Note: Descriptions are shown in the official language in which they were submitted.
2 ~ IJ
-- 1 --
59912.564
Benzimidazoles
The present invention relates to new henzimidazoles,
processes for their preparation and pharmaceutical
compositions containing them.
It has been found that certain novel benzimidazoles have
valuable pharmacological properties, in particular,
angiotensin-antagonistic activities.
Thus, viewed from one aspect, the present invention
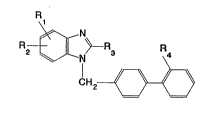
provides compounds of formula I:
R2- ~ ~ R3 R
~H2 1/~=\
(I)
(wherein
R1 denotes a hydrogen, fluorine, chlorine or bromine atom
or a C13-alkyl, C37-cycloalkyl, fluoromethyl,
difluoromethyl or trifluoromethyl group;
''
R2 denotes a phthalimino or homophthalimino group,
wherein a carbonyl group in a phthalimino group may be
replaced by a methylene group and a methylene group in a
homophthalimino group may be substituted by one or two
C13-alkyl groups,
or R2 denotes a 5-, 6- or 7-membered alkyleneimino group
(optionally substituted by one or two Cl3-alkyl groups)
2'7
-- 2
in which a methylene group is replaced by a carbonyl or
sulphonyl group,
or R2 denotes a maleic acid imido group optionally mono-
or disubstituted by a C13-alkyl group or by a phenyl
group, wherein the substituents may be identical or
di~ferent,
or R2 denotes a benzi.midazol-2-yl or 4,5,6,7-tetrahydro-
benzimidazol-2-yl group optionally substituted in the 1-
position by a C16-alkyl group or by a C35-cycloalkyl
group or in the phenyl nucleus thereof by a fluorine
atom or by a methyl or trifluoromethyl group,
or R2 denotes an imidazo~2,1-b]thiazol-6-yl,
imidazo[1,2-a]pyridin 2-yl, 5,6,7,8-tetrahydro-
imidazo[1,2-a]pyridin-2-yl, 3-chloro-5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl, imidazo[l,2 a]pyrimidin-2-
yl, imidazo[4,5-b]pyridin-2-yl, imidazo[4,5-c]pyridin-2-
yl, imidazo[l,2-c]pyrimidin-2-yl, imidazo[l,2-a]pyrazin-
2-yl, imidazo[l,2-b]-pyridazin-2-yl,
imidazo[4,5-c]pyridin-2-yl, purin-8-yl,
imidazo[4,5-b]pyrazin-2-yl, imidazo[4,5-c]pyridazin-2-yl
or imidazo[4,5-d]pyridazin-2-yl group, or a carbon-
attached pyrrolidine, piperidine or pyridine ring
wherein a phenyl group may be condensed on to the
pyridine ring via two adjacent carbon atoms and a
methylene group adjacent to the N-atom in a pyrrolidine
or piperidine ring may be replaced by a carbonyl group,
or R2 denotes an R7-NR6-CO-NR5- group wherein
Rs denotes a hydrogen atom or a C1s-alkyl,
cyclohexyl or benzyl group,
R6 denotes a hydrogen atom or a C16-alkyl, allyl,
cyclohexyl, benzyl or phenyl group,
2 ~
-- 3
R7 denotes a hydrogen atom or a C13-alkyl group, or
R6 and R7 together with the nitrogen atom between
them denote an unbranched cyclic C46-alkyleneimino
group or a morpholino group, or
R5 and R6 together denote a C23-alkylene group;
R3 denotes a C15-alkyl, C3s-cycloalkyl or C13-alkoxy
group; and
R4 denotes a tetrazolyl group substituted in the 1- or 2-
position by an Ra-CO-O-CH2- group, or an
Rb-CO-O-(RcCH)-O-CO-, RaO-CO- or RbO-CO-O-(RCCH)-O-CO-
group, wherein
Ra denotes a straight-chained or branched C16-alkyl
group or a Cs7-cycloalkyl, benzyl, l-phenylethyl,
. 2-phenylethyl, 3-phenylpropyl, methoxymethyl or
cinnamyl group,
Rb denotes a straight-chained or branched Cl6-alkyl
group or a Cs7-cycloalkyl, phenyl, benzyl, 1-
phenylethyl, 2-phenylethyl or 3-phenylpropyl group
and
Rc denotes a hydrogen atom or a methyl group,
or if
(i) R3 denotes an alkoxy group, R4 may also denote a
carboxy, lH-tetrazolyl or 2H-tetrazolyl group or, if
(ii) R2 is other than a l-methyl-benzimidazol-2-yl group
and R3 denotes a cyclopropyl group, R4 may also represent
a carboxy group or, if
2 ~ 2 7
-- 4 --
(iii) R2 denotes a butanesultam-l-yl group, R4 may also
denote a carboxy group or, if
(iv) R2 denotes a 1-methyl-5-fluoro-benzimidazol-2-yl
group and R3 denotes an ethyl group, R4 may also
represent a carboxy, lH-tetrazolyl or 2H-tetrazolyl
group)
and the tautomers and the salts thereof.
Examples of the definitions of groups R1, R2, R3 and R4
mentioned hereinbefore include the following:
R1 may denote a hydrogen atom or in the 4-position a
fluorine, chlorine or bromine atom, a methyl, ethyl, n-
propyl, isopropyl, cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl, cycloheptyl, difluoromethyl or
trifluoromethyl group;
R2 may denote a phthalimino, homophthalimi.no,
4,4-dimethyl-homophthalimino, 1-oxo-isoindolin-2-yl,
2-oxo-pyrrolidino, 2-oxo-piperidino, 2-oxo-
hexamethyleneimino, propanesultam-1-yl,
butanesultam-l-yl, pentanesultam-l-yl, maleic acid
imido, 2 methyl-maleic acid imido, 2-phenyl-maleic acid
imido, 2,3-dimethyl-maleic acid imido,
3-methyl-2-phenyl-maleic acid imido, 2,3-diphenyl-maleic
acid imido, piperidin-2-yl, piperidin-2-on-1-yl,
quinolin-2-yl, isoquinolin-l-yl, isoquinolin-3-yl,
pyridin-2-yl, benzimidazol-2-yl,
1-methyl-benzimidazol-2-yl, 1-ethyl-benzimidazol-2-yl,
1-n-propyl-benzimidazol-2-yl, l-isopropyl-benzimidazol-
2-yl, 1-n-butyl-benzimidazol-2-yl, l-isobutyl-
benzimidazol-2-yl, 1-n-pentyl-benzimidazol-2-yl, l-n-
hexyl-benzimidazol-2-yl, 1-cyclopropyl-benzimidazol-2-
yl, l-cyclobutyl-benzimidazol-2-yl, l-cyclopentyl-
benzimidazol-2-yl, 1-cyclohexyl-benzimidazol-2-yl, 5-
2 ~ 2 !~
-- 5
fluoro-l-methyl-ben~imidazol-2-yl, 6-fluoro-1-methyl-
benzimidazol-2-yl, 5~trifluoromethyl-benzimidazol-2-yl,
5-trifluoromethyl-1-methyl-benzimidazol-2-yl,
imidazo[l,2-a]pyridin-2-yl, 5,6,7,8-tetrahydro-
imidazo[l,2-a~pyridin-2 yl, 3-chloro-5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl, imidazo[l,2-a]pyrimidin-2-
yl, imidazo[4,5-b~pyridin-2-yl, imidazo[4,5-c]pyridin-2-
yl, imidazo[2,1-b]thiazol-6-yl, imidazo[l,2-c~pyrimidin-
2-yl, imidazo[l,2-a]pyrazin-2-yl,
imidazo[l,2-b]pyridazin-2-yl, imidazo[4,5-c]pyridin-2-
yl, purin-8-yl, imidazo[4,5-b]pyrazin-2-yl,
imidazo[4,5-c]pyridazin-2-yl, imidazo[4,5-d]pyridazin-2-
yl, aminocarbonylamino, methylaminocarbonylamino,
dimethylaminocarbonylamino, N-methylaminocarbonyl-
methylamino, N-(dimethylaminocarbonyl)-methylamino, N-
dimethylaminocarbonyl-ethylamino, N-
dimethylaminocarbonylisopropylamino, N-
(dimethylaminocarbonyl~-n-pentylamino, N-
methylaminocarbonylethylamino, N-methylaminocarbonyl-n-
pentylamino, N-methylaminocarbonyl-n-hexylamino, N-
methylaminocarbonyl-n-octylamino, N-
methylaminocarbonyl-cyclohexylamino, ethylamino-
carbonylamino, N-ethylaminocarbonyl-methylamino, N-
ethylaminocarbonyl-ethylamino, N-ethylaminocarbonyl-n-
hexylamino, N-ethylaminocarbonyl-n-heptylamino, N-
ethylaminocarbonyl-cyclohexylamino,
diethylaminocarbonylamino, N-(diethylaminocarbonyl)-
methylamino, N-(diethylaminocarbonyl)-ethylamino, N-
(diethylaminocarbonyl)-n-butylamino, N-
(diethylaminocarbonyl)-n-hexylamino, N-
(diethylaminocarbonyl)-n-octylamino,
isopropylaminocarbonylamino, N-isopropylaminocarbonyl-
methylamino, n-butylaminocarbonylamino, N-(n-butyl-
aminocarbonyl)-methylamino, N-(n-butylaminocarbonyl)-
ethylamino, N-(n-butyl.,~inocarbonyl)-isopropylamino, N-
(n-butylaminocarbonyl)~n-butylamino, N-(n-
butylaminocarbonyl)-n-h*xylamino, N-(n-
2~ ~yr~
-- 6
butylaminocarbonyl)-Acyclohexylamino,
N-(di-(n-butyl)-aminocarbonyl)-amino, N-(di-(n-butyl)-
aminocarbonyl)-methylamino, N-(di-(n-butyl)-
aminocarbonyl)-ethylamino, N-(di-(~-butyl)-
aminocarbonyl)-n-butylamino, N-(di-(n-butyl)-
aminocarbonyl)-n-hexylamino, N-(n-pentylaminocarbonyl)-
methylamino, N-(n-pentylaminocarbonyl)-ethylamino, N-(n-
hexylaminocarbonyl)-ethylamino, n-
hexylaminocarbonylamino, n-heptylaminocarbonylamino, n-
octylaminocarbonylamino, N-(n-hexylaminocarbonyl)-n-
butylamino, N-(n-hexylaminocarbonyl)-n-pentylamino, N-
(n-hexylaminocarbonyl)-n-hexylamino, N-(n-
hexylaminocarbonyl)-cyclohexylamino, di-(n-hexyl)-
aminocarbonylamino, N-(di-(n-hexyl)-aminocarbonyl)-
methylamino, N-((n-hexyl)-methylaminocarbonyl)-amino,
cyclohexylaminocarbonylamino, N-cyclohexylaminocarbonyl-
methylamino, N-cyclohexylaminocarbonyl ethylamino, N-
cyclohexylaminocarbonyl-n-butylamino, N-
cyclohexylaminocarbonyl-isobutylamino, N-
cyclohexylaminocarbonyl-n-pentylamino, N-
cyclohexylaminocarbonyl-n-hexylamino, N-
cyclohexylaminocarbonyl cyclohexylamino, N-(ethyl-
cyclohexylaminocarbonyl)-methylamino, N-(propyl-
cyclohexylaminocarbonyl)-methylamino, N (n-butyl-
cyclohexylaminocarbonyl)-methylamino,
allylaminocarbonylamino, benzylaminocarbonylamino, N-
benzylaminocarbonyl-isobutylamino,
phenylaminocarbonylamino, pyrrolidinocarbonylamino,
pyrrolidinocarbonyl-methylamino,
piperidinocarbonylamino,
hexamethyleneiminocarbonylamino,
morpholinocarbonylamino, 3,4,5,6-tetrahydro-2-pyrimidon-
l-yl, 3-methyl-3,4,5,6-tetrahydro-2-pyrimidon-1-yl, 3-
ethyl-3,4,5,6-tetrahydro-2-pyrimidon-1-yl, 3-n-propyl-
3,4,5,6-tetrahydro-2-pyrimidon-1-yl, 3-isopropyl-
3,4,5,6-tetrahydro-2-pyrimidon-1-yl, 3-n-butyl-3,4,5,6-
tetrahydro-2-pyrimidon-1-yl, 3-isobutyl-3,4,5,6-
2 ~ 2'~
tetrahydro-2-pyrimidon-1-yl, 3-n-pentyl-3,4,5,6-
tetrahydro-2-pyrimidon-1-yl, 3-n-hexyl-3,4,5,6~
tetrahydro-2-pyrimidon-1-yl, 3-cyclopentyl-3,4,5,6-
tetrahydro-2-pyrimidon-1-yl, 3-cyclohexyl-3,4,5,6-
tetrahydro-2-pyrimidon-1-yl, 3-cycloheptyl-3,4,5,6-
tetrahydro-2-pyrimidon-1-yl, 3-benzyl-3,4,5,6-
tetrahydro-2-pyrimidon-1-yl, 3,~,5,6-tetrahydro-2(lH)-
pyrimidon-l-yl, 3-methyl-3,4,5,6-tetrahydro-2~lH)-
pyrimidon-l-yl, 3-ethyl-3,4,5,6-tetrahydro-2(lH)-
pyrimidon 1-yl, 3-n-propyl-3,4,5,6-tetrahydro-2(lH)-
pyrimidon-1-yl, 3-isopropyl-3,4,5,6-tetrahydro-2(lH)-
pyrimidon-l-yl, 3-benzyl-3,4,5,6-tetrahydro-2(lH)-
pyrimidon-l-yl, 3-(2~phenylethyl)-3,4,5,6-tetrahydro-
2(lH)-pyrimidon-l-yl or 3-(3-phenylpropyl)-3,4,5,6-
tetrahydro-2(lH)-pyrimidon-l-yl group;
R3 may denote a methyl, ethyl, n-propyl, isopropyl, n-
butyl, isobutyl, tert.butyl, n-pentyl, l-methyl-butyl,
2-methyl-butyl, 3-methyl-butyl, cyclopropyl, cyclobutyl,
cyclopentyl, methoxy, ethoxy, n-propoxy or isopropoxy
group; and
R4 may denote a hydroxycarbonyl, methoxycarbonyl,
ethoxycarbonyl, n-propyloxycarbonyl,
isopropyloxycarbonyl, n-butyloxycarbonyl,
isobutyloxycarbonyl, tert.butyloxycarbonyl, n-
pentyloxycarbonyl, isoamyloxycarbonyl, n-
hexyloxycarbonyl, cyclopentyloxycarbonyl,
cyclohexyloxycarbonyl, benzyloxycarbonyl, 1-
phenylethyloxycarbonyl, 2-phenylethyloxycarbonyl, 3-
phenylpropyloxycarbonyl, methoxymethoxycarbonyl,
cinnamyioxycarbonyl, acetoxymethoxycarbonyl,
propionyloxymethoxycarbonyl, n-
butyryloxymethoxycarbonyl, isobutyryloxymethoxycarbonyl,
n-pentanoyloxymethoxycarbonyl, isopentanoyloxy-
methoxycarbonyl, pivaloyloxymethoxycarbonyl, n-
hexanoyloxymethoxycarbonyl, cyclopentanoyloxy-
2 ~ f~
-- 8 --
methoxycarbonyl, cyclohexanoyloxymethoxycarbonyl,phenylacetoxymethoxycarbonyl, 2-phenylpropionyloxy-
methoxycarbonyl, 3-phenylpropionyloxymethoxycarbonyl, 4-
phenylbutyryloxymethoxycarbonyl,benzoyloxymethoxycarbonyl, l-acetoxyethoxycarbonyl, 1-
propionyloxyethoxycarbonyl, l-n-butyryloxyethoxy-
carbonyl, l-isobutyryloxyethoxycarbonyl, l-n-
pentanoyloxyethoxycarbonyl, 1-
isopentanoyloxyethoxycarbonyl, l-pivaloyloxyethoxy-
carbonyl, 1-n-hexanoyloxyethoxycarbonyl, 1-
cyclopentanoyloxyethoxycarbonyl, 1-
cyclohexanoyloxyethoxycarbonyl, 1-
phenylacetoxyethoxycarbonyl, 1-(1-phenylpropionyloxy)-
ethoxycarbonyl, 1-(2-phenylpropionyloxy)-ethoxycarbonyl,
1-(3-phenylbutyryloxy)-ethoxycarbonyl, 1-
benzoyloxyethoxycarbonyl, methoxycarbonyloxy-
methoxycarbonyl, ethoxycarbonyloxymethoxycarbonyl, n-
propyloxycarbonyloxymethoxycarbonyl,
isopropyloxycarbonyloxymethoxycarbonyl, n-butyloxy-
carbonyloxymethoxycarbonyl, isobutyloxycarbonyloxy-
methoxycarbonyl, tert.butyloxycarbonyloxy-
methoxycarbonyl, n pentyloxycarbonyloxymethoxycarbonyl,
isoamyloxycarbonyloxymethoxycarbonyl, n-
hexyloxycarbonyloxymethoxycarbonyl,
cyclopentyloxycarbonyloxymethoxycarbonyl,
cyclohexyloxycarbonyloxymethoxycarbonyl,
benzyloxycarbonyloxymethoxycarbonyl, 1-
phenylethoxycarbonyloxymethoxycarbonyl, 2-phenylethoxy-
carbonyloxymethoxycarbonyl, 3-phenylpropyloxy-
carbonyloxymethoxycarbonyl,
cinnamyloxycarbonyloxymethoxycarbonyl, 1-
(methoxycarbonyloxy)-ethoxycarbonyl, 1-
(ethoxycarbonyloxy)-ethoxycarbonyl, 1-(n-
propyloxycarbonyloxy)-ethoxycarbonyl, 1-
(isopropyloxycarbonyloxy)-ethoxycarbonyl, l-(n-
butyloxycarbonyloxy)-ethoxycarbonyl, 1-
(isobutyloxycarbonyloxy)-ethoxycarbonyl, 1-
3 t~ ~ 7
g
(tert.butyloxycarbonyloxy)-ethoxycarbonyl, l-(n-
pentyloxycarbonyloxy)-ethoxycarbonyl, 1-
(isoamyloxycarbonyloxy)-ethoxycarbonyl, 1-(n-
hexyloxycarbonyloxy)-ethoxycarbonyl, 1-
(cyclopentyloxycarbonyloxy)~ethoxycarbonyl, 1-
(cyclohexyloxycarbonyloxy)-ethoxycarbonyl,
cyclopentylcarbonyloxymethoxycarbonyl, 1-
(benzyloxycarbonyloxy)-ethoxycarbonyl, 1~
phenylethoxycarbonyloxy)-ethoxycarbonyl, 1-(2-
phenylethoxycarbonyloxy)-ethoxycarbonyl, 1-(3-
phenylpropyloxycarbonyloxy)-ethoxycarbonyl, 1-
(cinnamyloxycarbonyloxy)-ethoxycarbonyl, cyano, lH-
tetrazolyl, 1-triphenylmethyl-tetrazolyl, 2-
triphenylmethyl-tetrazolyl, 1-acetoxymethyl-tetrazolyl,
2-acetoxymethyl-tetrazolyl, 1-propionyloxymethyl-
tetrazolyl, 2-propionyloxymethyl-tetrazolyl, 1-
butyryloxymethyl-tetrazolyl, 2-butyryloxymethyl-
tetrazolyl, l-isobutyryloxymethyl-tetrazolyl, 2-
isobutyryloxymethyl-tetrazolyl, l-pivaloyloxymethyl-
tetrazolyl, 2-pivaloyloxymethyl-tetrazolyl, 1-
ethoxycarbonyloxymethyl-tetrazolyl, 2-
ethoxycarbonyloxymethyl-tetrazolyl, 1-[1-
(ethoxycarbonyloxy)-ethyl]-tetrazolyl, 2-[1-
(ethoxycarbonyloxy)-ethyl]-tetrazolyl, 1-[1-
(cyclohexyloxycarbonyloxy)-ethyl]-tetrazolyl or 2-[1-
(cyclohexyloxycarbonyloxy)-ethyl]-tetrazolyl group.
Preferred compounds according to the invention include
those of formula I wherein
R1 denotes a hydrogen atom or in the 4-position a
chlorine atom, a Cl3-alkyl group or a trifluoromethyl
group;
Rz denotes a phthalimino or homophthalimino group,
wherein a carbonyl group in a phthalimino group may be
replaced by a methylene group,
2 ~ 2 ~
-- 10 --
or R2 denotes a 5-, 6- or 7-membered alkyleneimino group
in which a methylene group is replaced by a carbonyl or
sulphonyl group,
or R2 denotes a maleic acid imido grcup optionally mono-
or disubstituted by a methyl or phenyl group, wherein
the substituents may be identical or dif~erent,
or R2 denotes a benzimidazol-2-yl or 4,5,6,7-tetrahydro-
benzimidazol-2-yl group optionally substituted in the 1-
position by a C16-alkyl group or by a cycloalkyl group
or in the phenyl nucleus thereof a fluorine atom or a
methyl or trifluoromethyl group,
or R2 denotes an imidazo[2,1-b]thiazol-6-yl,
imidazo[1,2-a]pyridin-2-yl, 5,6,7,8-tetrahydro-
imidazo[1,2-a]pyridin-2-yl, 3-chloro-5,6,7,8-tetrahydro-
imidazo[1,2-a]pyridin-2-yl, imidazo[1,2-a~pyrimidin-2-
yl, imidazo[4,5-b]pyridin-2-yl, imidazo[4,5-c]pyridin-2-
yl, imidazo[l,2-c]pyrimidin-2-yl, imidazo[l,2-a]pyrazin-
2-yl, imidazo[1,2-b]pyridazin-2-yl,
imidazo[4,5-c]pyridin-2-yl, purin-8-yl,
imidazo[4,5-b]pyrazin-2-yl, imidazo[4,5-c]pyridazin-2-yl
or imidazo[4,5-d]pyridazin-2-yl group,
or R2 denotes an R7-NR6-CO-NRs- group wherein
Rs denotes a hydrogen atom or a C15-alkyl,
cyclohexyl or benzyl group,
R6 denotes a hydrogen atom or a C16-alkyl, allyl,
cyclohexyl, benzyl or phenyl group,
R7 denotes a hydrogen atom or a Cl3-alkyl group, or
R6 and R7 together with the nitrogen atom between
them denote a straight-chained C4 6-alkyleneimino
2 ~ 2 7
-- 11 --
group or a morpholino group, or
R5 and R6 together denote a Cz3-alkylene group;
R3 denotes a C15-alkyl, C3s-cycloalkyl, or C23-alkoxy
group; and
R4 denotes a tetrazolyl group substituted in the 1- or 2-
position by an R~-CO-O-CH2- group, or an
Rb-CO-O-(RcCH)-O-CO-, RaO-Co- or RbO-CO-O-(RcCH)-O-CO-
group, wherein
Ra denotes a straight-chained or branched C16-alkyl
group or a C57~cycloalkyl, benzyl, l-phenylethyl,
2-phenylethyl, 3-phenylpropyl, methoxymethyl or
cinnamyl group,
Rb denotes a straight-chained or branched C16-alkyl
group or a C57-cycloalkyl, phenyl, benzyl, 1-
phenylethyl, 2-phenylethyl or 3-phenylpropyl group
and
Rc denotes a hydrogen atom or a methyl group,
or, if
(i) R3 denotes an alkoxy group, R4 may also denote a
carboxy, lH-tetrazolyl or 2H-tetrazolyl group or, if
(ii) R2 is other than a 1-methyl-benzimidazol-2-yl group
and R3 denotes a cyclopropyl group, R4 may also represent
a carboxy group or, if
(iii) R2 denotes a butanesultam-1-yl group, R4 may also
denote a carboxy group or, if
(iv) R2 denotes a 1-me-thyl-5-fluoro-benzimidazol-2-yl
2 ~ 3 ~ 2 ~
- 12 -
group and R3 denotes an ethyl group, R4 may also
represent a carboxy, lH-tetrazolyl or 2H-tetrazolyl
group,
and the tautomers and the salts thereof.
Particularly preferred compounds according to the
present invention include those of formula I wherein
R1 denotes a hydrogen atom or in the 4-position a methyl
group;
R2 denotes an imidazo[l,2-a]pyridin-2-yl, 5,6,7,8-
tetrahydro-imidazo[1,2-a]pyridin-2-yl, 3-chloro-5,6,7,8-
tetrahydro-imidazo[1,2-a]pyridin-2-yl, 1-methyl-
benzimidazol-2-yl, 1-methyl-5 fluoro-benzimidazol-2-yl
or butanesultam-l-yl group;
R3 denotes a C24-alkyl group, a cyclopropyl group or a
C23-alkoxy group; and
R4 denotes a tetrazolyl group substituted in the 1- or 2-
position by an Ra-CO-O-CH2- group, or an
Rb-CO-O-(RcCH)-O-CO-, RaO-CO- or RbO-CO-O-(RcCH)-O-CO-
group, wherein
Ra denotes a straight-chained or branched C16-alkyl
group or a C57-cycloalkyl, benzyl, l-phenylethyl,
2-phenylethyl) 3-phenylpropyl, methoxymethyl or
cinnamyl group,
Rb denotes a straight-chained or branched C16-alkyl
group or a C57-cycloalkyl, phenyl, benzyl, 1-
ph~nylethyl, 2-phenylethyl or 3-phenylpropyl group
and
Rc denotes a hydrogen atom or a methyl group;
2 ~ 2 1
- 13 -
and the tautomers and the salts thereof.
More particularly preferred compounds according to the
invention include the following compounds of formula I:
4'-~(2-cyclopropyl-4-methyl-6-(4,5,6,7-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
biphenyl-2-carboxylic acid;
4'-[(2-ethoxy-4-methyl-6-(4,5,6,7-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
biphenyl-2-carboxylic acid;
4'-[(2-ethoxy-4-methyl-6-(4,5,6,7-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
2-(lH-tetrazol-5-yl)-biphenyl;
4'-[(2-cyclopropyl-4-methyl-6-(imidazo[1,2 -a ] pyridin-2-
yl)-benzimidazol-l-yl)~methyl]-biphenyl-2-carboxylic
acid;
4'-[(2-ethoxy-4-methyl-6-(imidazo[1,2-a]pyridin-2-yl)-
benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylic acid;
4'-[(2-ethoxy-4-methyl-6-(imidazo[1~2-a]pyridin-2-yl)-
benzimidazol-l-yl)-methyl]-2-(lH-tetrazol-5-yl)-
biphenyl;
1'-[(2-ethoxy-4-methyl-6-(1-methyl-benzimidazol-2-yl)-
benzimidazol-l-yl)-methyl]-2 (lH-tetrazol-5-yl)-
biphenyl;
4'-[(2-ethyl-4-methyl-6-(1-methyl-5-fluoro-benzimidazol-
2-yl)-benzimida 7. ol-1-yl)-methyl]-2-(lH-tetrazol-5-yl)-
biphenyl;
4'-[(2-ethyl-4-methyl-6-(1-methyl-5-fluoro-benzimidazol-
- 14 -
2-yl)-benzimidazol-1-yl)-methyl]-biphenyl-2-carboxylic
acid; and
4'-[(2-n-propyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-l-yl)-methyl] biphenyl-2-carboxylic acid;
and the tautomers and the salts thereof.
The present invention particularly relates to the
following compounds of formula I:
4'-[(2-ethyl-4-methyl-6-(1-methyl-5-fluoro-benzimidazol-
2-yl)-benzimidazol-1-yl~-methylJ-biphenyl-2-carboxylic
acid;
4'-[(2-n-propyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylic acid;
4'-[(2-cyclopropyl-4-methyl-6-(imidazo[1,2-a]pyridin-2-
yl)-benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylic
acid;
4'-[(2-cyclopropyl-4-methyl-6-(5,6,7,8-tetxahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
biphenyl-2-carboxylic acid; and
4'-[(2-ethoxy-4-methyl-6-(1-methyl-benzimidazol-2-yl)-
benzimidazol-l-yl)-methyl]-2-(lH-tetrazol-5-yl)-
biphenyl;
and the tautomers and the salts thereof.
Viewed from a further aspect, the invention provides a
process for the preparation of compounds of the
invention, said process comprising at least one of the
following steps:
2 ~
- 15 -
a) cyclising a compourld of formula II
R1
~Xl
R2~"~`Yl
(II)
(wherein
R1 and R2 are as hereinbefore defined,
one of the groups X1 or Y1 represents a group of formula
--NR5--CH2 ~--`~>
and the other group X1 or Y1 represents a group of
formula
- NH - C - R
wherein
R3 and R4 are as hereinbefore defined,
Rs represents a hydrogen atom or an R3CO- group, wherein
R3 is as hereinbefore defined,
Zl and Z2~ which may be identical or different, represent
amino, hydroxy or mercapto groups optionally substituted
by C16-alkyl groups, or
Z1 and Z2 together represent an oxygen or sulphur atom,
an imino group optionally substituted by a C13-alkyl
2 1 ~
group, or a C23-alkylenedioxy or C23-alkylenedithio
group) optionally formed in the reaction mixture, or a
corresponding hydroxylamine and if required reducing a
cyclized N-oxide thus obtained;
b) reacting a benzimidazole of formula III
~XN 3
H
,
(III)
(wherein
R1, R2 and R3 are as defined hereinbefore) with a
biphenyl compound of formula IV
Z3- CH2 ~
(IV)
(wherein
R4 is as hereinbefore defined and
Z3 represents a nucleophilic leaving group such as a
halogen atom, e.g. a chlorine, bromine or iodine atom,
or a substituted sulphonyloxy group, e.g. a
methanesulphonyloxy, phenylsulphonyloxy or p-
toluenesulphonyloxy group);
c) (to prepare a compound of formula I wherein R4
represents a carboxy group) converting a compound of
formula V
2 ~ 2 7
-- 17 --
~N R
~H2 ~r ~
(v)
(wherein
R1, Rz and R3 are as hereinbefore defined, and
R4' represents a group which may be converted into a
carboxy group by hydrolysis, thermolysis or
hydrogenolysis) by hydrolysis, thermolysis or
hydrogenolysis;
d) tto prepare a compound of formula I wherein R"
represents a lH-tetrazolyl group) cleaving a protecting
group from a compound of formula VI
N R~
CH2 '~
(VI)
(wherein
R1, R2 and R3 are as hereinbefore defined and
R4" represents a lH-tetrazolyl group protected in the 1-
or 3-position by a protecting group);
e) (to prepare a compound of formula I wherein R~
represents a lH-tetrazolyl group) reacting a compound of
formula VII
21 ~ ~ ~ 2 7
-- 18 --
2~CN~P~3 CN
(VII)
(wherein
R1, R2 and R3 are as hereinbefore de~ined) with hydrazoic
acid or a salt thereof;
f) (to prepare a compound of formula I wherein R4 denotes
a tetrazolyl group substituted in the l or 2-position
by an Ra-CO-O-CH2- group, or R4 denotes an RaO-CO-,
Rb-CO-O-(RcCH)-O-CO- or RbO-CO-O-(RcCH)-O-CO- group)
reacting a compound of formula VIII
R
2 ~N~R3 R4
CH2 --~ =
(VIII)
(wherein
R1, R2 and R3 are as hereinbefore defined and
R4~ denotes a carboxy, lH-tetrazolyl or 2H-tetrazolyl
group) with a compound of formula IX
Z4 Y2 (IX)
(wherein
Y2 denotes an Ra-CO-O-CH2-, Rb-CO-O-(RcCH)-,
RbO-CO-O-(RcCH)- or Ra~ group, wherein Ra~ Rb and Rc are as
2 ~ 2 ~
-- 19 --
hereinbefore defined ar.d
Z4 denotes a nucleophilic leaving group such as a halogen
atom, e.g. a chlorine or bromine atom, or, if Y denotes
an Ra~ group, Z4 may also represent a hydroxy group);
g) resolving a 1-, 3-isomer mixture of a compound of
formula I thus obtained into the 1- and 3-isomers
thereof;
h) converting a compound of formula I thus obtained into
a salt thereof, more particularly, for pharmaceutical
use, into a physiologically acceptable salt thereof with
an inorganic or organic acid or base, or converting a
salt of a compound of formula I into the free compound;
and
i) performing a process as defined in any one of steps
(a) to (h) above on a corresponding protected compound
and subsequently removing the protecting group used.
The cyclisation of step (a) is conveniently carried out
in a solvent or mixture of solvents such as ethanol,
isopropanol, glacial acetic acid, benæene,
chlorobenzene, toluene, xylene, glycol,
glycolmonomethylether, diethyleneglycol dimethylether,
sulpholane, dimethylformamide, tetraline or in an excess
of the acylating agent used to prepare the compound of
formula II, e.g. in the corresponding nitrile,
anhydride, acid halide, ester or amide, e.g. at
temperatures between 0 and 250C, but preferably at the
boiling temperature of the reaction mixture, optionally
in the presence of a condensing agent such as
phosphorusoxychloride, thionylchloride,
sulphurylchloride, sulphuric acid, p-toluenesulphonic
acid, methanesulphonic acid, hydrochloric acid,
phosphoric acid, polyphosphoric acid, acetic anhydride
or optionally in the presence of a base such as
potassium ethoxide or ~otassium tert.-butoxide.
2 ~ 2 r~
- 20 -
However, the cyclisation may also be carried out without
a solvent and/or condensing agent.
It is particularly advantageous to carry out the
reaction of step (a) by preparing a compound of formula
II in the reaction mixture by reducing a corresponding
o-nitro-amino compound, optionally in the presence of a
carboxylic acid of formula R3COOH, or by acylation of a
corresponding o-diamino compound. When the reduction of
the nitro group is broken off at the hydroxylamine
stage, the N-oxide of a compound of formula I is
obtained in the subsequent cyclisation. The rasulting
N-oxide is then converted by reduction into a
corresponding compound of formula I.
The subsequent reduction of the N-oxide of the compound
of formula I is preferably carried out in a solvent such
as water, water/ethanol, methanol, glacial acetic acid,
ethyl acetate or dimethylformamide with hydrogen in the
presence of a hydrogenation catalyst such as Raney
nickel, platinum or palladium/charcoal, with metals such
as iron, tin or zinc in the presence of an acid such as
acetic acid, hydrochloric acid or sulphuric acid, with
salts such as iron(II)sulphate, tin(II)chloride or
sodium dithionite, or with hydrazine in the presence of
Raney nickel, at temperatures between 0 and 50C, but
preferably at ambient temperature.
The reaction of step (b) is conveniently carried out in
a solvent or mixture of solvents such as methylene
chloride, diethylether, tetrahydrofuran, dioxane,
dimethyl-sulphoxide, dimethylformamide or benzene,
optionally in the presence of an acid binding agent such
as sodium carbonate, potassium carbonate, sodium
hydroxide, potassium tert.-butoxide, sodium hydride,
triethylamine or pyridine, whilst the latter two may
simultaneously also be used as solvent, preferably at
temperatures between 0 and 100C, e.g. at temperatures
2~1
between ambient temperature and 50C.
In the reaction of step (b), a mixture of the 1- and 3-
isomers is preferably obtained which can if desired
subsequently be resolved into the corresponding 1- and
3- isomers, by crystallisation or chromatography using a
substrate such as silica gel or aluminium oxide.
In step (c), functional derivatives of the carboxy group
such as the unsubstituted or substituted amides, esters,
thiolesters, orthoesters, iminoethers, amidines or
anhydrides, the nitrile group or the tetrazolyl group
may, for example, be converted by hydrolysis into a
carboxy group, esters with tertiary alcohols, e.g. the
tert.butylester, may, for example, be converted by
thermolysis into a carboxy group and esters with
aralkanols, e.g. the benzylester, may, for example, be
converted by hydrogenolysis into a carboxy group.
The hydrolysis of step (c) is appropriately carried out
either in the presence of an acid such as hydrochloric
acid, sulphuric acid, phosphoric acid, trichloroacetic
acid or trifluoroacetic acid or in the presence of a
base such as sodium hydroxide or potassium hydroxide, in
a suitable solvent such as water, water/methanol,
ethanol, water/ethanol, water/isopropanol,
water/dioxane, methylene chloride or chloroform, at
temperatures between -10C and 120C, e.g. at
temperatures between ambient temperature and the boiling
temperature of the reaction mixture. When hydrolysis is
carried out in the presence of an organic acid such as
trichloroacetic acid or trifluoroacetic acid, any
alcoholic hydroxy groups present may simultaneously be
converted into a corresponding acyloxy group such as the
trifluoroacetoxy group.
If R4' in a compound oE formula V represents a cyano or
aminocarbonyl group, these groups may also be converted
2 ~ 2 ~
- 22 -
into a carboxy group with a nitrite, e.g. sodium
nitrite, in the presence of an acid such as sulphuric
acid, which may simultaneously also be used as solvent,
at tem~eratures between 0 and 50C.
If R4' in a compound of formula V represents for example
a tert.-butyloxycarbonyl group, the tert.-butyl group
may also be thermally cleaved, optionally in an inert
solvent such as methylene chloride, chloroform, benzene,
toluene, tetrahydrofuran or dioxane and preferably in
the presence of a catalytic amount of an acid such as p-
toluenesulphonic acid, sulphuric acid, phosphoric acid
or polyphosphoric acid, preferably at the boiling
temperature of the solvent used, e.g. at temperatures
between 40C and lOO~C.
If R4' in a compound of formula V represents, for
example, a benzyloxycarbonyl group the benzyl group may
also be hydrogenolytically cleaved in the presence of a
hydrogenation catalyst such as palladium/charcoal, in a
suitable solvent such as methanol, ethanol,
ethanol/water, glacial acetic acid, ethyl acetate,
dioxane or dimethylformamide, preferably at temperatures
between 0 and 50C, e.g. at ambient temperature, under a
hydrogen pressure of 1 to 5 bar. During hydrogenolysis,
other groups may be reduced at the same time, e.g. a
nitro group may be reduced to an amino group, a
benzyloxy group to a hydroxy group, a vinylidene group
to the corresponding alkylidene group or a cinnamic acid
group to the corresponding phenyl-propionic acid group,
or they may be replaced by hydrogen atoms, e.g. a
halogen atom may be replaced by a hydrogen atom.
Suitable protecting groups for use in step (d) include,
for example, the triphenylmethyl, tributyl tin or
triphenyl tin groups. The cleaving of a protecting
group used is preferably carried out in the presence of
a hydrohalic acid, preferably in the presence of
2~a~ 7
- 23 -
hydrochloric acid, in the presence of a base such as
sodium hydroxide or alcoholic ammonia, in a suitable
solvent such as methylene chloride, methanol,
methanol/ammonia, ethanol or isopropanol, at
temperatures between 0 and lOODC, but preferably at
ambient temperature or, if the reaction is carried out
in the presence of alcoholic ammonia, at elevated
temperatures, e.g. at temperatures between 100 and
150C, preferably at temperatures between 120 and 140C.
The reaction of step (e) is preferably carried out in a
solvent such as benzene, toluene or dimethylformamide,
at temperatures between 80 and 150C, preferably at
125C. Advantageously, either the hydrazoic acid is
liberated during the reaction from an alkali metal
azide, e.g. from sodium azide, in the presence of a weak
acid such as ammonium chloride, or the tetrazolide salt
obtained in the reaction mixture by reacting with a salt
of hydrazoic acid, preferably with aluminium azide or
tributyl tin azide, which are also conveniently prepared
in the reaction mixture by reacting aluminium chloride
or tributyl tin chloride with an alkali metal azide such
as sodium azide, is subsequently liberated by
acidification with a dilute acid such as 2N hydrochloric
acid or 2N sulphuric acid.
The reaction of step (f) is conveniently carried out by
esterification with a corresponding alcohol or with a
corresponding reactive derivative such as the halide,
conveniently in a solvent or mixture of solvents such as
water, methylene chloride, chloroform, ether,
tetrahydrofuran, dioxane or dimethylformamide or in an
excess of the acylating agent as solvent, optionally in
the presence of an acid activating or dehydrating agent
such as thionylchloride, with the anhydrides, esters or
halides thereof optionally in the presence of an
inorganic or tertiary organic base such as sodium
hydroxide, potassium carbonate, triethylamine or
2 ~ 2 ~1
- 24 -
pyridine, whilst the latter two may simultaneously be
used as solvent, at temperatures between -25 and 100C,
but preferably at temperatures between -10 and 80C.
In the reactions described hereinbefore, any reactive
groups present such as hydroxy or imino groups may be
protected during the reaction by means of conventional
protecting groups which are split off again after the
reaction.
By way of example, protecting groups for a hydroxy group
may include a trimethylsilyl, acetyl, benzoyl, methyl,
ethyl, tert.butyl, benzyl or tetrahydropyranyl group and
protecting groups for an imino group may include an
acetyl, benzoyl, ethoxycarbonyl or benzyl group.
The optional subsequent cleaving of a protecting group
used is preferably carried out by hydrolysis in an
aqueous solvent, e.g. in water, isopropanol/water,
tetrahydrofuran/water or dioxane/water, in the presence
of an acid such as hydrochloric acid or sulphuric acid
or in the presence of an alkali metal base such as
sodium hydroxide or potassium hydroxide, at temperatures
between 0 and 100C, preferably at the boiling
temperature of the reaction mixture. However, a benzyl
group is preferably cleaved by hydrogenolysis, e.g. with
hydrogen in the presence of a catalyst such as
palladium/charcoal, in a solvent such as methanol,
ethanol, ethyl acetate or glacial acetic acid,
optionally with the addition of an acid such as
hydrochloric acid, at temperatures between 0 and 50~C,
but preferably at ambient temperature, and under a
hydrogen pressure of from 1 to 7 bar, but preferably
from 3 to 5 bar.
An isomer mixture of a compound of formula I thus
obtained may if desired be resolved by chromatography
using a substrate such as silica gel or aluminium oxide.
2 ~ ',,), 7
- 25 -
Moreover, the compounds of formula I obtained may be
converted into ~he ac:Ld addition salts thereof, more
particularly for pharmaceutical use into the
physiologically acceptable salts thereof with inorganic
or organic acids. Suitable acids for this purpose
include hydrochloric acid, hydrobromic acid, sulphuric
acid, phosphoric acid, fumaric acid, succinic acid,
lactic acid, citric acid, tartaric acid or maleic acid.
Furthermore, the new compounds of ~ormula I thus
obtained, if they contain a carboxy or lH-tetrazolyl
group, may if desired subsequently be converted into the
salts thereof with inorganic or organic bases, more
particularly for pharmaceutical use into the
physiologically acceptable salts thereof. Suitable
bases for this purpose include for example sodium
hydroxide, potassium hydroxide, cyclohexylamine,
ethanolamine, diethanolamine and triethanolamine.
The compounds of formulae II to IX used as starting
materials are known from the literature in some cases or
may be obtained by methods known from the literature.
Thus, for example, a compound of formula II may be
obtained by alkylation of a corresponding o-amino-
acylamino compound with a compound of formula IV. The
o-amino-acylamino compound required for this is
preferably obtained by reduction of a corresponding o-
l nitro-acylamino compound which in turn may be obtained
by nitration of a corresponding acylamino-acetophenone,
subsequent conversion of the resulting corresponding o-
nitro-acylamino-acetophenone into the corresponding ~-
bromo-acetophenone, su~sequent cyclisation of the ~-
bromo-acetophenone witr~ a corresponding acid amide and
subsequent reduction c~ the nitro group. Before the
reduction of the nitrc group, an oxazol-4-yl compound
thus obtained may be nverted into the corresponding
2 ~ ) ?,J~7
- 26 -
imidazol-4-yl compound by means of a corresponding
amine, preferably ammonia, under pressure, or an
imidazol-4-yl compound unsubstituted in the l-position
obtained in this way may be converted by alkylation into
a corresponding imidazol-4-yl compound alkylated in the
l-position.
A starting compound of formula III may be obtained by
reduction and cyclisation of an o-nitro-acylamino
compound as described hereinbefore.
Starting compounds of formulae V, VI, VII and VIII may
be obtained by reacting a compound of formula III with a
corresponding compound of formula IV.
The new compounds of formula I and the physiologically
acceptable salts thereof ha~e valuable pharmacological
properties. They are angiotensin-an~agonists,
particularly angiotensin-II-antagonists.
By way of example, the following compounds:
A = 4'-[(2-ethyl-4-methyl-6-(1-methyl-5-fluoro-
benzimidazol-2-yl)-benzimidazol-1-yl)-methyl]-
biphenyl-2-carboxylic acid;
B = 4'-[(2-n-propyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylic
acid;
C = 4'-[(2-cyclopropyl-4-methyl-6-(imidazo[1,2-a]-
pyridin-2-yl)-benzimidazol-1-yl)-methyl]-biphenyl-2-
carboxylic acid;
D = 4'-[(2-cyclopropyl-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[1,2-a]pyridin-2-yl)-benzimidazol-1-yl)-
methyl]-biphenyl-2-carboxylic acid; and
2 ~ 't~ 2 ~
- 27 -
E = 4'-[(2-ethoxy-4~methyl-6-(1-methyl-benzimidazol~2-
yl)-benzimidazo~-l-yl)-methyl]-2-~lH-tetrazol-5-yl)-
biphenyl,
were investigated for their biological activities as
follows:
Description of method: anqiotensin-II-rece~tor bindinq
The tissue (rat's lung) is homogenised in Tris buffer
(50 mMol Tris, 150 mMol NaCl, 5 mMol EDTA, pH 7.40) and
centrifuged twice for 20 minutes each time at
20,000 x g. The finished pellet is resuspended in
incubation buffer (50 mMol Tris, 5 mMol MgClz, 0.2% BSA,
pH 7.40) 1:75, based on the moist weight of the tissue.
Each 0.1 ml of homogenate is incubated for 60 minutes at
37C with 50 pM [125I]-angiotensin-II (NEN, Dreieich,
Germany) and increasing concentrations of the test
substance in a total volume of 0.25 ml. The incubation
is ended by rapid filtration through glass fibre filter
mats. The filters are each washed with 4 ml of ice cold
buffer (25 m~lol Tris, 2.5 mMol MgCl2, 0.1% BSA, pH 7.40).
The bound radioactivity is measured in a gamma-counter.
The corresponding IC50 ~alue is determined from the
dosage-activity curve.
Substances A to E show the following IC50 values in the
test described:
Substance IC50 [nM]
_ 7.4
C 1 7
D 1.3
3.3
In view of their pharmacological properties, the new
~a~-~2 l
- 28 -
compounds and the phy~iologically acceptable salts
thereof are suitable for the tr~atment of hypertension
and cardiac insufficiency and for treating ischaemic
peripheral circulator~ disorders, myocardial ischaemia
(angina), for the prevention of the progression of
cardiac insufficiency after myocardial infarct and for
treating diabetic nephropathy, glaucoma,
gastrointestinal diseases and bladder diseases.
Furthermore, the new compounds and the physiologically
acceptable salts thereof are suitable for treating
pulmonary diseases, e.g. lung oedema and chronic
bronchitis, for preventing arterial rP-stenosis after
angioplasty, for preventing thickening of the vascular
walls after vascular operations, arteriosclerosis and
diabetic angiopathy. Because of the effect of
angiotensin on the release of acetylcholine and dopamine
in the brain, the new angiotensin antagonists are also
suitable for alleviating central nervous system
disorders, e.g. depression, Alzheimer's disease,
Parkinson's syndrome and bulimia, as well as disorders
of cognitive functions.
Thus, viewed from a further aspect the present invention
provides a pharmaceutical composition comprising a
compound of formula I or a physiologically acceptable
salt thereof together with one or more physiologically
acceptable carriers or excipients.
Viewed from a still further aspect the invention
provides the use of a compound of formula I or a
physiologically acceptable salt thereof for the
manufacture of a therapeutic agent having an
angiotensin-antagonistic activity.
In particular, the inv~ntion provides the use of a
compound of formula I or a physiologically acceptable
r~ 9 r~
-- 29 --
salt thereof for the manufacture of a therapeutic agent
for the treatment of hypertension and cardiac
insufficiency and for treating ischaemic peripheral
circulatory disorders~ myocardial ischaemia (angina),
for the prevention of the progression of cardiac
insufficiency after myocardial infarct and for treating
diabetic nephropathy, glaucoma, gastrointestinal
diseases and bladder diseases.
More particularly, the invention provides the use of a
compound of formula I or a physiologically acceptable
sal~ thereof for the manufacture of a therapeutic agent
for treating pulmonary diseases, for preventing arterial
re-stenosis after angioplasty, for preventing thickening
of the vascular walls after vascular operations,
arteriosclerosis and diabetic angiopathy, or for the
treatment of depression, Alzheimer's disease,
Parkinson's syndrome, bulimia and disorders of cognitive
functions.
Viewed from a yet still further aspect the invention
provides a method of treatment of the human or non-human
animal body for the treatment of hypertension and
cardiac insufficiency and for treating ischaemic
peripheral circulatory disorders, myocardial ischaemia
(angina), for the prevention of the progression of
cardiac insufficiency after myocardial infarct and for
treating diabetic nephropathy, glaucoma,
gastrointestinal diseases and bladder diseases, said
method comprising administering to said body a compound
of formula I or a physiologically acceptable salt
thereof.
More particularly, the invention provides a method of
treatment of the human or non-human animal body for
treating pulmonary diseases, for preventing arterial re-
stenosis after angioplasty, for preventing thickening of
2 ~ 2 7
- 30 -
the vascular walls after vascular operations,
arteriosclerosis and ~iabetic angiopathy, or for the
treatment of depression, Alzheimer's disease,
Parkinson's syndrome, bulimia and disorders of cognitive
functions, said method comprising administering to said
body a compound of formula I or a physiologically
acceptable salt thereof.
The dosage required to achieve these effects in adults
is appropriately, when administered intravenously, 0.5
to 100 mg, preferably 1 to 70 mg, and, when administered
orally, 0.1 to 200 mg, preferably 1 to 100 mg, 1 to 3
times a day. For this purpose, the compounds of formula
I prepared according to the invention, optionally in
conjunction with other active substances such as, for
example, hypotensive agents, ACE inhibitors, diuretics
and/or calcium antagonists, may be incorporated together
with one or more inert conventional carriers and/or
diluents, e.g. with corn starch, lactose, glucose,
micro-crystalline cellulose, magnesium stearate,
polyvinylpyrrolidone, citric acid, tartaric acid, water,
water/ethanol, water/glycerol, water/sorbitol,
water/polyethyleneglycol, propylene-glycol, cetylstearyl
alcohol, carboxymethylcellulose or fatty substances such
as hard fat or suitable mixtures thereof, in
conventional galenic preparations such as plain or
coated tablets, capsules, powders, suspensions or
suppositories.
Examples of additional active substances which may be
used in the combinations mentioned above include
bendroflumethiazide, chlorothiazide,
hydrochlorothiazide, spironolactone, benzothiazide,
cyclothiazide, ethacrinic acid, furosemide, metoprolol,
prazosin, atenolol, propranoloI, (di)hydralazine-
hydrochloride, dlltiazem, felodipine, nicardipine,
nifedipine, nisoldipine, nitrendipine, captopril,
~ a ~?~rl
- 31 -
enalapril, lisinopril, cilazapril, quinapril, fosinopril
and ramipril. The dosage of these active substances is
conveniently 1/5 of the lowest dose normally recommended
up to 1/1 of the normally recommended dosage, that is
for example 15 to 200 mg of hydrochlorothiazide, 125 to
2000 mg of chlorothiazide, 15 to 200 mg of ethacrinic
acid, 5 to 80 mg of furosemide, 20 to 480 mg of
propranolol, 5 to 60 mg of felodipine, 5 to 60 mg of
nifedipine or 5 to 60 mg of nitrendipine.
The following non-limiting Examples are provided to
illustrate the invention. Unless otherwise specified,
all percentages and ratios are by weight, other than
eluant or solvent ratios which are by volume.
- 32 -
Example 1
4'-[(2-Ethyl-4-methyl-6~ methyl-5-fluoro-benzimidazol-
2-yl)-benzimidazol-1-yl)-methyl]-biphenyl-2 carboxylic
acid
a) 2-Ethyl-4-methyl-6-(1-methyl-5-fluoro-benzimidazol-
2-yl)-benzimidazole
A mixture of 4.5 g (21 mMol) of 1-methylamino-2-amino-4-
fluoro-benzene-dihydrochloride and 4.3 g (21 mMol) of 2-
ethyl-4 methyl-benzimidazol-6-yl-carboxylic acid is
stirred for four hours at 140C in 100 g of
polyphosphoric acid, then stirred into about 300 g of
ice water and made alkaline with concentrated ammonia
solution. The crude product precipitated is suction
filtered, dried and then purified by column
chromatography (300 g of silica gel; methylene
chloride/ethanol = 95:5).
Yield: 3.1 g (48% of theory),
Rf value: 0.24 (silica gel; methylene chloride/ethanol =
19 : 1 )
b) Tert.butyl 4'-[(2-ethyl-4-methyl-6-(1-methyl-5-
fluoro-benzimidazol-2-yl)-benzimidazol-1-yl)-
methyl]-biphenyl-2-carboxylate
616 mg (5.5 mMol) of potassium tert.butoxide are added
to a solution of 1.55 g (5 mMol) of 2-ethyl-4-methyl-6-
(l-methyl-5-fluoro-benzimidazol-2-yl)-benzimidazole in
1 30 ml of dimethylsulphoxide and the resultin~ mixture is
stirred for 15 minutes at ambient temperature. Then
1.9 g (5.5 mMol) of tert.butyl 4'-bromomethyl-biphenyl-
2-carboxylate are added and stirring is continued for a
further 20 hours at ambient temperature. The mixture is
then stirred into about 80 ml of saturated sodium
chloride solution, the crude product precipitated is
suction filtered and purified by column chromatography
(150 g silica gel; eluant: methylene chloride/ethanol =
2 ~ ~ O c~
- 33 -
98:2).
Yield: 1.4 g (50% of theory),
Rf value: 0.47 (silica gel; methylene chloride~ethanol =
19:1)
c) 4'-[(2-Ethyl-4-m`ethyl-6-(1-methyl-5-fluoro-
benzimidazol-2-yl)-benzimidazol-1-yl)-methyl]-
biphenYl-2-carboxYlic acid
A solution of 1.4 g (2.4 mMol) of tert.butyl 4'-[(2-
ethyl-4-methyl-6-(1-methyl-5-fluoro-benzimidazol-2-yl)-
benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylate and
15 ml of trifluoroacetic acid in 30 ml of methylene
chloride is stirred for 14 hours at ambient temperature,
then concentrated by evaporation, the residue is mixed
with about 30 ml of water and made alkaline with 2N
sodium hydroxide solution. After extracting twice with
30 ml of diethyl ether, the a~ueous phase is acidified
with 20% citric acid. The crude product precipitated is
suction filtered and purified by column chromatography
(100 g silica gel; eluant: methylene chloride/ethanol =
96:4).
Yield: 850 mg (69% of theory~,
Melting point: 246-248~C
C32H27FN402 (518.60~
Calculated: C 74.11 H 5.25 N 10.80
Found: 73.95 5.34 10.80
Mass spectrum: m/e = 518
Example 2
4'-[(2-n-Propyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-l-yl)-methyl]-biphenyl-2 carboxylic acid
Prepared analogously to Example 1 from tert.butyl 4'-
[(2-n-propyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-1-yl)-methyl]-biphenyl-2-carboxylate and
trifluoroacetic acid.
Yield: 87% of theory,
2~49Q~ ?J 7
Melting point: 269-271C
C29H3lN304S (517-65)
Calculated: C 67.2~ H 6.04 N 8.12
Found: 67.56 6.12 8.28
Rf value: 0.39 (silica gel; methylene chloride/ethanol =
9 : 1 )
Mass spectrum: m/e = 517
Exam~le 3
4'-[(2-Cyclopropyl-4-methyl-6-(imidazo[1,2-a]pyridin-2-
yl)-benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylic
acid
Prepared analogously to Example 1 from tert.butyl 4'-
[(2-cyclopropyl-4-methyl-6-(imidazo[1,2-a]pyridin-2-yl)-
benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylate and
trifluoroacetic acid.
Yield: 50% of theory,
Melting point: 245-248C
C32H26N402 (498-59)
Calculated: C 77.09 H 5.26 N 11.24
Found: 76.88 5.37 11.30
Rf value: 0.63 (silica gel; methylene chloride/ethanol =
9 : 1 )
Mass spectrum: m/e = 498
Example 4
4'-[(2-Cyclopropyl-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
biphenyl-2-carboxylic acid
Prepared analogously to Example 1 from tert.butyl 4'-
[(2-cyclopropyl-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
biphenyl-2-carboxylate and trifluoroacetic acid.
Yield: 53% of theory,
2 ~L ~ 2 1
- 35 -
Melting point: 310-312C
C32H30N402 (502.62)
Calculated: C 76.47 H 6.02 N 11.15
Found: 76.23 5.97 10.85
Rf value: 0.17 (silica gel; methylene chloride/ethanol =
9 : 1 )
Mass spectrum: m/e = 502
Example 5
4'-[(2-Ethyl-4-methyl-6-(1-methyl-5-fluoro-benzimidazol-
2-yl)-benzimidazol-1-yl)-methyl]-2-(lH-tetrazol 5-yl)-
biphenyl
- a) 4'-[(2-Ethyl-4-methyl-6-(1-methyl-5-fluoro-
benzimidazol-2-yl)-benzimidazol-1-yl)-methyl]-2-
cyano-biphenyl
616 mg (5.5 mMol) of potassium tert.butoxide are added
to a solution of 1.55 g (5 mMol) of 2-ethyl 4-methyl-6-
(l-methyl-5-fluoro-benzimidazol-2-yl)-benzimidazole in
30 ml of dimethylsulphoxide and the mixture is stirred
for 15 minutes at ambient temperature. Then 1.5 g
(5.5 mMol) of 4'-bromomethyl-2-cyano-biphenyl are added
and the resulting mixture is stirred for a further 20
hours at ambient temperature. Then the mixture is
stirred into approximately 80 ml of saturated sodium
chloride solution, the crude product precipitated is
suction filtered and purified by column chromatography
I (150 g of silica gel; eluant: methylene chloride/ethanol
= 97:3).
Yield: 1.9 g (76~ of theory),
Rf value: 0.43 (silica gel; methylene chloride/ethanol =
19 : 1 )
2~3 ~2'~
b) 4'-[(2-Ethyl-4-methyl-6-(1-methyl-5-fluoro-
benzimidazol-2-yl)-benzimidazol-1-yl)-methyl]-2-(lH-
tetr zol-5-yl)-biphenyl
A solution of 1.9 g (3.8 mMol) of 4'-[(2-ethyl-4-methyl-
6-(1-methyl-5-~luoro-benzimidazol-2-yl)-benzimidazol-1-
yl)-methyl]-2-cyano-biphenyl, 4.1 g (76 mMol) of
ammonium chloride and 4.9 g (76 mMol) of sodium azide in
30 ml of dimethylformamide is heated to 140C for 15
hours, then a further 2.0 g of ammonium chloride and
2.4 ~ of sodium azide are added and the mixture is
heated for another 4 hours to 140~C. Then the solution
is stirred into about 80 ml of saturated sodium chloride
solution, the crude product precipitated is suction
filtered and purified by column chromatography tl50 g of
silica gel; eluant: methylene chloride/ethanol = 19:1).
Yield: 1.25 g (61% of theory),
Melting point: 267-269C
c32H27FN8 (542-60)
Calculated: C 70.84 H 5.02 N 20.65
Found: 70.52 5.04 20.82
Rf value: 0.60 (silica gel; methylene chloride/ethanol =
9 : 1 )
Mass spectrum: m/e = 542
Example 6
4'-[(2-Ethoxy-4-methyl-6-(1-methyl-benzimidazol-2-yl)-
benzimidazol-l-yl)-methyl]-2-(lH-tetrazol-5-yl)-biphenyl
a) 4'-[(2-Ethoxy-4-methyl-6-(1-methyl-benzimidazol-2-
yl)-benzimidazol-1-yl)-methyl]-2-(2-triphenylmethyl-
tetrazol-5-yl)-biphenyl
To a solution of 570 mg (1.86 mMol) of 2-ethoxy-4-
methyl-6-(1-methyl-benzimidazol-2-yl)-benzimidazole in
20 ml of dimethylsulphoxide are added 224 mg (2.0 mMol)
of potassium tert.butoxide and the mixture is stirred
for 15 minutes at ambient temperature. Then 1.11 g
2 ~ 2 ~
- 37 -
(2.0 mMol) of 4'-bromomethyl-2~(2-triphenylmethyl-
tetrazol-5-yl)-bipheny'l are added and the mixture is
stirred for a further 3 hours at ambient temperature.
Then the mixture is stirred into about 50 ml of
saturated sodium chloride solution, the crude product
precipitated is suction filtered and purified by column
chromatography (100 g silica gel; eluant: ethyl
acetate/petroleum ether = 4:1).
Yield: 860 mg (59% of theory),
R~ value: 0.56 (silica gel; ethyl acetate/petroleum ether
= 4:1)
b) 4'-[(2-Ethoxy-4-methyl-6-(1-methyl-benzimidazol~2-
yl)-benzimidazol-l-yl)-methyl]-2-(lH-tetrazol-5-yl)-
biPhenylA mixture of 830 mg (1.06 mMol) of 4'-[(2-ethoxy-4-
methyl-6-(1-methyl-benzimidazol-2-yl)-benzimidazol-1-
yl)-methyl]-2-(2-triphenylmethyl-tetrazol-5-yl)-
biphenyl, 2.5 ml of lN sodium hydroxide solution and
20 ml of ethanol is stirred for 2 hours at 80C. The
solution is then evaporated down, the residue is mixed
with about 30 ml of water and made slightly acidic with
glacial acetic acid. It is then extracted three times
with about 20 ml of methylene chloride, the combined
organic extracts are washed with 20 ml of water and
concentrated by evaporation. The crude product thus
obtained is purified by column chromatography (50 g
silica gel; methylene chloride/ethanol = 97:3).
Yield: 430 mg (75~ of theory),
Melting point: 194-197~C
C32H2sNs (540.60)
Calculated: C 71.10 H 5.22 N 20.73
Found: 69.99 5.36 20.54
Mass spectrum: m/e = ~70
~c~a~1~ 2 ~
- 38 -
Example 7
4'-[(2-Ethoxy-4-methyl-6-(imidazo[1,2-a]pyridin-2-yl)-
benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylic acid-
hydrate
-
Prepared analogously to Example 1 from tert.butyl 4'-
[(2-ethoxy-4-methyl-6~(imidazo[1,2-a]pyridin-2-yl)-
benzimidazol-1-yl)-methyl]-biphenyl-2-carboxylate and
trifluoroacetic acid in methylene chloride.
Yield: 72% of theory,
Melting point: 207-209C
C3~H26N403 x H20 (520.60)
Calculated: C 71.52 H 5.42 N 10.76
Found: 71.22 5.37 10.76
Rf value: 0.36 (silica gel; methylene chloride/ethanol =
19 : 1 )
Example 8
Mixture of
4'-[(2 n-Propyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-l-yl)-methyl]-2-[1-(pivaloyloxymethyl)-
tetrazol-5-yl]-biphenyl and
4'-[(2-n Propyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-l-yl)-methyl]-2-[2-(pivaloyloxymethyl)-
tetrazol-5-yl]-biphenyl
A solution of 400 mg (0.74 mMol) of 4'-[(2-n-propyl-4-
methyl-6-(butanesultam-1-yl)-benzimidazol-1-yl)-methyl]-
2-(lH-tetrazol-5-yl)-biphenyl, 0.16 ml (1.1 mMol) of
chloromethyl pivalate and 194 mg (1.1 mMol) of potassium
carbonate-dihydrate in 10 ml of dimethylformamide is
stirred for 14 hours at ambient temperature, then
stirred into about 50 ml of saturated sodium chloride
solution and extracted three times with about 20 ml of
methylene chloride. The combined or~anic extracts are
washed with water and evaporated down. The crude
2 1
- 39 -
product thus obtained is purified by column
chromatography (50 ~ silica gel; eluant: methylene
chloride/ethanol = 98:2).
Yield: 400 mg (82% of theory),
Melting point: amorphous
C3sH41N704S (655.80)
Calculated: C 64.10 H 6.30 N 14.95 S 4.88
Found: 63.99 6.22 14.80 5.03
Rf value: 0.46 (silica gel; methylene chloride/ethanol =
19 : 1 )
~=,~
Mixture of
4'-[(2-n-Propyl-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
2-[1-(pivaloyloxymethyl)-tetrazol-5-yl]-biphenyl and
4'-[(2-n-Propyl-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[1,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
2-[2-(pivaloyloxymethyl)-tetrazol-5-yl]-biphenyl
Prepared analogously to Example 8 from 4'-[(2-n-propyl-
4-methyl-6-(5,6,7,8-tetrahydro-imidazo[1,2-a]-pyridin-2-
yl)-benzimidazol-1-yl)-methyl]-2-(lH-tetrazol-5-yl)-
biphenyl and chloromethylpivalate.
Yield: 75% of theory,
Melting point: 203-205C
C38H42N8O2 (642.80)
Calculated: C 71.00 H 6.59 N 17.43
Found: 70.85 6.63 17.43
Rf value: 0.43 (silica gel; methylene chloride/ethanol =
19 ~
Mass spectrum: m/e = 642
2rl
- 40 -
Example 10
4'-[(2-n-Propyl-4-methyl-6 (5,6,7,8-tetrahydro-
imidazo[1,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
2-[l-(cyclohexyloxycarbonyloxy) ethyloxycarbonyl]-
biphenyl
-
A solution of 504 mg (1.0 mMol) of 4l-[(2-n-propyl~4-
methyl-6-(5,6,7,8-tetrahydro-imidazo[1,2-a]pyridin-2-
yl)-benzimidazol-1-yl)-methyl]-biphenyl-2-carboxylic
acid, 600 mg of l-(cyclohexyloxycarbonyloxy)-ethyliodide
and 350 mg of potassium carbonate in 25 ml of
dimethylsulphoxide is stirred for 14 hours at ambient
temperature, then stirred into about 70 ml of saturated
sodium chloride solution and extracted three times with
30 ml of ethyl acetate. The combined organic extracts
are washed with water and evaporated down. The crude
product thus obtained is purified by column
chromatography (100 g silica gel; eluant: methylene
chloride/ethanol = 98:2).
Yield: 325 mg (48% of theory),
Melting point: 162-164C
C41H46N4O5 (674.85)
Calculated: C 72.97 H 6.87 N 8.30
Found: 72.63 6.77 8.17
Rf value: 0.52 (silica gel; methylene chloride/ethanol =
19 : 1 )
Mass spectrum: m/e = 674
Example 11
4'-[(2-n-Propyl-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[1,2-a]pyridin-2-yl)-benzimidaæol-1-yl)-methyl]-
2-(pivaloyloxymethyloxycarbonyl)-biphenyl
Prepared analogously to Example 8 from 4'-[(2-n-propyl-
4-methyl-6-(5,6,7,8-tetrahydro-imidazo[1,2-a]pyridin-2-
yl)-benzimidazol-1-yl)-methyl]-biphenyl-2-carboxylic
3 ~ ~
- 41 -
acid and chloromethylpi~alate in dimethylformamide.
Yield: 76% of theory,
Melting point: 142-144C
C38H42N4O4 (618.79)
Calculated: C 73.76 H 6.84 N 9.09
Found: 73.60 6.92 9.17
Rf value: 0.40 (silica gel; methylene chloride/ethanol =
19 : 1 )
Mass spectrum: m/e = 618
Example 12
4'-[(2-n-Propyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-1-yl)-methyl]-2-(pivaloyloxymethyloxy-
carbonyl)-biphenyl
Prepared analogously to Example 8 from 4'-[(2-n-propyl-
4-methyl-6-(butanesultam-1-yl)-benzimidazol-1-yl)-
methyl]-biphenyl-2-carboxylic acid and
chloromethylpivalate in dimethylformamide.
Yield: 70% of theory,
Melting point: Oil
C3sH41N36S (631.80)
Calculated: C 66.54 H 6.54 N 6.65 S 5.08
Found: 66.21 6.67 6.54 5.34
Rf value: 0.49 (silica gel; methylene chloride/ethanol =
19 : 1 )
Mass spectrum: m/e = 631
Example 13
4'-[(2-n-Propyl-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
2-[1-(ethoxycarbonyloxymethyloxy)-carbonyl]-biphenyl
Prepared analogously to Example 8 from 4'-[(2-n-propyl-
4-methyl-6-(5,6,7,8-tetrahydro-imidazo[1,2-a]-pyridin-2-
yl)-benzimidazol-l-yl)-methyl]-biphenyl-2-carboxylic
2 7
- 42 -
acid and 1-(ethoxycarbonyloxy)-methylchloride in
dimethylformamide.
~ield: 38.5% of theory,
Melting point: 123-125~C
C3lH40N405 (620.76)
Calculated: C 71.59 H 6.50 N 9.03
Found: 71.57 6.58 9.03
Rf value: 0.33 (silica ~el; methylene chloride/ethanol =
19 : 1 )
Mass spectrum: m/e = 620
Example 14
4'-[(2-Ethoxy-4-methyl-6-(imidazo[1,2-a]pyridin-2-yl)-
benzimidazol-l-yl)-methyl]-2-(lH-tetrazol-5-yl)-biphenyl
r
Prepared analogously to Example 6 from 4'-[(2-ethoxy-4-
methyl-6-(imidazo[1,2-a]-pyridin-2-yl)-benzimidazol-1-
yl)-methyl]-2-(2-triphenylmethyl-tetrazol-5-yl)-biphenyl
and sodium hydroxide solution in ethanol.
Example 15
4'-[(2-Ethoxy-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
2-(lH-tetrazol-5-yl)-biphenyl
Prepared analogously to Example 6 from 4'-[(2-ethoxy-4-
methyl-6-(5,6,7,8-tetrahydro-imidazo[1,2-a]pyridin-2-
yl)-benzimidazol-l-yl)-methyl]-2-(2-triphenylmethyl-
tetrazol-5-y~)-biphenyl and sodium hydroxide solution in
ethanol.
2 '~
43 -
Example 16
4'-[(2-Ethoxy-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
biphenyl-2-carboxylic acid
Prepared analogously to Example 1 from tert.butyl 4'-
[(2-ethoxy-4-methyl-6-(5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-l-yl)-methyl]-
biphenyl-2-carboxylate and trifluoroacetic acid in
methylene chloride.
Yield: 63% of theory,
Melting point: 238-240C
Rf value: 0.62 (silica gel; methylene chloride/ethanol =
9:1)
Example 17
4'-[(2-Ethyl-4-methyl-6-(butanesultam-1-yl)-
benzimidazol-1-yl)-methyl]-biphenyl-2-carboxylic acid
Prepared analogously to Example 1 from tert.butyl 4'-
[(2-ethyl-4-methyl-6-(butanesultam-2-yl)-benzimidazol-1-
yl)-methyl]-biphenyl-2-carboxylate and trifluoroacetic
acid in methylene chloride.
Yield: 68~ of theory,
Melting point: > 240C
C28H29N34S (503.60)
Calculated: C 66.77 H 5.80 N 8.34
Found: 66.57 5.69 8.30
Rf value: 0.36 (silica gel; methylene chloride/ethanol =
9 : 1 )
2 ~ ; 2~1
- 44 -
Example 18
4'-[(2-Ethyl-4-methyl-6-(3-chloro-5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl3-
biphenyl-2-carboxylic acid
Prepared analogously to Example 1 from tert.butyl 4'
[(2-ethyl-4-methyl-6-(chloro-5,6,7,8-tetrahydro-
imidazo[l,2-a]pyridin-2-yl)-benzimidazol-1-yl)-methyl]-
biphenyl-2-carboxylate and trifluoroacetic acid in
methylene chloride.
Yield: 43% of theory,
Melting point: 295-297C
C3lH29ClN402 (525-06)
Calculated: C 70.91 H 5.57 N 10.67 Cl 6.75
Found: 70.81 5.54 10.55 6.83
Rf value: 0.36 (silica gel; methylene chloride/ethanol =
9 : 1 )
2 ~ ~ ~ b ~
In the Examples of Pharmaceutical Formulations which
follow, any suitable compound of formula I, particularly
those wherein R4 represents a tetrazolyl group
substituted in the 1- or 2-position by an Ra~CO-O-CH2
group, or wherein R4 denotes a carboxy, lH-tetrazolyl,
RaO-C0-, Rb-CO-O-(RCCH)-O-CO- or RbO-CO-O-(RCCH)-O-CO-
group, may be used as the active substance:
Example I
Ampoules containing 50 mg of active substance per 5 ml
Composition
Active substance 50 mg
KH2PO4 2 mg
Na2HP04 x 2H20 50 mg
NaCl 12 mg
Water for injections ad 5 ml
PreParation:
The buffer substances and isotonic substance are
dissolved in some of the water. The active substance is
added and, once it has been completely dissolved, water
is added to make up the required volume.
Example II
I Ampoules containing 100 mg of active substance per 5 ml
-
Composition
Active substance 100 mg
Methyl glucamine 35 mg
Glycofurol 1000 mg
Polyethyleneglycol-polypropylene-
glycol block polymer 250 mg
2 ~
46 -
Water for injections ad 5 ml
Preparation:
Methyl glucamine is dissolved in some of the water and
the active substance is dissolved with stirring and
heating. After the addition of solvents, water is added
to make up the desired volume.
Example III
Tablets containing 50 mg of active substance
Composition
Active substance 50.0 mg
Calcium phosphate 70.0 mg
Lactose 40.0 mg
Corn starch 35.0 mg
Polyvinylpyrrolidone 3.5 mg
Magnesium stearate 1.5 mg
200.0 mg
Preparation:
The active substance, CaHPO4, lactose and corn starch are
uniformly moistened with an aqueous PVP solution. The
mass is passed through a 2 mm screen, dried at 50C in a
circulating air dryer and screened again.
I!
After the lubricant has been added, the granules are
compressed in a tablet making machine.
Example IV
Coated tablets containing 50 mg of active substance
2 ~J ~
- ~7 -
Composition
Active substance 50.0 mg
Lysine 25.0 mg
Lactose 60.0 mg
Corn starch 3~.0 mg
Gelatin 10.0 mg
Magnesium stearate 1.0 mq
180.0 ~g
Preparation:
The active substance is mixed with the excipients and
moistened with an aqueous gelatin solution. After
screening and drying, the granules are mixed with
magnesium stearate and compressed to form tablet cores.
The cores thus produced are covered with a coating by
known methods. A colouring may be added to the coating
suspension or solution.
Example V
Coated tablets containing 100 mg of active substance
Composition
Active substance 100.0 mg
Lysine 50.0 mg
Lactose 86.0 mg
Corn starch 50.0 mg
Polyvinylpyrrolidone 2.8 mg
Microcrystalline cellulose60.0 mg
Magnesium stearate 1.2 mg
350.0 mg
2 ~
- 48 -
Preparation:
The active substance is mixed with the excipients and
moistened with an aqueous PVP solution. The moist mass
is passed through a 1.5 mm screen and dried at 45C.
After drying, it is-s~reened again and the magnesium
stearate is added. This mixture is compressed into
cores.
The cores thus produced are covered with a coating by
known methods. Colourings may be added to the coating
suspension or solution.
Example VI
.~
Capsules containing 250 mg of active substance
-
Composition
Active substance 250.0 mg
Corn starch 68.5 mg
Magnesium stearate 1.5 mq
320.0 mg
Preparation:
The active substance and corn starch are mixed together
and moistened with water. The moist mass is screened
and dried. The dry granules are screened and mixed with
magnesium stearate. The final mixture is packed into
size 1 hard gelatin capsules.
Example VII
Oral suspension containing 50 mg of active substance per
5 ml
.
21 ~ ~ ~ 2 ~
- 49 -
Composition
Active substance 50.0 mg
Hydroxyethylcellulose 50~0 mg
Sorbic acid 5.0 mg
70% sorbitol 600.0 mg
Glycerol 200.0 mg
Flavouring 15.0 mg
Water ad 5.0 ml
Preparation:
Distilled water is heated to 70C. Hydroxyethyl-
cellulose is dissolved therein with stirring. By the
addition of sorbitol solution and glycerol the mixture
is cooled to ambient temperature. At ambient
temperature, sorbic acid, flavouring and active
substance are added. The suspension is evacuated with
stirring to remove any air. One dose of 50 mg is
contained in 5.0 ml.
Example VIII
Suppositories containing 100 mg of active substance
Com~position
Active substance 100.0 mg
Solid fat 1600.0 m~
1700.0 mg
.
Preparation:
The hard fat is melted. At 40C the yround active
substance is homogeneously dispersed in the melt. It is
cooled to 38C and poured into slightly chilled
suppository moulds.