Note: Descriptions are shown in the official language in which they were submitted.
WO 94/19358 ~ 9 PCT/EP94/00520
SUBSTlTUTED PHOSPHONATES, THE PROCESSES FOR
THEIR PREPARATION AND PHARMACEUTICAL CO~IPOSlTIONS
CONTAINING THEM
5 This invention relatcs to novd phosphonates substituted with a dialkyl phenol moiety
and the processes for their preparation. It further relates to pharmaceu~cal
compositions containing these compounds and their therapeutic use in diseases inwhich reactive oxygen radicals have been implicated and more sp cifically in thetreatment of atherosclerosis.
Reactivc oxygen species are involved in a number of pathologies, pharmacologicaland clinical evidencc have been firmly established in the following cases (Halliwell,
B. et al. "Role of Free Radicals and Catalytic Meta'l Ions in Human Disease: an
Ovennew", Methods Enzymol. 1~6, 1-85, 1990):
"~
- inflammato~y and immunologic injuries (autoimmune diseases, rheumatoid
arthrids),
- ischemia/reperfusion injury,
- radiadon injury,
20 - premature ageing,
- Pa*inson's and Alzheimer's diseases,
- cancer and and-cancer treatments,
- condidons associated with impaired blood circuladon such as intermittent
claudicatdon, excessive platelet aggregadon, myocardial infarction and
2s atherosclerosis.
In these pathological situadons andoxidant products would be useful as therapeudc
agents. The tests perfo~med by the inventors show that the phosphonates of formula
a) through their dialkyl phenol and phosphonate moiedes display potent andoxidant
30 activides and therefore offer this therapeudc potendal.
,
In the particular case of atherosclerosis, it is now clearly proven that cholesterol
camed in LDL is the most atherogenic form of plasma cholesterol. On the other
hand current rçsearch shows that the uptake of oxidized LDL by macrophages leads35 to the formation of lipid-laden foam cells, which is the first step in the development
of atherosclerosis. Numerous epidemiological studies have now firmly establishedthat high blood cholesterol is a major risk factor for coronary heart disease. Based on
the above, it can be postulated that ~e combination of a cholesterol lowering
Wo 94/19358 ~ pcTEwDoos2o
regimen with an antioxidant treatment might be more effective than either one. Adrug which possesses the dual hypocholesterolemic and antioxidant property couldtherefore be highly effective in the treatment of atherosclerosis.
5 The phosphonate compounds (I) of this invention inhibit markedly the synthe~is of
cholesterol in human cell lines similarly to the HMGCoA reductase enzyme
inhibitors (lovætatin, simvastatin) which are potent hypocholesterolernic drugs in -
man. The combination of their antioxidant and hypocholesterolemic activities confer
to the phosphonates of this invention the potential for treating diseases associated
10 with elevated cholcsterol levels and pathological lipid oxidation. Furthermore, the
lipophilicity of thcse compounds predicts that they will become incorporated in the
LDL and protect these particles against the damages caused by oxidative species.
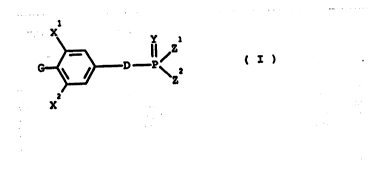
The generic structure of the compounds of the present invention is represented by
15 formula (I)
~D--P~ ( I )
where ''
- Xl, x2 identical or different are straight or branched Cl to C6 alkyl groups,
-YisOorS,
zl, z2, identical or different~ are:
- OR where R is H, a straight or branched Cl-C6 alkyl group,
- NRlR2 where Rl, R2, identical or different are H or a straight or branched Cl- C6 alkyl group,
zl, z2 together may form a C2-Cg alkylidenedioxy group,
- G is OH or a bioprecursor thereof;
2~
j WO 94/19358 PCT/EP94/00~20
D is a saturated or unsaturated Cl-Cl 1 alkylene chain in which one or more of the
methylene groups can be replaced by a sulphur atom, an oxygen atom, a carbonyl
group; optionally one or more methylene groups can be subs~tuted by one or more
halogen atoms (F, Cl or Br), Cl-6 alkyl, phenyl, hydroxy or
s acyl oxy groups,
and salts, sohates and hydrates thereo
Preferably, Xl and x2 are identical and are butyl gr~ups, in par~cular ~-butyl
groups. .:
Prefe~ably, Y is oxygen.
Preferably Zl and z2 are identical, in particular O~ in which R is H, or a s~aight or
5 branched Cl 6alkyl group. More prefcrably, zl and z2 are idenacal OR groups inwhich R is Cl 6aLlcyl, in particular methyl, ethyl or i-propyl.
PreferaUy, G is OH.
20 Suitable bioprecu~sors of the group OH as defimed for G include, for example, oR3
groups where R3 is a straight or branched Cl-C6 alkyl group, a perfluorinated
Cl-C6aL~yl group, a substituted or unsubsdtuted phenyl group, a subsntuted or
unsubstituted benzyl group, suitable biopr~cursors can also be a R3-C(o)o- group, a
R3O-C(o)o- group, a R3NH-C(o)o- group, a R3C(o)oCH20- group, a R3-So20-
25 group wherc R3 is defined as abovc.
P~efo~bly, D is A-C(O)-B,A-CH(OH~-B,A-C~H2-B, (C~H2)r(CH=CH)n~(C~H2~t or
S-(CH2)t, where
- A is (CH2)t, (CH=CH)n-C~H=CX3, (CH2)t-CHX3, S-(CH2)t-(CH=CH)n,
S-CX4X5,
(CH~H)n-C~l=CH-C(O)-CHX3, (CH2)p-CH-~H-C(o)-CHX3,
(CH2)t-C(o)-CHX3, S-(CH2)t-(CH-CH)n-C(o)-CHX3, S-CX4XS-C(o)-CHX3,
(CH~H)n-CH=CH-C]H(oH)-CHX3, (CH2)p-CH=CH-CH(oH)-CHX3,
35 (CH2)rC]H(oH)-CHX3, S-(CH2)r(CH=CH)n-CH(oH)-CHX3,
S-CX4XS-CH(oH)-CHX3
wo 94~9358 ~ PcT/EPs4lon~20
where n is zero, 1 or 2, t is a number f~m O to 4, p is a number ~om 1 to 3,
- X3 is H, a s~aight ~r branched a~l Cl-C6 group, a substituted or unsubs~tuted
5 phenylgroup,
- X4, XS identical or different are H, a s~ght or ~ranched Cl-C4 alkyl group,
- B is CH2. CH-X6, X6-C-X7, where x6 and X7 identical or different are halogen
10 atoms (F,Cl, Br), straight Qr branched Cl-C6 alkyl groups, a substituted or
unsubsatuted phenyl group;
when A is (CH2)t, (C~H-~H)n-CH=CX3. (CH2)t-CHX3, S-(CH2)t~ H)
S-CX4~5, then B is also CH=CH-(CH2)p, ~H-CH-CHX6, CH=CH-CX15X7,
5 whe~e p and X6, X7 are defimed as above.
PrefeIably, D can also be A'-CH(O-C~X8~B' whe~e A' is (CH2)t,
(CH~)n-~H=CX3, (CH2)rC~3X3, S-(CH2)r(CH=CH)n, S-CX4X5, B' is CH2,
CH X6, X6-C-X7, CH~H-(CH2)p, CH~l CHX6, CH=CH-CX6X7 where t, n, p,
20 X3, X4, X5" x6 and X7 are as described above, and X8 is a sahlIated or unsa~ra~ed
Cl-C6al~1 or alkenyl chain.
. :
Suitable sal~s included within the scope of formula (I) indude, for example,
corresponding salts of the group OR (in z1/Z2), for examplc salts formed with aL~ali
2s metal atoms such as sodium or potassium.
~ .
The prescnt invention also relates to the processes used for prcpanng substituted
30 phosphonates (I).
D iS A C(O)-B
35 A is (CH2)t. (CH=CH)n-(~=CX3. (CH2)t-CHX3. S(C~H2)t-(CH=CH)n or
s-cx4xS
.
~ ,,.. , . ,. ,. . . . . :
--~ wo 94/19358 ~ 4 ~ 9 PCT/EW4/00520
B is CH2, CH-X6 or C-X6X7
`
The proccdure described in Fig. 1 p. 10 consists in reacting the commercially
available alkylphosphonate m with a suitable base such as n-butyllithium or ~ithium
s diisopropylamide. The lithium anion of compound m thus formed is then reacted in
sin.~ with the appropriate ester II to give the substituted phosphonates (I). The
reaction is carried out in an ether solvent such as dimethoxyethane or tetrahydrofuran
(1~:), preferably in THF, at a temperature between -78C and room temperature
(25C).
A second procedure descnbed in Fig 2 p. 11 consists in condensing the unsaturated
aldehyde IV with the starting compound ketophosphonate V using titanium
tetrachloride and N-methyl morpholine as condensadon agents. The reaction is
camed out in an ether solvent such as tetrahydrofuran, dioxane or dimethoxyethane,
preferably 1~ at a temp~ure between -30C and the boiling point of the solvent
(66C in the case of 1~;). A compound of formula (I) where A is
(CH=CH)n-CH=CX3 is obtained. Compounds of formula (I) where A is
(CH2)rCHX3 can be preparcd by reacting the starting compound ketophosphonate
V with an excess of a base or combination of bases. The bases are sodium hydride,
sodium allcoxides, n-buql lithium or lithium diisopropylamide. The anion of
ketophosphonate V thus folmed is then reacted with the halide VI, where Hal = Br o~
Cl. The reacdon is ca~ied out in tetrahydrofuran, dimethoxyethane, dioxane,i
benzene or toluene. The temperature of the reaction varies between 0C and the
boiling point of the sol~ent.
:
Examples 1, 2, 3 and 4 further illustrate the expenmental aspects of the processdescribed in Fig 2. - ` - - `
Conceming condensation reactions, in the case where B is a CH2 group, i.e. when
the carbon alpha ~o the phosphonate functional group has two protons, in addition to
the main reaction pToduct formed by monocondensation at tho gamma positdon, a
side pToduct is also formed by double condensation with the dialkyl phenol groups at
the alpha and gamma positions (see cx 1 and 2). In the case where B - CHX6 or X6-
C-X7, e.g. wben two p~tons are not a~ailable at the alpha position, the compoundfoTmed by monocondensadon at the Bamma posidon is the sole reaction p~duct (see
ex 4 and 10).
WO 94/19358 ?,~Q0409 PCTIEP94/00520
Likewise, when the alpha position is completely unsubshtuted (B~H2), in additionto ~e ~ reac~on compound formed by monoaddi~:ion~ these is also formed a side
product occ~g by double addi~ons at ~e alp)ta and gamma posiaons (see ex 3).
5 A is (CE~-CH)n-CH=CH-C(o)-C~X3f (CH2)p-CH=C~I-C(O)-CHX3,
(CH2)rC(O)-CHx3~ S(cH2)r(cE~-cH)n-c(O)-c~EIx3 or
S-CX4XS-~(o~ X3
10 B is CH2, CH-X6 or C-X6X7
The p~cess described in Fig. 3 p. 12 consists in condensing an ester of formula VII
with the dianion of ketophosphonate V as dle gamma position. The dianion is
5 generated by stepwise reaction of V with an equiYalent of sodium hydride and an
excess o a stronger base, E.g. n-butyl lidlium or lithium diisopropylamide (IDA) in
te~hydrof~an at a tempe~ature betwcen -30 and 30C. The excess of dianion is
then reacted wi~ ~he este~ V~ at a tempe~ re between -70 and 30~C to yield dle
ketophosphonate (I) according to ~ig. 3.
Compounds (I) which possess two ketonc groups in dleir strucnlres m~y in solution
be in tautomcnc equilibrium with cnol forms. The di~etone and enol forms of
compounds (I) are integral part of this invention.
A is (CH2)t, (CH~l)n-CH~3, (CH2)rCHX3 or S(CH2)t~ H CH)n or
S-CX4X5
B is CH=CH-CX6X7 or CH~CH-(CH2)p
Thc procedure for prepanng compounds of formuls (I) where D is A-C(O)-B, where
A is ~CH2)t, (CH=CH)n-C~H=CX3, (CH2)t-CHX3,
S(CH2)t-(CH=CH)n or S-CX4X5, B is CH=CH-CX6X7 or CH=CH-(CH2)p consists
35 irl reacting an aldehytle of formula VIIIa or VIIIb
.
WO 94/1~3~8 ~ PCTtEP94t00520
OF1C--CX X--P ~, OHC--~C~2 ) p--P ~ 2
VIIIa VIIIb
Y~dhapho~phorusreagent wh~ch may beaphosphonatecompound offornnular~ or
aphosphoniu¢~s~t ~f fonnu~a X:
~ R
~A C--CH2--PO3 (Alkyl ) 2
IX
~ 8 +
~A--C--CH2--P ~C6 ~5 ) 3 Br
X X
0 ~ ercac~io~iscamed outin an ethersolvent,such asd~nethoxyethane or
tetrahydrofuranin presence ofa basesuch assodiurn hyd~de or~thium
diis~propylannne. - :
.
D is A~C H(O H)-B
- -
Thc kct~pho~phonatcs of fornnula ~ pleviously descr~bed can be seduced to the
coqresponding hydroxyphosphonate delivatives. The reduction can be camed out
with complex hydridc reagents such as sodium borohydride, lidlium borohydride,
sodium bis (2-methoxyetlhoxy) alu~num hydride, sodi~m trime~hoxyborohydride~
20 sodium cyanoborohydride.
Suitable solvents include ether, tetrahydrofu~an, toluene, methanol, ethanol,
isopropanol. ~Prefered reduction conditions are sodium borohydride in methanol at a
tempcrature between -20C and 65C.
wo 94/193~82 ~ ~ ~ 4 0 ~ PCT/EP94/00520 -`,
D IS A'-CH(O-CO-X8)-B'
Thc above-mcntioned hydroxyphosphonates can bc esterified to the co2responding
5 acyloxj-phosphonatc dcrivativcs by employing known proccdurcs. Suitable reaction
conditions involvc heating the hydroxyphosphonatcs with an appropriate acid
anhydride (X8-CO)20 or an appropriatc acid chloride X8-CO-Cl in prescnce of a
tertiary aminc, cg. triethyl aminc or pyridine. The reaction temperature can rangc
betwccn 0C to the boiling point of the acylating agent.
~o
D is A CH2-B, (cH2)t-(cH=cH)n-(cH2)t~ S-(CH2)t
Thc kctophosphonates (I) can be rcduced to the corresponding alkylphosphonates
and alkenylphosphonatcs by reduction of the p-toluenesulfonylhydrazone derivatives
15 with sodium borohydride, sodium cyanoborohydride or catechol borane.
Phosphonic acids of structure (I) where Zl=Z2=OH can be prepared from the
co~responding phosphonate esters by reaction vith bromotrimethyl silane to produce
bis (tnmcthylsilyl) phosphonates which are reacted in sin~ widl water or methanol.
The starting compounds allcylphosphonates m are commcrcially a~ailable. Thc
starting compounds l~etaphosphonatcs V arc preparcd according to known literaturc
mcthods: E. J. Corcy and G. T. Kwiatlcowsld, J. Am. Chem. Soc, 2Q. p. 6816-6821
(1968) and F. Mathey and P. Savignac, Tet~ahedron ~, p. 649-654 (1978).
3 8 1l 1 ` 8 Il Zl ~
X - CH2--C--L + M CH2--P\ 2 ~ X --CH2--C--CH2--P\ 2
V
L = Cl or OEt M = Li or Cu
30 The structures of new compounds of formula (I) arc determincd by infrared (IR),
mass (MS) ind nuclcar magnetic resonance (NMR) spcctroscopies. The purity of thecompounds is verificd by elcmental analysis and standard chromatographic methods: `
tilin layer chromatography, gas liquid chromatography or high performance liquidchromatography.
---) WO 94/19358 2 ~ 1 8 ~ O g PCTIEPs4/OOQO
The abbreviations used in this pa~ent application are as follows:
In the tables n- is normal, i- is iso-, sec is secondary-, t is terdary. In the NMR
spectra, s is singlet, d is doublet, t is triplet, m is muldplet. The temperatures are
5 measured in degree Celsius and the meldng points a~e unco~ected.
The present invendon will be further described by ~e examples 1 to 22 which are
typical of the syndledc procedures used.
wo 94/19358 ~ 409 " PCT/EP94/00520 - )
O = V
~ ")X
'rX
X J3` N X C
V -- :
~>
I
h
C ~ U~
~ ~ ~X X
C., C;
E- .L) ~1 :
_ O
C~
I ~
:> N N ~ 5
H X
11
_ :
0~
i~l H
-'X~X
V
--~ WO 94/19358 21 1 8 gl O 9 PCTIEP~4/00520
o X
:~
æ v
~ /
_~ ~ H
~=
m
m
i~ I o= I
C S \ /
X
Z =CI)
, I ,~ + O
:I: V
C
m N
~o ~ ~ 1
5 ~ XJ~ x ~, xJ~ X
~X ~X
~X~
1 1
WO 94/19358~7'" ~ r~ ~ `! PCT/EP~4/00520 "
4~g
,
T
o= I .
~_X ~ ~X
o
I
xJ~ X U
V :C :
~ ~,
~ . , T
O ¦N
. U ~
i- ~N ~
,
= tt
r
X 3:
~C O
O O
C
X N X
l2
--~ WO 94/19358 ~118 ~ ~9 ~ PCT/EP94/00520
~ ,.
Dimethvl 1.5-bis(35~i-tert-butvl-4-hvdrox~henvl)-3-ox~l~,oentadien-2-vl
~hosohonate
and
Dimethvl ~(3.5-di-tcrt-butvl~hvdrox~hen~1~-2-oxo-3-buten-1-Yl ~hos,ohona~
Titanium tctrachloride (114.5g, 0.6 mol) was added dropwise with stimng to 300 ml
of dry tetrahydrofuran CTh~) kcpt under nitrogcn at -20C. Solid 3,5-di-tert-butyl-~
hydroxy benzaldebyde (58.7g, 0.25 mol) was added, followed by 200 ml THF then
dimethyl 2-oxopropylphosphonatc (50 g, 0.3 mol) was added. Finally N-methyl
morpholinc (121.7g, 1.2 mol) was introduced slowly and the reaction mixture was
s~rred at room tempcrature for 1 h. Cold water (200 ml) was introduced and tbe
mixture was extracted with 1000 ml diethyl ether. Thc ether fraction was washed
with watcr until neutral pH, dried over magnesium sulfate and evaAoorated in vacuo.
Purification was carried out by chromatography on silica gel using a 98/2
chloroform/methanol mi~cturc.
~he first compound (5.5 g,4%) to dute from the column was identi~led as dimethyl1,5-bis(35~i-tert-butyl~hydroxyphenyl)-3-ox~l,~pentadien-2-yl phosphonate.
` t--Bu ~ t-Bu
H~CEI=CEI--C--C=CH ,~OH
PO3Me2
t--Bu t--Bu
C3sHslO6P Theor. %C 7Q21 %H 8.59 %P 5.17
25 Found %C0.96 %H8.55 ~` %P5.31
mp = 190-191C.
IR tKBr): 3620 cm-l: OH, 1610: C=O,1580: C=C, 1430 and 1420: t-Bu, 1240:
30P-O, 1020: P-O-C
MS: ~/e = 599: M++1,598: M+,488: M+-H-PO3Me2, 367 tl00%)
WO g4/19358 PCT/EP94/00520 - `
NMR (CDC13) ;'
= 7.75 (d, J = 26 Hz, lH): Ph-C~I=C-P
7.57 (d, J = 16 Hz, lH): Ph-C~
7.3 and 7.23 (2 s,2H cach): m. H
6.60 (d, J= 16Hz, lH): Ph-CH~
5.55 and 5.51 (2s, lH each): O~
3.82 (d, J = l lHz, 6H): P-O-C~13
1.40 and 1.34 (2 s, 18H each): t-C4~g
The second compound (36 g, 38 % yield) was iden~fied by IR, MS and NMR as
dimcthyl 4-(3,5-di-te~t-bu~14-hydroxyphenyl)-2-oxo-3-buten-1-yl phosphonate
t--Bu
EI~CEI=CEI--C --CEI2 --PO3 Me2
t-BU
mp = 107-109 (ligroin, 80-95 frac~on)
IR (KBr): 3420 cm-l: OH, 1650: C=O, 1590: CzC, 1420: t-Bu,1260: P=O and
1030: P-O-C
MS (m~e): 382 M~, 367: M+-Me,272: M+-HP03Me2,259:M~-CH2P03Me2,
151: M+-co-cH2-po3Me2
.
N~ (CDC13)
' ~ = 7.61 (d, J z 16Hz, lH): Ph-C~lzCH
7.42 (s, 2H): m. H
6.73 (d, J = 16 Hz, lH): Ph-CH~l
5.62 (s, lH): O~l
3.81 (d, J = llHz, 6H): P-O-C~I3
3.35 (d> J = 22Hz, 2H) C~2-P
1.40 (s; 18H): t-C4~1g
C2~H315P Theor % C 62.81 % H 8.17 % P 8.10
Found. 9Co C 62.63 % H 7.97 % P 8.24
--I Wo 94/19358 2 1 1 8 ~1 0 9 pcTlEp94loos2o
~2i~yLl.s-bi~ t~-hutvl~ ~x~1~4-Dentadie~-2-yl
and
Titanium tctrachl~ride (137.1 g, 0.72 mol) was added dropwise tO 300 ml dry THF
at -15C. 3,5-Di-tert-butyl 4 hydroxybenzaldehyde (70.4 g, 0.30 mol) was added
10 followed by died~yl 2-oxo~opylphosphonate (70 g, 0.36 mol). N-methyl morpholine
(145.8 g, 1.44 mol) was introduced and the rcaction mixture was stirred at room
temperature for 2 h. Work up was camed out by addition of 200 ml water and 800
ml diethyl ether. The etha phase was washed with water to neu~al pH, dried over
MgSO4 and cvaporated. The residue was chromatographed on silicagel using 98/2
15 C~HC13/MeOH as duent.
The first product (15 g, 8% yield) to dute was identified as diethyl 1,5-bis(3,5-di-
tert-butyl~hydroxyphenyl)-3-oxo-l,~pentadien-2-yl phosphonate.
t--Bu t--Bu
H~CH=CH--C--C=CH ~OH
PO3 Et 2
t-~su t-Bu
C37H5sO6P Thcor. % C 70.90 % H 8.84% P 4.94
Found 96 C 71.18 % H 8.67% P 4.75
25 mp - 174-175C
IR: (KBr): 3620 cm-l: OH, 1640: C=O, 1585: C=C, 1430 and 1415: t-Bu,
1230: P=O and 1030: P-O-C
30 MS: mle = 626: M~, 488: M+-HP03E~2, 395 (100%)
i~8 ~9i ;
wo 94/19358 2 i PCT/EP94/00520
NMR (CDC13)
= 7.7 (d, J - 26 Hz, 2H): Ph-~lCC-P
7.59 (d, J = 16Hz, 2H): Ph~C~
7.30 and 7.25 (2s, 2~I each): a~om. H
6.63 (d, J = 16 Hz, 2H): Ph-CH~
5.54 and 5.4812s. lH each): O~ -
4 18 (q~int., J = iHz, 4H): P-O-C~I2-CH3
1.40 and 1.34 (2s, 18H each): t-C4~1g
1.34 (t, J= 7 Hz, 6H): P-aC~2-C~3
The second product was identified as diedlyl 4-(3,5-di-te~t-butyl~hydroxyphenyl) -
2-oxo-3-buten-1-yl phosphonate (44 g, 36% yield) ;~
t--Bu
O
~ 11 ' ,:
H~CH=C:H C --CH2 PO3Et2
t--Bu
mp = 11~111C (rccrystallized from ligroin 80-95 fraction).
.
IR (KBr): 3620 cm~l: OH, 1680:C=O, 1590: C=C, 1430 and 1415: tl3u, 1240: P=O
and 1230: P-O-C
MS (m/e): 411: M+1, 410: M+, 272: M+-H-PO3Et2, 259: M+-CH2PO3Et2, 57
(100%) tBu+
Nl~ (CD(~3)
~ = 7.61 (d, J = 16 Hz, lH): Ph-~=CH
7.40 (s, 2H): arom. H
6.74 (d, J = 16Hz, lH): Ph-CH~
5.6 (s, lH): OEI.
4.16 (m, 4H): P-O-C~2-CH3
3.33 (d; J = 23 Hz, 2H): ~EI2-P
1.45 (s, 18H): t-C4~19
1.34 (t, J = 7Hz, 6H): P-O-CH2-C~3
16
-! wo 94/1935~ 8 4 D Y PCT/EPs4/oos20
C22H3sOsP ~eor% C 64.37 % H ~.S9 % P 7.55
~ Found% C 64.52 % H 8.6S % P 7.36
s ~m~l~
and
e~v~ 3.5-di-t~hvdrs~1~-2-Q2~1-~ts!l~bQsPh~l~
Sodium hydride (1.4~ g of a 60% dispersion in mineral`oil, 36.25 mmol) was
suspended under nitrogen in hexane, this latt~r was pipe~ted out and replaced by 60
ml dry T~. Diethyl 2~xopropylphosphonate ~7.0 g, 36 mmol) was introcluced. The
mixn~re was s~rred at room temperature for 30 min then was cooled to 0 ~C then n-
bu~l lithium (37.5 ml of a 1.6 M solution, 60 mmol) was aclded. Finally a soluaon
of 3,5-cli-tert-butyl~-hydroxybenzyl bromide (9.0 g, 30.1 mmol) in 40 ml THF wasadded and the reaction mixture was s~ed at room temperan~re ovemight. Work-up
was ca~ied out by par~ti~n between 150 ml 15~c HCl and two fractions of 1~0 ml
ether. The ether phase was washed with so~ium bicarbonate and satuIated sodium
20 chloride to ~eutral pH. After d~ying and solvent evap~ration, the crude mixture was
purified by column chromatography (SiO2, 8/2 CHC13/AcOEt).
The first compound (1.~ g, 8% yield) to elute was die~hyl 1,5-bis(3,5-di-~ert-butyl-
~hydroxy-phenyl)-3-oxo-2-pentyl phosphonate, mp = 60 61C.
t--Bu t-Bu
)~ 8 ,~(
HO~CH2--CH2 C--CH--CH2 ~OH
PO3 Et2
t--Bu t--Bu
IR (KBr): 3620 cm-l: OH, 1710: C=O, 1430: t-Bu, 1230: P=O and 1010:
P-~C
MS:m/e= 630: M+, 493: M+-HP03Et2
17
WO 94/19358 ? '~ , ~ PCT/EP94100520 --
NMR (C~DC13)
o = 6.89 (2s, 2H each): arom. H
5.09 and 5.03 (2s): O~
4.1 (m,4H): P-O-C~2-CH
S 3.5 and 3.25 (2m,2H): Ph-(~I2-CH-P
3.04 (txd, lH): Ph-CH2-C~(P~CO
2.9 - 2.4 (2m,4H): Ph-c~a2-c~l2
1.14 and 1.39 (2s,18H cach): t-C4~1g
1.32 (2t, J=7Hz, 6H): P-O-CH2-
The sccond compound was dicthyl 4-(3,5-di-tert-butyl 4- hydroxyphenyl)-2-oxo-1-
butyl phosphonate: 2.2 g (18% yicld) of an oil was obtained which slowly solidified
upon standing.
t--Bu
~CH2-CE12--C--C~2 PO3Et2
t--Bu
IR (~m): 3620 cm~1: OH, 1710: C=O, 1430: t-Bu, 1250: P=O, 1010: P-O-C
MS m/c= 412: M+, 274: M-HPO3Et2
, ..................................... :
N~: (C~DC13)
o = 6.98 (s, 2H): arom. H
5.1 (s, lH): oH
4.14 (m,4H): P-O-CH2CH
3.05 (d, J=25Hz, 2H): CEl2-p
2.94 (m,2H): Ph-C~I2-CH2
2.83 (m, 2H): Pb-cH2-c~l2
1.42 (s, 18H): t-C4Hg
1.32 (t, J=7Hz, 6H): P-O-CH2-C~13
-- I Wo 94/19358 21 1 8 4 ~ 9 PCT/EP94/~0~20
~mQ~ '
Diethvl ~(3.5 di-tert-bd~-~b~h~-1 .l~im~hvl-2~ bu~n-l-vl-
t--Bu
o CH3
~ 11 1
H~CH=CH - C--C--PO3 Et2
(~H3
~--Bu
Dicthyl 1,1-dimethyl-2-ox~pr~pylphosphonate (bp = 60~/0.2 mbar) was synthesized
acco~ding to V. Roussis and D.F. Wiemer, J. Org. Chem. ~, p. 627-631 (1989).
To 20 ml dry THF kept at 0C were a~ed sequentially TiC14 (1.72 g, 9 mmol), 3,5-di-tert-buty1-4-hydroxybenzaldehyde (0.9 g, 3.8 mmol), diethyl 1,1 dimethyl-2-
oxopropylphosphonate (1.0 g, 4.5 mmol), N-methyl mo~pholine (1.82 g, 18 mmol)
then thc seaction mixt was s~rcd for 1 h at room temperan~re. Work up ca~ied
out in the usual manner gave an oil which was purified by column chromatography
(SiO2, 8/2 CHCl31AcOEt). An amount of 1.1 g (68% yield) of th~ atle compound
was obtained. Mp = 91-92C (petroleum cther 40-60).
KBr): 3440 cm-1: OH, 1670: C=O, 1590: C=C, 1430 and 1410: t-Bu, 1210:
P~ and 1030: P-O-C
MS: mle= 439: M~ + 1, 259: M+-C(CH3)2-P03Et2, 57 (100%): tBu :~
N~: (CDC13)
= 7.66 (d, J = l~Iz, lH): Ph-~=CH
7.43 (s, 2~I): aro~ H
7.26 (d, J = 16Hz, lH): Ph-CH~
5.55 (s, lH): OH
4.15 (m, 4H): P-O-~12-CH3
1.~0 (d, J = 17H~ 6H): -C(C~3)2-P
1.46 (s, 18H3: t-C4~19
1.32 (t, J = 7Hz, ~H): P-aCH2-~3
C24H39O5P ~alc. % C 65.73 % H 8.96 % P 7.06
Found % C 66.00 % H 8.98 % P 6.82
19
WO 94/19358 PCT/EP94/00520
o9
, ~ t~ ~a~u~
Dho~
t-BU
~\ 11 .
HO~CH--C----C --C~2 PO3 Et2
CH3
t-Bu
Diethyl 2-oxobutylphosphonate (bp = 90/0.6 mbar~ was prepared according to F.
Mathey and P. Savignac, Tetrahed-~n 34, p. 649-654 (1978).
To 20 ml dry T~IF ke~t at 0C were added successively TiC14 (1.82 g, 9.6 mmol),
3,~i-ter~-butyl~-hydroxybenzaldehyde (0.94 g, 4 mmol), diethyl 2-
oxobutylphosphonate (1 g~ 4.~3 mmQl) and N-methyl morpholine (1.86 g, 18.4
mmol). The reacaon mixture was s~red at ~oom temperature for 30 min then was
heated to reflux for 1 h. After the usual wor~-up and extraction ~e crude reacaon
~ture was purified by column c}~romatography (SiO2, 8/2 CHC13/AcOEt). A
small amount of unrcacted benzaldehyde was first collected ~en 0.35 g (21%) of ~e
dtle compound was obtai~ed. Mp = 97-101C (petroleum e~er 4~60).
IR (KBr): 3400 cm-1: OH, 1680: C=O, 1610 and 1590: C=C, 1440: t-Bu, 12S0: P=O
and 1020: PO-C
MS: m/e = 424: M+, 409: M t-CH3~ 367: M+-tBu, 286: M~-HP03Et2, 57 (100%):
~Bu+
NMR (CDC13):
~ = 7.56 (s, lH): Ph-~l=C
2s 7.33 (s, 2~): arom. H
5.5 (s, lH): O~
4.16 (m, 4H): P-O-C~2-CH3
3.Sl (d, J=22Hz, 2H): ~2-P
2.1 (s, 3H): ~13
1.46 (s; 18H): t-C4~g
1.32 (t, J= 7HZ, 6H): P-O-CH2-~3
wo 94/19358 211~ 4 ~ 9 PCTIEPg4/00520
~23~37O5P C alc. % C 65.07 ~O H 8.79 % P 7.30
Found % C 65.35 % H 8.58 ~ P 7.51
~mu~
thyl9~i-tert-~utvl~c yll 3 (n ~Q~3-~
t--Bu
HO~ CH=C C CH2 PO3 Me2
r n-C4 Hg
t--Bu
A mixture of 3,5-di-tert-butyl~hydroxybenzaldehyde ~3.65 g, 15 mmol), dimethyl
2-oxoheptylphosphonate ~4.0 g, 18 mmol), rlCl4 (6.84 g, 36 mmol) and N-methyl
mospholine (7.27 g, 72 mmol) in 80 ml dry TH~ was refluxed as desc~ibed in
example 7. Purification by column chromatography (SiO2, 8/2 CHCl3/AcOEt) gave
1.2 g (20% yield) of the tide compound. Mp - 82-85 (petrolcum ether 4~60)
M~: m/c 439 M~ 1, 438 M+, 381 M+-C4Hg, 57 (100%): tl3u
NMR (CDC13)
7.48 (s, lH): Ph~ =C(C4Hg)
7.32 (s, 2H): arom. H
5.5 (s, l~ O~
3.81 (d9 J = llHz, 6H): P-O-C~13
3.52 (d, J = 22Hz, 2H): ~2-P
2.~7 (m, 2~I): ~I2-C3H7
1.5-1.4 (m, 4H) CH2-~ )2-CH3
1.5 (s, 18H): t-C4~19
0,94 (t, J = 7Hz9 3~): (CH2)3-C~3
, ~ . - . .
WO 94/19358 sj~ ~ PCl'/EP94/00520 --
Me~ 3.s~ u~l-~byd~henyL~-2~Q~ -3-bu~n
PhQS~
t-BU
H~CH=C--g --CH2 --PO3 Et2
t-BU Ib
The procedure desclibed in example S was employed, using as the phosphonate
~eagcnt diedlyl 2-oxo-3-phcnylpropylphosphonate (bp=150/0.5 mbar). The ~itle
10 compound was isolated ~y column chromatography under ~e usual condî~ons
~SiO2, 8/2 CHCl~/AcOEt) at ca 18% yqeld.
.,
mp=139-143C
15 IR (KBr): 3500: OH, 1640: C=O, 1610 and 1~90: C=C, 1430: t-Bu, 1230: P=O,
1020: P-~C.
MS: mlc=486 (100%) M~, 348: M+-HPQ3Et2, 335: M+-CH2P03Et2
20 N~:(CDC13)
= 7.70 (s, lH): Ph-~~
7.45 - 7.25 (m, SH): (~6~5
6.97 (s, 2H): aro~ H
5.46 (s, lH): OH
4.15 (m, 4H): P-0-~2-CH3
3.33 (d, J=22Hz, 2H): C~12-P
1.32 (t, J=7Hz, 6H): P-O-C~H2-C~3
1.23 (S3 18H): C4~19
C2gH3g0sP Calc. % C 69.11 % H 8.08 % P 6.37
Found %C69.37 %H8.11 %P6.61
wO 94/19358 2 I I 8 4 0 9 PC rEW4/oos20
12is~hyL4 (~di-tcrt-~1-4~by=h~-l-methyl-2-oxQ 3-bu~n~
S ::
t-Bu
HO~CH=CH ~ C--CH --PO3 Et2
CH3
t--Bu
Diethyl l-methyl-2-oxopropylphosphonate was prepared according to Mathey and
Savignac as cited as example S. ~
' '-
A mixture of 3,5-di-test-butyl-~hydroxybenzaldehyde (7.19 g, 29 mmol), diethyl 1-
methyl-2-oxopropylphosphorlate (8.20 g, 35 mmol), TiCl4 (13.47 g, 71 mmol), N- ::
methyl mo~pholine (14.34 g, 140 mmol) in 150 ml dry TH~ was reacted at room
temperan~re for 1 h, then at reflux temperature for 18 h. After work up, column
lS chromatog~aphy (SiO2, 8/2 CHC13/AcOEt) gave 4.7 g (38%) of the title compound.
mp~2-94C : i
NMR (CDC13) : .
~ = 7.63 (d, J - 16Hz, lH): Ph-C~=CH .
7.42 (s, 2~1): a~om. H
6.87 (d, J = 16Hz, lH): Ph-CH~
5.6 (s, lH): O~ ::
4.15 (m, 4H): P-O-C~2-CH3
3.~3 and 3.46 (n~o q, J = 24Hz and 7Hz, lH): C~I-P
1.5~1.43 (two d, J = 7Hz~: ~-(P)-~3
1.46 (s, 18H): C4~g
1.32 (two ~, J ~ 7Hz): P-~CH2C~3
23
WO 94/1s358 PCT/EP94/00520
Dimethyl q-(3.~ tert-butvl-~hvd~ )-2-Q2~-bute~-1-yl ~ ,ahQn~
t--Bu
H(~CH CH --C CH2--PO3 Me2
t--Bu
S Under ni~gen ahnosphere dime~yl methylphosphonate (3.17 g, 25.6 mmol) was
added at -60 to a solution of n-butyllithium (16 ml of a 1.6 M solution in hexane, 25.6
mmol) in 1~ ml anhydrous THF. The reac~on mixturc was stirred at -50 for 30 min to
allow fo~ complcte fo~mation of the lithium anion (slight ~rbidity~. The m~ix~e was
again cooled to -60 and a solution of e~yl 3,5-di-tert-butyI-~hydroxycinnamate (2.6
10 g, 8.5 mmol) in 20 ml dry THF was added. The resulhng orange-colored mixture was
left to stir at room temperanlIe (25C) for 18 h. Hydrolysis was caIIied out by adding
10 ml of a lO~o HCl sc~ludon and the produc~ was extracted into ether. After ~ngove~ MgSO4, e~e~ was evapor~ed to yield a yellow solid. RecIystallization in 40 - 60
petroleum ether gave 3.0 g (92% yield) of dimethyl ~(3,~ te~-bu~yl~
15 hydroxyphenyl)-2-ox~3-buten-1-yl phosphonate.
mp=107-109C
The ~de compound can also be obtained by using as the bases a mixture of n-BuLi and
20 LDA ~ithium diisopropylamide). To a solu~on of n-butyllithium (16ml of a 1.6 M
solution, 25.6 mmol) in 20 ml dry l~ was added at -60C diisopropyl amine (0.86 g,
8.5 mmol). The resulting mixture was st~red at 40 for 15min, then dime~yl
methylphosphonate (2.11 g, 17 mmol) was added. After 15 min, ethyl 3,5-di-tert-butyl-
4-hydroxycinnamate (2.6 g, 8.5 mmol) was added and the rea~tion mixture was stirred
25 at room temperat~e (25C) fo~ 15 h. Work up by addition of 10% HCl and extraction
into ether gave 3.10 g (95%) of dimethyl ~(3,5-di-tert-butyl~hydroxyphenyl)-2-ox~
3-butcn-1-yl phosphonate.
mp=107-109C
30 The compound p~epared by dther of these n~o variant p~ocesses has spectroscopic data
idenical to thiose of the same product described in example 1.
24
, i1 ' . ~
~) WO 94/19358 2I 18 4 09 PCT/EPg4/00520
C2bH3105P Theor. 9~oC 62.81 %H 8.17 96P 8.10
Pound %C 62.56 %H 8.21 %P 8.24
; 5 ~ ~m~ll~
Diethv! ~f3 S-di-tert-butvl~hvdroxv~henvl)-l.l-dime~vl-2-oxo-3.5-hexadien-1-vl
~hos~hona~e `
..
t--Bu ~ `
)~ 8 1CH3
~; HG~CH=CH--CH=CH C C--PO3Et2
CH3 `
t--Bu
: ' ~`.
To 15 ml dry THF kcpt at 0 were added rlcl4 (1.23 g, 6.47 mmol), 3,5-di-tert-butyl-
4hydm~ cinnamaldehydc (0.7 g, 2.67 mmol), diethyl 1,1 dimethyl-2-
~; ~io~_ (0.72 g, 3.24 mmol) and N-methylmorpholine (1.31 g, 12.97
~ . Thc leaction mixture was sti~rcd for 1 h at 20C then 1 h at 30C. Work-up
gare a da~ oil which was purified by column chromatography (SiO2, 8/2
C~IC13/AcOEt). 650 mg (52%) of yellow crystals werc obtained, mp = 13~133C
:
NMR (CDC13)
o = 7.40 (d x d, J = 11 and 15Hz, lH): Ph-CH~-CH=CH
7.2 (s, 2H): arom. H - ~- ` ` `
6.87 and 6.85 (2 d, J = 15Hz, 2H): Ph-~=CH-CH~
6.75 (d x d, J = 11.5 and 16Hz, lH): Ph-CH=CH-C~H
- 5.4(s, lH): OH
4.1 (m, 4H): P-O-C~12-CH3
1.40 (d, J = 16Hz, 6 H): C(C~I3)2
1.39 (s. 18H): t-C4Hg
1.24 (t, J = 7Hz, 6 H): P-O-CH2-C~3
MS: tn/e~: M+, 326: M+- HP03Et2, 285 (100*): M+- CMe2-P03Et2
C26H41SP Calc- % C 67.22 % H 8.90 % P 6.67
Found % C 66.68 % H 8.56 % P 6.21
wO 94/19358 ~Q~ 4~9 ; PCT/EP94l00520 -~ ~
`:
t--Bu
~ 11 .
H~CH2-CH2- C --CH2 PO3Me2 ;~
t--Bu
A solution of dimethyl lithiomethylphosphonate was prepared under nitrogcn by
adding dimethyl mcthylphosphona~c (3.2 g,25.6 mmol) to a solution of n-
butyllithium (16 ml of a 1.6 M soludon in hexane, 25.6 mmol) in 15 ml dry THF at - `
60C. To this solution was added at -60 a solution of ethyl 3,5-di-tert-butyl-~hydroxyhydrocinnamate (2.6 g, 8.5 mmol) in 20ml THF. The resulting solution was
stirrcd at -60 for 30 min and then was allowed to reach r~om temperature (25C)ovemight. 25 ml 10% HCl was addcd and the mixture was ex~racted into ether. The ;~
residue of the eth~ phase was purified by column chromatography (SiO2,8/2
- CHC13/AcOEt) to yield a white solid. Recrystallizadon in petroleum ether gave 2.1 g
(64% yield) of thc dde compound.
mp=78-79C
MS: m/e=384: M+, 284: M+-HP03Mc2,57 (100%): tBu+
N~ (CDC13)
~ = 6.98 (s, 2H): arom. H
5.06 (s, lH): OH
3.75 (d, J = 11.5Hz, 6H): P-O-C~I3
3.08 (d, J = 22.5Hz,2H): (~12-P
2.90 (m, 2H): Ph-c~2-cH2
2.81 (m, 2H): Ph-CH2-C~12
1.41 (s~ 18H): t-C4~1g
26
WO 94/19358 21 18 ~ ~ 9 PCT/EP94/00520
~2, :
Diethvl ~(3~-di-tert-butvl-4-hv~henyl~-æ4-di~-s-hex~n-l-vl ~ho~hon~
t Bu
)~\ i~) ~) .'
HO~\ /)--CH=CH C --CH2 C -- CH2 PO3 Et2
~ `:
t-Bu
Diethyl 2-oxopr~pylphosphonate (3.4 g, 17 mmol) was added at room temperan~re
under nitrogen to a suspension of sodium hydride (0.82 g of a 60% dispension, 201~ mmol) in 35 ml dry T~IF. The mixture was sti~ at room temperature for 60 min,then diisopropylan~ine (1.71 g, 17 mmol) was added at 0, followed by n-
butyllithium (21 ml of a 1.6 M solution, 34 mm~l). After 30 min at 0, the mixture
was cooled to -60 and a solution of ethyl 3,5~ tert-butyl-~hydroxycinnamate (2.6
g, 8.5 mmol) in 25 ml T~ was induced dropwise. The resulting mixture was sdrred
at 0 fc~r 2 h, at 25C for 1 h, hydrolyæd with 60 ml H20 whereupon it separatedinto two phases. Thc aqueous phase was acidified with 10% HCl and was extracted
wi~ two 100 ml po~ons of ether. The ether extracts were pooled with the T~
phase, dried and cvaporated. The residue was purified by column ch~omatography
(SiO2, 8/2 C~HC13/AcOEt) to yield a viscous olange oil. Recrystallizadon in ligroin
gave 2.25 g ~60% yield) of the title compound, mp=109-1 10C
NI~ (CDC13)
= 7.60 (d, J=16Hz, lH): Ph-C~=CH
7.37 (s, 2H): arom. H
6.35 ~d, J= 16Hz, lH): Ph-CH~
5.83 (s, lH): CO-(~=C-OH
5.54 (s, lH): phenol O~
4.17 (m, 4H): P-O-C~12-CH3
3.02 (d, J = 22Hz, 2H): C;~12-P
1.46 (s, i8H): t-C4~g
1.34 (t, J = 7Hz, 3H): P-O-CH2-~3
MS: m/e 452 M+, 314 M+ - HP03Et2, 57 (100%) tBu
~ -~~ . .
WO 94/19358 . PCT/EPg4/00520 - `~
E~ ..
t~BU
O O
~\ ~1 il ~
HO~C:H2 -CH2--C--CH2 C CH~--PO3 Et2
/
t Bu
Diethyl 2-oxopropylphosphonate (2.02 g, lO mmol) was added at room temperature
to sodium hydride (0.48 g of a 60% dispersion, 12 mmol) suspended ~ 30 ml T~.
10 Aftc~ 30 min the mixn~re was cooled to 0 and diisopropylamine (1.01 g~ 10 mmol)
and n-butyllithium (13 ml of a 1.~ M solution, 21 mmol) we~e added. After 30 min,
the mixture was cooled to -60 snd ethyl 3,5~i-tert-butyl-4-hydroxy hydrocinnamate
(1.6 g, 5.2 mmol) dissolved in 15 ml THF was added. The ~h~re was left to react
at -60 for 15 min then at 0 fo¢ 2 h and 25C for 1 h and was hydrolyzed with 10%
5 HCl and ex~wted with e~er. Column chromatography (SiO2, 8/2 CHCl3/AcOEt)
gave 1.0 g (42%) of ~he tide compound as a colorless oil.
. ~ ................................ .
(CDC13) ~ ; r
~ ~ 6.98 (m, 2H): arom. H
5.70 (s, lH): CO-C~CC-OH
5.09 (s, lH): phenol OH
4.15 (m, 4H~: P-O~ 2-CH3
2.92 (d, J = æ.~ 2H): ~I2-P
2.85 (m, 2H): Ph-~2-CH2
2.59 (m, 2H): Ph-cH2-(~I2
1.43 (s, 18H): t-C4~Ig
1.33 (t, J = 7Hz, 6H): P-~CH2-C~3
MS: 455 M+ +1, 454 M+, 436 M+ -H20, 57 (100%) t:Bu
wo 94/193~8 21 I 8 4 0 9 PCT/EP94/00520
E~l!l .
Dimethvl ~f3~$ di-te~-~u~l-~h~drox~henvl2-~d~oxo-~-h~n-1-vl
~b~ na~
:
t--Bu
O O
/~\ 11 11
HO~ ~CH=CH C CH2 ~ C --CH2 PO3 Me2
r
t--Bu
The procedure ~s~ibed in example 12 was followed using dime~yl 2-ox~
pr~pylphosphnnate to give the title compound at ?9% yield, yellow solid with
lo mp = 14~-146C.
~ .
,2-r4-(3 5-~-butvl~-hvdrox~nyl~2-ox~3-bu~n-1-vU
15 f2-ox~1.3.2~ioxaDhosphorinan)
t -Bu
11~~
H~CH=CH--C --CH2 P
t-Bu
Under nitrogen atmosphe~e, 2-methyl-2-ox~1,3,2-dioxaphosphorinan (2.62g, 19.2
mmol) dissolved in THF~dioxane (30ml each) was added to an equimolar amount of
n-butyllithium in 35ml TH~ at -60C. The mixture was stirred at -60C for 30 minthen a solu~on of ethyl 3,5-di-tert-butyl-4-hydroxycinnamate (2g, 6.4 mmol) in
20ml TH~ was added. The resulting mixture was sti~Ted at -60C for 30 min and left
to attain room temperature (25C) over 16 h. After the usual work up, column
chrornatogra~hy (silicagel, 98/2 CHC13~MeOH) gave 0.76g of the title compound
(30%).
mp = 158-161C
MS: m/e = 394 M+, 57 (100%): tBu
29
WQ 94/19358 . PCT/EP94/00520 -" ~
4~ ~
. ~ '
Nl~ (CD~13): ,
- 7.65 (d, J= 16Hz, lH): Ph~
7.42 (s, 2H): arom. H
6.74 ( d, J = 16Hz, lH): Ph-CH~
5.7 (s, lH): QH
4.5-4.4 (largc m, 2H): P-O-C~2
3.4~ (d, J = 22~z): ~I2-P
2.1 and 2.0 (2m, 2H): P-~CH2-C~2
1.45 (s, 18H): t-C4Hg
~am~2~
t--Bu
)~ 8
HO~ C CH=CH CH2 --PO3 Et2
t--Bu
To a suspcnsion of 730mg 60% sodium hydride (18 mmol) in 20ml THF was added
3;0g t8.4 mmol) of dimethyl 2-(3,5-di-tèrt-butyl-4-hydroxyphenyl)-2-oxo-1
20 e~ylphosphonatc in 25ml THF. Thc mixture was s~ed for 30 min then it was
cooled to 0 and 3.1g tl8 mmol) die~hyl foqmylmethylphosphonate was added. The
resulting mixture was sti~red at 0C for 1 h then at 25C for 16h, par~tioned into
cther and watcr and the o~ganic phasc was evaporated. Purificadon by means of
column chromatography (silicagd 98/2 dichlo~omethane/methyl t-butyl ethcr) gave
25 550mg (16%) of the dde compound.
mp = 68-70C
MS=m/e: 410 M+, 39~: M+-CH3, 272: M+-HP03E~2,
233 (1~0%) M+-CH=CH-CH2P03Et2
-! W094/l9358 21I8~09 PCTIEP~4/00520
NMR: CDC13
= 7.83 (m, 2H): arom. H,
7.1 (dd, 1=15 and 4.5Hz, lH), Ph-CO~
6.9 (m, 2H): Ph-CO-CH~
5.7 (s, lH): OH
4.15 (m, 4H): P-O-C~2-CH3
2.86 (dd, J=23 and 7Hz): C~2-P
1.48 (s, 18H): t-C4Hg
1.34(t, 6H): P-O-CH2-C~3
~ime~vl ~(3.5-di-~ert-butvl~hy~roxv ~henvl)-2-hvdroxv-3-buten-1-vl
~ ~.
lS
t-Bu
)~ f
HO~ CH=CH CH CH2 PO3Me2
r
t--Bu
To 100 ml of a methanol solution of dimethyl 4-(3,5-di-tert-butyl~hydroxyphenyl)-
2-oxo-3-buten-1-yl phosphonate (19g, 5 mmol) cooled to -10C were added 0.75g
20 sodium borohydride. The reacting mixture was stirred at -10C for 1 h then at room
- temperature (2SC) for 2 h. Wor~ up was carried out by addi~on of 100ml ether and
60ml sodium bicarbonate soludon. Thc ether phase was washed with brine, dried
over MgSO4 and e~raporated to yield 1.6g (85%) of ~e tide compound.
.;!-
25 mp = 122-123C -
MS (m/e)= 384:M+, 366: M+-H20, 256: M+-H2~HP03Me2
Wo 94l19358~ ~ 3 PCTEP94/0052U
NMR (C~DC~3): .
7.21 (s, 2H): arom.;H
6.59 (d, J = 16Hz, i`H): Ph-C~H,
6.08 ~dd, J = 16 and 6Hz, lH): Ph-CH-{~
5.26 (s, lH): OH (phenol)
4.7 (rn, lH): C~-OH
3.78 (2xd, 6H): PO3~2
3.4 (hump, lH): CH-O~
2.15 (distorted dd, 2H): C~2-P
1.44 (t, 18H): t-C4Hg
Dimethvl ~r3.s di-tert-blltvl~hvdrox~henvl~-2-ox~3^butcn-1-vl
.~iono~ho~honate
t--Bu
O S
/=\ 11 11 ,
HO--~\ /; CH=CH C --CH2 P (OMe) 2 :
~~ .
t--Bu
Undcr nitrogcn atmosphcrc dimethyl methylthionophosphonate (3. lg, 25 rnmol) was20 added at -60C to a soludon of n-butyllithium (16ml of a 1.6M soludon, 25.6 rnmol)
in 50ml lL~. Thc mixture was st~ed at -60C for 15 rnin then a solution of ethyl3,5~-tert-butyl 4 hydro~cy cinnamate (2.5g, 8.2 mmol) in 20 ml T~ was added.
Thc resulting mi~cture was sturcd at -60C for 30 min and at room temperature
(25C) for 2 h. After the usual hydrolysis and wo~ up, the compound was purified25 by column chromatography (SiO2, 9/1 CHCl3/AcOEt) and rec~ystallization in
CHC131petro1eum ether to yield 1.7g (52%) of a yellow solid, mp = 88-90C
MS: mle = 398: M+, 259: M+-CH2P(S)(OMe)2, 125: P(S)(OMe)2,
57 (100%)
~. ~.. .
`` WO 94/19358 2118 4 09 PCTIEP94/00520
NMR (CDC13~
~= 7.60 (d, J - 16Hz, lH): Ph-C~=CH
7.42 (s, 2H): aro~ H
6.75 (d, J = 16Hz, lH): Ph-CH=C~
5.6 (s~ lH): O~
3.78 (d, J = 14Hz, 6H): P(S)-~3
3.56 (d, J = 20Hz, 2H~ 2-P
1.46 (s, lg~ t-C~g
~ ~2(iH31O4PS Theor. %C 60.28 %H 7.84 %P 7.77 %S 8.04
Pound %C60.58 %H 8.04 %P8.05 %S 8.30
~m~2
t--Bu
HO~5 CH2 C--CH2 PO3 Me2
t--BU : :
To a T~ solu~on o~ dimethyl lidliomethylphosphonate (24.6 mmol) kept at -60C
20 was added a solu~on of e~yl 3,5 di-tert-bu~rl~hydroxyphenyl ~ioacetate (2g, 6.2
mmol) in l5ml T~. The resul~ng mixnIre was stim2d at -60C for 30 min and was
allowed to reach r~om tempe~re (25C) over 4 h. Afte~ addition of l~ml 10% HM~
thç reaction mix~e wæ pardtioned between ether and water. The residue after
evapo~don OI the orgal~ic phase was purified by column chromatography (SiO2, ~/22s CHC13/AcOEt) and re~ystallization in petroleum e~er to give O.95g of dle dtle compound (38%).
mp = 63-65C
MS: m/e 402: M+, 251: M+-(CO-CH2-P03Me2), 57
WO 94/19358 ?,~,~,Q~ 4~3 PCT/EP94/00520 -`
NMR (CDCl3~
= 7.21 (s, 2H): arom H
5.3 (s, lH): OH
3.75 (d, J ~ 1 lHz, 6H): P-O-C~13
3.72 (s, 2H): S-C~2-CO
3.33 (d, J = 22Hz, 2H): C~2-P
1.41 (s, 18H): t-C4~1g
~m~2Q
,Dimethvl 3-f ~di~:~l~hvdrox~henvlthio)-3~imç~d-~ 1-~l
~hos~hona~
t-Bu
)~ CH3 8
H~S--C--C - CH2 PO3 2
t-Bu
'. 15
i, To 10ml of a T~ solutioD of dimethyl lithiomethylphosphonate (68.2 mmol) kept at
60C was added a solution of e~yl 2-(3,5-di-lcrt-butyl~hydroxyphcnyl-thio-2-
mcthylpropionate (6g 17.1 mmol) in 15ml THF. The resulthg mixture was sti~ed at
-60C for 30 min and was allowed to reach room temperature (25C) over 4 h. After
addition of 15ml 1096 HCI, the reaction mi1cture was partitioned between ether and
water. The residue after evaporadon of thc organic phase was purified by column
chromatography (SiO2, 8n CHC13/AcOEt). Rec~ystallization in pe~leum ether
gave 4.16g of the title compound (57%).
mp = 105-106C ~ ~
2s
MS: m/e 430: M+, 279: M+-(CO-CH2-P03Me2), 194 (100%), 57
N~ (CDC13)
~ = 7.15 (s, 2H): arom H
5.38 (s, lH): OH
3.80 (d; J = 1 lHz, 6H): P-O-C~13
3.46 (d, J = 22Hz. 2H): C~2-P
1.41 (m, 24H):t-C4~1g + C(C~13)2
- WO 94/19358 21 1 ~ 4 0 9 - ~CT~P94/00~20
~mR~
t-Bu
~,
HO~CH=CH-CH~CH PO3Me2
t-Bu
A mix~ure of dimethyl ~(3,5-di-tert-butyl~hydroxyphenyl) 2-hydroxy-3-buten-1-
yl phosphor~ate ~2.3g, 6 mmol) in 1.70ml acetic anhydride and 1.26ml triethylamine
was stirred at 60C for 3h. Following hydrolysis with lOml H20, the reac:tion
lo mixture was ex~acted with 20ml diethyl ethcr. The e~her phase was extracted with
10% HCl and d~ied over MgSO4. The residue after evaporation was purifiled by
column chromatography to give l.Og (46%) o~ the title compound.
mp= 113-115C
MS: m/e: 366 M~, 351 M+-Me, 57 (tBu)
NMR (~DC13)
= 7.32-720 (m, 3H): arom H + CH~H-~ P
6.82 (lH,d)~ ~-CH~I-P
6.74-6.66 (lH, m) C~H~-CH=CH-P
5.67 (lH, dd, J = 16.6 and l9.~Hz): CH=CH-CH~-P
3.75 (d, 6H): Po-(~EI3
1.46 (19H, s): t-C4~g
WO 94/19358 , . PCT/EP94/00520 `"`,
12im~h~4 (3.5~ tert-butvl ~ hvdrox~ 2-f4 ~en~n~vln~ u~l
~QS~hopate
0 :~
t-Bu 1 ¦
)~ o--C--CH2-CH2-CH~CH2
H~cH2 -CH2 CH -cH2--Po3 Me2
t--~u
To a mixture of acetyl~pentcnoate (0.66g, 4.6 mmol) and triethylamine (0.24ml,
1.7 mmol) was added 0.50g (1.37 mmol) of dimethyl ~(3,S-di-tert-butyl~
10 hydroxyphenyl)-2-hydroxy- l-butyl phosphonate. The reaction mixture was kept at
60C for 6 h, cooled and extracted in~o diethyl ethe~ and water. The residue of the
dried organic phase was purified by column chromatography (SiO2, 7/3
CHC13~Mc~yl-tert-butyl ethcr) to yield OAg of a colorless oil (62%).
MS: m/e: 468: M+, 368: M+- CH22H-CH2-COOH, 258: 368 - HP03Me2
N~ (CDC13) t'
= 6.94 (s, 2~: arom H
5.85 (m, lH): C~H2~-CH2-CH2-COO
5.18 (m, lH): Ph-CH2-CH2-~-CH2-P
5.0 - S.l (m, 2H): C~I2-cH-cH2-cH2-coo
5.15 (s, lH): OH (phenol)
3.72 (2xd, 6H): P3~2
2.55 (m, 2H~: Ph-cH2-cH2-cH-c~2-p
2s 2.41 (m, 4H): CH2~-c~2-c~l2
1.43 (s, 18H): t-C4Hg
36
`) wo 94/19358 21 1 8 4 0 9 PCT/EP94tO0520
I:a}2~L PHC)SPHONATES OF FORMULA (I)
X~ y
)~ ,I,Z
~D--P~ ( I )
Comp G xl, x2 D _ zl z2 mp ~C) I~Jcroar;lysi5
1 HO ~BuCH=C~-C(0~ 2 O OMe 107-109 C20H3105P
2 HO ~BuCH=~H~(0~H2 OEt 110~111 C22H3505P
3 HO ~BuC~H_C~H-C(~)~C~H2 O O~Pr121 - 122 C24H390sp
4 HO tBuC~H=~H-C~O)~C iH2 ChiBu6~i7 C26H4305P
HO ~BuC~H=~H-C(O)-C(cH3)2 O OMe 125-126 cæH3sosp
6 HO ~BuC~H=c~H-c(o)-c(c~H3)2 O OEt 91 -92 ~24H3905P
7 ~IO ~BuC3H=CH-C(O~C(C~3)2 O O~Pr138-139 C26H4305p
8 HO ~BuC~H=C~H-C(03-CH(C~H3) O OMe 144-145 C21H330sp
9 HO ~BuCH=C~H-C(O)IC~H(C~H3) O OEt 92-94 C23H3705P
EK~ tBuC~H=dC~C~H3~C(~)~ClH2 O OMe 131-133 C21H3305p11 HC) ~BuC~=d~(C~H3)~(0~5~H2 O OEt 97-101 C23H3705P
12 H~ tBuCH=C(nC4Hg~C(O) CH2 O OMe 82-85 C24H3905P
13 HO tBuCH=C(nCsH1 l)-C(O)~H2 O OMe 67-72 C25H4105P
14 HO tBuCH=C~Ph~C(O) CH2 O OMe 147-149 C26H3505P
HO tBuCH=C(Ph) C(O) CH2 O OEt139-143 C2gH3905P16 HO ~uCH2 CH2~(0) CH2 o OMe 78-79 C20H3305P
17 HO tBuCH2-CH2~(0~CH2 O OEt oil C22H3705P
18 HO tBuCH2-CEI2~(0) C(CH3)2 O OMe 85-87 C22H3705P
19. HO IBuCH2~ -C(O~C(cH3)2 O OEt 64 66 C24H4105P
HO IBuCH~H-CH=CH~(O) CH2 C) OEt oil
21 HO tBuCH=CH-CH=CH~(O~C(CH3)2 O OMe142-146
22 HO tBuCH=CH-CH=CH-C(O)-C(CH3)2 O OEt130-133 C26H4105
23 HO tBu C(O~CH2 O OMe108-111 C18H2905
24 HO tBuCH=CH-C(O)-CH2 O NMe2178-179 C22H3703
HO IBuC(O)-CH2-C(O)-cH2 O OMe108-109 C20H3106
26 HO tBuCH=CH-C(O~CH2~(0)-CH2 O OMe145-146 C22H3306
27 HO tBuCH=C~-C(O~CH2-C(O)-CH2 O OEt109-110 C24H3706
28 HO lliuCH2-CH2-C(O) CH2-C(O)-CH2 O OEl oil C24H3906
37
wo s4tls3s8 PCT/EP94/00520 f- ~ ~
4~9 . I
I:ak~ PH(:)SPHONAl'ES O~; FORMULA (I) (cont.)
x
P~ ( I ~
Comp. G xl,x2 y zl z2mp(C) M~c~analysu
.. . __ .... . " . _ _ .......... . .
29 HO ~BuCH2~2~(0) CH2~(o)~H2 O OMe oil C22H3506P
HO tBuCH=C(C2Hs)-C(O~CH~ O OMe 128-132 C22H3505P
31 HO sBuCH=CH-C(O)-CH2 O OMe oil C20H3 l 5P
32 HO s~uCH=CH-C(O~H2 O OEt oil C22H350'iP
33 MeO tBuCH=CH-C(O)-CH2 O OMe 73 75 C21H335P
34 HO tBuCH=CH-C(O)~H2 S OMe 88-90 C20H3104P
HO tBuCH=CH-C(O)-CH2 O-(~H2)30 156-lfil C21H3l05
36 HO lBuC(O)-CH=CH~H2 O OMe oil C20H3los
37 HO tBuC(O) CH=CH~H2 O OE~ 65-70 C22H3505
38 HO tBuC(O)-CH2 C)OEt 112-115 C20H3305
39 HO tBuC(O)-CH2 OOiPr 127-131 C20H3705
HO tBuS-CH2-C(O)-CH2 O OMe 62~5 C19H3105
41 HO tBuS-CH2-C(O)-CH2 (~OEt 76-78 C21 H3505
42 HO tBuS-C(CH3)2-C(O) CH2 o OMe 105-106 C21H3505
43 HO tBuS-C(CH3)2-C(O~cH2 O OEt 8~81 C23H3gOs
44 HO tBuS-CH2-C(~CH2-C(o)-cH2 O OMe oil C21H3306
HO tBuS-CH2~(0) CH2-c(o)-cH2 O OEl oil C23H3706
46 HO tBuCH-CH-CH(OH)-CH2 O OMe 122- 123 C20~335
47 HO tBuCH2-CH~H(OH) CH2 o OMe 88-91 C20H3505
48 HO IBuCH(0H) CH2 O OMe 128-133 C18H310s
49 HO IBuCH(OH)-CH2 CH(OH)-CH2 OOMe 129-132 C20H3506
HO tBuS-CH2-cH(OH) CH2 oOMe 1~108 Cl9H335
51 HO ~uCH=CH-CH=CH OOMe 113- 115 C22H3506
52 HO tBu(CH~)2-CH(O-CO-cH3) CH2 OOMe oil C22H370
53 HO tBu(cH2)2-cH(o-co-ncsHl l)~H2 OOMe oil C26H450
54 HO ~u(CH2)2-cHlo-co-(cH2)2~H=cH23-cH2 oOMe oil C25H416
HO SBuS-CH2-CH(O-CO~H3)-CH2 OOMe oil C21H3506
56 HO ~BuS-CH2 CH(O-CO-nC5Hll)-cH2 OOMe l~il C25H430
38
`) WO 94/193~8 PCT/EW4/00520
2118~og
A. ~i:~=~
I
1) lron i,nduced,~eroxide fo~mation in rat li~hQm~en~
Wis~r raes were euthanasied by cthe~ inhiala~on. The liversi were dissected out and
', homogenised with a poeter homogeneiser in 4 volumes of phosphate buffer (4C,
pH 7.4). After cen~ifugation at 2000 ~pm for 10 min the supema~ant obtained was
o kept ae 4C.
A mixeure containing 0.2 ml of liver homogenate, 1.7 ml of phosphate buffer was
incubated with 0.1 ml of a 2mM PeS04 soludon to induce peroxide foImaeion
according to the method descnbed by A. T. Quintanilha et al., Ann. N. Y. Acad. ,:
lS Sci., ~, 32~7, 1982.
Compounds to be tested for antioxidant ac~vity were dissolved in DMSO or
cehanol and added iLn a volume of 5 111 to the incubation mixture. Stock solu~ion of
the compounds were diluted scquen~ally to obtain final concen~ation of 0.5, 1, 2,
- 20 5 and 5 pM. Oxidation was pe~fon~ed at 37C for 2 hours and was stopped by the
addition of 20 pl of a 2* ethanolic solution of BHT. The generated peroxides
meas~d as malondialdehydc fonnjation were quantitated according to the method
of Yagi ("Iipid Pe~xidcs in Biology and Medicine" p. 223-242, 1982, Ed. K.
Yagi, Academic Press ~c.) u~g dle Thioba~bituric Acid Reaction and with
2s 1,1,3,3,-telrame~oxy~ropa~ie as standard. Results aIe given as concentranon in
pmoUI which inhibits malondialdehyde formation by 50% (IC50).
..,. ~ . .... ..
All ~e tested compounds have IC50's between 0.5 and 5 ~M and are more active
~an Probucol, vitamin E and Vitamin C Bu~l hydroxytoluene (13HT) has an IC50
of 3.3 ~M in ~is assay. Compound of foImula (I) are thus useful for the treatment
of disease sta~es jD which oxygeD re~c~ve species ale involved.
39
wo 94/19358 Q~ ~9 PCT/EP~4/00520 --!
T~ble 2
. ...... ~
Compound lC50 (pM)_
Probucol ~ 25
~ita~n E _ > 25
Yitam~n C _ ~ 25
BHT _ 3.3
, , _l, ~.01
2 _ 3~20
_ 3 _ _ 1~37
4 1~41
_ _
1~0__
_ 16 4~89
17 3~03
_
1 2~85
3 ~ _ 1~88
~ ~ 3~18
~ ~38
7 . ~ . . t ^ ' ' 2 06
18 3~56
i9~ ~ 2~49
_
12 ~ 1~71
3~48
4 3~07
20 ~ 2~44
24 ~ 4~55
25 ` ~_ 2~71
26 0~68
27 0.70
28 1.0
32 2~78
33_ 5~0
34 2~91
2~84
`~ WO 94/19358 21 1 810 9 PCTIEP94/00~20
2)~
Plasma was obtained after low speed centrifuga~on of blood from donors. I,DL
(d=1.006 1.C63 glml) were isolated by preparative ultracentrifugation in a salt
s solu~on (NaBr, KBr). The isolated LDL fraction was dialysed against phosphate
buffer.
LDL oxida~on was pe$fo~med a~eording to Ester~auer et al. (Continuous
Monit~g of in Vitro Oxidation of Human Low Density Lipoprotein, Free Rad.
Res. Commun. ~, 67-75, 1989). Briefly the LDL suspension (50-2001~g protein/ml)
was distributed in quartz cuvettes and kep~ at 37C then a solution of CuC12 wasadd~d at a final concentration of 511M. The increase in optical density at 235 nm
was recorded using a W-visible spec~ophotometer. The time course of oxidation
was recorded over a penod of 8 hours at 10 min intervals. Compouncls to be tested
we~e dissolved in ethanol and added at the final concentraaon of 0.1 ~M. Controls
~eceived ethanol only. The lag phase is pr~longed by the p~esence of antioxidants.
This medlod was validated with Probueol and vi~-E as reference antioxiclants.
The prolongation of the lag phase was uscd to quantify ~e antioxidant activity of
the compounds tcsted, this pr~longation W2~ expressed in percent of ~e value
measured in absence of exogen an~oxidant (controls).
` ~,,, J` , ,`
The phosphonates of Formula (I) as noted in TaUe 3 prolong the lag phase
compared to control. An inhibitoIy activi~ on LDL oxidation is thus demonstrated2~ which is clearly supe~ior to that of Prt)bucol and vitamin E. Since these two
antioxidants have been shown to be anti-atherosclerotic in animal models, the
therap~utic poten~ial of phosphonates of Fo~mula (I) is ob~rious.
41
wo 94/193~ 9 PCT/EP94100~20 .--
Table 3
C~-: _nll Lag pbase
% con~ol
. ~ . . _ _
~obuccl _+ 7
Vitamin E _ 0
1 _ ~ 1 45
~ _ _ 7
3_ ~ + 172
4 _ _ ~ 74
_ ~ 233
16 _ l 159
17 ~195
11 _ _+212
13 _ + 191
~ 147
6 . + 129
`~. . . ~ ~
18 _ ~ 223
19 ~ 157
: . 9 __ + 1 12
179
:~ 10 + 126
14 138
: 20 ~- + 38
24 16
+ 1 18
26 ~
27 + 42
28 ~
32 + 80
33 + 14
~ 8
+ 118
42
2118409
- ~ wo 94/193~8 PCT/E:Pg4/00520
B. ~
The human intes~nal cell line CaCo~ cells (ATCC HTB37) was used to study the
effect of compounds of formula (I) on choles~erol syndlesis. The cells were grown
S in 6 wells dishes (Falcon) in 2 ml of Dulbecco's m~dified Eagle cult~re medium
(I:)MEM) supplemented with 20% fetal calf serum (Flow). Cells were maintained
at 3PC in a 5% CO2 a~nosphe~e and the labelling expenment was done 8 days
after cell pla~ng. To ~e culture were added lû pl of ~e ethanol solu~on of the
compounds to be tested. Control wells ~eived lOpl of ethanol alone. One hour
later 0.7 yCi of 14 C-acetate 53.4 mCilmmol was added, labelling was connnued
for 4 hours and w~s stopped by washing ~e ccll layer with chilled P~3S. The cells
wcre collected in 2ml of O.O1 N NaOH and 1 ml of PBS. Lipids were extracted by
the Folch method and separated on silica gd TLC plates developed in petroleum
ethes: diethyl ether: acetic acid (70:30: O.5). After exposition to iodine vapors, ~e
bands coiTespondiT~g to cholesterol and choleste~yl esters were scrapped off and~dio~ctivity was measur~d in a liquid scin~llator countcr.
l~ne amount of radioactivity inco~ted in cholestcrol and cholesteryl esters in
presence of compounds to be tested was compared to that of the control cells.
HMGCoA r~ductase inhibito~s such as simvastatin (lyM) s~ved to validate the
measuremcnt of 14C-acetate inco~poration in ~holestcrol and cholesteryl csters.
All thc compounds tcstcd inhibited cholestcrol and cholesteryl est~s synthesis.
-- Most of the compou~ds inhibi~ed choles~e~yl ès~ers synthesis by more than 50%
CTablc 4~. Phosphonates of fo~mula (I) display an inhibitory activity on choleste~ol
and cholestayl estcrs and can be considcrd as therapcutic agcnts in thc treatmcnt
of hgpe~lipidania and ather~sclerosis.
43
WO ~4lls35B , PCTIEPg4/00~20 --~
Table 4
_ ~
Choleteryl
(::ompounds Choleterol ester~
% %
Simvasta~in 96_ __ 70.
_ _ _ _ _ _
~ _ -5? - __
___ 3 _ -i55 -84
. 4_ _ _ 21 80
11 -34 -80
__ __ .. .
_13 ~9 _ 84
. --26 j _
-22 _ -75
19_ _~46 -83
9 _ -49 _ 81_ .
i - 14 26 ~ 84 _
_
1 ~ 24 ` ` 1 1 5 r ~l ~ -60
_ _
33 - 54~ ; ~ -34
~0
wo 94/1g358 2 I 1 8 4 0 9 PCT/EPs4/00520
C. ~alci~ Fntr~ Rlockin~ activitv of ketoDhosDhonates
Expenments were performed on aor~c nngs from rnale Sprague -Dawley rats
(28~350g body weight) which were killed by stunning and exsan~nation.
Thoracic aortas were cleared of connective tissues and cut into ~ings of
approximately 2mm in length. Each ring was mounted under a resting tension of
2g and was equilibrated for 1 hour at 37C in a lO ml organ bath containing a
HEPES bufferRinger soludon (Buffer composition (mM): NaCl 139.0, KCl 5.0,
MgC12 3-7. ~Glucose 11.0, HEPES 5.0, pH 7.4) acrated with 95% 2:5% CO2.
~'
Maximal contractions we~e produced within 5-lO min exposure to 10pM
Phenylephnne. The dssues were then washed with a Ca+2 free HEPES buffer.
After 30 min the dssue was depolarised with KCl (60~I).One hundred ~ of the
vehicle (10% DMSO) or compound solution (1 yM) werc added S min later. The
final concentra~on of DMSO was 1%. The tissues ~n=2 per compound) were
further equili~ated for 15 min in the presence of compounds prior to cumulative ` -
addition of Ca+2 (0.1-30 mM). The contractions to cach concentration of calcium
are calculated as a percentage of the second phenylephrine contraction and the
EC30 (concentration of Ca2+ producing a contraction 30% of the phenylephrine
contraction) calculated. Thè potericy index of cach compound is expressed as theconcentration ~io (calcium drug EC30hehicle EC30), where a potency index ~1
indicates a c~npound effect.
The compounds of formula (~ are thus potentially useful in the treatment of
cardiovascular discases via their calcium eritry blocking activity. The pnmary
indicadons of these compounds would be the t~ent of atherosclerosis, angina
pocto~is, congesdve heart failure and hypertension.
Tabae 5
-
Effect`of compounds (I) on Ca+2 induccd
contraction of K+ depolarizcd rat aoqta
.
Compound EC30Ratio
.` 6 21.8
7 9.2
16 4.2
26 4.7
28 5.~ ~-