Note: Descriptions are shown in the official language in which they were submitted.
3418/OA/
Pharmaceutical preparations containing sulfonamides,
novel sulfonamides and processes for their production
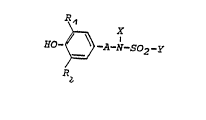
The present invention concerns pharmaceutical agents
which contain sulfonamides having the general formula I
~~ ~ _so2_y
in which
R1 and R2, which can be the same or different,
denote a hydrogen atom or a straight-chained or branched
alkyl residue with 1 to 4 c atoms,
.
A denotes a straight-chained or branched alkylene
chain with 1 to 5 C atoms,
X denotes a hydrogen atom or a C1-C4 alkyl residue,
and
- 2 ~ 7
Y denotes a straight-chained or branched alkyl
residue with 1 to 5 C atoms, an aralkyl or aryl
residue whereby the aryl residue can be substituted
once to three times in all possible positions on
the ring by halogen, trifluoromethyl, C1-C4 alkyl,
amino, C1-C4 acylamino, di(C1-C4) alkylamino or by
nitro ,
provided that A can also denote valency if Y does not
represent an aryl residue,
as well as their pharmacologically safe salts.
Rl and R2 preferably denote hydrogen, methyl or the
tert.-butyl group.
The bridge A is pre~erably -CH2-; -CH2 CH2-, or the
-CH2-CH(CH3)-- -CH2-ctCH3)2
X preferably represents an H atom or a methyl group.
An aralkyl group preferably denotes benzyl or phenethyl.
An aryl group preferably represented by phenyl.
An isopropyl, n-butyl, benzyl or phenyl residue is
particularly preferred for Y, whereby this can in turn
be substituted in all positions once to three times by
fluorine, chlorine, methyl, amino, acetylamino,
dimethylamino or nitro.
-
Benzene sulfonamides with an analogous structure whichdecrease atherogenic lipids are described in the
European application EP-A-384 279.
In EP-A 4011, EP-A 255 728 and EP-A 325 245
hydroxyphenyl compounds having the general formula I are
described as intermediate products for the production of
pharmacologically active phenoxyalkyl carboxylic acids
without there being a statement about their own
pharmacological action.
Compounds of formula I with A=valency are described in
other documents such as for example in US-A 3,737,316,
FR-A 2,309,524 and FR-A 2,193,216 without a statement
about a pharmacological action.
The compounds of the present invention have a strong
antioxidant activity. The lipo~hilicity of this
antioxidant group causes an acc:umulation of the
compounds in the atherogenic low-density lipoprotein
(LDL) and an e~fective protection of the sensitive
components of the LDL against reactive oxygen species.
This results in a substantial reduction of the LDL
influx into the macrophagic foam cells since a
prereguisite for the pathologically increased uptake of
atherogenic LDL into the atheroma ~ells is their
oxidative modi~ication.
Antioxidants are substances which - in general - cause a
considerable delay in the oxidative processes in a
product to be protected. Probucol~ is an antioxidant and
potent antiatherosclerotic agent which has a
hypolipaemic ef~ect in various animal species and in
humans. It is a sterically hindered alkyl phenol which
~ g~'~J1
accumulatas in LDL. It has been shown in animal
experiments that Probucol blocks the oxidative
modification of LDL in the arterial wall and directly
prevents atheroma formation because of the antioxidant
activity t~. Steinberg et al., Amer. J. Cardiol. 57,
16 M (1986)).
The disadvantages of Probucol~ are the low absorption of
the substance as well as the extremely long retention
time in body tissue; the excretion of Probucol mainly
takes place via the faeces (see M.N. Cayen, Pharmacol.
Ther. 29, 157 (1985)).
In addition compounds of the formula I lower the plasma
lipids by blocking the intestinal absorption of
cholesterol which results in a reduction of the
intrahepatic pool of free cholesterol and
correspondingly decreases the secretion of the dietary-
dependent lipoproteins from the liver into the plasma.
The inhibition of cholesterol absorption is due to the
inhibition of the acyl-coenzyme A-cholesterol
transferase (ACAT) react~on. AC,AT catalyses the
esterificatior1 of cholesterol in the enterocytes which
is necessary in order to package cholesterol in the
chylomicrons and to introduce them into the blood - -
circulation via the intestinal lymph and thoracic duct.
The substances are readily absorbed and inhibit the
~CAT-dependent esterification of free cholesterol not
only in the enterocytes but also in the cells of the
atheroma itself. They thereby prevent their degeneration
into xanthoma cells caused by overloading with
cholesterol ester.
~ 5 ~ ~ Jt
The compounds of formula I are used as pharmaceutical
preparations in particular as antiatherosclerotic agents
because of their stabilizing effect on lipoproteins.
Furthermore they act antibiotically - in particular
anti-bacterially -, anti-inflammatorily,
cytoprotectively as well as anti-asthmatically. However,
they can also be used as inhibitors of reperfusion-
dependent lipid peroxidation and as stabilizers of the
"lung surfactant factor".
The production of compounds of formula I is
characterized in that an amine having the formula II, in
which Rl, R2, A and X have the meanings stated above is
reacted with sulfonic acid chlorides having the formula
III in which Y also has the meaning stated above. This
is usually carried out at room temperature in a
chemically inert solvent such as CH2C12, toluene or such
like, preferably in the presence of an acid-binding
reagent such as e.g. pyridine, triethylamine (e.g.
analogously to F. Muth in Houblsn-Weyl, Vol. 9, p. 613).
R.~
X 1Ol e.g. pyridine
~o~ A-NH Cl-l-Y >
~ O - HCl
R~
II III
Alternatively sulfamides which are also substituted at
the sulphone group can be alkylated with suitably
substituted aralkyl halides to produce compounds having
the formula I.
- 6 ~ 7
If the compounds obtained in this way having the
formula I do not represent the final product, the
residue X (e.g. CH3) can be additionally introduced at
the sulphonamide nitrogen, if desired using alkyl
halides (e.g. methyl iodide).
The required starting materials II and III are in
general known from the literature, or can be produced in
an analogous way by the usual methods (for detailed
information see the examples).
If individual reaction products are not produced in
sufficient purity, the crude products can be purified by
crystallization or column chromatography.
The present invention also concerns new sulfonamides
having the formula I'
W- ~--A--N--SCIz - Y ( I l )
R2
in which
Rl and R2 denote in each case a tert. butyl group,
A denotes a straight-chained alkylene chain with 1 to
5 C atoms or the group -CH2-CH(CH3)-
X denotes a hydrogen atom or a Cl-C~ alkyl residue,
and
Y denotes an aralkyl or aryl residue whereby the aryl
residue can be substituted once to three times in
all possible positions on the ring by halogen,
trifluoromethyl, Cl-C4 alkyl, amino, C1-C4
acylamino, di(Cl-C~) alkylamino or by nitro,
: as well as their pharmacologically sa~e salts.
The following compounds of formula I' are particularly
preferred:
4-chlorobenzene-sulfo-(3~5-di-tert.butyl-4-hydroxy)-
phenethylamide (example ld)
4-chlorobenzene-sulfo-(3,5-di-tert.butyl-4-hydroxy)-
benzylamide (example 2)
N-methyl-4-chlorobenzene-sulfo-(3,5-di-tert.butyl-4-
hydroxy)-benzylamide (example 3)
~,
Benzylsulfo-(3,5-di-tert.butyl-4-hydroxy)-benzylamide
(example 5.6)
2,4,6-tris-isopropyl-benzene-sulfo-(3,5-di-tert.butyl-4-
hydroxy)-benzylamide (example 5.10)
2-[benzylsulfo-(3,5-di-tert.butyl-4-hydroxy)]-
phenethylamide (example 5.13)
8 ~9~ 7
2-[2,4,6-tris-isopropyl-benzene~sulfo-[3,5-di-
tert.butyl-4-hydroxy]-phenethylamide (example 5.17)
2-[3-trifluoromethyl-benzene-sulfo-(3,5-di-tert.butyl-4-
hydroxy)]-phenethylamide (example 5.18)
3-[2,4,6-tris-isopropyl-benzene-sulfo-(3,5-di-
tert.butyl-4-hydroxy)]-phenylpropylamide (example 5.25)
4-fluorobenzene-sulfo-(3,5-di-tert.butyl-4-hydroxy)-
benzylamide (example 5.27)
2-[4-fluorobenzene-sulfo-(3,5-di-tert.butyl-4-hydroxy)]-
phenethylamide (example 5.28)
2-[benzylsulfo-(3,5-di-tert.butyl-4-hydroxy)]-
phenylpropylamide (example 5.32)
2-[(4-chlorobenzene)-sulfo-(3,5-di-tert.butyl-4-
hydroxy)]-phenylpropylamide (example 5.37)
The compounds of formula I can be reacted with the
corresponding bases in order to prepare salts with
physiologically tolerated organic or inorganic bases
such as for example sodium hydroxide, potassium
hydroxide, calcium hydroxide, ammonium hydroxide,
methylglucamine, morpholine, triethylamine or
ethanolamine. Mixtures of the acidic compounds with a
suitable alkali carbonate or hydrogen carbonate also
come into consideration.
For the production of pharmaceutical preparations the
compounds having the general formula I are mixed in the
9 ~r~t~ r~
usual way with suitable pharmaceutical carrier
substances, axomatics, flavourings and dyes and are for
example formed as tablets or coated tablets or are
suspended or dissolved in water or oil such as e.g.
olive oil with the addition of corresponding auxiliary
agents.
The substances having the general formula I can be
administered orally and parenterally in liquid or solid
form. Water, which contains stabilizing agents,
solubilisers and/or buffers which are usually used in
injection solutions, is preferably used as the injection
medium. Such additives are e.g. tartrate buffer or
borate buffer, ethanol, dimethyl sulfoxide, complexing
agents (such as ethylenediamine tetraacetic acid), high-
molecular polymers (such as liquid polyethylene oxide)
~or the regulation of viscosity or polyethylene
derivatives of sorbitol anhydrides.
Solid carrier materials are e.g. starch, lactose,
mannitol, methyl cellulose, talcum, highly-dispersed
silicic acid, higher molecular fatty acids (such as
stearic acid), gelatine, agar-agar, calcium phosphate,
magnesium stearate, animal and ~egetable fats or solid
high-molecular polymers (such as polyethylene glycols).
Suitable preparations for oral application can contain,
if desired, flavourings and artificial sweeteners.
The administered dosage depends on the age, health and
weight of the recipient, the extent of the disease, the
type of further treatments which may be being carried
out at the same time, the frequency of treatments and
the typa of desired effect. The daily dosage of the
active compound is usually 0.1 to 10 mg/kg body weight.
- 10 ~ 8ll7
Preferred compounds within the scope of the present
invention, apart from those of formula I mentioned in
the examples, are the following:
N-methyl-4-chlorobenzene-sulfo-(3,5-di-tert.butyl-4-
hydroxy)phenethylamide
N-methyl-4 fluorobenzene-sulfo-(3,5-di-tert.butyl-4-
hydroxy)phenethylamide
N-methyl-4-chlorobenzene-sulfo-(3,5-di-tert.butyl-4-
hydroxy)phenylpropylamide
Example
a) ~-hydroxy-3,5-di-tert.butyl-benzylchloride
52.8 g (0.256 mol) 2,6-di-tert.-butylphenol is
dissolved in 200 ml n-heptane, 250 ml 37 per cent
formalin solution and 500 ml concentrated
hydrochloric acid are added, it is flushed with
nitrogen and stirred for 8 h at the reflux
temperature. After cooling the organic phase is
separated off, the aqueous phase is extracted with
n-heptane and the combined heptane phases are
washed with water. After drying with Na2SO4, it is
evaporated in a vacuum and the residue is processed
further as the crude product.
Yield: almost quantitative (crude product)
Lit.. Neureither, J.Org.Chem. 28, 3486 (1963)
b) 4-hYdroxy-3~5~di-tert.butylbenzylcyanide
A solution of 104.5 g (0.41 mol) 4-hydroxy-3,5-di-
tert.butylbenzylchloride and 155 ml ethanol is
added dropwise within 1 hour to a 80-85C hot
mixture of 38.4 g (0.71 mol) sodium cyanide, 50 ml
water and 72 ml ethanol. Subsequently it is kept
for a further 3 h at the reflux temperature, cooled
down and inorganic material is removed by
aspiration. The liquid phase is evaporated, water
is added to the evaporation residue and it is
extracted with ether. The ether phase is dried
(Na2SO4), evaporated and crystallized by addition
of ligroin.
Yield: 61.1 g (61 % of theory)
Melting point: 109-110C
analogous to Fuson and Rabjohn, org. Synth. Vol.
25, 66.
c) 3l5-di-tert.butyl-4-hydroxv-phenethylamine
In a shaker autoclave 10 g Raney nickel and 100 ml
liquid ammonia are added to 30 g (0.12 mol) 3,5-di-
tert.butyl-4-hydroxy-benzylcyanide in 400 ml
methanol while deep cooling and subsequently
hydrogenated for 16 h in a hydrogen atmosphere at
80C and 140 bar. After releasing the autoclave the
catalyzer is filtered off and the crude product is
freed of solvent in a vacuum. The residue is again
dispersed in ether/water, the ether phase is
separated and dried over MgSO4. After aga~n
evaporating green, wax-like crystals remain.
Yield: quantitative (crude product).
-
- 12 ~ $ ~ ~7
d) 4-chl~orobenzene-sulfo=~3,5~di-tert.buty1~4-
hydroxy)-phenethylamide Ia
_=
A solution of 2.1 g (10 mmol) 4-chlorobenzene-
sulfochloride dissolved in 10 ml dichloromethane is
added dropwise at room temperature to a solution o~
2.49 g (10 mmol) of the amine obtained in section
c) in 30 ml dichloromethane and 0.81 ml (10 mmol)
pyridine. After stirring for 12 h at room
temperature it is poured onto 1 N HCl, the organic
phase is separated, washed and dried with Na2SO47
After removing the solvent in a vacuum the residue
is purified by filtration over silica gel using
hexane/ethyl acetate (3:1) as the mobile solvent.
After evaporation the product thus obtained is
recrystallized from cyclohexane.
1.4 g colourless crystals, melting point: 122-23C.
Example 2
4-chlorobenzene-sul~o-(3L5-di-tert.butyl-4-
hydroxy~benzylamide Ib
1.55 g (8 mmol) 4-chlorobenzene-sulfochloride dissolved
in 10 ml dichloromethane is added dropwise at room
temperature to a mixture of 1.9 g (8 mmol) 3,5-di-
tert.butyl-4-hydroxy-benzylamine (prepared analogously
to : Houben-Weyl, "Methoden der Organischen Chemie" Vol.
11/1, p. 502, from 3,5-di-tert.butyl-4-hydroxy-
benzaldoxime) and 0.63 ml (8 mmol) pyridine in 20 ml
dichloromethane. After stirring for 24 h at room
temperature it is poured onto 1 N HCl, the organic phase
- 13 - ~ ~ 9 ~
is separated, dried with Na2SO4 and the solvent is
removed in a vacuum. The crude product thus obtained is
recrystallized from toluene.
1.3 g colourless crystals, melting point: 119-21C.
~xample 3
N-methyl-4-chlorobenzene-slllfo-L3 5-di-tert.butyl-4-
hydroxy)-benzylamide Ic (X = CH3)
==
1~5 g (4 mmol) 4-chlorobenzene-sulfo-(3,5-di-tert.butyl-
4-hydroxy)benzylamide Ib is added to a suspension of
==
0.1 g (4 mmol) sodium hydride in 10 ml THF. After
stirring for 30 min, 0.31 ml (5 mmol) methyl iodide is
added dropwise and stirred again for 3 h at room
temperature. Subsequently it is poured onto l N HCl, the
organic substance is extracted with ether. After drying
and evaporating the ether extract, an oily crude product
is obtained which is purified on a medium pressure
column (silica gel; mobile solvent heptane/ethyl acetate
9:1~. After removing the solvent the product solidifies
to form crystals. 0.35 g yellow crystals, melting point:
140-43C.
Example 4
a) 3-(3,5-di-tert.butyl-4-hydroxyLehenylpropylamine
In a shaker autoclave 10 g Raney nickel and 100 ml
liquid ammonia are added to 77.8 g (0.3 mol~ 3-
(3,5-di-tert.butyl-4-hydroxy)phenylpropionitrile
_ (produced according to: DOS 2 240 609 (1972),
~9~7
HOECHST company) in 500 ml methanol while deep
cooling and subsequently hydrogenated for 16 h in a
hydrogen atmosphere at 80C and 140 bar. After
releasing the autoclave, the cataly~er is filtered
off and the crude product is freed of solvent in a
vacuum. The residue is dispersed again in
ether/water, the ether phase is separated and dried
over MgSO4. After evaporating again a semi-solid
mass remains which slowly completely solidifies.
70.2 g greenish crystals (crude product).
b) 4-chlorobenzenesulfo-3-(3~5-di-tert butyl-4-
hydroxy~phenylpropylamide Id
==
2.04 ml (15 mmol) triethylamine is firstly added to
a suspension of 3.95 g (15 mmol) of the amine
obtained above in section a) in 50 ml toluene and
then 3.2 g (15 mmol) 4-chlorobenzenesulfochloride
dissolved in 10 ml toluene is added at room
temperature. After stirring for 12 hours at room
temperature 50 ml iced water is added, the organic
phase is separated, dried and the solvent is
removed in a vacuum. The crude product is purified
- by filtration over silica gel (hexane/ethyl acetate
` 1:1 as mobile agent).
1.9 g brownish crystals, melting point: 152C.
Example 5
one can obtain the following compounds in an analogous
manner to the compounds mentioned in Examples 1, 2, 3,
or 4 (see table):
- 15 ~ 8~
Table
.. _ _ ..... __ ..
- Example Y A m.p. [C]
. _ ..
5.1 ~3C ~C~2)3- valency 115-18
5.2 (C~3)2cX- valency 160-61
5.3 ~ -C~2- va.lency 123-24
5,4 H3C-(C~I2)3- -CH2- 74-75
5.5 ~C~3)2cH- -CX2- 114-15
~ 16 -
Table I continued
. .. _ _ _ _ -.. _
Exampl e Y A m . p . [ C ]
.. ___ _", . _ ... __
5. 6 ~--C~I2~ -~I2- 118-19
5 . 7 02N-~ -CH2~ 144-45
5 . 8 H2N- e~ ~CH2 - o i 1
5 . 9 H3COC-N~ -CH2- 174-76
5 .10 ~ -CH2- 145-4 6
5 .11 H3C- (CH2 ) 3~ -CH2-C~2- 109-10
5 . 12 ( C}I3 ) 2 CE- -CH2 -C~2-- 81- 8 2
5 .13 ~CH2 -CK2-CH2- 163-64
5.14 02N_ ~3 CH2-CX2- 147-48
5 .15 H2N- ~ -CX2-CR2-- oil
5 . 16 H3COC-N-e3 -C~2 -CH2 17 2 -7 8
5 .17 ~ -CH2-CH2 - 159 -6 0
5 .18 ~ -CH2 -CH2 - 9 4 ~9 5
5.~9 ~I3C-(CX2) 3~ -(C}~2) 3~ oil
5 . 2 o ( CH3 ) 2 CH- - ( CH2 ) 3 ~ oi 1
5 . 21 ~--CH2 ~ - ( C}I2 ) 3 oi 1
5 . 2 2 02N ~9 - ( CH2 ~ 3 - oi l
- 17
Table I continued
~ .
Example Y A m.p. [c]
. ~
5.23 H2N- ~ -(CH2)3- 122
5.24 H3COC-N- ~ -(CH2)3- 164
5.25 ~ -(CH2)3 144
5.26 H3C- ~ -(CH2)3- 129-30
5.27 ~ F -CH2- 131-32
5.28 /~ ,-F -CH2-CH2- 118-20
5.29 ~rCH3 ,l 121-22
CH3
5.30 n-Butyl- -CH2-CH- 126
5.31 i-P~opyl- " 113-21
5.32 Benzyl- " 147-48
5.33 ~ ~ N02 " 139-40
5.34 ~ NH2 " 149-51
5.35 ~ N-COCH3 92-93
5.36 / ~ " 128-29
5.37 ~ Cl " 139-40
_ n-Butyl CH2-(CH3)2C- 90-91
. ._ _
- 18 -
s~
~ . ~
Example Y A m.p. [C]
.. _ . ~
5.39 Benzyl- " 143-44
5.40 ~N02 180~81
5.41 ~ ~NH2 " 147-48
5.42 ~N-C-CH3 " 195 - 98
5.43 ~~ " 177-79
5.44 ~Cl " 168-70
- 19 - ~n~
Example 6 Pharmacological investi~ations
The compounds of formula I cr I' are antioxidant
sul~onamide analogues and inhibitors of cholesterol
ester formation in the arterial wall for the treatment
of arteriosclerosis.
a) The cytogenesis of atheromatosis
Hypercholesterolaemia and syndromes o~
hypercholesterolaemia/hypertriglycerideaemia are
risk factors for premature arteriosclerosis. The
oxidative modification of lipoproteins in the
arterial wall plays an important role in the
pathogenesis of the arteriosclerotic lesion itself.
It leads to the release of mediators which act
chemotactically and to the migration of
macrophagocytic cells from the blood into the
arterial wall. In the vessel wall there is a
particularly intensive unregulated uptake of
oxidatively changed low-density lipoprotein ~LDL)
by macrophages. The concomitant increase in
cholesterol in~lux and the load on cellular
metabolism leads to an increase in intracellular
cholesterol, an increase in the reacylation of
cholesterol and storage of cholesterol esters which
in turn lead to the formation of large cytoplasmic
cholesterol ester depots and to the degeneration of
these macrophages ("foam cells"). The foam cells
are the cellular substrate of the atheroma and
create the macroscopically visible strips of fat
from which the fibrous plaque lesions develop as
the disease progressas.
- 20 ~ 9~7
b) The basis for an antiatherosclerotic therapy_in
disturbances of fat metabolism
~n antiatherosclerotic therapy oriented towards
causal relationships is ideally based on three
pillars- 1. a decrease in the pathologically
increased level of lipid in plasma by diet and/or
drugs, 2. an effective antioxidant protection in
particular for the strongly atherogenic LDL in
order to reduce the oxidative LDL catabolism in the
atheroma and 3. an inhibition of the formation of
cholesterol esters in the foam cells of the early
fat strip lesions.
Some agents which lower serum cholesterol act by
contracting the hepatic pool of free cholesterol
and increasing the catabolism of the atherogenic
LDL cholesterol: ion excha;ngers interfere for
example with the enterhepatic cholesterol
circulation, cause a depleltion of bile acids in the
hepatocytes and stimulate the u~take of choles~erol
from the plasma compartment by an increased hepatic
uptake of cholesterol via LDL and the hepatic apo
B,E receptors (LDL receptors). The blockers of
cholesterol synthesis, e.g. Lovastatin, act in a
similar manner. They decrease hepatic cholesterol
and also cause an increased uptake of LDL
cholesterol from the plasma by means of an increase
in LDL receptors.
Blockers of cholesterol absorption also cause a
lowering of the hepatic cholesterol pool by
reducing the exogenous dietary load of cholesterol.
Examples of this are the so-called acyl CoA:
cholesterol acyl transferase (ACAT) inhibitors
- 21 ~ 7
which suppress the cholesterol efflux via
chylomicrons in the enterocytes by inhibiting the
estarification of free cholesterol. In the liver
these inhibitors of cholesterol ester formation in
addition increase free cholesterol and endogenous
hepatic cholesterol biosynthesis is indirectly
inhibited by end-product inhibition. In the foam
cells of the atheroma the blocking of cholesterol
ester formation causes increased free cholesterol
to be incorporated into the "reverse cholesterol
transport pathway" of the high-density lipoproteins
(HDL) and as a consequence this produces an
additional direct antiatherogenic effect.
Antioxidants are substances which - when viewed in
general - cause a substantial retardation oP the
oxidative processes in a product which is to be
protected. A potent antiatherosclerotically
effective antioxidant is Probucol which, apart from
its antioxidant action, has a hypolipidaemic action
in various animal species and in humans. It is a
sterically hindered alkyl ,phenol which accumulates
in LDL. It has been shown in animal experiments
that Probucol blocks the substantially increased
oxidative modification of :LDL in atherosclerotic
lesi~ns of the arterial wall and as an antioxidant
strongly inhibits atheroma formation by retarding
the catabolism of LDL cholesterol in the foam
cells.
- 22 - ~9~ ~r~
c) Amino substitu ~L~henols with an antiatherogenic
actlon
The compounds of formula I and I' fulfil all three
aforementioned requirements for an effective
pharmaceutial agent against premature atheromatosis
and progressive arteriosclerosis since they act
hypolipidaemically (Table 1) and exhibit a direct
antiatherosclerotic activity in the atheroma by
inhibition of cholesterol ester formation (Table 2)
as well as by their antioxidative activities in LDL
(Table 2~ and in opposition to cellular
lipoxygenases (Table 2).
d) Dose-dependent effect of the compound ~rom example
5.28 on the serum cholesterol level in rats with
dietary induced hypercholesterolaemia.
Method:
SD rats weighing 220-250 ~ received a standard diet
for 4 days which was enriched with 2 %
cholesterol/0.2 % cholic acid. The animals were
; intubated daily between 8 and 9 o'clock and
received the stated single doses via a stomach tube
in 2 ml vegetable oil. S-cholesterol was determined
on day 5 (test kit ~rom Boehringer Mannheim Co.).
- ~3
Table 1:
Dose S-Cholesterol % Controls Statistics
(mg/kg/d) (mg %)
0248 +/- 36 100
5187 +/- 23 75 p < 0.05
15120 ~/- 15 48 p < 0.05
50114 +/- 14 46 p < 0.02
10084 +/- 8 33 p < 0.01
. _ .
Mean +/- SEM, n=6 animals/group, statistics: U-test
e) In vitro effect of the compounds o~ formula I
and I'
Inhibition of cholesterol ester formation in
macrophages (according to Huber et al. (1990) Free
Rad. Res~ Comms. 8, 167-173):
Human plasma LDL was isolated by sequential
ultracentrifugatioll at preselected densities from
the plasma of healthy male non-smokers and
incubated for 5 hours at 37C in F-10 medium + 1 ~M
Cu(II) to deplete its lipophilic antioxidants. The
formation of cholesterol esters in P 388 D.1
macrophages under the influence of conditioned
human plasma LDL was measured after incubating the
cells (2 x 196/ml) for 18 hours in F-10 medium + 1
~M Cu(II) + 50 ~g/ml LDL + 5 % FCS and after
extracting the neutral fats by thin layar
chromatography.
Reference substance: 10 ~M Probucol, 25-42 % (n=7)
_ inhibition of cholesterol formation.
- 24 -
2~968~7
Stabilization of plasma LDL (according to Huber et
al. (1990) Free Rad. Res. Comms. 8, 167-173):
LDL (125 ~g protein/ml) was oxidized at ~2C in
20 mM Tris-Cl pH 8, 150 mM NaCl, 10 ~M CuCl2 with
and without the test compound (dissolved in
ethanol, final ethanol concentration 0.5 %). In
each case 3 molecules of test substan~e were used
per L~L particle. The oxidation was monitored
continuously by photometry at A 234 by measuring
the diene formation. The so-called "lag phase" is
defined as that time interval which is given by the
intersection of the tangent to the curve during the
propagation reaction of the lipid peroxidation with
the time axis.
Reference substance: Probucol, 3 molecules/LDL
particle prolong the lag phase of LDL by 10 ~.
Inhibition o~ soya bean liPoxy~enase (15-
li~oxygenase) w~ LDL as substrate (according to
Cathcart et al. (1991) J. Lipid Res. 32, 63-70):
LDL (100 ~g protein/ml) was incubated at 37C in
20 mM Tris-Cl pH 8.3, 150 mM NaCl with and without
the test substance (dissolved in ethanol, final
ethanol concentration 0.5 %) with 5000 U/2 ml soya
bean lipoxygenase (Sigma, Munich): The oxidation
was monitored continuously by measuring the dienes
at A 234. The stated IC50 value is that
concentration of the test compound which leads to a
halving of the diene formation in the test mixture
which is linear over time.
.
- 25 - ~ 7
Reference substance: Probucol, IC50 value=20 ~M.
Table ?:
_ ._ ___
Compound Inhibition of Stabilization Inhibition of
example) cholesterol ester of plasma LDL: 15-lipoxy-
formation inincrease in genase-mediated
macrophages"lag phase" LDL oxidation
(P388D.1) % IC50 (~M)
IC50 (~M) 10 ~M %
_ . ~
2 2.50 -78~7 44 0.2
3 -54.9 40 2
4b -49.2 0.2
5.10 7 -60.3 99 5
5.17 -78.0 50 3
5.19 -43.8 0.09
5.2~ 1.20 -92.6 25 <0.1
5.31 -33.9 18 0.2
- . . :
' ' -