Note: Descriptions are shown in the official language in which they were submitted.
WO 92/06967 PCT/US91/06863
209032.
-1-
THERAPEU'TTCALL~USEFUL 2-AMINOTETRA1,,IN DERIVATIVES
The present invention is related to new 1,2,3,4-tetrahydro-2-naphthylamines,
to
processes for preparing such compounds, pharmaceutical preparation of such
compounds
arid the use of such compounds in manufacture of a pharmaceutical preparation.
Psychiatric diseases are thought to be due to dysfunctions in monoaminergic
neuronal systems, particularly those involving serotonin (5-HT) and dopamine
(DA).
Anxiety is associated with increased activity in 5-HT systems. In animals
where
5-li'T has been depleted, benzodiazepine anxiolytics are not active in anti-
anxiety assays
that they otherwise are effective in. Seronotin neurons have autoreceptors
that, when
activated by agonists, depress firing rates o:f 5-HT cells. These receptors
are of the 5-
HT1A subtype. 5-HTIA agonists are anxiolytic. Buspirone is a marketed 5-HT1A
agonist that is an anxiolytic. Gepirone is another 5-HT1A agonist with
clinically
demonstrated anti-anxiety activities.
Depression is a psychiatric condition thought to be associated with decreased
5-
HT release. Many anti-depressants potentials the effects of 5-HT by blocking
the
termination of activity through reuptake into nerve terminals. 5-I3T1A
agonists can can
activate postsynaptically; they thus may also b~e anti-depressants. Gepirone
has already
been demonstrated to have ameliorative effects on some depressive endpoints in
some
patients.
Serotonin is also involved in the regulation of feeding and sexual behavior
and
in cardiovascular regulation. Thus, 5-HT1A agonists may be useful in treating
overeating and sexual dysfunction. These com~oounds have been shown to alter
feeding
and sexual behavior in animals. They may also be useful in the treatment of
obses-
sive/compulsive disorders, alcohol abuse and violent behavior. 5-FiTlA
agonists are also
known to depress sympathetic nerve discharge and thus lower blood pressure.
Thus,
they may be useful in treating hypertension, congestive heart failure (by
reducing
cardiovascular afterload) and heart attack (be removing sympathetic drive to
the heart).
Schizophrenia is thought to be due to hyperactivity in DA systems. Thus,
currently available anti-psychotics are DA antagonists. Dopamine autoreceptors
depress
DA neuron firing rates, DA synthesis and release. Thus DA autoreceptor
agonists can
WO 92/06967 ~ PGT/U591/06863
-
also be expected to be anti-psychotics. DA agonists are also useful for
treating
Parkinsonism, a disease caused by degeneration of DA neurons, and
hyperprolactinemia,
since DA agonists depress prolactin release.
Dopamine autoreceptor antagonists are a new class of drugs that increase
release
of DA by releasing the DA neuron from autoreceptor control. Thus, these drugs
can !~e
expected to be useful in conditions treatable with amphetamine and other
similar
stirnulants which directly release DA. However, DA autoreceptor agonists will
be much
milder stimulants because, rather than directly releasing DA, they simply
increase the
release associated with the normal DA activity by releasing the cell from
autoreceptor
control. Thus, DA autoreceptor antagonists can be expected to be useful in
treating
overeating, attention deficit disorders, psychiatric, cognitive and motor
retardation in
demented and elderly patients, and in treating nausea and dizziness with space
travel.
The compounds of the present invention have a variety of effects at 5-HT1A and
DA receptors, and offer a variety of utilities associated with those
activities.
Clinically, 5-HT1A agonists have also demonstrated anxiolytic properties. The
drug, Buspirone, is the only currently available marketed 5-HT1A agonist
having
anxiolytic activity. This compound antagonizes dopamine receptors at the same
dose it
stimulates 5-HT1A receptors. A similar drug, Gepirone, also has dopamine
antagonist
properties. These dopamine antagonist properties reduce the clinical utility
of these
compounds however because long term treatment with dopamine antagonists can
produce
tardive dyskinesia.
The search for new CNS active compounds is focused on finding compounds with
selective 5-HT1A receptor agonist effects without detrimentally influencing
central
dopamine receptors.
Drugs acting on central dopamine transmission are clinically effective in
treating
a variety of central nervous systern disorders such as parkinsonism,
schizophrenia, and
manic-depressive illness. In parkinsonism, for example, the nigro-neostriatal
hypofunction can be restored by an increase in postsynaptic dopamine receptor
stimula-
tion. In schizophrenia, the condition can be normalized by achieving a
decrease in
postsynaptic dopamine receptor stimulation. Classical anti-psychotic agents
directly
block the postsynaptic dopamine receptor. The same effect can be achieved by
inhibition
of intraneuronal presynaptic events essential for the maintenance of adequate
neuro-
transmission, transport mechanism and transmitter synthesis.
WO 92/06967 ~ ~ ~ ~ ~ ~ ~ pCT/US91/06863
-3-
In recent years a large body of pharmacological, biochemical and
electrophysical
evidence has provided considerable support in favor of the existence of a
specific
population of central autoregulatory dopamine receptors located in the
dopaminergic
neuron itself. These receptors are part of a homeostatic mechanism that
modulates nerve
impulse flow and transmitter synthesis and regulates the amount of dopamine
released
from the nerve endings.
Direct dopamine receptor agonists, like apornorphine, are able to activate the
dopamine autoreceptors as well as the post synaptic dopamine receptors. The
effects of
autoreceptor stimulation appear to predominate when apomorphine is
administered at low
doses, whereas at higher doses the attenuation of dopamine transmission is
outweighed
by the enhancement of postsynaptic receptor stimulation. The anti-psychotic
and anti-
dyskinetic effects in man of low doses of apomorphine are likely due to the
autoreceptor-
stimulator properties of this dopamine receptor agonist. This body of
knowledge
indicates dopamine receptor stimulants with a high selectivity for central
nervous dop-
amine autoreceptors would be valuable in treating psychiatric disorders.
The following documents could be important in the examination of this
application.
Arvidsson, L.-E., et al., J. Med. Chem., ~, 921 (1981), describes hydroxy-2-
aminotetralins where the amine is substituted with one n-propyl, one benzyl or
two n-
propyl substitutents. The 5-, 6-, and 7-hydroxy compounds are described as
active
central dopamine-receptor agonists and the 8-hydroxy compound is described as
a central
5-HT receptor agonist devoid of dopamine receptor stimulating activity.
Arvidsson, L.-E., et al., J. Med. Chem., ~, 45 (1984), describes 2-aminotetra-
Tins where the amine is substituted with one or two methyl, ethyl, n-propyl, i-
propyl, n-
butyl, or benryl substituents. The 2-piperidinyltetralin is also described.
Several of
these compounds were found to be potent 5-HT agonists devoid of dopamine-
mimetic
effects.
Asrvidsson, L.-E., et al., J. Med. Chem., ~Q, 2105 (1987), describes 8-hydroxy-
1-methyl-2-(di-n-propylamino)tetralins. These compounds were 5-~iT receptor
agonists.
The Arvidsson, L.-E. et al 8-hydroxy and 8-methoxy tetralin compounds are also
disclosed in Derwent documents 00389J/47, 94981D/S 1 and 045535J.48.
McDermed, et al., J. Med. Chem., ~, 362 (1975) describes 5,6 dihydroxy-2-
CA 02090321 2001-09-26
-4-
aminotetralins. In addition, the 5,8 and 7,8 disubstituted compounds are also
disclosed.
The amine can be a mono or di substituted with simple alkyl groups, benzyl
groups
alkylalkoxy groups or the amine can be a 5 or 6 membered hydrocarbon or
heterocyclic
amine. These compounds are indicated to have dopaminergic properties although
certain
compounds are reported to be inactive.
McDermed, et al., J. Med. Chem., ~Q, 547 (1976) describes 5-,
6-, or 7-hydroxy-2-dipropylaminotetralins. These compounds are described as
dopaminergic compounds.
Rusterholz, et al., J. Med. Chem., IQ, 99 (1976) describes 5,8 disubstituted-2-
aminotetralins with the amine being substituted with hydrogen, methyl, or
cyanopropyl
groups. Some of these compounds are potent prolactin inhibitors and believed
to be
dopamine agonists.
Ames, et al., J. Chem. Soc. 2636 (1965) describes the preparation of a large
number of compounds, where the aromatic ring is substituted by methoxy,
ethozy, n- or
iso-propoxy, or n-, sec- or tent-butoxy group in the 5 or 8 position and the
amine is
substituted by hydrogen or alkyl groups having 1-4 carbon atoms. The compounds
are
indicated to be prepared for pharmacological testing. However, no utility or
pharmaco-
logical activity is yet known for the compounds just mentioned.
EPO Publication No. 343,830 discloses 2-amino 1,2,3,4-tetrahydronapthalenes as
selective inhibitors of serotonin reuptake. It has a publication date
subsequent to filing date
of the parent application of this case
German Patent DE 2 803 582 describes 2-aminotetralins where the aromatic ring
is substituted on the 5,6,7 or 8 position with the group Rl, where RI is
hydrogen,
alkanoyl having 1 to 20 carbon atoms or a group -CO-(CH~a R~, n is a number 0
to 5,
R~ is a phenyl group with substituents as defined further, R2 is hydrogen,
hydrozy,
halogen or alkylsulfonylamino, R3 is hydrogen, R4 is hydrogen, CH20H, CH20-CO-
Rg
or CHZ-O-CO-(CH~ri R7 with further definition and RS and R6 are hydrogen,
alkyl or
aryl or aralkyl groups further defined or RS and R6 are together an alkylene
with 4 to
6 carbon atoms. The compounds are disclosed as having pharmacodynamic activity
in
particular a stimulating effect on alpha- and beta-adrenoceptors and dopamine
receptors.
Among the compounds described are compounds having the group RIB in the 8
position
and having R2 or R4 other than hydrogen.
Great Britain Patent 1,377,356 describes 2-aminotetralins where the aromatic
ring
WO 92/06967 PGT/US91/06863
2~9~321
-5-
is substituted on the 5, 6,7 or 8 position by R1, where R1 is hydrogen or
methyl, the
aliphatic ring is substituted by R2, where R2 is alkyl having 1-6 carbon
atoms, and the
amine is substituted by R3, where R3 is hydrogen or alkyl having 1-6 carbon
atoms are
described. Such compounds are stated to possess analgesic activity. 1,1-
Dimethyl-2-
(N,Ia1-dimethylamino)-7-hydroxytetralin is mentioned as one example of a
compound
covered by the patent. This compound is also described in Chem. Ab., 79:
146294b as
having analgesic and intestinal movement accelerating actions.
J. Pharm. Sci., ~7, 880-82 (1978) describes the compound I-methyl-2-(cyclopro-
pylamino)-5-methoxytetralin and indicates the compound possess local
anesthetic activity.
Derwent documents 58,247B/32, 40 378A/23, 83-729388/32, 83-729387/32,
29348D/17 and 06733V/OS refer to 8-carboxyamino tetralins. Additional
07633V/OS
refers to 8-amido and 8-alkylamido tetralin.
EPO patent application EPO 270 947 (1988) discloses 8-hydroxy and 8-methoxy-
tetralins.
EPO patent application EPO 272 534 (1988) discloses aminotetralins including
8-amido compounds.
The references cited herein are disclosures describing work related to the
invention:
Hjorth, S.; Carlsson, A; Lindberg, P.; Sanchez, D.; Wikstron, H.; Arvidsson,
L.-E.; Hacksell, U.; Nilsson, J.L.G., J. Neural Transm., 1982, 55, page 169.
Mellin, C.; Bjork, L.; Karlen, A.; Johansson, A.M.; Sundell, S.; Kenne, L.;
Nelson, D.L.; Anden, N.-E.; Hacksell, U., J. Med. Chem., 1988, ~, gage 1130.
Cossery, J.M.; Gozlan, H.; Spampinato, U.; Perdicakis, C.; Guillaumet, G.:
Pichat, L.; Hamon, M., European J. Pharmacol., 1987, pages 140-143.
WO 92/OG967 ~ ~~~,~ PCT/US91/06863
SUMMARY OF THE INVENTION
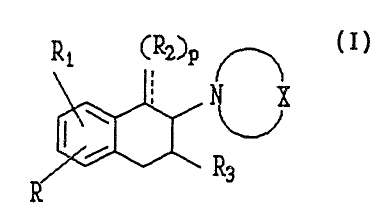
A compound having the formula I
wherein R is hydrogen or halogen,
wherein R1 is
(a) -hydrogen, (k) -CSNRgRb
(b) -OR4, (1) -OS02CF3
(c) -SR4,
(d) -CONRSR6,
(e) -CN,
(f) -het,
(g) -C(O)het,
(h) -CF3
(i) -S02NRSRb
(j) -s-oxazolyl
wherein R2 and R3 are independently
(a) -hydrogen,
(b) -(C1-Cg) ~Yl~
(c) -(C2-Cg) alkenyl,
(d) -(C2-Cg) alkynyl,
(e) -(CH~m (Cg-Cg)cycloalkyl,
(f) -(CH~m (C3-Cg)cycloalkenyl,
(g) -(CH~m aryl,
-(CH~t"COZR4,
(i) -(CH~m OR4,
2s wherein R4, RS and R6 are independently
(a) -hydrogen,
(b) -(C 1_C~alkyl,
(c) -(C I-C~alkenyl,
(d) -(Cg-Cg)cycloalkyl
and wherein X is (CH~n
m = 0-4
n = 4-8
p = 0,1
WO 92/06967 PCT/US91/06863
2~9032~
and pharmaceutically acceptable acid addition salts thereof;
with the provisos that when R1 is hydroxy or methoxy and R2 is hydrogen NBC
cannot
be piperidino, piperazino or homopiperazino.
Selected compounds of this invention possess selective pharmacological
properties
and are useful in treating central nervous system disorders including anti-
depression
symptoms, anti-psychotic symptoms, anxiolytic symptoms, panic attacks,
obsessive-
compulsive disturbances, senile dementia, emotional disturbances related to
dementia
disorders, and stimulation of sexual activity. Selected compounds of this
invention are
also useful to alleviate aggressive behavior, confusional delirious states and
impotence.
Selected compounds of this invention are further useful as anti-diabetic, anti-
obesity,
anti-hypertensive agents and for treating sexual impotency. Compounds of this
invention
are also useful as antitussive agents.
Processes for preparation of these compounds, their pharmaceutical use and
pharmaceutical preparations employing such compounds constitute further
aspects of the
invention.
An object of the invention is to provide compounds for therapeutic use,
especially
compounds having a therapeutic activity in the central nervous system. Another
object
is to provide compounds having an effect on the 5-HTIA receptor in mammals
including
man. A further object of this invention is to provide compounds having an
effect on the
subclass of dopamine receptors known as the D2 receptor.
nFTatI.FD DF~CRIP'TION OF THE INVENTION
The compounds of this invention are identified in two ways: by the descriptive
name and reference to labelled structures contained in appropriate charts. In
appropriate
situations, the proper stereochemistry is also represented in the charts.
In this document the parenthetical term (Cri Cm) is inclusive such that a
compound of (Cl-Cg) would include compounds of one to 8 carbons and their
isomeric
forms. The various carbon moieties are defined as follows: Alkyl refers to an
aliphatic
hydrocarbon radical and includes branched or unbranched forms such as methyl,
ethyl,
n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, n-pentyl,
isopentyl, neo-pentyl,
n-hexyl, isohexyl, n-heptyl, isoheptyl, and n-octyl.
Alkoxy as represented by -ORl when R1 is (C1-Cg) alkyl refers to an alkyl
radical which is attached to the remainder of the molecule by oxygen and
includes
branched or unbranched forms such as methoxy, ethoxy, n-propoxy, isopropoxy, n-
inn
WO 92/06967 ~ ,Z J~J w PCT/US91/0686~
~v
_g_
butoxy,isobutoxy, sec-butoxy,t-butoxy,n-pentoxy,isopentoxy, neo-pentoxy, n-
hexoxy,
isohexoxy, n-heptoxy,isoheptoxy, and n-octoxy.
Alkenyl refers to a radical of an aliphatic unsaturated hydrocarbon having a
double bond and includes both branched and unbranched forms such as ethenyl, 1
methyl-1-ethenyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 2-
methyl-1
butenyl, 1-pentenyl, allyl, 3-pentenyl, 4-pentenyl, 1-methyl-4-pentenyl, 3-
methyl-1
pentenyl, 3-methyl-allyl, 1-hexenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, 1-methyl-
4
hexenyl, 3-methyl-1-hexenyl, 3-methyl-2-hexenyl, 1-heptenyl, 2-heptenyl, 3-
heptenyl,
4-heptenyl, 1-methyl-4-heptenyl, 3-methyl-1-heptenyl, 3-methyl-2-heptenyl, 1-
octenyl,
2-octenyl, or 3-octenyl.
Cycloalkyl refers to a radical of a saturated cyclic hydrocarbon such as
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, or cyclooctyl.
Het refers to a five atom heterocyclic ring containing nitrogen, carbon and in
some cases oxygen. It includes 2-pyrrolyl, 2-oxazolyl, 2-imidazolyl, 2-
oxazolinyl, 2
imidazolinyl.
Halogen refers to bromine, chlorine or fluorine.
It will be apparent to those skilled in the art that compounds of this
invention do
contain chiral centers. The scope of this invention includes all enantiomeric
or dia-
stereomeric forms of Formula I compounds either in pure form or as mixtures of
enantiomers or diastereomers. The compounds of Formula I contain 1-3
asymmetric
carbon atoms in the aliphatic ring moiety, including the ring carbon atoms
adjacent to
the nitrogen atom. The therapeutic properties of the compounds may to a
greater or
lesser degree depend on the stereochemistry of a particular compound. Pure
enantiomers
as well as enantiomeric or diastereomeric mixtures are within the scope of the
invention.
Both organic and inorganic acids can be employed to form non-toxic pharmaceuti-
cally acceptable acid addition salts of the compounds of this invention.
Illustrative acids
are sulfuric, nitric, phosphoric, hydrochloric, citric, acetic, lactic,
tartaric, palmoic,
ethanedisulfonic, sulfamic, succinic, cyclohexylsulfamic, fumaric, malefic,
and benzoic
acid. These salts are readily prepared by methods known in the art.
The compounds of this invention may be obtained by one of the following
methods described below and outlined in the appropriate charts.
Chart A
Substituted 2-tetralone A-1, is subjected to reductive amination whose
procedures
WO 92/06967 PCT/US91/06863
~0903~1
are known in the art in step 1. In step 2, when R1 is methoxy A-2 may be
demethylated
via procedure known in the art to yield A-3.
In Step 1 of Chart B, the phenol B-1 is reacted with trifluoromethanesulfonic
anhydride in the presence of a solvent according to methods well known in the
art to
yield the triflate B-2. The phenol B-1 can be prepared from the appropriately
substituted
tetralones by the process depicted in Chart A.
In Step 2, a solution of B-2 in a solvent mixture such as methanol/DMF is
reacted
with carbon monoxide gas, palladium acetate, triethylamine and 1,3-
bis(diphenylphos-
phino) propane to form the carboxylic acid methyl ester B-3.
In Step 3, the methyl ester B-3 is hydrolyzed with sodium hydroxide in
methanol.
The resulting acid B-4 is then coupled in step 4 with ammonia in the presence
of
diethylcyanophosphonate and triethylamine in a solvent such as DMF to yield
the
carboxamide B-5.
Chart C
In step 1, acid chloride C-1 is treated with ethylene in the presence of
aluminum
trichloride to obtain tetralone C-2. C-2 is reductively aminated with the
appropriate
amine to yield C-3. C-3 is then dissolved in a solvent such as THF and reacted
in the
presence of t-butyllithium and trimethylsilyl isocyanate to yield amide C-4.
Chart D
In step 1, ketal D-2 is generated by stirring tetralone D-1 with ethylene
glycol in
the presence of an acid catalyst. In step 2, the 8-trifluoromethyl compound D-
3 is easily
obtained by heating a mixture of D-2, copper(I) iodide, sodium
trifluoroacetate and N-
methylpyrrolodone to 160°C. In step 3, hydrolysis using aqueous acid
gives tetralone
D-4 which is reductively aminated in step 4 using the known procedure of
mixing the
appropriate amine acetic acid, and sodium cyanoborohydride to yield D-5.
s~ ~
In step 1, substituted 2-tetralone E-1 is alkylated at the 2-position to
produce E-2
by reaction with an alkyl halide utilizing base in accordance with alkylation
methods well
known in the art. In step 2, E-2 is subjected to reductive amination to
produce E-3.
When R1 is methoxy. In step 3, E-3 is demethylated via procedures well known
in the
art to yield E-4.
WO 92/06967 ~~~,~ PCT/US91l06863
-10-
In step 1, substituted tetralone F-1 is reacted with dimethylcarbonate in the
presence of base such as LDA to produce F-2. In step 2 F-2 is reacted with
alkyl halide
in the presence of base to produce F-3. In step 3, F-3 is decarboxylated to
produce F-4.
In stt;p 4, F-4 is subjected to reductive amination to produce F-5. F-5 is
demethylated
when Rl is methoxy to yield F-6.
C
In step 1, bromotetralone G-1 (C-2) is reductively aminated using typical
conditions with the appropriate amine to obtain G-2. In step 2, G-2 is treated
with t-
butyllithium followed by dimethylformamide to obtain aldehyde G-3. This
aldehyde is
condensed in step 3 with "TOSMIC" under typical conditions to obtain the
oxazole G-4.
In step 1, substrate H-I is treated with t-butyllithium followed by sulfur
dioxide
to obtain sulfonic acid H-2. In step 2, H-2 is treated with sodium hydride to
obtain H-3
followed by treatment with N-chlorosuccinimide in step 3 to obtain
sulfonylchloride H-4.
IS Treatment of H-4 with ammonia in step 4 gives H-5 which is hydrolysized
with aqueous
acid to H-6 in step 5. In step 6, reductive amination using typical conditions
using the
appropriate amine gives H-7.
,~ art I
Bromo compound I-1 (C-3) is treated with t-butyllithium followed by trimethyl-
silylisothiocyanate to give I-2.
Chart J
In addition compounds of J-1 can be converted to compounds of Formula I
wherein Rl is arylcarbonyl by the process illustrated in Chart J. In Step 1 a
solution of
J-1 is reacted with the pyrrole-adduct in a solvent such as toluene in the
presence of
ethylmagnesium bromide to yield J-2.
In Step 2, J-2 is reacted with an appropriate amine in the presence of acetic
acid,
platinum oxide and absolute ethanol under a hydrogen atmosphere to yield
compound J-
3.
Methods for preparing compounds of Formula I wherein R1 is hydrogen, -OR6,
or -SR6 are illustrated by the processes illustrated in Charts A, E and F. In
each of
these processes, 2-tetralone derivatives are utilized as the starting
material.
Methods for conducting reductive amination are well known in the art and any
such methods may be used in the procedures described above. One such method
WO 92/06967 2 0 9 0 3 2 ~ P~/US91/06863
-11-
involves reacting the tetralone with an amine in the presence of sodium
cyanoboro-
hydride and glacial acetic acid in tetrahydrofuranlmethanol.
The 8-amido compounds B-S or C-4 can be converted to the corresponding 8-
cyano compounds by reacting with a "Burgess salt" utilizing conditions well
known in
the art. The Burgess salt can be prepared by the procedure described in
Organic
~, ~, page 40.
In clinical practice the compounds of the present invention will normally be
administered orally, rectally, or by injection, in the form of pharmaceutical
preparations
comprising the active ingredient either as a free base or as a
pharmaceutically acceptable
non-toxic, acid addition salt, such as the hydrochloride, lactate, acetate,
sulfamate salt,
in association with a pharmaceutically acceptable carrier. The use and
administration to
a patient to be treated in the clinic would be readily apparent to a person of
ordinary skill
in the art.
In therapeutical treatment the suitable daily doses of the compounds of the
invention are 1-2000 mg for oral application, preferentially 50-500 mg, and
0.1-100 mg
for parenteral application, preferentially 0.5-50 mg.
The compounds of this invention where Rl is in the 8 position in the aromatic
ring are very selective 5-HT1A receptor agonists having little or no
dopaminergic
activity. The ICSp ratio of dopamine D2 to 5-HT1A in vitro binding data shown
in Table
1 for one compound of this invention, demonstrates the selectivity for the 5-
HT1A
receptor. The compounds of this invention also have been shown to have high
oral
potency and a long duration of action. Both these features are beneficial to
effective
clinical treatment.
The utility of the compounds of this invention to treat central nervous system
disorders is shown in behavioral, physiological and biochemical tests. The
methods are
given as follows:
Binding: Inhibition of 8-OH-DPAT binding in a bovine brain homogenate.
Potency is given as nM dose required to inhibit 50~ of DPAT binding (ICg~.
This test
measures ability to bind to 5-hydroxytryptamine (5-HTIp) receptor.
Hypothermia: Starting with a dose of 30 mglkg, four mice are injected
subcutaneously with test compound. Twenty minutes later, the number of animals
whose
body temperature has decreased by 2°C. or more are counted. If all four
animals reach
criteria, the drug is considered "active", and subsequent readings are taken
at 60 and 120
CA 02090321 2002-O1-14
-12-
minutes after drug. The time for last statistically significant drug affect on
mean body
temperature is indicated in minutes. For all "active" compounds, doses are
lowered by 0.5
log intervals until a dose which does not lower body temperature by
2°C. in any animal is
found. Potency is given as mg/kg ED50 (dose required to depress temperature in
two of four
mice) as measured by Spearman-Karber statistics.
Sympathetic Nerve Discharge (SND): The i.v. mg/kg dose causing a 50%
depression
in SND in chloralose anesthetized cats and the maximum inhibition of
sympathetic activity
observed in the dose range tested (0.001-1.0 mg/kg i.v.).
BP SND/MAX: The blood pressure of the chloralose anesthetized cats in percent
control at the dose causing 50% depression in SND and the maximum reduction in
blood
pressure as percent of the control blood pressure in the same animals observed
in the dose
range tested (0.001-1.0 mg/kg i.v.).
WO 92/06967 PCT/US91/06863
24~032.~
-13-
CNS and anti-hypertensive biological data are shown in Tables 1 and 2
respectively.
TABLE I
CNS BIOLOGICAL DATA
5-HTiA Binding Hypothermia
Example Compound
No No ICgO (nM) EDSp(mg/kg)
2 2-1 26 2.3
( > 1000 Ic50(nm at
D2 receptor)
4 4-1 10.9
3 3-1 4
1 1-3 103.4 2.3
TABLE II
ANTI-HYPERTENSIVE BIOLOGICAL DATA
Serotonin SND Assax
Max. Decr. % BD Max
Example Compound SND ED50 SND % (at SND Decr.
No. No. Control ED50) BP
2 2-1 > . 296 - 71.0
1 1-3 0.62 31.0 75.0 68.7
The compounds of this invention are useful as anti-diabetic, anti-obesity,
anti-
hypertensive and anti-tussive agents. While all of the compounds do not have
all of
these pharmacological activities the utility of a particular compound can be
determined
by one skilled in the art utilizing the following tests.
CA 02090321 2001-09-26
-14-
Anti-diabetic
A. Testine For Blood Glucose Lowering In the KKAy Mouse
All KKAy mice used for screening are produced and selected by methods outlined
by T. Fujita et al., Diabetes, 32, pp. 804-10 (1983). The screening is done in
groups
of six animals per group.
Pre-treatment non-fasting blood glucose (NFBG) samples are measured five days
prior to the start of a screening run by previously described methodologies.
These blood
sugar values are used to place animals into gmups with equal mean blood
glucose
concentrations and to eliminate any mice with a NFBG value < 250 mg/dl. On day
0,
compounds chosen to be run are incorporated into ground mouse chow (PurinaTM
SO15).
Compounds are included at a rate of I mg/gram of chow. Generally, 300 g of
drugs
containing diet is prepared for each group. Mice receiving ground chow only
are the
negative control.
Each screening run also uses ciglitazone (T. Fujita et al., supra) as a
positive
control (0.5 to 1.0 mg/gram chow).
Initial body and food weights are taken on day one. Food is placed in a crock
which contains an adequate amount to last for the length of the study. In
order to
acclimate the mice from pelleted mouse chow to ground mouse chow, they are fed
the
ground chow for nine days prior to use in the screen. On day four of
treatment, a
NFBG sample is again measured, as well as food and body weights. Food
consumption
measurements are used to determine an average mg/kg dose the mice received
over the
testing period, and to evaluate the compound's effect on food consumption.
Acceptance and activity are determined by the following criteria:
A. Negative Control
This group must not show a significant change (p < .OS) from pre- to post
treatment. If there is a significant decrease in blood sugar, the run is not
valid.
B. Positive Control
This group must show a significant depression in blood sugar mean levels
from pre- to post-treatment. A lack of activity in this group would also
invalidate the
run.
C. Negative Control vs. Positive Control
This contrast must be significant. It is a further assurance that both
control groups performed as expected.
WO 92/06967 PCT/US91/06863
-15-
D. Compound
A compound's activity is based on several criteria:
1. A significant decrease in blood sugar mean levels from pre- to
post-treatment.
2. Negative control vs. compound: This contrast allows one to
determine if these groups are dissimilar, which is required for the compound
to be
considered active.
II. Anti-obesity. Activity
Upjohn Sprague-Dawley rats are housed individually and given food and water
~ 1_i~bitum. Food consumption is measured daily. The animals are orally dosed
with 100
mg/kg or 200 mg/kg of the compound in Tween 80. Controls receive an equivalent
volume of (0.25) of Tween 80. If the daily food consumption of the treated
animals is
in the range of 4 grams less than that of the control animals the compound is
considered
to have anorexic activity.
ExRgrimental Procedures
Without further elaboration, it is believed that one skilled in the art can,
using the
preceding description, practice the present invention to its fullest extent.
The following
detailed examples describe how to prepare the various compounds and/or perform
the
various processes of the invention and are to be construed as merely
illustrative, and not
limitations of the preceding disclosure in any way whatsoever. Those skilled
in the art
will promptly recognize appropriate variations from the procedures both as to
reactants
and as to reaction conditions and techniques.
g~~r, tra ion 1 7-(1-hexahydroazepinyl)-5,6,7,8-tetrahydronaphthalene-1-
trifluoromethyl-
sulfonate (B-2, Chart B)
Asolutionof2.lg7-(1-hexahydroazepinyl)-5,6,7,8-tetrahydro-l-naphthalenoland
4.06 g pyridine in 100 mL CH2C12 was stirred and cooled to O~C under a
nitrogen
atmosphere. The trifluoromethanesulfonic anhydride (4.32 g) was added dropwise
over
a 20 min period. The yellow solution was warmed to room temperature and
stirred for
45 min. TLC aliquot showed no starting material present. the reaction was
quenched
with satd. NaHC03 to pH > 8. The mixture was extracted with methylene
chloride.
The organic layers were washed with water, brine, dried (MgSO~, filtered and
concentrated to yield an oil. Flash chromatography on 1 kg silica gel eluting
with
hexane/ethyl acetate (1:1) (collecting 45 mL fractions) yielded a pale yellow
WO 92/06967 '~~ ~~ PCT/US91/t168F.'i..
-16-
oil (3.05 g, 95 % )
Preparation 2 7-(I-Hexahydroazepinyl)-5,6,7,8-tetrahydronaphthalene-1-
carboxylicacid
methyl ester (B-3, Chart B)
A solution of 2.9 g 7-(1-hexahydroazepinyl)-5,6,7,8-tetrahydronaphthalene-1-
trifluoromethylsulfonate and 2.13 mL triethylamine in 9 mL methanol and 27 mL
DMF
was degassed with nitrogen through a syringe for 10 min. Carbon monoxide was
then
bubbled through the solution for 10 min. During this time, a solution if 172
mg
palladium acetate and 379 mg in 7 mL DMF was dissolved and degassed with
nitrogen
for 10 min. This solution was added to the reaction , heated to 70~C and
carbon
monoxide gas bubbled through overnight. An aliquot was treated with satd.
NaHCO3
and EtOAc and showed no starting material. Nitrogen was bubbled through the
solution
and then quenched with satd. NaHC03. The mixture was extracted with ethyl
acetate
(3 X 500 ml) and the combined organic layers were washed with brine, dried
(MgS04),
filtered and concentrated to yield an oil. Flash chromatography using 400g
silica gel and
eluting with hexane/ethyl acetate (1:1) to yield an oil (1.56 g, 71 %).
~r~aration3 7-(1-Hexahydroazepinyl)-5,6,7,8-tetrahydronaphthalene-1-
carboxylicacid
(B-4, Chart B)
A mixture of 1.56 g 7-(1-Hexahydroazepinyl)-5,6,7,8-tetrahydronaphthalene-1
catboxylic acid methyl ester , 1.57 mL 12 N NaOH, 1.57 mL water in 10 mL
methanol
was refluxed (70-80~C) overnight. TLC showed no starting material remaining.
The
mixture was neutralized with 6 N HCl to pH 5-6 and concentrated to dryness
using
toluene and methanol. A white solid was recovered and used crude.
P~aration 4 8-bromo-2-tetralone (C-2, Chart C)
Substitute 2-bromophenylacetylchloride in the procedure detailed in A.H. Horn,
C.J. Grol, D. Dijkstra, and A.H. Mulder, J. Med. Chem. 21, 825 (1978).
~aration 5 8-bromo-2-(spiro-1,3-dioxolan-2-yl)tetralin (D-2, Chart D)
8-Bromo-2-tetralone (29g), ethylene glycol (24g), p-toluenesulfonic acid
(O.Sg),
and benzene (250 ml) were heated to reflux with azeotropic removal of water
for 16 hr.
The solution was cooled and extracted with aq. sodium carbonate, water, and
then brine.
The solution was dried over anhydrous sodium sulfate and the solvent removed
under
vacuum.
Preparation 6 8-trifluoromethyl-2-(spiro-1,3-dioxolan-2-yl)tetralin (D-3,
Chart D)
8-Bromo-2-(spiro-1, 3-dioxolan-2-yl)tetralin( 12.4g), sodium trifluoroacetate
(25 g),
WO 92/0696? ~ ~ ~ ~ 3 ~ 1 PCT/US91/06863
-17-
copper (I) iodide (17.5g) and N-methyl pyrrolidone (368 ml) were heated under
nitrogen
to 160°C and maintained there for 4 hr. The solution was cooled and
ether and hexane
werE: added. The slurry was filtered through diatomaceous earth and the
elutant was
washed with water (3X) and brine. The solution was dried over anyhdrous sodium
sulf <tte and the solvent removed under vacuum. Flash chromatography was
performed,
eluting with ether/hexane (1:9) giving 9.9g of a pure liquid.
Pyaration 7 8-trifluoromethyl-2-tetralone (D-4, Chart D)
8-Trifluoromethyl-2-(spiro-l,3-dioxolan-2-yl)tetralin (9.9g), water (15 ml),
THF
(120 ml), and 2 N aq. HCl (12 ml) were heated to 50°C for 15 hr. This
solution was
cooled and extracted with ether, washing the organic layer with aq. sodium
bicarbonate
and then brine. Drying over anhydrous sodium sulfate and solvent removal
afforded a
clear liquid.
Pr~aration 8 1,2.,3,4-Tetrahydro-2-oxo-l-(2-propenyl)-naphthalene (E-2, Chart
E) and
1,2,3,4-Tetrahydro-2-oxo- 1,1-di-(2-propenyl)naphthalene
To a solution of 7.3 g (50 mmol) 2-tetralone in 75 mL THF in a 3-neck round-
bottomed flask, equipped with a gas inlet and septum, was added 36.? mL LDA
(55
mmol, 1.5 M in cyclohexane) at -30°C under a nitrogen atmosphere. The
solution was
allowed to warm to 0°C over a 30-minute period and 5.6 mL (65 mmol)
allyl bromide
was added. TLC analysis was used to monitor the reaction. After stirring for
24 hours
at room temperature, the reaction mixture was quenched with 10% sodium
bisulfate to
pH 2-3. After removal of THF under reduced pressure, the mixture was extracted
with
ethyl acetate (2 X 1 L) and the combined organic layers were washed with
brine, dried
(MgSO~, filtered and concentrated in vacuo. The crude product was purified by
liquid
chromatography on 800 g of silica gel 60 (230-400 m), eluting with 1 L of
hexane,
followed by 5 L of 5 % ethyl acetate/hexane, and collecting 40 mL fractions.
Fractions
65-82 gave 3.1 g (3390 of pure 1,2,3,4-Tetrahydro-2-oxo-l-(2-propenyl)-
naphthalene
as a light yellow oil.
1HNMR (CDCl3, TMS): 7.27-7.16 (m, 4H); 5.81-4.95 (m, 3H); (s, 3H); 3.54-
2.45 (m, 7H).
IR (film): v ~ 1717, 1640 and 1582 crri 1.
MS: M+ 186, other ions at m/z 168, 145, 128, 117.
TLC (Silica Gel GF): Rf = 0.51 in hexane/ethyl acetate (4:1).
on.
WO 92/06967 ~~~~ ~ PCT/US91/0686~
-18-
Fractions 41-64 gave 4.2 g (37%) of pure and 1,2,3,4-Tetrahydro-2-oxo-l,1-di-
(2-
propenyl)naphthalene as a colorless oil
P~aration 9 1,2,3,4-Tetrahydro-8-methoxy-2-oxo-l-(2-propenyl)-naphthalene and
1,2,3,4-Tetrahydro-8-methoxy-2-oxo-l-di-(2-propenyl)-naphthalene (E-2, Chart
E)
To a solution of 8.8 g (50 mmol) 8-methoxy-2-tetralone in 250 mL THF in a
three-neck round-bottomed flask, equipped with a gas inlet and septum, was
added 40
mL LDA (60 mmol, 1.5 M in cyclohexane, at -30°C under a nitrogen
atmosphere. The
solution was allowed to warm to 0°C over a 30-minute period and 6.5 mL
(75 mmol)
allylbromide was added. TLC analysis was used to monitor the reaction. After
stirring
the mixture at room temperature for three hours and at 40°C for one
hour, the reaction
mixture was quenched with 10% sodium bisulfate to pH 2-3. After removal of THF
under reduced pressure, the mixture was extracted with ethyl acetate (2 X 1 L)
and the
combined organic layers were washed with brine, dried (MgSO~, filtered and
concentrated in vacuo. The resulting oil (about 3b/22b=4 by LC purification in
a small
scale run) was used without purification in the next step. For the analytical
purpose the
small amount of the crude product ( < 1 g) was purified by liquid
chromatography on 185
g of silica gel 60(230-400 m), eluting with hexane/acetone (19:1). Fractions
homoge-
neous by TLC were combined and concentrated in vacuo. Pure title compounds
were
isolated as a light yellow oil.
Physical data for 1,2,3,4-tetrahydro-8-methoxy-2-oxo-2-(2-propenyl)naphthlene:
1HNMR (CDC13, TMS): 7.21-6.76 (m, 3H); 5.73-4.87 (m. 3H); 3.82 (s, 3H);
3.88-3.82 (m, 1H); 3.32-2.43 (m, 6H).
IR (film):v ~X 1712,1640, 1586 cm 1.
MS: Calcd for C14H1602: 216.1150.
Found: 216.1151.
Analysis: Calcd for C14H1602~ C, 77.75; H, 7.46.
Found: C, 77.56; H, 7.68.
TLC (Silica Gel GF): Rf = 0.32 in hexane/acetone (4:1)
Physical data for 1,2,3,4-tetrahydro-8-methoxy-2-oxo-l-di-(2-propenyl)-
naphthlene:
1HNMR (CDGI3, TMS): 7.22-6.73 (m, 3H); 5.44-4.77 (m, 6H); 3.85 (s, 3H);
4.0-2.52 (m, 8H).
IR (film): v ~x 1712, 1639 and 1582 cm-1.
WO 92/06967 2 ~ ~ ~ ~ ~ ~ PCT/US91/06863
-19-
MS: Calcd for CI~H2~02: 256.1463.
Found; 256.1470
Analysis: Calcd for CI~H2002: C, 79.65; H, 7.86.
Found: C, 79.56; H, 8.29.
TLC (Silica Gel GF): Rf=0.46 in hexane/acetone (19:1).
Preparation 10 1,2,3,4-Tetrahydro-5-methoxy-2-oxo-l-(2-propenyl)-naphthalene
(E-2,
Chart E) and 1,2,3,4-Tetrahydro-5-methoxy-2-oxo-l,l-di-(2-pro-penyl)-
naphthalene
To a solution of 5.3 g (30 mmol) 5-methoxy-2-tetzalone in 45 mL THF in a
three-neck round-bottomed flask, equipped with a gas inlet and septum, was
added 22
mL LDA (33 mmol, 1.5 M in cyclohexane, at -30°C under a nitrogen
atmosphere. The
solution was allowed to warm to 0°C over a thirty-minute period and 3.4
mL (39 mmol)
allylbromide was added. TLC analysis was used to monitor the reaction. After
five
hours of stirring, the reaction mixture was quenched with 10% sodium bisulfate
to pH
2-3. After removal of THF under reduced pressure, the mixture was extracted
with ethyl
acetate (2 X 1 L) and the combined organic layers were washed with brine,
dried
(MgSO~, filtered and concentrated in vacuo. The resulting oil was purified by
liquid
chromatography on 800 g of silica gel 60 (230-400 m), eluting with 1 L of
hexane and
5 L of hexane-ethyl acetate (19:1), and collecting 40 mL fractions. Fractions
45-87 gave
2.5 g (32.5 %) of pure 1,2,3,4-tetrahydro-5-methoxy-2-oxo-l, l-di-(2-propenyl)-
naphthalene as a near colorless oil and fractions 88-140 gave 1.07 g (16.5"6)
of pure
1,2,3,4-tetrahydro-5-methoxy-2-oxo-l-(2-propenyl)naphthalene as a light yellow
oil.
1HNMR (CDCl3, TMS): 7.23-6.77 (m, 3H); 5.75-4.97 (m, 3H); 3.85 (s, 3H);
3.52-2.49 (m, 7H).
IR (film): v ~x 1717, 1641 and 1586 cm-1.
MS: M+ 216, other ions at m/z 175, 159, 147.
TLC (Silica Gel GF): Rp = 0.42 in hexane-ethyl acetate (4:1).
$~paration 11 1,2,3,4-Tetrahydro-8-methoxy-1-(cyclopropylmethyl)-2-oxo-
naphthalene
(E-2, Chart E)
To a solution of 3.52 g (20 mmol) 8-methoxy-2-tetralone in 50 mL THF in a
three-neck round-bottomed flask, equipped with a gas inlet and septum, was
added 14.3
mL LDA (22 mmol, 1.5 M in cyclohexane) at -30°C under a nitrogen
atmosphere. The
solution was allowed to warm to 0°C over a thirty-minute period and 2.4
mL (24 mmol)
allyl bromide was added. TLC analysis was used to monitor the reaction. After
stirring
WO 92!06967 ~~~~~ PCT/US91/06863
-2o-
for two hours, the TLC analysis appeared to show little progress. The reaction
mixture
was therefore treated with 1.1 mL (12 mmol) allylbromide and the mixture was
heated
to reflux for 72 hours. The reaction mixture was quenched with 10% sodium
bisulfate
to pH 2-3. After removal of THF under reduced pressure, the mixture was
extracted
with ethyl acetate (2x 1 L) and the combined organic layers were washed with
brine,
dried (MgS04), filtered and concentrated in vacuo. The resulting oil was
purified by
liquid chromatography on 400 g of silica gel 60 (230-400 m), eluting with
hexanelace-
tone (9:1) and collecting 40 mL fractions. Fractions homogeneous by TLC were
combined and concentrated in vacuo to give 4.5 g (97.8%) of pure title
compound as a
near colorless oil.
1HNMR (CDCl3, TMS): 7.20-6.75 (m, 3H); 3.91 (t, J=7 Hz, 1H); 3.81 (s,
3H); 3.33-1.62 (m, 6H); 0.64-0.09 (m, SH).
IR (film): v ~x 1711 ctri 1.
MS: Calcd for ClgHlg02: 230.1307.
Found: 230.1290.
Analysis: Calcd for ClgHlg02: C, 78.23; H, 7.88.
Found: C, 77.93; H, 8.06.
TLC (Silica Gel GF): Rf = 0.46 in hexane-acetone (4:1).
Pre»aration 12 1,2,3,4-Tetrahydro-8-methoxy-2-oxo-1-naphthalene-carboxylic
Acid
Methyl Ester (F-2, Chart F)
To a solution of 17.6 g (0.1 mol) 8-methoxy-2-tetralone in 200 mL THF in a
three-neck round-bottomed flask, equipped with a gas inlet and septum, was
added 86.7
mL LDA (0.13 mol, 1.5 M in cyclohexane) at -30°C under a nitrogen
atmosphere. The
solution was allowed to warm to 0°C over a thirty-minute period and
84.3 mL (1.0 mol)
dimethylcarbonate was added. After refluxing for 24 hours (bath temperature
70°C), the
TLC analysis indicated no starting material remaining. The reaction mixture
was
quenched with 1 N HCI to pH 2-3. After removal of THF under reduced pressure,
the
mixture was extracted with methylene chloride (2 X 1 L) and the combined
organic
layers were washed with brine, dried (MgS04), filtered and concentrated in
vacuo. The
resulting oil was purified by flash chromatography on 1 Kg of silica gel 60
(230-400 m),
eluting with 1 L hexane, 2 L 10 %, 8 L 20 % ethyl acetate/hexane and
collecting 500 mL
fractions. Fractions 7-9 gave 0.5 g (2%) of a yellow oil which was shown to be
1,1-
dicarbomethoxy product by 1HNMR. Fractions 11-22 afforded 21.1 g (90%) of pure
WO 92/Oib967 ~ ~ ~ ~ ~ ~ ~ PCT/US91/06863
i
-21-
title compound as a yellow oil.
IHNIViR (CDC13, TMS): 7.28-6.77 (m, 3H); 4.72 (s, 1H); 3.80 (s, 3H); 3.72-
2.17 (m, 7H).
IR (film): v ~x 1750, 1718 and 1588 cni 1.
MS: M+ 234, other ions at m/z 202, 191, 174, 147, 131, 115, 103, 91.
Analysis: Calcd for C13H1404~ C~ 66.65; H, 6.02.
Found: C, 66.49; H, 5.93.
TLC (Silica Gel GF): Rf = 0.33 in hexanelethyl acetate (3:1).
~~aration 13 1,2,3,4-Tetrahydro-8-methoxy-2-oxo-3-(2-propenyl)-1-naphthalene-
carboxylic acid methyl ester (F-3, Chart F)
A solution of 10.2 g (43.5 mmol) 1,2,3,4-tetrahydro-8-methoxy-2-oxo-l-
naphthalene carboxylic acid methyl ester in 108 mL of THF in a three-neck,
round-
bottomed flask, equipped with a dropping funnel, was added dropwise 63.8 rnL
(95.7
mmol) of LDA (1.5 M in cyclohexane) at -30°C to -40°C under a
nitrogen atmosphere.
The solution was allowed to warm to 0°C and 6.0 mL (69.6 mmol) of
allylbromide was
added. After stirring the mixture for one hour at room temperature, TLC
analysis
showed no starting material remaining. The reaction was quenched with 3N
hydrochlo-
ric acid to pH 2-3 and extracted with ethyl acetate (2x 1 L). The combined
organic
layers were washed with brine, dried (MgSO~, filtered, and concentrated in
vacuo. The
resulting oil was purified by liquid chromatography on 800 g silica gel 60
(230-400 m),
eluting with hexane-acetone (3:1), and collecting 40 mL fractions. Fractions
36-63 gave
10.3 g (8? 90) of pure title compound as a yellow oil.
1HNMR (CDC13, TMS): 7.27-6.76 (m, 3H); 5.89-5.02 (m, 3H); 4.75, 4.59
(two s, 1H); 3.80, 3.81 (two s, 6H); 3.32-1.64 (m, SH).
IR (film): v ~ 1751, 1717 and 1589 cm-1.
MS: M+ 274, other ions at m/z 242, 233, 214, 201, 187, 173, 159, 145.
Analysis: Calcd for C16H1804: C, 70.05; H, 6.61.
Found: C, 69.73; H, 6.65.
TLC (Silica Gel GF): Rf = 0.34 in hexane-ethyl acetate (3:1).
~aration 14 1,2,3,4-Tetrahydro-8-methoxy-3-(2-propenyl)-2-oxo-naphthalene (F-
4,
Chart F)
To a solution of 10.3 g (37.6 mmol) of 1,2,3,4-tetrahydro-8-methoxy-2-oxo-3-(2-
propenyl)-1-naphthalene carboxylic acid methyl ester in 26.3 mL of DMSO and
1.1 mL
~1~1
WO 92/45967 Og~~ PCT/US91/0686~
-22-
of water was added 1.9 g (45.1 mmol) of lithium chloride. The reaction mixture
was
he<~ted at 125°C (bath temperature) for five hours. TLC analysis showed
no starting
material remaining. The mixture was cooled to room temperature and extracted
with
ethyl acetate (1 L). The organic layer was washed with 10% aqueous calcium
sulfate
(an efficient way of removing DMSO from organic layer), dried (MgS04),
filtered and
concentrated in vacuo. The crude product was purified by liquid chromatography
on 800
g silica gel 60 (230-400 m), eluting with hexane-ethyl acetate (3:1), and
collecting 40
mL fractions. Fractions 26-53 gave 7.65 g (94%) of pure title compound as a
yellow
oil
1HNMR (CDC13, TMS): 7.18-6.74 (m, 3H); 5.95-4.95 (m, 3H); 3.82 (s, 3H);
3.70-2.08 (m, 7H).
IR (film): v ~X 1756, 1710 and 1589 cm-1.
MS: M+ 216,other ions at m/z 185, 174, 159, 146, 134, 115, 104.
Analysis: Calcd for C14H1602v C~ 77.75; H, 7.46.
Found: C, 77.21; H, 7.65.
TLC (Silica Gel Gf): Rf = 0.53 in hexane-ethyl acetate (3:1).
Pr~aration 15 8-Aminosulfonyl-2-(spiro-l,3-dioxolan-2-yl)tetralin. (H-2, Chart
H)
Magnesium (3.83 g, 0.158 mol) was covered with dry tetrahydrofuran (250 mls),
and 8-bromo-2-(spiro-l,3-dioxolan-2-yl)tetralin (28.29 g, 0.105 mol) was
added. A few
crystals of iodine were added, and the mixture was heated to reflux on the
steam bath
until the reaction became exothermic. The reaction was stirred at ambient
temperature
until the reaction subsided. The reaction mixture was refluxed gently on the
steam bath
for an additonal 40 minutes. The Grignard solution was removed from the excess
magnesium via needle stock and cooled to -15°C. Sulfur dioxide gas was
bubbled
through the solution for 30 minutes. The mixture was diluted with diethylether
and
washed with dilute hydrochloric acid and brine containing sodium bicarbonate.
The
solution was dried (MgSO~, and the solvent was removed under vacuum to leave
the
sulfinic acid as an off white solid (27.26 g).
(H-3, Chart H) Sodium hydride (5.3 g, 50% in oil, 0.11 mol) was washed twice
with hexane, and covered with dry tettahydrofuran (400 mls). A solution of the
sulfinic
acid (26.38 g, 0.104 mol) in dry tetrahydrofuran (300 mls) was added via
needle stock.
The mixture was stirred at room temperature overnight and then heated at
reflux for 15
minutes. The mixture was diluted with diethylether, and the precipitate was
filtered
WO 92/06967 Pf,:T/US91/06863
2903 ~1
-23-
while blowing argon over the surface of the compound. The compound was washed
several times with diethylether, and dried under vacuum leaving the sodium
sulfinate as
a solid (26.77 g).
(H-4, Chart H) A suspension of the sodium sulfinate (26.77 g, 0.0969 mol) in
methylene chloride (400 mls) was cooled in ice, and N-chlorosuccinimide (13.75
g,
0.103 mol) was added. The mixture was stirred at room temperature for 2 hours.
Diethylether was added, and the mixture was washed with water and brine. The
solution
was dried (MgSO~, and the solvent was removed under vacuum to leave the
sulfonyl
chloride as an amber solid (23.3 g).
(H-5, Chart H) A solution of the sulfonyl chloride (23.3 g) in tetrahydrofuran
(80
mls), was added to an ice-cooled solution of ammonium hydroxide (100 mls) in
acetone
(500 mls). The cold bath was removed, and the mixture was stirred for 2 hours.
The
solvent was evaporated, and the residue was pardoned between 4:1
diethylether/tetrahy-
drofuran and brine. The solution was washed twice with 2 % hydrochloric acid,
sat.
sodium bicarbonate, and brine. The solution was dried (MgS04), and the solvent
was
removed under vacuum to leave the sulfonamide as a tan solid (19.8 g). A
sample (0.75
g) was crystallized from ethyl acetate/hexane to give off white crystals of
the
sulfonamide (0.68 g, m.p. 127-128°C).
Per ,raration 16 8-Aminosulfonyl-2-tetralone (H-6, Chart H).
8-Aminosulfonyi-2-(spiro-l,3-dioxolan-2-yl)tetralin (18.36 g, 0.0682 mol) was
dissolved in acetone (400 mls), and p-toluenesulfonic acid (1.85 g, 9.7 mmol,
14 mol
percent) was added. The mixture was stirred at room temperature for 21 hours.
Saturated sodium bicarbonate (50 mls) was added, and the solvent was removed
under
vacuum. The residue was diluted with water and cooled in ice. The precipitate
was
filtered, washed with water, and dried under vacuum. The compound was boiled
in
ethyl acetate (350-400 mls) until most of the solid dissolved and then
filtered. Hexane
was added, and crystallization occurred leaving the ketone as an orange solid
(10.34 g,
m.g. 173-175°C).
WO 92/06967 ~C~~ ~ PCTlU591/0685~
-z4-
P~aration 17 8-Carbomethoxy-2-[spiro-2-(1,3-dioxolyl)]tetralin (J-1, Chart J)
8-Trifluoromethylsulfonyl-2-[spiro-2-(1,3-dioxolyl)]tetralin (62 g, 183.4
mmol)
was placed in a round bottom flask with palladium acetate (2.88 g, 7 mold),
bis(diphenylphosphino)propane (6.81 g, 9 mol°k), diisopropylamine (70.3
ml, 2.2 eq.),
methanol (183 ml) and dimethylsulfoxide (550 ml). The flask was thoroughly
flushed
with carbon monoxide, which was subsequently bubbled through the solution. The
solution was heated to 70 ° and stirred for four hours. The solution
was cooled and 400
ml. of methylene chloride and 800 ml of ether added. This solution was washed
with
water (4 x 500 ml) and brine (400 ml), and dried over anhydrous sodium
sulfate.
Solvent removal in vacuo and filtration through a plug of flash silica gel (6
cm x 30 cm)
with ethyl acetate/hexane (25:75), followed by solvent removal afforded 39 g
of the title
compound (85 % yield) as an oil.
Pret~aration 18 (1,2,3,4-Tetrahydro-2-oxonaphthalene-8-yl)(2-pyrrolyl) ketone
(J-2,
Chart J)
Pyrrole (3.17 ml) was dissolved in toluene (40 ml} and cooled to
0° while
ethylmagnesium bromide (15.2 ml of a 3 M solution in ether) was added. This
solution
was allowed to warm to 25° and stirred for 30 minutes. A solution of
methyl 1,2,3,4-
tetrahydrospiro-2-[2-(1,3-dioxolane)]naphthalene-8-yl-carboxylate (5.15 g,
20.8 mmol)
dissolved in toluene (20 ml) was added and the solution refluxed for 24 hours.
The
solution was cooled and quenched by the addition of saturated aqueous ammonium
chloride. Ether (100 ml) was added and the solution extracted. The organic
layer was
washed with water (2 x 100 ml), saturated aqueous sodium bicarbonate (50 ml)
and brine
(50 ml). Drying over anhydrous sodium sulfate and solvent removal in vacuo
afforded
a dark oil. This was placed in a solution of acetic acid/THF/water (3:1:1) and
heated
to 50° for five hours. After cooling, the solvent was removed in vacuo
and the residue
placed on a flash silica gel column (3 cm x 40 cm) and eluted with ethyl
acetate/hexane
(40:60) (added methylene chloride to dissolve crystallized product off the
column.
Solvent removal afforded 3.7 g of the title compound (74 % yield) as light
yellow needles
(m.p. 174°C).
~ ~ -Octahydro-l-(1,2,3,4- tetrahydro-8-methoxy-2-naphthalenyl}-azocine
hydrochloride (A-2, Chart A).
To a solution of 1.76 g (10 mmoI) 8-methoxy tetralone and 5.66 g (50 mmol)
heptamethylenamine in 30 mL MeOH/THF (1:1) was added HOAc dropwise to adjust
WO 92/06967 PGT/US91/06$63
-25_
the pH to 4-S. The reaction mixture stirred for 15 minutes under N2, then 1.26
g (20
mmol) NaCNBH3 was added. When the reaction was complete by TLC (24 h), 1 N
NaOH (25 mL) and H20 (200 mL) was added to quench the reaction. The solution
was
extracted with CH2C12 (2 X 500 mL) and the combined organic layers were washed
with
brine, dried (MgS04), filtered and concentrated in vacuo. The resulting was
purified
by liquid chromatography on 400 g of silica gel 60 (230-400m), eluting with
hexane/ace-
tone (5:1). Fractions homogeneous by TLC were combined and concentrated in
vacuo
to give pure compound as an oil. The HCl salt was formed by using a MeOH/HCl
solution. The title compound was recovered as a white solid by
recrystallization using
EtOAc/MeOH (2.65 g, 86%): mp. 211-213°C.
1HNMR (CDCl3, TMS): 7.13(t, 1H); 6.69 (t, 2H); 3.81 (s, 3H); 3.54 (m, 3H),
3.31-3.18 (m, 3H); 2.94-2.67 (m, 4H); 2.17 (m, 2H); 2.1-1.46 (m, 8H).
IR (mull): v ~x 2529, 2503, 1585, 1470, 1463, 1453, 1251 crri 1.
Analysis: Calcd for C1gH27NO~HC1: C, 69.769; H, 9.108; N, 4.521.
Found: C, 69.6; H, 9.24; N, 4.65.
utilizing a procedure similar to that of Example 1 but substituting the
appropriate starting
materials there is obtained
Hexahydro-1-( 1,2,3,4-tetrahydro-8-methoxy-2-naphthalenyl)-1H-azepine
hydrochloride white solid: m.p. 236°-237°C.
1HNMR (CDC13, TMS): 7.12-7.04 (t, 1H); 6.71-6.64 (q, 2H); 3.81 (s, 3H);
3.03-2.76 (m, 7H); 2.45 (q, 1H); 2.0 (m, 1H); 1.62 (m, lOH).
IR (mull): v ~ 3000, 2600, 2550, 1590, 1480 cni 1.
Analysis: Calcd for C17H~NO~HC1: C, 69.017; H, 8.859; N, 4.735.
Found: C, 68.91; H, 9.04; N, 4.67.
Hexahydro-l-(1,2,3,4-tetrahydro-2-naphthalenyl)-1H-Azepine
Hydrochloride as a white solid: m.p. 243-245°C - Compound 1-3.
Example (1-Hexahydroazepinyl)-5,6,7,8-tetrahydronaphthalene-1-carboxamide (B-
5,
Chart B) - Compound 2-1.
Gaseous ammonia was bubbled through a solution of 7-(1-Hexahydroazeginyl)-
5,6,7,8-tetrahydmnaphthalene-1-carboxylic acid and 2.25 mL Triethylamine in 30
mL
DMF. After 10 min, 1.75 mL diethylcyanophosphonate was added and the solution
was
stirred overnight with ammonia bubbling through the solution. Direct spot TLC
showed
no starting material remaining. The reaction mixture was evaporated and
dissolved in
WO 92/06967 ~~'~~~'~ PCTJUS91/06~.'1,
~O J
-26-
mL methanol. This mixture was flash chromatographed on 200 g silica gel
eluting
with chloroform first, followed by chloroform/4M NH3 in methanol (95:5).
Homoge-
nous samples were combined and concentrated to yield 1 g of a solid.
Crystallized from
aceG~ne. (Mpt. = 141~C)
5 ~~~,ple 3 (+)-1,2,3,4-tetrahydro-2-pyrrolidino-8-bromo-tetralin (C-3, Chart
C) -
Compound 3-1.
8-Bromo-2-tetralone (40 g., 177 mmol) and pyrrolidine (3 ec~ were dissolved in
methanol (350 mL) with bromocresol green as an indicator. Hydrochloric acid (3
ec~
in methanol was added. Pyrrolidine (2 ec~ was added and the solution stirred
at CPC for
10 30 minutes. Sodium cyanoborahydride (2 ec~ was added and the solution
stirred for
another hour at 0°C. It was stirred at room temp for an hour then the
solvents were
removed under vacuum. Ether was added and sodium carbonate (sat'd, act. The
ether
layer was washed with brine, dried over sodium sulfate, and stripped of
solvent under
vacuum. After chromatography, the yield of pure oil was 29 g, 58.5%. The
racemate
(21.6 g) and D-tartaric acid (1 ec~ were dissolved in acetonitrile (1800 mL)
and methanol
(50 mL). The resulting white solid was recrystallized from acetonitrile until
a constant
optical rotation was achieved for the salt, [a]25= +34.07°, 165.0-
166.5°C mp. Yield
of the title compound as an oil was 2 g, [a]ZD= +61.27°.
~a~p,[e 4 (+)-1,2,3,4-tetrahydro-2-pyrrolidino-naphthalene-8-yl-carboxamide (C-
4,
Chart C) - Compound 4-1.
(+)-8-Bromo-2-pyrrolidinotetralin (1.84 g, 6.57 mmol) was dissolved in
tetrahydrofuran (13 mL) and cooled to -78°C. t-Butyllithium (1.7 M in
hexane, 2.1 ec~
was added. After 10 min, trimethylsilylisocyanate (2.5 ec~ was added. After 10
min,
the bath was removed and the solution allowed to warm to room temp. Sodium
carbonate (sat'd, act was added and the product extracted with
ether/chloroform. 1fie
organic layer was washed with water, brine, and then was dried over sodium
sulfate.
It was stripped of solvent under vacuum to yield white solid.
Recrystallization from
acetonitrile gave 0.63 g (39.390) of the hydrochloride of the title compound
as white
needles, 220.0- 224.0 °C, [a]~ _ +90.62° (228.5-230.5° mp
for the hydrochloride
salt).
Utilizing a procedure similar to that of Example 4, but substituting the
appropriate
bromotetralin there is obtained
(-)-1,2,3,4-tetrahydro-2-pyrrolidino-naphthalene-8-yl-carboxamide (mp 228.5-
WO 92/06967 PCT/US91106863
_2~- 2Q9fl~~~
230. s°c)
(+)-1,2,3,4-tetrahydro-2-(1-piperidinyl)-naphthalene-8-yl-carboxamide (mp
229°C)
(-)-1,2,3,4-tetrahydro-2-(1-piperidinyl)-naphthalene-8-yl-carboxamide
.s (mp 229°C).
The compound (+)1,2,3,4-tetrahydro-2-(1-pyrrolidinyl)naphthalene-8-yl-
carboxamide and its salts, processes for preparing said compounds and methods
of
employing said compounds in therapy represent the best mode of carrying out
the
invention.
Image
Image
WO 92/06967 PCT/US91/068(~3
a ~~~''~ -30-
~Q J
CHART B
oa g 1
I
i
B-1
N g 2
B-2
c~~3 NN g
I~
i
B-3
cooH
N X
B-4
CONHZ
N
I
i
B-S
WO 92/06967 PGT/US91/06863
-31-
&, ar
0 1 ~ 0 2
ci
C-1 C-2
Br
g 3 coNa2 N
U I
C-3 C-4
WO 9Z/06967 ~ PCT/US91/0684,~
_32_ ,
CHART D
Br
2
0
0
D-1 (G2) D-2
~,3 3 CF3 4
0~ 0
0
D-3 D-4
~3 ~"'1
N g
D-5
Image
WO 92/06957 PGT/US91/0685Z
-34-
~ ~~J . HART F
oo2cH3
0 1 i o 2
--.. I w _-,.
i
R R
F-1 F-2
OOZCH3
R1 ~ 3 R1
w o w 0 4
-r ( ~ -
R R3 8 R3
F--3 F-4
R1 g ' H
s
.--
R R3 t R3
F-5 F-s
WO 92/06967 PCT/US91/06863
~~~~32~
-35-
1 ~ 2
o -~" ~ ,.
i
G-1 (GZ) G-2 (C-3)
0/~ N
cso N g ' N
~i ~ ~i
G-3 G-4
WO 92/06967 PGT/US91/06g~
-36-
H
I ,. °~ 1 0'~ a
-. I ~ o --r
H-1 (D-2) H-2
o so2c~
I ~ ~ 3 0~ 4
y 0 -
i
H_3 H_4
s HH2
0 6
0
I ~ --. I ~ --.
i
H 5 H-6
i
H-7
Image
WO 92/06967 PCT/US91/068~,
-38-
0
CHART J
~~3
1 tt ~ 0 2
o --~.
i
i
J-1 J-2
o /-'
N
N
J-3