Note: Descriptions are shown in the official language in which they were submitted.
WO93/21010 21 181 15 PCT/US93~0~70 ~,
,'
Le~ses With
~igh Impact Resistance and-~igh Scratch Resistance
This invention is in the area of composit~ lens
structures, and in particular is a polymer-polymer composite
lens structur_ exhibiting high impact resistance and high
scratch resistance. This in~ention also includes fast curing
polymeric composltions that are suitable for casting into
ophthalmic lenses, lenses prepared from these compositions,
and apparatus for the production of lenses. ~-
Bac~ground of the In~ention
The invention disclosed herein addresses two `
problems in the area of the production of ophthalmic lenses.
The first problem is the need to pro~ide a lens structure
that has both high impact resistance and high scratch
resistance. The second problem is the desire on the part of
retail eyewear outlets to be able to produce plastic lenses
on-site for customers, instead of merely grinding plastic
lens blanks to a desired prescription. The art in these
areas are considered below in order.
~,
High Scratch Resistant, High Impact~Resistant
Ophthalmic Lenses -- - ~
Oph~halmic (prescription) glasses and both
prescription and nonprescription sun glasses ha~e `
traditionally been prepared using inorganic glass as the
lens material. Recently, organic polymers ha~e been ~`
introduced as an alternative lens material. Currently, both
inorganic glass and organic polymers-p~ay a major role in the
prescription and nonprescription lens market. ;~ ;
Glass is considered a pristine optical-quality
3~ material and is extremely scratch-reaistant. However, glass
is heavy and is easily shattered. Tempering (either by
thermal or chemical processes) improves the impact
resistance, but at the expense of lengthening process time
and escalating production COSt. Plastics perform better ln
impact resls~ance, and are lighc weight. However, ehe
WO93/21010 2 ~ P~T/US93/0~70
~ scratch resistance of plastics is inferior to glass. A
marriage of the two in the form of a ~ront plano glass wafer
with a back polymer layer has been used as a means to obtaln
the desired qualities of both materials. The outer glass ~.
layer is convex outward, where the majority of the scratch '
"incidences" would normally occur. Hence the structure
offers protection against scratch. The inner plastic layer
enhances the overall shatter resistance of the composite. '
The glass-plastic interface has to be chemically coupled to
ensure strong adhesion over the complete service temperature
range (from sub-zero to above standard room temperature). A
significant disadvantage of the glass/plastic composite is
that firm attachment of the two, especially in a stress-free
and defect-free manner, is exceptionally difficult. Further, , -
the inherently divergent thermal expan~ion properties of j
glass and most plastic materials make such composites prone ¦
to thermoelastic stresses and potential failures during !
thenmal~cyc~ling asd ~shocks.
;In~general, scratch resistance and impact resistance ¦
20~ is~ difficult~to attain in the same polymeric material. The I ;
~'5,'''"~ former~attribute~requires~a hard material with great internal
cohesive energies, while the latter requ~res an elastomeric
behavio~r ~i.e~., elastic~ity~under sharp impulse stress loading
conditions)~ It is~èxtrèmely~difficul~t, if not impossible, !:
~'25~ to opti'jmize both~s~cratch~resistance and impact resistance in
one;materi-al. - ~
U.S.~Patent`No~. 4,544,572 to Sandvig, et al., ~ -
'~ discIoses a polymeric ophthalmic lens that has a thin (50
m~c'rons or less)~ abrasion-resistant polymeric coating. The~ ' `
'30- ~ lens~is prepared by~applying a layer of a composition
-comprising a maeerial that contains ethylçnically unsaturated .
: groups oo~~he~first face of a lens mold, reacting the
composi~tion to a dry film, filing the mold with an organic
mat~erial capable~of solidification, and then hardening the
3~5~ orgànic material.~ This process is time consuming in that the
:first layer must be partially cured before injection of the
back layer hardenable material. As stated in the patent, it
. ~ , .
WO93/21010 2118 ~ PCT/US93/0~70
can take up to 16 hours to prepare ane lens. Further, this
method does not provide a means to impart contour shaplng tO
the front layer.
There remains a need for a lens material that has
high impact resistance and high scratch resistance, and that
does not exhibit thermoelastic stress and failure during
thermal cycling and shock. There is also a need for a
process for the preparation of a lens composite with high
impact resistance and high scratch resistance ~hat does not
impart significant stress and/or defects into the material
during adhesion of the layers. Further, there is a strong
need to provide a method to prepare high impact resistant,
high scratch resistant lenses that can be accompli~hed in a
relatively short timeframe, and that provides a means to
impart contour shaping to the front wafer of ~he lens.
Rapid Preparation of High Ouality Plastic Ophthalmic Lenses
Many of the plastic ophthalmic lenses sold today by
optical dispensers such as retail èyewear outlets are made by
machining the desired prescription into the back face of a
semi-finished lens blank made from diethylene glycol
bis(allyl carbo~ate) resin, also known as CR-39. These
blanks are manufactured off-site by casting the starting
monomer for CR-39 between a set of glass moids held together
by a flexible gasket and restraints. The mold assembly is
initially heated in an oven using a precise:cure schedule.
During the subsequent polymerization step,- the liquid resin
is converted into a glassy solid. Shrinkage- of up to 16
percent of the ma~erial occurs during polymerization and
cro~slinking. The molds must be designed to account for the
shrinkage, so that the lens blank has`the desired front
curvature. The complexity of de3ign--ls~:increased if, instead
of a semi-finished lens blank, a finished~lens is desired in
which both the front and back surfaces have defined
curvatures. Another disadvantage in preparing CR-39 lenses
is that they require cure schedules of as long as sixteen
hours.
WO93/21010 2 1 1 8 1 1 5 PCT!USg3/0~70
--4--
Casting lenses from polymerizable compositions on- '
site would be preferable to a retail eyewear outlet over
machining lens blanks if problems associated with shrinkage
of the polymerizable material during casting and the long
cure time could be solved. One advantage of casting on-site
is that the equipment needed for casting is less expensive
than the lens generators and polishing instruments used in
lens machining. Second, the casting process is cleaner and
generates less waste than the machining process. In
addition, the cost of the finished lens to the eyewear outlet ~`
using a casting process may be lass than that when the lens '
is prepared by machining a lens blank, particularly for ~-'
aspheric, multifocal, and progressive lenses. -
CR-39 is unsuitable as a material for casting into
lenses in one hour processing laboratories because of its ~`
slow reaction rate. It would be of great benefit to have a
materia~l that~maintains~most of the desirable properties of I '
CR-39,~such~as good abrasion resistance, chemical resistance, ?
mpact resistance, clarity and generally.superior optical
~20 properties, yet~polymerizes in a short amount of time. It
would~also~be~of~ben fit to have an apparatus that can be s;
used~to~p ~ e~lenses on-site in a short amount of time. '~`
Urethanes~have been used in coatings for ophthalmic
lenses~ U.S.~Patent~;NQ. 4,800,l23 to ~oekeler discloses a ''
'~25~ scratch-~resistant coating~prepared from a po}ymerizable
co'mposition~that~ includes~at~least one polyfunctional monomer
having~three~or more-~a ~ l~oloxy groups per molecule, and at
~ lea-t onQ~N-vinyl;imido~group containing monomer. U.S.
'~' Patent No. 4,435,450`to'Coleman discloses a method for
3,0 ~ applying~ abra,sion~resistant thin polyurethane coatings that `'
includes forming a hydroxy-ter~inated prepolymer which is ~"
subs-quent1y-cross~linked using a relatively non-volatile
triisocya~n~ate,~and appl-ying the material by flow coating onto
a glass or lens. '-~'-~~ `~'
35~ ` U.S.~ Patent;No~ 4,912,185 to Toh discloses a cross-
~ linkable casting composition for ophthalmic lenses that
'?,"'~ inc~ludes (~A) a polyoxyalkylene glycol dimethacrylate or
: " ~
?
SUBSTITI.JTE SHEET - ```
21~
WOs3/210l0 PCT/US93/0~70
-5-
diacrylate, (B) at least one polyfunctional cross-linking
agent, and (c) up to 40~ by wPight of a uret~ane monomer ;~
having from two to six terminal acrylic or methacrylic
groupS~ ~rpolymeriZable composition disclosed in the '185
patent was designed to be used in conjunction with
traditional methods for the preparation of ophthalmic lenses,
wherein the polymerizable solution is poured into numerous
molds, cast with blanket radiation, heated and then removed.
The method of casting requires the use of a low viscosity
polymerizable solution to minimize the problems that result
from air entrapment. The '185 p~tent states that the
viscosity of the polymerizable solution should not exceed
approximately 200 cps at 25C. The polyoxyalkylene qlycol
diacrylate or dimethacrylate, present in the polymerizable
composition in a range of 40 to 60% by weight, functions as a
viscosity reducing agent for the composition. The
polyoxyalkene moieties are based on ethylene oxide or
propylene oxide repeating units, with ~; to 11 alkylene oxide ! ,
repeating units preferred, as shown below.
O o O o
CE~ -CHC0-~CH~CH20)~-CCH-CH2 CH2~CHCO-(CH2C~CH~O)o-CCH~CH~
ethylene oxide based system propylene oxide based system
Methacrylate terminated polyoxyalkylene glycols are
preferred over acrylate terminated polyoxy~lkylene-glycols in
the '185 patent because they have lower reactivities than the
acrylate counterparts, which, using the traditional casting
process, reduces surface aberration and intern~al stress. The
patent indicates that this composition can be fully cured by
two to four passes under a W lamp followed by one hour of
heat treatment at 100 degrees C.
Japanese Patent No. 61064716 (Chem. Abstract
105:192198b) discloses an impact resistan~ optical resin
prepared by polymerizing acrylate or methacrylate, adducts of
monoepoxide and brominated bis-phenol, poly-isocyanate and
SUBSTlTUTE SHEET
21l~
WO93/21010 PCT!US93/0~70
other unsaturated compounds such as styrene or
divinylbenzene.
An advance ln the art of polymerizing shrinkable
materlals is disclosed in U.S. Patent Nos. 5,114,632 and
5,llO,514 to David S. Soane. Briefly, polymerizable material
is introduc~d b~tween two mold halves, one of which is, or
both are, constructed of a material that transmits energy,
either thermal or W . Stress related voids in the polymeric
material are eliminated by causing the partially polymerized
material to polymerize in a differential fashion along a
mo~ing front, so that the material ahead of the moving
polymer zone r^mains liquid, and the material that the front
has passed is solidified. In a typical method, the moving ~;~
front is a slit through which W or thermal energy is
lS transmitted. The still-liquid material ahead of the moving i
polymer zone can then flow freely, at a rate that equals the ¦ ~;
rate of shrinkage, and a void-free, reduced stress polymeric
network is produced. Using this process, lenses can be cast : ;
in a way to prevent cavitation, or voids caused by the ¦ ~.
shrinkage of material during polymerization. This method is
referred to below as l'sequential polymerization. Il :~
Accordingly, it an the object of the present
invention to provide a polymerizable composition that can be
polymerized into a lens that maintains or exceeds the
mechanical and optical properties associated with CR-39, yet
has a faster cure rate- than-CR-39.
It is still another object of this invention to
provide a polymeric material that can be sequentially
! polymerized in~to a finished product in less than one hour,
preferably less than thirty minutes.
It is another object of the present in~ention to .
provide a polymerizable ~omposi~ion for the production of
high quality ophthalmic lenses that is suitable for u e in .
combination with the apparatus and method for sequential
polymerization disclosed in U.S. Patent Nos. 5,llO,514 and ,
5,114,632. 1~ ;
2 ~ '3
WO93/2101~ -7- PCT/US93/0~70
It is still another object of the present invention
to provide an apparatus for the production of lenses usinq
the sequential polymerization method. ---
It is also an object of the present invention to
provide a lens material with high impact resistance and highscratch resistance.
It is another object of the presant invention to
develop a composite lens that can be prepared in a short
process time, and preferably, on site, at an eyewear outlet.
It is another object of the present invention to
provide a lens with high impact resistance and high scratch
resistance that does not have si~nificant internal stresses
or defects.
It is a further object of the present invention to
provide a lens material that does not exhibit thermoelastic
stress and failure during thermal cycling and shock.
It is another object of the present invention to
provide a pretinted or pretintable composite lens wherein the
front and back materials can be impregnated with different
dyes.
It is another object of the present invention to
provide a composite lens that incorporates progressive or !
multifocal prescription features while the overall exterior
contour remains smooth.
_
~u~ary of the Invention
~ .
In one embodiment, the invention is a composlte lens
that includes a front scratch-resistant polymeric wafer and a
back impact-resistant polymeric layer. The unique structure
~ allows maximal design flexibility, and is easily and
relatively quickly manufactured. The design allows
simultaneous optimization of impact and scratch resistance.
It also provides other desirable features such-as-easy pre-
tin~ing, uniform or controlled ~radient, coloring~, and the
possibility of built-in anti-reflective characteristics.
The lens structure disclosed herein can be
manufactured quickly and easily on site, for example, at an
9UUBST~UTE SHEEl
WO93/21010 2 1 1 8 1 1 5 PCT/US93/0~70
-8-
eyewear outlet, ~y polymeri~ing or otherwise adhering the
back impact resistant layer onto a premanufactured front
wafer. The front premanufactured wafer can be made with any
desired contour on the inner concave surface (the surface ~;
that interfaces with the convex surface of the back layer). ~
The composite lens can incorparate progressi~e or multifocal ;~-
prescription features while the overall exterior contour
remains smooth. The progressive multifocal corrections are
afforded by elaborate internal interface contour design. ~
Either layer, or both layers, of the polymer-polymer :`-
composite lens can be pretinted as desired, witX the same or
different dye, in the same or different amounts. In one
embodiment, one layer is pre-loaded with the desired
dyestuff, and the other used to fine-tune the shade and
color.
In another embodiment, a front premanufactured layer ¦
can be used that has an anti-reflective coating.
The invention also includes a polymerizable 'I
composition, and the polymer formed thereby, that is useful I -
as ~he impact resistant or scratch resistant material in the
poIymer-polymer composite lens, or alternatively, can be used
alone as a fast-curing material for`the preparation of
plastic lenses on-site by a commercial retail eyewear outlet.
The polymerizable composition includes: ¦
a) between 20 and 90 weight percent, and
-- preferably, at least 50- weight percent, of urethane, epoxy,
or polyester oligomers end~terminated with acrylate or
methacrylate (or mixtures of acrylate and methacrylate);
j b) -between 5 and 50 tO 80 percent, preferably
between 10 and 40 percent,-by weight of an optional diluent,
such as a hydrocarbon diol end terminated with acrylate or
methacrylate, or mixtures thereof, or a crosslinkable tri-,
tetra-, or poly- acryIate or methacrylate, or mixtures
thereof; and
c) conventional optional additives, including but
not limited to free radical initiators, W absorbers, mold
211~
WO93~1010 PCT/US93/0~70
_9_
release agents, stabilizers, dyes, antioxidants, and wetting
agents.
This polymerizable composition can be cast using W
radi~tio~ ~o produce an optically transparent object with low
haze that has impact and abrasion resistance approximately ;~
equal to or better than CR-39. In one embodiment, the
polymerizable composition has a viscosity of greater than 200
cps . `~
In a preferred embodiment, this polymerizable
composition is cast using the sequential polymerization
method, as described in more detail below, in a time ranging
from lO minutes to 30 minutes depending on the polymerizable
composition, initiator concentration, and W intensity
employed. Relatively high viscosity polymerizable solutions
can be cast usin~ the sequential polymerization method since
the fluid can be introduced into the mold cavity without
entrapping air using a procedure such as that illustrated in
Figure 2. The ability to use high viscosity polymerizable
solutions allows flexibility ïn choosing the kind and
concentration of monomer and oligomer. Higher oligomer
content can be used to impart superior impact resistance to
the lens. Diluents that have high functionality (and thus
viscosity) can be used to impart superior abrasion
~-~ resistance.
In par~icular, urethane, epoxy, or poLyester
acrylate or methacrylate oligomers (or mixtures thereof) are
selected that impart desired abrasion and impact resistance
to the lens and reduce the amount of shrinkage tha~ occurs
during polymerization, because the ratio of non-reacting to
reacting components is high. These oligomers have a
relatively high viscosity, typically between one-a~d one
hundred megapoise at room temperature when undiluted~
Therefore, these oligomers were not appropriate for use in
the traditional manufacture of lenses using blankat
radiation, in morè than minor amounts. For example, U.S.
Patent No. 4,912,185 to Toh indicates that tetraacrylic
urethane monomers can be present in the polymerizable
SUBSTITUTE S~EET
WO93~21010 2 118 115 PCT/US93/0~70
- 1 o ~
composition for a lens using classical technology at up to 40
percent by weight of the composition. In the polymerizable ;
composition disclosed herein, the urethane and/or epoxy
acry~an~r~sr~methacrylate oligomers is preferably at least 50% `~
by w~ight of the polymerizable composition.
A diluent such as a hydrocarbon diol diacrylate or
dimethacrylate is included as necessary for viscosity 1~-
red~ction, so that the solution can be cast between molds. ~-
The dilùent can also impart desired mechanical properties to
the final product, such as hydrophobicity and abrasion
resistance. Since the diluent p~rticipates in the
polymerization rPaction, no solvent is evaporated. The
diluent has a significantly lower molecu}ar weight (typically `
less than 600) than the oligomers (400-9000 weight average
molecular weight), and therefore shrinks more on a per-volume
basis during polymerization. Typical concentrations of the
diluent in the polymerizable composition are less than 50% by
weight, preferably, between lO and 40% by weight. ,~
The polymerizable compositions can also be used in
2~ the preparation of materials other than ophthalmic lenses,
such as plastic and glass laminates and specialty optics or 1i
lenses.
This invention also includes an apparatus that can
~e used for the preparation-of ophthalmic lenses in retail
eyewear outlets, using the-sequential polymerization method. ; `
- Bri-f D-scr~p ffon-of the Figur-s
Figure l is a schematic side cross sectional view of
a first embodiment of a carriage system for use in the
sequential polymerization of~ a polymerizable composition into -
an ophthalmic lens. _
Figure 2 is a schematic side cross sectional view of
a portion of the carriage system embodiment of Fig. 1,
illustrating the proced~re for syringe filling of the lens
mold.
SUBSTITUTE SHEET
`.
WO93/21010 ~ ~i 81 1 ~ PCT/USg3/0~70
Figure 3 ls a schematic slde cross sectional view of
another portion of the carrlage system embodiment of Fig. l,
with lens mold rotated 180 degrees, positioned ln front of a
movable W source.
Figure 4 is a schematic side cross sectional view of
an apparatus for the preparation of a polymer-polymer lens
compo~ite.
Detailed De-acription of the I~e~tion
As used herein, the term ~hard monomer" or ~hard
material" refers to a monomer or material that polymerizes to
form a polymeric material that is below its glass transition
temperature at the temperature of use (typically room
temperature).
As used herein, the term "soft monomer" or "sof~
moiety" refers to a monomer or moiety that on polymerization
~orms a material that is above its glass transition .
temperature at the temperature of use (typically room
temperature).
As used herein, the term "aryl" refers to phenyl,
phenyl subst1tuted with alkyl or halogen, naphthalene or
naphthalene substituted with alkyl or halogen, or hig-her .- j
aroma~ics, either unsubstituted, or substituted with alkyl or
halogen. .
As used herein, the term "alkyl acrylate" refers~to- :
H C=CHCO2R, wherein R is a s~raight, branched, or cyclic alkyl :
group, preferably Cl to C~0, and specifically includes methyl,
ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl, pentyl-i
cyclopentyl, isopentyl, neopentyl, hexyl, isohexyl, .
cyclohexyl, 3-methylpentyl, 2,2-dimethylbutyl, and 2,3- _ .
dime~hylbutyl, and other longer chain homologues.
As used herein, unless otherwise indicated, the term
alkyl refers to a straight, branched, or cyclic alkyl group,
preferably C, to C20, and specifically includes methyl, ethyl,
propyl, isopropyl, butyl, isobutyl, t-butyl, pentyl,
cycloven~yl, lsopentyl, neopen~yl, hexyl, isohexyl,
WO93/21010 21 1~ PCT/US93/0~70
-12-
cyclohexyl, 3-methylpentyl, 2,2-dimethylbutyl, and 2,3-
dimethylbutyl and other long chaln homologues.
As used herein, the terms diacrylate and
dimethacrylate include mixtures of acrylate and methacrylate.
As used herein, the term (meth)acrylate refers to
either acrylate, methacrylate, or a mixture of acrylate and
methacrylate.
As used herein, the term "alkyl methacrylate" refers ;~
to H~C=C(CH3)CO~R, wherein R is a straight, branched, or cyclic :
- 10 alkyl group, preferably C~ to C20, and specifically includes
methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl,
pentyl, cyclopentyl, isopentyl, neopentyl, hexyl, isohexyl,
cyclohexyl, 3-methylpentyl, 2,2-dimethylbutyl, and 2,3-
dimethylbutyl, and other long chain homologues.
As used herein, the term "aryl" or "aromatic" refers ¦
to phenyl, phenyl substituted with alkyl, halogen,
naphthalene or naphthalene substituted with alkyl or halogen,
or higher aromatics, either unsubstituted, or substituted
with alkyl or halogen. ¦
The ~erm aralkyl refers to an aryl group with an
alkyl substituent.
The term alkaryl refers to an alkyl group that has
an aryl substituent. - j
The term alkenyl, as referred to herein, and unless
otherwise specified, refers to a straight, branched, or
cyclic (in the case of C5 ~) hydrocarbo~ of C2 to C20 with at
least one double bond.
As used herein, the term oligomer refers to a
compound with repeating units, of weight average molecular
weight ranging from 400 to 9000, and preferably, between 800
and 2500. _
. _ -
As us~=~ herein, a diluen~ is-a compound that is used
to reduce the viscosity of a material, and typically has a
viscosity of less than 600 CPS, and preferably, less than 150
cps at room temperature.
As used herein, the term aliphatic refers to an
alkyl, alkenyl, or alkynyl group of Cl to C~0.
WO93/21010 211 8~ 13- PCT/US93/0~70
As used herein, the term "chain polymerization"
refers to a polymerization process in which there is a series
of reactions each of which consumes a reactive particle and
produces another, similar particle. The reactive particles
S are radicals, anions, or cations. The polymerization of
reactive particles occurs without elimination of a small
molecule (as in during a typical condensation reaçtion).
Chain polymerization is typically carried out with
ethylenically unsaturated monomers.
As used herein, the term "monomer" refers to the
small reactive molecules that are subsequently joined
together to form a polymer.
As used herein, the term "unsaturated hydrocarbon
polymer" refers to a polymer that consists essentially of
carbon and hydrogen atoms, and that includes alkene (vinyl)
groups in the polymer.
As used herein, the term "oligomer" refers to a
polymer with 20 or less repeating units.
As used herein, the term "high impact resistant
material" refers to a material that will.withstand sudden
imposition of sudden force without fracture, and that passes
the U.S. Food and Drug Administration's requirement for
impact resistance for ophthalmic lenses (the standard drop-
ball test).
As used herein, the term "high scratch resistant"
material refers to a material that will withstand abrasion
without substantial surface deterioration. A typical "
abrasion test consists of applying a known vertical pressure~ - -~``
on the sample, while moving a steel-wool pad laterally
against the surface. Visible scratches are then counted
! after, a number of rubbing movements. A high scratch
resistant material will exhibit only a few scratches after
this process.
. .
I. Polvmer-Polvmer Lens Com~osite
:
The invention as disclosed includes a polymer-
polymer lens composite that exhibits superior impact and
8~)B5TITUTE SHEET
WO93/21010 ~ llS PCT/US93/0~70
-14-
scratch resistance. The front wafer ls a very hard,
scratch-resistant polymeric material, and the back wafer is a
high impact resistant polymeric material. Since the thermal
expansion properties of most plastics are similar,
thermoelastic stress of the polymer-polymer composite lens is
greatly reduced over a glass-polymer composite. Even if
minor stresses are induced by extreme temperatures, polymers
seldom fail catastrophically by virtue of their tendency to
yield, i.e., elastically deform (or even plastically deform).
In the case of the polymer composites disclosed herein, the
strain field accompanying the thermoelastic stress field
(both being time and temperature dependent) almost never
exceeds the ultimate strain (failure limit) of either the ;~
front or the back material. This critical distinction
between glass-polymer and polymer^polymer lenses is a key
aspect of this invention. ! `
By separating the refinements into front wafers and
back layers, individual attributes including cost, -
transparency~ hardness for the front wafer, impact resistance
for the back layer, machinability, tint, grindability, and
refractivity-dependent interface design can be optimized.
The front polymeric wafer can be mass manufactured
with uniform thickness at low cost, by either resin transfer
molding or casting (for thermosets) or injection molding (for
thermoplastics), using known procedures.
The back layer of the lens comp~site must be rigid
and machineable (for grind-polish). However, it must also
exhibit significant elastomeric characteristics in order to
!: ~endow the composite structure with adequate impact
resistance. It has been discovered that a marshmallow-
toothpick model super polymeric network, as disclosed in more
detail below, is an ideal material for tl~e~b:ack layer. Many
other block copolymers, interpenetrating networks, graft
copolymers, random copolymers, and even homopolymers that are `
elastomeric in nature yet sufficiently rigid, can be selected
as well, as long as they are sufficiently optically
transparent and dimensionally stable.
WO93/21010 211(~ ~ 15 PCT/US93/03470
-l5-
The use of a polymer-polymer lens composite provides
a number of advantages other than individual optimization of
properties of the front and back wafers and ease of
fabr'ication.
The polymer-polymer composite structure can be made
such that only the front layer or the back layer is colored,
or alternatively, both layers are colored. Dye chemistry for
polymers'is well known to those skilled to the art.
As an example, the front wafers can be tinted as
~l0 generally known to those skilled in the art after the wafers
have be~en produced (but before ,ncorporation into the
composite) to proYide either uniform or gradient tinting.
Since-the~front~wafer is typically uniform in thickness, the
resulting lenses can appear uniformly colored when viewed '~
from the front. These mass produced tinted front wafers are
~incorporated into the polymer-polymer composite by any of the,
methods~desoribéd below. The back layer can be reserved for 1'
c ~ ure~formation,~ i.e., prescription. Alternatively, the
1ay-r~iican;~b- tinted diff-rently from the~ front to give a
20~ ihost ~of~colors~and shades.
The~polymer-po~1ymer composites described herein'can
be~ dy-d in a slmil~r fà~hion to that of standard lenses (such
as~ CR-39)~ for~example, by immersion in one or more of a wide
variety or~dye-baths.~ The dye baths are typically maintainèd '
25'~ ait~ elev~ated~temperature,~usually at or slightly below the
boiling~point~o~ the~bath. The finished lenses are dipped
into ~th~cho--n~b~thJ~;~for~a~fixed period~ e.g., 2 minutes, to
achi-ve~th~ désired~color. Longer times are employed for `-
darker tints. I f a gradient-color is desired, then the
~30 l-qses~are~periodically withdrawn half-way from the bath so
only half of the lens is richly tinted, whereas the other - ' ~'
half~is -lightly tinted. Thermoset precursors (commonly i'
iguid-like)l can also be loaded with dyes, W absorbers
.... . . . . .
y~ (e.g.~ melànin or synthetic dyes), antioxidants, mold release ~~~~~ ¦-
3-5~ ~ agents,~-tc., before resin transfer molding into the final
waf~er shape.
8U~STIT~TE ~HEET
~,
t -
WO93/21010 2 1 1 ~ 1 1 5 PCT/US93/03470
-16-
Another-advantage of the~-~composite structure is the
11thickness" of the back layer. Since the ~ront preformed
wafer already possesses a degree of thickness greater than -~
that of a mere coating, the back layer can be relatively thin ,
and still effectuate prescription and shatter resistance,
especially when a high-impact-resistant material is employed.
Since the back polymeric layer can be thin, the dwell time of
W irradiation used to initiate polymerization of the layer -i
can be relatively short. In addition, heat removal and `-~
temperature control accompanying exothermic
polymerization/curing reactions are less troublesome with a
thin layer. The speed of polymerization àllows the on-site
production of lenses in optometric outlets.
The front wafer (for example, an epoxy) and the back
layer (for example, a polymer prepared as in Example 1) may j
be slightly mismatched in refractive index. The front wafer
can be largely uniform in thickness, and therefore contribute
little to prescription. Alternatively, curvature can be
incorporated into its shape design as discussed in detail
below.
In one embodiment, the front wafer has an anti-
re~lective coating on its convex surface. Reflectivity is
- measured in terms of percentage of light (intensity)
reflected relative to the incident light (intensity). A
number close to 0~ reflectivity is ideal, while a number
close to 100% would give a shining surface with much glare.
... . I .
Anti-reflective coatings are used to reduce the amount of
light that is reflected off of a lens surface. This is
achieved by depositing a dielectric film with a specific
thickness and refractive index on the desired surface. The
coating thickness determines the wavelength of light that is
affected and is on the order of a-q~arter wavelength.
Generally, for ophthalmic applications, the wavelength chosen .
is in the yellow-green portion of the visible spectrum where
the eye is most sensitive. At wavelengths on either side of
the yellow-green region, the amount of the reflected light
increases. To improve efficiency usually more than one film
W093~21010 ~ S PCT/US93/0~70
-l7-
is deposited on the surface. For multi-layer systems, a
combination of hlgh and low refractlve index coat~ngs are
used. Zirconium dioxide, titaniu::. dioxide, and zinc sulfide
are commonly used high refractive index layers while cerium
fl11oride and magnesium fluoride often serve as low refracti~e
index layers. With a properly applied multl-layer coating,
light transmission may be increased from 92~ to 99.5%. The
coatings are applied by a process known as vacuum deposition.
Firms that apply antireflective coatings on ophthalmic lenses
include VM Products and Silor, both of which are located in
California.
The face of the front wafer that subsequently
becomes the interface between the front and the ba~k (the
front wafer concave surface) can alternatively have carefully
introduced diffraction patterns (mimicking the moth's eye). `~
The patterns can be in the master mold and an imprint left on!~
the sample after injection molding or resin transfer molding,
similar to how compact disks are made. These patterns (after
accounting for the refractive index discrepancy) can produce
full-fledged anti-reflection effects, in a manner akin to the
working principles of a moth's eye. The moth's eye has
naturally engineered diffraction patterns, so light
reflecting back from differer~ _ocales interfere
destructively. Hence, the ne- reflectivity is appreciably
lower than that from the otherwise smooth int~rface.
Bifocal and multifocal polymer-pol~Ler composite
lenses can be easily produced by use of a Fresnel-like front
wafer, stacked with a cur~ature forming back layer. The back
layer can be polymerized and then ground/polished, or it can
be produced with a known curvature by use of a specific mold
half.
Bifocal, multifocal, progressive, and/or asti~matic~
lenses can also be prepared from the polymer-polymer
composite by employing a high refractive index wafer in the
front, with a low-refractive index material in the back
layer, or vice versa. In this way, the outside surfaces of
the front wafer (the convex surface) and back layer (the
WO93/21010 21~ PCT/USg3/0~70
concave surface) of the composite can be-spherical. The
progressive, bifocal, cylindrical, aspheric or other complex
requirements can be incorporated through intricate shapes and
contours at the inter~ace between the front and back layers.
Those skilled in the art of optical calculations can readily
design the desired surface container with the aid of computer
simulation programs. These refractive-index internally-
complex designs can be used to achieve demanding vision-
correction without the use of a thick lens, producing more
comfortable eyewear.
The back and/or front layer can be polymerized in a
way to prevent cavitation, or voids caused by the shrinkage -
of material during polymerization, using the sequential ,~
polymerization process and apparatus disclosed in U.S. Patent
Nos. 5,114,632 and 5,110,51~. Briefly, the partially -:
polymerized material is inserted between two mold halves, one
of which is, or both are, constructed of a material that
transmits energy, either thermal or W . Stress related voids
in the polymeric material can be eliminated by causing the
partially polymerized material to polymerize in a
differential fashion along a moving front, so that the
material ahead of the moving polymer zone remains liquid, and
the material that the front has passed is-solidified. In a -
typical method, the moving front is a slit through which W
or thermal energy is transmitted. The still-liquid material
ahead of the moving polymer zone can then flow freely, at a
- rate that equals the rate of shrinkage, and a void--free,
reduced stress polymeric network is produced.
I The polymer-polymer lens composite described herein
is distinct from lenses prepared by polymerization around an
"insert". Insert technology involves covering both the front
and back sites with photo or thermally cured-material.
Insert technology does not allow for the individual
optimization of desirable attributes in the front and back
layers, since both ma~or exposed surfaces are of the same
material. A stiff surface surrounding a soft core is not
WO93/21010 ~ 115 PCT/US93/0~70
- 19 -
impact resistant, as crack-initiation originates from the
surface.
Hardenable materials useful for the front preformed
polymeric wafer and back polymeric layer are described in
detail below. Other suitable materials are disclosed in U.S.
Patent No. 4,544,572, incorporated herein by re~erence.
1. Composit~on of the H~gh Impact ~es~stant Polymeric Layer
The back polymeric layer should exhibit an impact
resistance of at least that of C~-39, a well-known lens blank
material. The material performance can be tested using the
well known FDA l'Drop-ballll test. The material should be able
to withstand the impact of a standardized steel ball dropped
from the same height as that endured by a CR-39 lens of
similar size and thickness. It must be dimensionally stable
at the service temperature, generally room temperature.
Polymers that fulfill these requirements are known to those
skilled in the art, and include bloc~ copolymers,
interpenetrating networks, graf~ copolymers, random
copolymers, and even homopolymers that are elastomeric in
nature yet sufficiently rigid. The block or graft segments
should be so small that they are on the order of molecular
dimension so that they do not scatter light. A preferred --
high impact resistant macromolecular network is described in
detail below. A preferred polymeric material for the impact
2;5 resistant layer is the polymerlzable composition described in
detail in Section II. Another example of a polymeric
mateFial that i9 suitable material for this purpose is
described below.
It has been discovered that an appropriaee material
for the back layer is a macromolecular network that includes
stiff members interconnected by soft, elastomeric, `~
multifunctional crosslinking sites. Polymerization is
controlled 90 that the stiff monomers primarily self- -
polymerize to create members with controlled length that are
attached to one another via chemical bonds on the soft
crosslinking bridges. The crosslinking bridges (or cores)
WO93/t1010 2 1 18 1 15 PCT/US93!0~70 :-
-20- -
are randomly dispersed in space, providing the shock-
absorbing capacity for the overall rigid yet impact
resistant, and optionally, transparent optical, material.
Machina~ y (polish and grind) of the hard plastics is
retained, while lntroducing resilience to the polymer
networks.
The macromolecular network is prepared by mixing the
soft joint material (the crosslinking substance) with the
hard monomers or a hard material and a free radical ~
in~tiating agent, or other suitable polymerization initiator. ;
It is important that the hard mo~omers or hard material and
the soft joint material are completely miscible, forming a
homogeneous solution. The mixture is allowed to partially
polymerize into a honey-like or molasses-like consistency
(typical viscosity ranging from lO0 centipoise to lOoO poise) ~`
with vigorous stirring, at which point the material is poured ;
into a mold or cast onto sheets and polymerization completed
without agitation.
A key aspect of this method is the pre-cast ;~
polymerization or "prepolymerization" step, which is employed ;
to ensure true dispersion (molecular level dispersion) of the
hard and soft reactants. Since the soft elements are multi-
functional, the prepolymerization step effectively ties up
the majority of the soft reactants so subse~uent segregation
of the hard and soft reactants not possibIe. -
$he polymeric rigid members (struts) are joined on
both end~ by soft, polymeric multifunctional crosslinking
sites ~soft joints). The invention as disclos`ed-also
includes macromolecular networks yielding hard-plastics
wherein the rigid framework is articulated, branched, or
random coiling. Liquid crystal (rigid-rod)~polymers can be
used as the stiff segments. Liquid crystal struts with
artificial molecular bends are articulated.-~-High glass
transition amorphous polymers ase generally-random coiling.
In addition to these topographically linear rigid members,
the rigid l'struts" may themselves be further crosslinked by
rigid crosslinkers.
~ BSTITU~E SHEET
WO93/21010 ~ 5 PCT/US93/0~7
-21-
In general, transparency of a clear mat~rial is
affected when a heterogeneous material is introduced tha, has
a size comparable to or greater than the wavelength of
visible light. Since the crosslinking sites are molecular ln
dimension in the macromolecular network described herein,
there is no appreciable light scattering and therefore no
ad~erse effect on the transparency of the material. As long
as the crosslinking molecules do not self aggregate to
dimensions comparable to a wavelength of light, sample
transparency is guaranteed.
The hard and soft segments can be random,
alternating, block, or graft in their sequence distribution
and spatial arrangement. Random, or alternating, copolymers
are generally single-phased, and thus transparent in their
pure form except for possible optical absorption bands. 1 v
~lock and graf~ copolymers are generallr multi-phased, and
the phase-separated domain ~ize must be made small in order
for the material to retain transparency. In all cases, the
ultimate mechanical properties represent a compromise. The
polymeric supernetwork described herein combines the best of
; mechanical and optical properties.
a). Descript~on of Rigid Framewor~
Hard monomers or hard materials are chosen for the --
rigid framework portion of the macromolecular network that,
once polymerized, give rigid transparent plastics with a
glass transition temperature above the temperature of u~e ~ -
(typically ambient temperatures) and with good optical
properties. The monomer is in general one that polymerizes
!- through a chain mechanism, such as an alkene derivative. A
preferred monomer is methylmethacrylate~ Other alkene ;~
deri~atives include other alkyl methacrylates, - _
alkylacrylates, allyl or aryl acrylates and methacrylates,
cyanoacrylate, styrene, ~-methyl styrene, vinyl esters,
including vinyl acetate, vinyl chloride, methyl vinyl ketone,
vinylidene chloride, acrylamide, methacrylamide,
acrylonitrile, methacrylonitrile, acrylic acid and
WO93/21~10 2 1 1 8 1 1 5 PCT/US93/0~70
-22-
methacrylic acids. Mixtures of monomers can also be used in
- the polymerization process.
Partially halogenated or perhalogenated hard
monomers, including fluorine containing monomers, can also be
used in the rigid framework, lncluding but not limited to
fluorine containing methacrylates and acrylates, such as Cl to
C7 partially or fully fluorinated esters of methacrylic or
acrylic acid, for example, 2,2,2-tri~luoroethyl methacrylate,
trifluoromethyl methacrylate, 2,2,2,3,4,4,4-heptafluorobutyl
methacrylate, and 2,2,2,2',2',2'-hexafluoroisobutyl
methacrylate.
Acrylate-terminated or otherwise unsaturated
urethanes, carbonates, and epoxies can also be used in the
rigid framework. An example of an unsaturated carbonate is
allyl diglycol carbonate (CR-39). Unsaturated epoxies
include, but are not limited to, glycidyl acrylate, glycidyl ¦ :
methacrylate, allyl glycidyl ether, and 1,2-epoxy-3-allyl
propane.
Bisphenol-A-bis-2-hydroxypropylmethacrylate, and
bisphenol-A-bis-2-hydroxypropylacrylate can also be used as
hard monomers. In addition, allyl terephthalate, allyl
isophthalate, aryl terephthalate or isophthalate, or acryl
isophthalate or terephthalate can be usPd. -
Preformed polymers that have ethylenically
unsaturated groups can also be made more impact resistant by
the methods described herein. Acrylate-terminàtëd novolacs
can be used as or in the rigid framework of the polymeric
macromolecular network. Polyurethanes, polymeric epoxies,
and polycarbonates that have been derivatized to include
acrylate, methacrylate, or other unsaturated functional :
groups are well known and commercially available. Examples
of commercially available photocurable~materials are the line
of Synocure products sold by Cray Valley Products (for
example, Synocure 3101, a diacrylate derivative o~ bisphenol-
A, and Synocure 3134, an aliphatic urethane diacrylate), and
the Epon products sold by Shell Corporation (for example,
Epon 1001 and Epon 828, which are both diacrylates of the
I
~VO93/21~10 2 ~ PCT/US93~0~70
-23-
diglycidyl ether of blsphenol-A). Vinyl-termlnated liquid
crystalline polymers can also be used.
Poly(carbonyldioxy-l,4-phenyleneisopropylidene-l,4-
phenylene) is sold under the trade names Lexan, Makrolon, and
Merlon. This polycarbonate has good mechanical properties
over a wide temperature range, good impact and creep
resistance, high transparency, and good dimensional v
stability. Unsaturated derivatives of this polymer, such as
the allyl or acrylate derivatives o~ poly(carbonyldioxy-l,4-
phenyleneisopropylidene-l,4-phenylene) can be made more
impact resistant by reacting the polymer with a soft moiety
as described herein.
Optical grade epoxies with terminal unsaturation
include those made from l,2-propylene oxide, l,2-butylene
oxide, 1,2-epoxydecane, 1,2-epoxyoctane, 2,3-epoxynorbornane,
1,2-epoxy-3-ethoxypropane, 1,2-epoxy-3-phenoxypropane,
- oxetane, l,2-epoxy~5-hexene-l,2-epoxyethylbenzene, l,2-epoxy-
l-methoxy-2-methylpropane, perfluorohexylethoxypropylene
oxide, benzyloxypropylene oxide, and mixtures of these.
Mixtures of hard monomers can be used in the
preparation of the macromolecular network. For example,
methylmethacrylate can be polymerized in combination with
alkylacrylate or arylacrylate, such as methylacrylate or --
ethylacrylate.
The hard monomers can be mixed in any desired ratio,
as long as the components remain compatible and miscible. ~~
Acrylates can be mixed with methacrylates over the entire
composltion range as long as the esters are compatible,
i~ typically of comparable length. Acrylates generally
polymerize more rapidly than methacrylates using either
photochemical or thermal initiation. _ ¦ "
Additionally, preformed polymers with terminal or~ -
internal unsaturation can be copolymerized with hard monomers
in the presence of a soft moiety with ethylenic unsaturation, -
3S to form a material with high impact resistance.
In an alternative embodiment, inert polymers can be
added to the starting mixture, tO thic~en the mixture, for
WO g3/21010 ~ 1 1 5 PCT/US93/0~70 ~
-24-
ease of handling, to reduce the tota-l reaction time, or for
other reasons. The inert polymer1c material can be any
polymer, and can be used in any amount, that does not
adversely affect the desired properties of the final
material. Inert polymers in general are polymers that do not
react with other components in the reaction solution. In one
embodiment, an inert polymer of the hard monomer or hard
material is added to the polymerization solution. For
example, if methyl methacrylate is used as the hard monomer
in the macromolecular network, polyme~hylmethacrylate can be
added to the polymerization solution.
Additives such as W absorbers, tinting agents, and
anti-oxidants can also be added to the polymerization mixture
to obtain the desired properties of the final product. See,
e.g., R.~. Seymour Ed., "State of the ~rt; Additives for
Plastics". Academic Press, New York, 1978. ~;
b.) Descr~ption of Soft Joints
A polymer or oligomer should be chosen for use as
the soft joints of the macromolecule that has a low glass
transition temperature (ranging from below room temperature `
to as low as obtainable), that provides a soft, pliable
material when homopolymerized, is stable to high and low
temperatures, and is compatible with and soluble in the
copolymerizing agene. The polymer or oligomer used for the
soft joints must be of a slze that does not scatter light,
and therefore is less than approximately 100 nanometers, and
optimally, no larger than approximately 10 nanometers in
order of magnitude.
Examples of suitable polymers for the soft joints
include ~inyl substituted siloxanes, allyl~substituted
siloxanes, acrylate terminated or substituted siloxanes, and
partially or perfluorinated derivat ves o~.vinyl substituted
siloxanes, allyl substituted siloxanes,~~r-acrylate
terminated or substituted siloxanes. For example,
polydimethylsiloxane has a glass transition temperature of
-123C, which is the lowest known polymeric glass transition
temperature. When one of the two methyl groups attached to
W093/~l~10 21 I ~ I 1 5 PCT/Us93/0~70
-25-
the silicon atom is replaced with a vinyl group, a reactive,=
multifunctional polymer, polyvinylmethylsiloxane (PVMS) is
produced. Thls polymer is soft and can copolymerize with a
variety of monomers through well established free radical
chemistry. The Si-O bonds are quite flexible and provide a
cushion effect on impact. Poly~inylperfluoromethylsiloxane
can also be used as the soft joint material.
Hydrocarbon polyunsaturated (multi-functional)
compounds (both homo- and copolymers, and especially
oligomers) can also be used as the soft joints in the -
macromolecular network. Hydrocarbon llnsaturated compounds
can be produced, among other ways, by the polymerization of
conjugated dienes such as butadiene, isoprene, and
chloroprene. Two different types of polymerization reactions ;~
occur with 1,3-dienes. One type invol~es the polymerization
of one or the other of the double bonds in a typical chain
polymerization. In a second pathway, the two double bonds
act ~1n~concert~to yield an allylic radical that can react -
with a successive monomer at either carbon 2 or carbon 4 i '
~2~0~ 1,2-polymerization and 1,4-polymerization, respectively).
; The 1,2-polymer has pendant unsaturation, whereas the 1,4-
polymer has unsaturation in the polymer backbone. All of
- ~ these various types ~f polymerization products of conjugated
dienes can be used~to prepare the polymeric network described
herein. ~ `
: , : :,
To ensure optical transparency while using a finite - -- -
amount of soft~segments, molecular compatibility is critical.- -
Compatibility is usually optimized by using low molecular
; weight soft moieties.
c.)~End Chai~ Tet~ers -
; The macromolecular network prepared as described
above can be further crosslinked by including a small ~~ ~~~`~-
difunctional or multifunctional reactive molecule, or mixture
of small di- or multifunctional molecùles.
~35 ~ Crosslinking agents for hard monomers that are
polymerized by a chain process are known to those skilled in
the~art, and include tri- or tetrafunctional acrylates or
,., :
WO93~21010 ~ 5 PCT/US93/0~70
-26-
methacrylates, divinylbenzene (DVB), alkylene glycol and
polyalkylene glycol diacrylates and methacrylates, including
ethylene glycol dimethacrylate and ethylene glycol
diacrylate, vinyl or allyl acrylates or methacrylates, -
divinylbenzene, diallyldiglycol dicarbonate, diallyl maleate,
diallyl ~umarate, diallyl itaconate, ~inyl esters such as
divinyl oxalate, divinyl malonate, diallyl succinate,
triallyl isocyanurate, the dimethacrylates or diacrylates of
bis-phenol A or ethoxylated bis-phenol A, methylene or
polymethylene bisacrylamide or bismethacrylamide, including
hexamethylene bisacrylamide or hexamethylene
bismethacrylamide, di(alkene) tertiary amines, trimethylol
propane triacrylate, pentaerythritol tetraacrylate, divinyl
ether, divinyl sulfone, diallyl phthalate, triallyl melamine,
2-isocyanatoethyl methacrylate, 2-isocyanatoethylacrylate, 3-
isocyanatopropylacrylate, l-methyl-2-isocyanatoethyl
methacrylate, and 1,1-dimethyl-2-isocyanaotoethyl acrylate. i -
Particularly useful are tetraethylene glycol diacrylate,
tetraethylene glycol dimethacrylate, triethylene glycol
diacrylate, triethylane glycol iimethacrylate, hexanediol
dimethacrylate, hexanediol diacrylate, and other high alkane
(including but not limited to C4 tO Clo) diol diacrylates or
~ dimethacrylate~. These bifunctional molëcules are spaced by
a relatively long bridge between the acrylates or
methacrylates, providing flexibility.
The crosslinking agent is added to the- hard monomers
;~ or polymers and soft joints prior to the initial
prepolymerization step. The amount of crosslinking agent
added will determine how tightly crosslinked the final
network is. The crosslinking agent can be used in any amount
that produces the desired results. It is typically added in
an amount ranging from O.l~ to less tha~ ~0~-by weight.
In one embodiment, an acrylate^terminated
polybutadiene (PB) is used as the soft joint material. The
P~ can be any molecular weight that provides the desired
results, typically from lO0 to lO0,000. A mixture of the
hard monomers methyl methacrylate and benzyl acrylate (or in
I
WO93~21010 ~ I 18115 PCT/US93/0~70
-27-
general aromatic esters of acrylates or methacrylates) is
useful for polymerization with PB, because aromatic acrylates
and methacrylates elevate the refractive index of the base
polymeric material in such a way as to match, or approximate,
that of PB. As a nonlimiting example, a high impact material
can be prepared by mixing 38~ benzylmethacrylate with 62
MMA. This mixture is then mixed with PB (13,000 MW) at a 73%
to 27~ by weight ratio, respectively. This solution is
homogeneous. With a slight addition of photoinitiator, it
can be prepolymerized in such a way that PB no longer phase
separates in the subsequent casting step. Very thin (1 mm)
disks of this material endure the FDA dropball test up to 8
times (i.e., 400 inches) the height. re~uired by FDA (50
inches) without cracking (even crazing). In contrast CR 39
sheets at comparable thickness fractured often before
reaching the height (50 inches) required by the FDA. To this
basic formulation can be added tetraethylene glycol
diacrylate or dimethacrylate, triethyleneglycol acrylate or
~- methacrylate, or hexanediol diacrylate to improve scratch
resistance. The samples are transparent after 20~ addition
of the crosslinkers.
d). Proparation of the Networ~
To prepare a macromolecular network in which the
;~ soft joints are homogeneously dispersed throughout the
polymer, it is impo~rtant~that the hard monomer and the soft
component be continuously~and efficiently stirred during the
initial stages of polymerization. If the soft component and
the rigid component are simply mixed and left to polymerize
in a ! static, ~uiescent cavity, the two components will tend
to phase separate during polymerization. Phase separation
d ~ before polymerization causes hazy or opaque products. In
addition, the two components typically have different
densities. If left alone in a quiescent cavity for a long
time, the heavier component will migrate to the bottom, and
~5 on polymerization, a product will be produced that has a
composition gradient in the direction of the gravitational
~ field. When polyvinylmethylsiloxane and methylmethacrylate
:~:
;,~'-
WO93~21010 2 1 1 8 11~ PCT/US93/0~70
are combined, the PVMS collects near the bottom of the
container. Polymerization of this stratified mixture
produces a product that has a softer lower portion topped by
a rigid upper portion.
In a pr~ferred embodiment, in a first stage of
polymerization, the two components in any desired ratio are
continuously stirred while polymerization is initiated.
Inert polymer can be included in the polymerization mixture
as desired, to thicken the reaction mixture, to reduce the
reaction time, or for other reasons. This prepolymerization
step can be accomplished in an open vessel such as a beaker,
exposed to the atmosphere, or preferably, under an inert gas
such as ~. Polymerization is allowed to proceed with
continuous stirring until an incipient copolymer and partial
network is formed. The viscosity of the partially-
polymerized reaction solution increases to the point that
phase separation and stratification does not occur when the
solution is poured and then left undisturbed for a long time.
The partial polymerization step that includes stirring during
the early stages of polymerization before final mold filling
and completion of polymerization, ensures that phase
separation and sedimentation are totally suppressed,-either
.
by ~irtue of the slow kinetics, or for thermodynamic reasons
since the incipient copolymer are structurally similar and
~25 uniform everywhere in the sample.
- In the second stage of polymerization, the -- ;
partially-polymerized material is poured into the final
static and quiescent mold cavity, for example, a mold lens,
to form the final object, that should be clearj transparent,
and without composition gradient. The requirements of exact
dimensions, shape, and cur~ature preclude injection, _
transfer, or compression molding. The resulting mat-erial may
or may not be tightly attached tO one or both sides of the
mold, depending on whether a laminate or a-pure plastic
product is desired.
As an example, under intense W irradiation,
mixtures with from approximately 90% methylmethacrylate and
WO93~21010 2 1 1 8 .1 ~ ~ PCT/US93/0~70
-29-
10~ polyvinylmethylsiloxane (weight average molecular weight
ranging from 300 to an upper limit that is low enough to
ensure optical transparency), up to virtually ali -
methylmethacrylate, with a trace of PVMS provide a
transparent plastic with improved fracture resistance. The
more PVMS used, the greater the improvement of fracture
resistance. When a 90~ methylmethacrylate and l0~
polyvinylmethylsiloxane plastic network prepared as described
in Example l is projected with great velocity against a hard
concrete surface, the material recoils a large distance
without shatter, chipping, or fracture. E~en a 2 millimeter
sheet prepared from 90% methylmethacrylate and l0~
polyvinylmethylsiloxane passes the U.S. Food and Drug
Administration's requirement for impact resistance for
ophthalmic lenses (the standard drop-ball test).
The copolymèrization of hard a~d soft monomers give¦ -~
final materials with intenmediate properties, depending on
their composition. A compromise is generally reached when
the product polymer is neither extremely rigid nor 1~
unnecessarily soft. ! `
Any ratio of components can be used in the
macromolecular network thàt produces the desired results.
The~prepolymerization step and the final polymerization can ~
be accomplished at any temperature that produces the desiredi `
product, and typically ranges from ambient temperature to the
- boiling point of the lowest boiling component. The ~`
prepolymerization step typically takes from approximately a .
few minutes to a few hours. Optical clarity can be maximized
by i~nsuring vigorous agitation, minimizing trapped air~during
agitation, and by allowing the prepolymerization step to
proceed to the point that the soft joints are homogeneously `
and permanently distributed throughout the partially
polymerized network.
The co~ple~ion of polymerization is preferably
carried out in an inert atmosphere if done in an open mold
and free radical reactions are occurring. It is known that
oxygen inhibits free radical polymerization, and gives rise
WO93/21010 2 1 l 8 ~15 PCTIUS93/0~70
-30-
to extended polymerization times. If closed molds are used
to form the article, the mold should be made from an inert
material that has non sticking properties such as
poly(tetrafluoroethylene), silicone rubber, polyethylene,
polypropylene, and polyester. Glass and metallic molds can
be used if a suitable mold-releasing agent is used. If it ls
desired to use the high impact plastic material as a
laminate, the mold may actually be comprised of, or may
include, the material to which the laminate is attached.
This final step of polymerization can be carried out
in a method to prevent cavitation, or voids caused by the
shrinkage of material during polymerization, using the
seguential polymerization process and apparatus illustrated
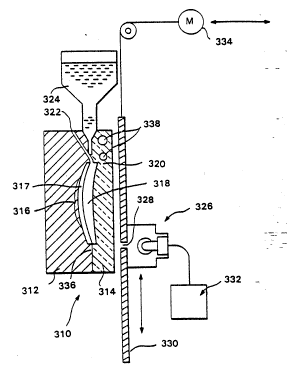
in Figure 4. Referring to Figure 4 a mold body 310 is shown
in cross-section. The mold body is designed specifically ~or
an ophthalmic lens that has convex and concave surfaces. The
device 310 shown in Figure 4 is formed of at least two parts
312 and 314, brought together to fonm a cavity 316. Ca~ity
316 is formed having the shape of the precision lens that is
desired to be molded. In the cavity is optionally inserted
preformed lens 317; that has a convex surface that will
become the convex surface of the finished lens, and a concave
surface that interfaces with the convex surface of ~he back
polymeric layer 318. It should be understood that the
apparatus illustrated in Figure 4 can be adapted such that
the preformed wafer ultimately forms the back layer of the
lens and front layer is polymerized in situ. As is usual
with a mold, a gate 320 provides access to the mold body 310
when the first and second part are engaged. Communicating -
with gate 320 is a reservoir 324 which is utilized to feed
raw material to cavity 316 through gate 320. Reser~oir 324
is represented in Figure 4 as a hopper-like device. A vent
322 may also be included to facilitate the filling of cavity
316. It should be understood that other means for providing
raw material to ca~ity 316 through gate 318 may be
advantageously used. For example, it may be appropriate to
provide raw material to cavity 316 under pressure.
WO93/210l0 211~ PCT/US93/0~70
.'' '
-31- ;
Mold body 310, as can be seen in Figure 4,
necessarily has one part, in the case illustrated, part 314,
that is transparent to a source of energy. A source of ;
energy 326 is movable relative to mold body 310 and includes
a focusing means such as gate 330. The source of energy 326
..:ay be drawn across the second part 314 by means of a two-way
motor 334. Source of energy 326 is selected according to the ~
material to be molded. For example, i~ the monomers (the ;"
reaction mixture or polymer precursor) provided to the mold
cavity 316 from reservoir 320 are to be polymerized by heat,
then source of energy 326 is appropriately a heat source
which is focused through an opening 328 in focusing gate 330.
Opening 328 is preferably designed to focus a plane of energy
on second part 314. The plane of energy is substantially
normal to the movement of focusing gate 330. AlternativelyI ;~.
i~ the monomers utilized in cavity 316 are polymerized by an ¦
ultra violet source or other light source, then source of
energy 326 may be a light of the proper wave length. Again, i-~
se~ond part 314 is of necessity transparent to the wave
lengt;~ of light utilized in source of energy 326 in the event
polymerization takes place under the imposition of a light ;
source. In the event that polymerization takes place as a
result of the imposition of heat, second part 314 is
appropriately thin and made of material that has little or no
insulative qualities. It may also include passages 338 for
cooling. These passages may be selectively used so that a
time-dependent temperature gradient will be maintained. -
Movement of focusing gate 330 relative to mold body
310 is controlled so that source of energy 3Z6 scans across
the mold body 310 starting at the closed end 336 of cavity
316 and moving toward gate 320.
The reaction mixture which is contained in reservoir
324 is constantly resupplied to cavity 316 through gate 320
thus as polymerization occurs at the lower end or closed end
336 of mold 310 the shrinkage that occurs and would
eventually appear as a void is immediately replenished by the
reaction mixture or mixture of polymers contained in
WO93/21010 21 l~ PCT/US93/0~70
-32-
reservoir 324. It is of course understood that the reactlon
mixture is highly mobile and flows readily to fill the volume
lost due to shrinkage of the part of the mixture that has
already undergone reaction. The instantaneous replacement of
the space formed by shrinkage by unreacted material ensures a
final piece that is defect free and distortionless. The
movement of the energy source 326 relative to the mold body
310 must, of necessity, start with opening 328 in focusing
gate 330 moving from closed end 336 to gate 320 in a manner
such that polymerization takes place at a steady rate from
the closed end to the gate end.
In the event the source of energy 326 is by the
nature of the monomer a heat source, movement of the focusing
gate across the mold body 310 must be at a rate that does not
permit heat transmission through ~econd part 314 at a rate
faster then polymerization is taking place. That is, as the
heat source of energy 326 moves upwardly, second part 314
will absorb heat and conduct that heat inwardly to cavity 316
where polymerization takes place. The portion of second part
314 above opening 328 must be kept cool which may be
accomplished by circulating a cooling fluid through passages
338 so that the upper portio~ of part 314 remains cool in -
relation to the lower portion of part 314 thereby providing
differential heating of mold cavity 316.
Mold 310 is clamped together in a co~ventional
manner with reservoir 324 in the position shown. Reservoir-
324 is filled with the reacting mixture in this case a -
monomer, a mixture of monomers or a monomer/crosslinker
mixture loaded with an initiator and/or other catalysts, such
that the material will easily flow into cavity 316. It is
important to ensure that cavity 316 is fully filled with the
reacting mixture before polymerization is attempted. ~~ ~
Accordingly, it may be appropriate to provide a vent 322 to
the mold cavity 316. In the event a vent is employed, lt
should be closed and plugged before polymerization takes
plac-. Closing ehe vent will assist in drawing a ditional
WO93/21010 ~ 11 3 ~ 1 5 PCT/US93/0~7
-33-
reaction mixture into cavity 316 during polymerization rather
than permitting air to enter the mold.
Once mold cavity 316 is filled, the source of energy
322 may be activated and focusing gate 324 moved relative to
mold body 3l0 thereby imposing either heat or light, as
appropriate, to the mold body in a differential manner.
Should heat be the source of energy, then it may be
appropriate to activate cooling passages 338 at the upper end
of the mold body to ensure that heat conduction through the
mold body will not initiate polymerization in the upper ~
portion of the mold before the focusing gate 324 tra~erses ~;
the entire face of the mold. ,~
Once focusing gate 330 has completed its passage and
polymerization is complete in the mold body 310, then the
mold structure can be taken apart and the molded precision
par~ removed. ¦
Any W or thermal free radical initiator known to
those skilled in the art for free radical polymerization can
be used to initiate this process. Examples of W and j
thermal initiators include benzoyl peroxide, acetyl peroxide,
lauryl peroxide, azobisisobutyronitrile, t-butyl peracetate,
cumyl peroxide, t-butyl peroxide, t-butyl hydroperoxide,
bis(isopropyl)peroxydicarbonate, benzoin-methyl ether, 2,2'^
azobis(2,4-dimethylvaleronitrile), tertiarybutyl peroctoate, Z
phthalic peroxide, diethoxyacetophenone, and tertiarybutyl
-~~~ peroxypi~alate, diethoxyacetophenone, 1-hydroxycyclohexyl
phenyl ketone, 2,2-dimethyoxy^2-phenyl-acetophenone, and
phenothiazine, diisopropylxanthogen disulfide. An example of
a commercial product that provides non-yellowing products is
Irgacure 184 (sold by Ciba Geigy Corporation, l-
hydroxycyclohexyl phenylketone).
Any amount of initiator can be used that produces
the desired product. Typically, the amount of initiator
varies from 0.l~ to 5~, by weight of hard monomer, and is
preferably in the range of 0.5~ to 3%.
!
j .
WO93/21010 2 I 1 8 I 1 5 PCT/US93/0~70
-3~-
Example 1 Preparation of Macromolecular Ne~work of
Methylmethacrylate a~d Polyvinyl~ethylsiloxa~e.
Methylmethacrylate (94~ by weigh~),
polyvinylmethylsiloxane (5~ by weight, M~ 300-S00), and 1~ W
photoinitiator (Irgacure) were mixed under W light (300-400
nm) while stirring to provide strong agitation, until the
mixture reached a consistency of that of honey or molasses.
The solution was poured into a closed lens mold, and
polymerization carried out sequentially as described above
and illustrated in Figure 4 with W light to provide a clear,
transparent material that is highly shatter resistant.
The refractive index of the material prepared as in
Example 1 is relatively high (approximately 1.51), and falls
between common hydrocarbon polymers and inorganic glass.
!
Example 2 Preparation of Macromolecular Network fro~
Methylmethacrylate, S~yre~e, Divi~ylbenze~e,
and PolyY~nylmethyl~loxane
Methylmethacrylate (90~ by weight), styrene (5~ by
weight), divinylbenzene (0.5~) and poly~inylmethylsiloxane
- 20 (3% by weight), and 1.5~ W photoinitiator
(cyclohexylbenzoint are mixed under W light (300-400 nm)
with strong agitation, until the mixture reaches a
consistency of that of honey or molasses. The solution is
poured into a closed lens mold, and polymerization carried -
out sequentially as above and illustrated in Figure 4 with Wlight to provide a clear, transparen~ material that is highly.
shatter resistant.
Alternatively, e~erything other than the
divinylbenzene is first mixed and polymerized under W until
the desired consistency is reached. Then di~inylbenzene i-s~
added to the mixture before pouring into the mold.
WO93/21010 2 1 1 8 ~ 1 ~ PCT/US93/0~70
Example 3 Preparation of ~igh Impact Polycarbonate
Allyldiglycol carbonate (CR-39) is stripped of its
inhibitors by passing the liquid monomer through an
absorption bed (basic alumina). It i9 then mixed with PVMS
in a ratio of 98~ carbonate to 2~ PVMS by weight. The
material is then polymerized as described in Example 1.
Example 4 Preparatio~ of ~igh Impact Optical Grade Epoxy
Synocure 3101 (95~ by weight) is mixed with 4.5~ by
weight of polybutadiene, and 0.5~ initiator, and polymerized
as described in Example 1.
!
Example 5 Preparat~on of Macromolecular Network of
Methylmethacrylate, Methylacrylate and
Poly~inylmethylslloxane.
Methylmethacrylate ~42.5~ by weight), and
methylacrylate (42.5~ by weight) were mixed, and
polyvinylmethylsiloxane (4~ by weight, M~ 300-500), and 1% W
photoini~iator (Irgacure) were mixed under W light (300-400
nm) while stirring to provide strong agitation, until the
mlxture reached a consistency of that of honey or molasses.
The solution was poured into a close~d lens mold, and
polymerization carried out sequentially as described above
- and in Example 4 with W light to provide a clear,
transparent material that is highly shatter resistant.
~- - Example 6 Preparation of Mac~omolecular Network of
Methylmet~acrylate and Poly~i~ylmethylslloxa~e
Wlth Ethyle~eglycol Dimethacrylate as
Crossl~n~i~g Agent
., I
~ Methylmethacrylate (92% by weight),
- polyvinylmethylsiloxane (5~ by weight), ethyleneglycol ~}
= 30 dimethacrylate (1~ by weight) and (2~) W photoinitiator
~~ (Irgacure) are mixed, and then reacted under W light with
strong agitation, until the mixture reached a consistency of
that of honey or molasses. The solution is poured into a
closed lens mold, and polymerization carried out sequentially
as described above with W light to provide a clear,
transparent material that is highly shatter resistant.
WO93/21010 2 1 l X l 1 S PCT/US93/0~70
-36-
Alternatively, ethyleneglycol dimet~acrylate can be ~
added after the prepolymerization step.
Example 7 Comparison of the Physical Prope~ties o~
Traditlo~al Polymerq wlth a ~lgh Impact
~eRistant Polymer
The density, refractive index, Abbe number, flexural
modulus, CTE (Coefficient of Thermal Expansion), glass
transition temperature, and percent ~isible light
transmission of a macromolecular networ~ prepared as in
Example l (referred to as S-5) were compared to a
polycarbonate thermoplastic (Lexan), and a polycarbonate l
thermoset prepared from allyldiglycol carbonate (CR 39) .
obtained from PPG Industries. The results are provided in :
Table l.
= _ ~
' ~ ,
WO 93/21010 2 1 1 8 ~ 1 ~; PCr/US93/03470 `
o
a~ ~,
,
a~ O
Cq ,, . U~ S 0 o~
m ~ ~ o J~ ~ "
cq ~ ~ u~ ;:
~ n U~
E-~
U `'
J~ ~
~ '.`
O ~1 a~
E ~ d' a~ o ,~
~ a) ~ U) o
E~ X
~q ~:
.,
o
o ,:
_1 0 ~ ~ C~
,~ D X ~
o O
~ .
_
,, ~ o
E o
~ o
. :,
-, ~a
X to .,1
tJ H ~ _ ~;
Ei h
, - ~
_- ~ ~ æ ~P. v
0~) n h a) X -- -
:~ ~ a) Q ~~ cn
E~~ ~ ~ ~ C) E~ o~O
WO93~21010 2 1 1 8 1 1 ~ PCT/US93/0~70
2. Compositlon of the Front ~igh Scratch-Resistant
Waf er
The front wafer can be prepared from any polymer
that exhibits a scratch resistance of at least that of bare
(uncoated or untreated) CR-39. Any of the materials
described above for use as the rigid component of the high
impact resistant macromolecular network (without the soft
moiety and with or without end tethers), that exhibit the
desired scratch resistance can be used in the front plano
wafer. Alternatively, the polymeric composition described in ;
Section II. can be used.
In the typical case, the front wafer is a preformed
wafer of at least 100 microns and more typically, from
typically 0.5 mm to 1.5 mm.
Traditional scratch resistant polymers include CR-
39, polymethylmethacrylate, and polycarbonate. CR-39 has
long been the material of choice. It is offered by PPG
~; Industries and is essentially the polymer of allyl diglycol
carbonate (although it may also include mold release agents,
dyestuffs, and/or antioxidants). It is currently the market
leader for lens materials. Polymethylmethacrylate (PMMA) is
` ~ less scratch resistan~, but has been used extensi~ely in
precision optics.
Polycarbonaees such as poly~carbonyldioxy-1,4-
25~ phenyleneisopropylidene-1,4-phenylene) ha~e a higher
refracti~e index and~impact resistance than CR39, but ~-~
typically exhibit less scratch resistance than CR39.
The scratch-resistant front plast~cs may also be
seleated from a nu~ber of other transparent, high-performance~
engineering thermoplastics, including, but not necessarily
~-~ exhaustively, poiyetherimides, polyimides, polyethersul~ones,
polysulfones, polyethyleneterephthalate, and other amorphous ~=
(random copolymer) polyamides, polyesters, and urethanes.
Nonlimiting examples include the polymers illustrated below
:
,~"
WO93/21010 2 1 ~ PCT/US93/0~70
-39-
Alternatively, transparent, high-performance
engineering thermosets, including but not limited to epoxies
or bismaleimides, may be used. Thermosets require resin
transfer molding to shape into plano (flat) wafers, in -
contrast to thermoplastics that are injection molded. Many
hardeners (crosslinkers) for thermosets exist and are known,
including aliphatic and aromatic amines and anhydrides.
Homopolymerized epoxies can also be used. Rigid, transparent ~;
epoxy encapsulants that are useful as the front wafer
material have long been formulated for integrated circuit
protection and for co~ering ~EDs.
III. Preparat~o~ of the Polymer-Polymer Compogite `
The polymer-polymer lens composites described herein
can be prepared by a variety of methods that are ideal for a
wide range of applications. The invention includes a method 1 ;
for the rapid, on site, preparation of a wide variety of high
impact resistant, high scratch resistant lenses by eyewear
manufacturers and retailers.
Lens molds typically include a front metal or glass
mo_d and a back W transparent or heat transmitting mold, as
illustrated in Figure 4. These conventional molds can be
used to produce the polymer-polymer composites. One of skill
in the art, gi~en the disclosure herein will be able to
prepare the composite by using one of the methods set out
below using traditional lens molds, or by other known
~- -methods. The composites can be prepared by polymerizing a
~ back layer onto a preformed front wafer, polymerizing a front
wafer onto a preformed back layer, or by attaching a
preformed front wafer onto a preformed back wafer.
The surface of either layer of the composite can be
_ modified by glow discharge to change the surface
~~ hydrophobicity or hydrophilicity, to obtain good antifogging
properties, or other desired properties.
WO93/21010 ~ PCT/USg3/0~70
-40-
Attachment of a Prior Prepared Front Wafer to a Prior
Prepared Back Wafer
In one embodiment, the polymer-polymer lens
composite can be prepared by attaching a prior-prepared front
scratch resistant polymeric wafer and a prior-prepared back
impact resistant wafer with an adhesive. In a preferred
embodiment, the front is a scratch resistant polymer and the `~
back is an impact resistant polymer. The two layers can be
adhered with any adhesive material that is known to those
skilled in the art for adhering polymer-polymer or polymer-
glass composites. In a preferred embodiment, the two layers
are adhered with a partially polymerized impact resistant
material as described in detail above, that is polymerized ln
9~ by the sequential polymerization method described above.
lS The front and back layers can have different
cur~atures, 80 either positive or negative lenses can be
made.
Ex~mple 8 Attachment of a Pr~or Prepared Front ~ens wlth
a Prior Prepared ~ack Lens
The front wafer is a CR-39 bifocal flat-top 1 mm
wafer. The back layer (also CR-39) is glued to the front '
wafer by in-situ sequential-polymerization of the partially
polymerized material thoney-like consistency) of the material
described in Example l.
Polvmexization of Back Layer onto Preformed Front Wafer ,
In an alternative embodiment, the polymer-polymer
lens composite can'be prepared by in situ polymerization of
the back polymeric layer onto a prior-prepared front -
polymeric wafer. This procedure is illustrated in Examples
9-l1. ~ ;`
Example 9 Preparation of ~ens with CR-39 Fro~t Wafer and
P~MS/MMA Bac~ Wafer -
A CR-39 "flat top" (bifocal) thin front wafer
(thickness approximately l.2 mm, and diameter approximately
~35 75 mm~ is used as the front wafer. ~ehind the wafer is
WO93/21010 21 I 8 ~ ~ S PCT/US93/0~70 ~
~ formed a cavity with center spacing on the order of 1 mm.
Into the cavity is inserted a partially polymerized mlxture
of 7~ PVMS and 93% MMA. The material is then polymerized as
described above and illustrated in Figure 4. The back mold -
is a clear, W-transparent fused silica precisely curved
piece, so that when the lens is finished, it has the correct
prescription.
Example l0 Preparatio~ of Le~s with ~poxy Front Wafer and
PVMS\MMA Back Layer.
An epoxy (novolac cured with dianhydride) thin front
wafer (plano 6 curvature, l mm thick, 71 mm diameter) is used
as the front wafer. On top of the wafer is formed a cavity
with-center spacing on the order of 1 mm. Into the cavity is
inserted a partially polymerized mixture of 3~ PVMS, 96% MMA,
and 1% EGDMA (ethylene diglycol dimethacrylate). The !
material is then polymerized as described above, and
illustrated in Figure 4. The back mold is a clear, W -
transparent fused silica precisely curved piece, so that when
the lens is finished, it has the correct prescription. ~
,,.
Example ll. Preparation of Lens w~th PET (polyethylene
terephthalate) Front Wafer a~d
PVMS/MA Mothylacrylate Back Layer
A PET thin front wafer (plano 6 curvature, 1 mm
- - thic~, 71 mm diameter) is used as the front wafer. On top of
- the wafer is formed a cavity with center spacing on the order
of 1 mm. Into the cavity is inserted a partially polymerized
mixture of 5~ PVMS and 95~ MMA. The material is then
-- polymerized as described above, and illustrated in Figure 4.
~~ The back mold is a clear, W -transparent fused silica
precisely curvec ,iece, so that when the lens is finished, it
-~ ~-~~ has the correct prescription.
I
~118~1S
WO93/21010 PCTiUS9~/0~70
-42-
III. Fast Curing Polymeric Compositions For Ophthalmic
~enses and Ap~aratus for Preparina Lenses ~ -
A polymerizable composition is disclosed for use in the
pre~,,aration of ophthalmic lenses that can be cured into a high
quality, impact and abrasion resistant material in thirty
minutes or less using the sequential polymeriz~tion method. The
polymerizable composition disclosed herein can also be
polymerized using conventional methods and apparatus for
polymerization known to those skilled in the art. The
composition includes at least 50% by weight of urethane, epoxy,
or polyester oligomers (or mixtures thereof) end terminated with
acrylate or methacrylate (or mixtures of acrylate and
methacrylate~, and an optional diluent, such as hydrocarbon diol
end terminated with acrylates or dim~thacrylates, or a low
lS molecular weight crosslinkable tri-, tetra-~ or poly-acrylate or 'I
methac~ylate~
l. Oligom-rs
Proper se~ection of~-the oligomer is important to
obtaining the desired physical propertie~ of the resulting lens
as the oligomer is the predominant component by weight in the
polymerizable composition. Polymers prepared,from acrylate and
methacrylate terminated oligomers are known for their
outstanding optical and mechanical properties. Because they can
be tailored to obtain desired mechanical properties by blending
~25 various materials, they are candidates for numerous applications
~- including coatings, adhesives, medical plastics, lenses, fiber
optics and glazing materials.
i It hasibeen~discovered that three types of oligomers
are preferred for the preparation of ophthalmic lenses usin~ the
~30 sequential polymerization method: urethanes end terminated with
~ acrylate or methacrylate (or mixtures thereof), and epoxies or
,, polyesters that are end terminated with acrylate or methacrylate
(or mixtures thereof). In g`eneral, urethane oligomers impart
toughness and abrasion resistance to the final lens, while epoxy
~35 and polyester oligomers impart hardness and chemical resistance.
8UBSrltUTE SHE~T
.
WO93/21010 2 i ~ PCT/US93/0~70
-43-
preferred embodimen~, the ollgomers used ln the manufactur-
of lenses have molecular weights ranging from 400 to 9000,
but preferably between 800 and 2500. High molecular weigh~
~ligomers can produce a lens with too much flexibi~lty, while
s iow molecular weight oligomers can produce a lens that is tOO
rigid with low impact resistance. The functionality
(acrylate or methacrylate) of the oligomers can range from
two to six. The oligomers should comprise between 20% and
90% by weight of the final formulation, preferably greater
than 50~ of the composition, and more typically, between 50%
and 75~ by weight in the composition.
a) ~rethane Acrylates
Polyurethanes are a general class of polymers that
contain at least two -NHCOO- linkages in the backbone of the
polymer, optionally along with other functional groups in the
backbone such as esters, ethers, urea and amides. Polymers
prepared from urethane oligomers exhibit good abrasion
resistance, toughness, flexibility for impact resistance,
cla_ity, and stain resistance. These propertles, which have
made urethanes useful in the coatings industry, are also
important attributes of ophthalmic products.
There are a wide ~ariety of ways known to those
skilled in the art to prepare urethane polymers. Urethane
prepolymers are typically reaction products of aliphatic or
aromatic- po~yols, polyesters, or polyethers of diverse
composition with a stoichiometric excess of diisocyanate.
Typically, the number of terminal hydroxyl groups of the
polyol, polyester, or polyether is two or greater. The
terminal-hydroxyl groups react with the diisocyanate to
produce ur~thane linkages, and the resulting prepolymer
becomes-end capped with isocyanate groups. Depending on the
- stoichiometric ratio of NCO/OH groups, the urethane linkage
can also be incorporated into the backbone of the isocyanate
terminated oligomer. Different urethanes can be obtained by
changing (1) the diisocyanate, (2) the polyol, polyester, or
polyether, or (3) the NCO/OH stoichiometric ratio. For a
WO93/21010 2 1 1~ 1 1 5 PCT/US93/~70
-44-
description of urethane oligomers and polymers, see Frisch,
K.C., Applied Polymer Science (eds. J.K. Cra~er & R.W. Tess),
Chapter 54, p. 828, ACS, ORPL, Washington, 1975.
Examples of suitable diisocyanates include 4,4~-
S diphenylmethane diisocyanate (MDI, avaiiable from ICI
Polyurethanes Group, West Deptford, New Jersey; PBA 2259 (a
more stable water dispersible version of MDI also a~ailable
from ICI Polyurethanes Group); 3-isocyanatomethyl-3,5,5-
trimethylcyclohexyl isocyanate ~IPDI, or isophorone
'~ diisocyanate, available from Huls America, Inc.); 2,4- and
2,6-toluene diisocyanate (TDI); ethylene diisocyanate,
trimethylene diisocyanate, tetramethylene diisocyanate,
hexamethylene diisocyanate, octamethylene diisocyanate,
decamethylene diisocyanate, cyclohexyl diisocyanate,
methylenebis-(4-cyclohexylisocyanate), phenylene
diisocyanate, diphenylether-4,4'-diisocyanate, 2,2,4-
trimethylhexamethylene diisocyanate, xylene diisocyanate,
tetramethyl xylene diisocyanate, polyether diisocyanate,
polyester diisocyanate, polyamide diisocyanate, dianisidine
~0 diisocyanate, 4,4'-diphenylmeehane diisocyanate, toluidine
diisocyanate, and dimer acid diisocyanate (a diisocyanate
prepared from the reaction product of two unsaturated
~ carboxylic acids).
-~ Urethane prepo}ymers are made radiation curable by
adding acrylate or methacrylate groups to the prepolymer.
This is typicaIly accomplished by reacting the isocyanate
terminated oligomer with hydroxy substituted acrylates or
methacrylates. Examples include but are not limited to 2-
hydroxyethyl (meth)acrylate, 2-hydroxypropyl (meth)acrylate,
dodecyloxyhydroxypropyl (meth)acrylate, and glycerin
~-~ (meth)acrylate. Higher (meth)acrylate functionality can be
obtained by reacting the isocyanate terminated oligomer with
compounds such as pentaerythritol tri~meth)acrylate, which
contains approximately one equivalent of hydroxyl and three
(meth)acrylate groups per mole of compound. Alternatively,
acrylate or methacrylate esters that include other functional
groups that can react with an isocyanate can also be used,
, ,
WO93/21010 2 ~ PC~US93/0~70
-45-
such as-epoxy containing compounds such as glycidyl acrylate
or methacrylate, or amino containing esters such as
aminoalkyl or aminoaryl acrylate or methacryla~e.
A nonlimiting example of a difunctional or
trifunctional urethane prepolymer is shown below:
O O O ',
p~ ~ ~ Nnl ~ A ~ ~
.
wherein P is an aliphatic or aromatic polyether, polyes~er or
polyol, R is the residue of the diisocyanate to which the
isocyanate moieties are attached, n = 2 or 3 and A is the
aliphatic (typi~ally alkyl) or aromatic ester portion of the
hydroxylated acrylate or methacrylate used to end-cap the
oligomer. Urethane (meth)acrylates containing between two
and six acrylate or methacrylate functional groups are
readily avai}able in industry.
The properties of the acrylate or methacrylate
terminated oligomers depend on the backbone structure. Alkyl .
esters and ethers yellow less and are more stable to light
than aromati~c esters and ethers. However, aromatic esters --
and ethers impart hardness to the composition, and posse~s a
higher refractive index than their alkyl counterparts, which
20 i9 desirable to reduce lens thickness for a gi~en
- prescription. Alsol polyester based urethane acrylates or
methacrylates are generally harder than polyether based
systems because polyesters provide a more polar bond
structure and a more basic oxygen with which stronger dipolar
and hydrogen bonding interactions can occur with the urethane
segments. ~
Hydroxy-terminated polyester starting materials are
often prepared from dicarboxylic acids or anhydrides,
including but not limited to adipic acid, phthalic anhydride,
and dimerized linoleic acid, with monomeric glycols and
triols. Examples of glycols include ethylene glycoi,
WO93/21~10 2 1 1 8 1 1~ PCT/US93/0~70
-46-
propylene glycol, l,2-butylene glycol, l,4-butylene glycol, --
l,6-hexylene glycol, trimethylolpropane, glyce~ol, and l,2,6-
hexanetriol.
Widely used polyether diols and polyols used ro S-
S produce uretAane ollgomers include poly(oxypropylene)glycol,
poly (1,4-oxybutylene)glycol, random copolymers of alkylene
oxides and copolymers of tetrahydrofuran and alkylene oxides.
Depending on the diisocyanate monomer, polyol group and
stoichiometric ratio, urethane oligomers with widely
different mechanical and chemical properties result.
Branched oligmers based on branched polyols,
polyesters, or polyethers are also useful in the
polymerizable composition.
Since the urethane methacrylate or acrylate has a
functionality greater than one, the resulting material after
polymerization is a thermoset rather than a thermoplastic
ma~erial. The material cannot be reprocessed once it is
~ cast, but has the advantage of significant chemical
; ~ resistance and thermo-mechanical stability. An important
~actor that affects the mechanical properties of thermosets
is the crosslink density of the network. Increasing the
density, which is achieved by either decreasing the molecular
weight between acrylate groups or increasin~ the
functionality of the oligomerj in general leads to a higher
- 25 Tg and a more abrasion resistant system. However, loss of
~ flexibility, which causes poor impact resistance and greater
~- - shrinkage during casting, is also a result of high crosslink
density.
Examples of suitable commercial acrylate or
.
methacrylate terminated urethanes that can be used in the
polymerizable composition disclosed herein include but are
_,
~ - not limited tO urethane acrylates 2264, 284, 4881, 4a66, 8301
and 8804 from UCB Radcure, urethane acrylates CN955, CN960,
CN961, CN963 and CN970 from Sartomer Company, and urethane
, ~ ~
~ 35 acrylate NR2075 from Imperial Chemical Ind.
,,~ .
WO93/21010 2 1 18 ~ ~ 5 PCT/US93/0~70
-47-
- b) Epoxy a~d Polyester Acrylates
Epoxy and polyester acryla~es and m~thacrylates ar~
also useful ~ligomers for inclusion in a fas~ curing
polymerizabie solution for ophthalmic lenses because polymers
prepared from these materials exhibit deslred properties such
as hardness, chemical resistance, and high refractive index.
Polymers prepared from these monomers can be less flexible,
and thus less impact resistant, than the urethane systems.
Aromatic epoxy and polyester acrylates and me~hacrylates have
poorer light stability than alkyl urethane acrylates or
methacrylates. In a preferred embodiment, epoxy and
polyester acrylates do not replace, but are instead used in
any suitable combination with urethane acrylate or
methacrylate oligomers. In one embodiment, epoxy and or
polyester acrylates or methacrylates comprise from 0~ up to
50~ by weight of the total oligomer content.
Epoxy acrylates are typically o~tained by reacting
epoxide functionalities with acrylic acid, methacrylic acid,
or a mixture thereof, to form an esterified acrylate or
methacrylate resin. The reaction is shown below for a
difur.__ional epoxy terminated resin:
.
.
O C~ O
CH2~P~2 ~ 2 CF~2=1 -CO-H
R
o -- OH C~H O
11 1 1 ~
CH~ C CL ~ 2- ~ -P- ~ -CH2-C~C-CH=~ H2
~ --- R
wherein P represents an aliphatic or aromatic chain that
optionally includes heteroatoms such as oxygen, nitrogen, and
sulfur and functional groups in the backbone such as amide,
ester epoxy, ether, and amino; R is H (in the case of
WO93/21010 2 ~ 5 PCT/US93/0~70
-48-
~ acrylate) and C~3 (in the case of methacrylate). The acrylate
or methacrylat~ group then undergoes normal free radical
polymerization. Typical epoxies used include aliphatic or
aromatic glycidyl ethers, epoxy phenol novolac, epoxy cresol
novolac, polyamine or polyamide modified epoxies,
cycloaliphatic epoxy resins, and others. A portion of the
epoxy moieties can remain unesterified. The final epoxy
acrylate or methacrylate oligomeric composition can include
~meth)acrylates, epoxies, esters, and acids.
Epoxy acrylates are disclosed in Kirk-Othmer,
Encyclopedia of Chemical Technology, 2nd 3d. Vol. 8, pages
294-312 by John Wily & Sons, Inc., New York (1965),
incorporated herein by reference.
Polyester acrylates are prepared by esterification
of polyesters having an excess of hydroxyl groups using
acrylic or methacrylic acid. Preparation of the hydroxy
terminated polyesters are usually obtained by reacting acids
such as adipic acid, phthalic anhydride, isophthalic acid,
azelaic acid, or dimeriæed linoleic acid, with monomeric
-~ 20 glycols, triols and ~-caprolactone. Alkyl glycols and triols
; can be based on, as a nonlimiting example, ethylene,
~; propylene, l,2-butylene, l,4-butylene, and l,6 hexylene
glycol. Triols used, for exampl-, include
trimethylolp~opane, glycerol, and l,2,6 hexanetriol. More
highly branched systems can also be used to provide greater
- - -- c~osslinking density. Polyester acrylates can impart both
- elastic and rigid properties to the final product. As an
.
example, polyesters that include aromatic acids such as
- phthalic anhydridé or isophthalic acid impart rigidity and
.
~- 30- temperature resistance to the final product. Also, highly
branched systems impart rigidity, increased chemical and heat
~~resistance, hardness and low elongation.
2. Dilue~t
Low viscosity reactive diluents are included in the
polymerizable composition to improve the processability of
the final resin. Since the diluents are incorporated into
WO93/21010 2 ~ i 8 1 1 ~ PCT/US93/03470
-49-
the -lens, they should-be selected appropriately to lmpart the
desired characterlstlcs such as hydrophobicity, abrasion
resistance and impact resistance. The diluents can be
monofunctional, difunctional, or multi-functional, wherein -
the term "functional" is used to refer to groups that are
reactive on curing with radiation, such as acrylate and
methacrylate.
In general, acrylates are preferred over
methacrylatPs for use in both the oligomeric component and
the diluent component of the polymerizable composition,
because acrylates cure more quickly than methacrylates,
reducing processing time.
The diluent should be chemically compatible with the
urethane acrylate or methacrylate, or epoxy acrylate or
methacrylate used in the polymerizable composition. The
diluent is considered compatible if pha3e separation does not
occur on polymerization of the composition.
In one embodiment, the polymerizable composition
includes a diluent of the structure:
: 11 11
CH,=CRCO-(X)-OCR=CH~
hydrocarbon diol series
.
wherein R is independently H or methyl, and X is a straight
or branched alkane of C2 ~o ~ Hydrocarbon diol and branched
hydrocarbon diol based diacrylates-and dimethacrylates are
- preferred over polyoxyalkylene glycol diacrylates or
dimethacrylates. The hydrocarbon diol acrylate series of
diluents, which includes but is not limited tG ethylene
3~ glycol diacrylate and dimethacrylate, l,4-butane diol ~:
diacrylate and dimethacrylàte, l,6-hexane diol diacrylate and
dimethacrylate, decamethy~ene diol diacrylate and
dimethacrylate, and neopentyl glycol diacrylate and
dimethacrylate, are more hydrophobic than the polyoxyalkylene
glycol based systems. They are also superior in withstanding
chemical attack from polar solvents such as alcohols, which
are frequently used as cleaning iolu~ions. In addition,
WO93~21010 PCT/US93~0~70
21181i~
-50-
butanediol and hexanediol diacrylate and dimethacrylate in
particular impart good hardness and abrasion resistance
without sacrlficing i.~pact resistance. They also exhibit
good light stability and are low in -,-iscosity. Typical
concentrations of the diacrylate or dimethacrylate diluent
are between 0 and 50~ by weight, and preferably between 2
and 20~ by weight.
In another embodiment, multi-functicnal acrylates
and methacrylates are included in the composition to provide
a strong thermoset network. These higher functional systems
impart good abrasion resistance to the final lens product.
Examples include tri-, tetra-, penta- and hexa- acrylated and
methacrylated aliphatic or aromatic monomers that can be
ethoxylated, and include, but are not limited to, ethoxylated
trimethylolpropane tri(meth)acrylate, propoxylated
trimethylolpropane tri~meth)acrylate, ethoxylated
pentaerythritol tetra~meth)acrylate, pentaerythritol
~` tri~meth)acrylate, glyceryl propoxy tri(meth)acrylate,
- dipentaerythritol penta~meth)acrylate, dipentaerythritol
hexa~meth)acrylate, trimethylolpropane tri(meth)acrylate,
~- pentaery~hritol tetra~meth)acrylate, di-trimethylolpropane.
The ethoxylated and propoxylated monomers, that can include
any desired amount, but typically from three to nine-moles of
~thoxylation, provide increased flexibility, reduced
shrinkage, and lower toxicity at the expense of reduced Tg
a-n~ higher viscosity.
3. I~itiator
Any W or thermal free radical initiator or mixture
-- o~ initiators known to those skilled in the art of free
.
~; 30 radical polymerization can be used to initiate
~~~ bolymerization. Mixtures of the photoinitiators are
sometimes preferred since they can in certain cases provide a
more efficient production of radicals. The initiator should
be non-yellowing, have a broad absorption spectrum if it is a
W initiator, and good curing efficiency. It should also be
non~oxic and have low odor. Concentrations of the initiator
WO93/21010 ~llt~ ~ l S PCT/US93/0~70
in t~ polymerizable composition typically range from 0.1 tO
5~ by weight, although any amount can be used that provides
the desired product. A relatlvely low concentration of
initiator, between 0.1 to 0.8~ by weight, is preferred to
reduce yellowing.
There are a number of non-yellowing commercially
available W initiators. Examples include but are not
limited to Irgacure 184 (1-hydroxycyclohexyl phenyl ketone),
and Darocur 2959 or 1173 sold by Ciba Geigy Corporation, and
KIP 100F (2-hydroxyalkyl phenone) sold by Fratelli Lamberti
Esacure. KIP 100F and Darocur 2959 and 1173 are liquids,
that are readily miscible with the other componen~s of the
polymerizable composition. Irgacure 184 is a white powder
with extremely good absorbance and non-yellowing properties.
Other W and thermal initiators include
benzophenone, trimethylbenzophenone, isopropylthioxanthone,
and ethyl 4-~dimet~ylamino)benzoate, benzoyl peroxide, acetyl
peroxide, lauryl peroxide, azobisisobutyronitrile, t-butyl
peracetate, cumyl peroxide, t-butyl peroxide, t-butyl
hydroperoxide, bis(isopropyl)peroxydicarbonate, benzoin
methyl ether, 2,2'-azobist2,4-dimethylvaleronitrile),
tertiarybutyl peroctoate, phthalic peroxide,
diethoxyacetophenone, and tertiarybutyl peroxypivalate,
diethoxyacetophenone, 1-hydroxycyclohexyl phenyl ketone, 2,2-
dimethoxy-2-phenyl-acetophenone, phenothiazine, and
diisopropylxanthogen dis-ulfide.
4. Inhibitors
' Inhibitors are optionally added to the polymerizable
composition to inhibit polymerization under nonmal storage
conditions, by acting as radical scavengers. Any inhibitor
known to those skilled ~~n the art can be used in any
effective concentration. The most common inhibitors are
hydroquinone (HQ) and hydroquinone monomethylether (MEHQ).
HQ has been found to increase yellowing at high
concentrations while MEHQ does not. Inhibitor levels should
be minimized since they retard the speed of the initiation
2118115
WO93/2~010 PCT/US93/0~70
-52-
and~propagation process during polymerization. Typical
concentrations in the final formulations are optimally
between 0.002 to 0.2 weight percent.
5. ~V Stabilizers
Stabilizers can be used to pre~ent changes in lens
- properties with time. These include W absorbers (W A),
hindered light amine stabilizers (HALS) and antioxidants
(AO). W As preferentially absorb incident W radiation,
thereby preventing the radiation from reaching the casted
polymer. Examples include Tinu~in 328, Tinuvin 900, and
Tinuvin 1130 from Ciba Geigy. HALS do not function by
absorbing W radiation, bu~ inhibit degradation of the casted
polymer by binding with free radicals. Examples include
Tinu~in 292, and Tinuvin 144 from Ciba Geigy. AOs also
terminate free radicals, particularly those associated with
peroxy radicals. They are not generally used as light
stabilizers. Examples include Irganox 1010 and Irganox 1076
from Ciba Geigy.
The lens material can also be protected from W
radiation after casting, by applying an anti- W coating or by
dipping the lens in a suitable solution.
6. Other Addltives
Internal mold releases can be added tO the
polyme~izable~composition to improve releasability from the
molds, but- arë not required, and if possible, should be
avoided as they can reduce clarity. Examples of release
agen'ts include butyl stearate, dioctylphthalate, Zelec UN and
Zelec NE sold-by E.I. DuPont NeMours and Company. Other
addit1ves, such as dyes and wetting agents, can also be
includ~ds.--
7. Proce~s of Polymerization
The problems of shrinkage and lengthy cure time
associated with the traditional casting process for
ophthalmic lenses are solved by casting the polymerizable
WO93/21010 2 ~ PCT/US93/0~70
composition disclosed herein using the sequen~ial
polymerization technique as described in detail above. Uslng
sequential polymerization, mold deslgn is scraightforward.
The costly experimentation required to engineer a mold that
accounts for resin shrinkage is avoided. The sequential
process is easily adapted to either radiation or thermal
curing. Radiation curing is preferred because it is mor~
convenient and in general requires a shorter cure time.
Radiation curing can be performed at moderately elevated
temperature to further reduce polymerization time.
The apparatus for the production of a lens from a
polymerizable composition preferably includes a carriage
frame; a conca~e (or convex) mold that allows the
transmission of energy that is capable of initiating
polymerization attached to the carriage frame; a moving stage
that can be driven across the carriage frame; a means for
moving the stage across the carriage frame; a convex (or
concave) mold, wherein the convex (or concave) mold is
attached to the moving stage, and wherein the ~ ~.vex (or
~20 conca~e) mold can be moved adjacent to the transparent
concave (or con~ex) mold to define an internal cavity there
between, the cavity corresponding to the precise dimensions
of the lens; a means for introducing polymerizable
composition into the internal cavity; a source of energy for
;~ 25 transmission through the concave (convex) mold in a
-~ sequential manner; and a means for-sequentially exposing the
polymerizable material to the energy~soùrce beginning at a
point opposite to that wherein the polymerizable composition
- , is introduced, and proceeding to the point wherein the
polymerizable composition is lntroduced.
~- One embodiment of a method and apparatus for the
- preparation of a lens by se~e~tial polymerization is
-~ illustrated in Figures l to-3. Figure l illustrates a
carriage system that holds a concave l0 mold that forms the
convex surface of the finished lens and a convex mold 20 that
forms the concave sùrface of the finished lens. In a
preferred embodiment, the convex mold 20 is attached to a
WO93/21010 2 ~ PCT/~S93/0~70
-54-
moving stag~ 30 while the concave mold 10 is snapped into a
hc --r that s part of the carriage frame 40. Experiments
hav~i shown tha~ the abrasion resistance of the face closer tO
the lamp source (i.e., the front mold) can be greater than
the face away from it. Since most ophthalmic retail outlets
have the capability of coating the concave surface of the
lens, this embodiment is preferred. The opposite arrangement
(i.e., convex mold attached to the stage, with the concave
mold attached to the holder) is also possible. The mold
attached to the holder shc,uld be made of a material that is
W transparent, such as BK-7 glass. Note that the back mold
20 does not need to be W transparent. Therefore, metal,
non- W transmitting glass, or even plastic molds may be
utilized. While glass or metal molds provide a longer usage
life if not mishandled, they are extremely expensive and can
easily be damaged. Plastic molds, particularly those that
may be injection molded, are inexpensive to produce.
Materials such as polystyrene, polyester tPET or PBT),
polya~etals, polyfluorinated alcohols, teflon, polyamides,
polysulfones, polyimides, etc. that possess hardness, heat
stability, and low surface energy to allow release of the
finished part are possible mold candidates for this process.
This mold 10, attached to the carriage frame, can be enclosed
by an outer ring that serves as a goniometer indicating the
degree of rotation about an axis. This featurei is necessary
~ for ~on-sphe-rlcal molds. Rotation is required with respect
- to the mold attached to the stage 20 to dial in the desired
cylinder orientation particularly when aspheric, multi-focal,
or progressive lenses are being fabricated.
.
-- - A lead screw 50 drives the stagc 30 forward and
bac~ward along a guide rod 60. A distance indicator 70
_. I
informs-Ehe user of the location of the stage.
. .
The carriage forms a cavity between the two molds
10,20 that is filled with the fluid polymerizable
composition. Figure 2 is a schematic side cross sectional
view of a portion of the carriage system embodiment of Fig.
1, illustrating the procedure for filling of the lens mold.
WO93/21010 2 1 1 ~ PCT/US93/0~70
~efore the molds are brought=together, a flexible gas~et lO0
made of an inert material such as flexible PVC, silicone, or
rubber, is fitted around mold lO. A rigid c1amp llO is then
attached around the gasket to provide support. The stage 30 `
is then positioned such that the molds are separated by a
desired distance. A brake or locking system is then employed
so that even under high pressure (between 30 and 50 psi), the
stage i9 fixed in its locked position. Any suitable locking
system, such as a back stop or even a brake, can be used. In
a preferred embodiment, the inside of the cavity other than
the W transparent mold lO is black or lined with an anti-
reflective coating to prevent light scattering.
The polymerizable resin ls contained in a reservoir
80 and can be introduced into the mold cavity by a number of
methods. Mechanical methods, such as a motorized piston, may
be employed. However, an easier and preferred system is to
use gas pressure to drive the fluid (see Figure 2). A piston
may be added to separate the gas from the fluid. However, if
the gas is in contact with the fluid, it is preferred that
- 20 the gas be inert ~for example nitrogen or helium), to
minimize dissolved oxygen in the composition. The reservoir
is connected to the mold cavity by a fle~ible plastic hose
-~ which ends with a valve 85 and a tapered tip 90. The tapered
tip is pushed through a small ho}e-in the bottom of the
gasket and resins introduced. Fluid flows from the bottom of
the cavity to allow bubble free- fill-l~g. Air is allowed to
escape through a tiny vent hole 120 at the top of the gasket.
~fter the cavity is filled, a cap 130 i9 screwed on to plug
the lair vent sealing the cavity.
3C The carriage is ~ransferred to a curing station for
se~uential polymerization. Figure 3 is a schematic side
cross sectional view of another~portion of the carriage
system embodiment of Fig. l, with lens mold rotated l~0
degrees, positioned in front of a movable W source. The
curing station consists of a long wave W light source (250
to 400 nm) lS0, that preferably emits collimated light that
is a~ached to a moving stage 160. A colored glass filter
WO93/21010 2 ~ 1 8 1 1~ PCT/US93/0~70
5~-
l65 that allows U.v. light tO pass but retard3 the
transmission of visible and IR radiation is added _3 minimize
radioactive heating of the resin by the light source. The
stage is driven by a lead screw 170 attached to a motor 180
and drive system 190. The motor is preferably connected to a
control system, such as a computer, that sets and varies scan
rates as desired. A slit 200 of adjustable vertical opening
of between 0.25 and 2.0 inches, attached to a frame 210
provides a plane band of W light. The frame is attached to
~he moving stage 160 to allow the light source and slit to
move as a single unit. In the preferred embodiment, during
sequential polymerization, movement of the light source/slit
assembly relati~e to the carriage assembly is controlled such
that the plane band of W light scans across the carriage
starting at the top of the ca~ity 230 and moving toward the
bottom 220 where the resin line is located.
The opposite arrangement wherein the plane band of
W light scans across the carriage starting at the bottom of
the cavity 220 and moves towards the top 230 is also feasible
if the carriage 140 is rotated in such a manner that the
resin feed source is repositioned to the top of the carriage.
However, this method requires an additional rotation step
over the preferred-arrangement. Also, if air in the cavity t
is not completely removed during the filling stage, rotation
of the cavity after fllling may induce the trapped air
bubbles to rise-up the cavity during cure which would produce
a defecti~e lens. An alternate arrangement that does not
involve rotation is to move the carriage and fix the light
source and slit arrangement.
Other similar schemes may be envisioned to produce
the sequential_e~fect. Instead of a slit, a curtain may be
. _ _ . ... .
lowered (or~ra~ èd) first exposing W light to the area
opposite the feed port. The curtain is moved until the
entire lens is exposed. Note that for this arrangement, the
W exposure time is not constant throughout the sample, but
depends on position. Another possible arrangement is to
continually open a slit starting from the center of the lens.
WO93/21~10 2 ~ ~ 8 ~ ~ ~ PCT/US93~0~70
-57-
Here, the central ~ortion of the lens will have the longest
exposure to the W light. A major disadvantage of thls
scheme is that two feed ports will be required at opposite
ends of the direction the slit opens. Only one port wi'l be
required if instead of an increasingly expanding slit, an
expanding hole is employed. This may be accomplished using
an iris diaphragm. With the diaphragm, the initial W
exposure area is a small circular hole at the center of the
lens assembly. This exposure area is radially increased by
opening the diaphragm. By continuously opening the
diaphragm, the entire lens assembly can be fully exposed.
Since the edge will be the final area -xposed to W ligh~,
only one port is necessary for this process. The expansion
rate will require adjustment depending on the reactivity of
lS the sample, the W intensity, and the thickness of the part
being irradiated. The lens assembly may be held ~erticaliy
or even horizontally during the curing process.
; The fluid polymerizable composition, that is
contained in the reser~oir 240, is constantly resupplied to
the ca~ity. A known positive pressure or farce, typically
between 20 and 50 psi, is applied to the syringe during the
polymerization step. The optimal pressure is dictate~ by the
flow arrangeme~lt, system ~iscos ty, and cure rate. Thus, as
polymerization occurs in the region exposed to the light, the
shrinkage that occurs is immediately replenished by
additional polymerizable composition. The po-lymerizable
composition is highly mobile and flows readily to fill the
volume lost during shrinkage of the part of the mixture that
has'already p~olymerized. The nearly instantaneous
replacement of the space formed by shrinkage with unreacted
fluid ensures a final object that is virtually defect free
and distortionless.
In an optional embodiment, after sequential
polymerization is completed, a post cure step can be carried
out wherein the entire mold cavity is exposed to blanke~ W
radiation. To ensure that defects do not appear, post curing
should be carried out only when the entire lens is at a
WO93~1010 2 1 1 8 1 ~ ~ -58- PCT/US93!0~70
sufficiently advanced stage of cure that shrinkage is minimal.
Post curing is pre~erably performed while the article is still in
the mold to prevent oxygen inhibition of the curing process. At
the comFIE~ of the curing process, the mold structure can be
taken apart and the precision cast part removed.
The equipment described above can be used to produce
spherical, progressive and aspheric lenses. The f inal lenses can
optionally be tinted with dye or anti- W agents after the
polymerization process is complete.
Finished spherical lenses of ~4 mm diameter with a
-2.0 diopter power were prepared using a variety of
embodiments of the polymerizable composition described herein,
using the sequential polymerization method illustrated in Figures
. 1 to 3. The initiator and other additives were added to the
diluent and stirred vigorously. This mixture was then added to
the oligomers and the sample heated carefully to approximately, 50
degrees C in a water bath, and stirred vigorously for between two
and three hours (stirring can range from thirty minutes to three
hours), taking care to ensure that the oligomer is completely
incorporated in the solution. The resin was then degassed in a
vacuum oven to remove dissolved gas. The resulting polymerizable
fluid was poured into the reservoir and introduced between two
glass molds that were separated with a center distance of 2 mm
(see Figure 2)-. -The glass molds were constructed of BK-7 glass
that transmits-~ong wave W radiation. The molds were coated
with an external release agent. The fluid was retained between
the molds using a flexi~le PVC gas~et and c}amp assembly.
Pressure of between~~25-and-35 psi was maintained on the fluid
during the entire curing process. The composition was
se~uentially polymerized using mercury vapor lamps of between 160
and 300 W and a horizontal slit assembly as described above and
in U.S. Patent No.~-5,114,632. The slit opening was varied from
0.5 to 1.0 inches.-- Se~uential polymerization time varied ~rom 8
SUBSTITUTE SHEET
WO93/21010 2 1 1 ~ PCT/US93/0~70
-59-
to 27 minutes. ~Initiator concentration was varled from 0.2
to C., weight percent.
Example 12 Preparation of Plastic Ophthalmic Le~s
A mixture of 50 percent by weight Radcure 284
urethane diacrylate, 20 percent by weight Radcure 8301
urethane hexaacrylate, 29.6 percent by weight ethoxylated
trimethyolpropane triacrylate, and 0.4 percent by weight KIP
lOOF initiator was prepared and cast as described above,
using a 3/4 inch slit size. The sample was sequentially
irradiated for 22 minutes.
Example 13 Preparation of Plastic Ophthalmic Lens
~ mixture of 75 percent by weight Radcure 284
urethane :iacrylate, 24.6 percent by weight hexane diol
diacrylate, and 0.4 percent by weight Darocur 1173 initiator
lS was prepared, as cast as described above. A 3/4 inch slit
size was used with an irradiation time of 22 minutes.
Exam~le 14 Preparation of Plastic Opht~almic ~e~8
A mixture of 75 percent by weight Sartomer 963E75
urethane diacrylate, 24.6 percent by weight pentaerythritol
~0 triacrylate, and 0.3 percent by weight Irgacure 184 initiator
was prepared, and cast as described above. A one inch slit
size was used with an irradiation time of 18 minutes.
'
Exa~ple l5 Preparation of Plastic~Ophthal~ic Less
A mixture of 37.5 percent by weight Radcure 264
urethane triacrylate, 37.5 percent by weight Sartomer 963E75
urethane diacrylate, 24.7 percent by weight hexane diol, and
0.3 percent by weight Darocure 1173 initiator was prepared,
and cast as described above. A one- lnch slit size was used
with a run time of 18 minutes.
Example 16 Preparation of Plastic Ophthalmic Lens
A mixture of 40 percent by weight Radcure 284
urethane diacrylate, 40 percen~ by weight Radcure 264
WO93J21010 2 1 1 8 ~ 1 S PCT/US93/0~70
-60-
urethane triacrylate, l9.7 percent by welght trimethylol
propane triacrylate, and 0.3 percent by wei~ht Irgacur~ 184
initiator was prepared, and cast as described above. A one
inch slit size was used with an irradiation time of 18
minutes.
Example 17 Preparation of Piastic Ophthalmic Le~s
A mixture of 37.5 percent by weight Radcure 284
urethane diacrylate, 37.5 percent by weight Radcure 264
urethane triacrylate, 24.7 percent by weight ethoxylated
trimethyolpropane triacrylate, and 0.3 percent by weight
Darocure 1173 initiator was prepared, and cas~ as described
above. A one inch slit was used with an irradiation time of
18 minutes. ~
Example 18 Preparation of Plastic Ophthalmic Lens
A mixture of ~3 percent by weight Radcure 284
urethane diacrylate, 37 percent by weight Radcure 264
urethane triacrylate, lO percent by weight ethoxylated
- trimethyolpropane triacrylate, 9.7 percent by weight
trimethyolpropane triacrylate, and 0.3 percent by weight KIP
lOOF initiator was prepared, and cast as described. A one
inch slit was used with an irradiation time of 18 minutes. - -
-: !
Example l9 Preparation of Plastic Ophthalmic Lens
A mixture of 40 percent by weight Radcure 284
urethane diacrylate, 40 percent by weight Radcure 264
urethane triacrylate, l9.8 percent by weight ethoxylated
pentaerythritol tetraacrylate, and 0.2 percent by weight
Irgacure 184 initiator was prepared, and cast as described
; above. A 0.75 inch slit size was used with an irradiation _ -
time of lS minutes. ~= --~~~~~`
30 Exampl8 20 Preparation of Pla~tic Ophthalmic Le3~
A mixture of 37 percent by weight Radcure 284
urethane diacrylate, 33 percent by weight Radcure 264
urethane triacrylate, 29.~ percent by weight ethoxylated
WO93/21010 2 1 1 8 11 5 PCT/US93/0~70
6l-
pentaerythritol teraacrylate, and 0.2 percenc by weight
Irgacure 184 initiator was prepared, and cast as described
above. A 0.75 inch slit size was used with an irradiation
time of 15 minutes.
E~ le 21 Preparation of Plastic Ophthalmic Lerls
A mixture of 60 percent by weight Radcure 284
urethane diacrylate, lO percent by weight polyester acrylate,
29.6 percent by weight ethoxylated trimethylolpropane
triacrylate, and 0.4 percent by weight Irgacure 184 initiator
was prepared, and cast as described above. A 0.75 slit size
was used with an irradiation time of 18 minutes.
Example 22 Evaluation of Le~ses Prepared i~ Examples 12-21
The lenses prepared in Examples 12-21 were evaluated
for impact and abrasion resistance. A ~ikon lensometer was
used to evaluate the optical power of the lenses. The
opticaI powers of all of the lenses were within l/8 diopta of
the specified power (-2.0 diopta) and no cylinder was found
- throughout the lens.
The lenses were subjected to abrasion testing using
the Bayer test (ASTM F-735), which is based on a haze reading
of an abraded lens. The results of the abrasion test are
- presented in Table 2, which indicates the difference in
abrasion resistance between the test lens and CR-39.
-~ Impact resistance was evaluated using the FDA drop
- ~5 ball test. FDA regulations re~uire that lenses not crack
when impacted with a 5/8" stainless steel ball dropped from a
- : heiglht of 50 inches. All of the lenses prepared as described
herein easily pass this test. The lenses were also tested by
- dropping progressively heavier balls from the 50 inch height
~30 until the lens cracked. Table 2 indicates the relati~e
.
increase in ball weight over the 5/8" FDA ball required to
crack the lens. The greatest weight used was 8 times heavier
than the 5/8" ball. Some formulations did not crack even
under this weight.
WO93/21010 2 1 1 ~ 1 ~ 5 PCT/US93/0~70
-62-
Tabl~ 2
Oligomer Diluent Abrasion Impact Test
Ex~ Name wt~ Name wt% (x CR39) ~x FDA)
12 8301 20 ETMPTA 30 0.9 5.8
284 so
,
13 284 75 ETMPTA 25 1.3 ~8
. . ~
14 963E75 75 PETA 25 0.8 3.4
264 37 HD 25 1.2 4.1
- 963E75 37
; v ~ U 11~1~1A G v~
264 40 `.
.. _ . . .. ..
17 284 37 ETMPTA 25 1.8 ~8
264 37
. . .
18 284 43 TMPTA lO l.5 6.9
-~ ~ A ~ ~ m~
L~r l ~ l V
. _. .. _ , _ _ _ . _
19 284 40 EPETA 2û 1.9 ~8
264 40
_ _ . _ . . _ . _ . . .
284 37 EPETA 30 1.6 ~8
264 33
- G 1 6 i~ U ~ LlVl~ l~ ~ U
770 lO
83ûl - Radcure Urethane hexaacrylate
284 - Radcure Urethane diacrylate dilu~ed with HD
264 - Radcure Urethane triacryla~e diluted with HD -
963-E75 - Sartomer Urethane diacrylate diluted with ETMPTA
~ ~ V ~ v.~ C ,vv ~ y c~ c ~
hydroxyethylmethacrylate _--- .
TMPTA - trimethylol propane triacrylate --
ETMPTA - ethoxylated trimethylol propane triacrylate - ~
PETA - pentaerythritol triacrylate
EPETA - ethoxylated pen~aerythritol tetraacrylate
HD - 1,6 hexane diol diacrylate
WO93/21010 2 ~ PCT/US93/0~70
-63-
This invention has been described with reference to its
- preferred embodiments. Variations and modifications of the
invention described herein will be obvious to those skilled in
the art from the foregoing detailed description of the invention.
It is intended that all of these variations and modifications be
included within the scope of the appended claims.
~: .
.,
, ~ , - .
~, .
,,
8UBSTITUTE SHEET