Note: Descriptions are shown in the official language in which they were submitted.
WO92/15695 21~ ~ 7 !1 PCT/DK92/00064
Process for the C-terminal modification of peptides having
a C-terminal penultimate proline residue
.
The present invention relates to a process for the C-
terminal modification of peptides, especially for the
preparation of peptides having a C-terminal proline amide
or N-substituted amide, e.g. human or salmon calcitonin or
their analogs.
The use of biologically active peptides for pharmaceutical
purposes and in agriculture has increased the importance
of being able to synthesize such compounds in bulk-scale.
Three methods are available: (a) chemical synthesis, (b)
enzymatic synthesis, and (c) fermentation with genetically
manipulated microorganisms. While the methods (a) and (b),
or a combination of them, are the preferred for short
peptides it becomes more and more apparent that long
peptides in the future will be produced through exploita-
tion of the advances in the methods dealing with recombi-
nant DNA. However, these methods do not permit a number of
modications such as incorporation of D-amino acids and C-
terminal amide or N-substituted amide groups which may be
of importance for the biological activity. A subsequent
enzymatic modification is therefore highly desirable and
such reactions have only been studied to a limited extent.
An enzyme has been described which~catalyses the hydroxyl-
ation of C-terminal glycyl residues which subsequently
decomposes leaving the penultimate residue amidated (US
Patent No. 4.708.934, EP 308067A, DK Application No.
4489/88). This glycine oxidase enzyme which is dependent
on Cu2~, 2 and ascorbate as cofactors is considered to be
the enzyme responsible for in vivo formation of peptide
amides. It has been utilised for amidation of peptides in
small scale but as it exhibits low activity its applicab-
.
. . . . . . . , . -
... ... : .. . , :
21~1 ~7i~
W O 92/15695 PC~r/DK92/00064
ility for large scale work is still questionable. Theenzyme, as isolated from natural sources like rat
medullary thyroid carcinoma is very costly.
S In the above-mentioned EP 308067 a number of similar a-
amidating enzymes of natural origin capable of
specifically cleavin~ C-terminal Gly are described. It is
stated that their amidation activity is based upon the
conversion of short D-amino acid containing substrates,
and that activities vs. any physiologically relevant
substrates in L-substrate had not been demonstrated.
In application W0 90/08194 (Tanaka et al) an ~-amidating
enzyme from the s~in of Xenopus laevis (a frog) is
described. Tanaka et al have produced this enzyme
biosynthetically, but its a-amidating activity is also
conditioned by the presence of C-terminal Gly.
Amidation may also by achieved by protease-catalysed
condensation reactions using an amino acid amide or
peptide amide as nucleophile. The yields of condensation
reactions are generally low even in the presence of
organic solvents unless the product precipitates in the
reaction mixture and this is often not the case with long
peptides. In addition, the precursor peptide may exhibit
- poor solubility in such media. However, serine or thiol- -
protease catalysed transpeptidation reactions may be
carried out in hi~h yield but it is a prerequisite that
the enzyme exhibits specificity for a peptide bond close
to the C-terminus. Endopeptidases are not generally
suitable since they usually will cleave at other positions
in the peptide chain as well. Serine carboxypeptidases, on
the other hand, exhibit strict specificity for the C-
terminal peptide bond and are able to catalyse the
exchange of the C~terminal amino acid with an amino acid
amide, added to the reaction medium to compete as nucleo-
. ;' , ' .' ';. ~ . , , , ., . ,.: .
.:' .: ' '. ' ' " '''. " . . .. , ~ . ,
WO92~15695 PCT/DK92/000~
2 ~ ~ ~ D 7.;~
-- 3
phile with water.
This property of serine carboxypeptidases was realized bya group of researchers at Carlsberg Research Center and
lead to a large family of patents assigned to the present
assignee based on DK application No. 1443/79 represented
by EP Patent No. 17485, US Patent No. 4.806.534 and its
parent US patent No. 4.339.534 and International Applica-
tion W0 80/02151, which lead to a number of patents i.a.
~K Patent No. 155613 and JP Patent No. 1.489.494. ~hese
patents were based on the at that time surprising finding
that exopeptidases were suitable as catalysts for
enzymatic peptide synthesis, while the prior art dealt
exclusively with endopeptidases. Dependent on the nature
of the reactants (substrate and nucleophile components),
and the reaction conditions, particularly the pH, serine
and thiol carboxypeptidases may catalyze peptide synthesis
by chain elongation or by transpeptidation. The preferred
enzyme is carboxypeptidase Y (CPD-Y) from yeast.
The underlying and subsequent research has been further
described in a number of articles (Ref. 1-8), which
together with the above-mentioned patents are all
incorporated by reference.
The general principle of enzymatic peptide synthesis by
transpeptidation in the presence of serine or thiol
carboxypeptidases is disclosed in US Patent No. 4.806.473
and its parallel Danish Patent No. 155613. With particular
reference to the production of peptide amides these
patents generally disclose and claim the production of
peptide amides A-B-NH2 where A represent an N-terminal
protected amino acid residue or an optionally N-terminal
protected peptide residue and B-represents an L-amino acid
residue, by reacting as substrate component an optionally
N-terminal protected peptide A-X-OH, where A is as defined
.. . .. . .
.: , ~ . . . . -
:. - . . , .j",.i : ' ;
, . . - ' ' ' '
21~107~ 1
W092~15695 PCT/DK92/000~
- 4 - !
above and X represents an amino acid, with a nucleophile
(amine) component H-B-NH2 in the presence of an L-specific
serine or thiol carboxypeptidase enzyme from yeast, or of
animal, vegetable or microbial origin in an aqueous solu-
tion of dispersion being a pH from 5 to 10.5. As furtherexplained in Ref. 1 the preferred pH is about neutral if
the formation of a peptide amide is desired.
Further experiments are disclosed in Ref. 1-5 which
support the pioneer character of these early patents and
the general applicability of serine carboxypetidases as
catalysts for C-terminal modification of peptides.
In particular, Ref. 3 compared the reactivity of various
nucleophiles in the exchanges of C-terminal amino acid
residues in peptides, and i.a. concluded that ammonia was
an applicable nucleophile in transacylation reactions in a
manner equivalent to the above-mentioned use of amino
acids H-B-NH2. Thus Z-Ala-Ala-O~ was reacted with H-Gly-
NH2 and with NH3 resulting in the formation of Z-Ala-Gly-
NH2 and Z-Ala-N~2 in coupling yields of 100% and 75~,
respectively. In comparison the reaction with H-Gly-OMe
also lead to the formation of Z-Ala-Gly-OMe in a yield of
75%.
So already before the publication of the above patent
family it was obvious to a person skilled in the art that
ammonia was an applicable nucleophile in C-terminal
modification of peptides along with amino acid amides and
amino acid esters.
In ref. 2 was described the use of a series of primary
amines other than a-amino carboxylic acids as nucleophiles
in carboxypeptidase catalyzed coupliny to amino acid
3S esters. The nucleophiles included ammonia, hydrazine and
N-alkyl or other substituted derivatives of these.
- - - , ................ , . . , - . ................. : . .
: . . . :. . , ' . . . , :' :'.. ~ .
. .. . . : ~ . - ., . . . . . :
W092/15695 2 ~ ~ ~J ~ 7~ PCT/DK92/000~
However, the only substrates used were particular N- a
protected amino acid esters, viz. BzAlaOMe, and no
transpeptidation reactions were attempted.
Application WO 91/18998 assigned to the present assignee
describes a process for the preparation of derivatives of
growth hormone releasing factor GRF(1-29)NH2 and analogs
thereof by serine carboxypeptidase catalyzed trans-
peptidation.
In this process a substrate component of the formula
GRF'-Met-Ser-X
wherein GRF' denominates the native GRF(1-26) sequence or
analogs thereof including GRF(n-26) fragments , where n is
from 1 to 8, and X is an uncharged hydrophilic:acyclic -
amino carboxylic acid residue having the side chain of at
least the size of a methyl group, is reacted with H-Arg-
NH2 as nucleophile component in the presence of an L-
specific serine or thiolcarboxypeptidase enzyme from yeast
or of animal, vegetable or other microbial origin in an
aqueous solution or dispersion having a pH of from 6 to 9,
and if necessary the desired N-terminal (1-(n-1)) fragment
is coupled chemically or enzymatically.
Preferred amino acids X are Ala, Thr, Ser, Asn or Gln.
In this application, which is incorporated herein by
reference, a general discussion of the transpeptidation
principles and the competing reactions are given.
To recapitulate the essence of the above observations,
incorporation of C-terminal amide groups in peptides by
transpeptidation in the presence of a serine carboxy-
peptidase using the proper amino acid amide as the
- , . ' . .: . ~ : .. : .
.. ~ .,... : : : .
.
WO ~6~ 7 ~ PCT/DK92/oOO~
6 --
nucleophile as broadly described and claimed in US patent
no. 4,806,473 and the other family members is a very
appropriate method virtually applicable for any peptide.
Also as shown already in Ref. 3, it could be expected that
ammonia would be a suitable nucleophile in such amidation
reactions.
~ owever, the process is not always sufficiently selective
and necessitates purification procedures in order to
remove products of various side reactions in particular
when longer peptides are used, in which case the optimal
reaction conditions for suppressing the side reactions are
difficult to establish.
In the early articles by the original inventors published
shortly after filing of the above patent applications,
attempts were made to analyze the influence of the C-
terminal (leaving group) amino acid and the penultimate
amino acid. Thus in Ref. 1 Breddam et al using Leu-NH2 as
the nucleophile and Gly, Ala, Ser, Val, Leu and Phe as the
leaving groups suggested on the basis of the obtained
yields that only in cases where the leaving group is one
of the smallest amino acids, i.e. Gly, Ala or Ser is the
reaction successful, and at least for the simple sub-
strates tested there was no dependence on the penultimate
residue (being Ala, Phe and Gly).
In Ref. 3 Breddam et al using Gly-NH2 as the nucleophile
and Z-Ala-X as the substrate, where x was Gly, Ala, Ser,
Arg, Pro, Lys, Asn, His, Val, Met, Phe and Asp modified
the earlier statement to the effect that when using Gly-
- NH2 as the nucleophile, the yield is strongly dependent on
the nature of the C-terminal (leaving group) amino acid.
The yields varied from 10 to 100~ with the lowest yields
obtained with substrates where a hydrophobic acid (Val,
., - , . . .
- , . ., ' :' , - ,. , ,,.: . '
'' : , . : .
W092/1569~ ~ i Q ~ ~ 7 ~ PCT/DK92/000~
Met, Phe) serves as leaving group. It should be noted that
the yields with basic acids (Arg, Lys) are comparable to
the yields with the hydrophilic acids (Ala, Ser), Lys
being even better than Ser.
As for the penultimate amino acid residue of the peptide
substrates the influence was investigated using a series
of N-blocked dipeptides with different penultimate amino
acids (Ala, Val, Leu, Ile, Phe and Val) as the leaving
group. Using Gly-NH2 as the nucleophile, it was apparent
that the coupling yield which varied from 45% for Ile to
5~ for Phe is dependent on the penultimate amino acid
residue, but no obvious trend could be found.
In Ref. 4 some o~ the experiments underlying US patent no.
4,645,740 and its family members were discussed. Here
porcine insulin Ins-Pro-Lys-Ala was reacted with i.a. Thr-
NH2 and it was concluded that Ins-Pro-Lys-OH was a better
substrate that Ins-Pro-Lys-Ala-OH, since Ins-Pro-Thr-NX2
was formed in greater yields than Ins-Pro-Lys-Thr-NH2. By
inference Lys in this reaction was a better leaving group
than Ala. Also a significant oligomerization under
formation of Ins-Pro-Lys-Thr-Thr-NH2 occurred.
These results were further confirmed in Ref. S using Bz-
Lys-Ala-OH as a model peptide alongside with porcine
insulin. The conclusive message was that or the future
use of CPD-Y (the serine carboxypep~idase used in the
experiments) in transpeptidation reactions it is important
to be aware of the possibility that side products may be
formed.
Besides the above investigations of the applicability of
serine carboxypeptidase in C-terminal modifications of
insulin, a further experiment with amidation of longer
peptides using CPD-Y as a catalyst has been reported.
"
....
2~ 07 ~ 1
WO9V15695 PCT/DK92/000
-- 8
Thus in EP-B2-197794 and the parallel US patent no.
4,709,014 (Tamaoki) human calcitonin-Leu peptide was
reacted with ammonia as the nucleophile using CPD-Y as the
catalyst under conditions otherwise similar to those used
by Breddam et al in Ref. 3.
Tamaoki obtained S-sulfonated human calcitonin amide in a
yield of 24.7~, leaving 57~ unreacted substrate and 17.2%
non-amidated side products.
The S-sulfonated calcitonin was reduced with glutathione
to give a mature human calcitonin, but no yield is stated.
In its more general aspects the Tamaoki patents which are
incorporated by reference disclose a process for the pre-
paration of a peptide having a C-terminal proline amide,
which comprises reacting in aqueous solution a peptide
substrate having C-terminal Pro-Leu, Pro-Ile, Pro-Val or
Pro-Phe with carboxypeptidase Y in the presence of
ammonia.
Without in any way wanting to endorse the statements made ~-
by Tamaoki, it should be mentioned that he claims that
contrary to the findings of Breddam et al in Ref. 3, where
a preference for hydrophilic C-terminal amino acids as
leaving groups is expressed, the use of hydrophobic amino
acids (Leu, Ile, Val and Phe) gives better yields than
Gly, when Pro is the penultimate amino acid.
Nevertheless the yields of the amidation products in
Tamaoki's examples using Cbz-Ala-Pro-X-OH as the sub-
strate, where X is Leu, Leu, Val, Phe and Ile, were only
35,1%, 43~, 15,4%, 13,4~, and 22,6~, respectively. The
remainder was - to the extent reported - unreacted
starting material and non-amidated side-products Cbz-Ala-
Pro-OH.
.- :
, .. . , . .
,:' '.'~ , '' , : '
~092/15695 21 ~1 0 7 `~ PCT/DK92/000~
g
Summing up what has been said above, previous literature
on carboxypeptidase catalyzed transpeptidations using
amino acid amides as nucleophiles has shown no obvious
trend in the influence of the penultimate amino acid
residue, as stated in Ref. 3. As for the influence of the
leaving group likewise varying results have been found.
Thus, in the above-mentioned reference, small hydrophilic
leaving groups, or even larger positively charged ones,
gave the best yields on Z-Ala-X models, while in W0
91/18998 large uncharged hydrophilics were preferable for
transpeptidation on R-Met-Ser-X substrates. Likewise,
using ammonia as a nucleophile, Ref. 3 indicates superior
yields for Z-Ala-X substrates using Ala as leaving group.
Using peptides of type R-Pro-X Tamaoki states a particular
group of hydrophobic leaving groups to be unique for the
reaction with ammonia, viz. Leu, Phe, Ile and Val, for
which reasonable, albeit modest yields were obtained, in
contrast to using Gly as a leaving group or Ala as
apparent from Tamaoki's JP priority application no.
72705/1985, where no reaction product was formed.
The present invention is based on the surprising finding
that in peptides of the above type, which may serve as
models for i.a. Calcitonins, a different group of amino
acid residues is able to act as good leaving groups in
carboxypeptidase catalyzed reactions with ammonia,
hydrazine or substituted derivatives thereof, ensuring
both speediness and high yields, usually far superior to
the ones reported in the a~ove-mentioned Tamaoki patent.
Consequently, the invention relates to a process for the
preparation of C-terminally modified peptides of the
general formula
Peptide-Pro-NH-R
,, , . : ,
.
2 ~
WO92~15695 PCT/DK92/000~ _~
-- 10 --
wherein R is selected from hydrogen, hydroxy, Cl-C6 al~yl,
hydroxy Cl 6 alkyl, C6 9 aralkyl or R is a group NHRl,
wherein Rl is hydrogen, Cl 6 aralkyl, C6 9 aralkyl or a
group C0-R2, wherein R2 is selected from NH2, Cl 6 alkyl
and C6 9 aralkyl.
Cl 6 alkyl encompasses straight chain or branched alkyl,
e.g. methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl
tert.butyl, pentyl and hexyl. A preferred alkyl group is
ethyl.
C6 9 aralkyl encompasses e.y. phenyl-Cl 3 al~yl, e.g.
benzyl, phenylethyl and phenylpropyl.
lS The alkyl and aralkyl groups may be substituted with one
or more inert substituents, e.g. halogen (F, Cl, Br, I),
hydroxy or nitro.
The process according to the invention is characterized by
reacting a substrate component of the general formula
Peptide - Pro - X
wherein X is an amino acid having an uncharged or
positively charged side chain comprising at least two
carbon atoms and further comprising at least one hetero
atom selected from N, 0 and S, with a nucleophile
component NH2-R, wherein R has the above meaning in the
presence of an L-specific serine or thiolcarboxypeptidase
enzyme from yeast or of animal, vegetable or other
microbial origin in an agueous solution or dispersion
having a pH of from 7.5 to lO, and if desired converting a
reaction product wherein R is different from hydrogen into
a peptide amide.
, .. . . . . . . .
.: . -- . -,
: . , ,: . ,
- : : . ~ : ,, : :
-. : . . : . .. .
~092/15695 2 ~ 7 ~ PCT/DK92/000~
For reasons of record it is noted that among the
applicable nucleophiles R = H is ammonia, R = NHRl,
wherein Rl = H is hydrazine, R = NHCOR2, wherein R2 is NH2
is semicarbazide.
The applicable group of amino acid leaving groups X spans
a wide range of hydrophilicity - from the hydrophobic
tryptophan and tyrosine over methionine and its protected
derivatives, e.g. sulphone, Met(o), histidine and
threonine to the hydrophilic glutamine, asparagine,
arginine and lysine using the scale of Hopp & Woods (Ref
13). They share the common structural property of having
large (at least C2) side chains, which carry at least one
hetero atom chosen among N, 0 and S, said side chains
being uncharged or positively charged.
Using ammonia as the nucleophile, the process of the
invention is suitable for the production of various
peptide hormones of the calcitonin type, e.g. human
calcitonin having the amino acid sequence:
r S S - I
Cys-Gly-Asn-Leu-Ser-Thr-Cys-Met-Leu-Gly-Thr-Tyr-Thr-Gln-
5 lO
Asp-Phe-Asn-Lys-Phe-His-Thr-Phe-Pro-Gln-Thr-Ala-Ile-Gly-
15 20 25 .
Val-Gly-Ala-Pro-NH2
30 32
A particularly interesting calcitonin having a 20 times
greater potency is salmon calcitonin having the amino acid
sequence:
, - - ~ . : , . ~ . . , . :
.
.
. .. : . . . .. . . . ,.. ,, .. ~ . ~ :
. . ': ' .. '' ' : . . :: .
W092~ ~Q 1 Q 7-~ PCT/DK92/000
~ - 12 -
r-S -- S -~
Cys-Ser-Asn-Leu-Ser-Thr-Cys-Val-Leu-Gly-Lys-Leu-Ser-Gln-
5 10
Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Thr-Gly-
15 20 25
Ser-Gly-Thr-Pro-NH2
32
Also calcitonin from other natural species, e.g. eel,
chicken, ovine, bovine, porcine and murine calcitonin may
be prepared by the process according to the invention.
The struc~ures of these calcitonins are described in US
patent No. 4,652,627 (Kempe et al.) which is incorporated
by reference. Kempe also discloses such calcitonins having
a D-amino acid substituent in position 31. They can also
be made by the process according to the present invention.
If hydrazine is used as nucleophile, the process may be
used to produce the corresponding hydrazides, e.g.
calcitonin hydrazides. If it is desired to convert these
further by chemical methods, e.g. the azide method, it is
necessary first to protect N-terminal and possible side
chain amino groups in the substrate, by e.y. ~oc, before
the reaction. The protected azide may then be reacted with
ammonia to form a protected peptide amide, from which the
protective groups may be removed in a manner known per se
to form the desired peptide amide, e.g. calcitonin.
It has also been found that in contrast to the statements
made by Tamaoki better yields are obtainable using as the
nucleophile elevated concentrations of ammonia in free
base form, particularly in the presence of oryanic acids,
preferably acetic acid or formic acid, but not equal
yields using lithium hydroxide as base in adjustment of
... ..
~, . ~ . , . . . - , . . ..................... ,-, . .
~, . ,, , :. . . , - ::
: . , .... :
W092/lS695 2 1 ~ ~ ~ 7 ~~ PCT/DK92/0~ ~
NH4Cl. Without wishing to be bound by any particular
theory, it may be envisaged that the sterical effects of
the large residues in combination with hydrogen bonding or
salt or electronic effects often possible from hetero
atoms may cause the action, but the magnitude, influence
or exact nature of these interactions are not yet clear.
Finally, it apparently was not possible to keep the
substrates preferred by Tamaoki soluble in the human
Calcitonin precursor, necessitating an oxidation of the
disulfide bridge before the transpeptidation and a
subsequent reduction before the desired Calcitonin could
be obtained. Using the analogous substrates in the
preferred embodiment of the present invention, it has been
possible in many cases to keep the substrates in solution
and thus performing directly a one-step reaction instead
of Tamao~i's three-step reaction provided that guanidinium
hydrochloride is added.
However, if for technical reasons, it is desirable to use
an open chain or solubilized intermediate, this is also
possible in the process according to the invention.
Using ethyl amine or semicarbazide as nucleophiles,
compounds containing a C-terminal proline N-ethyl amide or
proline semicarbazide may be prepared according to the
process. As examples of such peptides containing these
groups of biological interest are e.g. the nonapeptide
Luteinizing Hormone Releasing Hormone analogue drugs,
Leuprolide~, see GB patent No. 1.434.694, and Buserelin,
see US patent No. 4.263.282, having C-terminal Pro-NEt and
Goserelin, see US patent No. 4.100.274, having C-terminal
Pro-SEM.
The applicable carboxypep~idases in the process of the
invention are L-specific serine or thiol carboxy-
, , ,: ,:
W092/~ 7 ~ PCT/DK92/000
- 14 -
peptidases. Such enzymes can be produced by yeast fungi,
or they may be of animal, vegetable or other microbial
origin.
A particularly expedient enzyme is carboxypeptidase Y from
yeast fungi (CPD-Y). This enzyme is described in the
earlier patents i.a. with reference to Johansen et al
(Ref. 10) who developed a particularly expedient purifica-
tion method by affinity chromatography on an affinity
resin comprising a polymeric resin matrix with coupled
benzylsuccinyl grups. CPD-Y, which is a serine enzyme is
available in large amounts and displays relatively great
stability. Further details are given in Ref. l.
The native CPD-Y is a well characterized serine
carboxypeptidase. A comparison with other such
carboxypeptidases is given in Ref. 7. These were also from
other sources than yeast or genetically or chemically
modified types. Another CPD-Y homologous serine
carboxypeptidase from yeast, KEX 1, is described in Ref.
14 and further characterized in Ref. 15. A combination of
chemical and genetic modification of a yeast
carboxypeptidase is described in Ref. 16.
CPD-Y is easily isolated from baker's yeast after
autolysis (Ref. lO) or from the medium of genetically
manipulated yeast cells (Ref. 11) as applied in example
18. ~his enzyme has a different glycosylation and
molecular weight, but has proved to be equally useful in
the native form. The cost of the enzyme is rather low and
the procedure described here therefore seems to be a
valuable alternative to the use of the much more rare
glycine oxidase.
In addition to CPD-Y, which is the preferred enzyme at
present, the process of the invention is feasible with
' ~ ' , .
.' . .' ~ :
, ~
,, ' ~
W092/tS695 21 ~ 1 0 7~ PCT/DK92/000
- 15 -
other carboxypeptidases from other sources than yeast,
such as those listed in the following survey:
Enzyme Origin
Fungi
Carbo~ypeptidase(s) from Penicillium janthinellum
Carboxypeptidase(s) from Aspergillus saitoi
Carboxypeptidase(s) from Aspergillus oryzae
Plants
10 Carboxypeptidase(s) C Orange leaves
Orange peels
Carboxypeptidase CN Citrus natsudaidai Hayata .
Phaseolain French bean leaves
Carboxypeptidases M from Germinating berlay
15 Carboxypeptidases W from Wheat bran
Carboxypeptidases from Germinating cotton plants
Tomatoes
Watermelons
8romelain(pineapple)powder
The close relationship between a number of the above
carboxypeptidases is discussed by Kubota et al (Ref. 12)
and further expanded in Ref. 7.
As stated above the process of the invention may be
carried out at pH 7.5 to 10.0, preferably at pH 8.5 to
9.5, most preferably from 9.0 to 9.5. Accordingly, it is
necessary for the enzyme to have sufficient stability in
alkaline media during the reaction period. The preferred
pH-value, which is often within a very narrow range,
depends upon the enzyme used and the substrate employed.
For CPD-Y, a favourable pH for most substrates is about
9.2. -
The preferred agents for pH-adjustment in starting
solutions containing NH3 are low molecular carboxylic
. . . . . .. , .. , . ~ ,.:. ; ., .: . - . . . . .: .... .
2lal~7~
W092/l5695 PCT/DK92/00064
.
- 16 -
acid, preferably acetic acid or formic acid and good
results have also been obtained using some ammonium salts,
e.g. NH4N03.
The reaction is carried out in an aqueous reaction medium
which, if desired, may contain up to 25% by volume of an
organic solvent not including possible organic pH-adjust-
ment agents. Preferred organic solvents are dimethyl
formamide and dimethyl sulfoxide, but also alkanols, e.g.
methanol and ethanol, glycols, e.g. ethylene glycol or
polyethylene glycols, triethylene glycol dimethyl ether,
glycerol, alkanoic acids, e.g. acetic acid, tetra-
hydrofurane, dioxane and dimethoxyethane may be used.
Preferably only small amounts, e.g. 2-12~, of organic
solvent are used.
The selection of the composition of the reaction medium
depends particularly upon the solubility of the reaction
components and the reaction products involved and upon the
stability of the enzyme. These can be affected by addition
of urea and/or detergents. Examples are anionic, e.g.
pentanesulphonic acid, zwitterionic, e.g CHAPS0, nonionic,
e.g. Brij 35 or Tween 20 and cationic, e.g. guanidinium
hydrochloride.
Stabilization of the enzyme might also be brought about by
addition of carbohydrates, e.g. mannitol or proteins, e.g.
BSA.
Naturally this variety of additives may also affect the
course and synthetic ratio of the reaction.
The reaction medium may also comprise a component that
renders the enzyme insoluble, but retains a considerable
part of the enzyme activity, such as an ion exchanger
resin. Alternatively, the enzyme may be immobilized in
.,: ,
. :, . ,, : , . .
W092/l5695 ~ 7 ~t PCT/DK92/00064
- 17 -
known ~anner, e.g. by bonding to a matrix, such as a
cross-linked dextran or agarose, or to a silica, polyamide
or cellulose, or by encapsulating in polyacrylamide,
alginates or fibres. Besides, the enzyme may be modified
by chemical means to improve its stability or enzymatic
properties.
The addition of a chelating agent e.g. EDTA to the
reaction medium is often not necessary.
However, the medium preferably contains a gelation
inhibiting a~ent, e.g. guanidium hydrochloride.
The concentration of the two participants in the reaction
lS may vary within wide limits, as explained below. A
preferred starting concentration for the peptide substrate
is 0.1-5.0 mM, preferably 0.2-l.0 mM, in parti~ular about
0.5 mM, and when the nucleophile is ammonia, it is
preferably added as a saturated solution or on liquid
20 form, the concentration is 4.0 to 12.0 M, preferably 4.3 -
9.7 M, in particular 5 - 8 M. This is in contrast to
Tamaoki, who only used 4.5 M solution and claimed this to
be optimal.
For many of the other nucleophiles, e.g. benzyl amine, a
much lower concentration, viz. 0.1 M may be used.
Generally concentrations of l.0 to 4.0 M, preferably 2.0
to 3.0 M are used.
It is not necessary to protect the N-terminal or possible
side chain amino acids or carboxylic groups in the
substrate during the reaction with the nucleophile.
However, it may be necessary if the reaction product, e.g.
a hydrazide, is to be used for further reactions. By the
same token it may be desirable to protect one of the amino
groups in hydrazine e.g. with Boc. ~ -
.- . . . ,, -, . . . .
- : ........................ . .. ~ . . . ..
, . . . .
,, . . ' . , . . ' ,: , ", . . ' ' ~ ' . : , :
2 1 0 ~
W092/l5695 PCT/DK92/00064
- 18 -
The enzyme activity may vary as well, but for CPD-Y the
concentration is 5 - 50 ~m, preferably 5 - 20 ~m. The most
advantageous activity depends i.a. on the substrate chain
and concentration, the nucleophile concentration, the
reaction time, the reaction temperature, the pH, and the
presence of organic solvents and/or salts.
According to the invention the reaction temperature is 20
to 40C. An appropriate temperature will usually be about
33 to 39C, preferably about 37C, taking into account
due consideration for enzyme activity and stability.
Similar variations occur for the reaction time which
depends very much upon the above-mentioned reaction
parameters, especially the enzyme concentration. The
standard reaction time in the process of the invention is
about 1 - 5 hours.
As for the pressure, the reaction is preferably carried
out in closed vessels at a pressure of 1 - 3 bar,
preferably l - 2 bar.
~he abbreviations of amino acids, amino acid derivatives
and peptides are according to Guidelines of the IUPAC-IUB
Commission on Biochemical Nomenclature and the amino acids
are on L-form unless otherwise stipulated.
The following additional abbreviations are used: HOAc,
acetic acid; Bz, N-benzoyl, Boc, tert.butyloxycarbonyl;
~MF, N,N-dimethylformamide; EDTA, ethylene diamine
tetraacetic acid; GRF, yrowth hormone releasing factor;
HPLC, high performance liquid chromatography: SEM,
semicarbazide, TFA, trifluoroacetic acid; TGME,
triethylene glycol dimethyl ether; THF, tetrahydrofuran:
Z, carbobenzoxy; CHAPSO, 3-[(3-cholamidopropyl)-
.. ,: ~ . " .. , : , . - -
WO92/15695 21 ~ PCT~DK92/00064
- 19 -
dimethylammonio~-2-hydroxy-1-propane-sulfonate.
Before the process of the invention will be illustrated by
examples, starting materials, methods of measurement, etc.
will be explained in general terms.
General procedure for examples 1 - 19
The reactions were performed on four different groups of
test substrates, wherein X designates the amino acid
leaving group:
Tripeptides of formula Z-Thr-Pro-X-OH and Z-Ala-Pro-X-OH
served as short model substrates for salmon and human
calcitonin, respectively. Substrates of formula: H-Leu-
His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Thr-Gly-Ser-Gly-
Thr-Pro-X-OH served as longer model substrates:for salmon
calcitonin, corresponding to residues 16 - 32 elongated by
X, and henceforth referred to as SAL (16-32)-X. These
substrates were obtained by chemical synthesis, by
employing liquid and solid phase methods at conventional
state of the art level, e.g. as described in appln. WO
91/18998. Finally, human calcitonin elongated by one amino
acid residue can be obtained by an enzymatic coupling of
the free amino acid to the corresponding calcitonin methyl
ester using CPD-Y catalysis in aqueous medium (see example
13 below) or by standard solid phase methods followed by
formation of the disulfide bridge by cyclization.
Following HPLC purification all shortened substrates were
found to be more than 95~ pure by HPLC in systems similar
to the ones used for monitoring the reaction, and for
selected specimens including the longest peptides, amino
acid analysis or gas sequence analyses were performed and
proved the correct identity. Met-sulfone substrates were
obtained by oxidizing the corresponding methionine
substrates e.g. by treatment with hydr~gen peroxide. By
::, . ,, . . ., : . . ..
- . . . . . . . .
.. . . .
, ........... . . . . , , -
.. ~ . . . . .
wo 92/l56925~ ~ ~ 0 7 ~ PCT/DK92/00064
- 20 -
synthetical methods similar to the ones described above
the corresponding amide products, byproducts and hydro~
lysis byproducts were synthesized as HPLC reference
substances.
Reaction monitoring, product identification and determina-
tion o f product yield were performed by means of reverse
phase HPLC (Waters 6000 A pumps, automated gradient
controller, UK 6 injector) on a C18 NOVA PAK column
(Waters, RCM) using suitable gradients of elution systems
containing 50 mM triethylammonium phosphate, pH 3.0 or
7.0 from 0~ to 80~ acetonitrile with a flow of 2 ml/min.
Elution was monitored by means of a UV detector (Waters
480) at 230 nm, 254 nm or 278 nm.
The products were identified by amino acid analysis of
fractions from the HPLC analysis, which corresponded to
the assumed product peak and/or by HPLC comparison with a
chemically synthesized reference product. In all cases,
HPLC systems were employed where the products could be
clearly separated from other compounds. Sometimes HPLC at
several pH-values were performed.
Concentrated ammonia stock solutions were made to form the
bulk of the reaction media. Normally at room temperature
and at atmosphere pressure of around 1 bar, they were made
basically in either of the two ways: either from a
concentrated solution of ammonia in water which was mixed
with a concentrated or aqueous acid solution or from an
ammonium salt which following dissolution in water was
mixed with a solid alkali base. Following cooling to room
temperature and if necessary dilution by water p~ was
adjusted using the relevant acid or base. Thus 10 ml of
25~ aqueous ammonia mixed with either 6.5 ml of glacial
acetic acid (HOAc) or 3.7 ml of concentrated formic acid
yields solution of 8 M and 10 M ammonia/ammonium
.........
~ . .
, ~ . , .
. ' ' . ':
,
W092/t5695 21 ~ 0 7 PCT/DK92/U00
- 21 -
concentrations when adjusted to pH 9 . 2 with concentrated
acid, and likewise at the same pH a 4.5 M ammonia/ammonium
solution may be obtained by mixing about 26.7 g NH4Cl and
8.1 g NaOH in 100 ml water. Ammonia concentrations are
later corrected for decreases as a result of the addition
of other solvents, reactants or additives.
Most reactions were performed in closed vessels, which
were normally closed at room temperature. pH recorded was
thus the value measured at this temperature and the
initial reaction pressure was usually above 1 bar by an
unmeasured margin smaller than 1 bar, i.e. 1 - 2 bar.
Unless otherwise mentioned, all experiments were carried
out at a volume of 1 ml, or sometimes 0.5 ml, in
Eppendorph plastic micro test tubes with safety lid loc~,
inserted into an Eppendorph 5427 thermomixer. Other tubes,
i.e. glass tubes, have shown similar results. In a typical
short model peptide reaction a 10 mM solution in an
organic solvent, water or mixtures of these was made up
and poured into a tenfold volume excess of 2 - 10 molar
ammonia solution at the appropriate pH for reaction
containing desired salt or additive like guanidium
chloride. In case of CPD-Y the reaction was initiated by
addition of a suitable amount of dry 20~ w/w CPD-Y on
citrate or a similar 333 uM CPD-Y solution of purified
Carboxypeptidase Y produced by Carlbiotech Ltd. A/S,
Copenhagen. Recombinant secreted CPD-Y was a kind gift
from Dr. Jaeob ~. Winter of Carlsberg Research Center
and was purified as described in example 18. An
appropriate volume was added to initiate the reaction at
correct molarity of enzyme, normally 5 - 30 ~M. The
reaction mixture was then shaken in the thermomixer for
duration of the experiment, usually from 1 - 5 hours,
while aliquots of 10 to 20 ul were extracted from HPLC
analysis at regular intervals. It was checked that no
precipitation, i.e. loss of UV absorptive groups in the
.. . ,, . . ~ . , .................................. . -
.
W092/1 ~ ~ Q ~ ~ PCT~DK92/000
- 22 -
analysis, occurred. In some instances aliquots were
withdrawn and high amounts of organic solvent added to
stop the reaction, or the reaction was quenched by
addition of concentrated HCl.
During the time course of reactions, these were monitored
for substrate consumption, amide product obtained, primary
hydrolysis byproduct and amide byproduct formed by
secondary aminolytic cleavage of the proline residue in
the primary hydrolysis byproduct or amide product. The
relative molar percentages of these four types of
compounds, substrate, amidated product, primary hydrolysis
byproduct and amidated byproducts were calculated from the
relative area counts obtained by integration of the HPLC
W absorptions obtained at 254 or 230 nm, respectively,
using appropriate correction factors for difference in
absorption. These are listed in the examples as substr.,
yield, hydr. and oth., respectively. In the case of longer
substrates than tripeptides, the latter term has been
calculated to include also possible secondary hydrolysis
and aminolysis products, and for the human Calcitonin also
byproducts formed by known chemical side reactions in the
long peptide, i.e. a, ~-shifted aspartic acid or oxidized
methionine produced during the course of reaction, which
could also be separated in the systems employed. It was
not attempted to counter the oxidation reaction by
addition of reductive additives.
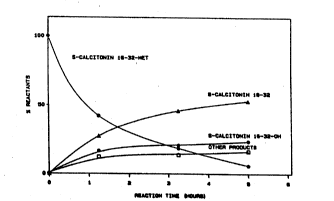
The time course of a typical reaction according to example
8 is illustrated in fig. l.
The relative amount of amidated product is in itself a
valid parameter for the feasibility of the process. To
help optimize reactions, a further variable was introduced
and calculated. Thus, RATI0 is calculated from the
following formula:
- . ,
.
. . , - ' . , ., ' ~
- : : .. : . , . . . . : - ..
WO92~15695 21 1~1 0 7 ~ PCT/DK92/00064
- 23 -
lOO*YIELD
RATIO =
(100 - SUBSTR.)
which indicates the catalytical efficiency in forming
product from substrate and may also be indicative of the
feasibility of introducing a recycling step in which
unreacted substrate is recovered and put through a renewed
reaction.
This value is also listed in the tables as are the ammonia
compounds SALT which were adjusted by the pH adjusting
agent PHADJ to obtain the final ammonia concentration
CNH3. Other additives are listed as ADD and their
concentrations as CADD. The temperature for performing
the experiment is listed as TEMP.
Finally, the initial substrate is often listed in the
examples as a core peptide named PEPTIDE with a separate
leaving group listed as LEAVING and the initial substrate
ccncentration as CPEPTIDE. .
.. :- . , . . . . , .. .: ,..... . . . . . . .
' : : . :: . . ,
., . . ~ . ,, , . . - .
. .: : .
21~o7!1 1
W O 92/~5695 PC~r/DK92/00064 _ ~.
24
Example 1
Scale amidation of Z-Thr-Pro-Met-OH
In a lOO ml glass flask, 25 mg Z-Thr-Pro-Met-OH was dis-
solved in l.O5 ml DMF, and 50 ml 8 M ammonia/ammonium
solution was added, wherein pH had been adjusted to 9.2
using glacial acetic acid. The glass flask was placed in
a stirred water bath thermostated at 37C and following
the addition of O.78 ml of a 0.3 mM solution of Carboxy-
peptidase Y to initiate the reaction, the flask was
sealed with a plastic screw-cap lid and left in the wa-
ter ~ath for 75 minutes, after which HPLC showed 76~ of
the amidated Z-Thr-Pro-NH2 product, 20% of the hydro-
lysis byproduct Z-Thr-Pro-OH and 4~ remaining substrate.
Example 2
Amidation of Z-Ala-Pro-Met-OH
O.Z3 mg Z-Ala-Pro-Met-OH was placed in an Eppendorph
plastic tube and dissolved in 40 ~l of DMSO and 945 ,ul
of 8 M ammonia/ammonium solution, in which p~ had been
adjusted to 9.2 using glacial acetic acid, was added,
followed by addition of 95 mg of guanidinium chloride.
The reaction was initiated by addition of 15 ~1 of a 0.3
mM solution of Carboxypeptidase Y and was shaken in the
Eppendorph mixer thermostated at 37C for the duration
of the experiment, following closing of the lid lock.
After 1290 minutes, HPLC showed 85~ of the amidated
product Z-Ala-Pro-NH2, 2~ of the hydrolysis byproduct Z-
Ala-Pro-OH, 1% of the amide byproduct Z-Ala-NH2 and 12% ,,
unreacted substrate.
- - , - . . . . .
, ., . . . . , . : ,' , ',
.., -- ., . ,., : , : :
, , . - ~ . , . ' ' , ' ' ' '
WO92/15695 2 1 ~ . ~ o~ 1 PCT/DK92/00064
Example 3
Amidation of Z-Thr-Pro-Met-OH
. _
0.46 mg Z-Thr-Pro-Met-OH was placed in an Eppendorph
plastic tube and dissolved in 20 ~1 of nMso and 965 ~1
of 1~ M ammonia/ammonium solution, in which pH had been
adjusted to 9.2 using concentrated formic acid, was
added. The reaction was initiated by addition of 15 ~1
of a 0.3 mM solution of Carboxypeptidase Y and was
shaken in the Eppendorph mixer thermostated at 37C for
the duration of the experiment, following closing of the
lid lock. After 90 minutes, HPLC showed 88% of the
amidated product Z-Thr-Pro-NH2, 12~ of the hydrolysis
byproduct Z-Thr-Pro-OH and no remaining substrate.
Example 4
Amidation of Z-Thr-Pro-Met-OH
0.46 mg Z-Thr-Pro-Met-O~ was placed in an Eppendorph
plastic tube and dissolved in 20 ~1 of DMF and 965 ,ul of
4.5 M ammonia/ammonium solution, in which pH had been
adjusted to 9.2 using solid sodium hydroxide and
ammonium chloride, was added. The reaction was initiated
by addition of lS ~1 of a 0.3 mM solution of
Carboxypeptidase Y and was shaken in the Eppendorph
mixer thermostated at 37C for the duration of the
experiment, following closing of the lid lock. A~ter 143
minutes, HPLC showed 74% of the amidated product Z-Thr-
Pro-N~2, 10% of the hydrolysis byproduct Z-Thr-Pro-OH
and 16~ remainin~ substrate.
-.
- ....... , . ~ : . . . .. . .
'. . '. ' ... . .. .' ' ' ' ' ., . : :" ~ .. : ,
21a ~ ~7!'l
W092/l569~ PCT/DK92/00064 ,_
26
Fxample 5
Amidation of_Z-Thr-Pro-Met(O)-OH
S 0.46 mg Z-Thr-Pro-Met~O)-OH, in which (O) designates the
side chain sulfone, was placed in an Eppendorph plastic
tube and dissolved in 905 ~1 of 8 M ammonia/ammonium
solution, in which pH had been adjusted to 9.2 using
glacial acetic acid, was added, followed by addition of
95 mg guanidinium chloride. The reaction was initiated
by addition of 15 ~1 of a O.3 mM solution of
Carboxypeptidase Y and was shaken in the Eppendorph
mixer thermostated at 37C for the duration of the
experiment, following closing of the lid lock. After 90
minutes, HPLC showed 33~ of the amidated product Z-Thr-
Pro-NH2, 8~ of the hydrolysis byproduct Z-Thr-Pro-OH, 2~
of the amide byproduct Z-Thr-Pro-NH2, and 57~ remaining
substrate.
Example 6
Amidation of salmon calcitonin(16-32)-Thr-OH
1.0 mg SAL(16-32)-Thr-OH was placed in an Eppendorph
plastic tube and dissolved in 20 ,ul of DMSO and 435 ~1
of 8 M ammonia/ammonium solution, in which pH had ~een
adjusted to 9.2 using glacial acetic acid, was added.
The reaction was initiated by addition of 45 ~1 of a 0.3
mM solution of Carboxypeptidase Y and was shaken in the
Eppendorph mixer thermostated at 37C for the duration
of the experiment, following closing of the lid lock.
After 300 minutes, HPLC showed 51~ of the amidated
product R-Thr-Pro-NH2, 24~ of the hydrolysis byproduct
R-Thr-Pro-~H, 16~ of other byproducts and 9% remaining
substrate.
~n the above, R designates the salmon calcitonin 16-30
.. : . . . . . , :
. : . , ......... .: . . - ~ . . ................ :: :
' - : :' . .~ . : ' , , , : :
,, : : : ~ , ,
WO92/1569~ 2 3 ~ PCT/DK92/00064
27
sequence.
Example 7
Amidation of salmon calcitonin(16-32 )-Tyr-OH
1.0 mg SAL(16-32)-Tyr-OH was placed in an Eppendorph
plastic tube and dissolved in 20 ~ul of DMSO and 43S ,ul
of 8 M ammonia/ammonium solution, in which pH had been
adjusted to 9.2 using glacial acetic acid, was added.
The reaction was initiated by addition of lS ~1 of a 0.3
mM solution of Carboxypeptidase Y and was shaken in the
Eppendorph mixer thermostated at 37C for the duration
of the exReriment, following closing of the lid lock.
After 1050 minutes, ~PLC showed 44~ of the amidated
product R-Thr-Pro-NH2, 13~ of the hydrolysis ~yproduct
R-Thr-Pro-OH, 31% of other byproducts and 12~ remaining
substrate.
In the above, R designates the salmon calcitonin 16-30
sequence.
~:
-:
, ~ . . , , . ~ .
, ~ , . . .
,. , : .
.. ;. ~
2~ 3~ ~
WO92/15695 PCT/DK92/000~
. ,_
2~
Example 8
Amidation of various salmon calcitonin fragment SAL(16-
32)-X peptides to ~ield SAL(16-32)-NH2.a)
Leav- CPEP- CPD-Y Time Yield Substr. Hydr. 0th. Ratio
ing TIDE
(X) (mM) (~M) (min) (%) (~
10 Metb) 0.5 20 1200 33 28 1920 46
MetC) 1.0 20 300 53 6 2417 56
Thr 1. 0 30 300 5l 9 2515 56
Tyr 1.0 30 180 36 19 36 9 44
Tyr 0.5 10 1050 44 12 1431 50
_
a) Reaction conditions: 7.7 M ammonia/ammonium, PHADJ:
HOAc, pH 9.2, 37C in Eppendorph mixer, 4% DMSO
b) No DMSO added, 1 M guanidinium chloride.
c) Time course of reaction is illustrated in Figure 1.
:
: 35
.... .
WO92/15695 2 ~ ~ ~ O ~ .~ PCT/DK92/000~
29
Example 9
Amidation of various Z-Thr-Pro-X peptidesa)
Leav- Sol- CPD-Y Time Yield Substr. Hydr.Oth. Ratio
ing vent
(X) (~M) (min) (%) (%) (%) (~) (%)
Met Water 5 60 78 3 19 080
10 Met(O) Water 5120 33 57 9 276
Met(O) 4% DMS0 S 180 16 81 2 2 81
Thr Water 45 85 58 27 15 079
Thr 4~ DMS015380 60 16 22 271
~hr 4% DMS0b) 15 250 60 8 293 65
15 ~rp 4~ DMS0b) 10 95 77 6 170 82
Arg 4~ DMS0~) 5 1200 57 4 337 59
Arg 4% DMS0b)C) 5 130 26 15 600 30
Lys 4% DMS0b) 5255 56 34 10 085
Lys 4% DMS0b)C) 5 190 41 26 340 55
20 Asn 4~ DMS0b) 17 150 3S 60 5 0 88
Glud)4% DMS0b) 17 1380 0 94 6 0 0
'
a) Reaction conditions: 1 mM peptide, 7.7 M CNH3
(ammonia/ammonium), PHADJ: HOAc, pH 9.2, 37C in
Eppendorph mixer, 1 M guanidinium chloride.
b) No guanidinium chloride added.
c) pH 7.8.
d) Comparison example. .:
" ' . . ~, ' '
:, , ~ ' ' ' ' ' ' `' ' .' , ' . ~ '
... ..
2 ~ 7 ~
W092/15695 PCT/DK92/00064
Example lO
Amidation of Z-Thr-Pro-Met in various solvents and
ammonia/ammonium mixturesa) _ _
Sol- CNH3 Salt+ Time Yield Su~str.Hydr.Oth. Ratio
vent PHADJ
(M) (min) (%)
-
Dioxaned) 7.7 NH3+ 180 70 14 16 O 82
HOAc
Dioxane ) ) 4.3 NH4Cl+ 1020 21 79 - - lOO
NaOH
Dioxane ) ) 4.0 NH4Cl+ 60 64 14 20 2 75
NaOH
DMSO 4 . 3 NH4Cl+150 70 21 9 0 88
NaOH
.
a) Reaction conditions: lmM peptide, 4~ solvent, 5 ~M
CPD-Y, pH 9.2, 37C in Eppendorph mixer.
- 25
b) 3.3 ~M CPD-Y added.
c) 33 ~M CPD-Y added.
d) Dioxane was peroxide free grade.
, ,' ~' ' ~ .' ~ ' ' :
.,, . : . .
:~ , ~ . . . .
WO 92/15695 2 1~ ~ 0 7 !~l PCT/DK92/000~
31
Example 1 1
Amidation of Z-Thr-Pro-Met at various concentrations of
DMSO )
DMSO CNH3 CPD-Y Time Yield Substr. Hydr. 0th. Ratio
Conc.
(~) (M) (,uM) (min) (~
104 7.7 575 78 0 22 0 78
8 7.0 5120 77 8 14 0 84
12 6.9 101200 72 16 9 2 86
12 6.9 lOb) 1242 68 14 15 2 80
_
a) Reaction conditions: SALT/PHADJ: NH3/HOAc, p~ 9.2, 1 --
mM peptide, 37C in Eppendorph mixer.
b) O.S mM peptide.
:
.: :. . :~
21~1 ~71
WOg2/1569j PCT/DK92/000
32
Example 12
Amidation of Z-Thr-Pro-Met in various ammonia/ammonium
mixtures containing DMFa)
CNH3 SALTtPHADJ Time Yield Substr. Hydr. 0th. Ratio
(M) (min) (%) (%) (%) (~) (%)
3/H2S4 70 2373 0 3 88
6.2 NH3/HC1 70 80 9 11 0 88
9-7 NH3/HCH 90 88 o 12 0 88
7.7 NH3/HOAc 39 78 4 18 0 81
7.7 NH3/HOAc 75 76 4 20 0 79
7,7 NH3/HoAc 68 79 3 lS 3 81
7-7 NH3/HAC 40 83 6 11 0 88
4.3 NH3/HOAc 53 68 0 32 0 68
4.3 NH4Cl/LIOH188 4345 9 3 78
4.1b) NH4Cl/NaOH 3281 5 14 0 85
4.1 NH4Cl/NaOH143 7416 10 0 88
3/NH4C1 14070 20 10 0 88
a) Reaction conditions: 1 mM peptide at pH 9.2, 5 ~M
CPD-Y, 2~ DMF, 37~C in Eppendorph mixer.
b) 20 ~M CPD-Y added.
: ,
WO92/15695 21 ~ 1 0 7 ~ PCT/DK92/000~
Example 13
Synthesis of human calcitonin (1-32)-Met-OH by enzymatic
pe~tide synthesis
_
About 2 mg of human calcitonin a carboxylic methyl ester
containing some impurities was dissolved together with
89 mg of L-Methionine and 95 mg of guanidinium chloride
in 850 ,ul H20 and 40 ,ul DMSO and pH was adjusted to 8.8
using 2 M sodium hydroxide in a pH-stat vessel thermo-
stated at 37C. The reaction was performed in this ves-
sel. The reaction was initiated by addition of 15 ~ul of
a 0.3 mM CPD-Y solution and was complete within 30 min.
The mixture was adjusted to pH 2.5 and applied to a re-
verse phase C18 ~PLC column, from which ~he product waseluted using a 0.1~ TFA/agueous acetonitrile gradient to
yield 1.2 mg (60%) upon drying under nitrogen.
Amino acid a~alysis (ratio)
-
Asp + Ala (5.1), Glu (1.9), Ser (0.9), Gly (4.0), His
(0.6), Thr (4.9), Pro (2.1), Tyr (1.1), Val (0.9), Met
(1.9), Cys (1.2), Ile (1.0), Leu (2.3), Phe (3.2).
The chemical breakdown of cysteine and the confirmed
presence of two methionine residues is noted.
, :. - . .: ~ : . :
.
W092/~ 0 7 `~ PCTJDK92/00064
34
Example 14
Amidation of various Z-Ala-Pro-X peptides )
5 Leav- Sol- CPD-Y Time Yield Substr. Hydr.Oth. Ratio
ing vent
(X) (~M) (min) (~
Met 2% DMFb)2.5150 67 31 0 2 97
10 Met 4~ DMSO 5 235 82 17 2 0 98
Gln Water 20 100 24 41 35 0 41
Gln 4% DMSO20 1200 30 48 23 0 S6
~is Water 20 1140 25 26 46 3 33
His 4~ DMSO20 1200 20 60 20 0 51
15 Thr 4% DMSOC) 25 1440 17 76 6 1 71
Tyr 4% DMSO17 1320 45 17 38 0 54
~rp 4~ DMSO50 180 68 17 15 0 82
Arg 4% DMSO17 264 17 83 0 0 100
Lys 4% DMSO17 386 28 72 0 0 100
20 Glye) 2% DMFC)d) 50 1440 0 100 0 0
Gly ) 4~ Dioxane 200 1260 0 100 0 0 0 :
a) Reaction conditions: 1 mM peptide, 7.7 M CNH3
(ammonia/ammonium), PXADJ: HOAc, pH 9.2, 1 M
guanidinium chloride, 37C in Eppendorph mixer.
b) No guanidinium chloride added.
c) No guanidinium chloride added and 4.3 M CNH3,
PHADJ/SALT: NaOH/NH4Cl.
d) 25C.
e) Comparison example.
"'' " ' ''` ' ' ' ' ' . . , ' '.. ' ' .' ' ' ' .'
' ' , . ' " ' , '. . ' , , ' ' ~ ~'
', ' . . ' ' ........ ' ." ' ' '' , ' , " '. .. ., '
'
W092/1569~ 2 ~ PCT/DK92/000
Example 15
Amidation of Z-Ala-Pro-Met at various pH values and
concentrationsa)
pH CPEP- CNH3 Time Yield Substr. Hydr.Oth. Ratio
TIDE
(mM) (M) (min) (%) (%) (%) (%) (%)
10 9 5 0.5 8.81360 55 39 6 0 91
9.2 0.5 7.71290 85 12 2 1 97
9.2 0.2 7.81120 70 27 2 0 97
9.2b) 5,0 7.781 72 22 o 6 92
a) Reaction conditions: SALT/PHADJ: NH3/HOAc, 1 .M
guanidinium chloride, 5 ~M CPD-Y ~ additional 10 ,uM
after 1100 minutes, 37C in Eppendorph mixer.
b) 16 ,uM CPD-Y initially.
, . . . . .............. .
.. . . . ..
, ~ , , ~, .
, . , : .
. .~ ~ "'' ' ' '
2 ~ 7 i
WO92/15695 PCT/DK92/00064
-
36
Example-16
Amidation ~f Z-Ala-Pro-Met-OH in different solvents at
various temperaturesa)
Sol- Temp. CPD-Y Time Yield Substr.Hydr.Oth. Ratio
vent
(C) (~M) (min) (%) (~) (%) (~) (%)
10 4~ Dioxaneb) 3733 1260 62 26 12 0 84
4% DMSO 37 20 1200 68 0 32 0 68
4% DMSO 25 20 1265 57 0 43 57
10~ Glycerol 37 20 1260 22 0 0 78 22
10~ Glycerol 25 20 1260 42 0 0 58 42 -
15 10~ TGME 37 20 1200 13 87 0 0 100
10~ TGME 25 20 1260 27 73 0 0 100
. .
a) Reaction conditions: 4.2 M CNH3, SALT/PHADJ:
NH4Cl/NaO~, pH 9.2, 1 mM peptide.
b) 0.7 mM peptide, peroxide free dioxane.
30`
.
- , . . ., . . . . .. - . -
. .
W092/15695 2 i ~ 7 !1 PCT/DK92/000
Example 17
Amidation of Z-Ala-?ro-Met and Z-Thr-Pro-Met in various
ammonia/ammonium mixturesa)
_
Pep- CNH3 Salt+ pH Time Yield Substr.Hydr.Oth. Ratio
tide PHADJ
-MetOH (M) (min) (~ ) (%) (%) ~%)
~ =
10ZThrPro 7.9 NH3+ 8.2 85 48 0 52 0 48
HOAc
ZAlaPro 4.3 NH3+ 9.2 67 55 4 39 2 57`
15HOAc
ZAlaPro 4.3 NH4C1~ 9.2225 41 45 14 0 74
NaOH
ZAlaPro 4.5 NH3+ 9.2120 17 83 0 0 100
20b) NH4N03
ZAlaPro 7.5 NH3+ 9.292 43 38 19 0 70
b) NH4N03
25ZAlaPro 9.0 NH3~ 9.2 40 8 92 0 0 100
b)c) NH4N03
a) Reaction conditions: 1 mM peptide, 5 ~M CPD-Y, 37C
in Eppendorph thermomixer.
b) 17 ~M CPD-Y, 4~ DMSO
c) 30C
~: , . ' . ~ . , ' ,. : `
:.' ' : ; ' :
: ~ : , . . :
W~ 7 -1 Pcr/DK92tooo64
3~
Example 1 8
Amidationa) for ZThrProMetOH and ZAlaProMetOH catalyzed
by different forms b)c) of carboxypeptidases from yeast
in different ammonia/ammonium mixtures adjusted to pH
9.2 with acetic acid. _ _ _
Peptide CPD Enzyme Time Yield Substr.Hydr.Oth. Ratio
-Met (~M) form (min) (~) (%) (~
1 0 - . . . . . ._
ZThrProd) 2 NE ~ lOO 76 1 19 4 77
ZThrProd) 2 SRb) 65 73 4 20 3 76
ZAlaProe) 3 NEb) 75 99 1 0 ) O ) 100
ZAlaProe) 2 SRb) 80 99 1 0 ) of) 100
--
a) 1 mM peptide, 37C, salt: NH3, PHADJ: HOAc
b) NE = Native Extracted CPD-Y prepared and purified
according to
Johansen, J.T., Breddam, K. and Ottesen, M. (1985)
Carlsberg Res. Commun. 41, p. 1-14
c) SR = Secreted Recombinant CP~-Y expressed and
prepared according to
Nielsen, T.C., Holmberg, S. and Peterson, J.G. (1990)
Appl. Microbiol. Biotechnol. 33, p. 307-312
and purified as under b).
d) 2~ DMF CNH3: 4.3 M
e) 4~ DMSO CNH3: 7.5 M
f) Traces.
- . " , . ~ , , , ,,.,: ,. . -
.. , , . , .. , :, . , .
''`''-' - ~ '' , . ' ' ' .~ .`, ' . ~ '- ... -, .,
WO92t1569~ 2 .t ~ , PCT/DK92/00064
39
Example 19
CPD-Y catalyzed amidation of ZAlaProThr-OH in
ammonia/ammonium mixtures pH-adjusted with acetic acida)
and further- containing various anionic and nonionic
deterqents at 0.5~ (w/v).
Detergent Time Yield Substr.Hydr.Oth. Ratio
(min) (~ ) (%) (%) (%)
. _ . _ . . .
0.5~ N-Lauroylsarcosine 412 31 26 43 0 42
0.5% Octanesulfonic
Acid 3007 80 13 0 34
15 0.5% Brij 35 3004 84 12 0 27
0.5~ Tween 20 30018 33 49 0 27
_
a) Reaction conditions: 8.0 M CNH3, PHADJ: HOAc, pH 9.2, ~-
20 lmM ZAlaProThr, 4% DMSO, 17 ,uM CPD-Y, 37C in
Eppendorph thermomixer.
2 1 ~ 1 0 7 ~ ~
WO92~15695 PCT/DK92/00064
.
Example 20
CPD-Y catalyzed amidation of ZAlaProMet-OH in
ammonia/ammonium mixtures pH-adjusted with acetic acida)
and further containing various zwitterionic or anionic
detergents at 5~ (w/v).
i
DetergentTime Yield Substr.Hydr.Oth. ~atio
(min) (~) (%) (~) (%) (~)
5~ CHAPSO 35 60 40 O 0 100
5~ Pentanesulfonic
Acid 1200 89 1 10 0 89
-~
a) ~eaction conditions: 8.0 M CNH3, PHADJ: HOAc, pH 9.2,
lmM ZAlaProMet, 4% DMSO, 17 ~M CPD-Y, 37C in
Eppendorph thermomixer.
. . . ,, _ _, .. ~ .. . . . ... ... .... . .. . . . . . . . . . .. . . ..
.
- ... , . . ., . . ~ ..
.,: . ' , ; '. ' ' ' -. " ' ' .. ", . ' , . - : . ,'
W092/1569~ 21 ~ :~ 0 7 ` PCT/DK92/00064
41
Example 21
CPD-Y catalyzed amidation of ZAlaProTyr-OH in
ammonia/ammonium mixtures pH-adjusted with acetic acida)
and further containin~ various additives.
-
CADD Additive Time Yield Su~str.Hydr.Oth. ~atio
(M) (min) (~ ) (%) (~) (%)
0.5 Mannitol 249 37 58 S 088
2.0 Urea 144 8 92 0 0100
1.0 Guanidine hydro- 198 8 90 Z 0 80
chloride
--
a) Reaction conditions: 8.0 M CNH3, PHADJ: HOAc, pH 9.2,
lmM ZAlaProTyr, 4% DMSO, 5 ~M C~D-Y, 37~C in
Eppendorph thermomixer.
.- , . I . . :, .
''- ' ~' ",' ' ' ~' ' .
2 ~ 7-~
WO92/1569~ PCT/DKg2/00064
42
Example 22
CPD-Y catalyzed amidation of various peptides of
structure Z~laPro-X-OH in various acetic
S acid/ammonia/ammonium mixturesa) in the pH range 8.5 to
9.0
Leaving pH CNH3 Time Yield Substr. Hydr. 0th. ~atio
(X) (min) (%) (~
Arg 8.8 7.5 360 36 52 12 0 75
Arg 9.0 7.7 360 31 45 24 0 57
Thr 8.5 7.0 60 27 32 41 0 40
Thr 8.8 7.5 205 42 36 23 0 65
Thr9.0 7.7 325 45 20 36 0 56
Trp 8.5 7.0 85 29 6 6S 0 31
Trp 8.8 7.5 155 53 4 42 0 56
Tyr 8.8 7.5 400 46 11 43 0 51
- ~yr 9.0 7.7 350 43 13 44 b 49
----
a) ~eaction conditions: 1 mM ZAlaPro-X, 1 M guanidine
hydrochloride, 4~ DMSO, 17 ~M CPD-Y, 37C in
Eppendorph thermomixer.
' -- -
.- . . - . . . . ~ . .. , : . ... . . .
, , .. : . , . . . : . .
,, : . . , . ;: , ,'. ' ' ,".. ,", , . ,: ' . ' .
. :, . . ...
W092/15695 21~ 7 ~ PCT/DK92/000~
43
Example 23
Amidation of human calcitonin 1-32-Met-OH to form human
calcitonin amide
1.1 mg of cyclic human calcitonin 1-32 elongated by
methionine acid was directly dissolved in a glass test
tube in 20 ,ul of DMSo, 0.47 ml of a 25~ (w/w) ammonia
solution p~ adjusted to 9.2 by glacial acetic acid was
added to give an unclear solution. 47 mg guanidinium
chloride was then dissolved herein. The mixture was
thermostated to 37C in an Eppendorph thermomixer, after
which the reaction was initiated by addition of O.3 mM
CPD-Y to yield a CPD-Y concentration of about 5 ~M.
Following shaking of the closed reaction vessel for
about half an hour at 37C, the solution was shown to
contain 42~ human calcitonin amide by HPLC as well as
21~ of the hydrolysis byproduct calcitonin free acid,
the remainder being unreacted substrate and some
breakdown products.
, . . . . .. .
-- . ~ , .. ,, , ,, , ~ .: , , ' . :
,:. . , , , . . . . . - :
2 1 ~
WO 92/15695 PCl'/DK92/00064 ~,~
44
Example 24
Amidation of human calcitonin 1-32-~hr-0~ to form human
calcitonin amide
S
1.9 mg of cyclic human calcitonin 1-32 elongated by
threonine acid was directly dissolved in a test tube in
40 ~1 of DMS0 and 0.92 ml of a 25~ (wJw) ammonia
solution pH adjusted to 9.2 by glacial acetic acid and
96 mg guanidinium chloride was added to the solution.
The mixture was thermostated to 37C in an Eppendorph
thermomixer, after which the reaction was initiated by
addition of 0.3 mM CPD-Y to yield a CPD-Y concentration
of about 8 ~M. Following shaking of the closed reaction
vessel for forty minutes at 37C, about 1/3 of the
substrate had been converted and the solution was shown
to contain 16% human calcitonin amide by HPLC at several
pH values as well as only traces of the hydrolysis
byproduct calcitonin free acid, the remainder being 67
unreacted substrate and some brea~down products.
,
, : . ' . ., -' ., . .. : . ,: . :
, : , .. : : . ', . .': : . .: :
W092/tS695 2 1~ 1 ~ 7 l PCT/DK92/000~
Example 25
Amidation of human calcitonin 1-32-Tyr-OH to form human
calcitonin amide
1.8 m~ of cyclic human calcitonin 1-32 elongated by
tyrosine acid and further containing a little calcitonin
acid was directly dissolved in a test tube in 40 ~1 of
DMS0 and 0.94 ml of a 25% (w/w) ammonia solution pH
adjusted to 9.2 by glacial acetic acid and containin~ 96
mg guanidinium chloride was added to the solution. The
mixture was thermostated to 37C in an Eppendorph
thermomixer, after which the reaction was initiated by
addition of 0.3 mM CPD-Y to yield a CPD-Y concentration
of about 20 ~M. Following shaking of the closed reaction
vessel for about half an hour at 37C, 37~ of the
substrate had been converted and it wàs shown by HPLC
that 26~ of this had been converted to human calcitonin
amide, i.e. a total yield of about 10%, as well as only
a little more of the hydrolysis byproduct calcitonin
free acid, the remainder being 63% unreacted substrate
and some breakdown products.
. . - . . ~ .,
. . - - , .
- ~
W092/~ 7 ~ PCT/DK92tO00
46
Example 26
CPD-Y catalyzed amidation of ZThrProMet-OH with various
substituted amines of structure H2N-R to give products
of structure ZThrProNH-R )
Amine -R Conc.Time Yield Subst.Hydr.Oth.Rat.
(M)
Ethyl
amine 2CH3 2.0 120 16 31 52 0 24
2-Ethanol
amine -CH2cH2H 3-0 90 59 15 26 0 69
Hydrazine -NH2 2.0 75 80 5 15 0 84
Semi-
20 carbazideb) -NHCONH2 2.0 75 69 24 8 0 90
Benzyl
amineC) -CH2C6H5 0.1 34 54 35 8 3 83
a) ~eaction conditions: 1 mM ZThrProMetOH, 5 ~M CPD-Y,
4~ DMSO, pH 9.2, pH adjusted with acetic acid, 35C -
in Eppendorph Thermomixer.
b) Used as hydrochloride, pH adjusted with NaOH.
c) 100 ,uM CPD-Y, pH adjusted with HCl, reaction in pH-
stat cup. ''
;
3 5 ' 1 . .
'' ' .' ,, . .... , " '' ', '' ' ''' ' ~ ~: ` ", " ',' ' ' , . .', . " ,''
,,, ', ', ' :' ., '' ' ',. '' ' ' ~' '. . '' ' ' ', :, '''" ' ",~ ' '
W092/t5695 21~ v ~ ' PCT/DK92/000~
- 47 -
i
REFERENCES:
I
1. Breddam, K., Widmer, F. & Johansen, J.T. (1980~ t
Carlsberg Res. Commun. 45, 237-247
2. Widmer, F., Breddam, K., and Johansen, J.T., (1981),
Carls~erg Res. Commun., 46, 97-106.
3. Breddam, K., Widmer, F. & Johansen, J.T. (1981)
Carlsberg Res. Commun. 46, 121-128
4. Breddam, K., Widmer, F. & Johansen, J.T. (1981)
Carlsberg Res. Commun. 46, 361-372
5. Breddam, K., Johansen, J.T. & Ottesen, M. (1984)
Carlsberg Res. Commun. 49, 457-462
6. Breddam, K. (1985) Carlsberg Res. Commun. 50, 309-323
15 7. Breddam, K., (1986), Carlsberg ~es. Commun. 51, 83-128
8. Breddam, ~. (1988) Carlsberg Res. Commun. 53, 309-320
9. ~reddam, K. & Ottesen, M. (1984) Carls~erg Res.
Commun. 49, 473-481
lO. Johansen, J.T., Breddam, K. & Ottesen, M. (1976)
Carlsberg Res. Commun. 41, 1-14
11. Nielsen, T.L., Holmberg, S. & Petersen, J.G.L. Appl.
Microbiol. Biotechnol (1990), 33, 307-312
12. Kubota et al. Carboxypeptidase CN (1973), J. Biochem.
74, no. 4, 757-770
25 13. Hopp & Woods, Proc. Natl. Acad. Sci. USA, 78,p. 3824-
3828 (1981)
14. Dmochouska, A., et al., (~987), Cell, 50, 573-584
15. Cooper, A & Bussey H., (1989), Molecular and Cellular
Biology, 9, 27~6-2714
30 16. Bech, L. M., & Breddam, K., (1988), Carlsberg Res.
Commun., 53, 381-393
, - : . ., . ~ :
'; '' , ', , ' ' . ~ ., :.. , ~. '',, , '' ''