Note: Descriptions are shown in the official language in which they were submitted.
2~73~
iv-~tLvcs~ h-cir ~
The present invention relates to N-sulfonyl-2-oxoindole derivatives, their
S pr~paration and the pharmaceu~ical cosnposi~ions in which they are presen~.
International patent applica~ion WO 91/01 3C16 descTibes Z-oxoindole
derivatives which are useful for the treatmen~ of senile dcmentia. These
compounds have the fonm~la
R"~
1~ O
COR"4
in which
lR"1 is hyd~ogen, a halogen, an alkyl or an alkoxy;
- R"2 is hydrogen or a lower alkyl;
15 - R"3 is an alkyl, a cycloalkylmethyl, a benzodioxanylmethyl, or an optionally
substituted benzyl; and
- R"4 is a 1~propylbutyl, a pyridyl or an optionally substituted phenyl.
Several patent applications have recently described families of compounds
with a non-peptide structure which are active on the vasopressin and/or ocytocin20 receptors. European applications EP 382185 and EP 444 945, international
application WO 91/05 549 and, more particularly, Japanese patent application JP
03/127732 can be cited in this respect. The latter describes indole-3-plopionic
acid derivatives of the fonnula:
~ ~ R " ' l~ 4
:: 25 R " '
~ ~ .
~, -
21~73~
in which
- R"'1 is hydrogen, an aikyl, an alk~nyl, a phcnylalkyi, a tetrahyd~ofuryl, a
alkoxycarbonyl, an alkaxycarbonylalkyl, a carboxyalkyl or an alkanoyl;
- R"'2 is hydrogcn, a hydroxyl, an alkoxy, an allsyl, a phenylalkyl, a
S phenylalkoxy or a halogcn;
- R"'3 is a hydrogen, an alkoxy, a ~ree or substituted amirlo group or an arnino acid residue;
- R"'4 is hydrogen, an alkyl or a phenylalkyl; and
- R"'s is a benzoyl, a phenyl, an alkyl, a phenylalkenylcarbonyl, a
thienylcarbonyl, ~ phenylsulfonyl, a pyridylcarbonyl or an imidazolylcar~onyl,
it being possible ~or the phenyl and alkyl ~oups of the substituent R"'s to be
substituted.
These compounds are vasopressin antagonists.
Patent US 4,8()3,217 clairns hapalindolinones obtained by fermentatinn,
which are vasoprexsin antagonists. These compounds have the following fonnula:
CH3 R
CH2 ~ CH ~
~C,J~ J~; o CH3
in which R is H or Cl.
The N-sulfonyl-2-oxoindole derivatives accoIding to the present inve~ion
have an ~ffinity for the vasopressin and ocytocin receptos~.
Vasopressin is a horrnone known for its antidiuretic effe~t and its eiYect in
regulating the arterial pressure. It stimulates several types of receptols, namely
V~ a, Vlb~ and V2, and thus exerts cardiovascular, hepatic, antidiuretic and
platelet-aggregating effects and effccts on the central and peripheral nervous
systems. Va~sopressin receptor antagonists can affect the regulation of the central
and peripheral circulations, especially the coronary, renal and gastric circulatiuns,
as well æ the regulation of hydration and the release of adrenocorticotrophic
hormone (ACI H). Non-peptide agonists of vasopressin can advantageously
2~ar~3~8
r~placc vasopressill or its analt)gs in thc trcatnlcnt o~ ciiabctcs insipidus; thcy can
also bc usccl in th~ trcatrncnt of ~nur~,sia and in ~hc rcgulation of hcmostasis;
treatm~nt of hcmophilia al1~1 of Von Willcbrand's synclrom, antidotc to platelet-
aggrcgating agents; Dlllg ll~v~stigation, 1990, 2 (S~lppl. S), 1-47. Thc vasopr~ssin
S reccptors, like thc ocytocin r~cepto~s, arc also found on thc smooth muscl~ of the
utems. Ocytocin has a pepticic structure similar to that of vasopressin. Its receptors
are also found on myoepith~lial cells of thc mammary gland and in the central
nervous system (Presse médicale, 1987, l6 (l()), 481-4gS, J. Lab. Clin. Med.,
1989, 114 (6), 617-632, and Phannacol. Rev., 1991, 43 (1), 73-108). This
10 hormone is involved in palturition, lactation and sexual behaviour.
Thus the compounds according to the invention are useful especially in the
treatmeîlt of complaints of the ccntral and peripheral nervous systerns, the
cardiovascular system~ ~he renal sphcre and the gastric sphere and in diso~ders of
sexual behavior, ;n humans and animals.
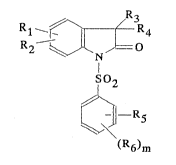
The present invention relates to compounds of the formula
R~
R ~ / JR
R2 ~,1. N ,L--O
SO
l 2 (I)
- ¦--R5
(R6)m
in which
- Rl and R~ are each independently a hydrogen, a hydroxy, a Cl-C4~cl1-
20 halogenoalkoxy, a halogen, a C1-C4-alkyl, a trifluoromethyl, a C1-C7-
alkoxy, a C1-C4-polyhalogenoalkoxy, a C2-C4-~hydroxyalkoxy, an ~-
methoxyalkoxy in which the alkyl is C2-C4, a C2-C4-~-aminoalkoxy which
is free or substituted by one or two C1-C4-alkyls; a C3-C7-cycloalkyloxy; a
cycloalkylmethoxy iD which the cycloalkyl is C3-C7; a phenoxy; a benzyloxy;
25 a C1-C4-alkylthio; a phens~lthio; a nitro; an amino which is free or substituted
by one or two C1-C4-alkyls; a cyano; a C1-C4-acyl; a C1-C4-acyloxy; a
.
2~ ~PI34~
Cl~C~_alk~lsul~onallli(lo; a phcn~lsulfonamido; a Cl-C4-~lkylamido; a Cl-
C4-.1lk~ ycarbollylanlino; a urcido which is unsubstitutcd or substituted by a
phcnyl or by OllC or two Cl-C4 alkyls;
- R3 an(l R4 ar~ cach indepcndcntly a (:1-C6-alkyl, a C3-C~-cycloalkyl, a
5 phcnyl, a bcnzyl, a cycloalkylmcthyl in which the cycloalkyl is C3-C7;
or
- R3 and R4 ~ogether fonn ~ group ~(CH~)pX(CH~)q;
or
- R3 and R4 together with thP carbon atom to which they are bonded, fonrl an
10 optionally fused, saturated or unsaturated C3-C1o hydrocarbon ring which is
unsubstituted Ol substituted by one or mnre Cl-C7-alkyl groups Ol by a C3-
Cs-spirocycloalkyl;
or else
- R1 and R4 each have one of the above meanings and R2 is located in the 4-
1~ position of the indole and fonns a group (CH ~)3 with R3;
- Rs and R6 are each independently a hydrogen, a halogen, a C1-C7-alkyl, a
trifluoromethyl, a cyano, a nitro, an amino which is free or substituted by one or
two C1-C7-alkyls; a hydroxyamino; a hydroxy; a carboxy; a group OR7; a
group SR7; a C1-C7-acyl; a Cl-C7-alkoxycarbonyl; a phenoxycarbonyl; a
20 benzyloxycarbonyl; a carbamoyl substituted by groups R'6 and R'16; a
thiocarbamoyl which is free or substituted by one or two Cl-C7-alkyls; a
sulfamoyl; an alkylsulfamoyl or a dialkylsulfamoyl in which the alkyl is C1-
C7;-a group SO2R'7; an alkylsulfonamido in which the alkyl is C1-C7; a
group COR'7; a group NRgRg; a group CO-NH-CH(Rl~)-COR12; if
25 appropriate, the phenyl group forming part of the substitutent Rs and/or R6 can
be unsubstituted or monosubstituted or polysubstituted by a C1-C7-alkyl, a
trifluoromethyl, a methoxy, a halogen, a sulfamoyl, an alkylsulfamoyl in which
the alkyl is Cl-C4, a carboxy, an alkoxycarbonyl in which the alkyl is C1-C7,
a C1-C7-acyloxy or an imidazolyl;
30 - R'6 and R"6 are each independently hydrogen, a C1-C7 alkyl which is
unsubstituted or substituted by R"'6, a phenyl, a pyridyl, a methylpyridyl, a
piperidin-4-yl, a methylpiperidin-4-yl; or R'6 and R"6 form, with the nitrogen
atom to which they are bonded, a heterocycle selected from piperazine and
piperidine;
21(~73~
~ R"'6 is ~1 hyclro,Yy, a cy~no, a carl~o.Yy which is free or esterifi~d by a C1-C7-
alks~l or by a bcnzyl; a phenyl; a pyridyl; a methylpyridyl; an amino whieh is
free or substitutcd by onc ~r two C~ -C7-~lkyls;
- R7 is a Cl-C7-alkyl, a phenyl, a b~nzyl, a C3-C7-eyeloalkyl, a C2-C4-
S alkenyl, a C1-C7-~1)-halogenoalkyl, a C1-C7-polyhalogenoalkyl, a Cl-C7~
acyl, a C1-C7-~-carboxyalkyl which is free or est(~rified by a Cl-C4-alkyl or
by a benzyl, a C~-C7 ~-aminoalkyl in which the amino group is ~ree,
substituted by one or two C1-C4-alkyls or in the folm of an ammonium ion;
- R'7 is a piperazin-1-yl group which is unsubstituted or subslituted in the 4-
10 position by a group R"7, a piperidino group which is unsubstituted orsubstituted in the 4-position by a group R"'7, an a7etidin-1-yl group which is
unsubstituted or substituted in the 3-position by a group R"'7, a pyridyl group
which is unsubstituted or substituted by a methyl;
- R"7 is a C1-C4-alkyl, a phenyl, a benzyl, a C1-C4-acyl;
15 - R"'7 is R"~ or an amino which is free or carries a protecting group;
- R8 and R~ are each independently a hydrogen, a C1-C7-alkyl, a phenyl or a
benzyl; Rg can also be a C1-C7-acyl, a Cl-C7-thioalkyl, a
cycloalkylcarbonyl in which the cycloalkyl is C3-C7, a cysloalkylthiocarbonyl
in which the cycloalkyl is C3-C7, a Cl-C4-~-aminoacyl, a (:1-C4-~-
20 hydroxyacyl, a C1-C4-~-benzyloxyacyl, a phenoxycarbonyl, a
thienocarbonyl, a pyridylcarbonyl, a methylpyridylcarbonyl, a C1-C4-
alkoxycarbonyl, a benzoyl, a group C-CH(~10)-NR11R 11~ a group
CH(R10)~23~11, a group (CH~)tCOR1~ a group CO(CH2)tCOR12, a
carbarnoyl which is unsubstituted or substituted by a phenyl or one or two Cl-
2S C4-alkyls,
m is 1 or, when R6 is a halogen, a C1-C7-alkyl or a C1-C7-alkoxy, m can
also be 2, 3 or 4 or else (R6)m can be m substituents having different meanings
selected from halogen, C1-C7-alkyl or C1-C7-alkoxy;
p and 9 are each integers, it being possible for their sum to vary from 3 to 6;
30 - t is an integer which can vary from 1 to 5;
- X is oxygen, sulfur or a group NR13;
:: - R1o is hydrogen, a C1-C4-alkyl or a benzyl;
- R11 and R'11 are each independently hydrogen or a C1-C4-alkyl;
.
:
. : .
210 ~ 3 l~ g
~ R12 is a hydroxy, a Cl~Cd,~-alkoxy or an amino which is unsubs~ituted or
substitutcd by one or lwo C1-C4 alkyls;
~ ~13 is hyd~ogen, a C1-C~-alkyl, a phenyl, a benzyl, a C1-C4-acyl, a C1-C4-
alkoxycarbonyl or a c~rbamoyl which is unsubstituted or substituted by one or
Stwo Cl-C4 alkyls;
as well as ~heir possible salts.
If a compound ascording to the invellt~on has one or more asymmetAc
carbons, the invention ineludes all the optical isomers of this compound.
The salts of the compounds o~ formula (I) according to the present invention
10include ths)se with mineral or organic acids which permit a suitable SeparatiOD or
~ystalliza~ion of the compounds of ~ormula (I), such as picric acid, oxalic acid or
an optically active acid, for example a mandelic acid s)s a camphoslllfonic acid, and
mineral or organic acids which form physiologically acceptable salts sllch as the
hydrochloride~ hydrobromide, sulfate, hydrogensul~ate, di-hydrogenphosphate,
15maleate, ~umarate or naphthalene-2-sulÇonate.
The salts of the compounds of fo~nula (I) also include the salts with or~9anic
or mineral bases, for e~cample the salts of alkali rnetal or -alkaline ear~h metals,
such as the sodium, potassium and calcium salts, sodium and potassium salts being
preferred, or with an amine such as trometamol, or else the salts of arginine, Iysine,
20or any physiologically aeceptable amine.
According to the present invention, halogen is understood as meaning an
atom selected from fluorine, chlorine, bromins and iodine, preferably fluorine or
chlorine. Amino-protecting group is understood as meaning a ~oup such as, for
example, tert-butoxycarbonyl, ben~yloxycarbonyl or benzyl.
2SAccording to the present invention, optionally fused condensed9 saturated OI
unsatu~ated C3-C1o hydrocarbon ring is understood as meaning various
hydrocarbon nngs with a monocyclic9 dicyclic or tricyclic structure, for example a
cyclobutane, a cyclopentane, a cyclohexane, a cycloheptane, a cyclooctane9 an
indane, a hexahydroindane, an adamantane, a norbornane, a norbornene, a
30dihydrophenalene, a tricyclo[S.2.1.02,6]decane or a tricyclo[5.2.1.02~6]dec-8-ene.
The cornpounds of formula (I) in which Rl is in the 5-position of the indole
and R2 is hydrogen are preferred compounds.
The compounds of formula ~I) in which Rl is a chlorine atom or an ethoxy
group in the 5-position of the indole and R2 is hydrogen are preferIed compounds.
2~73~8
Thc compounds of ~ormul~ (I) in which R3 an~l R4 togcthcr with thc carbon
to which they are bonded, form a C3-Clo hy~lrocasbon ~ng are prefcrred
compounds; particularly pr~ferrcd compounds ~rc thosc in which R3 and R4,
together with the carbon to which they are bonded, ~orm a cyeloheptane, an
S adamantane, a tricyclo[s.2.l.o2~6]dec-g-e~le s~r a cyclohexane which is
unsubstihlted or substituted by a C3~ s-spirocycloalkyl or by one or two Cl-
C7-alkyl ~oups.
l~e compounds of fonnula ~I~ in which lR3 and R4, together with the ca~bon
ts~ wh;ch they are bondçd, form a pipencline-4 or N-methylpiperidille-4 ring arealso preferred.
The compounds of fonnula (~) in which Rs and R6 are each a methoxy are
prefeIred compounds. Li!cewisi, the compounds in which Rs in the 2-position is amethoxy and R6 in the 4-position is a C1-C7-af~ylamino, a C1-C4-dialkyluseido
or an alkoxycar~onylalkylcarbamoyl in which the alkyl groups are C1-C7 are
preferred compound~.
rhe following abbreYiations are used in the description and in the examples.
DCM: dichloromethane
Ether: ethyl ether
Iso ether: isopropyl ether
Boc: tert-butoxycarbonyl
Me, MeO: methyl, mcthoxy
Et: ethyl
Pr, iPr, nPr: propyl, isopropyl, n-propyl
Bu, iBu? tBu: butyl, isobutyl, ter~-butyl
2S Ph: phenyl
Bz: benzyl
Ac: acetyl
AcOEt: ethyl acetate
AcOH: acetic acid
MeOH: methanol
Etl:)H: ethanol
~; DMF: dimethylformamide
THF: tetrahydrofuran
DMSO: dimethyl sulfoxide
::
:
2~73~
DIPEA: diisopropylcthylaminc
IEA: tricthylamine
TF~: tAfluoroacetic acid
TMEDA: tetr~nethylethylcncdi~mine
s M.p.: melting point
Salin~ solution: satu~ated aqueous sodium chloride solution
TLC: thin layer chromatography
HPLC: high pressure liquid cblomatography
Aqueous hydrochloric acid: dilute hydrochloric acid~ about 1 N
RT: room temperature
The present invention fu~her relates to the method of preparing the
compounds accolding to the invention, characterized in that:
a benzenesulfonyl halide of the formula;
::
Hal~02~R~5
VI~ m
~:: in whlch R's and RVI are respectively either Rs and R6 as defined above for ~1)9 or
plecursor groups of Rs and R6 is reacted with a 2-oxoindole disu~stituted in the: ~ :: : 3 position, of the fonnula:
20:
R3
R' 7 ~ D`N -- (II)
~: : H
m~which R'l and~R'2 aro~respectivoly either R1 and R2 as defined above for (1~, or
: precursor groups of 12 1 and R2, and R3 and R4 are as defined above for (1),
25~ - either, if R'1=R1, R'2=R2, R's-Rs and RVl=R6~ the r~sulting compound of
formula (~ is isolated; ~
orj if any one of the groups R'1, R'2~ E~'s and RVI is respectively a precursor
group of R1, R2,:: Rs and/or R6, the compound obtained is subjected to a
subsequent treatment in order to prepare the compound of fonnula (1~ by
;~:
.
. . .
21~73'18
conversion of any one of thc grouys R'l, R'2, R's and RVl to R1, R2, Rs and
R6, rcspectivcly.
The rcactioll is calr~ed out in an anhydrous solvent such as DMF or THF, in
the presencc o~ a metal hydride such as~ for example, sodium hydnde, or in the
presence of an alcoholate such as potassium tert-butylate.
The 2-oxoindoles (ll~ can be prepared using different procedures. Some of
the~ compounds are novel and form part of the invention.
Compounds (II) in which E~ll and/or R'2 are a halogen and R3 and R4,
together with the carbon So which they are bonded, fonn a spirocyclobutane, a
spirocyclohexane or a spi~ocycloheptane are known, for example in D. W.
Robertson et al. J. Med. Chem., 1987, 30 ~5), 824 829. Also, 5-chloro-3-
spirocyclopentaneindol-2-one is described in US Patent 3,94~,451.
To prepare the compounds ~II) in the case where ~3 and R4 are together or
separately a hydrocarbon group, it is possible to use the Brur~er reaction desc~ibed
by R.F. Moose and S.G.P. Plant in J. Chem. Soc., 1951, 3475-3478; which leads tothe preparation of compounds (Il) in which CR3R4 is a cyclopentane or a
cyclohexane.
This reaction is carried out by cyclizing a phenylhydrazide derivative of the
fonnula:
~ 2~LNH NH 1 1 ~H~
in which R'l and R'2 are as defined above for (II), and R3 and R4 have the
meanings indicated above for (1), for example by heating in ~he presence of
calcium oxide and quinoline.
~ccording to the same authors, the phenylhydrazide derivative (IV) is
obtained by reacting a hydrazine derivative of the fonnula:
:~'
N H~H2
: R' (V)
210 ~ 3 4 8
in which R'1 and IR'2 havc thc mcanings ;ndicated ab~vc for ~11), wi~h an acid
halide of the fonnula:
H~ CR3R4
l l (VI)
o
in whieh R3 and R4 have the meanings indicated above fos ~1).
According to a particular embodiment, if R3 and R4 together with the
carbon to which they are bonded, fonn a fused polyGyClic hydsocar~, for
example norbornane or norbornene, the reaction is carried out by the method
described by J. Wolff et al., Tetrahedron, 1986, ~ (15), 42~7-4272: first of all, a
lithium salt of the compound (IV3 is prep~ed by reaction with a lithium rea~rlt
such as n-butyllithium, in an inert solvent such as THF, at low temperature, and~hen the cyclization is effeeted by heating jD a solvent such as naphthalene or
prehnitene (1,2,3,4-tetramethylbenzene).
The compounds (Il) in which R1= R2= H and CR3E~4 is adamantane are
s describet in I. Fleming et al., J. Chem. Soc., Perkin Trans I, 1991, ~, 617-626.
Thus, the ~mpounds (I~ in which R3 and R4, together with the carbon atom to
. ~ ~ whieh they are bonded, form an adamantane and R1 and R2 aIe other than
hyd~ogen, are novel and form part of the invention. l'h~y can be prepared by themethod described above.
The hydrazine derivatives (V) are lcnows~ or are prepared by known rnethods.
The same applies to the acid halides (Vl).
A 2 ~oxoindole disubstituted in the 3-position (II) can also be prepared from
a 2-oxoindole of the fonnula:
R2~ ~ (V
: ~ : 25 : H
:~ ~
in which R'l and R'2 are as defined above for (II), by using variou~ methods.
;: : For example, the method described by A.S. Kende and J.C. Hodges in Synth.
Commun., 1982, 12 (1), 1-10, involves the addition of an alkyla¢ing agent in an
appropriate solvent. Thus, to prepare a compound (Il) in which R3 - , the
~ ~ reaction is carried out in THF at -75- C, in the presence of TMEDA, by addition of
::
2 1 ~
an alkyllithium such as butyllithium, followcd by rcaction with a halide of the
fomlula R3Hal; if R3 and R4 are diffcrent, the alkylating reaction can be
performed in two steps with 2 different alkyl halides of the formulae R3Hal and
R4Hal. To p~epare a compound (Il) in which R3 and R4 together form a group of
s th~ fomlula -(CH2)n-, in which n varies from 2 to 7, the reactant uscd is a
compound of fonnula Z(CH2)rlZ, in which Z is an electroYI-accepting group such
as a halogen, preferably bromine or iodine, a tosyloxy group or a mesyloxy group.
lhe compounds of fonnula (II) in which R3 and R4 are each independerltly
an alkyl or a phenyl are known. ~or example, paten~ DB 3 300 522 des~ibes 5-
0 alkoxy-3,3,-dimethylindol-2-ones.
Thc compounds of ~ormula (II) in which R3 and R4, together with the carbon
to which they are bonded, form a C4-C8 hydroc~rbon ring substituted by one
more C1-C7-alkyl groups or by a C3-Cs-spirocycloalkyl are prepared in the
samc manner. ~ese compounds are novel and fonn part of the invention.
If R3 and R4 together fonn a ~(CH2)pX(CH~q_ ~oup, in which p, q and X
are as defined above for (I), a 2-oxoindole disubstituted in the 3-position, of
fonnula (II~, can be prepared ~om a 2-oxoindole unsubs~ituted in the 3-position
(VI~ by reaction with a compound of the fonnula:
Z~CH2)p-X~CH ~)q~Z (VIIr)
in which Z is as defined above and X, p and q are as de~med above for (1). The
~eaction is carried out irl the presence of an alcoholate, for example potassium tert-
butylate, in an anhydrous solvent such as, for example, THF.
If X is a nitrogen atom substituted by a C1-C4-acy1, a C1-C4-
alkoxycarbonyl or a C1-C4-alkylcarbamoyl, the substitution on X can be effected
either on the 2-oxoindole derivative (Il) or on the ~mal compound (I) starting from
a compound in which the nitrogen atom (X = NH) is not substituted.
The compounds (I) in which X = NH a~e preferred compounds according to
theinvention.
Thus, if X is a nitrogen atom substituted by a C1-C4-alkoxycarbonyl, the
~Irst step is to prepare a compound (Il) or (I) in which X is NEI, which is thenreacted with the appropriate chloroformate to give the desired compound (II) or (~).
In the same way, a Cl-C4-alkyl isocyanate is reacted with a compound (II) or (I)
.
2~073~
in which X = NH to g~ve a 2-oxoindole dcrivativc (II) o~ a compound (I) in whichX is a nitrogen atom substituted by an alkylcarbamoyl. An acid chloride s)r an
anhydride is reacted with a compound (Il) or (1~ in which X - N~ in order to
prepare a compound of fonnula (I) in which X is a nitrogell atom substituted by an
5 acyl,
The compounds (Il) in which R3 and R4 together with the ~bon to which
they are bonded, ~onn a pyrrolidine, N-alkylpyrrolidine, pipendine or N-
alky1piperidine ring are described by M. J. Komet in J. Med. C}lem., 1976, ~, (7),
~92-899.
In particular, the horsfiline of the formula:
~ Me
Me O ~ ~
is an alkaloid described in A. Jossang et al., J. Org. Chem., 1991, ~6 (23)7 6527-
~5 6530.
T~e compounds (Il) in which R3 and R4, together with the carbon to which
they are bonded, fonn a group ~(CH2)pX(CH2)q~ in which p and q are integers
whose sum can vaIy from 3 to 6 and X is oxy~n, sulfur or a ~oup NR13, R13
being a Cl-C4-acyl, a benzyl, a Cl-C4-alkoxycarbonyl or a carbamoyl which is
20 unsubstituted or substituted by one or two C1-C4 alkyls, are novel and fo~n part
of the invention.
To prepare a compound of fonnula (I~) in which R3 and R4, together with
the carbon to which they are bonded, form a tr~cyclo[S.2.1.O2~63decane or a
tncyclo[5.2.1.O2~6]dee-8-ene, a compound of formula (VII') or a compound
25 ~" respectively, of the formulae
.
Z H2~ Z~H
(VII)' (VII)"
210734~
in which Z is as ~cfin~d ~bove, is rcac~cd with a compound of formula (VII).
Cs)mpounds (Vll)' and (Vll)" substitutcd by onç or more Cl-C4-allcyl groups are
used to prcpare compounds (Il) in which said carbocycles are substituted.
To prepare a compound (Il) in which R3 and ~, together with the carbon to
s which they are ~onded, forrn a~ indane or a hexahydroindane, a compound ~VII~'
or a compound (VIII)" respectively~ of the formulac
~H2~¦ Z~H2
Z~H~ Z~H
(~III)' (VIIl)~
o in which Z is defined as indicated above for (VII~, is reacted with a compound
~VII). Compounds (VIII)' and ~YIII)" substituted by one or mo~e Cl-C~-alkyl
groups are used to prepare compounds (Ir) in which the indane or lhe
hexahydroindane are substituted.
The compounds (Il) in which R3 and R~ together with the carbon to which
they are bonded; folln a tricyclo[S.2.1.02~6]decane, a tricyclo~S.2.1.02~S~dec-8-
~; ~ ene, an indane or a he~ahydroindane which are unsubstituted or substituted by one
or more C1-C4-alkyls, are novel and ~orm part of the invention.
If R3 and R4 each are a phenyl, the method described iD E~elv. Chim. Acta,
1946, ~2. 415-432, can be used to prepare a compound (II).
The 2-oxoindole derivatives (VII) are known or are prepared by known
methods. An example which may be cited is J. V. RajanBabu in J. Org. Chem.,
1986, ~, 1704-1712.
The compounds of formula (Il~ which carry certain substituents R'1 and R'2
on their benzene rnoiety are used as precursors for the preparation of compounds of
2s; Ionnula ~ which car~ other substituents R'1 and R'2. For example, the
compounds (I~ in which R'1 and/or R'2 = H can be nitrated with the conventional
reagents; they car~ also be acylated by reaction with an acid chlolide of formula
RCOCI, in which R is a C1-C4-alkyl, in the presence of a Lewis acid such as
alumimum chloride, in order to prepare a compound (II) in which R'1 and/or R'2 =30 COR. A compound (Il~ in which R'1 is an amino group is prepared by catalytic
, .
~:,
7 3 ~ ~
hydrogcnation of a compound (Il) in which R'l is a nitro glOUp and R12 is
hydrogcn.
The compounds of thc formula
~N l~
in which
- R1 and R2 are each independen~ly a hydrogen, a hydr~xy, a C1--C4-~-
halogenoalkoxy, a halogen, a Cl-C4-alkyl, a trifluoromethyl, a C1-C7~
alkoxy, a Cl-C4-polyhalogenoalkoxy, a C2-C4-~-hydroxyalko~cy, an ~-
metho~yalkoxy in whif h the alkyl is C2-C4, a C2-C4-~-aminoalk~y whi~h
is ~e or substituted by one or two C1-C4-alkyls, a C3-C7-cycloalkoxy, a
cycloalkylmethoxy in which the cycloalkyl is c~q, a phenoxy, a benzyloxy,
a Cl-C4-alkylth;o, a phenylthio, a nitro, an amino which is free o~ substituted
by one or two Cl-C4 alkyls, a cyano~ a C1-C4-acyl, a C1~4-acylo~y, a
C1-C4-alkylsulfonamido, a phenylsulfonamido, a Cl-C4-alkyla;nido, a Cl-
~: C4-alkoxywbonylarnino or a ureido which is unsubstituted or substitlated by a
phenyl or by one Ol two C1-C4-alkyls; and
R3 and R4, together with the carbon to which they are bonded, form
~: 20 . an adamantane,
an indane or a hexahydroindane which are unsubstituted or substituted by
one or more C1-C7-alkyl groups,
: . a tricyc~o[5.2.1.02~6]decane or a tlicyclo[5.2.1.02~6~dec-8-ene which are
unsubstituted or substituted by oDe or more C1-C7-alkyl groups, or
: 25 . a C4-C~ hydrocarbon ring substituted by one or more C~ alkyl groups
or by a C3-Cs-spirocycloalkyl; or else
R3 and R4 together form a group ~(CH2)p~X(CH2)q~ in which p and q are
integers whose sum can vary ~om 3 to 6 and X is oxygen, sulfur or a ~oup
NR139 R13 being a phenyl, a benzyl, a C1-C4-acyl, a C1-C4-alkoxycarbollyl
~or a carbamoyl which is unsubstituted or substituted by one or two C1-C4-
alkyls,
with the limitation that if CR3R4 is adamantane, R1 and R2 are other than
hydrogen,
:, .
,
,~ '
12~73~8
a~e 3~0v~:1 and ~onr~ part of the invcntion.
The compounds of thc fonnula
N (II)"
H
s
in which
- R1 is a hydroxy, a Cl-C4~ halogeDoalkoxy, a halogerl, a C1-C4-a~yl, a
trifluorQmethyl, a C1-C7-alko~y, a C1-C~-polyhalagenoalkoxy, a C~-C4-CI~
-hydr~xyalkoxy, an ~o-methoxyallcoxy in which ~he alkyl is C2-( 4, a C2-C4-
~3-amino~lkoxy which is ~ee or substituted by onc or two Cl-~4-alkyls, a
C3 q-cycloalkoxy, a cycloalkylmethoxy in which ~he cycloalkyl is c3-q, a
phenoxy, a ~nzylo~y, a C1~C4-alkylthio, a phenyl~hio, a nit~, an a3nino
which is ~e or substituted ~y one or two Cl-~4-alkyls, a cyano, a C1-C4-
acyl, a 3:1-C4-acyloxy, a Cl-C4-alkylsulfonamido, a phellylsulfollamido, a
C~ -alkylamido, a C1-C4-alkoxycarbonylamino or a ureido which is
unsubstituted or substituted by a phenyl or by vne or two C1-C~-alkyls;
- R3 and R4 tog~ther ~orm a group -(CH2)p X(CH2)q~; or
- R3 and R4, together with the carbon to which they are bonded, foIm an
optionally fused, saturated or unsaturated C3-C1o hydrocarbon nng which is
~: 20 unsubstituted or substitutcd by one or morc S~ C4-alkyl groups or by a C3-
~: Cs-spirocycloalkyl;
- p and q are each an integer, it being possible for theîr sum to vary ~om 3 to 6;
X is oxygen, sul~r Ol a group NR13; and
- R13 is hydrogen, a C1-C4-alkyl, a phenyl, a bcnzyl, a C1-C4-acyl, a C1-C4-
25 allcoxycarbonyl or a carbamoyl which is unsubstituted or substituted by one or 2Cl-C4-alkyls,
with the limitation that
- if Rl is methoxy, CR3R4 is other than a pylTolidine-3 which is unsubstituted or
N-substituted by a C1-C4-alkyl, and if R1 is a halogen, CR3R4 is other than a
30 pentane,
~: :
are novel and form part of the invention.
2a,3,4,5-Tetrahydrobenz[c,d]indol 2(1H)-one of the fonnula
~ ' .
2107~
16
is commercially available; its derivatives are known OI are prepared by k~ow~
methods.
S The ber~enesulfonyl halides (II~) are known and are prep~ed by known
me~hods. Thus, ~r example, 4-dimethylaminobenzene~ulfonyl chloride is
prepared according to C.N. Sukenik et al., J. Amer. Chem. Soc., 1977, 99~ 851-
858. More generally, the benzenesulfonyl halides (1l1) in which the substituent Rs
is a din~ethylamino group are knowsl or are prepared by known methods; p-
berlzyloxybenzenesulfonyl chloride is prcpared according tv ~uropean patent
application EP 229 566.
The alkoxybenze~esulfonyl chloride is prepared ~om the ~odium
;: aLlcoxybcnzenesulfonate, which is itself prepared by reacting an alkyl halide with
sodium hydroxybenzenesulfonate.
2~4-Dimetho~ybcnzenesulfonyl chloride is prepared according to J. Am.
Chem. Soc., 1952, 74, 2û08.
The halogenoalkoxybenzenesulfonyl chlorides can be prepared according to
paterlt US 2 540 057.
The benzenesulfonyl halides of the formula
S02CI
~Alk
YRV
:": ~: : : ::
in which
Alk is a C~ alkyl;
25: - Y is O or S; and
: - ~
:. .
21~7~ 1~
l7
~ RV is a C1-5~7-alkYl, a C3-C7-cycloalkyl, a (:2-C4-alkcnyl, a C1~C7-~-halogcnoalkyL a C1-C7-polyhalogcnoalkyl, a bcn~yl, a Cl C7-acyl or a Cl-
C7-~-carboxyalkyl esterified by a Cl-C4-alkyl or by a bcnzyl,
are novel and form part of the invention.
S lllese compounds are preparf d according to O. Hofmann et al. in Liebigs
Ann. Chem., 1982, 287-297. Benzene compounds carrying the subseituents YRV
and OAlk iD the 1- and 3-positions are reacted with trimethylsilyl chlorosulfonaSe
in a solvent such as OCM, at RT. ll~e method of R. Passerini et al. in Gazz. Chim.
Ital., 1960, 90, 1277-89, is then applied and this is followed by neutralization, for
example with allcali metal carbonate, and then by reaction with a hali~e such asPOCl3 to give the desired benzenesulfonyl halide.
The benzenesulfonyl halides ~ in which the substituent R's is an
alko~ycarbonyl, a phenoxycarbonyl, a benzyloxycarbonyl, an alkylthio, a
phenylthio~ a benzylthio or a group SR7, R7 being as defined for ~I), are prepared
accordiYlg to Col. ~zechoslov. Chem. Commun., 1984, 49, 1184, from an aniline
derivative substituted by the same grouping R'5, said aniline derivative itself being
obtained ~om the colTesponding nitrated derivative.
The nitroberlzoic acid derivatives are known; the corresponding alkyl and
phenyl estPrs are obtained by subjecting this acid to an appropnate esterification
reactioD.
The benzenedisulfonyl dihalides (III, R's = S02Hal) are known or are
prepared by known methods. For example, 2,4-dimethoxybenzene-1,5-disulfonyl
dichloride is descnbed in R.J.W. Cremlyn, J. Chem. Soc. C, 1969,1344.
The halogenoalk~xybenzenesulfonyl chlorides (III, R's = o~-
halogenoalkoxy) are used to prepare compounds according to the invention in
which the substituent R5 is an ~-aminoalkoxy which is unsubstituted or
substituted by one or two alkyls, according to the following equation:
-O-Alk'-Hal + NHR~Rg --> -OAlk'-NRgRg
3~
in which Alk' is a Cl-C4-alkyl.
For certain meanings of the substituents Rl, R2, Rs and/or Rs, the
compounds (1~ according to the invention can be prepared from a precllrsor of
for nula (I)' substituted by a group R'l, R'2, R's and/or RVI~ called a precursor
group of R1, R2, Rs and/or R6
21~07~8
~hc dcscription wtlich follows dcscribcs the p~cparatiorl of the cornpounds of
formula (I) carrying substitucnts Rl an~Vor Rs; thc same mcthods apply to the
preparation of the compounds in which the substituents R2 and/or R6 are defmed
as indicated for R1 and Rs.
S I~e compounds (1) in which Rl and/or R5 a~e a hydroxy can ~e obtained by
the catalytic hydrogenation of a compound of fonnula (I)' in which R'l and/or R'$
are a benzyloxy, for cxample irl the preseDce of palladium-on-charcoal.
The c~mpounds (I)' in which R'l and/or E~'s are a hydroxy c~n be used to
prepare compounds (I3 in which R1 and/or Rs are an alkoxy by reaction with an
alkyl halide in the presence of a base such as a metal hydride or an alkali metal or
aikaline earth metal carbonate like K2C03 or Cs~CO3, in a solvent such as THF orI)MF. ~ikewise, the compounds of formula (I) in which Rl an(Vor Rs aIe an ~-
aminoalkyl~7~y are prepared by reacting an ~-chloroalkylamine wi~h the
compounds in which R'1 and/or R's = OH; similarly, the sompounds in which Rl
and/or Rs aN an ~-hydroxyalkoxy are preparcd by reaction with a chloroalkyl
alcohol; in the particul~r case of the preparation of a compound (I) in which Rlandlor Rs = O(CH2)20E~, it is also possible to react ethylene carbonate with a
compolmd ~1)' in which R'l and/or R'5 = OH.
The compounds of foDnula (I) in which R1 and/or Rs are an acyloxy are
obtained by reacting an acid halide or an anhydride with a compound (~' in whichR'1 and/or R'S are a hydroxy.
To prepare compounds of formula (I) in which R~ Vor R5 are a
monoalkylamino or a dialkylamino, the compounds of fonnula (1~' in which R'l
and/or R's are an amino can undergo reductive alkylation. If R'l and/or R's are an
amino, it is also possible to perform a nitrosation, for example in the presence of
nitrous acid or an alkyl nitrite, to prepare a compo~md (I) in which Rl and/or Rs
are a diazonium salt; reactions krlown to those skilled in the art then afford the
compounds (I) according to the invention in which Rl and/or Rs are a cyano, a
halogeno or a Cl-C4-thioalkyl. Finally, compounds (1) in which Rl and/or Rs are
3û one of the groups of the forrnulae RCONH-, ROCONH-, RNEICONH- and
RSO2NH-, in which R is a C1-C4-alkyl, can be prepared by conventional
reactions starting ~om compounds (I)' in which R'l and/or R's = NH2
The compounds of formula (I)' in which the substituent R'S is a
phenoxycarbonyl can be used to obtain the compounds (I) in which Rs is a
phenylcarbamoyl or an alkylcarbamoyl by reaction with an aniline or an
alkylamine. A substituted aniline or an alkylamine substituted on the alkyl can be
2ln73~s
19
used to obtain compounds of formula (I) in which Rs is a phcnylcarbamoyl or,
rcspcctivcly, an alkylcarbamoyl substitutcd on thc alkyl.
The compoun~s of formula (I)' in which R'5 is a ben~yloxycarbonyl can bc
uscd to obtain thc compounds (~) in which Rs is a carboxy by catalytic
S hydrogenation. Reaction with a thionyl halidc givcs the compounds of ~onnula (I)
in which RS is a balogenocarbonyl. Such compounds arc used to prepare
compounds of formula (I) in which Ks is an N-substituted carbamoyl by reactior
with a substituted amine.
The eompounds of formula (I) in which Rs is a group COR"7 are prepared
from corresponding compounds (1~' in which R~5 is a phenoxycarbonyl by reaction
with a substituted piperazine or azetidine.
A compound (1~' in which K~5 is a nitro group can be used to obtain a
compound ~1) in which R5 is an amino group by catalytic hydrogenation, for
example in the presence of platinum oxide; other compounds in which the amino
group is substituted can then be prepared by using reactions well known to thoseskilled in the art.
For example, if it is desired to obtain a compound (1~ accolding to the
invention in which Rs is a group NR~3Rg, Rg being an optionally substituted
ber~oyl, the henzoyl chloride in which the phenyl carries ~he appropriate
substituent is reacted with a compound (1)' in which R's is an amino group, in the
presence of an amine such as triethylamine. For example, 4-chlorosulfonylbenzoylchloride can be reacted in order to prepare a compound (~' in which R's is a 4-
chlorosulforlylbenz3mido group, after which a compound (I) in which the
substituent Rs is a 4-sulfamoylbenzamido group or a 4-alkylsulfamoylbenzamido
group is obtained by reaction with ammonia or a Cl-C4-alkylamine respectively.
In the same way, if it is desired to prepare a compound (I) in which Rs is a
group NRgR~, Rg being a Cl-C7-acyl, the appropriate anhydride is reacted with
a compound (I)' in which R's is an amino group, in the presence oP an amine suchas triethylamine.
Ln another preparative example, a compound (I) in which Rs is an
alkylsulfonamido gr~up is obtained by reacting an alkylsulfonyl halide with a
compound (1~' in which R's is an amino group.
The compounds of fonnula (I)' in which R's is an amino group are also
useful for the preparation of compounds in which this amino group is substitutedby a group (CH2)t-COR12. In this case, a compound of the formula Hal-(CH2~t-
~ COOAlk, in which Hal is a halide, for example bromine, and Alk is a Cl-C4-
21073 ~
2U
alkyl, is rcacted with (I)~ in thc presenc~ of cuprous chloridc; if appropriate, the
r~sulting cstcr is convertccl io thc acid or an amide. The reaction of a lactonc, such
as butyrolactone or valcrolactonc, with a compoun~l (I)~ in which ~'5 is an amino
can be used to pr~pare thc compound (I)' in which R'5 = NHCO~CH2)tCOzH,
5 whcre t = 2 or 3.
ln the same way, the compounds of formula ([) in which Rs is an amino
grollp substituted by a group CH(R1o)CO2R1 1 are prepared by reacting a
compound of the formula Hal-CH(Rlo)C02Rll with the corresponding
compolmds (I)' in which the substituent R's is an amino.
A compound (13 in which R5 is an amino group substituted ~y an
alkoxycarbonyl or a phenoxycarbonyl is prepared by reacting an alkyl or phenyl
chlorofonnate with a compound (1~' in which the substituent R's is an amino.
A compound of formula ~1) in which Rs is a ureido is prepared by reacting
ammonia with a compound of fonnula (I)' in which R'5 iS an amino ~oup
substituted by a phenoxycarbonyl; a compound of fonnula (13 in which ~5 is N-
phenylureido or N-alkylureido or N,N-dialkylureido in which the alkyl is C1-~4
is prepared by reacting an aniline or a C1-C4-monoalkylamine or -dialkylamine
with such a compound of fonnula (I)'.
A compound (I) in which R5 is a carbamoyl which is unsubstituted or
substituted by one or 2 alkyl groups is prepared by reacting an appropriate amine
with a compound (1~' in which the substituent R'5 is an amino, in ~he presence of
phosgene.
It is also possible to prepare a compound (I) in which Rs is an amino group
substituted by an alkylcarbamoyl or by a phenylcarbamoyl by reacting an alkyl orphenyl isocyanate with a compound (1)' in which the substituent R's is an amino.Furthermore, a compound (I) in which Rs is a sul~amoyl ~oup which is
unsubstituted or substituted by a Cl-C4-alkyl is prepared by reacting ammonia oran alkylamine with a compound (I~' in which R's is a halogenosul~onyl ~çroup.
The compounds of formula (I)' which are useful as precursors for the
preparation of compounds of formuîa (1) are included in fonnula (1) aDd form part
of the invention.
Among the compounds of formula (I), the compounds of formulae (IX), (X),
(XI), (XII) and (XIII) below, which are useful for the preparation of other
compounds of formula (1~, are preferred compounds according to the invention.
Thus one subject of the prcsent invention consists of the compounds of the
formula
2~3~8
~1
N
S~ (LX)
s
COOH
i~ which ~1~ R2, R3, R4 and Rs are defined as indicated above for (1), and theirfimctional derivatives such as their esters.
S Another subject of ~he present invention consists of the compounds of the formula
R2 ~ O
- ~ S O 2
R5
NH2
in which Rl, R2, R3, R4 and Rs are defined as indicated a~ove for ~I), and their10 salts where appropriate.
Yet ~other subject of the present invention consists of compoul~ds of tbe
folmula
.
~::
:~ :
:~ :
2 ~ O r j~ 3 !1 ~
R~ oR4
~0~
~¦- R~;
OH
in which R~, R2,, R3, R4 and Rs are defimed as indicated above for (I).
Another sub~ect of the present invention consists of compounds of the
S ~onnula
HO R3
\ ,'-'` ~ R
: S2 (XIl)
~,~--R5
(R6)m
jD which R3, E24, Rs, R6 and m are de~med as indicated above for (I).
Yet another subject of the present invention consists of the compounds of the
10 fonnula
:: : :
~3 ~7~
1~ 1 ~ ` Ni~l
2 ~Jr /~
S 0~ ~XIIl)
~R6~m
in which R1~ R2, R5, R~ and m are defined as inclicated above for (1~.
The affinity of the compounds according to the invention for the vasopressin
S receptors was determined in vitro by using the method d~ scribed in C.J. Lyllch et
al., J. BioL Chem., 19.85, 260(5), 2B44-2851. This method consists in studyiDg the
displacement of tritiated vasopressin bound to the Vl sites of rat liver memb~anes.
The concentr~tions of the compounds accarding to thc is~vention which inhibit the
binding of tritiated vasopressin by 50% (ICso) are low, rangillg up to 10-7 M.
The affinity of the compounds (I) according to the invention for the V2
receptors was measured on a bovine kidney membrane preparation using a method
adapted ~m P. Crause et al., Molecular and Cellular E~ndocrinolo~y, 1982, 28,
529-541, and ~om F.L. Stassen et al., J. Phannacol. Exp. ll~er., 1982, ~3, 50-54.
The compounds according to the invention inhibit the binding of tritiated
arginine-vasopressi~ to the receptors of the membrane preparation. The ICs~
values of the compounds according to the invention are low, ranging up to 10-9
The activity of the compounds according to the invention as V2 receptor
antagonists was demonstrated by the adenylate cyclase activity assay peronned by
a method adapted from M. I~burthe et al., Molecular Pharmacol., 1986, 29, 23-27.A bovine kidney membrane preparation is used and each product is incubated ~or
~: 10 minutes at 37-C, by itself or in the presenee of AVP (arginine-vasopressin) at a
concentration of 3.10-8 M. The cyclic AMP (çyclic adenosine monophosphate)
produced is measured by radioimmunoassay. 7'he concentration which causes a
50~o inhibition (ICSo) of the stimulation of adenylate cyclase induced by 3.10-8:: M AVP is detenDined. The ICso values detennined are of the order of 10-7 M,
rangirlg up to 10-8 M.
2~ ~10 7 3 !i 8
Thc activity of the ~ompo~lnds according to the in~cntion, administered
orally, ~s V~ rcc~ptor agonists or antagonists is ~valuated in hypcrhydratcd rats
(OFA strain, SpraKuc~ awlcy) trcated with vasoprcssin.
Lik~wis~, the affinity of th~ c~mpounds (I) according t~ thc invcnti~n for the
S ocytocin rcceptors was dctcrmin~d in vitro by the displacemcnt of a radioiodinated
ocytocin analog bound to thc r~ccptors of a gCSt~tiDg rat mamma~y gland
membrane preparation, using a tcchnique similar to that described by J. lEland et al.
in Eur. 3. Pharmacol., 1987, 147, 197-207. The ICso values of the compounds
according to the invention reach 10-8 M.
The compounds accûrding to the invention ar~ active aftsr administration by
various routes, especially orally.
No sign of toxicity is observed with these compounds at the
pharmacologically active doses.
Thus the compounds according to the invention can be used in the treatment
15 or prevention of Yarious vasopressin-dependent or ocytocin-dependent
complaints, especially cardiovascular complaints such as hypertension, cardiac
insufficiency or coronary vasospasm, in particular in smokers, cardiac ischemia,hemostatic disorders, especially hemophilia, and Vsn Willebrand's syndrome;
complaints of the central nervous system, for sxarnple cerebral edemas, depression,
20 anxiety, psychotic states and memory disorders; complaints of the renal system,
such as renal vasospasm, necrosis of the renal co~tex, hyponatremia and
hypokalemia; and complaints of the gastric system, such as hepatocirrhosis, ulcers,
the pathology of vomiting, for ~xample nausea, travel siclcness or else the
syndrome of inappropriate secretion of antidiuretic hormone (SIA.DH), diabetes
25 insipidus andl enuresia. The compounds according to the invention can also be used
in the treatment of disorders of sexual behavior; in women, the compounds
according to the invention can be used for the treatment of dysmeno~hea or
premature labor.
The present invention further relatcs to pharmaceutical compositions
30 containing an effective dose of a compound according to the invention, or of a
phannaceuti~lly acceptable salt, and suitable excipients.
Said excipients are chosen according to the pharmaceutical fonn and the
desired mode of administration.
~~ the pharmaceutical compositions of the present invention for oral,
35 sublingual, subcutaneous, intramuscular, intravenous, topical, intratracheal,mtranasal, transdermal or rectal administration, the active principles of fo~mula (I)
~J~73~8
abo~c, or thcir salts where appropnate, ccm bc administe~cc~ to animals and humans
in unit forms of adlllinistr~tion, mixcd with c(~nvcntional pharmaccutical ~rriers,
for thc prophylaxis or treatmcnt of the above clisordcrs or diseascs. Thc appropriate
unit folms of administration include fonns for oral administration, such as tablets,
S gela~in capsules, powders, granules and solutions or suspensions to be taken orally,
forms for sublingual, buccal, intratracheal or in~ranasal administration, fonns fc~r
subcutaneous, intramuscular or intravenous administration and for3ns ~or rectal
administration. For topical application, the compounds accordin~s to the invention
can be used in creams, ointments or lotions.
To obtain the desired prophylactic or therapeutic effect, thc dose of active
principle can vary between 0.01 and 50 mg per kg of body weight per day.
Each unit dose can contain f~om a.s to 1000 mg, preferably from 1 to 500
mg, of active ingredients in combination with a pharmaceutical carrier. This unit
dose can be administered 1 to 5 times a day so aS to adrninister a daily dosag~ of
0.5 to 5~0 mg, preferably 1 to 2500 mg.
If a solid composition in the ~orm of tablets is prepared, the main active
ingredlent is mixed with a pharmaceutical vehicle such as gelatin, starch, lactose,
magnesilJm stearate, talc, gum arabic or ~he like. The tablets can be coated with
sucrose, a cellulose deriva~ive or other appropriate substances or they can be
treated so as to have a prolonged or delayed actiYity and so as to release a
predetennined amount of active principle continuously.
A preparation in the ~onn of gelatin capsules is obtained by mixing the active
ingredient with a diluent and pouring the resulting mixture into soft or hard gelatin
capsules.
A preparation in the fonn of a SyIup or elixir or for administration in the
form of drops can contain the active ingredient in combination with a sweetener,which is preferably calorie-free, and methylparaben and propylparaben as
antiseptics, as well as with a flavoring and an appropnate color.
Water-dispersible granules or powdels can contain the active ingredient
mixed with dispersallts or wetting agents or with suspending agents such as
polyvinylpyrrolidone, as well as with sweeteners or taste correctors.
Rectal administration is effected using suppositories, which are prepared
with binders melting at the rectal temperature, for example cacao butter or
polyethylene glycols.
Parenteral administration is effected using aqueous suspensions, isotonic
saline solutions or sterile and injectable solutions which contain pha~nacologically
2~ 073~8
compatiblc dispcrsants and~or wctting agcnts, for example ptopylene glycol or
butyl~nc glycol.
Thc a~tivc principlc can also be fonnula~cd as microcapsules, if appropriate
with one or more carriers or additives.
S Apart f~om the products of fonnula (I) above or one of the ph~ma~utically
acceptable sal~s, the compositions of the present invention can contain other ac~ive
principles which may be useful in the treatment of the disorde~s or diseases
indicated above.
Thus the present invention fu~her relat~s to pharmaceutical compositions
containing several active prineiples in association, one of which is a compound
accoIding to the in~ention.
Thus, according to the present inv~ntion~ it is possible to prepare
pharmaceutlcal compositions containing a compound which i5 a V1 ~eceptor
antagonist in assaciation with a compound which acts on the renin angiotensin
system, such as a converting enzyme inhibitor, an angiotensin ~ antagonist OT a
renin inhibitor. They can also be associated for example with a periph~ral
vasodilator or a calcium inhibitor. Such compositions will ~e uscful in paIticular in
the treatment of hypertension or cardiac deficiency.
~0 Preparationof2- xQindoles
Preparation 1:
4,6-Dimethyl-3-spirocyclohexaneindol-2-one
This compound is prepared accordiDg to M~le and Plant in J. Chem. Soc.,
1951, 3475.
A mixture containing 15 ml of quinoline and 10 g of calcium o~ide is
refluxed under an ineIt atmosphere and S g of the 3,5-dimethylphenylhydrazide ofcyclohexanecarboxylic acid (Il, R'l~ R'2 = CH3, CR3R4 = cyclohexane) are added
over 30 minutes. The reaction medium is cooled and then poured into an
ice/~ydrochloric acid mixture. Extraction is carried out with ethyl acetate and the
extract is washed with nolmal hydrochlolic acid and with water until the washings
are neutral, and then dried and concentrated under vacuum to gi~e a brown solid.Trituration in iso ether gives the expected cornpound.
M.p. = 223-C.
3S The indol-2-one delivatives desclibed in Table 1 below are obtained by
following the same procedure and varying the starting hydrazide.
.
~7 2~L073~8
Thesc compounds arc purificd by chromatography on a silica column using
DCM as thc cluent or hy chromatography on an alumina column using DCM or iso
cttlcr as thc clucnt.
S TABLE I
r-- ll4
~ _ . ~ ~
R'1 R'2C~3~4 M p. C
_ ~ ~.
5-CI Hcyclobutane 191
5-CI Hcyclopentane 189
S-CI Hcyclohexane 186
H Hcyclohexane 123 124
5-CH3 Hcyclohexane 164
S-CE130 Hcyclohexane 226
6-CI Hcyclohexane 16$
CF30 Hcyclohexane 164
Hcyclohexane __160
Preparation 2:
10The 3-spirocyclohexaneindol-2-one described in Table 1 above can also be
obtained by aikylation of the indol-2-one using the method described below.
A soiution of 30 g of indol-Z-one in 900 ml of THF is kept at -40-C under a
nitrogen atmosphere and 1û1 g of potassium tert-butylate are addcd. The tempe-
rature is allowed to rise to O-C over 1 hour, the mixture is then cooled to -60-C
15and a solution of 52 g of 1,5-dibromopentane in 50 ml of THF is added dropwise.
~fter 30 minutes at -60C, the temperature is allowed to rise to RT, 30 ml of water
,
are then added and the solvent is evaporated off under reduced pressure. The
residue is taken up in 500 mi of DCM and ~no ml of water, the insoluble materialis thcn filtered off and the organic phase is separated off, washed with 100 ml of
20 water, dried over magnesium sulfate and evaporatcd under vacuum. The residue is
.~ ~
~:
'
''
2~ 2:L~73~8
chromatographccl on silica using a cyclohcYanc/ethcr rnixturc as thc clucnt to give
thc cxpectc~ compound, which is rccrystallizcd from heptallc.
m=34g.
M.p. = 1~3-~24'C.
S A similar procedure can be applied star~ing from other indol-2-ones and
other alkylating agents.
By way of example, among the starting compounds of fonnula (VII), S
chloroindoi-2--one is described by Bright in j. ~n. ~hem. Soc., 1~56, 79, 221,
and by RajanE~abu in J. Org. Chem., 1986, 51, 1704. 4-Chloroindol-2-s~ne can be
prepared from 2-chloro-6-nitrotoluene by the method desc~bed in J. Am. Chem.
Soc., 1956, 78, 221.
S-Methoxyindol-2-one is prepared from 4-rnethoxyaniline by the method
described in J. Am. Chem. Soc., 1974, 96, 5512. ~ the same way, various indol-
2-ones are prepared from the appropriate aniline derivative.
Preparatjon 3:
S-Ethoxyindol-2-one
A - 3-Thiornethyl-5-ethoxyindol-2-one
23.6 g slf ethyl thiomethylacetate in 6() ml of DCM are added to a solution,
cooled to about -70-C, of 12.5 g of chlorine in 400 ml of DCM. After stirring for S
minutes at the same temperature, a solution of 4-ethoxyanilin~ ~48.3 g) in 120 ml
of DCM is added. The mixture is stirred for one hour at about 70C, 39.3 ml of
triethylamine are added and the resulting mixture is left to warm up to room
temperature. 200 ml of water are added and the organic phase is decanted, dried
over magnesium sul~ate and evaporated under reduced pressure. llle residue is
takerl up in S00 ml of isopropano~ and 2û ml of concentrated hydrochloric acid.
The mixture is stirred for about 16 hours at room temperature and filtered and the
precipitate is separated off. The filtlate is concentrated under reduced pressure to
~; give the expected product.
B - S-Ethoxyindol-2-one
Ihe above solid, in 1500 ml of ethanol, is dethiomethylated in the presence
of 100 g of Raney nickel ~80 to 100 m2 per g), under reflux, for 3 hours, under a
;` ~ nitrogen atmosphere. The mixture is filtered on talc, the mateIial on the filter is
rinsed with lM00 ml of ethanol and the filtrate is concentrated under reduced
3S pressure. 16 g of the expected product are isolated after recrystaliization from
toluene.
'
'9 2~7~
M.p. = lS6'C.
Thc following arc isolcltcd in thc samc manner star~ing from ~he
corrcsponding anilillcs:
S-bcnzyloxyindol-2-onc m.p. - 152-C
S S-n-propylindol-2-onc m.p. = 136-C
S-eshylindol-2-one m.p. = 152-C
5-(2,2,2-trifluoroethoxy)indol-2-one m.p. = 145-C
The compounds of fonnula (Il) describrd below are obtained by following
the technique described in Preparation ~ and varying the starting indol-2~one
10 derivative and the alkylating reagent.
TA33LE
R3
R'~ L R (II)
H
: 15
~ ~ -. _
~: : R~1 R~2 CR3R4 M.p C Alkylating reagent
~ . . .. _ , .
: S-CI E [cyclohexane 186-189Br~CH2)s Br
S-CI Hcycloheptane 202Br(CH2)6Br
5-Cl H4,4-dimethyl 180TSo(cH2)2c(cH3)2
cyclohexane -(CH2)2oTs
S-CI : H2-hexahydroindane 223 cis~1,2-
diiodomethylcyclohexane
~ S-CH30 H4,4-dimethyl 202TsO (CH2)2C(CH3)2-
: : cyclohexane -(CH2)2-oTs
: : 5-Cl H~-indane 228a,a'-dibromomethyl
:: : ~ : orthoxylene
:S-CI H` C(CH3)7 160 CH3I
S-CI HC(CH ~CH3)2 156 CH3CH2I
:: S-CI HC(n Pr) ~ 158 nPrI
S-CI HC(iBu~ ~ 164 iBuI
.
.
2~073~8
S-CI HN-mcthyl-4- 260Cl(CEI232N(CH3?-
pipcridine -(CH2)
S-CI H4-tctrahydro - 2231(CH2)20~CH2)2
pyrannc
4-Cl Hcyclohexane 215Br(CH2)sBr
S-BzO Hcyclohcxane 162Br(CE~2)5B~
H HC(CH2C6Hs)2 206C6HsCE~2Br
5-CI HC(n-pentyl3~ 142CH3(cH2)~Br
5-Cl H2,3-dihydro _
phenalene-2 ~/J\~
5-13zO H4,4-dimethyl 1$4TsO(CH2 )2C(CH3)2-
cyclohexane (~H2)2OTs
5-CI H4-spirocyclopent~ne 202 ~ (CH2)2OTs
cyclohexane (~2~2OTs
5-nPr Hcyclohexane 151Bl(CH2~5Br
5-EtO HN-tBu-4- _/ (CH2)2Br
. . ...................... tBu~ \
: plpendme (CH2)2Br
5-CI HN-Bz-4- 165/ (CH2)2Br
piperidine Bz ~
(CH2)2Br
S-CI HN-phenyl-4- 188 / (
~: ~ pipendine (CH2)2cl
S-CI H\C ~ l 300~ CH20S02CH
CH20S02CH
~ ~ :
S~EtO H4,4-diethy5 132TSo(cH2)2c(c2H5)2
I : cyclohexane -(CH2)2oTs
~;~ : S-EtO Hcyclohexane 163Br(CH2)sBr
5 -EtO H4,4-dimethyl 17~TSo(cH2)2c(cH3)2
cyclohexane -(CH2)2oTs
31 21073~
~-EsO ~l cycl~h~pt~ne 139 13r(CH?)~r
S-Et }I ~,4-clirncthyl I fi()TsO(CH2)2~(CH3)2~
cyclohcx~nc -(~H2)~0Ts
5-CF3CH~0- ~1 4,4--~in~cthyl 164 ditto
cyclohexane
H H 4,4-dilllcthyl 169 di~to
cyclohcxane
_ _ ~_ ____
E~e~:
3-Spiroadamantaneindol-2-one
This compound is prepared according to 1. Fleming et al., Tetrahedron
S Letters, 198'', ?053-''056, from 2-bromoaniline and adamantan-2-one.
Preparation 5:
5-Chloto- 3,3-diphenylindol-?-one
This compound is prepared by the method described in Helv. (:him. Acta,
1946, 79, 41S-431, by the reaction of benzene with 5-chloroisatin in the presence
of aluminum chloride.
M.p. = 281-C.
S-Nitro-3-spirocyclohexaneindol-2-one
This compound is prepared by the method described in J. Am. Chem. Soc.,
1945, 67~ 499, by the nitration of 3-spirocyclohexaneindol-2-one.
M.p. = 19?-C.
5-Nitro-3-spiroadamantaneindol-2-one is prepared in the same manner
starting from 3-spiroadamantaneindol-2-one.
M.p. > 260''C.
5-I~itro-3-spiro(4,4-dimethyl)cyclohexaneindol-2-one is also prepared.
M.p. = 195C.
::
_reparation 7:
S-Amino-3-spirocyclohexaneindol-2~one
This compound is prepared by the method described in J. Chem. Soc., 1951,
3475, by the reduction of 5-nitro-3-spirocyclohexaneindol-2-one, prepared
abovc.
3~ 21~3~
~ p.- 17~5C~.
5-Amino-3- spiroadamantanc is prcparccl in ~hc samc rmanner.
~.p. = 245-C.
S Preparaeion 8:
S-Fiuoro-3-spirocyclohexaneindol-2-one
A - S-~iazonium-3-spirocyclohexaneindol-2-onP tetrafluoroborate
A solution containing 4 g of S-amino-3-spirocyelohexaneindol-2~one in
9.2 ml of 6 N hydrochloric acid is cooled ~o O-C and 2.27 g of sodium nitrite in 2.6
ml of water are added, fi~llowed by 2.54 g of sodium tetrafluoroborate in 9 ml of
water. After stirring for S minutes, the precipitate is filtered off and washed with a
5% solution of tetrafluoroborate, with 3 ml of methanol cnoled to about 0-C and
then with S ml of ether. The salt obtained is dried under vacuum at RT in the
presence of phosphorus pentoxide.
lS
B- S-Fluoro-3~spirocyclohexaneindol-~-one
1 g of the compound obtai~led in s~ep A is placed in 5 ml of xylene and
heated at about 115-C for 2 hours. The mixture is cooled to RT, the precipitate is
filtered off and rinsed with toluene and 0.1 g of active charcoal is added to the
filtrate. After filtration, the solvent is evaporated off under reduced pressure ~o give
0.45 g of the expected compound, which is recrystallized from pentane.
~.p.= 114~
Preparation 9:
S-~yano-3-spirocyclohexaneindol-2-one
4.78 g of potassium cyanide and 4.95 g of cuprous cyanide are dissolved at
RT in 40 ml of DMSO. The solution is cooled to about 15-C and 4.15 g of the
diazonium salt obtained in step A of the previous preparation are added.
After stirnng for 30 minutes at RT, 100 ml of water and 100 ml of ether are
added and the organic phase is then separated off, dried over magnesium sulfate
and evaporaeed under reduced pressure. The residue is chromatographed on silica
using a cyclohexas~e/ether mixture as the eluent to give the expected compound~
which is recrystallized from heptane.
rn = 1.4 g.
~I.p. = 216-C.
33 21Q73~8
Prc~p~j~
S--Chloro-3-spiroacl~n~antanein(lol--~-onc
1 g of the p-chlorophenylhy~rclzidc of adamantanc-2-carboxylic acid is
dissolvcd ancl 2.5 ml of a solution of n-buty!lithium (1.6 M in hexane) are added at
5 -40DC. After stirring for S minutes, the mixture is concentratcd under vacuum with
the temperature bcing kep~ below 3()^C. 30 ml of 1,2,3,4-tetramethylbcnzene are
added and ~he mixture is refluxed for 1 hour. It is concentrated under reduced
pressure, the residlle is taken up in normal hydrochloric acid, extraction is carried
ou~ with ether and the extract ;s washed, dried ansl conccntsated under vacuum.
10 The oil obtained is chromatographed on a silica column using l)CM as the eluent
to give û.3 g of the expected product in the fonn of a wax, which is crystallized
from iso ether.
M.p. = ~49~C.
Pre~aration 11:
5-Chloro-3-cyclohexyl-3-methylindol -2-one
The method described in Synth. Commun., 1982, 12(1), 1-10, is used to
prepare 5-chloro-3-cyclohexylindol-2-one as an intermediate, and the expected
compound is then obtained by reaction with methyl iodide.
Preparation 12:
5-Acetyl-3-spirocyclohexaneindol-2-one
2.56 g of acetyl chloride and then 8.25 g of anhydrous aluminum chloride are
added to a solution, cooled to 5-C, of 4 g of 3-spirocyclohexaneindol-2-one in 35
25 ml of 1,2-dichloroethane. The mixture is refluxed for 2 hours, the solvent isevaporated off under reduced pressure and the medium is hydrolyzed with 50 g of
ice and e~tracted with ethyl acetate.
The organic phase is washed with water, dried over magnesium sulfate and
then evaporated under reduced pressure. The residue is chrornatographed on a
30 silica column using a mixture of heptane and ethyl ether as the eluent to give 3.6 g
of the expected product.
M.p. = 192-C.
The benzenesulfonyl chlorides described in the Table below were prepared
using the procedure described.
:~
3~
2:~07~
~Z
OCH3
~,J
Y R~,~
_
Y RV M.p. C
, . ~ . _
S CH3 8S
O CH2BZ 95
O CH2CO2Et 89
O (CH-2)3Br
Starting from the various 2-oxoindoles described above and appropriate
: S benzenesulfonyl chlorides, the compounds according to the invention were
prepared using the procedures reported in the Examples below.
EXAMPL~ 1
S-Chloro-1-(2-methoxy-4-nitrobenzenesulfonyl)-3-
spirocyclohexaneindol-2-one
A mixture containing 0.7 g of 5-chloro-3-spirocyclohexaneindol-2-one
and 70 mg of sodium hydride in 7 ml of THF is stirred under nitrogen at RT for 30
minutes. 0.7 g of 2-metboxy-4-nitrobenzenesulfonyl chloride is introduced and
stirsing is maintained at RT for 20 hou~s. The mixture is concentrated under
15: ~ ~ vacuum, the residue is taken up in 30 ml of water, extraction is calIied out with
;: ethyl acetate and the extract is washed with water and then dried and concentrated
to give~: 1.1 g of the expected compound, which crystallizes ~om iso ether.
M.p. = 18~-C.
20 ~ ~ E~AMPLE2
1-(4-Amino-2-methoxybenzenesulfonyl)-5-chloro-3-
:: spirocyclohexaneindol-2-one
~ ~.
3s ~ 3 ~ ~
0.8 g of thc compouncl obtaincd in thc prrvious Exarnplc is reduccd with
hydrogcn un(lcr normal prcssurc at RT for 20 hours in 10 ml of acetic acid, in the
prescncc of 30 mg of platinum oxide. The rcaction medium is filtcred, thc filtrate is
conccntratcd, thc residuc is takcn up in a watcr/ethyl acetatc mixture and the
S organic phas~ is washed with water, dned and concentrated. Thc yellow foam
obtained is chromatographed on alumina using DCM as the eluent to give 0.2 g of
the expected product.
M.p. = 173-C.
EXAMPLE 3
S-Chloro- 1-[4-(2-methylphenylcarboxamido~-2-
methoxybenzenesulfonyl]-3-spi~cyclohexaneindol-2-one
A mixture containing 0.2 g of the compound prepared in the previous
Example, 0.5 ml of triethylamine, 5 ml of DCM and 0.1 g of orthotollloyl chloride
15 is stirred at RT for 48 hours. It is concentrated undcr vacuum, the residue is taken
up in a water/ether mixture and le~t to decant and the organic phase is washed with
a saturated solution of sodium hydrogencarbonate and then with water, dried and
concentrated under vacuum to give 250 mg of a solid, which is chromatographed
on silica using DCM as the eluent to give ().1 g of the expected product
:~ 20 M.p. = 192-C.
EXAMPLE 4
6-Chloro-1-(2,4-dimethoxybenzenesulfonyl)-3-spirocyclohexaneindol-
2-one
A mixtuIe containing 0.15 g of 6-chloro-3-spirocyclohexaneindol-2-one
and 15 mg of sodium hydride in 2 ml of THF is stirred for 30 minutes at RT undernitrogen; 0.15 g of ~,4-dimethoxybenzenesulfonyl chloride is introduced and
stsning is maintained at RT for 20 hou~s. The mixture is concentrated under
vacuum, the residue is taken up in 30 ml of water and extracted with ethyl acetate
30 and the extract is washed with water, dried and concentrated under ~acuum. Ihe
product obtained is recrystallized from iso ether.
M.p. = 147-C.
EXA~PLE 5
Acid fumarate of S-chloro-1-[4-(3-
dimethylaminopropoxy)benzenesulfonyll-3-spirocyclohexaneindol-2~one
36 21~73
A) 4-(3-Bromopropoxy)bcnzcncsulfonyl chloridc
A mixturc containing 23 g of sodiLIm 4-hydroxybcnzcncsulfonate dihydratc,
7 g of potassi~lm hydroxidc pcll~ts (85%), 30 ml of watcr, 50 ml of absolute
cthanol, ~0 g of l,3-dibromopropanc alld 3.4 g of t~trabutylammoslium
S hyclrogensulfatc is rcfluxed fi)r 3 hours. Thc reaction medium is concentratcdunder vacuum, taken up in cthanol and concentrated once again. Thc residue is
taken up in hot mcthanoi. The insoluble material is filtFred of~, thc filtrate is
conc~ntrated and the residue is triturated in ether to give 22.5 g of a white solid.
120 ml of phosphoms oxychloride and 16 g of phosphorus pentachloride are added
10 to this solid and the mixture is stirred for 20 hours at RT and then refluxed ~or 1
hour. The reaction medium is concentrated under vacuum, the residuc is then taken
up in an etherlwater mixture and the organic phase is decanted and washed with asaturated solution of sodium hydrogencarbonate. After drying and concentration,
the expected product is obtained in the ~onn o~ a yellow oil.
B) 1-~4-(3-Bromopropoxy)berlzenesulfonyl]-5-chloro-3-
spirocyclohexaneindol -2-one
A mixture containing 1.2 g of S-chloro-3-spirocyclohexaneindol-2-one
and 0.16 g of sodium hydride in 6 ml of TlHEi is stirred at RT for 30 minutes under
20 nitrogen. 1.6 g of 4-(3-bromopropoxy)benzenesulfonyl chloride are then added.After 20 hours at RT, the reaction medium is concentrated under vacuum, the
residue is taken up in a water/ethyl ether mixture and decanted and the organic
phase is washed with water, dried and concentrated. The oil obtained is purified by
chromatography on silica using iso ether as the eluent. The expected product is
25 obtained in the fonn of an oil, which crystallizes from iso ether.
~n=lg.
M.p. = 123-C.
~; C) Acid fumarate of S-chloro-1-[4-~3-
30 dimethylaminopropoxy)benzenesulfonyl]-3-spirocyclohexaneindol-2-one
A mixture containing O.S g of the product obtained in the aboYe step, O.S g of
potassium iodide and 20 ml of a 33% solution of dimethylamine in methanol is
stirred at RT for 20 hours. The reaction medium is concent~ated and taken up in 10
ml of water and, after trituration, the insoluble material is separated off and treated
35 with 10 ml of 3 N hydrochloric acid. A gum is formed which is dissolved in 30 ml
of wann water, and the solution is filtered on paper and then rendered alkaline by
2~073~
thc addi~ion of 1 N sodium hydroxide. Thc insolub3c matcrial is cxtractcd with
cthcr and the cxtract is washcd, (Iricd an(l thcn conccn~ratcd to give a yellow oil.
This is dissolvecl in 10 ml of acctonc, and 0.1 g of fumaric acid is ild(iccl to thc hot
SOlUtiOII.
S The expected product precipitatcs at 20-C.
m = 240 mg.
M.p. = 16BC.
EXAMPLE 6
5-Chloro-1-(2,4-dimethoxybenzenesulfQrlyl)-3-spiroadamalltaneindol-2-
one
A mixh3re containing 0.2 g of 5-chloro-3-spiroadamantaneindol-2-one
and 20 mg of sodium hydnde in 3 ml of THF is stirred for 30 minutes at RT under
a nitrogen atmospllere. 0.18 g of 2,4~dimethoxybenzenesulfonyl chloride is addedand stirring is maintained at RT for 20 hours. The reaction medium is concentrated
under vacuum, the residue is taken up in 30 ml of water and extracted with etherand the extract is washed with water, dried and concentrated urlder vacuum The
wax obtained crystallizes from 15 ml of iso ether.
m=240mg.
M.p. = 152-154-C.
EXAMPLE 7
5-Chloro-1-(2,4-dimethoxybenzenesulfonyl)-3-spirocycloheptaneindol-
2- one
A solution containing 0.156 g of potassium tert-butylate and 0 33 g of 5-
chloro-3-spirocycloheptaneindol-2-one in lS ml of THF is cooled to -40-C un-
der an inert atmosphere. The temperature is allowed to rise to about 10-C over 1hour, the solution is then cooled to about -40-C, a solution of 0.335 g of 2,4-di-
methoxybenzenesulfonyl chloride in 15 ml of THF is added dropwise and the
3 0 mixture is stirred at RT for 2 hours. The solvent is evaporated off under reduced
pressure and the residue is then taken up in 30 ml of DCM and 30 ml of water. I~e
organic phase is separated off, washed with 15 ml of water, dried over magnesiumsulfate and evaporated under vacuum. The oil obtained is evaporated under va-
cuum using a cyclohexane/DCM mixture as the eluent to give the expected com-
pound, which recrystallizes from heptane.
38 21D7348
m - 0.51 g.
M.p. = 13S-C.
E~CAMPLB 8
2,4-Dimethoxy-1-benzenesulfonyl-2a-methyl-2a,3,4,5-tetrahydrobenz[c,
d]indol-2-one (I: R1 = H, -R2-R3 = -(CH2)3-, R4 = CH3, R5 = R6 = OCH3~
2a,3,4,5-Te~rahydrobenz[c,d]indol-2-ollc is commercially available. With
the temperature maintained at -40~C and under a nitrogen atmosphere, a solution
containing 0.7 g of this compound and 1.36 g of potassium tert-butylate in 40 mllû of anhydrous ~F is prepared.
The temperature is allowed to rise to about ()-C, the solution is then cooled to-60C and a solution of 0.57 g of msthyl iodide in 20 ml of THF is added; the
medium is maintained at -10C for 30 minutes, with stirring, and then cooled to
about -40-C and a solution of 0.96 g of 2,4-dimethoxybenzenesulfonyl chloride in10 ml of THF is added. After stirnng for 16 hours at RT, the solvent is evaporated
off under reduced pressure and the residue is taken up in 30 ml of DCM and 30 mlof water; the organic phase is scp~rated off and then dried over magnesium sulfate
and evaporated. The oil obtained is purified by chromatog~aphy on silica using acyclohexane/DCM mixture as the eluent to give the expected product, which is
recrystallized from a cyclohexane/AcOEt mix~ure (9S/S; v/v).
M.p. = 160~C.
The compounds according to the invention collated in Table 3 below were
prepared from the 2-oxoindoles described above by following the procedure
described in the above Examples.
TABLE 3
R~ r f R4
R2~ O
S2
b ~
(R6)m
:,
21~73~8
39
Unless indicated othcrwise in thc Table bclow (~), R2 = H ancl m - 1
~ _ _ _._.~m ~ _ ~_ ~__
Ex. Rl CR3~4 R5 (~6)m M.p. ~C
~ _~ _ ~
9 S-CI C(C6Hs)2 3-MeO 4~ eO 178
10 5-N02 cyclohexane 3-MeO 4-MeO 157
11 S-CI cyclohexane 4-MeO H 112
12 S-CI C(CH3)2 3-MeO 4-MeO 110
13 5-NH2 cyclohexane 3-MeO 4-MeO 171
14 S-CN cyclohexane 2-MeO 4-MeO 148
S-CI cyclohexane 4-MeO2,3,6-triMe 188
6 S-CI C(Pr)2 2-MeO 4-MeO 186
: 17 S-CI indane-2 2-MeO 4-MeO 182
18 5-CI C(iBu)2 2-MeO 4-MeO 184
19 5-CI N-rncthyl 2-MeO 4-MeO 142
plperidlne-4
: ~ 20 S-CI C(Et)2 2-MeO 4-MeO 190
2t S-F cyclohexane 2-MeO 4-MeO 149
22 5-CI 4-tetrahydro 2-MeO 4-MeO 142
pyranne
23 S-CI 4,4-dimethyl 2-~leO 4-MeO 118
cyclohexane
`:
: :
2~ 073~
'~()
24 S-CI2-heYahy~lro 2 -McO 4-McO 89
25 4 -CIcyclohc,Y.Illc 2-~fcO 4-McO 150
26 5-CIcyclohcxane 3-MeO 4-MeO 152
27 5-CIcyclohcxane 4-Me ff 150
28 Hcyclohexa~e 3-MeO 4-MeO 107
29 5-Mecyclohexane 3-MeO 4-MeO 171
30 S-MeOcyclohexane 3-Me t) 4-MeO 124
31 S-CIcyclohexane 2-MeO 4-MeO 149
32 5-CIcyclohexane 4-CI H 154
33 S-CIcyclobutane 3-MeO 4-MeO 111
34 5-CIcyclopentane 3-MeO 4-MeO lO6
5-CIcyclohexane 4-MeO 2-CI 174
36 S-CIcyclohexane 4-N02 H 172
37 S-CIcyclohexane 4-CN H 198
38 5-CIcyclohexane 4-MeO 2-N02 147
39 S-CIcyclohexane 4-CF3 H 139
5-CIcyclohexane 4-CF30 H 134
41 5-CIcyclohexane 4-MeO 2-NH2 150
41 ~1~73~3
42 ~-CE13 cyclohcxane 4-MeO 2-McO 165
~ R~ fi-
43 S-CI cyclohexanc 3-Mc 4-BzO 127
44 S-CI cyclohexane 4-iPr 2,6-iPr 172
S-CI cyclohexane 2-CF3 H 154
46 S-CI cyclohexane 2-MeO CH~ C}13 215
47 S-CI cyclohexane 4-MeO /~co~ 193
4B S-CI cyclohexane 2 MeO 4-CE~3OCO 120
49 S-CI c~ 2-MeO 4-MeO 184
H adamantane 2-MeO 4~MeO 172
51S-MeO 4,4-dimethyl2-MeO 4-MeO 152
cyclohexane
52 5-CI cyclohexane2-MeO 4-CH3SO2NH 131
S3 S-CI cyclohexane2-MeO ~ ~ 240
~CONH
54 5-CI cyclohexane2-Me S-F lS3
S5 S-CI cyclohexane2CC23O-5-CF3CH2O 175
56 S-CI cyclohexane2-MeO /~N~Co 218
C~13
'
"' .
2~07~8
~2
57 5-CIcyclohexane 2-MeO /~ ~ ~o 165
`~
58 5-CI cyclohcxane S-soH22_2,4-~li MeO 270
595-BzOcyclohcxalle 2-MeO 4- MeO 15~
60S-BzO 4,4-dimethyl 2-MeO 4-MeO 142
cyclohexane
61 5-CI cyclohexane 2-MeO cll3o~c 192
o
62 5-CI cyclohexane 2-MeOC}~3 N~JN -~ 158
64 H C(CH2C6~5)2 3-MeO 4-MeO 146
655-CH3COcyc!ohexane 3-MeO 4-MeO 122
66 S-CI C(n-pentyl)2 2-MeO 4-MeO 140
67 5-CI C(CH2C6H5)2 2-MeO 4-MeO 185
685-H2N cyclohexane 2-MeO 4-MeO 230
69 5-CI 4-spirocyclo- 2-MeO 4-MeO 154
: pentane
70 5-CI cyclohexane 2-MeO 5-MeO 116
~715-nPrcyclohexane 2-MeO 4-MeO 138
72S-EtO N-tBu-4- 2-MeO 4-MeO 95
pipendino (o22s)
73 5-CI N-Bz-4- 2-MeO 4-MeO 76
~:~ pipel~dine (0 5
~3 21073~
74 5-CIN-phcnyl-~- 2-McC) 4-McO 163
piperidinc
S-CI~yclohcxanc 2-EtO 4-EtO :123
76 S-CI ~ C ~ 2-Me(:) 4-McO 190
77 S-EtO4,4-diethyl 2-McO 4-MeO 129
cyclohexanc
78 5-Etocycloheptane 2-MeO 4~MeO 130
79 5-EtOcyclohexane 2-MeO 4-MeO 134
8û S-EtO4,4-dimethyl 2-MeC) 4-MeO 160
cyclohexane
81 5-Et4,4-dimethyl 2-MeO 4-MeO 166
cyclohexane
: : :
82 S-EtO4,4-dimetbyl 2-MeO 4-NC)2 110
cyclohexane
~;:
~: 83 S-EtO4,4-dimethyl 2-MeO 4-NH2 230
~: cyclohexane
: :
: : 84 5-NO24~4-dimethyl 2-MeO 4-MeO 102
: cyclohexane
:85 5-NH24,4-dimethyl 2-MeO 4-MeO lB0
cyclohexane
~ :
~ 86 ~; 5-4,4-dimethyl 2-MeO 4-MeO 169
1~ CF~CH~Ocyclohexane
_ __ ,, . . ., _
21 073 ~8
~s~
EXAMPLE 87
1-(2,~~Dinlctho,Yybcllzcllcxulfonyl)- 3-(4,4-~imcthylspirocyciohc;~sane)-5-
hydro~yindol-2-onc
3.51 g of thc cs)mpollnd prcparcd in Examplc 60 are stirrctl at 50C for 1
S hour, under a hydrogcn atmospherc, with 0.5 g of 10% pall~dium-oll-charcoal in150 ml of cthanol. The catalyst is filtered off on talc, the material on the filter i5
rinsed with DCM and the filtrate is evaporated undcr reduced pressure to give 2.8 g
of the expected eompound, which is recrystallized from a cyclohexane/AcOEt
mixture ~90/1û; v/v).
M.p. = 22û-C.
EXAMPLE 8B
1 -(2,4-Dimethoxybenzenesulfonyl)-5-hydroxy-3-spirocyclohexaneindol-
2-one is prepared in the same manner starting from the S-benzyloxy derivative
15 described in Example 59.
M.p. = L96-C.
EXAMPLE 80 bis
(2,4-Dimethoxybenzenesulfonyl)-3 -(4,4 -dirnethylspir~cyclohexane~-5-
20 ethoxyindol-2-one
This compound, already describèd in Example 80, can be prepared by
another methorl starting from the homologous 5-hydroxy compound. 0.6 g of the
compound prepared in Example 87 is sti~red at RT for 16 hou~s, under an inert
atmosphere, with 0.19 g of anhydrous potassium carbonate and 0.315 g of ethyl
25 iodide in 11 ml of DMF. The solvent is evaporated off under reduced pressure and
30 ml of AcOEt and 30 ml of water are added. The organic phase is washed with
water, dried over magnesium sulfate and then concentrated under reduced pressure.
0.45 g of the expected product is obtained by crystallization ~om
cyclohexane.
M.p. = 160-C.
The compounds described in Table 4 below are prepared in the same manner.
:
.
21073~3
rAst E 4
R4
s
R R5
}:~ _ R5 R6 M.p.'C
. _ _ .
89 S-nPrO cyclohexane 2-MeO 4-MeO 139
S-nPrO 4,4-dimethyl 2-MeO 4-MeO 158
; cyclohexane
91 5 iPrO 4,4-dimethyl 2-MeO 4-MeO 154
cyclohexane
_ ~ . cyclohexane 2-MeO 4-McO 155
- :
EXAMPLE 93
5-Acetoxy- 1 -(2,4-dimethoxybenzenesulfonyl)-3-spirocyclohexaneindol-
2-one
0.5 g of the compound prepa~d in Example 88, 2.5 ml of isopropenyl acetate
10 and 0.165 g of potassium carbonate in 2.5 ml of toluene and 0.3 ml of DMF are
heated at about 65-C for 15 hours. Afler cooling, 10 ml of water and 15 ml of ethyl
acetate are added and the organic phase is decanted, washed with water, dried over
magnesium sùl~ate and concentrats~d under reduced pressure. 0.51 g of the
expected compound, containing 0.5 mol of cyclohexane, is isolated by
15 crysiallization from a cyclohcxane/ethyl ace~ate mixture.
:::
~ .
46 21073~
M.p. = 116-C.
EXAMPLE 9~
1 -~2,4-Dimethoxybf~nzcncsulfonyl)-5-(2-hydroxyethoxy~-3-
spirocyclohcxaneindol~2-onc
0.5 g of the compound prepared in Example 88, 0.5 g of ethylene carbonate
and 0.272 g of anhydrous potassium carbonate in 1.25 ml of DMF arc heated at
about 7QC ~or 40 hours. After cooling, 10 ml of water and 15 rnl of ethyl acetate
are added and the organic phase is decantsd, washed with wa~er, dried over
magnesium sulfate and concentrated wlder reduced pressure. The oily residue is
chromatographed on a silica column using a cyGlohexane/AcOEt mixture (70/30;
v/v) as the eluent to give 0.5 g of ~he expected product, which is recrystallized
from a heptane/DCM mixture.
M.p. = 170-C.
lS
EXAMPLE 95
5-(2-Dimethylaminoethoxy)-1-(2,4-dimethoxybenzenesulfonyl)-3-
spirocyclohexaneindol-2-one
0.6 g of the compound prepared in Example 88 is heated at about 40-C for 16
hours, under an inert atmosphere, with 0.32 g of N,N-dimethyl(2-
chloroethylamine~ and 1.76 g of cesium carbonate in 7.2 ml s)f acetone and 2.4 ml
j ~ of DMF. lhe salts are filtered off and 20 ml of water and 20 ml of AcOEt are
added to the filtrate. The organic phase is decanted, washed with water, dried over
magnesium sulfate and concentrated under reduced pressure. The residue is
chromatographed on silica using a DCM/MeOH mixture (9/1; v/v) as the eluent to
give 0.6 g of the expected product, which is recrystallized f~om a cyclohexane/iso
ether mi~ture.
M.p. = 122~C.
EXAMPLE 96
5-Chloro-1-(2,4-dimethoxybenzenesulfonyl)-3-~spiropiperidine-4)indol-
2-one
This reaction is perfonned according to J. Org. Chem., 1984, 49, 2795-2799.
0.75 g of 1-chloroethyl chlorofonnate is added at 0-C to a solution of 1.31 g of the
compound descnbed in Example 73 and 0.32 g of 1,8-bis-
dimethylaminonaphthalene in 22 ml of 1,2-dichloroethane. The mixture is
. .
L~ 7 2 ~ 4
refluxcd for about ~0 n~inutcs ancl conc~ntratcd undcr rcducc~l prcssurc to a
volume of about 10 ml, and ~2 nll of mcthanol arc ~hen adclcd. Aftcr rcfluxing for
50 minutes, thc rcaction mcclium is conccntrat~d unclcr rcduccd pressure and theresidllc is chromatographcd Oll a silica column using a 13CM/MeO~I mixture (95/5;
S v/v) as the cluent. 1.16 g of the expccted product arc isolatecl and recrystallized
from a mixturc of cyclohcxane anci ethyl acetate
M.p. = 172-C.
EXAMPLE 97
3~ Acetylspiropiperidine-4)-S-chloro-1-(2,4-
dimethoxybenzenesulfonyl)indol-2-one
0.086 ml of acetyl chloride is a;dded to a solution, cooled to about 0'C, of
0.35 g of the compound prepared in the previous Example and 0.23 ml of
triethylamine in S ml of DCM. The mixture is st~rred for one hour at 20-C, 5 ml o
15 water are added, the organic phase is decanted, washed with water, dried overmagnesium sulfate and concentra~ed under reduced pressure and the residue is
chromatographed on a silica column using a DCM/MeOH mixture (99/1; v/v) as
the eluent. 0.29 g of the expected product is isolated in the fonn of the
hemihydrate.
M.p. = 107-C.
EXAMPLE 98
5-Chloro-1-(2,4-dimethoxybenzenesulfonyl)-3-(N-
methoxycarbonylspiropiperidine -4)indol -2-one
~is compound is prepared from the one obtained in F,xample 96 by
reaction with methyl chloroformate.
M.p. = 147-C.
~XAMPLE 99
1-(3,4-Dimethoxybenzenesulfonyl)-5-propionamido-3-
spirocyclohexaneindol-2-one
A solution of 0.144 g of propionyl chloride in 3 ml of DCM is added to a
solution, cooled to about 0-C, of 0.5 g of the compound described in Example 13
and 0.167 ml of triethylamine in 10 ml of DCM. The mixture is stirred for 2 hours
35 at 20GC, 20 ml of water are then added and the organic phase is decanted, washed
with water, driod over magnesium sulfate and concentrated under r~duced
48 2~73~8
pressure. 0.5 ~ of thc e:~pectecl produet is isolate(l after reerystalliz~tion from a
hcptan~/AeOE~t mi;Yture (95/5; v/v).
M.p. = 158-C.
S E~5AMPLE 100
I -(3~4-Oimethoxybenzencsulfollyl)-S-~N-mcthylllrcido)-3-
spirocyclohcxaneirldol-2-one
0.15 g of methyl isocyanate is added to a solution, cooled to about O~C, of
0.5 g of the compound described in Example 13 in lO ml of DCM. After stirring
10 for about 16 hours at Rr, 20 ml of water are added and the organic phase is
decanted, washed with watcr, dried over magnesium sulfa~e and concentrated
under reduced pressure. 0.5 g of the expected product is isolated after
recrystallization from a mixture of heptane and ethyl acetate.
M.p. = 214-C.
The compound descnbed in the following Example is prepared in the same
manner.
EXAMPLE 101
(3,4-Dimethoxybenzenesulfonyl)-S-(N-phenylur~ido)-3-
20 spirocyclohexaneindol-2-one
M.p. = 124~C.
:
EXAMPLE 102
5-Dimethylamino-1-(2~4-dimethoxyben7enesulfonyl)-3-
25 spirocyclohexaneindol-2-one
A mixture of 0.5 g of the compound described in Example 68 with 0.5 ml
of a 35% solution of formaldehyde and 0.12 g of sodium cyanoborohydride in 10
ml of acetonitrile is stirred at RT, under a nitrogen atmosphere, and the pH is
adjusted to about 6.5 with a few drops of acetic acid. After 48 hours at 20~, the
30 solvent is evapora~ed off under reduced pressure and 20 ml of an approximately 2
N aqueous solution of sodium hydroxide and 20 ml of DCM are added. The
organic phase is decanted, washed with water and dried o~er magnesium sulfate
and the solvent is evaporated off under reduced pressure. The residue is
chromatographed on a silica column using a cyclohexane/ethyl acetate mixture
3s (80!20; v/v) as the eluent. 0.27 g of the expected product is isolated.
M.p. = 167-C.
21073~8
~9
EXAMPLE 103
1-( ,4-l)imethoxybcnzcncsulfonyl)-5-cthylthio-3-
spirocyclohcxaneindol-2-onc
This compo~lnd is p~ep~red according to J. Chem. Soc., Chem. Commun.,
1980, 16, 756. A mixturc of 2.95 g of diethyl disulfide ancl 0.386 g o~ isopcntyl
nitnte is heated to about 80~C, under an inert atmosphcrc, and 0.8 g of the
compound prepared in Example 68 is added. The medium is stirred for one hour at
~O-C and then concentrated under reduced pressure. The residue is
chromatographed on a silica column using a DCM/cyclohexane mixture (80/20;
lQ v/v) as the eluent. Ihe expected product is isolated after crystallization from
cyclohexane.
M.p. = 123-C.
EXAMPLE 104
5-Chloro-l-f4-(dimethylamillomethylcarboxamido)-2-
methoxybenzenesul~onyl]~3-spirocyclohexaneindol-2-one
A) S-Chloro~1-[4-(chloromethylcarboxamido)-2-
methoxybenzcnesulfonyl] -3-spirocyclohexaneindol-2-one
0.2 g of thç compound prepared in Example 2 is placed in 4 ml of DCM
and 0.5 g of TEA at RT and 0.1 g of chloroacetyl chloride is added. After stirring
for 20 hours at RT, the mixture is concentrated under vacuum. The con-~entrate is
` extracted with ethyl acetate~ the extract is washed with water and a solution of
sodium carbonate and the residue is thcn chromatographed on silica using a
mixture of DCM and AcOEt as the eluent to give 0.15 g of the expected product.
B) S-hloro-1-[4-(dimethylaminomethylcarboxamido)-2-
methoxybenzenesul~onyl]-3-spirocyclohexaneindol-2-one
The compound obtained in the previous step (150 mg) is stirred at RT for
20 hours in 20 ml of a 33% solution of dimethylamine in ethanol. Extraction is
carried out with AcOEt and the extract is washed with N sodium hydroxide and
then water. The residue is chIomato~raphed on silica using AcOEt as the eluent to
give 0.025 g of the expected product.
; M.p. = 173-C.
~, ~
. - .
21 0 rl 3 ~ g
s(~
EXAMPLE 105
I- ~4-(4--Sulfamoylphcnylcarbo,Yamido)-2-mcthoxybellzcncsul~nyl] -5-
chloro-3-spirocyclohcxaneindol -2-one
4-Chlorosul~nylbcnzoyl chloridc is prepared according to Chem. Ber.,
1~41, 271.
0.2 g of thc compound prepared in Example 2 is brought into contact with
0.5 g of TEA in 5 ml of DCM; 0.13 g of 4-chlorosulfonyibenzoyl chlorid~ is
added and the mi~sture is stirred for 20 hou~ at RT. It is concen~rated under
vacuum, the concentrate is taken up in THF, and 10 ml of aquf~-ous ammonia are
added. Stirring is continued for a fur~her ~0 hours at RT and the mixture is
concentrated under vacuum. ~he residue is extracted with ether and the extract is
washed with water, dried over sodium sulfate and then chromatographed on silica
using AcOEt ~s the eluent to give the expected product.
M.p. - 238-242~C after recrystallization from AcOEt.
13XAMPLE 106
1-[4-(3-Sulfamoylphenylcarboxamido)-2-methoxybellzenesulfonyl~-5-
chloro-3-spirocyclohexaneindol-2-one
A) 3-Chlorosulfonylbenzoyl cllloride
This compound is prepared according to patent US 3 290 370. 11 g of
chlorosulfonic acid are heated to 60-C and 8 g of phenylchloroform are added
dropwise. After heating for 2 hours at 130-C, ~he mixture is distilled to give 1 g of
thé expected product.
B.p. = 120-125-C under 0.5 mm Hg.
; 25
B) 1-[4-(3-Sulfamoylphenylcarboxamido)-2-methoxybenzenesulfonyl]-
5-chloro-3-spiIocyclohexaneindol -2-one
~; 210 mg of the compound prepared in Example 2 are placed in 10 ml of
DCM with 220 mg of the compound obtained in the previous step and 200 mg of
TEA, the mixture is stirred overnight and the solvents are then evaporat d off
under~vacuum. The residue is taken up in 20 ml of THF and 20 ml of aqueous
ammonia and the mixture is stirred for 6 hours at RT. The solvents are dAven offunder vacuum and the residue is then taken up in AcOEt and water. Extraction is
carried out with AcOEt and the ex~ract is washed with water and then
chromatographed on silica using an AcOEt/cyclohexane mixture (50/50; v/v~ as
the eluent to give the expected product.
~:
, .
5, 2:3 07~
p. = 176~.
EXAMPLE 107
l-[4-(2-Carboxyphc1lylcarboxamiclo)-~-methoxybenzenesulf'onyl~~S-
S chloro-3-spirocyclohexaneindol-2-one
The preparation is carricd out aceording to J. Hetcrocycl. Chem., 1974,
997-100û.
A mixture containing 0.2 g of the compound prepared in Lxample 2, with
0.5 ml of TEA and 160 mg of phthalic anhydride is stirred at 60-C for 3 hours. It is
concentrated under vacuum and trsated with normal hydrochloric acid, The
precipitate formed is filtered off and treated with a 10% solution of sodium
carbonate; a precipitate forms again, the aqueous phase ,is decanted and the
precipitate is treated with 10% AcOH. The precipitate is filtered off and then
washed with 10% AcOH followed by isopropyl e~hcr and recrystallized ~om iso
ether to give the expected product.
m=O.lSOg,
M.p. = 157-158-C.
EXAMPLE lU8
1-[4-(Benzyloxymethylcarboxamido~-~-methoxybenzenesulfonyl]-S-
chloro-3-spirocyclohexaneindol-2-one
This compound is prepared by the procedure described in Example 3 by
reacting bènzyloxyacetyl chloride with the compound prepared in Example 2.
M.p. = 143~C after recrystallizativn from iso ether.
EXAMPLE 109
5-Chloro-1-[4-(hydroxymethylcarboxamido)-2-
methoxybenzenesulfonyl]-3-spirocyclohexaneindol-2-one
This compound is obtained by hydrogenating the compound prepared in
3 0 ~ the previous Example, under the pressure of a water column, in the presence of 5%
palladium-on-charcoal in an EtOH/AcOEt mixture.
M.p. = 202~C.
EXAMPLE 110
5-Chloro-1-[4-(imidazol-l-ylphenylcarboxamido)-2-
methoxybenzenesulfonyl]-3-spi~cyclopentaneindol-2-one
, . .
5~ 2~73~8
A) Ethyl cstcr of ~-(imidazol~ yl)hcnzoic acid
A mixturc containing 35 ~ of 4-fluorob~nzoyl chlori~c in S() ml of lr)O
cth~nol is rcfluxcd for IS rninutcs. 35 g of thc cthyl cst~r of 4-fluorob~nzoic acid
obtaincd are mixcd with 22 g of imidazolc and 61 g of potassium carbonate in 35
S ml of DMSO. Thc rnixture is heatcd for 18 hours at 120-130C and SOO ml of iccd
water are then addcd. A prccipitat~ fo~s and the cxpccted product crystallizes
fiom iso ether.
M.p. ~ 98-C.
lQ B) Lmidazol-1-ylbenzoyl chloride
S g of the ester obtained in step A are refluxed for 2 hours in 20 ml of water
and 20 ml of sodium hydroxide solution. The reaction medium is washed with
ether and then acidified (pH 2) with concentrated hydrochlonc acid. The
precipitate ~ormed is filtered of~ and then washed with iso ether. S g of the acid
obtained are brought to the reflux temperature in 35 ml of thionyl chloride. Theprecipitate formed is filtered off and then washed with iso ether to give the
expected acid chloride.
M.p. = 243-C.
C) S-Chloro-1-[4-(imidazol-1-ylphenylcarboxamido)-2-
methoxybenzenesulfonyl]-3-spirocyclopentaneindol-2-one
A mixture containing 210 mg of the compound prepared in Example 2 and
200 mg of the acid chloride prepared in stcp B in 10 ml of DCM and 1.S ml of
TEA is stirred at RT for 1 h and then refluxed ~or 3 hours. The reaction medium is
extracted with DCM and then washed with water and an aqueous solution of
sodium hydroxide. After evaporation of the solvents, the residue is
c~romatographed on silica using a DCM/ methanol mixture as the eluent. I~e
expected product is recrystalli7ed from iso ether.
m=O.OlOg.
M.p. = 14S-C.
EXAMPLE 111
S-Chloro-1-[2-methoxy-4-(phenoxycarboxamido)benzenesulfonyl]-3-
spirocyclohexaneindol-2-one
This compound is prepared by reacting phenyl chloroformate with the
~ ~ compound prepared in Example 2.
:~:
53 2 1 ~
M.p. = 2()9-C aftcr recrystallization from iso cthcr.
EX~MPLE 112
S-Chloro-1-[4-(N-methylur~ido)-2-m~thoxybenzencsulfonyll-3-
5 spi~ocyclohexaneindol-2-one
140 ~g of th~ compouncl obtained in ~he prcvious Examplc arc mixed with
~` S ml of ethanol, S ml of DCM an~l S ml of a 33% solution of methylamine in
ethanol. After one hour at RT, the solvents arc driven off and the residue is tben
chromatographed on silica using a I)CM/MeOH mixture as the ellJent. Thc product
obtained is recrystallized f~om iso ether.
M.p. = 254C.
EXAMPLE~ 113
S-Chloro-1-(2-methoxy-4-ureidobenzenesulfonyl)-3-
spirocyclohexaneindol-2-one
A mixture containing 200 mg of the compound prepared in Example 111
with S ml of 20% aqueous ammonia, 5 ml of ethanol and 5 ml of DCM is stirred
for 1 hour at RT. After filtration of the reaction medium and evaporation of thesolvents, the expected product is crystallized ~om iso ether.
M.p. = 228-C.
lEXAMPLE 114
S-Chloro-1-[4-(N-o-tolylureido)-2-methoxy]-3-
spirocyclohexaneindol-2-one
A mixture containing 250 mg of the compound prepared in Example 2, 10
ml of xylene and 80 mg of orthotoluyl isocyanate is refluxed for 18 hours. A white
precipitate fonns and is filtered off. The reaction medium is extracted with ether
and the extract is washed with water and then chromatographed on silica using a
DCM/MeOH mixture as the eluent. The expected product crystallizes from iso
ether.
M.p. = 182~C.
EXAMPLE 115
Benzyl 4-(5-methoxy-2-oxo-3-spirocyclohexaneindol-1-yl)sulfonyl-
3-methoxybenzoate
5~ 2~73`1~
6() nlg of so~lium hy~ri(lc ~rc poured in small portions into ;~ mixtur~
containing 500 mg of 3-spirocyclohcx~nc-5 mcthocyindol-2-onc in 5() ml o~
THF. Aflcr 30 minutcs a~ RT, 8()0 mg of b~nzyl 3-mcthoxy-4-chlorosulfonyl-
benzoatc chloride arc added and the nlixture is stirrcd for 2 hours at RT. Tbe
S mcdium is concen~ratcd and taken up in AcOEt and the mixturc is washcd with
water, dned over sodium sulfate and concentratcd. The resi(lue is chromatographed
on silica using DCM as thc elucnt.
NM~ (at 250 M[Hz in DMSO):
l.2-1.8 ppm: 10H: cyclohe~yl
3.6 ppm and 3.8 ppm: 2 x 3H: 2 x VCH3
5.4 ppm: 2H: CO2-CH2-C6Hs
6.8-8.2 ppm: 11H: aromatic protons
E~AMPLE 116
4-(3-Spiroeyclohexane-5-methoxy-2-oxoindol-1-yl)sulfonyl-3-
methoxybenzoic acid
600 mg of the compound prepared in the previous Example are placed in
50 ml of AcOEt and hydrogenated at RT and atmospheric pressure in the presencc
of 140 mg of palladium-on-charcoal to give 310 mg of the expected acid, which
is recrystallized from a hexane/ ~thanol mixture (70/30; v/v).
M.p. - 210-C.
EXAMPLE 117
5-Chloro-1-[4-(N-(ethoxycarbonylmethyl)carbamoyl)-2-me~hoxy]-3-
spirocyclohexaneindol-2-one
450 mg of ethyl glycinate hydrochloride in 20 mg of sodium methylate are
placed in methanol. 200 mg of the compound described in Example 60 in Sû ml of
DCM are added and the mixture is stirred at RT for 48 hours. It is cxtracted with
DCM and the extract is washed with water, dried, concentrated and then
chlomatographed on silica using DCM/MeOH (99.5/0.5; v/v) as the eluent.
M.p. = 164-C.
EXAMPLE 118
1 -(4-Carbamoyl-2-methoxybenzenesulfonyl)-5-chloro-3-
spirocyclohexaDeindol-2-one
;
5~ 2 lO73~1g
30() m~ ot the compound dcscribcd in E~xample G0 arc mixed with 5 ml of
30% aqucous ammonia, 10 ml of cthanol and 10 ml of DCM. Aftcr 1 hour at RT,
thc mixture is conc~ntr~tcd ~md cxtsact~l with DCM and the cxtract is washed with
water, dned, concentra~ed an(l thcn chromatographed on silica USillg DCM/McOH
(99/1; v/v) as the eluent to givc 109 mg of thc cxpectcd product.
M.p. = 160~C.
EXAMPLE 119
5-Chloro-1-[2-methoxy-4-(N-(2~methoxycarbonylethyl~carbamoyl)~-
3-spirocyclohexaneindol-2-one
A mixture comprising 320 mg of the compound described in Example 60
and 2 g of methyl aminobispropionate in 3û ml of tetramethylbenzene is refluxed
for 30 minutes. It is extracted with AcOEt and the extract is washed with a 1 N
solution of hydrochloric acid, dried over sodium sulfate and concentrated. The
residue is chromatographed on silica using DCh~eOH (99/1; v/v~ as the eluent
to give 100 mg of the expected product.
M.p. = 147-C.
EXAMPLE 120
1-[4-(3-(N-Boc)aminoazetidin-1-ylcarbonyl)-2-
methoxybenzenesulfonyl]-S-chloro-3-spirocyclohexaneindol-2-one
A mixture containing 300 mg of the compound prepared in E~sample 60,
900 mg of 3-(N-Boc)aminoazetidine, 1 ml of triethylamine, 10 ml of DCM and
10 ml of methanol is stirred at RT for 1 hour. It is concentrated and extracted with
25~ ethyl acetate and the extract is washed with a 1 N solution of hydrochloric acid,
dried over sodium sulfate and concentrated. The expected product is obtained after
chrornatography on silica using DCM/MeOH (99/1; v/v) as the eluent.
M.p. = 136-C.
EXAMPLE 121
1-[4-(3-Aminoazetidin-1-ylcarbonyl~-2-methoxybenzenesulfonyl]-5-
chloro-3-spirocyclohexaneindol-2-one
A~mlxture containing 160 mg of the compound prepared in the previous
xample and 3 ml of TFA in 10 ml of DCM is stirred for 31) minutes at RT. The
35 ~ ~ reaction medium is concentrated and crystallized from iso ether and the crystals are
flltered off~and dried. The product obtained is dissolved in 10 ml of water and then
.: .
: :
~6 21~7~
I() ml o~ I N sodillm hyclro,~ide; thc SOIutiOll iS c~tractcd with DCM ansJ thc
c.l~tract is washcd with wcltcr, clric(l ovcr so(iium sulfatc and concelltratccl. Thc
cxpcct~i product is obt~nllccl aftcr chrom~tography Oll silica using DCM/McOH
(96/4; v/v) as the clucnt.
S M.p.- 14SC.
EXAMPLE 122
S-Fthoxy-1-[4-(3-dimethylaminopropoxy)-3-
methoxybenzenesulfonyl~-3-spirocyclohexaneindol-2-one hydrochloride
A) S-Ethoxy-1-[4-(3-hromopropoxy)-3-methoxybenzellesulfonyl~-3-
spirocyclohexaneindol-2-one
A mixture containing 0.5 g of S-ethoxy-3-spirocyelohexaneindol~2 one,
S ml of THF and 0.07 g of sodium hydride is stirred at 20'3C for lS minutes, 1.65 g
of 4-(3-bromopropoxy)-3-methoxybenzenesulfonyl chloride are then added and
~he resulting mixture is stirred for 20 hours at Rl'. It is concentrated under vacuullI
and exsracted with etheF and ~he extract is washed with wa~er and then a 10%
solution of sedium carbonate. The expected product crystallizes from pentane andis then r~crystallized from iso eth~r.
M.p. = 114-118~C.
B) S-Ethoxy-1-[4-(3-dimethylaminopIopoxy)-3-
methoxybenzenesulfonyl~-3-spirocyclohexaneindol-2-one hydrochloride
The compound obtained in the previous step is mixed with 7.5 g of a 33%
solution of dimethylamine in ethanol and placed in 10 ml of THF. After stirring for
2S 3 hours, the mixture is concentrated under vacuum and taken up in 1() ml of water
and the resulting mixture is extracted with ether. The ether phase is treated with 20
ml of 2 N hydrochloric acid, after which solid potassium carbonate is added to
render the solution alkaline to pH 9. The oil whieh precipitates is extracted with
DCM. The expected product crystallizes from ether.
M.p. = 135~138-C.
EXAMPL~ 123
1-[4-Aminosulfonamido-2-methoxybenzenesulfony]-S-ehloro-3-
spirocyclohexaneindol-2-one
0.3 g of the compound prepared in Example 2 is placed in 4 ml of DCM in
the presence of O.S g of TEA, and 0.3 g of aminosulfonyl chloride, prepared
21073~
accordin~ to Chem. Ber.~ 1958, 91~ 1339-1341~ is a(ldcd. Aftcr stirring for 2 days
at RT, the mcdium is conccntnatcd undcr vacuum and cxtracted with ether and the
c~ract is washed with watcr. After drying, thc rcsidue is chromatographcd on
silica using DCM alld thcn AcOFt as thc clucnt to give thc cxpccted product~
S which crystallizes from ethcr.
M.p. = 205-208C.
E~fAMPLES 124 and 125
1 -(4-Dimethylamino-2-methoxybenzenesulfonyl)-5-methoxy-3-
10 spirocyclohexalleindol-2-one and 1-(4-methylamino-2-
metho~ybenzenesulfonyl)-5-methoxy-3-spirocyclohexaneindol-2-one
50~ mg of 1-(4-amino-2-methoxybenzenesulfonyl)-5-methoxy-3-
spirocyclohexaneindol-2-one are mixed with 1 ml o~ a 37% aqueous solution of
formaldehyde, 10 ml of ace~onitrile and 430 mg of sodiunn cyanoborohyd~ide, and
15 û.12 ml of acetic acid is then added. The temperature of the medium rises and the
medium is cooled in an ice bath. Two products of different polarity are formed in
succession. 1 ml of an aqueous solution of fonnaldehyde, 300 mg of sodium
cyanoborohydride and 0.12 rnl of acetic acid are added to the medium. The
mlxture is stilred for 1 and a half hours, poured into iced water and then extracted
20 with AcOFt. ~e extract is washed with water, dried and roncelltrated to give 2
products, which are separated by chromatography on silica using DCM/AcOEt
(98i2; Viv) as the eluent.
M.p. = 210-C (Ex. 124).
M.p. = 170-C (Ex. 125).
:~ TABLE 5
S 2 (I)
R5
:
~ R6
:
, . ~ .
5~ 2~073~8
Unless indicatcd othcrwis~, th~ substitucnt R6 is in thc 4-position ~n~l m = 1.
... _.. _ ~ ,,
Ex R1 R2 R5 R6 M.p. C
~ . . ~ . .
126 Cl H 2-MeO~N--~ 210
127 Cl H 2-MeOMeO~CON 192
2-MeO /~ CON~I -
12~ Cl H ~CF3 188
129 Cl H 2-MeO/<~CONH - 146
;~ OMe
; 130 Cl H 2--MeO /~CH2CONH 190
Me
131 Cl H 2--MeO /~-CONH- 147
OCOCH3
132 Cl H 2-MeOCH3CONH- 230
2--MeO Me
33 ~ Cl H ~/~ CONN- ~S
134 Cl H 2-MeOHO2C(CH2)2- 2ûS
CONH-
:
: :
S~ 2~073~
135 Cl H H ~ ~ ~ 180
136
137 ~Cl H~ 2-MeO ~ C~N~-
138 MeO H2-MeO /~MCONH- 245
139 MGO H2-MeO ~CONII-
140 Cl H~ 2-MGO I /~CON~
141 Cl H2-MeO Me~ 140
142 Cl H2-MeO Me ~CONH 225
MGO H2 MeO ~lGocH2coNH- 161
144 ~MeO H2-MeO tBuCH2CONH- 209
'
~,o 21~73 ~3
145 1 ~tO ~ 2-M~0 <~ CON~- 223
Me
146 EtO H 2-MeO Mc \ 136
, N~fl2CONH
147 Cl H 2-MeO Me \ 226
/ N-CONH-
Me
148 CH30 H 2-MeO Me \ 190
/ N-CONH-
: ~ 149 EtO H 2-MeO Me \ 192 / N-CC)NH
Me
150 EtO H 2-MeC) Me ~ 160
Et/ N-C~NH-
151 EtO H 2-MeO N-CONH- 168
: Et /
~: 152 EtO H 2-MeQ Me \ 137
: : ~ / N-CONH-
, Pr
:~ : 153 Cl H 2-MeO /~\ 157
MeCONH \ N~
:154~: Cl ~ H 2-MeO \ N~CH2)2-NHC 163
: ~ Me
i55 Cl H 2-MeO Me\ 192
~ .
::~: :: ~ :
:
::~ ` ~ :
,
fi' 21 ~73 ~ ~
156 Cl H 2-McO M~N so 231
157 Cl H 2-McO H ~.06
158 Cl H 2-MeO /~ 226
Me -N\ JN -S 2 ~
159 Cl H 2-M[eO Bz' 117
160 MeO H 2-MeO 2~- 188
161 Cl H 2-MeO BzOCO- NMR
162 ~ Cl H ~ 2-MeO ~ ~Nl~CO~
163 MeO H 2-MeO NH2- 188
164 MeO H 2-MeO MeO- 172
165 MeO H 2-MeO /~o-CO- 162
166 ~ McO H 2-McO ~NHCO 198
167 EtO H 2-MeO ~I2N- 177
168 ~ MeO ~6-MeO 2-MeO MeO- lB3
6~ 21~73~8
169 EtO H 2-McO /\r~- NHCO- 15()
= Mc
170 EtO H 2-MeO BzOCO- 135
171 EtO H 2-MeO HOOC- NMR
172 3EtO H 2-MeO MeNHCO~ 239
173 EtO H 2-MeO S-MeO 131
174 EtO H 3-MeO MeO- 127
175 EtO H 2-MeO / 167
Me
176 EtO H 3-MeO 4,5-di-MeC) 130
177 EtO H 2-P~eO\/r \~ NHco- 195
~e
178 EtC) H 2--MeO/~\~cH2 NHCO 168
179 EtO H 2-MeO N20 160
180 EtO H 2-Me MeO 176
181 EtO H 3-McOCH2=cH-cH2o- 130
182 CF30 H 2-MeO MeO 127
::
~)3 21Q73~8
183 EIO ~ 2 MCO M~; 171
CHN H CO
184 EtO H 2~MCO EtOCOCH~NHCO 203
185 EtO H 2-MCO /~O~O-NH- 181
186 EtO H 2 MeO 4 5-d;-MCO 136
187 EtO H 2 Me 4-MeO 5-Cl
129
188 EtO H 2-MCO B~ N NHCO- 188
189 EtO H 2 M~O HO(C~2)2-NHCO 157
190 EtO H 2- H
117
MeOCO
191 EtO H 2-MeO Me\ 212
N~CH2)3~ -
~; ~ HCl
192 EtO H 2-MeO '~lLcoNH-
181
193 EtO H MeO- Et \
206
CH~ONH-
Et
194 EtO H 2-MeO BzOCOCH2NHCO
NMR
l 95 ~M
.
fi~ ~lO~f3~8
196 EtO H 2-MeO ~>~.OCO- 152
197 EtO H 2-McO Et / N~O 148
198 EtO H 2-MeO \ N~S 128
199 EtO H 2-MeO CN CH2NH-CO- 232
200 EtO H 2-MeO EtO2C~H2~O NMR
201 EtO H 2-l!~eO H02C-C~I2NH-CO- 137
202 Cl H 2-MeO ~Et)2N-CO-NH 194
203 EtO H 2-MeO C}CONH- 214
204 EtO H 2-MeO H2N(CH2)3O- 136-140
205 EtO ~ 2-MeO~H3)3N(CH2)3o- 145-~50
206 C6HsO H 2-MeO 4-MeO 130
: 207 E~O H 2-MeO O- CS~H- 210
; ~ ~ 208 DO H 2-McO `CH CON- 138
:
~: :
fiS 21073~
Thc NMR spectra arc run in DMSO at 200 MHz.
NMR of Examplc 161:
1.3-1.8 ppm: 10H: cyclohexyl
3.5 ppm: 3H: OCH3
5.3 ppm: 2H: O-cH2-c6Hs
7.2-8.2 ppm: 1 lH: aromatic proeons
NMR of Example 171:
1.15 ppm: 3H: CH3
1~19-2 ppm: 10H: cyclohexyl
3.6 ppm: 3H: OCH3
4 ppm: 2H; OCH2-CH3
6.7-8.2 ppm: 6H: aromatic protons
;
NMR of Example 200:
2.2 ppm: 16H: cyclohexyl + 2CH3
3 ppm: 3H: NCE~3
4-4.4 ppm: 6H: aromatic prots3ns
6.8-8.2 ppm: 6H: aromatic protons
A resolution of the signals is observed; this is associated with the amide
: : isomerism.
EXAMPLF 21()
1-(4-Benzyloxy-2-methoxybenzençsulfonyl)-5-ethoxy-3-
:spirocyclohexaneindol-2-one
A) Potassium 4-benzyloxy-2 methoxybenzenesulfonate
3U This preparation is carried out according to K. Ho~ann et al., Liebigs Ann.
Chem., 1982, 282-297.
10.5 g of 4-benzyloxy-2-methoxybenzene are mixed at 5-C with 30 ml of
DCM, and 8 ml of tnmethylsilyl chlorosulfonate in 30 ml of DCM are added over
15 minutes at a temperahlre between 5 and 10~C; after stirring for 15 minutes, 50 g
; :
-~ .
()6 2 1 ~ 7 J !l ~
of icc are a(ldcd. Thc mixtllrc is washed with ethyl cthcr, treated with potassium
hy(lrogcllcarbollatc ~nd thCIl conccntr~tc(l un(ler vacuum. After dryillg, the rcsidue
is takcn up in lS0 ml of mcthanol. Thc insolublc matcrial is filtercd off ~t thc boil
and the c~pecte~l compound thcn crystallizes at 5-C.
M.p. ~ 300-C.
The st~ucture of thc compound is con~irmed by analysis of the NMR
spectrum.
B) 4-Benzyloxy-2-methoxybenzellesulfonyl chloridc
2.8 g of the compound prepared in the previous Example are mixed with 30
ml of POCl3 and the mixture is r~fluxed for 3 bours. It is concentrated under
vacuum, treated with 20 g of ice and extracted with ethyl ether and the extract is
washed with 30 ml of N sodium hydroxide and then water. The medium is
concentrated and the oil obtained is then triturated in 30 ml of iso ether. The
expected product (0.7 g) crystallizes.
M.p.=95~C.
C) 1-(4-Benzyloxy-2-methoxybenzenesulfonyl)-5-ethoxy-3-
spirocyclohexaneindol-2-one
l'his compound is prepared by the usual procedure. It crystallizes from iso
ether.
M.p. = 135-C.
~e structure of the compound is verified by analysis of the NMR spectmm
in 2 dimensions (NOESY: Nuclear Overhauser lEf~ect Spectroscopy).
The compound of the next Example is subsequently prepared by
debenzylation.
EXAMPLE 211
5-Ethoxy-1 -(4-hydroxy-2-methoxybenzenesulfonyl)-3-
spirocyclohexaneindol-2-one
M.p. = 209-C.