unequ:ivocal
means of guantifying killer yeast strains in mixed culture
ferments.
BRIEF DESCRIPTION OF ~HE DRAWINGS
Figure l illustrate the construction of the GUS vector.
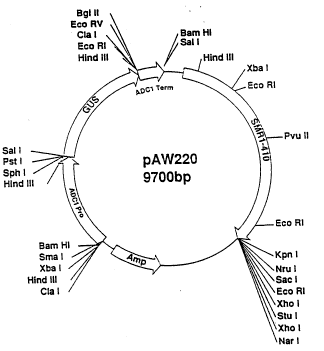
Flgure 2 is a map of pAW220.
Figure 3 illustrates the results of agar plate assay for
killer activity. The agar (ph 4.2, 0.003% methylene blue) is
seeded with an overnight oulture of strain 3AMC, and strains
to be tested for killer activity are patched onto the solid
media. llA is a known killer strain, and 2A i5 a known
sensitive strain. Strain 3AM displays an identical response
to killer llA, with a clear zone and methylene blue stained
border around the patch of growth.
Figure 4 illustrates the results of transverse
alternating field electrophoresis of chromosomes isolated
from strains 3AM and 3AMC.
Figure 5 illustrates the results of electrophoresis of
dsRNA species from killer strains llA and 3AM, and sensitive
strains 2A and 3AMC.
Figure 6 shows yeast growth (Panel A) and sugar
utilisation (Panel B) curves of strains 3AM (~) and 3AMC (o).
Figure 7 showg growth curves of control single
monoculture ferments and mixed culture ferments. Panel A:
Growth curves of control singlet monoculture ferments.
Symbols: ~ 3AM; o 3AMC; ~ 5A. Panel B: Growth curves of
WO91/l8986 PCT/AV91/002~9
mixed culture ferments. Symbols: ~ 3AM and 5A at an i~oculu~
ratio of 2:1; ~ 3AMC and 5A at an inoculum ratio of 2:1;
~ 3AM and 5A at an inoculum ratio of 1:1; ~ 3AMC and 5A at an
inoculum ratio of 1:1.
Figure 8 shows growth cur~es of each strain in mixed
culture ferments expressed as colony formi.ng units (cfus)~ml.
Panel A: Mixed ferment of 3AM(~) and 5~ (~) at an inoculum
ratio of 1:1. Panel B: Mixed ferment of 3AMC (o) and 5A (~)
at an inoculum ratio of 1:1. Panel C: Mixed ferment of 3AM
(~) and 5A (a) at an inoculum ratio of 2:1. Panel D: M:Lxed
ferment of 3AMC (o) and 5A (~) at an inoculum ratio of 2:1.
Figure 9 shows proportions of each strain in ~ixed
ferments expressed as percentage of the total yeast
population. Panel A: Mixed ferment of 3AM (o) and 5A (r) at
an inoculum ratio of 1:1. Panel B: Mixed ferment of 3AMC (o)
and 5A (a) at an inoculum ratio of 1:1. Panel C: Mixed
ferment of 3AM (o) and 5A (~) at an inoculum ratio of 2:1.
Panel D: Mixed ferment of 3AMC (o) and 5A (~) at an inoculum
ratio of 2:1.
Figure lO shows the time course in the proportion of
killer strain 3AM in the total population of a mixed culture
ferment with strain 5A for different inoculum ratios.
Symbols: ratio 3AM to 5A ~ 2:1; O 1:1; O 1:2; ~ 1:4.
DETAILED DESCRIP~ION OF ~HE INVENTION
Preferred embodiments are described below.
EXAMPLE I: A CONSTRUCTION OF MARKER SYSTEM FOR WINE YEAST
MATERIALS AND ME~HODS
S~rain~ and medla: Yeast strain AWRI 3A (also known as
AWRI 796) was obtained from The Australian Wine Research
Institute. E. co7i strain DH5a was used for the propagation of
all recombinant plasmids. Yeast growth media was YPD [1%
yeast extract (Difco), 2% bacto-peptone (Difco) and 2%
glucose] or SD [0.67% Bacto yeast nitrogen base without amino
acids (Difco) and 2% glucose~. Plasmid pWX509 was obtained
from G.P. Casey, Anheuser~Busch Co., St. Louis. Plasmid
pKLG4 was obtained from V. Walbot, Stanford Uni~ersity.
(Plasmid pKLG~ is also commercially available.)
WO9~/18986 PCT/AU91/00229
~r ~ J~
GUS-vector constructlon: Transformation of DHSa was
performed according to the method of Hanahan (30). Rapid
plasmid isolation was as described by Ish-Horowicz and Burke
(31). DNA fragments were isolated with a GENE CLEAN (BIO
101) kit following supplier's instructions.
Dephosphorylation of fragment ends was achieved by incubation
of DN~ with 1 unit of calf-intestinal a~kaline phosphatase
(Boehringer-Mannheim) for 30 minutes at 37 C in buffer as
recommended by supplier. DNA ligations were carried out
using T4 DNA ligase (Boehringer-Mannheim) in the recommended
buffer.
Yeast transformation and analysis: Yeast cells were
transformed usins the procedure of Ito et al. (32)
Transformants were selected on SD media containing
sulfometuron methyl (lO~g/ml). Total DNA was isolated from
transformants a~cording to the method of Davis et al . ( 33 ) .
Southern blotting and hybridizations were carried out by the
procedure of Southern (34) with minor modifications. The
membrane used was Hybond N~(Amersham), and DNA fixation was
achieved by exposing the membrane to W light for 5 mins.
Hybridizations were performed at 42 C in solutions consisting
of 4% polyethylene glycol 4000, 2 x SSPE, 1% SDS, 50%
formamide, 0.5% blotto and carrier DNA (0.5 mg/ml final
concentration). DNA probes were prepared with an
oligolabelling kit from Amersham.
Pulsad 4isld gel electrophoresis: Transverse
alternati~g field electrophoresis (TAFE) was carried out
using a Geneline (Beckman) unit. Yeast chromosomes were
prepared in a~arose plugs according to supplier's
recommendations. Electrophoresis was carried out at a
constant current of 150 mA; pulse times were 60 seconds for
18 hours, then 35 seconds for 6 hours.
Fermentation trial~: Rhine riesling must containing
200g/l reducing sugars was used. Fermentations were carried
out in 250ml conical flasks fitted with airlocks. The juice
was sparged with N2 gas prior to inoculation, and
fermentations were carried out at 18-C with agitation
WO9lt1B986 PCT/AU91/00229
~ 6
(approximately 100 r.p.m.) under anaerobic conditions.
Samples were remo~ed anaerobically during fermentation hy
needle and syringe through ports covered with rubber septums.
Samples were analysed for sugar level by refractometer
readings, and yeast growth by measuring spectrophotometric
absorbancy at 650 nm. Residual sugars and acetic acid
concentrations were determined with appropriate kits from
Boehringer-Mannheim. Measurement of sulfur dioxide in wine
was as according to RanXine and PococX (35). The alcohol
content was determined by near infra-red reflectance (36).
GUS as~ay~:
Enzyme as$ays: Yeast cells were grown to late log
phase, then lml was pelleted by centrifugation, and the cells
resuspended in GUS ext~action buffer (50mM NaPO4 pH7.0, lOmM
beta-mercaptoethanol, 10mM Na2EDTA, 0.1% sarcosyl, 0.1%
Triton X-100). Glass beads tsigma, 1000-1050 microns) were
then added to approximately half the volume, and the
suspension was vortexed for 10 mins at 1000 r.p.m. Aftèr
centrifugation, the supernatant was removed and used as the
cell extract.
1-50 ~l extract was added to 0.5 ml assay buffer (lmM
MUG in extraction buffer) and left to incubate at 37 C. At
various ~ime inter~als, 100~1 samples were removed from the
assay mix and added to 900~ stop buffer (0.2M Na2CO3).
Solutions were then assayed in a spectrophotofluorometer with
xenon lamp (Aminco SPF~125~), excitation 365 nm, emission 455
nm. Standard solutions of 4-methylumbelliferone tMU) (Sigma)
in the range of 100 nM to 1~1M were prepared for reference
values. l)lM MU corresponded to 100 relative units.
platQ a~s~ys: Yeast colonies were grown on solid YPD
media containing 50-100 ~g/ml x-gluc. After approximately 36
hours growth, a solution containing O.lM NaPO4 pH7.0, 1~
sarcosyl, 50 ~g/ml x-gluc and 0.7% agarose was poured as a
thin overlay on the plate and allowed to set. After 4-5
hours incubation at 37 C, a blue precipitate could be
detected in the transfoxmed colonie~.
WO91/~8986 PCT/AU91/00229
RESUhTS
GUS-vector Construct
The efficiency of the ~. coliy -glucuronidase gene as a
marker gene in yeasts is dependent upon maintenance of the
gene in a growing population and sufficient gene expression.
Expression of the GUS coding region was achieved by use
of the yeast alcohol dehydrogenase (ADC1) promoter and
terminator sequences. The GUS gene was isolated as a HindIII
frayment from the plasmid pRLG4. This wac: ligated into the
HindIII site of vector AAH5 (37). A clone with GUS coding
region in the correct orientation with respect to the ADC1
promoter was identified (plasmid pAW219). Digestion was
carried out with ~amHI. This digestion resulted in excision
of the GUS gene flanked with ADC1 expression signals (see
Figure 1).
Plasmid pWX509 (38) has been shown to integrate into
chromosome XIII of a wine yeast strain and be maintained
under fermentation conditions without adverse effects on
yeast performance. The GUS plus ADC1 signals cassette was
cloned into the BamHI site of vector pWXSO9 - giving rise to
plasmid pAN220 (Figure 2).
TR~NSFORMATION AND FERMENTATION TRIALS
The SMR1-410 gene on plasmid pAW220 is almost identical
in sequence to the 11Y_~ gene on the host chromosome - a
single base point mu~ation causes the herbicide resistant
phenotype. Therefore, sufficient homology exists between the
two sequences to target integration to the ilv 2 gene via
homologous recombination with SMR1-410. Recombination is
enhanced by digesting plasmid pAW220 with PvuII prior to
transformatlon, giving xise to a linear molecule with SMR1-
410 sequences at either end. The DNA ends are highly
recombinogenic, and integration is most likely to occur at
the P~uII site in the ilv 2 gene. The result of this event
will be two ilv 2 genes flanking the GUS cassette, one of
which will contain the SMR1-410 mutation conferring herbicide
resistance upon the transformed cell.
WO91/18986 PCT/AU91/002~9
PvuII digested plasmid pAW220 was introduced into wine
yeast straln AWRl 3A by the method of Ito et al (9).
Transformants were selected for resistance to the herbicide
sulfometuron methyl (10J~g/ml). A transformed colony
(designated AWRI 3AM) was then screened for the presence of
the GUS construct. Total DNA was isolated and digested with
PvuII, thereby releasing the GUS-vector constxuct intact from
the chromosomal DNA. A Southern hybridizat:ion was then
performed using the HindIII fragment from plasmid pKLG4 to
probe for the GUS sequence. A band of approximatel~ 9kb was
evident in the transformed strain, indicating the presence of
the GUS construct.
The chromosomal location of the GUS construct in the
transformed strain was determined. Intact chromoso~es were
prepared from strain 3AM, separated by pulsed field gel
electrophoresis on a TAFE unit and screened by Southern
hybridization for the GUS sequence. Results of this analysis
showed that the GUS construct has indeed i~tegrated
specifically into the chromosome (XIII) on which the ilv
gene is located.
The 3AM strain was used in a fermentation trial to
monitor the effects of transformation on the yeast
oenological properties. Three different transformants were
isolated and used to inoculate separate starter cultures.
Three colonies of control, un-transformed AWRI 3A yeast were
also inoculated into starter cultures. Each of these six
cultures was inoculated in duplicate into flasks of Rhine
Riesling grape juice at a concentration of 4 x lO6 cells/ml.
Fermentations were carried out under anaerobic conditions at
18 C. Samples were taken at regular intervals and assayed
for yeast growth (by measuring optical density at 610 nm) and
sugar content (by refractive index). Refractive indices were
averaged for both control ferments ~nd for transformant
ferments, and the two resulting sugar utilization curves were
plotted. There are no signifisant differences between the
fermentation curves of the control and transformed strains.
WO91/18986 PCT/AU91/00229
9 ~ J ~L
On completion of fermen~ation, pH, residual sugar~,
sulfur dioxide, alcohol and acetic acid concentrations were
measured for all twelve samples. Averages were calculated
for each parameter (see Table 1 below). Again, no
significant differences were evident between the two strai~s.
Finally, in order to test the stability of the GUS construct
in the total population, a s mple of yeast was recovered from
the finished 3~ ferments and assayed for GUS activity.
Approximately 500 colonies were analysed by the x-gluc plate
method described below; all of these colonies were positive
for GUS activity. ~his result indlcates that the in~roduced
cons~ruct is maintained in a growing yeast population under
ferm~ntation stress, and in the absence of selectio~ by
sulfometuron methyl.
~1
~--~!
p~ 3-32 13-~ 14.24 la.o ,
F~es.d~zl~ 1.43 ¦1.90 lû~60 1~-45
St)2 (to i) 131. 123.~ 1 ~0~ 10.~8
.
Sc)2 (fre~ ¦1 33 1.67 ¦1.13 1~.31
~_
Alc~hal ¦t2.~2 It2 t7 ¦1.6~ 10.23
I
# Variance ratio between the two strains
* F Prob. indicates the probability of the associated
variance ratio
AW~1-3A - Control unmodified yeast
AWR1-3AM - Modified yeast
WO91/18986 PCT/AU91/00229
s.~J ~ 10
GUS Assays
~-glucuronidase cleaves the substrate x-gluc to release an
indoxyl derivative which, upon oxidation, yives rise to an
indigo blue dye GUS activity can be detected by the
presence of a blue precipitate. Initial incubations of
transformed yeast cells in growth media containing x-gluc at
37'C gave no indication of GUS activity over a three hour
period. One possible explanation for this observation is
that the yeast cells were unable to take up the x-gluc
substrate. Transformed cells were then vortexed for two
minutes in the presence of glass beads (to break the
permeability barrier~ prior to incubation with x-gluc. This
treatment resulted in the formation of a blue precipitate
within one hour incubation at 37 C. These results indicated
that the GUS construct had been successfully introduced into
the wine yeast strain, and was being expressed. However, the
x-gluc was not taken up by the cells nor could it dif~use
into the aells.
A method was devised with these results in mind to assay
for GUS activity in yeast colo~ies. Yeast cells were plated
out on PD media (containing x-gluc) at an appropriate
dilution to obtain single, well spaced colonies. After
incubatlon at 30 C for approximately 36 hours, a molten
solution of 0.7% agarose and 1% sarcosyl was poured over the
colonies and allowed to set. After incubation at 37 C for 4-
5 hours, a blue precipitate could be detected in the
transformed colonies. This method enables the proportion of
marked strain in a total yeast population to be determined
simply by calculating the proportion of blue to white
colonies.
For more rapid ~nalyses of GUS activity, a sample of
yeast cPlls can be incubated with x-gluc in the presence of
1% sarcosyl until a deep blue precipitate is formed. The
proportion of blue to white colonies can be determi~ed under
a microscope.
An alternative assay for GUS activity can be performed
WO91/18986 PCT/AU91/00229
; .s ~ *
using the MUG substrate; ~-glucuronidase cleaves this
compound to give rise to a fluorescent product. This assay
is very sensitive, and was used to determine the G~S en~yme
activity.
EXAMPLE II: AN APPLICATION OF M~RRER SYSTEM
BACKGROUND
Riller activity in yeasts was first reported in strains
of Sacch~xomyces cerevisiae in 1963 by ~evan and Makower (7).
Killer yeasts secrete polypeptide toxins which kill sensitive
strains of the same genus and less frequently, strains of
dif f erent genera (8, 9). Previous studies indicate that the
toxin of Saccharomyces is a protein which binds to a receptor
on the cell wall of the sensitive yeast, disrupting the
electrochemical gradient across the cell membrane and hence
the intracellular ionic balance (10, 11).
Production of the toxin and immunity to it are
determined by a cytoplasmically inherited double st~.anded
(ds) R~A plasmid, otherwise known as the M-genome (12), which
is found only in cells containing an additional ds~A species
designated the L-genome. Both types of dsRNA exist in virus-
like particles and require a protein encoded by the L-dsRNA
for encapsidation (12, 13).
Based upon properties of the toxin, killer yeasts have
been classified into eleven groups ~R1 through ~11) (14, 15).
Those-unique to Saccharomyces fall into the fi.rst three (K1,
~2 and R3). The Saccharomyces toxin is reversibly inactivated
at low pH (2.0) and the irreversibly inactivated at pH in
excess of 5.0 (lS). More specifically, the biological
activity of K1 is optimal ba~ween pH 4.6 and 4.8 while K2
shows optimal activity between 4.2 and 4.7 (16). Compared to
K1, the K2 toxin is stable over a wider pH range (2.8 to 4.8)
(17) and is therefore more relevant in wine fermentation.
Riller activity has been detected in yeasts isolated
from established vineyards and wineries in various regions of
the world, including Europe and Russia, South Africa and
WO91/18986 PCT/AU91/00229
~-~.r r~ r~
~v~ ~ 12
Australia. This widespread occurrence has prompted interest
in the oenological significance of killer wi.ne yeasts. In
theory, selected killer yeasts could be usecL as the
inoculated strain to suppress growth of undesirable wild
strains of S~ccharomyces cerevisiae during grape juice
fermentation. In addition, as killer interactions have been
reported to occur between yeasts of different genera (18,
l9), the possibility exists to genetically engineer broad
spectrum killer strains of yeasts such as Saccharomyces
cerevisiae ( 20 ) .
Previous studies have been conducted to assess the
efficiency of killer toxin on sensitive yeast strains.
However, reports have been contradictory on the expression of
killer activity under fermentation conditions (4, 2l, 22).
Attempts to determine the population kinetics of killer and
sensitive strains during wine ~ermentation have been
restricted because of the difficulty involved in identifying
two types of strains when grown ln mixed cultures. Approaches
used to date include i) choice of killer and sensitive
strains that can be distinguished by their growth rates (23)
or production of hydrogen sulfide (24~; ii) use of
auxotrophic and respiratory deficient mutants of killer
strains and appropriate plating conditions under which they
can be identified (25, 26, 27); iii) use of killer and
sensitive strains which can be distinguished by differences
in colony morphology (28); and iv) assaying colonies dlrectly
~or killer activity (29). All of these methods are limited ~y
the fact that the assays involved are laborious and time-
consuming, or that only killer strains with specific
characteristics can ba studied.
MATERIALS AND METHODS
strainB and medla: Sensitive Saccharomyces cerevisiae
strains AWRI 5A (also known as AWRI 138), AWRI 2A (AWRI 729),
a~d killer strain AWRI llA (AWRI 92F) were obtained from the
Australian Wine Research Institute. Generation of the marked
killer strain 3AM (AWRI 796) has been prevlously ~escribed
(39). Yeast growth media was YPD [1% yeast extract (Difco),
WO91/18986 PCr/AU91/00229
~ ,r
2% bacto-peptone (Difco) and 2% glucose].
Cur~ng of Killer ~traln 3AM: A culture of strain 3AM
was grown overnight in YPD at 28 C. Serial dilutions were
made in 0.9% NaCl and 0.1 ml aliquots (containing
approximately 100 cells) were spread on YP~ plates and
incl~bated at 37'C. After 48 hours incubation, single colonies
were selected at random and assayed for killer activity as
described below.
Assay for cur~d straln: YPD (containing 1% agar) was
sterilized by autoclaving at 120-C for 20 mins. After cooling
to 49 C, the med1um was buffered to pH 4.2 with 0.05M
tartrate buffer. Methylene blue (to 0.003% w/v) and killer
sensitive strain 5A (to 105 cells/ml) were added to the
medium prior to pouring the plates. Colonies isolated after
hea~ treatment were then patched onto these ~ssay plates and
incubated at 18'C for approximately 72 hours. Curing was
recognised by the absence of growth inhi~ition (clear zones)
and lack of blue stained cells around the patched colony.
dsRNA l~olation: The dsR~A extraction procedure was
essentially that described by Fried and Fink (40). Samples of
RNA were analysed by electrophoresis on 1.5% agarose slab
gels at a cons~ant current of 100 mA. Gels were stained with
ethidi.um bromide and photographed on a short wave UV light
box.
Pul~ed ~leld gel ele~trophoresis: ~ransverse
alternating field electrophoresis (TAFE) was carried out
using a Geneline (Beckman) unit. Yeast chromosomes were
prepared in agarose plugs according to supplier's
recommendations. Electrophoresis was carried out at a
constant current of 150 mA; pulse times were 60 seconds for
18 hours, then 35 seconds ~or 6 hours.
Fermentatlo~ trialB: Starter cultures were prepared
by inoculating 10 ml of ~PD medium contained in a conical
flask with a loopful of yeast and incubated with vigorous
aeration at 28 C. After 24 hours, the cell density was
determined by microscopic counts. Samples were used to
WO91~18986 PCT/AU91/00229
`...3-~ 14
inoculate Rhine Riesling must (200 ml) to a density of ~ x
106 cells/ml. The must contained 220 g/l reducing sugars and
had a pH of 3.l. Fermentations were carried out in 250 ml
conical flasks fitted with airlocks. ~he juice was sterilized
by membrane filtration prior to inoculation, and
fermentations were carried out at 18-C with agitation
(approximately lO0 r.p.m.). Samples were removed
anaerobically during fermentation by needle and syringe
through ports covered with rubber septums.
Samples were analysed for the progress of fermentation
by refractometer readings, and yeast growth by measuring
spectrophotometric absorbancy at 650nm. For analysis of the
proportion of marked strain in the yeast population, serial
dilutions were made o~ the samples in sterile 0.9% NaCl and
O.l ml aliquots were plated (containing 200-500 cells) on YPD
media. The plates were then assayed as described below.
GUS plate asBay~: Yeast colonies were grown on solid
YPD media for approximately 36 hours at 28-C. A solution
containing O.lM ~a2HPO4, pH 7.0, 1% sarcosyl, x-gluc (100-150
u~/ml) and 0.7% agarose was then poured as a thln overlay on
the plate and allowed to set. After 4 6 hours incubation at
37-C, a blue precipitate could be detected in the marked
colonies.
RESULTS
Curi~g of Strain AWRI 3AM
In order to specifically analyse the effect of killer
toxin in fermentations, an experiment was designed to compare
two isogenic strains which differ only in the presence of the
M-dsRNA genome and therefore in their ability to produce
killer toxin.
Killer strain 3AM had previously been marked with the
Escherichia coli GUS gene (39). This system allows the marked
strain to be readily identified in a mixed population by a
simple plate assay which results i~ the formation of a blue
precipitate in marked colonies. Strain 3AM was cured of its
M-dsRNA plasmid by heat treatment (41), the cured or
sensitive colonies being identified by killer plate assays. A
WO91/18986 PCT/AU91/00229
~ r~
zone of inhibition clearly evident around strain 3AM was
absent around 3AMC, indicating that strain 3AMC is not
producing killer toxin (Figure 3).
In order to verify that the isolated strain 3AMC is a
genuine derivative of strain 3AM, both strains were
karyotyped by pulsed field gel electrophoresis. Total
chromosomes were isolated and ~lectrophoresed on a ~ransverse
Alternating Field Electrophoresis (TAFE) system (Figure 4).
An identical electrophoretic pattern was obtained for both
strains, thus illustrating a common genetic background. As
strain 3AMC is derived from 3AM, it inherits the GUS gene and
therefore is also a mar~ed strain.
Finally, dsRNA species were isolated from strains 3AM
and 3AMC and analysed by standard electrophoresis techniques
(Figure 5). A band representing the M-dsRNA genome is present
in strain 3AM, and absent in strain 3AMC.
Fermentation trials were then perormed on stra:ins 3AM
and 3AMC to determine the effect of the curing procedure on
yeast growth and fermentation rates. Starter cultures of eaah
strain were inoculated in triplicate into flasks o Rhine
Riesling grape juice at a con~entration of 5 x l06 cell/ml.
Samples were taken at regular intervals and assayed for yeast
growth and progress of fermentation. The average readings for
each strain were plotted over time (Figure 6). There were no
significant differences in the growth or fermentation rates
between strains 3AM and 3AMC.
ANA~YSIS OF RILLER AC~IYITY DURING FERM~NTATION
Strains 3AM and 3AMC were analysed for killer activity
in Rhine Riesling juice by co-inoculating each strain with
the sensitive Sacch~romyces strain 5A. Control ferments of
each s~rain (3AM, 3AMC and 5A) as pure inoculums were al50
perormed. Each ferment was conducted in duplicate at 18'C
with gentle agitation under anaerobic conditions. GUS plate
assays were then performed to idPntify the mar~ed strain (3AM
or 3AMC). Colonies of the marked strain turn a deep blue
colour as a result of this assay, allowing simple
identification.
WO91/18986 PCT/AU91/00229
16
G~S plate assays were also performed on the control
ferments to confirm the validity of the assay. ~late asRays
on the control 5A ferment were consistently negative,
highlighting the absence of back~round GUS activity in
natural yeast cells. However, control 3AM and 3AMC ferments
gave values of between 99 - 100% for the marked straln count.
In other words, occasionally colonies of a marked strain did
not turn blue in response to the GUS assay. ~he frequency of
this occurrence was always less than 1% of the total plate
count, and did not increase over time. This background
reversion frequency was taken into account throughout the
analysis.
The following mixed culture ferments were carried out:
i) 3AM and 5A at an inoculum ratio of 1:1 respectively;
ii) 3AMC and 5A at an inoculum ratio of 1:1 respectively;
iii) 3AM and 5A at an inoculum ratio of 2:1 respectively; and
iv) 3AMC and 5A at an inoculum ratio o~ 2:1 respectively.
These mixed erments exhibited normal growth kinetics, as did
the three control f2rments (Figure 7).
The time course of growth (colony forming units per ml)
of each strain in the mixed culture ferments was plotted in
Figure 8. At inoculum ratios of 1:1, there was a notable
increase in the proportion of killer strain 3AM, whereas the
cured strain 3AMC failed to exert any dominance over the
- sensitive strain under otherwise id~ntical conditions.
Statistical analysis was used to test the null hypothesis
that the ratio o~ killer: sensitive cells remains 1:1
throughout the ferment. A "goodness of fit" test tnormal
test) rejected the null hypothesis, with p-value c~ 0.001.
However, identical analysis of the cured : sensitive strain
ferment supported the null hypothesis that the ratio of the
two strains remains at 1:1 throughout the ferment.
With an increased proportion of strains 3AM and 3AMC in
the inoculum (ratio 2:1), the dominating effect of strain 3AM
was more pronounced, whereas strain 3AMC again showed no
apparen~ chang~ in proportion over time. The domina~ce of
~train 3AM in mixed culture ferments was illustrated more
W091/18986 PCT/AU91/Q0229
17
clearly when the percentage of each strain was plotted over
the time of the ~erment (Figure 9). For an :inoculum ratio of
l:l, 3AM increased to approximately 80% after 3 days, but for
a higher inoculum ratio of 2:l, 3AM eventua:Lly accounted for
97% of the total yeast population. It is important to note
that the strain 5A persisted, albelt at low levels,
throughout the ferments. This contrasts with previous reports
of killer activity, where the sensitive strain was not
detectable by the end of the ferment (28, 29).
EFFECT OF INOCULUM RATIO ON KILLER ACTIVITY
Experiments were conducted to determine the lowest
inoculum ratio of killer to sensitive cells at which
significant killer activity could be observed. Mixed ~erments
of strain 3AM and 5A at inoculatio~ ratios of l:2 and l:~
respectively were carried out under conditions described
above.
At various times during the ferment, samples were
analysed for the proportion of strain 3AM in the total
population tFigure lO). An increase in proportion of strain
3AM was evident when it was inoculated at greater than 50% of
the total population. However, when it was less than 50% at
the time of inoculation, there was no change in proportion.
DISCUSSION
Previous studies have indicated 100% stability of the
GUS marker gene in strain 3A throughout fermentation ~39).
However, analysis of a larger sample of colonies in these
experiments has revealed an instability of the construct.
~his instability was detected in the control ferments which
were inoculated with monocultures of either of the marked
strains (3AM or 3AMC). Samples fxom these ferments gave rise
to colonies which respondPd negatively to the GUS plate assay
at a frequency of less than 1% of the total plate count.
Occasio~ally a colony which was sectored in its response to
the assay was detectad, suggesting either excision of the
gene by homologous recombination (45) or loss of ths gene
after mitotic crossing-over (46). ~he frequency of
instability did not increase over time during fermentation,
WO91/18986 PCT/AU91/00229
o,~r~
18
and could be directly quantified in the conl:rol 3AM and 3AMC
ferments.
This marking system has enabled a direct comparison to
be made between the inoculation efficiency of a killer strai.n
(3AM) and a cured derivative of the same strain (3AMC) in
fermenting grape juice. An electrophoretic karyotype of the
killer and cured strains confirmed a common genetic
constitution, where analysis of dsRNA species revealed the
loss of the M~dsRNA genome in the cured strain. No
differences were detected in the growth rates or fermentation
curves of strains 3AM and 3AMC. Therefore, by comparing
strain 3AM and 3AMC, differences in properties can ~e
attributed directly to the presence of the M-dsRNA plasmid
and hence to the production of the R2 killer toxin.
Under identical fermentation conditions, the cured
strain 3AMC remained at 50% of the total population while the
killer strain increased to 80%. The ability of strain 3AM to
domina~e 5A during ermentation is likely to be due to the
production of killer toxin by strain 3AM and not to a
differe~ce in respective growth rates favouring the killer
strain. We can csnclude, thexefore, that the killer toxin has
displayed significant activity under these fermentation
conditions. This result is of particular interest to the
oenologist since the R2 toxin produced by strain 3A is
reported to show maximum activity at pH 4.2 (17), which is
0.5 to 1 pH unit higher than generally found in grape musts.
In cases where killer activity has been reported in
fermenting grape juice, a discrepancy exists as to whether
effective killing action occurs when the proportion of killer
cells is less than 50% of a mixed culture ferment. Heard and
Fleet (28) did not observe killer action when the ratio of
killer to sensitive cells was approximately 1:7, whereas
others have rsported killer activity with killer to sensitive
cell ratios of 1:10 and lower (23, 25, 26).
Our results sho~ed that an increase in ratio of killer
to sensitive cells to approximately 2:1 resulted in a
pronounced dominance of the fermsnt by strain 3AM to 97% of
W~91/~898~ PCT/~IJ91/00229
the total mixed population by the end of the ferment.
However, with killer to sensitive cell ratio6 of 1:2 or 1:4,
no effective killer action was evident. It :is possible that
differences in either composition of medium, fermentation
conditions or strain sensitivity may account for
discrepancies in reports of killer toxin efEiciency.
The relevance of killer strains in wine making has been
the focus of attention in countries where selec-ted yeast
cultures are inoculated into musts to induce fermentation.
This focus has intensified since the observations that yeasts
which are naturally present in the must also play signif:Lcant
roles in supposedly "pure" culture fermentations (22, 42).
These natural yeasts include species from the genera
~loeckera, Candida, ~ansenul~ and S~ccharomyces. Riller
Saccharomyces wine yeast strains may be effective in
suppressing natural Saccharomyces yeasts during fermentation,
and the possibility exists to engineer broad range killer
yeasts to control strains from other genera. For these
reasons, further study is needed to determi~e a~propriate
fermentation conditions for effective ~iller acti~ity.
The GUS marking system provides a method which allows a
broad range of killer strains to be rapidly and unequivocally
identified in a mixed culture. This system can be employed to
gain a better understanding of killer activity during
fermentation.
DI~CUSSION AND CO~CLUSIONS
The GUS-vector construct described in this specification
can be introduced to a range of yeast strains by
transformation procedures, e.g. using the SMR1-410 gene as a
dominant selection marker. Results show that the construct
can be integrated into a specific site in the yeast genome
without disrupting essential functions or affecting the
fermentation performance of a wine yeast strain. Once
integrated into the genome, the construct is maintained in a
stable manner throughout the fermentation.
WO91tl8986 PCT/AU91/00229
X ~ 20
Assaying for the GUS marker can be achieved by
fluorimetry, spectrophotometry or by agar plate assay.
Although natural transport of x-gluc or MUG substrates did
not occur across yeast cell membranes in the time course of
experiments described here, this problem was ovarcome by
inducing artificial permeation in assay procedures. Methods
have been described which allow the proportion of marked
strain in a total yeast population to be determined.
The applications of this ~arker system will be in
monitoring inoculated strains under different fermentation
conditions. Recent investigations by Heard and Fleet (42,
43, 44) made with both inoculated and uninoculated grape
juices under a range of fermentation conditions suggest that
Saccharomyces strains are not necessarily the dominant
organism during vinification. 'rhese studies, along with the
lack o~ knowledge regarding incidence and importance of wild
strains of Saccharomyces in fermentiny grape juice, call for
detailed monitoring of inoculated strains under varying
oenological environments. The ~US system described in this
specification will enable a wide range of yeast strains to be
marked, providing the means for unequivocal identification
and monitoring during fermentation.
REFERENCES
1. Rankine, B.C. The importance of yeasts in determining
composition and quality of wines. Vitis 1:22-49 (1968)
2. Vezinhet, F. and Lacroix, S. Marquage genetique de
levures: Outil de controle des fermentations en souche
pure. Bull. de l'O.I.V. 643-h44: 759-773 (1984~
3. Loiseau, G., Vezinhet, F., Valade, M., Vertes, A.,
Cuinier, C. and Delteil, D. Controle de l'efficacitP du
levurage par la mise en oeuvre de souches de levures
oenologiques marquees. ~ev. Fr. Oenol. 106: 29-36
(1987)
WO9l/l8986 ~CT/AU91/00229
4. Delteil, D. and Ai~ac, T. Comparison of yeast
inoculation techniques by the use of a 'marked' yeast
strain. Aust. and N.Z. Wine Industry Journal, November
1988: 53-56
5. Jeffersen, R.A., Bur~ess, S.M., and Hlrsch, D.
~-glucuronidase from Escherichia coli as a gene fusion
marker. Proc. Natl. Acad. Sci. USA 83:8447-8451 (1986)
6. Jeffersen, R.A., Ravanagh, T.A., and Beven, M.W. GUS
fusions~ lucuronidase as a sensitive and versa~ile
gene marker in higher plants. EMBO J. 6:3901-3907
(1987)
7. Bevan, E. A., and M. Makower. ~he physiological basis of
the killer character in yeasts. In: Genetics Today,
Procedings o the 11th }nternational Congress of
Genetics, 1 (1963). S. J. Gert (ed.). pp 202-203.
8. Philliskirk, G., and T. W. Young. The occurrence of
killer character in ye~sts of various ~enera. Antonie
van Leeuwenhoek 41: 147-151 11975).
9. Tipper, D. J. and K. A. Bostian. Double-stranded
ribonucleic acid killer systPms in yeasts. Microbiol.
Rev. 48: 125-156 (1984).
10. De le Pena, Pl, Barros, F., Gascon, S., Lazo, P. S. a.nd
Ramos, S. Effect of yeast killer toxin on sensitive
cells of Saccharomyces cerevisiae. J. Biol. Chem.
256:10420-25 (lg81).
11. Skipper, N. and ~. Bussey. Mode of action of yeast
toxins: ener~y requirement for Saccharomyces cerevisiae
killer toxin. J. Bacteriol. 12~: 668-677 (1977).
WV91/18986 PCT/AU91/00229
12. sostian, R. A., Hopper, J . E ., Rogers , D . J . and Tipper,
D. J. Translational analysis of the killer-associated
virus-like particle dsRNA genome of Saccharomyces
cerevisi~e: M-dsRNA encodes toxin. Cell 1~: 403-414.
(1980).
13. Harris, M. S. Virus-like particles and double-stranded
RNA from killer and non-killer strains of S~ccharomyces
cerevisiae. Microbiology 1: 161 174 (1978).
14. Naumov, G. I. and T. Io ~aumova. Comparative gene~ics of
yeast co~munication. XIII. Comparative study of killer
strains of Saccharomyces from different collections.
Genetika ~:140-145 (1973).
15. Young, T. W. and Yagiu, M. A comparison of the k.iller
character in different yeasts and its classification.
A~tonie van Leeuwenhoek J. Microbiol. 44: 59-77 (1978).
16. Shimaz~, R., Adachi, T., Kitano, R., Shimazaki, T.,
To suku, A., Hara, S., and H. H. Dittrich. 1985. Killer
properties of wine yeasts and characterisation of killer
wine yeasts. J. Ferment. Technol. 63: 421-429.
17. Rogers, D., and E. A. Bevan. 1978. Group classification
of killer yeasts based on cross reactions between
strains of different species and origin. J. Gen.
Microbiol. 105: 199-202.
18. Radler, F., Pfeiffer, P., and M. Dennert. 1985. Riller
toxins in new isolates of the yeasts Hanseniaspora
uvarum and Pichia kluyveri. FEMS Microbial hett. 29: -
269-272 .
WO91/18986 PCT/AU91/00229
23 '24~ `J ~
19. Rosini, G. Interaction between klller strains of
H~nsenula anom~la var. anomala and Saccharcmyces
cerevisiae yeast species. Can. J. Microbiol. 31:300-2
( 1985) .
~0. Boone, C., Sdicu, A. M., Wagner, J., Degre, R., Sanches,
C., and H. Bussey. 1990. Integration of the yeast ~C1
killer toxin gene into the genome of ~arked wine yeasts
and i~s effect on vinification. Am. J. Enol. Vitic~ 41:
37-42.
21. Cuinier, C. and C. Gros. Enquete sur la repartition des
levures killers en France. Vivnes et vins 38: 25-27
(1983).
22. Lafon-Lafourcade, S. and P. Ribereau-Gayon. Developments
in the microbiology of wine production. Prog. Xnd.
Microbiol. 19:1-45 t1984).
23. Barre, P. Le mecanisme killer dans la concurrence entre
souches de levures. Evaluatio~ et prise en compte. Bull.
O.I.V. ~41-~2: 635-643 ~1984).
24. Rosini, G. Effet d'une le~ure "killer" de Saccharomyces
cerevisiae sur une souche de levure sensible de la meme
espece non productrice de H2S et selectionee pur la
vinification dans un milieu de culture mixte. Bull.
O.I.V 648-~9: 214-217 91985).
25. Hara, S., Iimura, Y. and F. Otsuku. Breeding of useful
killer ~ine yeasts. Am. J. Enol. Vitic. 32:28 33 (1980).
26. Hara, S., Iimura, Y., Oyama, H., Kozeki, T., Kitano, R.
and Otsuku, ~. Breeding of cryophilic killer wine
yeasts. Agric. Biol. Chem. 4~: 1327-1334 ~1981).
WO91/18986 PCT/AU91/00229
~ 24
27. Seki, ~., Choi, E.~. and D. Ryu. Construction o~ killer
wine yeast strains. Appl. Environ. Micro. 49: 1211-1215
t1985).
28. Heard, G.M. and Fleet, G. H. Occurrenc~ and growth of
killer yeasts during wine fermentation. Appl. a~d
Environ. Microbiol. ~:2171-2174 (1987~.
29. Longo, E., Velazquez, J. B., Cansado, J., Calo, P. and
T. G. Villa. Role of killer effect in fermentations
conducted by mixed cultures of Saccharomyces cerevisiae.
FEMS Micro~iol. Letters 71: 331-336 (1990).
30. Hanahan, D. Studies of transformation of ~scherichia
coli with plasmids. J. Mol. Biol. L66: 557-580 (1983)
31. Ish-Horowicz, D. and Bur3~e, ~.F. Rapid and efficient
cosmid cloning. Nucleic Acids Res. 9: 2989-2998 (1981)
32. Ito, H., Fukuda, Y., Murata, ~. and Kimura, A.
Transformation of intact yeast cells treated with alkali
cations. J. Bacteriol. ~ 163-168 (1983)
33. Davis, R.W., ~homas, M., Cameron, J.R., St. John, T.P.,
Scherer,S. and Radgett, R.A. Rapid DNA isolations for
enzymatic and hybridization analysis. In Methods in
Enzymol., Vol. 65, Gross, L. and Moldave, K. (eds.),
Academic Press Inc., New York, pp 404-411 (1980)
34. Southern, E.M. Detection of specific sequences among
DNA fragments separated by gel electrophoresis. J.
Mol.Biol. ~: 503-517 (1975~
35. Rankine, B.C. and Pocock, K.F. Alkalimetric
determination of sulfur dioxide in wine. Aust. Wine
Brew. Spirit Rev. 91: 62,64,66 (1972~
W091/18986 PCT/AU91/OD229
~r~
36. Anon. Instructions for the us~ of the rrechnicon 260
Infra-analyser. Bran & ~uebbe Australia.
37. Gustav, A. Expression of genes in yPast using the ADC1
promoter. Ln Methods in Enzymol., Vol 101,
(eds), Academic Press Inc., New York, pp 192-201.
38. Casey, G.P., Nei Xiao and Rank, G.H. A convenien~
dominant selection marker for gene transfer i~
industrial strains of Saccharomyces yeast : SMR1 encoded
resistance to the herbicide sulfometuron methyl. J.
Inst. Brew. ~4: 93-97 (1988)
39. Petering, J. E., Henschke, P. A. and hanyridge, P. The
Escherichia coli ~-Glucuronidase gene as a marker for
Saccharomyces yeast strain identification. Am. J. Enol.
Vitic, 42: 6-12 (1991).
40. Fried, H. M. and G. Fink. Electron microscope
heteroduplex analysis of "killer" double-stranded RNA
species from yeas~. Proc. Natl. Acad. Sci. U.S.A. 6~:
2846-284~ (1978).
41. Nickner, R.B. "Riller character" of Saccharomyces
cerevisiae: curing by growth at elevated temperatures.
J. Bacteriol. 111: 115-126 (1974).
42. Heard, G.M. and Fleet, G.H. Growth of natural yeast
flora during the fermen-tation of inoculated wines.
Applied and Environmental Microbiology SO : 727-728
( lg85)
43. Heard, G.M. and Fleet, G.H. Occurrence and growth of
yeast species during the fermentation of some Australian
wines . Food Technology in Australia 38 : 22-25 (1986)
WO91/18986 PCT/AU91/00229
~r~ 26
s ~ ~ ~
44. Heard, G.M. and Fleet, G.H. The effect:s of temperature
and pH on the gro~th of yeast species cluring the
fermentation of grape juice. Journal of Applied
Bacteriology 65 : 23-28 (1988)
45. Struhl, R., Stlnchcomb, D.T., Scherer, S. and R.W.
Davis. High-frequency transformation of yeast:
Autonomous replication of hybrid DNA molecules. Proc.
Natl. Acad. Sci. USA 76: 1035-1039 (1979).
46. Roeder, G.S. and S.E. Stewart. Mitotic recombination in
yeast. Trends in Genet. 4: 263-267 (1988).