Note: Descriptions are shown in the official language in which they were submitted.
wo 9zizo~z~ 2 ~ 8'~ ~ ~ J Pcroca9lioo~6o
CATAL'1ST AND PROCESS FOR REMOVAL OF SULPHUR
COMPOUNDS AND NITROGEN OXIDES FROM FLUID STREAMS
Field of Invention
The desirability of identifying an effective means
for removing sulphur compounds from fluid streams will be
readily appreciated. This invention comprises a novel
method and catalyst for effecting such removal and the
subsequent treatment of such sulphur compounds to produce
elemental sulphur. More particularly this invention is
1U applicable to the removal of hydrogen sulphide and other
sulphur compounds from sour natural gas, and other fluid
streams, and the conversion of the sulphur therein to
elemental sulphur.
By the same process applied in a different order,
the invention may be used to remove certain oxygen
compounds from gas streams, and particularly to remove
sulphur dioxide, sulphur trioxide, nitrogen trioxide,
nitrogen peroxide, nitrogen pentoxide and carbon dioxide
from flue gases.
Background of the Invention
Sulphur compounds are often considered to be
undesirable compounds in gas mixtures and other fluid
streams. The most common example of this is that of
natural gas containing hydrogen sulphide. Natural gas may
also contain as undesirable sulphur compounds, quantities
of carbonyl sulphide, carbon disulphide, mono and dialkyl
sulphides, alkyl-type disulphides and thiophenes.
The removal of such sulphur-containing compounds
from gas streams has been addressed by a number of methods
in the past. These methods generally rely on direct
reactions with the sulphur compounds, or proceed to first
separate the sulphur compounds from the gas stream by an
absorption stage. In the latter case, the sulphur and
other constituent elements of the absorbed compounds must
then be extracted, if the absorptive medium is to be
regenerated. A particularly desirable regenerative
process would be one which produces elemental sulphur from
the same reaction bed.
Various systems have bean explored with the view of
removing hydrogen sulphide from gas streams and producing
9V0 92/20621 ~ ~ ~ ~ ~ ~ P~CT1CA91 /00160
2 -
elemental sulphur. The Claus process, as currently
applied, is a complex mufti-stage system involving the
absorption of the hydrogen sulphide in an amine absorbent,
flashing off II2S from the amine, followed by the burning
of part of the hydrogen sulphide to sulfur dioxide, and
subsequently reacting the hydrogen sulphide with the
sulfur dioxide to produce sulphur as the final product as
elemental sulphur.
Tt would be obviously desirable to provide a
l0 method for removal of Hydrogen sulphide, and other sulphur-
containing compounds from a fluid stream at ambient
temperatures followed by the subsequent conversion at
moderate temperatures of the sulphur compounds into
elemental sulphur and other non-sulphur containing
decomposition products.
Flue gases generally include appreciable
quantities of oxides of sulphur, nitrogen peroxide and
carbon dioxide. It would be desireable to have a process
which effectively removes such compounds from flue gas,
and allows for their separation and subsequent
utilization.
Ob-iects of the Invention
Tt is therefore an object of the invention to
remove sulphur compounds from a fluid stream and recover
elemental sulphur therefrom. It is further an object to
. do so in the same reaction bed.
Tt is also an object of the invention to provide a
means which will allow removal and decomposition of
hydrogen sulphide from a gas stream, at temperatures below
the condensation point for sulphur and the separation of
the sulphur so produced, at a modestly elevated
temperature (circa 250oC - 600oC).
A further object of the invention is to remove
sulphur dioxide, nitrogen trioxide, nitrogen peroxide
nitrogen pentoxide and carbon dioxide, separately or
collectively from a gas stream, and then to convert the
sulphur dioxide to sulphur, convert the nitrogen peroxide
and other oxides to nitric oxide, and separately release
the carbon dioxide, nitric oxide and sulphur so produced
:Eor subsequent utilization.
WO 92/2062
PCT/CA91 /00160
- 3 -
These and other objects of the invention will
become apparent from the description of the invention and
claims thereto which. follow.
Summary of the Invention
In its most general aspect this invention
comprises a regeneratable catalytic composition comprising
a support having associated therewith a non-gaseous, non-
fluid substance capable of retaining and providing
reactive oxygen for reaction with oxidizable substances
brought into contact with such composition, and thereafter
capable of being replenished with.reactive oxygen by
exposure to a source of oxygen. As such the invention may
be characterized as a "regeneratable solid peroxide" -
type of composition, and includes methods by which such
composition may be employed
More particularly, this invention comprises a
specially prepared bed for absorbing sulphur compounds,
and particularly hydrogen sulphide or oxides of sulphur
from a fluid stream and subsequently decomposing such
compounds into elemental sulphur. This same bed may be
used to absorb oxides of nitrogen, and particuarly
nitrogen peroxide but excluding nitrous oxide, and absorb
as well carbon dioxide from a gas stream for subsequent
separate recovery.
A suitable bed for treating non-oxide compounds of
sulphur comprises a support adapted to accommodate or
absorb such non-oxide sulphur compounds therein, and
particularly hydrogen sulphide, which support contains an
alkali metal sulphide or selenide together with a sulphide
or sulphides, (or selenide/sj of metals showing polyvalent
and/or amphoteric character deposited therein, and has
been rendered thereby capable of providing internally
available "reactive oxygen", e.g. having peroxide-like
characteristics, after exposure to a source of oxygen,
The use of "and/or" in the above discussion, and
throughout this disclosure, is to be taken in its non-
exclusory sense. Thus, a mixture of both amphoteric and
polyvalent compounds may be used in place of either alone,
and a metal which is both amphoteric and polyvalent is
~0 intended to be included by this expression.
WO 92/20621 PC'd'/CA91/iD0160
20'8'~~~~ _
The reference to "reactive oxygen" is intended to
refer to oxygen in an elevated energy state whereby the
oxygen is available to react with the non-sulphur
component of the compounds being treated in some cases
05 even at ambient temperatures so as to release sulphur.
Amphateric metals are those metals which show a
capacity to react both with acids and bases.
Examples of amphoteric or polyvalent metal
sulphides ar selenides suitable for use in this invention
l0 include, amongst others, sulphides or selenides of metals
from the group consisting of zinc, manganese, 9.ron,
copper, cobalt, aluminum, vanadium, molybdenum, tin and
nickel as well as mixtures thereof. Examples of alkali
metals suitable for use in this invention include lithium,
15 potassium, sodium, rubidium and cesium, as well as
mixtures thereof.
A bed so constituted and suitably conditioned
according to this invention is also adapted to remove and
decompose sulphur compounds such as carbonyl sulphide,
20 carbon disulphide, mono and dialkyl sulphides, alkyl-type
disulphides, and thiophenes from a gas or liquid stream by ,
contacting such a stream with the aforesaid bed, at
ambient temperatures. This same bed is capable of~
absorbing oxygen-containing compounds to provide reactive
25 oxygen. Suitable compounds for this effect are sulphur
dioxide, sulphur, trioxide and nitrogen oxides, including
nitrogen trioxide, nitrogen peroxide and nitrogen
pentoxide but excluding nitrous oxide, i.e. the "higher"
oxides of nitrogen.
30 One method of preparing the bed is by:
(a) preparing in aqueous solution a mixture of an
alkali metal salt and a polyvalent and/or
aanphoteric metal salt;
(b) impregnating a support with the mixture described
35 in (a) above;
(c) drying the support after it has been so
impregnated;
(d) sulphiding (or seleniding) the impregnated support
at ambient ar higher temperatures by exposing it
40 to a gas stream containing a reactive sulphur (or
selenide) compound such as hydrogen sulphide,
wo ~zizobz a
Pcric~~~ioomo
pai=:~,
.. - 5 -
carbonyl sulphide or carbon disulphide, or their
selenide equivalents, which has the effect of
converting the metal and alkali salts to sulphides
or selenides;
(e) heating the impregnated support at an elevated
temperature to drive off excess sulphur, or
selenium so as to thereby form the bed in its pre-
oxygenated form; and then,
(f) exposing the bed to a source of oxygen such as, by
way of example, to atmospheric oxygen or an
oxygen-containing compound such as sulphur dioxide
or nitrogen peroxide, whereby reactive oxygen
becomes available within the. bed and thereby
. create the bed in its oxygen-activated farm,
This invention further comprises the production of
elemental sulphur by the method of exposing, at a
temperature below the vaporization point of sulphur, a
gas stream containing non oxide compounds of sulphur, and
particularly for example hydrogen sulphide, to the
oxygenated bed and then regenerating the bed. The be
d is
regenerated by first aPPlYing heat at a predetermined
elevated temperature or temperatures (such as in the ra
nge
of 250oC to 600oC) bed in the presence of a substantially
non-reactive sweep gas. This will drive off any residues
of the oxidized non-sulphur component of the sulphur
compound, this being in the case of hydrogen sulphide
water, and elemental sulphur thus purging the bed
of these
substances. The regeneration process is then completed
and the bed reconditioned by exposure of such bed to an
unreactive sweep gas containing a source of oxygen.
optionally, a source of o<<cygen may also be provided duri
ng
the initial purging step either as an alter
native to
subsequent treatment with oxygen, or in addition.
The amount of oxygen provided with the sweep gas
in the final step may range from a stoichiometric amo
unt
necessary to oxidize the sulphur compound to be
subsequently treated and release elemental sulphur, up to
. a concentration of about 25~, although this is not
necessarily limiting in all cases. Tn certain cases
~0 excess oxygen and highly oxidizing agents such as
hydrogen
WO 92/20621 PC1'/CA91/00160
- ,..°,
peroxide must be avoided to prevent damage to the bed.
This invention further comprises the method by
which the oxides of sulphur, particularly sulphur dioxide
and the oxides of nitrogen, particularly nitrogen peroxide
but excluding nitrous oxide, are removed from a gas stream.
This is arranged by permitting these compositions to be
absorbed within and impregnate a bed comprised of a
microporous support which contains an~~alkali metal
sulphide or selenide, and a sulphide'or selenide of metals
showing polyvalent and/or amphoteric character, which bed
has been depleted of reactive oxygen by exposure to a
reducing gas, preferably hydrogen sulphide. The bed, so
impregnated, is then exposed to a.reducing gas, such as to
a stream of hydrogen sulphide, whereby the absorbed
sulphur dioxide and hydrogen sulphide are converted to
water and elemental sulphur, and the nitrogen peroxide and
other nitrogen oxides are converted to nitric oxide.
These products are then purged from the bed by heating the
bed in the presence of a sweep gas, thus returning the bed
to a condition whereby it is ready to repeat the cycle.
By a further feature of the invention, the bed
containing the above referenced activating ingredients,
when depleted of internally available reactive oxygen, is
capable of absorbing at ambient temperatures quantities of
carbon dioxide. The absorption of carbon dioxide can be
carried-out either separately or in conjunction with the
absorption of the other referenced oxides. Once carbon
dioxide has been absorbed, it can be released and
recovered by heating the bed.
These and further features of the invention and
its various aspects will be apparent from the description
of the examples and test results set forth in the
following.
Summary of the Figures
Figure 1 is a graph showing the effect of
temperature on the rate of desorption of hydrogen sulphide
from a series of sample catalytic beds which have been
saturated with hydrogen sulphide.
Figure 2 shows the capacity of a bed according to
the invention to become loaded with hydrogen sulphide and
WO 92/20b21 ~ ~ ~ ~ ~ ~ ~ PCT/CA91 /001 g0
-
sulphur dioxide as a function of pressure.
Characterization o.f the Catalyst within the Bed
The active catalyst within 'the bed that provides
reactive oxygen is believed to be characterized by a
chemical having as its constituents a complex containing
the combination of an amphoteric and/or polyvalent metal
(hereinafter referred to as the "metal"), an alkali metal,
(hereinafter referred to as the "alkali"), sulphur or
selenium and the capacity to retain an active oxygen-
lU containing moiety that contains an available reactive
oxygen group. This complex should preferably be formed
within a microporous support having a relatively high
surface area and a microporosity adapted to receive the
sulphur or oxide compound to be decomposed.
Alumina is considered a preferred support because
of its high surface area. Also, it is believed, without
limiting the invention as demonstrated, 'that alkali metal
incorporated into the support to form the active complex
will react with alumina to form an alkali aluminate and
facilitate bonding of the active complex to the carrier.
Alumina may thereby provide an etchable substrate upon
which active sites may be more readily formed.
The process of solvent extraction using methylene
chloride, when applied to an activated catalyst containing
manganese and potassium sulphides on alumina (Alcoa ~S-
lU0), showed the following extracted constituents:
free manganous sulphide - 51~ (by weight)
free potassium sulphide - 18~
other constituents including
3o potassium aluminate and - 31~
potassium hydroxide
An attempt to utilize methanol on the same catalyst
produced inconclusive results as the constituents were
apparently modified by the methanol as a solvent (perhaps
by hydrolysis of the manganous sulphide) as was indicated
by a change in colour of the solution from green to brown
shortly after extraction.
Tt has been found 'that 'the catalyst is capable of
decomposing a small poxtion of absorbed hydrogen sulphide
without the addition of oxygen during the decomposition
WO 92/20621
P~,T/CA91 /00160 '
- a;. ~.,
heating phase. The activity of the catalyst under such
conditions, however, declines rapidly. It is believed
that the catalyst is intrinsically capable of supplying
small amounts of oxygen, but that this capacity is rapidly
depleted. This belief is supported by the observation
that exposure of the catalyst to a reducing atmosphere
causes catalytic decompositianal activity to drop to
virtually zero.
The provision of oxygen to the catalytic bed,
either while decomposition is occurring or upon
regeneration of the catalytic bed has been found necessary
to preserve or restore the activity of the catalyst. Thus
while oxygen may be consumed in the decomposition cycle,
it is readily restorable by exposure of the catal~,' t
thereafter to a source of oxygen in either molecu:ar or
compound foran.
The ability of a microporous support, impregnated
with the components which form the active catalyst, to
absorb certain oxygen compounds has a separate utility. A
bed, prepared in accordance with the invention, will
absorb not only molecular oxygen, but else sulphur
dioxide, sulphur trioxide and nitrogen oxides, excluding
nitric oxide. All of these compounds axe capable~of
producing the reactive oxygen which is characteristic of
the invention.
This ability of the catalyst to become oxygen-
activated with such compounds allows a catalytic bed,
' prepared in accordance with the invention, to be used to
absorb such compounds from flue gas. The bed, once
saturated, may then be purged of such compounds by
exposure to hydrogen sulphide, followed by heating in the
presence of a sweep gas. In the case of sulphur dioxide,
this compound is decomposed into water and elemental
sulphur. Thus a major pollutant in flue gas can be
effectively removed from flue gas and converted to a
valuable commodity.
Preparation of the Catalytic Bed - Method 1
Catalytic beds were prepared by two alternate
methods. The first method commenced by dissolving a
WO 92/20621 c~ PCT/CA91/~D0160
_ g
predetermined amount of the alkali sulphide (sodium or
potassium) in water sufficient to form the ultimate
desired loading on the support and optionally boiling the
solution. To this solution a molar equivalent amount of
an amphoteric and/or polyvalent metal sulphide was added
and the solution was boiled again until the volume was
reduced to a point short of saturation. Then the support
(generally in the form of Alcoa alumina spheres, #S-100)
which had been dried by being heated to 250oC for 4 hours
l0 was added to the hot solution and mixed until all the
solution was absoxbed into the support. The partially
prepared catalytic bed was then dried (using a nitrogen
gas flow at 400oC) and cooled. The catalytic bed was then
sulphided by exposure to a stream of 10~ hydrogen sulphide
in nitrogen or methane at ambient conditions until
hydrogen sulphide was detected in the effluent and for at
least one hour thereafter. It was then purged of excess
sulphur by heating in a nitrogen gas flow at 400-500°C for
a period of 0.5 to 1.0 hours to drive off free sulphur.
The partially prepared catalytic bed can also be
sulphided by exposure first to a stream of l0~ hydrogen
sulphide in nitrogen or methane at 400°-500°C for 4 hours
and then to a stream of nitrogen or methane at 400°-500oC
to remove any excess sulphur. Some tests were run in
which the conditioning gas was a 50/50 mixture of hydrogen
sulphide and hydrogen and the active metal in the catalyst
was manganese. This change in the nature of the
conditioning gas considerably reduced its activity for the
sample catalyst so prepared.
Preparation of the Catalyst - Method 2
A second method of preparing the catalytic bed was
as follows. A sulphate, chloride or nitrate of a
polyvalent and/or amphoteric metal was dissolved in an
aqueous solution. The mixture was then heated to ensure
rapid dissolution. (This, as above, is considered an
optional step.)
The solution was then impregnated on a previously
dried alumina support (Alcoa S-100, 1/4 in. spheres) and
the impregnated support dried.
wo 9zizo~z~ Ycrica,~~ioo~bo
2~8'~'~6~
'10 -
A molar equivalent or greater amount of an alkali
metal sulphide was then prepared in an aqueous solution
arid impregnated on the support. Again, heating was
optionally employed to effect rapid dissolution.
The impregnated support was 'then heated to a
temperature of 125°C for a period of 2 hours in order to
fix the active ingredients within the support. This
was followed by a washing of the impregnated support with
water until all available alkali sulphate, chloride or
nitrate had been flushed from the support. The
impregnated support was then dried at 125°C.
It is believed that at this stage most of the
sulphate, chloride or nitrate originally impregnated has
become converted to a sulphide of the amphoteric and/or
polyvalent metal. The available sulphate, chloride or
nitrate salts of the alkali metal were washed out of the
support because they were not believed to contribute to
the activity of the catalyst and were thought to reduce
the availability of active sites within the support. The
catalyst could be prepared without this step and still be
capable of producing some decomposition of hydrogen
sulphide. However, it is believed that the catalyst would .
generally show reduced activity without this step.
A stoichiometric amount of the alkali metal
sulphide was then prepared in an aqueous solution and
impregnated on the carrier a second time. The impregnated
support was finally dried at 125°C, and sulphided and
purged of excess sulphur as described in Method 1.
Preparation of the Catalytic Bed - Further Alternate Methods
The above process has been carried-out with a
variety of amphoteric and/or polyvalent metals in the form
of sulphates, chlorides or nitrates and, it is believed,
may be carried-out with any soluble salts of such metals
including zinc, iron, vanadium, copper, nickel,
molybdenum, aluminum and manganese. zt is believed that
an active catalyst would be produced when these methods
are carried out with all amphoteric and/or polyvalent
metals. Tt is further believed that these methods would
be effective in producing an active catalyst whether
sulphide or selenide salts of all amphoteric and/or
WO 92/20621 ~ ~ g ~ ~ ~ ~ PCT/CA91/001b0
- 11 -
polyvalent metals are used. Where less soluble compounds
are employed, it may be appropriate to employ a basic
aqueous solution in order to facilitate dissolution. A
sufficiently basic solution can be created by adding
alkali hydroxide to the solution of the amphoteric and/or
polyvalent metal salt and boiling this mixture,
Method 2 described above has been followed using
either sodium or potassium as the alkali element. Tt is
believed that lithium, rubidium or cesium sulphides may
also be substituted for the elements sodium or potassium,
and still form an active catalyst using either methods.
It is further believed that selenium may be
substituted for the sulphur in the alkali sulphide arid
still produce an active catalyst.
Based on sample tests, a satisfactory standard of
performance for the catalyst in terms of both absorptive
and decomposing capacity can be obtained with an
approximate 1:1 molar ratio between the metal and alkali
components, and a similar 1:1 molar ratio where an alkali
hydroxide is additionally employed.
Absorptive capacity for hydrogen sulphide is
maximized for various metal sulphides at different levels
of impregnation of the support. For example, this occurs
between the 0.5~ to 2.5~ loading (by weight) range for a
catalyst incorporating a zinc sulphide/sodium sulphide
mixture deposited by Method 1 on the Alcoa carrier (S-100
spheres).
Preparation of the Catalytic Bed - Activation with Oxyaen
The bed may be activated in conjunction with the
sulphiding steps by exposing it, as an optional first
step, at ambient or higher temperatures to an unreactive
gas containing hydrogen sulphide, followed by heat
treatment in an unreactive sweep gas at a temperature of
250oC-700oC containing an amount of oxygen as referenced
above. Alternately, after treatment with the sweep gas at
elevated temperatures the bed may be exposed to oxygen at
temperatures down to ambient conditions.
"Unreactive'° is used here and throughout in the
sense of~a gas that does not substantially react in this
4o system.
WO 92/20621 PCTJCA91/00160
_ 12 _
~~~~~65
It is most desirable that the activating gas
streams not contain appreciable amounts of compounds or
elements, such as hydrogen, which will have a major
reductive effect on the activity of the catalyst. It is
also important for the trea~tment~of non-oxide compounds of
sulphur that the catalyst be exposed by the conclusion of
the conditioning process to sufficient oxygen to ensure
that reactive oxygen will be available within the catalyst
to render it activated.
The source of oxygen may be either atmospheric or
molecular oxygen, or may be a compound such as sulphur
dioxide or nitrogen peroxide. All three of these sources
have been found to produce, within the catalytic bed, the
reactive oxygen which is a characteristic of the invention.
Sweetening Decomposition Purainq and
Reactivation Procedures
The procedure followed to verify and quantify the
production of sulphur from hydrogen sulphide was as
follows.
A sample of a catalytic bed that had been purged
of free sulphur and hydrogen sulphide by regenerating it
at 400°C under an unreactive sweep gas (nitrogen or
methane) and then activated by exposure to oxygen was
weighed while placed in a reaction tube. A measured
volume of unreactive gas containing a known percentage of
hydrogen sulphide was then passed over the catalyst bed at
a specific temperature, usually ambient, to remove the
hydrogen sulphide from the gas stream. This was
designated as the "sweetening" cycle. The length of
exposure was either that required to produce an indication
of hydrogen sulphide "breakthrough" at the exit end (as
measured by the blackening of standardized lead acetate
paper, or other standard methods), or some lesser period
of time. A run to breakthrough was said to have saturated
the bed. A run carried to a point short of saturation was
designated as a "partial run".
The catalytic bed in its tube was then weighed to
determine_ either the saturation loading of the bed, or the
partial loading of the bed, in terms of its absorption of
hydrogen sulphide.
1'CT/CA91 /00160
WO 92/20621
_ 13 _
Throughout all experiments, the catalytic beds
utilizing molecular sieves or alumina supports showed a
capacity in the foregoing sweetening phase of maintaining
the hydrogen sulphide level in the out-flowing stream
below the measurable threshold vis, 1 part per mi~.llion
prior to breakthrough.
The catalytic bed in its reaction tube was then
put through the purging phase by exposing the bed to an
unreactive sweep gas (nitrogen ar methane) at a specific
temperature above the vapourization point for elemental
sulphur for. a period of time. The bed may then be
reactivated by exposing it to a source of oxygen. This
may be dane, for example, by utilizing a sweep gas
containing oxygen at levels of 0.01 to 250. Oxygen may
also be supplied in the form of sulphur dioxide or
nitrogen peroxide. Alternately, reactivation by exposure
to a source of oxygen may be effected separately, after
the purging phase is complete.
Tt has been found that with certain metals, such
as manganese, that the catalytic bed deteriorates if
exposed to excessive levels of oxygen, e.g. over 10~.
This may, it is believed, be due to the formation of a
sulphate. The catalyst in such a case was restored to
activity on re~-exposure to hydrogen sulphide. However, it
is believed that the concentration of oxygen should
preferably be limited in order to avoid such deleterious
effects.
The sweep gas exiting the catalytic bed was caused
to pass through a portion of the reaction tube that was
maintained at room temperature. During this process, when
carried out with the bed at temperatures over about 250oC
- 300°C, sulphur consistently condensed on the inside
walls of a cooler, exit portion of the reaction tube in a
condensation zone. Sample tests with glasswool placed
downstream of such deposits indicated that further sulphur
could not be collected by condensation from the cooled
exiting gas stream beyond the condensation zone.
A further procedure followed in some experiments
was to collect the exiting sweep gas during the
regeneration step and then determine its hydrogen sulphide
WO 92/20621 PC.T/CA91/00160
- 14 - ,.:.:..,
concentrations by gas chromotography. As further
discussed below, little or no hydrogen sulphide was
detected in the regeneration phase when the catalyst bed
was only partially loaded with hydrogen sulphide, well
below the saturation level fox the bed. For higher
loadings and approaching saturatian, much more hydrogen
sulphide was detected in the regeneration stage of
treatment.
After sulphur ceased to. be forming further within
the cooler portion of the .reaction tube, the tube and bed
were reweighed. Comparisons of this weight with the
weight of the tube following sweetening showed that
virtually all of the sulphur remained in the system, up to
this point. Then heat was applied to the outside portion
of the reaction tube where sulphur had deposited and the
sweep gas flow was maintained. This procedure was
continued until all of the sulphur in the reaction tube
had been vapourized and carried out of the tube. The
reaction tube and bed were then reweighed.
The catalyst bed, for purposes of experimental
certainty, was then put through a super--purging phase by
performing the previous procedure at 400-500°C for 1-2
hours. This step was shown through tests at higher
temperatures to be capable of completely purging the
catalyst bed of remaining traces of free sulphur and
residual hydrogen sulphide.
The inclusion of amounts of oxygen in the sweep
gas during the super-purging phase was not found to be
essential if it had been previously present as part of the
earlier treatment. Apparently, if sufficient oxygen is
available during the normal purging phase, then the
catalyst is reactivated. However, no deleterious effects
occurred where oxygen was present on the super-purging
phase as well. If insufficient oxygen was present during
the purging or super--purging phases, then oxygen should be
supplied to the bed as a further step, which may be
carried out at room temperature.
Oxygen may be supplied to the bed either in its
molecular form, or in a compound such as sulphur dioxide
or nitrogen peroxide. Sulphur dioxide has been found to
WO 92/20621 ~ ? ~ ~ PCT/CA91 /00160
- 15 -
produce a much higher deposition of reactive oxygen within
the catalyst. The use of sulphur dioxide also increases
the absorptive capacity of the bed with respect to
hydrogen sulphide.
The exposure of alumina to sulphur dioxide would
normally be expected to result in the production of
aluminum sulphite. :Lf oxygen is present, as well, then
aluminum sulphate will likely farm. Where, however,
alumina has been 'treated by the deposition therein of the
combination of sulphide or selenide salts of amphoteric or
polyvalent metals combined with sulphite or selenide salts
of alkali metals, the tendency of the alumina to form
aluminum sulphite or sulphate is believed to be
significantly reduced.
From the foregoing procedures calculations were
made to determine the extent to which the hydrogen
sulphide was converted to sulphur. The quantity of
hydrogen sulphide absorbed in the catalyst bed was
calculated based both on the gas flow rate, and on the
2o gain in weight of the bed and tube during the sweetening
phase. The quantity of sulphur produced was obtained from
the heat-vaporization procedure. The actual quantity of
hydrogen sulphide decomposed was also determined by the
difference between the volume of hydrogen sulphide
absorbed by the catalyst, and the volume of hydrogen
sulphide collected by a gas bag during the regeneration.
Of these methods, the mass of sulphur vaporized off the
interior of the reaction tube was taken as the more
reliable measure of the minimum decomposition that had
occurred.
Absorption of Sulphur Dioxide and Other Oxygen Compounds
The procedure of utilizing the bed first to absorb
hydrogen sulphide followed by reactivation with sulphur
dioxide may be reversed or shifted in order. Thus, where
it is desired to remove sulphur dioxide from a gas stream
the bed is first purged of sulphur dioxide by exposure to
hydrogen sulphide, then purged of sulphur by heating in
the presence of an oxygen-free sweep gas. So prepared,
the bed will then readily absorb sulphur dioxide to the
limit of saturation. Once the bed has been saturated with
WO 92/20621 fC'T/CA91/00160
- 1~ -
sulphur dioxide, it may be again exposed to hydrogen
sulphide to purge it of the sulphur and water that is
thereby formed.
The bed will similarly absorb sulphur trioxide,
which can be converted to produce sulphur by the same
steps.
It has been found that when sulphur dioxide is
used as the source for oxygen, it is relatively
tenaciously contained within alumina-type supports. This
enables an activated bed to be prepared in one location,
and then transported to another. Similarly where the bed
is only partially saturated with hydrogen sulphide in the
sweetening cycle, the bed material is readily
transportable.
The bed, suitably depleted of oxygen has an
affinity to absorb not only the oxides of sulphur, but
also nitrogen peroxide and similar higher oxides (but not
nitrous oxide), and carbon dioxide. Further, the bed has
the capability of absorbing all of these classes of oxides
simultaneously.
Absorption of Nitrogen Peroxide
The source of oxygen may also be nitrogen
peroxide. This is a component often found in the products
of combustion and in flue gases.
When nitrogen peroxide is used as the source of
oxygen, subsequent exposure of the bed to hydrogen
sulphide results in the production of elemental sulphur,
water and nitric oxide - NO. When the catalyst is purged
of sulphur by heating, the nitric oxide evolves. This
nitric oxide can then be trapped downstream, after air-
axidation to nitrogen peroxide and then used for other
chemical reactions, such as the preparaton of nitrates.
The advantage of this cycle is that the bed can be
employed to first absorb the nitrogen peroxide, separating
it from a flue gas stream for subsequent recovery.
Combined Absorption of Oxides of Sulphur and Nitrogen
It has also been found that the catalyst can be
activated by mixtures of N02 and S02 in an air stream, at
ambient temperatures. When this catalyst is treated with
a stream of H2S and subsequently heated, sulphur, water
~vo 9zizo~z~ ~ ~ ~ '~ ~ ~ 5 Pcri~A~noomo
17
and nitric oxide all distill off.
Tests based on the activation of a 2(Na2S)/ZnS
form of catalyst deposited in S-100 Alcoa spheres (at to
loading, by weight) show a capacity for such a bed to
absorb up to 6~ by weight of sulphur dioxide, 9.1% by
weight of nitrogen peroxide and 5~ of carbon dioxide,
simultaneously. The gas stream used for this test
contained 10-12~ of C02; 4-6~ of 02; 1000-2000 ppm of S02
and 100-400 ppm of N02. These ratios are typical for a
flue gas. The absorption capacities for each of these
components do not appear to be substantially cross-
related.
Absorption of Carbon Dioxide
Another environmentally-undesirable component of
stack gases in carbon dioxide, because of the so-called
Greenhouse Effect. Tests show that catalysts prepared
according to the invention absorb carbon dioxide strongly,
without in any way affecting their ability to take up the
oxides of nitrogen and sulphur.
Specifically, the absorption of carbon dioxide has .
been demonstrated by tests effected with the catalyst in
its sodium sulphide/zinc sulphide form, with the sodium
sulphide to zinc sulphide ratio being 2 moles of sodium
sulphide to 1 mole of zinc sulphide. The catalyst was
coated one percent on alumina. From a stream containing
C02, 502, and No2, we have found that one metric tonne of
the catalyst takes up 60 kg of C02, 60 kg of 502, and 91
kg of N02.
The catalyst will retain 60 kg of C02 from the
stream at saturation, and continues to absorb S02
until 60 kg of this compound has been removed from the
stream. The absorption of N02 then continues until
91 kg of this oxide had been recovered. To prevent any
S02 from escaping under these conditions, an additional
catalyst bed would be placed downstream to strip out any
S02 leaving the first catalyst chamber by desorption.
When this exposed catalyst is treated with
hydrogen sulphide, the oxides of sulphur and nitrogen
react with the absorbed hydrogen sulphide, and convert the
sulphur oxides to sulphur and water. After reaction is
wo ~zizosz~ ~cricA9~ioo~bo
- 18 - ':y..:
complete, the catalyst contains elemental sulphur, nitric
oxide, carbon dioxide and water. Heat treatment, at
400°C, drives off the sulphur, nitric oxide, and carbon
dioxide. Each may be separately recovered downstream.
After the absorption stage is completed and the
absorbed components have been treated with hydrogen
sulphide, the spent catalyst is heated. Carbon dioxide
C02 will be the first substance to desorb, and can be
trapped by many standard methods. As the temperature
rises nitric oxide, NO will next come off, which
substance can be converted in air to N02. This is an
important a.ndustrial chemical when so isolated. It can be
converted readily to nitrates, which axe of importance for
the fertilizer industry, for example. Finally, a;:: the
catalyst temperature approaches 300oC, elemental ::sulphur
will begin to distill off as another important industrial
product. The bed may be re-exposed to a reducing gas.
The catalyst will then be ready for another absorption
cycle in the stack gas stream.
Desorption Runs - Effects of Physical Absort~tion of H2
Returning to the absorption of hydrogen sulphides,
from the results of the tests performed, it was determined
that hydrogen sulphide was believed to be both physically
and chemically absorbed within alumina-based catalysts.
Tests on a blank alumina support, containing no active
ingredients, indicated that virtually all absorbed
hydrogen sulphide could be driven out of such a support by
heating it to 350oC under a sweep gas for a period of time
of 30 minutes. Supports that had been impregnated with
ingredients to form the catalyst showed a tendency not to
have released as much hydrogen sulphide at that
temperature as did the blank support.
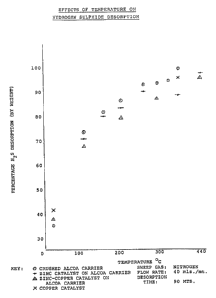
Figure 1 shows this effect in which a blank Alcoa
(S-100) alumina support is compared with catalysts
prepared by Method 1 with Zinc and Potassium sulphide;
Zinc, Copper and Potassium sulphides, and Copper and
Potassium sulphides all on the same type of S-100 support.
All beds-were loaded to saturation and then treated in the
WO 92/20621 ~~ ~ ~ ~ PCT/CA91/00160
- 19
sweetening phase for 90 minutes at various temperatures.
Figure 1 shows the percentage of the hydrogen sulphide
evolved, as a function of temperature after heating for 90
minutes at various temperatures.
Table 1 summarizes the~da~ta depicted in Figure 1
and adds the accumulated percent decomposition obtained
both after the 90 minute heating at a constant temperature
and after the final regeneration at 40ooC. These
percentages are based in both cases on conversion of
ZO sulphur, being the mass of sulphur vaporized divided by
the mass of sulphur available in the quantity of hydrogen
sulphide originally absorbed.
Table l
Effect of Heating at Various Temperature on
Hydrogen Sulphide Desorption and Decomposition
for Saturated Catalyst/Beds
Catalyst Heating Temp % Desorption % Sulphur Conversion
/Bed (oC) H2S After Total after
After Heatin Heatin Re eneration
Blan>C
Crushed
Alcoa
Support
$S-100 lBoC 35 __
100 73 __ -_
-_
150 82 __ -_
200 83 __ -_
250 93 __ --
300 93 __
325 , g4 __ --
--
350 100 __
Zinc -
Sodium 18 42
-- 1.6
Sulphides 100
70 -- 7.8
150 80
-- 10.3
200 83
-_ 17.2
250 90 1.6 10.2
300 87 3.3 10
2
350 g8 .
7
0
. 7.5
400 93 6.1
6.1
wo 9zizo~z~ Pcric~~noo~bo
_ 20 _ ,.
(able 1 Continued)
Zinc
Copper - 18 n/a -- 2.6
Sodium 68 ~ _-- 14.7
Sulphides 200 79 __ 9.8
300 81 3.2 10.8
350 94 5,3 6.3
400 96 3.2 3.2
Copper-
Sodium 18 42.I. __ g,2
Sulphides 350 95.7 1.1 1.5
(Heating Time: 90 minutes)
Partial Runs
The foregoing data on saturated catalyst beds give
a clear indication that decomposition is occurring
by the
quantities of elemental sulphur that are produced.
However, the decomposition effect is being masked~by
the
hydrogen sulphide that is being physically absorbed,
and then being desorbed without decomposing. The masking
effect of physically absorbed hydrogen sulphide can
be
., largely eliminated by exposing the catalyst to hydrogen
sulphide streams for periods of time less than that
necessary to saturate the bed. These are called 'partial
runs', zn such partial runs, the amount of hydrogen
sulphide evolved on regeneration was substantially
reduced. Correspondingly, higher percentage figures
for
the amount of available sulphur in the hydrogen sulphide
converted to elemental sulphur were obtained.
The catalyst, when used in association with
microporous supports such as alumina or zeolite, rapidly
absorbs hydragen sulphide. It may be that the rapidity
with which the hydrogen sulphide is absorbed permits
the
catalytic bed, at suitable flow rates, to saturate
progressively when e~cposed to a sour gas stream.
Tf the
sweetening phase is terminated with only a portion
of the
bed exposed (and saturated) with hydrogen sulphide,
then,
as heat is applied to the bed in the presence of a
sweep
gas absorbed hydrogen sulphide that may be desorbed
is
swept into a region of the bed containing unexposed
w0 92~zos11 ~ ~ ~ r~ ~~ ~ 5 PCT/CA91 /00160
- 2I -
catalyst. Consequently, a bed that is partially loaded to
saturation along only a portion of its length would be
capable, in the separation phase, it is believed, of
exposing virtually all of the hydrogen sulphide to
chemical-absorption leading to decomposition.
Thus, on whatever basis, it has been found that
with appropriately chosen partial loadings, it is possible
to obtain virtually 100% dissociation of the hydrogen
sulphide.
Tested Catalyst Variants
The dissociative capacity of different catalyst
formulations were tested and some of the results obtained
were as set out in Tables 2 and 3.
TABLE 2
CATALYST LOADING % SULPHUR CONVERTED
(including method (gms/100 gms (cumulative, at
of preparation) and as a % 400oC)
of saturation)
Zn-K-1C-1 0.6(20%) >90%
Zn-K-2W-1 0.7(23%) >80%
Cu-K-1W-2 1.4(100%) >70%
Mn-K-1C-1 0.6(20%) >90%
(Catalyst desig nation code:
Zn - K - 1C - 1
main alkali carrier: method of
amphoteric metal 1 - Alcoa preparation
or polyvalent 2 - ICI 1 - method
l
metal c - crushed 2 - method
2
w - whole using a
sulphate)
The data in Table 2 provides quantitative figures on the
extent of decomposition of hydrogen sulphide obtained,
stated in terms of the percent conversion to sulphur.
Table 3 lists combinations of further ingredients
all found to produce non-quantified but definite amounts
of elemental sulphur upon the consecutive exposure of the
WO 92/20621 PCT/CA91/00760
-
22 -
catalytic bed to a 10% hydrogen sulphide/90% nitrogen gas
stream at ambient temperature 18°C), followed by
regeneration of the catalyst at temperatures ranging from
350-400oC as previously described. A11 runs were carried
out using as a support the Alcoa alumina carrier No. S-
100. All of 'the samples listed in Table 3 were prepared
from sulphides in accordance with the procedure of Method
1.
The column entitled "Absarptive Capacity°'
indicates the percentage ratio of mass of sulphur absorbed
to the mass of catalyst, at 'the point where the catalyst
bed ceased to fully absorb further hydrogen sulphide (as
tested by the darkening of lead acetate paper at the
column exit).
TABLE 3
Metal Alkali Elemental Absorptive Capacity
Metal Sulphur (% sulphur loaded
Detected ner mass of catal~stl
Zinc Sodium yes 2.4
Zinc Potassium Yes 1.4
Iron Sodium Yes 2.4
Vanadim Sodium Yes 2.3
Copper (I) Sodium Yes 2rg
Copper (II) Sodium Yes 2.0
Copper (II)Sodium* Yes 2~4
Copper (II) Potassium Yes 2.2
Nickel Sodium Yes 2.9
Molybdenum Sodium Yes 2.3
Aluminum Sodium yes 2.7
Manganese Sodium Yes 2.g
Manganese Potassium yes 2.3
Cobalt Sodium Yes n/a
*2 moles of sadium
Tested Catalyst Variants -- Mixed Catalysts
A number of combined catalysts incorporating two
or three amphoteric and/or polyvalent metals have been
tested. Table 4 sets out tl~e absorptive capacity at room
WO 92/20621 ~ TCT/CA91/00760
- 23 -
temperature for all such catalysts based on the alumina
support, Alcoa No. S-I00. In all cases the catalyst was
prepared by Method 1 using a sulphide of the metal as the
initial salt. All components were incorporated into the
v 5 support in equal molar ratios.
TA7iLE n
Metal Components Alkali Absorptive Capacity
Component
(gms sulphur equivalent
' from ~I2S in 100
gms
catalyst)
l0
Iran & Zinc Sodium Sulphide 2.3
Irn, Copper & Sodium sulphide and
Zinc Sodium hydroxide2.2
Manganese & Zinc Sodium sulphide and
15 Sodium hydroxide
2.0
Manganese & Zinc Sndium sulphide 2.3
Manganese & Nickel Potassium sulphide 1.5
Manganese &
Molybdenum Potassium sulphide 1.7
20 Iron & Zinc Potassium~sulphide 1.2
In all of the cases listed in Table 4, sulphur was
observed to be evolved when the catalysts were regenerated
at a temperature of 400oC.
Description of Examples Usin~x Sulphur Dioxide
25 A two-to-one molar ratio of sodium sulphide to
zinc sulphide was deposited on S-100 Alcoa Alumina
Spheres. The amount of such components deposited was set,
for two different samples, at 1~ and 2~ by weight of the
final loaded support.
30 ane hundred grams each of the two classes of
catalyzed support, along with pure, crushed S-100 spheres
were then progressively loaded with sulphur dioxide at
roam temperature by exposure to a stream of 18~
WO 92/20621 PCT/~A91 /00160
24 -
concentration by volume of S02 in nitrogen; and then
exposed to a stream of methane containing 10% by volume of
hydrogen sulphide. The amounts of sulphur-equivalent
absorbed and then converted to sulphur are shown in Table
5 where a comparison to a blank alumina support is also
provided.
TABLE 5
So2 & H2S Loading and Regeneration Data
aior A1203, 1% and 2~% -- 2 (Na2S) :ZnS
under Saturation conditions
Run S02 H2S Total S % Con-
No. Bed Loading Loading Loading version
1. A12o3 3.3 5.3 8.6 77
(crushed)
2. A1203 3.1 5.3 8.4 72
(crushed)
3. A1203 3.1 5.2 8.3 66
(crushed)
4. A1203 3.4 5.1 8.5 75
(crushed)
i
..,
,, 5. 1% Catalyst 4.2 7.7 11.9 82
6. 1% Catalyst 4.7 6.9 11.6 82
7. 1% Catalyst 4.6 7.3 11.9 79
8. 1% Catalyst 4.6 7.3 11.9 79
9. 1% Catalyst 4.6 7.4 12.0 79
10.2% Catalyst 4.4 7.9 12.3 83
li.2% Catalyst 4.4 7.3 11.7 79
i2.2% Catalyst 4.6 7.2 11.8 78
13.2% Catalyst 4.3 6.5 10.8 80
512and H2S loading figures grams Sulphur per
are of
in
100g of catalyst.
xn order to determine he absorptive
if 't capacity
..; of the catalyzedsupportschangedover time,the 1% loaded
andblank aluminasamples of Table5 were
saturated
by
. 40 exposure to consecutive 1.9% sulphur
' streams dioxide,
of
WO 92/20621 ~ ~ ~'~ '~ 6 ~ 1PCT/~A91/00160
and 6.7% oxygen, both in methane, at room temperature, for
one hour each. These samples were then allowed to stand
at room temperature for 70 hours in sealed moisture-proof
containers.
5 Table 6 shows the data obtained when these aged
samples were exposed to hydrogen sulphide on 'the same
basis as previously. From Table 6 it is apparent that the
bare alumina absorbed a smaller quantity of sulphur
dioxide than in the earlier tests, after exposure to this
10 aging test, but the catalyzed beds was unaffected.
TABLE 6
Loading and Regeneration Data for the S02 Saturated Beds
After Soaking for 70 hrs at Room Temperature
15 Run S02 H2S Total
No. Bed Loading Loading S Loading Conversion
1. A1203 2.7 4.2 6.9 68
2. 1% catalyst 4.8 6.8 11.6 80
During the sweetening runs with beds activated
with S02 it was found that some sulphur dioxide was
evolving and finding its way into the effluent gas. As
much as 28% of the sulphur dioxide would become desorbed
at 150 psi. This is believed to be due to the highly
exothermic character of the reaction of hydrogen sulphide
and sulphur dioxide.
To reduce this effect, tests were run with the
beds only partially saturated with S02 (i.e.: to 75% of
80 capacity). Utilizing beds of catalyst one percent by
weight of 2(Na2S)aZnS on S--100 Alcoa spheres that had been
only partially saturated in this manner, a series of
sweetening and conversion cycles were run at varying
pressures. The results are set out in Table 7.
WO 92/20621 hC I'/CA91 /00160
- 26 -
TABLE 7
H2S Loading As A Function of Pressure For The
Partially S02-Loaded Beds
Run S02 H2S Total %
S
No. Pressure Loading Loading Loading Conversion
1. 150 psi 5.9 10.4 ~ 16.3 85
2. 150 psi 6.0 10.4 16.4 88
3. 80 psi 3.9 8.4 12.3 80
4. 80 psi 3.9 7.7 11,6 81
5. 40 psi 3.6 6.8. 10.4 77
6. 40 psi 3.8 6.6 10.4 77
1n the runs depicted in Table 7, no sulphur
dioxide was evolved until Gust before break-through of the
hydrogen sulphide occurred, and even then only trace
amounts were detected.
Table 7 shows sulphur conversion ratios that are
on the same order as those of Table 5. Further, the
increased absorptive capacity of the catalyzed support
under pressure is also shown.
The actual dependence of absorptive capacity under
a range of pressures was also determined using a 1%
loading of 2Na2S/ZnS deposited on S-100 supports that were
progressively saturated with sulphur dioxide and then'
saturated with hydrogen sulphide, both to the point of
breakthrough. The results are shown in Table 8. These
results are reproduced graphically in Figure 2.
TABLE 8
Loading As A Function of Pressure for
the Catalyst 2Na2S/ZnS
Run S02 H2S
No Pressure Loading Loading Total S Loading
(as % S) (as % S)
1. 14.? psig 4.6 7.3 11.9
2. 54.? psig 5.0 8.1 13.1
3. 94.7 prig 5.9 9.2 15.1
4. 164.7 psig 10.3 9,6 lg_g
WO 92/20621 ~ ~ ~ ~ PCT/CA91/00160
27
If we assume that the reaction occurring in the
catalyst between hydrogen sulphide and sulphur dioxide is
S02 + 2H?S > 3S + 2H20, then, expressed as weight of
sulphur, sulphur dioxide will oxidize twice its weight of
hydrogen sulphide. Thus we see~in Table 8 that above a
pressure of ca 100 psig, the loading of sulphur dioxide is
exceeding that required to oxidize the hydrogen sulphide
absorbed. Consequently, at higher pressures, it is
preferable that the catalyzed support be only partia7.ly
saturated with sulphur dioxide. This will avoid the
evolution of excess sulphur dioxide while still providing
a stoichiometrically sufficient amount of sulphur dioxide
to react with the hydrogen sulphide that can be absorbed.
Throughout the foregoing tests, during the sulphur
purging stags, tests for the presence of hydrogen sulphide
in the sweep gas were made. In the process described
which xelied on the depositing of molecular oxygen within
the catalyst, quantities of hydrogen sulphide were
released at this stage. By the process described herein
of activating the catalyst with sulphur dioxide, the
release of hydrogen sulphide from the catalyst can be
greatly reduced.
In the oxygen-activated process, it is believed
that the activation stage did not produce activated
sulphur sites at all possible locations within the micro--
porous support, to the exclusion of sites capable of
absorbing hydrogen sulphide. Consequently, during the
process of exposing hydrogen sulphide to the catalyst to
effect dissociation, considerable quantities of hydrogen
sulphide became absorbed without becoming dissociated.
In the oxygen-activated process, the catalyst was
cyclically exposed to the steps of being saturated with
hydrogen sulphide, then regenerated by purging it of water
and elemental sulphur (at 350oC), and then reactivated by
exposure to air (at 200oC). Due to the fact that some
hydrogen sulphide was merely absorbed within the catalyst,
this substance became released in the purge cycle,
contaminating the sulphur vapour being released and
causing exfoliation ~f such sulphur. These effects were
due to the presence and release of undecomposed hydrogen
WO 92/20621 PCT/CA9~/00760
~o~~~
2 8 - f:: --
o~ -
sulphide that was able to accumulate within the catalytic
support in the oxygen-activated process.
In the sulphur dioxide-activated process, aCtl.VatlOIl
of the catalyst is effected by exposing the micro-porous
support to sulphur dioxide. It~is believed that this
procedure is more efficient in forming active sites
that
are capable of dissociating hydrogen sulphide. This
greatly reduces the amount of hydrogen sulphide that
is
' absorbed and then released without being dissociated.
ZO When 'the catalyst has been activated by sulphur dioxide
virtually no hydrogen sulphide appears in the regeneration
phase.
The sulphur dioxide activation process relies upon
the formation of a highly reactive sulphite within
the
micro-porous support. To form this sulphite, a metal
must
be present within the support. Water must also be present
to allow the formation of the sulphite and the subsequent
dissociation of hydrogen sulphide.
The sulphur dioxide activated process is capable
of operating with a pure alumina support. The deposition
within this support of an amphoteric or polyvalent
metal
sulphide, together with an alkali sulphide, enhances
both
. the system's capacity to remove hydrogen sulphide~from
a
gas stream, and its efficiency in converting hydrogen
sulphide into sulphur.
With the deposition of a 1% loading by weight of
zinc sulphide and sodium sulphide (in a 2:1 molar ratio)
.r.v; on a micro-porous alumina support, the absorptive capacity
of the catalyst is enhanced 60% over that of pure alumina,
3o and the efficiency of conversion to sulphur is increased
by 50%. Tests have shown that the amount of sulphur
absorbed (in the form of hydrogen sulphide) is increased
from 8% by weight for pure alumina, to 12% by weight
with
the zinc and alkali sulphides present.
The quantitative runs to-date have utilized zinc
sulphide. It is believed that even better performance,
in
terms of absorptive capacity and dissociation efficiency,
will be obtained using manganese sulphide.
WO 92/20621 ~ ~ 1"~C.'T/CA31/00160
_ 29 -
Tests have been carried out with 1~ loading
ratios, by weight, for the metal and alkali sulphides on
an alumina support. It is believed that superior
performance will be obtained with a 2~ loading.
Decomposition of Other Sulphur Compounds
While tests have been carried out mainly on
hydrogen sulphide as the decomposed sulphide, it is
believed that the catalyst will be active in decomposing
organic-sulphur compounds such as carbonyl sulphide,
carbon disulphide, mono and d:ial.kyl sulphides, alkyl-type
disulphides and thiophene. It is also suitable for
removing all of the foregoing from a mixture of more
complex natural gas components in gaseous or liquid phase,
such as from butane or propane, and including, generally,
natural gas liquids.
Supgorts
The principal support used in testing has been
alumina in the form of Alcoa 1/4 or 3/4 inch spheres (#S-
100). Other supports tested for absorptive capacity
include alumina in the foam of Norton 5/16" rings (#6573),
Norton spheres (#6576); CIL Prox-Svers non-uniform
spheres, Davison Chemical molecular sieves (type 13x, 4-8
mesh beads), silica and char. The Alcoa support was
chosen as the preferred carrier due to its high absorptive
capacity, which was due, in turn, to its large surface
area (325m /gm).
The Alcoa support referenced is essentially
alumina that is reported as being in the gamma and
amorphous form. It is not believed that the type of
crystalline form in which the alumina may be found is of
significance to the dissociative capacity of the catalyst.
.Activity has been found where there is aluminum
present in the support. The presence of aluminum in the
support is relevant in that alumina will invariably be
formed. When preparing the catalyst, the alkali metal
WO 92/20621 PCT/C~191/00160
30 - t:."".
will attack the alumina and form alkali aluminate and
species containing available reactive oxygen. Thus the
aluminum-containing supports inherently are capable of
providing active centres necessary to support the activity
of the catalyst. Such supports'also provide an etchable
base upon which actively catalytic sites are thought to be
more likely to form.
Supports were tested for decompasition activity
when aluminum was not present. A distinct but non-
quantified showing of production of elemental sulphur
occurred on repeated cycles Of exposure of an oxygen
activated catalyst formed on a silica support, to a
continuous stream of loo hydrogen sulphide. This was
based upon manganese and sodium as the active metal and
alkali respectively. Due to the reduced surface area of
this latter carrier, only trace amounts of sulphur were
produced, and no quantitative measurements of
decomposition were made. However, this test demonstrated
that it is not essential that the support upon which the
catalyst is based contain aluminum.
The capacity of the support to fully absorb
hydrogen sulphide and/or other sulphur compounds is an
important feature when it is desired to remove all
significant traces of such compounds from a gas stream.
This characteristic is believed to be dominated by the
. support itself. When the production of sulphur is the
primary objective, the efficiency of absorption by the
carrier is less critical. In such cases supports may be
used that do not effect 100 absorption of hydrogen
sulphide prior to saturation.
Improved performance is also anticipated where the
metal and alkali sulphides are formed within the alumina
of the alumina support, rather than just being deposited
on the surface.
wo 9zi2osz~ ~ ~ 8 ~ 2 ~ ~ ~cric~~noomo
- 31 -
Recyclability of the Catalyst
The prepared catalysts were run 'through at least 4
cycles of absorption and regeneration before quantified
tests were carried out on 'them.' These initial cycles were
05 found appropriate to stabilize the catalyst and obtain
relatively consistent results in subsequent tests.
Generally, the activity of the catalyst in terms of its
decomposing capacity increased following these preliminary
recyclings.
No significant decline or loss of activity in
dissociative capacity of the catalyst has been found
despite a number of consecutive absorption and
regeneration cycles so long as replacement oxygen is
available. The absorptive capacity of the catalyst has
been shown to remain relatively unchanged through at least
30-40 cycles of absorption and regeneration.
Effects of Carbon Dioxide, Water and Heavy Hydrocarbons
and Decomposition on Hydrogen Sulphide Absox-tation
When carbon dioxide is present in the gas stream
it does not substantially affect the capacity of the
catalytic bed to absorb hydrogen sulphide, but is itself
absorbed. The presence of absorbed carbon dioxide within
the bed does not significantly affect the decomposition of
hydrogen sulphide.
When water is present in or exposed to the
catalytic bed as a vapour component in a gas stream, the
performance of the alumina-supported catalyst in terms of
absorptive capacity is somewhat enhanced. Water has not
been found, however, to have a significant effect on the
decomposing capacity of the catalyst.
When used to remove hydrogen sulphide from gas
streams containing high boiling point hydrocarbons,
contamination of the catalyst can occur. Prior scrubbing
of the gas stream has been found necessary to reduce the
effects of this problem.
WO 92/20621 IPCT/CA91/00160
20g'~~~~
r~
- 32 -
Pressure. Flow Rate and Sweep Gas Effects on
Absorptive Capacity for ~~droaen Sulphide
The absorptive capacity of the catalyst (in terms
of the ratio of the mass of hydrogen sulphide removed in
the absorption stage to the mass of the catalyst) is
relatively insensitive to the concentration of hydrogen
sulphide in the gas stream for concentrations of hydrogen
sulphide up to 10%. It rises, however, approximately
linearly with total pressure, up to at least 500 psig.
At modest flow rates, the rate of removal of
hydrogen sulphide by absorption in the case of alumina
carriers is relatively high, up to the point where the
catalyst bed has been nearly totally saturated with
hydrogen sulphide at ambient temperature and pressure.
Some tests were done with a 3 minute residence
time. Other tests were done with a 0.7 minute residence
time. Tn both cases Alcoa alumina carriers impregnated
with the necessary ingredients to form the catalyst were
capable, before saturation, of removing virtually 100% of
the hydrogen sulphide from the gas stream. The level of
hydrogen sulphide prior to breakthrough was below the
threshold of measurability, in both cases being below 1
ppm.
Throughout most of the laboratory tests based on
re-oxygenation, nitrogen or methane containing small
amounts of oxygen was used as the carrier gas in most
cases to re-activate the catalyst after the sulphur had
been driven--off using oxygen-free nitrogen or methane as
the sweep gas. In some tests effected using a source of
sour natural gas as the sweep and carrier gas, the
hydrogen sulphide absorptive capacity of sample catalytic
beds (based on the Alcoa carrier) was similar to that
obtained with the nitrogen as the background gas. While
duantitative measurements of decomposing capacity were not
made in these latter tests, visual examination of the
catalyst bed after exposure to sour natural gas and before
regeneration showed clear deposits of yellow sulphur.
From this it is concluded that the substitution of natural
gas for nitrogen or pure methane as the background gas and
as the sweep gas does not significantly decrease the
absorptive or dissociative capacity of the catalyst.
WO 92/20b21 ~ ~ ~ PCf/C X91/00160
- 33 -
Conclusion
The foregoing has constituted a description of
specific embodiments showing how the invention may be
applied and put into use. These embodiments are only
exemplary. The invention in its broadest, and more
specific aspects, is further described and defined in 'the
claims which now follow.