Note: Descriptions are shown in the official language in which they were submitted.
CA 02084582 2002-12-30
PROCESS FOR THE PRODUCTION OF MONO-N-SUBSTITUTED TETRAAZA
MACROCYCLES
The invention relates to a process f<,~r the production
of mono-N-substituted tetraazacyclododecane and
tetraazacyclotetradecane derivatives.
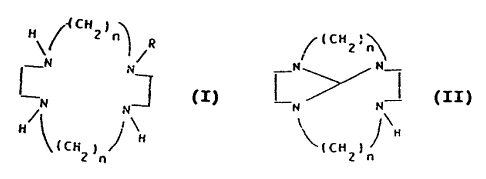
Mono-N-substituted tetraaza macrocycles of general
formula I
in which
(CHy)"
H ~ ~ R
N N
I),
N N
H H
tCH2)n
n stands for the numbers 2 or 3,
R stands for a (3-carboxylalkyl or (3-carboxylate alkyl,
~i-cyanide alkyl, ~i-carboxamidoalkyl, ~i-hydroxyalkyl,
aminocarbonyl, aminothiocarbonyl, ~-sulfamoylalkyl radical
or for a second tetraazacycloc~odecane or
tetraazacyclotetradecane molecule bound by a bis((3-
hydroxy)-alkylene chain, and c°:arboxy.l and hydroxy groups
are present optionally in protected form,
are important precursors of tz°i-N-carboxyalkyl, preferably
tri-N-carboxymethyl, substituted tetraaza macrocycles,
CA 02084582 2003-05-29
. _ 2 _
which are used as diagnostic agents and therapeutic agents
in the form of their complexes with metal ions of atomic
numbers 21 to 29, 31, 32, 38, 39, 42-44, 49 or 57-83 (see
European patent application publication no. 255471).
Because of their importance as key compounds for these
complexes, above all for the preferred NMR diagnostic
agents (Macrocyclic Chemistry Congress, Hamburg 1988),
production of mono-N-substituted tetraaza macrocycles has
been attempted in different ways, but without a
satisfactory method of synthesis previously having been
f ound .
For example, a statistical monoalkylation or
monoacylation of unsubstituted tetraaza macrocycles has
been described, which, however, is not suitable at least
for the production of sizable amounts of substance because
of the great excess of relatively costly initial amine to
be used, partially very expensive chromatographic
separation of the product from the initial material as well
as in most cases quite moderate yields. {see Kaden, Helv.
Chim. (Swiss Chem.) Acta 69, 2081 (1986); Kimura, J. Chem.
Soc. Chem. Commun. 1158 (1986); Kaden, Top. Curr. Chem.
121, 157 (1984); European patent applications no. 296522
and no. 353450].
If it is desired - in contrast to the above-described
statistical monosubstitution -- to perform a specific
monosubstitution, two variants are possible:
a) reaction of a tetraaza macrocycle, provided with
three nitrogen protecting groups, which was obtained by
statistical trisubstitution,
b) reaction of a tetraaza macrocycle, provided with
three nitrogen protecting groups, which was produced by
specific synthesis.
In the first-mentioned variant, the precursor carrying
the protecting groups (e. g., tosylate, benzoate) on three
nitrogen atoms is produced by statistical trisubstitution
of an unsubstituted tetraaza macrocycle, so that the above-
..-.r
~ 2084582
- 3 -
mentioned drawbacks of a statistical reaction, such as low
yields, separating problems (particularly, in the
production of sizable amounts of substance) also occur here
[see, e.g., Macrocyclic Chemistry Congress, Hamburg 1988].
After the subsequent specific monosubstitution to introduce
substituent R [Ciampolini, J. Chem. Soc. Chem. Commun. 998
(1984): Kaden, Helv. Chim. Acta 66, 861 (1983); Basefield,
Inorg. Chem. 25, 4663 (1986)], the protecting groups on the
three nitrogen atoms have to be removed, e.g., by alkali
metal in ammonia [Helv. Chim. Acta, 56, 2216 (1973); Helv.
Chim. Acta 59, 1566 (1976); J. Org. Chem. 53, 3521 (1988)],
lithium aluminum hydride [F. Voegtle; Liebigs Ann. Chem.
(1977), 1344], Red-A1~R~ [E. H. Gold, J. Org. Chem. (1972),
37, 2208], Na-Hg [M. Kellog, J. Org. Chem. 1984, 49, 110],
electrolysis [M. Hesse, Helv. Chim. Acta 71 (1988), 7,
1708] or hydrobromic acid/phenol/glacial acetic acid [N. G.
Lukyanenko, Synthesis, 1988, 355]. These processes of the
cleavage of the protecting groups are generally connected
with poor yields, limit the batch size with respect to the
2 0 amount o f reagent to be used ( a . g . , in the Na-Hg method )
and above all cannot be used in the case of substituents,
which carry sensitive groups (e. g., hydroxyalkyl).
If the procedure is performed according to variant b),
i.e., if it is desired to produce the tetraaza macrocycle
precursor carrying protecting groups on three nitrogen
atoms by specific synthesis, a start is made from two
reactants, which are cyclized according to methods known in
the literature [e.g., Richman, Org. Synthesis 58, 86
(1978); Atkins, J. Amer. Chem. Soc. 96, 2268 (1974)]; one
of the two reactants contains a protected nitrogen atom and
carries, on the chain end, two volatile groups (e. g.,
bromine, mesyloxy, tosyloxy, triflate or alkoxycarbonyl
groups), which are nucleophilically displaced from the
terminal nitrogen atoms of the second reactant, of a --
unlike the first reactant -- protected triaza compound.
- 4 -
.2084582
(If a reactant with two terminal ester groups is used,
the two amide groupings resulting by the cyclization --
preferably with diborane in THF -- have to be reduced. But
especially this cyclization variant is unsuitable for the
production of substantial amounts of substance, since this
reaction is to be performed in the highest possible
dilution, to avoid, e.g., polymerization reactions: see
Tabushi, Tetrahed. Lett. ~, 1049 (1977); Kaden, Inorg.
Chem. ~, 321 (1986). Aiso, the working up of the
subsequent diborane reduction -- again above all in greater
batches -- is not without problems.)
After cleavage of one protecting group, the thus
released imino grouping can be alkylated or acylated. As
an example, there can be mentioned the reaction of the
disodium salt of N,N',Na-tris-(p-tolylsulfonyl)diethylene
triamine [Ciampolini, J. Chem. Soc. Chem. Commun. 998
(1984)] with N-bis-(2-methanesulfonyloxy-ethyl)-
triphenylmethylamine in dimethylformamide at 80-150°C with
subsequent cleavage of the trityl group under acid
conditions. The yields of both reaction steps are
generally poor. Also, this variant bj is affected with the
drawbacks mentioned under a) regarding the cleavage of
three protecting groups coming from the second reactant.
Besides the previously presented process of the
statistical and specific monosubstitution, a specific ring
synthesis, in which desired substituent R already is
contained in one of the two reactants to be used in the
cyclization reaction, is also possible.
Besides the problems, already described above, of the
cleavage of the protecting groups, it has turned out that
the thus performed cyclizations generally take place with
smaller yields -- as compared to the reactions of the reac
tant provided only with protecting groups -- [see Atkins,
J. Amer. Chem. Soc. ~6, 2268 (1974); Richman, Org. Synthe
sis 58, 86 (1978); Fabbrizzi, Inorg. Chem. 25, 4131 (1986);
v
2084582
- 5 -
Gazetta, Chimica Italiana 115, 399 (1985)]. Further, the
reactants carrying substituent R first have to be specially
synthesized in a reaction sequence often comprising several
steps [see, e.g., Bulkowski, J. Org. Chem. 47, 412 (1982)].
Despite varied efforts, it therefore previously has
not been possible to find a satisfactory method of
synthesis for mono-N-substituted tetraaza macrocycles of
general formula I, which are to be considered as key
compounds for the tri-N-carboxyalkyl metal complexes being
used as valuable NMR and x-ray contrast media.
It has been determined, surprisingly, that a selective
monofunctionalization of mono-N-substituted tetraaza
macrocycles of general formula I '
/i CH2 ~~~
R
-N N
N N (I) r
H/ / N
~tC~~ 1
in which
n stands for the number 2 or 3,
R stands for a R-carboxylalkyl or Q-carboxylate alkyl,
R-cyanide alkyl, Q-carboxamidoalkyl, Q-hydroxyalkyl,
aminocarbonyl, aminothiocarbonyl, /3-sulfamoylalkyl radical
--
2084582
- 6 -
or for a second tetraazacyclododecane or
tetraazacyclotetradecane molecule bound by a
bis(B-hydroxy)-alkylene chain, and wherein carboxyl and
hydroxy groups are present optionally in protected form,
is achieved, if tetraazatricyclotridecane or
tetraazatricyclopentadecane of general formula II
I CH2 J
_N~N
~N N (II) ,
\ J H
'( CH2 )rt
is reacted with an a,8-unsaturated ester, amide or nitrile,
or an epoxide, isocyanate, isothiocyanate, aziridine or a
bisepoxide, with or without solvent at 0 to 220°C,
preferably room temperature to 210°C, within 1 to 48 hours,
preferably 5 to 12 hours, optionally at a pressure up to
100 atm. The thus obtained reaction mixture after cooling
to -20°C to 80°C, preferably 0°C to 30°C, is mixed
with a
mixture of water/organic solvent and stirred for 0.5 to 12
hours, preferably 0.5 to 3 hours at -20°C to room
temperature (e. g., about 25°C), preferably 0°C to room
temperature. The thus formed -- optionally to be isolated
-- intermediate products carrying a fonayl group on a
nitrogen atom are reacted by adding an inorganic base or an
acid at 0 to 150°C, preferably room temperature to 120°C,
within 1 to 72, preferably 6 to 24 hours, with stirring --
optionally followed by subsequent removal of protecting
groups in a way usual in the art -- to obtain the end
product of formula I, which can then be isolated in a way
known in the art, preferably as hydrochloride.
The tetraazatricyclotridecane or
tetraazatricyclopentadecane of general formula II used as
..: r';""-~.
~ ..a
~fe
a v'~
2084582
_ 7 _
intermediates are accessible according to methods known in
the literature, e.g., by reacting 1,4,7,10-
tetraazacyclododecane or 1,4,8,11-tetraazacyclotetradecane
with dimethylformamide-dimethylacetal (US patents 4,085,106
and 4,130,715), J. Am. Chem. Soc. ~0,, 6364 (1980), EP 292
689.
Advantageously, this reaction step is included in the
process according to the invention, without the
intermediates of general formula II having to be isolated
("one-pot reaction").
A special embodiment of the process according to the
invention is the production of compounds of general formula
I with
RZ R~
is
R meaning a -C-CH-A group,
R3
in which
R' stands for a hydrogen atom, a straight-chain or
cyclic C~-C6 alkyl, a phenyl or benzyl group -- in which the
phenyl or benzyl group can be substituted respectively by
1 to 2 chlorine, bromine, vitro, C~-C~ alkoxy, C~ Coo
aralkoxy, and/or Co2R~ radicals with R4 meaning a hydrogen
atom, a C~-C6 alkyl, phenyl or benzyl group,
R2 and R3, independent of one another, each stand for
R' or a COzR4 group,
RS
A stands for a CN, COzR4 or CON/ radical,
R6
in which RS and R6, independent of one another, each stand
for a hydrogen atom, a saturated or unsaturated, straight
chain, branched-chain or cyclic hydrocarbon radical with up
to 16 C atoms, optionally interrupted by 1 to 8 oxygen
atoms, or 1 to 3 phenylene or phenylenoxy groups, and
optionally substituted by 1 to 5 hydroxy groups, or 1 to 2
COZR4 radicals; for phenyl or benzyl radicals optionally
2084582
_8_
substituted by 1 to 3 hydroxy or C~-C6 alkoxy groups; or RS
and R6 together with the nitrogen atom stand for a saturated
or unsaturated 5- or 6-ring, optionally containing another
nitrogen, oxygen, sulfur atom or a carbonyl group, which
optionally is substituted by 1 to 3 C~-C6 alkyl radicals
optionally substituted by 1 to 3 hydroxy groups,
and optionally present hydroxy and/or carboxyl groups
optionally are protected,
characterized in that tetraazatricyclotridecane or
tetraazatricyclopentadecane is reacted with a feedstock of
general formula III
R~
R=
i ~ (III),
Rs
in which R', R2, R3 and A have the above-indicated meanings,
and optionally present hydroxy and/or carboxyl groups are
optionally protected, with or without solvent, preferably
aprotic solvents, such as, e.g.., benzene, toluene,
dichloromethane, tetrahydrofuran, dioxane, acetonitrile,
dimethylformamide, hexane or ether, are used as solvent, at
0°C to 210°C, preferably 50°C to 180°C (and in the
case of
the higher reaction temperature, the solvent used
optionally to dissolve the added feedstock of general
formula III was previously distilled off in a vacuum),
within 12 to 48, preferably 5 to 12 hours. The thus
obtained reaction mixture is cooled to -20°C to 80°C,
preferably 0° to 30°C, mixed with a mixture of
water/organic solvent, such as, e.g., methanol, ethanol,
isopropanol, tetrahydrofuran or dioxane, and stirred for
0.5 to 12 hours, preferably 0.5 to 3 hours, at
-20°C to room temperature, preferably 0°C to room
temperature. The thus formed -- optionally to be isolated
-- intermediate product carrying a formyl group on a
nitrogen atom is reacted by adding an inorganic base such
208 458 ~
_ 9 _
as, e.g., lithium hydroxide, sodium hydroxide, potassium
hydroxide, barium hydroxide or calcium hydroxide,
preferably sodium hydroxide and potassium hydroxide, or a
mineral acid, such as, e.g., hydrochloric, sulfuric or
hydrobromic acid, preferably hydrochloric acid, at O°C to
150°C, preferably room temperature to 120°C, within 1 to 72
hours, preferably 6 to 24 hours, with stirring --
optionally followed by subsequent removal of protecting
groups in a way usual in the art -- to obtain the end
product of formula I, which can then be isolated in a way
known in the art, preferably as hydrochloride.
If R' stands for a C~-C6 alkyl group, the methyl and
ethyl group is preferred. Other preferred radicals for R~
are the hydrogen atom and the optionally substituted phenyl
radical. As preferred substituents on the phenyl ring, the
nitro group, the C~-CT alkoxy radical, above all the methoxy
and ethoxy radical, and the C02R~ radical, can be mentioned,
with R4 being preferably hydrogen, methyl, ethyl, t-butyl or
benzyl.
As preferred radicals standing for RS and R6, hydrogen,
methyl, ethyl, 2-hydroxyethyl, 2-hydroxy-1-(hydroxymethyl)-
ethyl, 1-(hydroxymethyl)-ethyl, propyl, isopropenyl,
2-hydroxypropyl, 3-hydroxypropyl, 2,3-dihydroxypropyl,
butyl, isobutyl, isobutenyl, 2-hydroxybutyl,
3-hydroxybutyl, 4-hydroxybutyl, 2-, 3- and 4-hydroxy-
2-methylbutyl, 2- and 3-hydroxyisobutyl, 2,3,4-
trihydroxybutyl, 1,2,4-trihydroxybutyl, pentyl,
cyclopentyl, 2-methoxyethyl, hexyl, decyl, tetradecyl,
triethylene glycol methyl ether, tetraethylene glycol
methyl ether and methoxybenzyl group can be mentioned. The
amide radical can also be a heterocyclic 5- or 6-ring
formed with the inclusion of the amide nitrogen. As
examples, there can be mentioned: pyrrolidinyl, piperidyl,
pyrazolidinyl, pyrrolinyl, pyrazolinyl, piperazinyl,
morpholinyl, imidazolidinyl, oxazolidinyl, and
thiazolidinyl.
2084582
- 1U -
In substrate III, optionally present carboxyl and/or
hydroxy groups are present preferably in protected form.
As acid protecting groups, lower alkyl (e. g., Ct_~j,
aryl (e. g. , C6_~oj and aralkyl (e.g. , ou s, for
~-~zj ~' P
example, the methyl, ethyl, propyl, n-butyl, t-butyl,
phenyl, benzyl, diphenylmethyl, triphenylmethyl,
bis(p-nitrophenylj-methyl group as well as trialkylsilyl
(e. g., with C~-~-alkyl groups) groups, are suitable.
The cleavage of the protecting groups takes place
according to the processes known to one skilled in the art,
for example, by hydrolysis, hydrogenolysis, alkaline
saponification of the esters with alkali in aqueous
alcoholic solution at temperatures of 0 to 50°C, acid
saponification with mineral acids or in the case of, e.g.,
tert-butyl esters with the help of trifluoroacetic acid.
As hydroxy protecting groups, e.g., benzyl,
4-methoxybenzyl, 4-nitrobenzyl, trityl, diphenylmethyl,
trimethylsilyl, dimethyl-t-butylsilyl, and diphenyl-
t -butylsilyl groups are suitable.
The hydroxy groups can also be present, e.g., as
THP-ether, a-alkoxyethylether (e. g., with C~_~-alkoxy
groups), MEM-ether or as esters with aromatic or aliphatic
carboxylic acids, such as, e.g~., acetic acid or benzoic
acid. In the case of polyols, the hydroxy groups can also
be protected in the form of ketals with, e.g., acetone,
acetaldehyde, cyclohexanone or benzaldehyde.
The hydroxy protecting groups can be released
according to the methods in the literature known to one
skilled in the art, e.g., by hydrogenolysis, reductive
cleavage with lithium/ammonia, acid treatment of the ethers
and ketals or alkali treatment of the esters (see, e.g..,
"Protective Groups in Organic Synthesis," T. W. Greene,
John Wiley and Sons 1981).
Another special embodiment of the process according to
the invention is the production of compounds of general
formula I with R meaning a
~... t:
k
a: ,~
- 208 4582
- 11 -
OH
-CH-CH-Ra group,
R7
in which
RT and Ra, independent of one another, respectively
stand for a hydrogen atom, a C~-C~ alkyl radical, optionally
interrupted by 1 to 10 oxygen atoms, a phenylene,
l0 phenylenoxy or phenylenedioxy group, which optionally is
substituted by 1 to 3 C~-C6 alkyl, 1 to 3 trifluoromethyl,
1 to 7 hydroxy, 1 to 3 C~-CT alkoxy, 1 to 3 C~-Coo aralkoxy,
1 to 2 COZR~ and/or 1 to 2 phenoxy or phenyl groups
optionally substituted by 1 to 2 chlorine, bromine, nitro
or C~-C6 alkoxy radicals, and the optionally present hydroxy
radicals are optionally in protected form, characterized in
that tetraazatricyclotridecane or
tetraazatricyclopentadecane is reacted with-a feedstock of
general formula IV
0
(IVY ,
R' Ra
in which R~ and R8 have the above-indicated meaning and
wherein optionally present hydroxy and/or carboxyl groups
optionally are protected, with or without solvent,
preferably aprotic solvents, such as, e.g., benzene,
toluene, dichloromethane, tetrahydrofuran, dioxane,
acetonitrile, dimethylformamide, dimethylacetamide,
dimethylsulfoxide, hexane or ether are used as solvent, at
0°C to 220°C, preferably 50°C to 180°C (and in the
case of
the higher reaction temperature, the solvent optionally
used to dissolve the added feedstock of general formula IV
was previously distilled off in a vacuum) or in an
autoclave at an excess pressure of 1 to 100 atm. within 1
to, 48 hours, preferably 5 to 12 hours. The thus obtained
2084582
- 12 -
reaction mixture is cooled to -20°C to 80°C, preferably
0°C
to 30°C, mixed with a mixture of water/organic solvent,
such as, e.g., methanol, ethanol, isopropanol,
tetrahydrofuran or dioxane and stirred for 0.5 to 12 hours,
preferably 0.5 to 3 hours, at -20°C to room temperature,
preferably 0°C to room temperature. The thus formed --
optionally to be isolated -- intermediate product carrying
a formyl group on a nitrogen atom is reacted by adding an
inorganic base, such as, e.g., lithium hydroxide, sodium
hydroxide, potassium hydroxide, barium hydroxide or calcium
hydroxide, preferably sodium hydroxide and potassium
hydroxide, or a mineral acid, such as, e.g., hydrochloric,
sulfuric or hydrobromic acid, preferably hydrochloric acid,
at 0°C to 150°C, preferably room temperature to 120°C,
within 1 to 72 hours, preferably 6 to 24 hours, with
stirring -- optionally followed by subsequent removal of
the protecting groups in a way usual in the art -- to
obtain the end product of formula I, which can then be
isolated in a way known in the art, preferably as
hydrochloride.
Preferred radicals R7 and R8 are hydrogen, methyl,
ethyl, hydroxymethyl, 2-hydroxyethyl, 2-hydroxy-
1-(hydroxymethyl)-ethyl, 1-(hydroxymethyl)-ethyl, propyl,
isopropenyl, 2-hydroxypropyl, 3-hydroxypropyl,
2,3-dihydroxypropyl, butyl, isobutyl, isobutenyl,
2-hydroxybutyl, 3-hydroxybutyl, 4-hydroxybutyl, 2-, 3- and
4-hydroxy-2-methylbutyl, 2- and 3-hydroxyisobutyl, 2,3,4-
trihydroxybutyl, 1,2,4-trihydroxybutyl, pentyl,
cyclopentyl, 2-methoxyethyl, hexyl, decyl, tetradecyl,
triethylene glycol methyl ether, tetraethylene glycol
methyl ether and methoxybenzyl as well as
-CHZ-O-C~ ~ H22-OH ,
-CH2-O-C6H4-O- ( CHZCHZO ) Z-CH3 ,
-CH2-O-C6H4-O- ( CH2CH20 ) 3-CSH> > ,
3 5 -CHz-O-C6H4-O-C4H$-OH ,
- ( CHZCH20 ) 5-CH3 ,
I
2084582
- 13 -
-C9Iit8 OH,
-~18 ~H ~
-CH2-O-C6H~-O-CbHtt-COOH,
-CH2-O-C6H6-O-CbHg-O-CH2 CHOH-CHZOH,
- ( CH2CH20 j 3-CSHt ~ ,
-~2~-CtoHzo-COOH,
-CH2-O-C6H~-C 1,
-CH2-O-C6H4-NO2,
-CH2-O-CbH3C12.
-cHt-o-cbH6-cooH,
-CH2-O-CH2-CHOH-CH20H, rt/e ~ i
-CHOH-CH20H, Ne,,,~
-CH2-O-C6H4-O-CH2-COOH and
-CH2-O-C6H4-CSHt t .
In the use of volatile epoxides, such as, e.g.,
ethylene oxide or propylene oxide, the reaction is
performed in an autoclave.
In substrate IV, optionally present carboxyl and/or
hydroxy groups are present preferably in protected form, as
described above in the case of substrate III.
Another special embodiment of the process according to
the invention is the production of compounds of general
formula I with R meaning a -C-NHR9 radical,
a
X
in which
X means an oxygen or sulfur atom and
R9 means a phenyl, 1- or 2-naphthyl or straight-chain
or cyclic Ct-C6 alkyl group,
characterized in that tetraazatricyclotridecane or
tetraazatricyclopentadecane is reacted with a feedstock of
general formula V
R9-N=C=X ( V j ,
in which X and R9 have the above-indicated meaning,
~~.,,
. T
r zo 8 45~ 2
- 14 -
with or without solvent, preferably aprotic solvents, such
as, e.g., benzene, toluene, dichloromethane,
tetrahydrofuran, dioxane, acetonitrile, dimethylformamide,
hexane or ether, are used as solvent, at 0°C to 180°C,
preferably room temperature to 150°C (and in the case of
the higher reaction temperature, the solvent used
optionally to dissolve the added feedstock of general
formula V was distilled off previously in a vacuum), within
1 to 48 hours, preferably 5 to 12 hours. The thus obtained
reaction mixture is cooled to -20°C to 80°C, preferably
0°C
to 30°C, mixed with a mixture of water/organic solvent,
such as, e.g., methanol, ethanol, isopropanol,
tetrahydrofuran or dioxane, and stirred for 0.5 to 12
hours, preferably 0.5 to 3 hours, at -20°C to room
temperature, preferably 0°C to room temperature. The thus
formed -- optionally to be isolated --intermediate product
carrying a formyl group on a nitrogen atom is reacted by
adding an inorganic base such as, e.g., lithium hydroxide,
sodium hydroxide, potassium hydroxide, barium hydroxide or
calcium hydroxide, preferably sodium hydroxide and
potassium hydroxide, or a mineral acid, such as, e.g.,
hydrochloric, sulfuric or hydrobromic acid, preferably
hydrochloric acid, at 0°C to 150°C, preferably room
temperature to 120°C, within 1 to 72 hours, preferably 6 to
24 hours, with stirring to obtain the end product of
formula I, which can then be isolated in a way known in the
art, preferably as hydrochloride.
Still another special embodiment of the process
according to the invention is the production of compounds
of general formula I with R meaning a -(CHZ)2-NH-S02-R'°
radical,
in which R'° means a C~-C6 alkyl, -CF3 or a phenyl group
optionally substituted by a C~-C6 alkyl, chlorine, bromine
or nitro radical,
,, .
t,:
2084582
- 15 -
characterized in that tetraazatricyclotridecane or
tetraazatricyclopentadecane is reacted with a feedstock of
general formula VI
CH2
-SOZ-R1o (VI) ~
CH2
in which R~o has the above-indicated meaning,
with solvent, preferably aprotic solvents, such as, e.g.,
benzene, toluene, dichloromethane, tetrahydrofuran,
dioxane, acetonitrile, dimethylformamide, hexane or ether,
are used as solvent, at 0°C to 180°C, preferably room
temperature to 150°C (and in the case of the higher
reaction temperature, the solvent used was distilled off
previously in a vacuum), within 1 to 48 hours, preferably
5 to 12 hours. The thus obtained reaction mixture is
cooled to -20°C to 80°C, preferably 0°C to 30°C,
mixed with
a mixture of water/organic solvent, such as, e.g.,
methanol, ethanol, isopropanol, tetrahydrofuran or dioxane,
and stirred for 0.5 to 12 hours, preferably 0.5 to 3 hours,
at -20°C to room temperature, preferably 0°C to room
temperature. The thus formed -- optionally to be isolated
-- intermediate product carrying a formyl group on a
nitrogen atom is reacted by adding an inorganic base such
as, e.g., lithium hydroxide, sodium hydroxide, potassium
hydroxide, barium hydroxide or calcium hydroxide,
preferably sodium hydroxide and potassium hydroxide, or a
mineral acid, such as, e.g., hydrochloric, sulfuric or
hydrobromic acid, preferably hydrochloric acid, at 0°C to
150°C, preferably room temperature to 120°C, within 1 to 72
hours, preferably 6 to 24 hours, with stirring to the end
product of formula I, which can then be isolated in a way
known in the art, preferably as hydrochloride.
Preferred radicals R'° are the phenyl radical and
4-methylphenyl radical.
2084582
- 16 -
Another special embodiment of the process according to
the invention is the production of diners, i.e., compounds
of general formula I with R meaning a second 1,4,7,10
tetraazacyclododecane or 1,4,7,10-tetraazacyclotetradecane
molecule bound by a
bis(B-hydroxyj-alkylene chain -CHZ-CH-K-CH-CH2-
OH OH
in which R means a Co C~6 alkylene chain optionally
substituted by 1 to 6 hydroxy, 1 to 6 C~-C~ hydroxyalkyl, 1
to 8 C~-C~ alkoxy, 1 to 8 CT Coo aralkoxy, and/or 1 to 2
benzyloxy groups, and optionally interrupted by 1 to 6
oxygen atoms, 1 to 2 phenylene, phenylenoxy or
phenylenedioxy groups, and the optionally present hydroxy
groups optionally are present in protected form,
characterized in that tetraazatricyclotridecane or
tetraazatricyclopentadecane is reacted with a feedstock of
general formula VII
C~2 / H K ' / H2 (VII) ,
O O
in which K has the above-indicated meaning, and optionally
present hydroxy groups are optionally protected, with or
without solvent, preferably aprotic solvents, such as,
e.g., benzene, toluene, dichloromethane, tetrahydrofuran,
dioxane, acetonitrile, dimethylformamide, hexane or ether,
are used as solvent, at 0°C to 220°C, preferably 50°C to
180°C (and in the case of the higher reaction temperature,
the solvent used optionally to dissolve the added feedstock
of general formula VII was distilled off previously in a
vacuum) or in an autoclave at an excess pressure of 1 to
100 atm. within 1 to 48, preferably 5 to 12 hours. The
thus obtained reaction mixture is cooled to -20°C to 80°C,
preferably 0° to 30°C, mixed with a mixture of
water/organic solvent, such as, e.g., methanol, ethanol,
isopropanol, tetrahydrofuran or dioxane, and stirred for
0.5 to 12 hours, preferably 0.5 to 3 hours, at -20°C to
2084582
-1~-
room temperature, preferably 0°C to room temperature. The
thus formed -- optionally to be isolated -- intermediate
product carrying a formyl group on a nitrogen atom is
reacted by adding an inorganic base such as, e.g., lithium
hydroxide, sodium hydroxide, potassium hydroxide, barium
hydroxide or calcium hydroxide, preferably sodium hydroxide
and potassium hydroxide, or a mineral acid, such as, e.g.,
hydrochloric, sulfuric or hydrobromic acid, preferably
hydrochloric acid, at 0°C to 150°C, preferably room
temperature to 120°C, within 1 to 72 hours, preferably 6 to
24 hours, with stirring -- optionally followed by
subsequent removal of protecting groups in a way usual in
the art -- to obtain the end product of formula I, which
can then be isolated in a way known in the art, preferably
as hydrochloride.
Preferred binding links K are, for example,
-C2H4-
-CH2-
-CH2-O-CH2-
2 0 -CH2-O-CHZ-CH2-O-CH2-
-CHZ-O- ( CH2CH20 ) Z-CHZ_
-CHOH-
-CHOH-CHOH-
-CHOH-CHOH-CHOH-
2 5 -CHz-O-CH2-CHOH-CH2-O-CH2-
-CH2-O-C6H~-O-CH2-
-CH2-O-C4H$-O-CHZ-
-C (CH20H) Z-
-CH ( CH20H) -
3 0 -CHz-O-CbH4-O-C6H4-O-CH2_
-CHOH-CHOH-CHOH-CHOH-
-CH2-O-CHZ-CH ( CHZOH ) 2-CH2-O-CHZ-
-CH2-CH ( CH20CH3 ) -CH2-
-CH ( OCH3 ) -
3 5 -CH2-O-CH2-CbH4-CHZ-O-CHZ-
v.'~.
2084582
T
In contrast to substrates III to VI, which -- as
compared with feedstock II -- are reacted equimolarly to
any excess, preferably with 1.05 to 2.0 equivalents,
substrate VII is used in a deficiency of 0.5 to 0.3
equivalents.
In substrate VII, optionally present hydroxy groups
are present preferably in protected form, as described
above in the case of substrate III.
The above-described process according to the invention
is distinguished by high yields, a small number of reaction
steps, great variation range in desired substituents R,
problem-free performance of large batches (upscaling),
partially by possible dispensing with solvent, as well as
problem-free purification of the end products.
The following examples are used to explain the object
of the invention in more detail.
Without further elaboration, it is believed that one
skilled in the art can, using the preceding description,
utilize the present invention to its fullest extent. The
following preferred specific embodiments are, therefore, to
be construed as merely illustrative, and not limitative of
the remainder of the disclosure in any way whatsoever.
In the foregoing and in the following examples, all
temperatures are set forth uncorrected in degrees Celsius
and unless otherwise indicated, all parts and percentages
are by weight.
i~_:
..-r.
2084582
- 19 -
E X A M P L E 8
Reactions with compounds of general formula III:
R'
Ri
A
R3
Example 1
a) Mixture of 1- and 4-formamido-10-[(2-ethoxycarbonyl)-
ethyl]-1,4,7,10-tetraazacyclododecane
15.9 g (133.5 mmol) of dimethylformamide-
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
partially distilled off in this way. Then, 13.94 g (139.2
mmol) of acrylic acid ethyl ester is instilled under a
nitrogen atmosphere and heated slowly (within 30 minutes)
to 80°C. It is stirred for 12 hours at this temperature.
It is cooled in an ice bath to 0°C, and a mixture of 150 ml
of ethanol/20 ml of water is added. Then, it is stirred
for 30 minutes at room temperature. It is evaporated to
dryness in a vacuum and the residue is chromatographed on
silica gel (mobile solvent = ethanol/conc. aqu. ammonia =
10/1). After concentration by evaporation of the main
fractions, 31.71 g (91% of theory) of a yellowish oil is
obtained.
Analysis (relative to the anhydrous substance):
Cld: C 55.98 H 9.39 N 18.65
Fnd: C 55.91 H 9.43 N 18.59
b) 10-(2-Carboxyethyl)-1,4,7,10-tetraazacyclododecane
46.32 g (825.6 mmol) of potassium hydroxide is added
to 31.0 g (103.2 mmol) of the title compound of example la
in 150 ml of ethanol/150 ml of water and refluxed for 12
hours. It is cooled in an ice bath to 0°C. It is adjusted
with.6N hydrochloric acid to pH 6 and then concentrated by
2084582
- 20 -
evaporation in a vacuum. The residue is extracted with a
mixture of 300 ml of methanol/50 ml of methylene chloride
and filtered off from potassium chloride. The filtrate is
concentrated by evaporation in a vacuum and purified on a
reversed-phase column (RP 18/mobile solvent: gradient of
tetrahydrofuran/water).
Yield: 23.02 g (87% of theory) of a yellowish,
viscous oil, which solidifies after a short time
Analysis (relative to the anhydrous substance):
Cld: C 56.23 H 9.44 N 21.86
Fnd: C 56.17 H 9.51 N 21.83
Example 2
10-(2-Cyanoethyl)-1,4,7,10-tetraazacyclododecane
15.9 g (133.5 mmol) of dimethylformamide
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to room temperature. 9.24 g (174.15 mmol) of acrylic acid
nitrile is instilled under a nitrogen atmosphere and heated
slowly to 75°C. It is stirred for 9 hours at this
temperature. It is cooled to room temperature and a
mixture of 120 ml of methanol/30 ml of water is added. It
is stirred for 10 minutes at room temperature. Then, 13.93
g (348.3 mmol) of sodium hydroxide is added and it is
stirred for 24 hours at 40°C. It is evaporated to dryness
in a vacuum and the residue is extracted 3 times with hot
toluene (80°C). The organic phase is dried on potassium
hydroxide and concentrated by evaporation in a vacuum.
Yield: 23.28 g (89~ of theory) of a pale yellow oil,
which crystallizes with standing.
Analysis (relative to the anhydrous substance):
Cld: C 58.63 H 10.29 N 31.08
Fnd: C 58.57 H 10.34 N 30.96
A
2og45a~
- 21 -
Example 3
10-[(2-Phenyl-2-carboxy)-ethylJ-1,4,7,10-
tetraazacyalododeaane
15.9 g (133.5 mmol) of dimethylformamide
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to room temperature. 24.55 g (139.32 mol) of 2-phenyl-
vinyl acid ethyl ester is instilled under a nitrogen
atmosphere and slowly heated to 130°C. It is stirred for
12 hours at this temperature. It is cooled to room
temperature and a mixture of 150 ml of methanol/150 ml of
water is added. Then, it is stirred for 30 minutes at room
temperature. 52.11 g (928.8 mmol) of potassium hydroxide
is added and refluxed for 12 hours. It is cooled in an ice
bath to 0°C and adjusted with conc. hydrochloric acid to pH
7, then evaporated to dryness. The residue is taken up in
a mixture of 250 ml of methanol/50 ml of methylene
chloride. The precipitated potassium chloride is filtered
off and the filtrate is concentrated by evaporation in a
vacuum. The residue is chromatographed on silica gel
(mobile solvent - methyl-tert-butyl ether/methanol/conc.
aqu. ammonia = 6/2/1).
Yield: 28.27 g (76% of theory) of a vitreous solid
Analysis (relative to the anhydrous substance):
Cld: C 63.72 H 8.81 N 17.48
Fnd: C 63.64 H 8.93 N 17.37
Example 4
11-(2-Cyanoethyl)-1,4,8,11-tetraazacyclotetradecane
13.68 g (114.8 mmol) of dimethylformamide-
dimethylacetal (under nitrogen) is added to 20.0 g (99.83
mmol) of 1,4,8,11-tetraazacyclotetradecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
~~ i n
2084582
- 22 -
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to room temperature. 6.36 g (119.8 mmol) of acrylic acid
nitrile is instilled under a nitrogen atmosphere and heated
slowly to 75°C. It is stirred for 9 hours at this
temperature. It is cooled to room temperature and a
mixture of 120 ml of methanol/30 ml of water is added. It
is stirred for l0 minutes at room temperature. Then, 11.98
g (299.5 mmol) of sodium hydroxide is added and it is
stirred for 24 hours at 40°C. It is evaporated to dryness
in a vacuum and the residue is extracted 3 times with hot
toluene (80°C). The organic phase is dried on potassium
hydroxide and concentrated by evaporation in a vacuum.
Yield: 21.75 g (86~ of theory) of a pale yellow oil,
which crystallizes with standing
Analysis (relative to the anhydrous substance):
Cld: C 61.62 H 10.74 N 27.64
Fnd: C 61.53 H 10.84 N 27.52
For example, the compounds listed in the following
table are produced analogously.
CA 02084582 2004-07-21
- 23 -
TABLE la
H H
N ~ ~ N
CHO-\-N N/>-R H-N N-R
~.; J ~; ..~
H CHO
Ttmp. ('G) Ycdd
R time (b) Solvent ass (~) elementary analysis
O 100' G - 1,2 87 C 57,30 H 9,62 N 17,82 (cld)
~ ~II 12 h C 57,35 H 9,58 N 17.87
~O~
CO~Et 120' C. - 1,3 92 C 54,82 H 8,66 N 15,04 (cld)
12 h C 54,73 H 8,71 N 14,97
130' G - 1,5 84 C 63,80 H 8,57 N 14,88 (cid)
12 b C 63,75 H 8,50 N 14,84
CO~Et
N02 130' C. CH2C12 1,5 81 C 56,99 H 7,41 N 16,62 (cld)
12 b C 56,93 H 7,50 N 16,54
C02Et
n p 100' G - 1,2 93 C 53.11 H 9,29 N 25.81 (c1d)
CA 02084582 2004-07-21
- 24 -
TABLE lb
H
N
H-N N-R
~. ; J
H
bast/ exoas base Tmnp. Time Yield
R solvent (eq.) ('C.) (h) (g'o) elementary analysis
O KOH 8 reflex 12 83 C 57,75 H 9,69 N 2(1,T2 (cld)
~ ~ MeOH/H20 1:1 - C 57,68 H 9,78 N 20,67
/ V _OH
COON KOH 10 reflex 24 79 C 49,99 H &39 N 19 ~3 (c1d)
~COOH ~~~ la C 49.90 H 8.46 N t9,37
KOH 8 reflex 12 85 C 63,72 H 8,81 N 17,48 (c!d)
MeOH/HZO I:I C 63,65 H 8,87 N 17,39
COOH
H
N
H-N N-R
~. ; J
H
base/ excess Time Yield
base Temp.
R solvent (eq.) ('C.)(h) (%) elemrntary analysis
NOZ KOH 8 50' C. 24 79 C 55,88 H 7,45 N
19,16 (c1d)
MeOH/Hg0 1:1 C 55,8a H 7,52 N 19,08
COOH
NaOH 3 RT 24 89 C 54,29 H 10,35 N
28,78 (cld)
~ ' C 54,23 H 10,29 N 28,81
~Z MeOH/HZO I:I
2pgt~582
- 25 -
Reactions with compounds of general formula IV:
0
R~ Ra
Example 5
a) Mixture of 1- and 4-formamido-10-(6-hydroxy
2,2-dimethyl-1,3-dioxepan-5-yl)-1,4,7,10
tetraazacyclododecane
15.9 g (133.5 mmol) of dimethylformamide-
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
partially distilled off in this way. Then, 20.1 g (139.32
mmol) of 4,4-dimethyl-3,5,8-trioxabicyclo-(5.1.0)-octane is
instilled under a nitrogen atmosphere and heated slowly (1
hour) to 130°C. It is stirred for 12 hours at 120°C. It
is cooled to room temperature and a mixture of 120 ml of
methanol/30 ml of water is added. Then, it is stirred for
one hour at room temperature. It is concentrated by
evaporation in a vacuum and the residue is chromatographed
on silica gel (mobile solvent - methyl-tert-butyl
ether/methanol/aqu. conc. ammonia - 15/5/1). After
concentration by evaporation of the main fractions, 36.39
g (91~ of theory) of a pale yellow, viscous oil, which
crystallizes with standing, is obtained.
Analysis (relative to the anhydrous substance):
Cld: C 55.79 H 9.36 N 16.27
Fnd: C 55.82 H 9.29 N 16.20
b) 10-(6-Hydroxy-2,2-dimethyl-1,3-dioxepan-5-yl)-1,4,7,10-
tetraazacyclododecane
57. 0 g ( 1. 02 mol ) of potassium hydroxide is added to
35.0 g (101.6 mmol) of the title compound of example 5a in
_ ~r. .Z0 8 458 2
- 26 -
200 ml of methanol/50 ml of water and refluxed for 5 hours.
It is evaporated to dryness in a vacuum and the residue is
extracted 3 times with 200 ml of hot (80'C) toluene. The
organic phase is dried on potassium hydroxide and
concentrated by evaporation in a vacuum.
Yield: 31.5 g (98% of theory) of a pale yellow,
viscous oil, which becomes solid with standing
Analysis (relative to the anhydrous substance):
Cld: C 56.93 H 10.19 N 17.71
Fnd: C 56.87 H 10.25 N 17.63
Example 6
a) Mixture of 1- and 4-formaiaido-10-(2-hydroxypropyl)-
1,4,7,10-tetraazacyclododecane
15.9 g (133.5 mmol) of dimethylformamide-
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to 0°C. The residue is dissolved in 50 ml of toluene and
the solution is fed into an autoclave. 20.23 g (348.3
mmol) of propylene oxide is added and the autoclave is made
airtight. Then, it is heated for 24 hours to 100°C. It is
evaporated to dryness in a vacuum and the residue is taken
up in a mixture of 120 ml of methanol/30 ml of water.
Then, it is stirred for one hour at room temperature. It
is concentrated by evaporation in a vacuum and the residue
is chromatographed on silica gel. (Mobile solvent -
methanol/isopropanol/aqu. conc. ammonia = 10/5/1). After
concentration by evaporation of the main fractions in a
vacuum, 26.7 g (89% of theory) of a weak, yellow-colored
oil is obtained.
Analysis (relative to the anhydrous substance):
Cld: C 55.79 H 10.14 N 21.69
Fnd: C 55.72 H 10.19 N 21.61
z ~.._ -
2084582
- 27 -
b) 10-(2-Hydroxypropyly-1,4,7,10-tetraazacyclododecane
45.2 g (805.1 mmol) of potassium hydroxide is added to
26.0 g (100.63 mmol) of the title compound of example 6a in
250 ml of water and refluxed for 5 hours. It is evaporated
to dryness in a vacuum and the residue is extracted 3 times
with 200 ml of hot (80°C) toluene. The organic phase is
dried on potassium hydroxide and concentrated by
evaporation in a vacuum.
Yield: 22.02 g (95% of theory) of a weak, yellowish
oil, which solidifies after a short time
Analysis (relative to the anhydrous substance):
Cld: C 57.36 H 11.38 N 24.32
Fnd: C 57.30 H 11.43 N 24.28
Example 7
10-[2-Hydroxy-5-(2,2-dimethyl-1,3-dioxolan-4-yl)-
4-oxapentyl~-1,4,7,10-tetraazacyclododecane
15.9 g (133.5 mmol) of dimethylformamide-
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to 40°C. 24.04 g (127.7 mmol) of 2,2-dimethyl-4-(2',3'-
epoxy)-propoxy-methyl-1,3-dioxolane is instilled under a
nitrogen atmosphere and heated slowly (within one hour) to
110°C. It is stirred for 12 hours at this temperature. It
is cooled to room temperature and a mixture of 120 ml of
methanol/30 ml of H20 is added. Then, it is stirred for 30
minutes at room temperature. 65.1 g (1.16 mol) of
potassium hydroxide is added and refluxed for 5 hours.
Then, it is concentrated by evaporation in a vacuum and the
residue is extracted 3 times with 200 ml of hot toluene
(80°C). The combined organic phases are dried on potassium
hydroxide and evaporated to dryness in a vacuum. The
residue is chromatographed on silica gel (mobile solvent =
L~~.
c.~ -, :_
2084582
- 28 -
methanol/water/aqu. conc. ammonia - 8/2/1). The main
fractions are evaporated to dryness, the residue is
dissolved in 500 ml of hot toluene. It is filtered off
from insolubles (silica gel) and evaporated to dryness.
Yield: 36.41 g (87% of theory) of a pale yellow,
viscous oil
Analysis (relative to the anhydrous substance):
Cld: C 56.64 H 10.07 N 15.54
Fnd: C 56.53 H 10.13 N 15.49
Example 8
10-[3-(4-Nitrophenoxy)-2-hydroxypropyl]-1,4,7,10-
tetraazacyclododecane (as tetrahydrochloride)
15.9 g (133.5 mmol) of dimethylformamide
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
_. mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent
is distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to room temperature. A solution of 29.46 g (150.93 mmol)
of (4-nitrophenyl)-2,3-epoxypropyl ether in 100 ml of
methylene chloride is instilled under a nitrogen
atmosphere. Then, it is heated slowly to 120°C, and the
methylene chloride is distilled off (toward the end under
reduced pressure). It is stirred for 12 hours at 120°C.
It is cooled to room temperature and a mixture of 160 ml of
methanol/20 ml of water is added. Then, it is stirred for
minutes at room temperature. 50 ml of conc.
hydrochloric acid is added and refluxed for 12 hours.
Then, it is evaporated to dryness in a vacuum. The residue
30 is recrystallized from methanol/ether.
Yield: 48.27 g (81% of theory) of a yellow-colored
crystalline powder
Analysis (calculated for a CI-free compound):
Cld: C 55.57 H 7.95 N 19.06
Fnd: C 55.49 H 8.03 N 19.01
2084582
- 29 -
Example 9
a) 11-[3-(4-Nitroxyphenoxy)-2-hydroxypropyl]-1,4,8,11-
tetraazacyclotetradecane (as tetrahydrochloride)
13.68 g (114.8 mmol) of dimethylformamide
dimethylacetal (under nitrogen) is added to 20.0 g (99.83
mmol) of 1,4,8,11-tetraazacyclotetradecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to room temperature. A solution of 23.38 g (119.8 mmol) of
(4-nitrophenyl)-2,3-epoxypropyl ether in 100 ml of
methylene chloride is instilled under a nitrogen
atmosphere. Then, it is heated slowly to 120°C, and the
methylene chloride is distilled off (toward the end under
reduced pressure) . It is stirred for 12 hours at 120°C.
It is cooled to room temperature and a mixture of 150 ml of
methanol/20 ml of water is added. Then, it is stirred for
30 minutes at room temperature. 100 ml of conc.
hydrochloric acid is added and refluxed for 12 hours.
Then, it is evaporated to dryness in a vacuum. The residue
is recrystallized from methanol/ether.
Yield: 41.61 g (77% of theory) of a yellowish,
crystalline powder.
Analysis (relative to the anhydrous substance):
Cld: C 42.16 H 6.89 N 12.94 C1 26.20
Fnd: C 42.10 H 6.93 N 12.90 C1 26.08
Example io
a) Mixture of 1- and 4- and 8-formamido-11-[2-hydroxy-2
(2,2-dimethyl-1,3-dioxolan-4-yl)-ethyl]-1,4,8,11
tetraazacyclotetradecane
13.68 g (114.8 mmol) of dimethylformamide-
dimethylacetal (under nitrogen) is added to 20.0 g (99.83
mmol) of 1,4,8,11-tetraazacyclotetradecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
r- ~x
2084582
- 30 -
evaporation under reduced pressure. The residue is cooled
to room temperature. 17.27 g (119.8 mmol) of
2-(2,2-dimethyl-1,3-dioxolan-4-yl)-ethylene oxide is
instilled under a nitrogen atmosphere and then heated
slowly to 130°C. It is stirred for 12 hours at this
temperature. It is cooled to 0°C and a mixture of 160 ml
of methanol/40 ml of water is added, then it is stirred for
1 hour at room temperature. It is evaporated to dryness in
a vacuum and the residue is chromatographed on silica gel
(mobile solvent - methyl-tert-butyl ether/methanol/conc.
aqu. ammonia = 15/5/1).
Yield: 33.1 g (89% of theory) of a pale yellow,
viscous oil
Analysis (relative to the anhydrous substance):
Cld: C 58.04 H 9.74 ~ N 15.04
Fnd: C 58.13 H 9.61 N 14.92
b) 11-(2-Hydroay-2-(2,2-dimethyl-1,3-dioxolan-4-ylj-
ethyl]-1,4,8,11-tetraazacyclotetradecane
39.15 g (698 mmol) of potassium hydroxide is added to
32.0 g (87.22 mmol) of the title compound of example l0a in
200 ml of methanol/100 ml of water and refluxed for 5
hours. It is evaporated to dryness in a vacuum and the
residue is extracted 3 times with 200 ml of hot toluene
(80°C). The organic phase is dried on potassium hydroxide
and concentrated by evaporation in a vacuum.
Yield: 28.85 g (96% of theory) of a pale yellow,
viscous oil, which solidifies after a short time
Analysis (relative to the anhydrous substance):
Cld: C 59.27 H 10.53 N 16.26
Fnd: C 59.18 H 10.61 N 16.17
For example, the compounds listed in the following
table are produced analogously.
CA 02084582 2004-07-21
- 31 -
TABLE 2a
H H
N ~ ~ N
CHO-N N-R H-N N-R
~; J ~.; J
R CHO
Temp. excess Yield
('G)
R Tame (h) Solvent(epoxide) (qo)
elementary analysis
130' G - I,1 93 C 55,79 N 16,27 dd
12 h H 9,36 N 16,29
C 55,74 H 9,29
O
O
~V,
'~~
OH
120' C. - 1,1 91 C 61,69 N 15,99 cid
H 8,63 5
H
8 h C bI,61 N 1
8,67 ,94
~O
OH
120' G - 1,1 90 C 62,61 N 15,37 dd
H 8,85
O 8 h C 62,64 H 8,80 N 15,41
120' C. - I,I 92 C 56.17 N 14,56 CI
H 7,59 9,21 dd
8 h C 56,10 H 7.51 _ N 14,61
CI 9,14
~O
OH
~H3 120' G CH2C121,1 89 C 59,98 N 14,73 dd
H 8,48
8 h C 59,89 H 8,53 N 14,67
~O ,
OH
130' G - 1,1 87 C 62,04 N 12,06 ctd
H 8,68
24 h C 62,11 H 8,60 N 11,96
C02i,Pr
~O
OH
IZO' G - 1,1 93 C 56,93 N 17,51 dd
H 10,19
8 h C 55,86 H 10,25 N 17,46
O
OH
120' G - 1,1 87 C 59,97 N 18,65 dd
H 10,74
8 h C 59,92 H 10,68 N 18,71
OH
110' C: - 1,2 87 C 62.14 N 14,49 cid
12 h H 10,95 N 14,36
C 62,03 H 10,87
OH
C02i,Pr 110' G - 1,2 85 C 60.53 N 12.83 dd
H 8,31
12 h C 60,59 H 8,22 N 12,74
~O
OH
CA 02084582 2004-07-21
- 32 -
TABLE 2b
ezcas
Hale/ Bate Temp. Time Yeld
R solvent (eq.) ('C.) (h) (R'o) elementary analysis
KOH 10 reflwc 5 97 C 56,93 H 10,19 N 17,71 (cld)
MeOH/H20 4:1 C 56,88 H 10,25 N 17,63
O
~O
'O VH
NaOH 5 60' C. 24 97 C 63,32 H 9,38 N 17,38 (cld)
EtOH/HZ0 5.1 C 63,28 H 9,45 N 17,33
~O
OH
NaOH 5 60' C. 24 98 C 64,25 H 9,59 N 16,65 (c1d)
O ~ EtOH/Hz0 5:1 C 64,14 H 9,67 N 16,60
KOH 5 RT 48 97 C 57,21 H 8,19 N 15,70 CI 9,93 (cld)
EtOH/Hz0 8:1 C 57.14 H 8,27 N 15,62 CI 9,87
~O
OH
OCH3 KOH 5 RT 48 98 C 61,34 H 9,15 N 15,90 (c1d)
EtOH/HZ0 8:1 C 61,24 H 9,08 N 15,84
~O
OH
NaOH 2 RT 48 92 C 63,27 H 9,23 N 12,83 (ctd)
Dioxattl C 63,22 H 9,3I N 12,79
1 HBO 8~.1
CO2~,Pr .
O
OH
NaOH 10 rcflux 5 98 C 58,30 H 11,18 N 19,42 (ber.)
MeOlillIzO 4:1 , C 58,24 H 11,25 N 19,35 (cld)
O
OH
KOH 10 retlux 5 98 C 61,72 H 11,84 N 20,57 (ber.)
EtOH/Hz0 4:! C 61,67 H 11,91 N 20,49 (cld)
OH
KOH 10 rellux 5 97 C 63,64 H 11,81 N 15,63 (ber.) '
~O~ EtOH/Hz0 4:1 C 63;54 H 11,90 N 15,54 (cId)
OH
KOH 3 30' C. 48 93 C 61,74 H 8,88 N 13,71 (ber.)
EtOHIFFzO 4:1 C 61,66 H 8,95 N 13,62 (ctd)
~O _
OH
CA 02084582 2004-07-21
- 33 -
Reactions with compounds of general formula V:
R9-N=C=X
Example il
10-(N-Phenylcarbamoyl)-1,4,7,10-tetraazacyclododecane
15.9 g (133.5 mmol) of dimethylformamide-
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to 0°C. 16.6 g (139.32 mmol) of phenyl isocyanate is
instilled under a nitrogen atmosphere and heated slowly to
100°C. It is stirred for 12 hours at this temperature. It
is cooled to room temperature and a mixture of 160 ml of
ethanol/40 ml of water is added. Then, it is stirred for
10 minutes at room temperature. Then, 18.58 g (464.4 mmol)
of sodium hydroxide is added and it is stirred for 24 hours
at 40°C. It is evaporated to dryness in a vacuum and the
residue is taken up in 400 ml of water. The aqueous phase
is extracted 5 times with 200 ml of methylene chloride, the
organic phase is dried on magnesium sulfate and
concentrated by evaporation in a vacuum. The residue is
chromatographed on silica gel (mobile solvent - methyl-
tert-butyl ether/methanol/conc. aqu. ammonia = 6/3/1).
Yield: 29.1 g (860 of theory) of a pale yellow solid
Analysis (relative to the anhydrous substance):
Cld: C 61.83 H 8.65 N 24.03
Fnd: C 61.71 H 8.72 N 23.94
For example, the compounds listed in the following
table are produced analogously.
CA 02084582 2004-07-21
- 34 -
TABLE 3a
H H
N ~ ~ N
CHO-N N-R, H-N N-R
~; J ~.; J
H CHO
Temp. ('C.) excess Yield
R ~ Time (h) Solvent (aryl reagent) (%) elementary analysis
100' C. - 1,2 93 C 59,05 H 9,60 N 21,52 (cld)
12 h C 59,14 H 9,53 N 21,46
-CONH
100' C. - 1,2 89 C 65,02 H 7,37 N 18,96 (cld)
I8 h C 65,II H 7,30 N t8,89
-CONH
100' C. - 1,1 90 C 57,29 H 7,51 N 20,88 S 9,56 (cld)
12 h C 57.20 H 7,60 N 20,81 S 9,59
-CSNH
100' C. CHZCI~ , 1,1 87 C 62,31 H 7,06 N 18,17 S 8,32 (c1d)
l8 h C 62,35 H 7,01 N 18,08 S 8,27
-CSNH
CA 02084582 2004-07-21
- 35 -
TABLE 3b
H
N
H-N N-R
~. ; J
H
ezcGSS
Base/ Bax Temp. Tune Yield
R solvent (eq.) (~C.) (h) (9io) ekmrnt~ry aralysis
NaOH 4 40 24 96 C 60,57 H 10,50 N 23,55 (cld)
EtOH/HZO 4;1 C 60,48 H 10,58 N 23,50
-CONH
H
N
H-\N N,-R
~.. ; J
H
excess
Hale/ Bax Temp. Time Yield
R solvent (eq.) ('C.) (h) ('%) elemrntary analysis
NaOH 4 40 24 97 C 66,88 H 7,97 N 20,51 (cld) .
EtOH/H20 8:1 C 66,78 H 7,93 N 20,54
-CONH
NaOH 4 40 24 95 C 58,60 H 8,20 N 22,78 S 10,43 (cld)
EtOH/H~O 8:1 C 58,63 H 8,29 N 22,71 S 10,37
-CSNH
NaOH 4 40 24 96 C 63,83 H 7,61 N 19,59 S 8,97 (cld)
EtOH/H20 8:1 C 63,78 H 7,55 N 19,61 S 8,91
-CS)JFi
o~
CA 02084582 2004-07-21
- 36 -
Reactions with a compound of general formula VI:
CH2
N-SOZ-Rio
cH2
Example 12
a) Mixture of 1- and 4-formamido-10-[2-(p-
tolylsulfonylamino)-ethyl]-1,4,7,10-tetraazacyclododecane
15.9 g (133.5 mmol) of dimethylformamide-
dimethylacetal (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to 0°C. A solution of 25.19 g (127.71 mmol) of
p-tolylsulfonyl aziridine in 100 ml of toluene is instilled
under a nitrogen atmosphere and then stirred for 8 hours at
80°C. It is evaporated to dryness in a vacuum and the
residue is taken up in a mixture of 180 ml of ethanol/30 ml
of water. Then, it is stirred for 30 minutes at room
temperature. Then, it is evaporated to dryness.. The
residue is chromatographed on silica gel (mobile solvent =
methyl-tert-butyl ester/methanol/conc. aqu. ammonia
6/2/1) .
Yield: 40.61 g (88% of theory) of a vitreous solid
Analysis (relative to the anhydrous substance):
Cld: C 54.38 H 7.86 N 17.62 S 8.06
Fnd: C 54.31 H 7.93 N 17.58 S 7.99
b) 10-[2-(p-Tolylsulfonylamino)-ethyl]-1,4,7,10-
tetraazacyclododecane
20.12 g (503 mmol) of sodium hydroxide is added to
40.0 g (100.62 mmol) of title compound 9a in 180 ml of
ethanol/30 ml of water and refluxed for 12 hours. It is
evaporated to dryness and the residue is taken up in 100 ml
of water. The pH of the solution is brought to pH 10 by
CA 02084582 2004-07-21
- 37 -
adding 6N hydrochloric acid. Then, it is extracted twice
with 250 ml of hot toluene (80°C) . The organic phase is
dried on magnesium sulfate and concentrated by evaporation
in a vacuum.
Yield: 36.07 g (97% of theory) of a yellowish,
vitreous solid
Analysis (relative to the anhydrous substance):
Cld: C 55.26 H 8.46 N 18.95 S 8.68
Fnd: C 55.21 H 8.52 N 18.90 S 8.59
c) Mixture of 1- and 4-formamido-10-[2-
(methylsulfonylamino)-ethyl)-1,4,7,10-tetraazacyclododecane
Analogously to example 12a, methylsulfonyl aziridine
can be used instead of p-tolylsulfonyl aziridine.
Yield: 89% of theory
Analysis (relative to the anhydrous substance):
Cld: C 44.84 H 8.47 N 21.79 S 9.97
Fnd: C 44.76 H 8.53 N 21.73 S 9.90
d) 10-[2-(methylsulfonylamino)-ethyl]-1,4,7,10-
tetraazacyclododecane
Analogously to example 12b, the title compound of
example 12c can be used instead of title compound 12a.
Yield: 96% of theory
Analysis (relative to the anhydrous substance):
Cld: C 45.03 H 9.27 N 23.87 S 10.93
Fnd: C 44.96 H 9.34 N 23.98 S 10.85
CA 02084582 2004-07-21
- 38 -
Reactions with compounds of general formula VII:
CHZ CH-R-CH-CH2
O O
Example 13
a) Mixture of the bis-formamides of l,i~-(2,6-dihydroxy-4-
oxa-1,7-heptyl)-bis-[1,4,7,10-tetraazacyclododecane
15.9 g (133.5 mmol) of dimethylformamide-
dimethylacetai (under nitrogen) is added to 20.0 g (116.1
mmol) of 1,4,7,10-tetraazacyclododecane in 200 ml of
absolute toluene. It is refluxed slowly and the solvent is
distilled off in this way. Then, it is concentrated by
evaporation under reduced pressure. The residue is cooled
to 40°C. 7.25 g (55.7 mmol) of bis-[2,3-epoxypropylJ-ether
is instilled under a nitrogen atmosphere and heated slowly
to 120°C. It is stirred for 24 hours at this temperature.
It is cooled to room temperature and a mixture of 200 ml of
methanol/100 ml of water is added. Then, it is stirred for
one hour at room temperature. It is evaporated to dryness
and the residue is chromatographed on silica gel (mobile
solvent = methanol/isopropanol/conc. aqu. ammonia = 8/2/1).
Yield: 18.63 g (63% of theory) of a vitreous solid
Analysis (relative to the anhydrous substance):
Cld: C 54.32 H 9.50 N 21.11
Fnd: C 54.25 H 9.57 N 21.18
b) 1,1~-(2,6-Dihydroxy-4-oxa-1,7-heptyl)-bis-(1,4,7,10-
tetraazacyclododecane)
28.55 g (508.7 mmol) of potassium hydroxide is added
to
18.0 g (33.92 mmol) of the title compound of example 13a in
200 ml of methanol/100 ml of water and refluxed for 2
hours. It is evaporated to dryness in a vacuum and the
residue is extracted 3 times with 200 ml of hot toluene
(80°C). The organic phase is dried on potassium hydroxide
and concentrated by evaporation in a vacuum.
CA 02084582 2004-07-21
- 39 -
Yield: 15.62 g (97% of theory) of a pale yellow,
viscous oil, which solidifies with standing.
Analysis (relative to the anhydrous substance):
Cld: C 55.67 H 10.62 N 23.61
Fnd: C 55.61 H 10.68 N 23.56
For example, the compounds listed in the following
table are produced analogously.
CA 02084582 2004-07-21
- 40 -
TABLE 4a
i HO~H] i (CHO)
N ~ ~ N
(CHO)H-N N-R-N N-CHO(Il)
':J ~.rJ
H
Yield
Temp. Eqivalcnt (relative
('C.) to
R time Solvent diepozidediepozide)elementary
(h) analysis
OH I20' - 0,49 67 C 54,33 N 19.50
C. H 9.47 (chi)
24 h C 54,28 N 19,38
H 9,51
~/v O
OH
120' - 0,49 61 C 55,08 N 19,03
~O~O~ C. H 9,59 (cld)
24 h C 55,13 N 18,96
H 9,50
OH OH
120' - 0,49 60 C 55,79 N I8,59
~O~O~ C. H 9,70 (cId)
24 6 C 55,71 N 18,61
H 9,77 '
OH OH
120' - 0,45 59 C 59,38 H 8,97 N 18,47
C. C 59,31 H 9,05 (cld)
12 N 18,38
h
O
OH OH
130' - 0,45 58 C 55,27 H 9,28 N 19,10
C. 2 (cld)
h 55 N
12 C 19.02
,
1 H 9.37
OH OH
130' CH~Ch 0,49 61 C 57,8b H 8,74N 17,99
C. (cld)
24 C 57,79 H B,69 N 18,04
O h
CA 02084582 2004-07-21
- 41 -
TABLE 4b
H H
N ~ ~ N
H-N - N-R-N N-H
J;J J;J
H H
excess
Base/ BaseTemp.TimeMeld
R solvent (eq.)('C.)(h)(%) elementary
analysis
OH KOH 15 rcflux24 97 C 55,57 N 21,60(cld)
H 10,49
MeOH/Hz0 C 55,50 N 21,54
2:1 H 10,58
vvO
OH
KOH 15 reflex48 96 C 56,36 N 21,03(cId)
~O~O~ MeOH/HZ0 H 10,59 N 21,11
2:1 C 56,31
H 10,50
OH OH
KOH IS reflex48 98 C 57,11 N 20,49(cid)
~O~O~ MeOH/Hz0 H 10,69 N 20,44
2:1 C 57,04
H 1Q75
OH OH
KOH IS rcflux24 97 C 61,06 N 20,34(cld)
EtOH/HZ0 H 9,88 N 20,27
3:1 C 61,01
H 9,95
O
OH OH
KOH IS reflex48 96 C 56,58 N 21,11(dd)
~ 1 H 10,25 N 21
E C 56 06
OH/H 53 H
0 3 10
33
O z , ,
O : ,
t
OH OH
KOH 15 reflex48 98 C 59,34 N 19,77(c!d)
H 9,60 ~
~ E20H/Hz0 C 59,28 N 19,70
3:1 H 9,71
~O~O~
off ~~J/ OH