Note: Descriptions are shown in the official language in which they were submitted.
3 ;~
Case 600-7167
~mD P~ ~
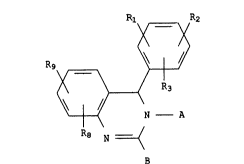
The invention relates to substitu~ed phenylquinazoline
derivatives. It concerns the ccmpounds of formula I
Rl R2
X~X
Rs ~ R3
N A
R8 N ~
B
wherein
~ 6 3 5
-2- 600-7167
Rl is hydrogen; alkyl of 1 to 3 carbon atoms; alkoxy of 1 to 3 carbon
atoms; trifluoromethyl; or fluoro or chloro;
R2 and Rg independently are hydrogen; alkyl of 1 to 3 carbon atoms; alkoxy
of 1 or 2 carbon atoms; trifluoromethyl; or fluoro or chloro;
R3 is hydrogen or alkyl of l or 2 carbon atoms;
either A and B together are a group of formula -C(R4R5)C(R6R7)-S- wherein
R4 is hydrogen; alkyl of 1 to 3 carbon atom~ optionally mono-
or independently di- or independently trisubstituted by
halogen of atomic number of from 9 to 35, whereby there is
at most 1 bromo; or phenyl optionally mono- or
independently disubstituted by alkyl of 1 to 3 carbon
atoms, alkoxy of 1 or 2 carbon atoms, or halogen of atomic
number of from 9 to 35, whereby there is at most 1 bromo;
R5 is hydroxy and R6 is hydrogen or
R5 and R6 together form a double (n) bond; and
R7 is hydrogen or alkyl of 1 or 2 carbon atoms;
or A is hydrogen and B i~ a group of formula -S-CHR7'-Rlo wherein
R7' is hydrogen or alkyl of 1 or 2 carbon atoms and
R1o is alkyl of 1 to 3 carbon atoms; alkenyl of 2 or 3 carbon
atoms; or -COORll wherein Rll is the re.3idue of an ester
group, with the proviso that R1o i~ other than methyl
when R7' is hydrogen;
R8 is hydrogen; alkyl of 1 to 3 carbon atoms; alkoxy of 1 to 3 carbon
atoms; trifluoromethyl; or halogen of atomic number of from 9 to 35;
in racemic or optically active, in free base or acid addition ~alt form,
hereinafter briefly named "the compound.~ of the inventionn.
When A and B together are a group of formula -C(R4R5)C(R6R7)-S-
it is the carbon atom carrying R4 and R5 which is bound to the nitrogen
atom.
2 ~ 9 ~ ~ ~, 3
-3- 600-7167
A compound of the invention in acid addition salt form preferably
is in pharmaceutically acceptable acid addition salt form. By thiC term is
meant an acid addition salt that ic physiologically acceptable, i.e. that
does not significantly increase the toxicity of the basic compound or
otherwise adversely affect its pharmacological activity. Examples are
salts with organic acids, e.g. the methanesulfonate, ethanesulfonate,
benzenesulfonate, p-toluenesulfonate, acetate, propionate, citrate and
tartrate salts, and salts with inorganic acids, e.g. the hydrochloride and
sulfate salts. Preferred are the pharmaceutically acceptable monoacid
addition salts. Although the co~pounds of the invention contain two
nitrogen atoms, only one is generally protonatable.
The residue of an ester group for significance R11 preferably is
the residue of a physiologically acceptable ester group. By this term is
meant a group which, together with the -COO- radical to which it is
attached, forms an ester group which is physiologically acceptable,
preferably a physiologically acceptable and hydrolyzable ester group, i.e.
a group which, together with the -COO- radical to which it is attached,
forms an ester group which i~ physiologically acceptable and hydrolyzable
under physiological conditions to yield a compound wherein Rlo i~ replaced
by -COOH and an alcohol which itself is physiologically acceptable, i.e.
non-toxic at the desired dosage level. The alcohol preferably is free of
centers of asymmetry. Examples of ~uch groups R11 are C1_3alkyl, n-butyl,
i-butyl, t-butyl and benzyl.
Subgroups of compounds of the invention are compounds wherein
- A and B together are a group of formula -C(R4R5)C(R6R7)-S-;
in a subgroup thereof (group Ia) R5 is hydroxy;
in a further subgroup thereof (group Ib) R5 and R6 together
form a double (n) bond;
- A is hydrogen and B is a group -S-CHR7'-R1o;
in a qubgroup thereof (group IIa) R1o i9 -COORll;
in a further subgroup thereof (group IIb) Rlo i9 other than -COORll.
2~3~3~3~
~4- 600-7157
Each compound of group Ib wherein no substituent contains a center
of asymmetry, each compound of group IIa wherein R7' is hydrogen and the
group -S-CHR7'-Rlo does not contain a center of a~ymmetry, and each
compound of group IIb wherein R7' is hydrogen or R7' and R1o are identical,
altogether has a single center of asymmetry (the ring carbon atom to which
the Rl-bearing phenyl group i~ attached), and, therefore, there are two
stereoisomer~ of each such compound. The two stereoisomers are designated
as the 5R and 5S enantiomers in the case of the compounds of group Ib and
the 4R and 4S enantiomers in the case of the aforementioned compounds of
groups IIa and IIb.
Each compound of group Ia wherein R7 i9 hydrogen and no further
substituent contain~ a center of asymmetry, altogether ha~ two centers of
asymmetry (the aforementioned ring carbon atom and the ring carbon atom to
which R4 is attached), and three, four or five centers of a~ymmetry when R4
contains one or two centers of asymmetry and/or R7 is Cl_2alkyl (the
aforementioned two ring carbon atoms and the ring carbon atom to which R7
is attached and/or any asymmetric carbon atoms in R4).
Each compound of group IIa wherein R7' i8 hydrogen and R
contains a single center of a9ymmetry or wherein R7' i9 Cl_2alkyl and R
i~ free of center~ of asymmetry, has two centers of a~ymmetry, and each
compound of group IIb wherein R7' is Cl_2alkyl and Rlo has a significance
that differ~ from that of R7' likewi~e has two centers of asymmetry (the
carbon atom to which the Rl-bearing phenyl group i~ attached and the carbon
atom to which R7' is attached or the center of asymmetry in Rll). Each
compound of group IIa wherein R7' is Cl_2alkyl and Rll contains at least
one center of asymmetry, or wherein R7' i9 hydrogen and Rll contains at
lea~t two centers of a3ymmetry, has three or more centers of a~ymmetry.
All stereoisomers of each compound of the invention are within the
scope of this invention. Preferred are those compounds containing one
center of acymmetry. Generally, insofar as the compounds of group Ib are
concerned, the 5R enantiomers and 5R,5S racemates are preferred over the
corresponding 5S enantiomers, with the 5R enantiomers being preferred over
the corresponding racemates. Also generally, insofar as the compound~ of
c3 5
-5- 600-7167
groups IIa and IIb having just one center of asymmetry are concerned, the
4R enantiomers and 4R,4S racemates are preferred over the corresponding 4S
enantiomers with the 4R enantiomers being preferred over the corresponding
racemates. Insofar as the compounds that contain more than one center of
asymmetry are concerned, these preferences repre~ent the preferred
configuration at the carbon atom to which the R1-bearing phenyl group is
attached.
Generally, the free bases of the compounds of group Ib are
preferred over the corresponding acid addition salts, and the acid addition
salts of the compounde of groups Ia, IIa and IIb are preferred over the
corresponding free bases.
R1 preferably is hydrogen. R2 preferably is hydrogen. R3
preferably is hydrogen. R4 preferably is hydrogen or alkyl of 1 to
3 carbon atoms, it more preferably is methyl or ethyl, especially methyl.
Rs and R6 preferably together form a double (n) bond. R7 preferably is
hydrogen. R7' preferably is hydrogen. R8 preferably is hydrogen or
chloro, especially hydrogen. When it i~ other than hydrogen the
substituent preferably is bound to the aromatic ring carbon atom in para
position to the nitrogen atom that is part of a double bond in formula I.
Rg preferably is hydrogen. Rlo preferably is alkyl of 2 or 3 carbon atoms,
alkenyl of 2 or 3 carbon atoms or -COORll~ wherein Rlla is alkyl of 1 to
3 carbon atoms, n-butyl, i-butyl, t-butyl or benzyl; R1o is more preferably
alkenyl of 2 or 3 carbon atom~ or ~COORlla, even more preferably vinyl or
-COORllb wherein Rllb is alkyl of 1 to 3 carbon atoms; Rlo is still more
preferably -COORll~; it is most preferably methoxycarbonyl or
ethoxycarbonyl, especially methoxycarbonyl.
When R4 ie optionally mono- or disubstituted phenyl it preferably
is unsubstituted phenyl.
A and B preferably are together a group of formula
-C(R4Rs)C(R6R7)-S-.
Alkyl of 1 to 3, or of 1 or 2 carbon atoms preferably is methyl.
Alkoxy of 1 to 3 or of 1 or 2 carbon atoms preferably is methoxy. Halogen
of atomic number of from 9 to 35, preferably is chlorine. Alkenyl of 2 or
3 carbon atoms preferably i9 vinyl.
~9~ 3~
-6- 600-7167
A further ~ubgroup of compound~ of the invention is the compounds
of formula Is
R~
R~, ~ Ia
N A~
N ~ B~
wherein
Rl~ is hydrogen, fluoro or chloro;
either A~ and B~ together are a group of formula -c(R4~R5)cHtR6)-s- wherein
R4~ is hydrogen; alkyl of 1 or 2 carbon atoms optionally
monosubstituted by bromo; or phenyl; and
R5 and R6 are as defined above;
or AJ is hydrogen and B~ is a group of formula -S-CH2-R1o~ wherein
Rloa i9 alkyl of 2 or 3 carbon atoms, alkenyl of 2 or
3 carbon atoms or -COORllJ wherein Rll~ is alkyl of 1 or
2 carbon atom~;
R8, ic hydrogen; alkyl of 1 or 2 carbon atoms; or halogen of atomic number
of from 9 to 35;
in racemic or optically active, in free base or acid addition salt form.
When A~ and B~ together are a group of formula -C(R4~R5)CH(R6)-S-
it i9 the carbon atom carrying R4~ and R5 which is bound to the nitrogen
atom. R4, preferably is methyl. R1o preferably is ethyl, vinyl or
methoxycarbonyl.
In a further subgroup of compounds of the invention R7 and R7' are
other than hydrogen. In a further subgroup Rlo i~ other than methyl. In a
further subgroup Rlo is oth~r than alkyl of 1 to 3 carbon atoms. In a
further subgroup R1o is -COORll. In a further subgroup Rl, R2 and R3 are
hydrogen. In a further subgroup R1, R2, R3, R~ and Rg are hydrogen.
~,~i J~ 3 3
-7- 600-7167
The compounds of the invention may be prepared by a proces3 which
comprises
a~ for the preparation of the compounds of formula I wherein
R5 and R6 together form a double (n) bond,
dehydrating a corresponding compound of formula I wherein
R5 is hydroxy and R6 is hydrogen;
b) for the preparation of the compounds of formula I as defined above
under formula I,
submitting to appropriate ring closure a compound of formula II
Rl R2
V~V
(\ /~
Rg ~ II
NH
R8 N
H
wherein the substituents are as defined above under formula I; or
c) for the preparation of the compounds of formula I wherein
A is hydrogen and B is a group of formula -S-CHR7'-R1o,
appropriately al~ylating a corresponding compound of formula II,
e.g. with a corresponding compound of formula III
X'-CHR7'-Rlo III
wherein X' is a reactive group and R7' and R1o are aq defined above;
and recovering the resultant compound3 of formula I in racemic or optically
active, in free base or acid addition salt form.
3 5
-8- 600-7167
The above process for the preparation of the compounds of the
invention may be effected in conventional manner.
Process variant a) (dehydration) is effected in an inert anhydrous
organic solvent such as 2-propanol or toluene. The temperature preferably
is from about 60 to about 120C, preferably from 80 to 83C or 100 to
112C. Particularly when R4 is hydrogen it may be indicated to convert to
the free base prior to dehydration.
Process variant b) (ring clo~ure) is e.g. effected with a compound
of formula
R4-CO-CHR7-X, preferably R4x-CO-CHR7-X, or of formula (R0)2CH-CHR7-X
wherein
R4 and R7 are as defined above,
R4X with the exception of hydrogen has the significance indicated above
for R4,
X i9 a reactive group, preferably halogen of atomic number of from 17 to
53, especially bromo, and
R i~ methyl or ethyl, preferably ethyl.
It thus include~ a preliminary alkylation ~tep. For the
preparation of the compounds wherein Rs is hydroxy and K6 i9 hydrogen, the
reaction, when a compound of formula R4-CO-CHR7-X is used, preferably is
effected at a temperature of from about -20 to about +50C, preferably
starting at about -20 to -10C, allowing to warm to 20-25C, and
completing the reaction at that temperature. The reaction preferably is
effected in an inert organic solvent such an ether solvent, especially
tetrahydrofuran or a mixture of diethyl ether and tetrahydrofuran.
ereferably an inert atmosphere is used. When a compound of formula
(R0)2CH-CHR7-X is used, preferably the temperature is from about 50 to
about 80C. The solvent preferably is a mixture of an ether solvent and
water. For the preparation of the compounds wherein Rs and R6 together
foDm a double (n) bond, preferably a compound of formula R4X-C0-CHR7-X is
used. When X is chloro, it i3 preferred to use a catalytic amount of
sodium iodide or, preferably, bromide. The temperature preferably is from
about 70 to about 80C. The inert organic solvent preferably is a
C2_4alkanol, especially 2-propanol. An inert atmosphere i~ preferred.
~ 3
-9- 600-7167
Process variant c) (al~ylation) preferably is effected with a
group R1o containing no substituent that would interfere with the reaction.
When R1o is a group -COOR11 which might so interfere, then it may be
indicated to effect the alkylation with a reagent containing a group
-COOR11 which does not interfere, and to transesterify to another group
-COOR11 after alkylation has taken place. X' preferably is halogen of
atomic number of from 17 to 53, preferably bromo. The alkylation reaction
preferably is effected at a temperature of from about 20 to about 75C.
Preferably an anhydrous inert organic solvent i9 used, especially an ether
solvent such a diethyl ether or especially tetrahydrofuran. Preferably the
reaction is effected in an inert atmosphere. Transesterification, if
indicated, may e.g. be effected in an inert aqueous organic solvent, e.g. a
mixture of water and a lower alkanol such as methanol or, especially,
ethanol; it is normally effected in two steps, saponification e.g. with a
strong inorganic base such as sodium or potassium hydroxide, at a
temperature of from about O to about 75C, preferably 20-50C, followed
by the addition of sufficient acid, e.g. 2 N hydrochloric acid, to lower
the pH to 2-4.5, at a temperature of 20-25C, isolation of the free acid,
and then esterification at a temperature of from about 20 to about 40C,
preferably 20-25C, with alcohol Rl1OH and a catalytic amount of a strong
acid, e.g. p-toluenesulfonic acid or hydrate thereof, in an inert anhydrous
organic solvent, e.g. an ether solvent such a~ tetrahydrofuran or alcohol
Rl1OH, if a liquid.
A compound of the invention may be recovered from the reaction
mixture and purified in conventional manner.
A com~ound in free base form may be converted in conventional
manner into a ~alt form and vice-versa. For the compounds of the invention
wherein R5 and R6 together from a double (n) bond the free base may be
recovered from a salt form e.g. by neutralization with an inorganic base,
preferably sodium bicarbonate, sodium carbonate or sodium hydroxide. The
temperature preferably is from about 15 to about 50C, preferably from 20
b ~ ~
-10- 600-7167
to 45C. It is effected e.g. in water or a mixture of water and an organic
solvent such as ethyl acetate. For the compounds wherein R5 is hydroxy and
R6 is hydrogen neutralization is e.g. effected in an organic base, e.g. a
tri-(Cl_3alkyl)amine, preferably triethylamine. The temperature is from
about 0 to about 25C, preferably from 20 to 25. An anhydrous organic
solvent preferably is used, preferably a hydrocarbon such as toluene. For
the neutralization of compounds wherein B is a group -S-CHR7'-Rlo,
e.g. either set of condition~ above may be used.
An acid addition salt form of a compound of the invention may be
recovered from the free base form e.g. by treating a solution or ~uspension
of the free base in a solvent such as a lower alkanol, e.g. methanol or
ethanol, or a mixture thereof with water, at, for example, from about 5 to
about 50C, preferably 10 to 40C, with, for example, 0.98-6 equivalents
of a pharmaceutically acceptable acid for, for example, 2 to 150 minutes,
optionally extracting the reaction mixture with an organic solvent that is
immiscible therewith, and concentrating or evaporating the reaction mixture
at reduced pressure to obtain the acid addition salt. Preferably, however,
irrespective of whether the acid is monovalent, divalent, trivalent, etc.,
0.98 to 2, preferably 0.99 to 1.02, moles of the acid per mole of the
compound of the invention in free base form is employed to obtain a
monoacid addition ~alt.
The compoundq of the invention, particularly tho~e wherein R5 is
other than hydroxy, may be obtained in optically pure form by utilizing
optically pure starting materials. Alternatively, a racemic form of a
compound of the invention may be converted into an optically active form in
conventional manner. Thus, each compound having one center of a~ymmetry
may be resolved into two optically active isomers by conventional
techniques. For example, it may be reacted with an optically pure
carboxylic acid having at least one center of asymmetry to form a mixture
of diastereoisomeric salts that may be separated e.g. by fractional
crystallization or column chromatography. After separation, the optically
pure salt may be reconverted to the corresponding compound in free base
3 ~
-ll- 600-7167
form by conventional means with retention of optical purity. Any compound
of the invention having two (or more) centers of asymmetry, e.g. the
compounds wherein R5 i9 hydroxy, may be separated into four (or more)
stereoisomers by conventional techniques. For example, diastereoisomers
may be separated by frastional crystallization, column chromatography,
preparative thin layer chormatography and HPLC, and each obtained racemate
may be resolved as set forth above.
If the compoundq wherein R5 is hydroxy are not easily separable,
they may be reacted with a carboxylic acid or, preferably, a reactive
carboxylic acid derivative such as a halide or anhydride, optionally
containing a center of asymmetry, to form the corresponding esters. The
esters may be separated as ~et forth aboqe and, after separation,
reconverted to the compounds of the invention with retention of optical
purity.
The compounds having one center of asymmetry are preferably
resolved by selectively precipitating one enantiomer from a solution
containing the D-(-)- or L-(+)-tartrate salts of a racemic compound having
just one center of asymmetry, i.e. from a solution containing substantially
equal amounts of the D-(-)-tartrate or L-(+)-tartrate salts of the 5R and
5S enantiomers of such a compound, or from a solution of such salts
enriched with one enantiomer. The precipitated ~alt contains at least
75 %, more preferably at least 98 % and especially at least 99.5 % of a
single enantiomer. The precipitated qalt is then neutralized to obtain the
correqponding free base having the same enantiomeric purity.
This procedure i8 particularly applicable to the compounds of the
invention wherein R5 and R6 together form a double (n) bond, especially the
compound of Example 2. Preferably, D-(-)-tartaric acid is utilized when it
iY desired to selectively precipitate a salt of the 5R enantiomer of these
compounds and thereby obtain the 5R enantiomer, and L-(+)-tartaric acid to
obtain the 5S enantiomer, particularly for the compound of Example 2.
Preferably, the precipitated tartrate salt of these compounds,
particularly when it is the compound of Example 2, is a monoacid addition
salt, i.e. a salt wherein one molecule of tartaric acid is associated with
each molecule of the compound, and it is the nitrogen atom that is part of
3 ~
-12- 600-7167
a double bond in formula I that is protonated, i.e. associated with the
tartaric acid. The precipitated tartrate salt may be solvated, i.e. the
salt may contain scme solvent. When the tartrate salt is precipitated from
methanol, particularly when it is a tartrate salt of the compound of
Example 2, it contains some methanol. Preferably, and particularly when
the compound is the compound of Example 2 and its tartrate salt i9
precipitated from methanol, it is precipitated at a temperature of
25-29C, e~pecially 27C.
Also preferably, and particularly when the compound i9 the
compound of Example 2 and its tartrate salt is precipitated from a
methanolic solution having a temperature as set forth in the preceding
paragraph, the solution has a maximum concentration of 0.3 M, preferably
0.21 M, of the two enantiomers together. In each case the minimum
concentration is preferably 0.1 M, more preferably 0.19 M. The most
preferred concentration is 0.20 M.
Resolution is preferably carried out by forming a solution of the
D-(-)-tartrate or L-(+)-tartrate salts of such a racemic compound,
selectively precipitating one enantiomer, and neutralizing the precipitated
enantiomer. The solution of the D-(-)- or L-(+)-tartrate salts of the
racemic compound is formed by, for example, reacting said racemic compound
with 0.98-2, preferably 1.0, mole of above acid per mole of the racemic
compound in, preferably, methanol, at a temperature of 30-60C, preferably
35-45C. The temperature of the methano~ic solution is initially as set
forth in this paragraph, and the temperature to which it is adjusted to
effect the selective precipitation and it~ concentration are as set forth
above. The precipitated tartrate salt is isolated and neutralized by
treatment with, for example, at least one equivalent of a base, preferably
an inorganic base such as sodium or potassium hydroxide, carbonate or
bicarbonate. If desired, the obtained single enantiomer is then isolated.
Preferably, the D-(-)- or L-(~)-tartrate salts are monoacid
addition salts of a 5R,5S racemate of a compound having ju~t one center of
asymmetry, preferably the compound of Example 2, the methanolic solution of
said salts has a temperature of 30-65C and a maximum concentration of
2 ~ 3 ~
-13- 600-7167
0.3 M of the two enantiomers together, and the temperature of the solution
is adju~ted to 25-29C to effect the selective precipitation. More
preferably, the methanolic solution has a temperature of 35-45C and a
concentration of 0.15-0.22 M, especially 0.20 M, prior to adjustment of the
temperature to 26.5-27.5C, especially 27C, to effect the selective
precipitation.
If necessary and/or desired, the precipitated tartrate salt or the
isolated free base may be recrystallized one or more times to obtain a
purer product.
A mixture of tartrate salts enriched with the non-precipitated
enantiomer may be obtained from the mother liquor and washings from which
additional quantities of the desired (i.e. precipitated) enantiomer may be
obtained, before, after or without isolating the undesired (i.e.non-
precipitated) enantiomer. Preferably, however, the mother liquor and
washings are combined and neutralized to obtain a mixture of the free bases
enriched with the other (i.e. undesired or non-precipitated) enantiomer,
the mixture of free bases is reacted with the other tartaric acid
[i.e. L-(+)-tartaric acid if the mixture of free bases is enriched with the
5S enantiomer and D-(-)-tartaric acid if it is enriched with the 5R
enantiomer] to form a mixture of tartrate salts, the undesired enantiomer
is selectively precipitated, the resulting mother liquor and washings are
combined and neutralized to obtain a mixture of free base~ now enriched
with the desired enantiomer and the original procedure is repeated to
obtain an additional quantity of the desired enantiomer. The "recycling"
may be repeated one or more times to obtain additional quantities of the
desired enantiomer.
Alternatively, the undesired enantiomer of the tartrate salts is
first precipitated, and a mixture of tartrate salts enriched with the
desired enantiomer is obtained from the mother liquor and/or the combined
mother liquor and washings. The mixture of tartrate salts enriched with
the desired enantiomer i~ then neutralized to form a mixture of free bases
enriched with the desired enantiomer which is used as the ~tarting material
in lieu of a racemate to obtain a larger quantity of the desired isomer
than had the racemate itself been utilized with no ~recyclingn.
~i i}~3~
-14- 600-7167
The starting materials may be obtained in conventional manner.
The compounds of formula II may e.g. be prepared by reaction of
2-aminobenzophenone or an appropriately substituted 2-aminobenzophenone
derivative with a reducing agent such as sodium borohydride and reaction of
the resultant ~-(2-aminophenyl)benzenemethanol derivative with e.g.
ammonium thiocyanate under acidic conditions.
The compounds of formula II may be prepared in optically active
form e.g. by reacting 2-aminobenzophenone or an appropriately substituted
2-aminobenzophenone derivative with hydroxylamine hydrochloride, reacting
the resultant 2-(hydroxyiminophenylmethyl)benzeneamine derivative with a
reducing agent such as powdered zinc in ammonium acetate and ethanol in the
presence of ammonium hydroxide, followed by salt formation of the resultant
racemic a-(2-aminophenyl)benzylamine derivative with either D-(-)- or
L-(+)-tartaric acid and fractionation of the resultant corresponding
diastereoisomeric ~-(2-aminophenyl)benzylammonium tartrate salts to give
the optically pure tartaric salts, conversion to the optically pure
a-(2-aminophenyl~benzylamine with e.g. sodium carbonate or bicarbonate, and
cyclization to the corresponding compound of formula II in optically active
form with e.g. thiophosgene and an organic base or l,1'-thiocarbonyldiimi-
dazole in an inert organic solvent.
Insofar as its preparation is not specifically described herein, a
compound used as a ~tarting material is known or may be prepared by known
methods starting from known compounds, e.g. as described in the Examples.
2~Q~6~
-15- 600-7167
The following Examples illustrate the invention. Temperatures are
in degrees Centigrade. m.p. s melting point; Me = methyl; Phe 5 phenyl;
Et = ethyl; b = in free base form. Optical rotation: c is given in g/dl.
~le 1: (R~ - and (S) -3-Methyl-5-phenyl-SH-thiazolo [2~ 3-bl quinazoline
[Formula I: A + B = -C(R4R5)C(R6R7)-S-, with the carbon atom carrying
R4 and R5 bound to the nitrogen atom;
R~, R2, R3, R7, R8, Rg = H; R4 = Me;
R5 + R6 = bond;
in R or, re3pectively, S configuration at C5;
in free base and hydrobromide salt form]
[Process variant a)(dehydration), and conversion of salt into free base]
a) Hydrobromide salt: A su~pension of 42 g of (5R)-2,3-dihydro-3-methyl-
5-phenyl-5H-thiazolo [ 2, 3-b] quinazolin-3-ol hydrobrallide ( R-i somer of
compound of Example 7) in 600 ml of 2-propanol i~ slowly heated to reflux
and refluxed for 2.5 hour~3, and the volume is reduced at reduced pre~sure
to obtain (R) -3~ethyl-5-~henyl-5~1-thiazolo 12, 3-b] quinazoline hydrob~allide .
The S enantier in hydrobromide salt form is obtained in
analogous manner.
b) Free base: 200 ml of saturated sodium chloride solution i9 added to the
product obtained under a) above, 300 ml of 2N sodium carbonate solution is
added, and the reaction mixture is stirred at 20-25 for 5 minutes and
extracted three times with methylene chloride. The methylene chloride
extracts are combined, dried over anhydrous ~odium sulfate, filtered and
evaporated at reduced pressure until the solution becomes cloudy.
Anhydrous diethyl ether is added with cooling at 0-5, and the resulting
solid is collected by filtration, washed with petroleum ether and vacuum
dried at 40 for 5 hours to obtain the Rtitle compound [m.p. 177-179;
[~]D25 = -181.1 (c = 1, CH30H)].
The S title compound i~ obtained in analo~ous manner [99.45 %
S product; m.p. 173-176; [a]D25 = + 151.8 (c = 1, CH30H)].
-16- 4 ~ ~ ~ b33 600-7167
E~ample 2~ 3-~ethyl-5-Phen~l-5~-thiarolol2~3-bl~uinazoline
[Formula I: as for Example 1;
in racemate form;
in free base and hydrochloride salt form]
[Process variant b~ (ring closure)]
a) Bydrochloride salt: 13.7 g of sodium bromide is added to a suspension
of 173.7 g of (+)-3,4-dihydro-4-phenyl-2(1~)-quinazolinethione (compound of
formula II) in 965 ml of 2-propanol, the resulting suspension is heated to
75 with stirring, 75.2 ml of 96 % chloroacetone is added over a 10 minute
period to the suspension stirred at 75-78, the addition being mildly
exothermic, and the resulting white suspension is stirred at 75 for
3 hours, the reaction mixture being stirred under nitrogen throughout. A~
much of the solvent as possible is removed by distillation at 40-45 and
175-l90 mmHg to obtain a thick stirrable slurry containing the title
compound in hydrochloride salt form, which is used further without
i301ation of the product.
b) Free base: 565 ml of ethyl acetate i9 added to the thick stirrable
slurry obkained under a) above, the resulting suspension is heated to 45
and, over a period of 10 minutes, a solution of 126 g of sodium bicarbonate
in 1.13 l of water is added dropwise with stirring at 35-45. The
resulting light beige su~pension is stirred at 45 for 1 hour, cooled to
20 and vacuum filtered. The filter cake is washed with two 625 ml
portions of warm (about 50) water and then with two 110 ml portions of
cold (about 0) ethyl acetate, and the damp solid is suspended in 2.1 1 of
acetonitrile. The suspension is heated to gentle reflux (about 81) to
obtain a solution, the solution is cooled to 10 over a period of about
15 minutes, and the resulting suspen~ion is stirred at 10 for 1 hour and
vacuum filtered. The filter cake is washed with 250 ml of cold (about 0)
acetonitrile, and the solid s collected and vacuum dried at 45-50 to
constant wei~ht to obtain the pale, light amber solid title co~pound in
free base forn (m.p. 184-185).
2 (~ a
-17- 600-7167
The starting material is obtained as follows:
a) 30.4 g of sodium borohydride pellets is added portionwise over a
5 minute period to a mixture of 301 g of 98 % 2-a~inQbenzophenone and
650 ml of 95 % ethanol stirred at 65, the addition being mildly
exothermic, and the reaction mixture is stirred at 65-70 for 1.5 hours to
obtain an off-white suspension, the reaction mixture being stirred under
nitrogen throughout. 550 ml of water is added over a period of 10 minutes
with stirring at 60-65, the addition being slightly endothermic, to
obtain a white mixture containing (~ -(2-aminophenyl)benzenemethanol
which is used further without isolation.
b) A solution of 125 g of ammonium thiocyanate in 250 ml of water is added
over a period of 20 minutes to the white misture obtained under a~ above,
stirred at 65-68, the addition initially being slightly exothermic. A
solution of 205 ml of 36 % hydrochloric acid in 400 ml of water is added
dropwise over a period of about 20 minutes with stirring at 65-70, the
addition being exothermic. The resulting bright white mixture is stirred
zt 65-70 for 2 hours, cooled to 45 and vacuum filtered through a Buchner
funnel having a polypropylene filter pad. The filter cake is washed with
three 500 ml portions of warm (50) water and then with two 300 ml portions
of 95 % ethanol. The resulting damp solid is suspended in a mixture of
270 ml of methanol and 300 ml of water, 800 ml of water is added, and the
suspension is stirred at 50 for 45 minutes and filtered at this
temperature through a Buchner funnel having a polypropylene filter pad.
The resulting filter cake is washed with two 250 ml portions of warm
(about 50) water and then with 150 ml of cold (about 5) methanol. The
solid is collected and vacuum dried at 40-45 to constant weight (about
16 hours) to obtain the 100 % pure ~ 3~4-~ihydro-4-phenyl-2(
quinazolinethione as a white solid (m.p. 226-229).
2a~ib3a
-18- 600-7167
xample 3: (R)- and (S)-3-methyl-5-phenyl-5~-thiazolo[2,3-bl-
quinazoline [S-(R~, R~)]-2,3-dihydrosybutanedioic acid ~alt
[Formula I: as for Example 1;
in R or, respectively, S configuration at C5;
in D-(-)-tartaric acid salt form]
[Conversion into vptically active salt]
A suspension of 185 g of title compound of ~ample 2 in 3.33 1 of
methanol is heated to 35-40, 100 g of D-(-)-tartaric acid is added
portionwise over a 5 minute period during which the temperature is
maintained at about 40. The resulting solution is cooled to 27 and
stirred at 27-28 for 2 hours, the reaction mixture being stirred under
nitrogen throughout. The suspension is filtered through a Buchner funnel
having a polypropylene filter pad, the filter cake is washed with 200 ml of
cold (about 0) methanol, and the solid is collected and vacuum dried at
40-45 to constant weight (about 16 hours) to obtain the 99.86 % pure
R-enantiomer as a white solid [m.p. 99 (dec.); []D25 = -108.51 (c = 1,
CH30H)].
An approximately 3:7 mixture of the R and the S enantiomer may be
obtained from the combined mother liquors and washings.
~ample 4: (R)-3~HethYl-5-Phenyl-5H-thiazolol2~3-~]quinazoline
[Formula I; as for Example 1;
in R configuration at C5;
in free base form]
[Conversion of salt into free base]
A solution of 56.3 g of sodium bicarbonate in 570 ml of water is
added dropwi~e over a 15 minute period to a suspension of 75.0 g of
3~!~3~
-19- 600-7167
title compound of ~ample 3, R-i~o~er in 875 ml of ethyl acetate stirred at
35, the resulting biphasic mixture is stirred at 35-40 for 1 hour, and
the (upper) organic phase is separated, washed three times with 100 ml
portions of warm (about 30-35) water and concentrated by vacuum
distillation at 45 and 100 mmHg to obtain a fluid residue. 375 ml of
n-heptane is added, and the mixture is cooled to -10, stirred at this
temperature for 15 minutes and vacuum filtered. The filter cake is washed
with 100 ml of cold (about 0~ n-heptane, and the solid is collected and
vacuum dried at 50-55 to constant weight ~a~out 16 hours) to obtain the
99.93 % pure title compound as a white solid [m.p. 177-179;
[]D25 = -181.1 (c = 1, CH30H)].
ample 5: (R)-3-~ethvl-5-phenvl-5H-thiazolo12,3-b]quinazoline
hydrochloride
[Formula I: as for Example 1;
in R configuration at C5;
in hydrochloride ~alt form]
[Conversion of free base into ~alt]
A solution of 1.42 g of anhydrou~ hydrogen chloride in 33.3 ml of
anhydrous diethyl ether is added dropwise over a period of 10 minutes to a
solution of 11.0 g of title compound of ~zample 4 in 300 ml of dry
tetrahydrofuran stirred at 5~, 300 ml of anhydrous diethyl ether is added,
the reaction mixture is stirred for 10 minutes, and the precipitate is
collected by filtration and vacuum dried to obtain the title compound
[m.p. 238-241].
2~9~3~
-20- 600-7167
~a~ple 6~ 7-Chloro-3-methYl-5-phenYl-5~-thiazolo[2,3-b]quinazoline
[Formula I: A + B = -C(R4R5)C~R6R7)-S-, with the carbon atom carrying
R4 and R5 bound to the nitrogen atom;
Rl, R2, R3, R7, Rg = H; R4 = Me;
R5 + R6 = bond;
R8 = 7-Cl (i.e. is separated by 4 carbon atoms from each of
the two nitrogen atoms in formula I);
in racemate form;
in free base and hydrobromide salt form]
[Process variant a) (dehydration) and conversion of salt into free base]
a) ~Ydrobromide ~alt: The title compound in hydrobro~ide ~alt for~ is
prepared analogously to Example la) starting from the compound of
~ample 8.
b) Free ba~e: The title compound in free ba~e form (m.p. 220-223) is
obtained analogously to Example lb), starting from the product of ~tep a)
above.
The title compound in free base and hydrobromide salt form is a
racemate that may be resolved to obtain the 5R _nd 5S Pnanti er~.
Alternatively, the two enantiomers may be prepared analogously to
Examples 7 and 8 starting from single isomers.
2~S3~
-~1- 600-7167
E~a~ple 7: (5R)- and (5S~-2,3-Dihvdro-3~methvl-5-phenYl-5~-thia
[2,3-b]~uinasolin-3-ol h~drobroJide
[Formula I: A ~ B = -C~R4R5)C(R6R7)-S-, with the carbon atom carrying
R4 and R5 bound to the nitrogen atom;
Rl, R2, R3, R6, R7, R8, Rg = H; R4 = Me;
R5 = OH;
in R or, respectively, S configuration at C5,
in hydrobromide salt form]
Process variant b) (ring closure)]
26.7 g of bro~oacetone dried over anhydrous sodium sulfate i~
added dropwise over a period of 5 minutes to a solution of 29.6 g of
(R)-3,4-dihydro-4-phenyl-2(1~)-quinazolinethione (compound of formula II)
in a mixiure of 400 ml of dry tetrahydrofuran and 170 ml of anhydrous
diethyl ether stirred at -20 to -10, and the reaction mixture is allowed
to warm 20-25 and stirred at this temperature for 64 hours, the reaction
mixture being maintained under nitrogen throughout. The resulting solid is
collected by filtration, washed with dry tetrahydrofuran, washed with
anhydrous diethyl ether and vacuum dried at 40 for 5 hours to obtain the
5R title compound [m.p. 162-163 (dec.); a previous batch melted at
168-170 (dec.)].
This compound may be a mixture of two diastereoisomers that may be
separated by conventional means to obtain the 3R,5R and 3S,SR enantio~ers.
The 5S title compound [m.p. 159-160~ (dec.)] is obtained in
analogous manner from the corresponding S starting material. It may be a
mixture of two diastereoisomers that may be separated by conventional meanQ
to obtain the 3R,5S and 3S,5S enantioners.
The R starting material (for the 5R title compound) is obtained as
follows:
a) 478.85 g of hydro~ylamine hydrochloride i9 added to 400 g of
2-aminobenzophenone in 3 1 of absolute ethanol, 470 ml of pyridine is
~ ~&~
-22- 600-7167
added, and the reaction mixture is gently refluxed for 16 hours, allowed to
cool and evaporated at reduced pressure to about 25 % of its original
volume. The resulting thick dark green oil is added to ethyl acetate, and
working up effected in conventional manner obtain
2-(hydroxy;minophenylmethyl)benzeneamine (m.p. 154-157; another batch
melted at 157-159).
b) 77 g of ammonium acetate is added to 285.63 g of product of step a)
above in 1.2 l of 95 % ethanol, 538 g of powdered zinc is added, 2.25 1 of
29 % ammonium hydroxide solution is added, and the reaction mixture is
warmed to 38, maintained at this temperature for 1.5 hours, gently
refluxed for 16 hours, cooled to 60 and filtered. The zinc is washed with
ethyl acetate, and the ethyl acetate washing iq added to the filtrate.
500 ml of 2N sodium hydroxide solution and 1 l of saturated sodium chloride
solution are added to the filtrate. The mixture is worked up in
conventional manner obtain crude (+)-~-(2-aminophenyl)-
benzylamine as an oil.
c) 165.1 g of D-(-)-tartaric acid i~ added to a solution of 217.8 g of
product of step b) above in 3.4 1 of 98.5 % ethanol stirred at 50. The
reaction mixture is refluxed for 30 minutes, 500 ml of 95 % ethanol is
added, and the mixture is refluxed for 3 hours and filtered hot (70-78).
The solid is washed with 400 ml of hot (70-78) absolute ethanol, washed
with petroleum ether and vacuum dried at 40 for 5 hours to obtain the
crude product. The crude product is added to 1.7 l of absolute ethanol,
the mixture is refluxed for 1 hour and filtered hot (70-78), and the
solid is washed with 250 ml of hot (70-78) absolute ethanol, washed with
200 ml of petroleum ether and vacuum dried at 40 for 5 hours to obtain
(R)-~-(2-ammoniumphenyl)benzylam~onium D-(-)-tartrate (94.58 % R product).
d) 500 ml of ethyl acetate is added to 80.27 g of product of step c) above
in 500 ml of water and 500 ml of saturated sodium chloride solution, 600 ml
of 10 % sodium bicarbonate solution is added, the reaction mixture is
stirred at 20-25 for 10 minuteq, the ethyl acetate layer is separated,
~9'3~3~
-23- 600-7167
and the aqueous layer is reextracted twice with ethyl acetate. The three
ethyl acetate layers are combined, dried over anhydrous sodium sulfate,
filtered and evaporated to dryness at reduced pressure to obtain
(R)-~-(2-aminophenyl)benzylamdne ~94.58 % R product).
e) 33.5 g of l,l'-thiocarbonyldiimidarole is added over a period of
20 minutes to a solution of 35.5 g of product of ~tep d) above in 500 ml of
HPLC grade methylene chloride stirred at -40 to -30, and the reaction
mixture is allowed to warm to 20-25 with stirring and stirred at 20-25
for 16 hours, the reaction mixture being maintained under nitrogen
throughout. The reaction mixture is extracted with a 1:1 mixture of
saturated sodium chloride solution and water, and the aqueous layer is
reectracted three times with methylene chloride. The four methylene
chloride layers are combined, dried oPer anhydrous sodium sulfate, filtered
and evaporated at reduced pressure until the solution becomes cloudy. The
cloudy solution i9 cooled to 0-5, and the resulting Qolid is collected by
filtration and vacuum dried at 40 for 5 hours to obtain ~R)-3,4-dihydro-
4-phenyl-2(1~)-quinazolinethione (more than 97.3 % R product;
m.p. 207-211). The filtrate is partially evaporated at reduced pressure
and cooled, and the resulting solid is collected by filtration and vacuum
dried at 40 for 5 hours to obtain a second crop.
The corresponding S ~tarting material (for the 5S title compound)
is obtained analogously (m.p. 205-209) using for step c) the following
conditions:
c') 202.6 g (1.35 moles) of L-(+)-tartaric acid is added to a solution of
26g.4 g (1.35 moles) of (~ -(2-aminophenyl)benzyla~ine in 5 1 of about
97.6 % ethanol qtirred at 50, and the reaction mixture is heated to reflux
over a period of about 30 minuteq (a precipitate forming at about 60),
refluxed for 30 minutes, cooled to 75 and maintained at this temperature
for 1 hour. 800 ml of absolute ethanol is added, and the solid is
collected by filtration (while warm), washed with 600 ml of hot (70-78)
) 3 5
-24- 600-7167
absolute ethanol, washed with 200 ml of petroleum ether and dried at 40
under high vacuum for 5 hours to obtain crude (S)-~-(2-am~oniumphenyl)-
benzylsmmonium L-tartrate (85.67 % S). The crude product is added to 2 1
of absolute ethanol, the mixture is refluxed for 1 hour and filtered while
hot ~70-78), and the collected solid is washed with 300 ml of hot
(70-78) absolute ethanol, washed with petroleum ether and vacuum dried at
40 for 5 hours to obtain 91.39 % S crude product. The crude product is
added with rapid agitation to 4.5 l of absolute ethanol, the mixture is
refluxed for 2 hours and filtered while hot (70-78), and the collected
solid is washed with 400 ml of hot (70-78) 95 % ethanol, washed with
petroleum ether and vacuum dried at 40 for 5 hours. The filtrates and
washings are combined, evaporated at reduced pressure to a total volume of
4.25 l and allowed to stand for 16 hours at 20-25. The resulting solid
is collected by filtration, washed with petroleum ether and vacuum dried at
40 for 5 hours to obtain 98.8 % S product (29.31 g). The filtrates and
washings are combined and evaporated at reduced pressure to about 50 % of
the original volume and allowed to stand for 16 hours at 20-25. The
resulting solid is collected by filtration, washed with petroleum ether and
vacuum dried at 40 for 5 hours to obtain a second crop (95.5 % S product).
2~ 3 ~ 35
-2S- 600-7167
Example 8~ 7-chlorn-2r3-c~lr~lro~3-methyl-5-pbenyl-5~-ehia
[2,3-b)q~ LulLinr3-ol bY~hlihn~mi
[Formula I: A + B = -C(F4R5~C(R6R7)-S-, with the carbon atom carrying
R4 and Rs bound to the nitrogen atom;
Rl, R2, R3, R6, R7, R9 = H; R4 = Me;
Rs = OH;
R8 = 7-Cl ~i.e. is separated by 4 carbon atoms from each of
the two nitrogen atoms in formula I);
in racemate form;
in hydrobromide salt form)]
[Process variant b) (ring closure)]
The title compound [m.p. 312-314 (dec.)] is obtained analogously
to Example 7, starting from (~)-7-chloro-3,4-dihydro-~-phenyl-2
quinazolinethione (compound of formula II). It is a mixture of
stereoisomers which may be separated by conventional means.
The starting material is obtained as follows:
a) 40.85 g of sodium borohydride is added portionwise to a solution of
250 g of 2-amino-5-chlorobenzophenone in 2.5 1 of HPLC grade methanol
stirred at 0 to 5 over a period of 4 hours, and the reaction mixture is
allowed to warm to 20-25 and stirred at this temperature for 16 hours,
the reaction mixture being maintained under nitrogen throughout. Ice and
sufficient acetic acid to adjust the pH to 6 are added, and working up
effected in conventional manner (~ -(2-A~dno-5-chlorophenvl)benzene-
methanol is obtained ~m.p. 104-107).
b) 85 ml of concentrated hydrochloric acid is added dropwise to 199.01 g of
compound of step a) above in 1.2 1 of water over a period of 30 minutes at
20-25, 70.3 g of ammonium thiocyanate is added in small portions, and the
reaction mixture is slowly heated to reflux with rapid agitation, refluxed
for 2.5 hours, allowed to cool to 20-25 and rapidly stirred at this
temperature for 16 hours. The resulting solid is collected by filtration,
washed with water, air dried and added to 3.5 1 of methanol. The mixture
3 3
-26- 600-7167
is refluxed for 30 minutes and allowed to cool to 20-25, and the solid is
collected by filtration, washed with petroleum ether and vacuum dried at
40 for 5 hours to obtain (~)-6-~hloro-3~4-dihydro-4-phenyl)-2(lH)-
quinazolinethione (m.p. 221-223). The filtrate i9 evaporated at reduced
pressure to about 50 % of its original volume, and the resulting solid is
collected by filtration, washed with petroleum ether and vacuum dried at
40 for 5 hours to obtain a second crop (m.p. 226-228).
~ample 9: Meth~ )-(6-chloro-3,4-~ih~dro-4-phenylquinazolin-2-~1-
thio)acetate hYdrobromide
[Formula I: A, R1, R2, R3, Rg = H;
B = -S-CHR7'-R1o (R7' = H; Rlo = -COOMe);
R8 = 6-Cl (i.e. is separated by 4 carbon atoms from each of
the two nitrogen atoms in formula I);
in racemate form;
in hydrobromide salt form]
[Process variant c) (alkylation)]
16.8 g of methyl bromoacetate is added to a solution of 15.0 g of
(~)-6-chloro-3,4-dihydro-4-phenyl-2(1~)-quinazolinethione [see Example 8,
step b)] in 150 ml of dry tetrahydrofuran stirred at 20-25, and the
reaction mixture is stirred at 20-25 for 48 hours, the reaction mixture
being maintained under nitrogen throughout. The resulting solid is
collected by filtration, washed with dry tetrahydrofuran, washed with
petroleum ether and vacuum dried at 40 for 5 hours to obtain the title
compound [m.p. 215-219 (dec.)]
The product is a racemate that may be resolv2d to obtain the 4R
and the 4S enanti er~. Alternatively, the two enantiomers may be prepared
from single isomers of the starting material.
3 ~
C~
~ ~ a~ a~ a) a) ~ ,t a~ ~ ~ a) c~
~ ~ n ~ ~
. O O O O O O O O O O O O O O O O O O
C~ a~ o o o c~ ~ o ~ ~ o ~
. ~ ~ a~ 0 o ~ ~~ O 1~ ~ ~ r~ u~ ~ ~ ~ ~ co ~o
o ~ ~ C~ ~ ~ ~ ~ _,
IIIIo~IIIIIIIIIIIII
O O O O C~ O O O O O O O O O O O O O O
u~ t~ o u~ ~r ~ oo ~ ~ r-- ~ ~ a
.. V~ ~
C . .
C I ~ _ ~ _ _ _ _ _ _ _
V~ _ _ _ _ _ _ N N N N N N N N N N
~; I D~ ~
~ ~ ~2 ~2 2~ 8 2, 2
o ~ ~ ~ d ~ ~ ~ d
a
~ ,
R~ O 3
a~ ~ ~: ~ YYY~ ~
Q. ~ ~ r r
a
~ s ~
c ~
~ol a) ~ ~ ~
c~
~ ~8
o ~ ~ ~
o
o 4~
o o
E~ ~ N ' N ~ N
t- ~ a
1_ ~ O ~1 N ~ ~ U~O r~l N 1~ ~ 11~ ~D 1` OD
o 3 ~~1 ~ N N ~ N N N N N N
~S!~63a
'~
o v ~
-------- ~ ~
ao a> o
'~ s a~
o o o o o ~5 ~ s
o u~
o o ~ ~ c~
c~ o ~
~ ~ :~
.c ~ ~
~ ~ c
~ r~
..
O
~: ~ ~ ~ a~ o ~ ~ ~
o~ ~ q~ o o o
~^ ~ ~ a v ~ _1
.r ~ ~ ~ V
c~ ~ u~ ~: ~ ~ s ~:: s
~ ~ ~ ~ ~ ~ o-~ o
s
~ ~ ~ ~ v
o~ ~ p u~ p ~
~ ,~ 3 ~ 3 ~
o~ ~ o ~.~ ~.,,
~ .,, V s ~ s ~
m ~ v p v p
c ~ '~ ~I) Y Y e~ ,8 C
~ ~ D ~ ~ V a) v a)
c: ~.~ Ro o Ro o ~a
O ~ V ~V ~-~
o ~1 ~U 9t 9~ R ~1 ~ .C ~ ~ C
~1r~ ~ ~ D V a~ v a
S ~IC ~ ~J Q Q Q ~ a~ P P
o~ 1~ y t~ V U~ O R O .a v
,
~ 1~ ~ ~ R Ll o 1
~ ~ Q, O O O O~ N N O
,~ r ~ v
a) ~ ~ ~ ~ ~ t~ a) c
S .~ V~ ~81S~ 6~SVC ~
~ 11 11 11 11 S ~ >1
H ~ e~ V S S
_I o o ~ ~D ~ 3 3 6
~ ~ n 0 6
O ~ ~ 6 n~
+ I _ _ ~ C
61 +
O C~
O rl a~ Il 11 11 11 ~1 6 t~l 6 V
U~ ~ 51 1~ O ~ O o
~ N ~ C~ V U~ V ~.)
C a~ C ~ c ~
r- ~ ~ .,~
~o ~ ~
1_ ~) ~ ~ O _I ~ ~ 1- 0 ~ V ~
g ~ ~ ~
~o ~
~.3.',~3~
-29- 6G0-7167
The compounds of formula I in racemic or optically active, in free
base or pharmaceutically acceptable acid addition salt form, hereinafter
briefly named "the agents of the inventionn, possess intere~ting
pharmacological activity. They are indicated for use as pharmaceuticals.
In particular, they raise the blood serum high density lipoprotein
[HDL; HDL-cholesterol; and apolipoprotein A-I (Apo A-I)] level. Many of
the compounds have the added benefit of lowering the blood serum total
triglycerides level.
This activity can be determined in conventional assay methods,
e.g. as follows:
a) Test A: In vivo HDL-cholesterol test:
-
The test is effected according to the method described in theliterature, e.g. in EP 528 146
Other conventional methods may be used to assay the blood serum
sampleQ for HDL-cholesterol and total triglycerides content.
b) Test B: Apo A-I test:
The test is effected as described in the literature, e.g. in
EP 528 146.
The above tests A and B indicate that the agents of the invention
are active at a dosage in the range of from about 10 mg/kg to abou~
200 mg/kg per day.
The agents of the invention are therefore indicated for use in
raising the blood serum high density lipoprotein (HDL; HDL-cholesterol and
apolipoprotein A-I) level and many of them al~o for lowering the blood
serum total triglycerides level and thus in the treatment of
atherosclero~is.
~0~3;3
-30- 600-7167
The precise dosage to be employed depends of course upon several
factors including the host, the nature and the severity of the condition
being treated, the mode of administration and the particular agent
employed. However, in general, satisfactory results are indicated to be
obtained at daily dosages in the range of from about 400 mg to about
2000 mg, conveniently administered in divided doses two to four times a
day.
The compounds may be administered by any conventional route, in
particular enterally, preferably orally e.g. in the form of tablets or
capsulec, or parenterally e.g. in the form of sterile injectable solutions
or suspensions.
The agent of the invention of Examples 1, 2, 3, 5 and 24,
particularly in the R optically active form, is preferred.
Thus e.g. the following activity has been determined:
HDL-
Average Duration cholesterolApo-I Total
Compound dose of level level triglyceride
treatment increaseincrease level decrease
(mg/kg/day) (days) (%) (%) (%~
Example 1, 83 8 402 62 15
R-isomer
(free base)
Gemfibrozil 53 8 211 88 15
.
It i~ therefore indicated that, for this use, the agent of
Example 1, particularly the R isomer may be administered at dosages lower
than those conventionally employed with gemfibrozil by similar modes of
administration, i.e. about 400-1000 mg/day orally.
~ù ~ 'J635
-31- 600-7167
Pharmaceutical compositions comprising an agent of the invention
together with at least one pharmaceutically acceptable carrier or diluent,
may be manufactured in conventional manner. They may be e.g. in unit
dosage form containing, for example, from about 100 mg to about 500 mg of
active compound.
The invention thus concerns a method for the prophylactic or
curative treatment of atherosclerosis comprising administering to a patient
in need of such treatment a therapeutically effective amount of an agent of
the invention.
It further concerns a process for the preparation of a medicament
against atherosclerosis which comprises mixing an agent of the invention
with at least one pharmaceutically acceptable carrier or diluent.
It further comprises the use of an agent of the invention for the
manufacture of a medicament against atherosclerosis.
The invention al30 concerns a pharmaceutical compo3ition
comprising an agent of the invention together with at least one
pharmaceutically acceptable carrier or diluent.
It further comprises an agent of the invention for use as a
pharmaceutical, particularly for use in the prophylactic or curative
treatment of atherosclerosis.