Note: Descriptions are shown in the official language in which they were submitted.
Wo 93/07472 P~r/uSs2/08363
`"` 211~13~
R~PID ASSAY FOR GOLD AND INSTRllMEN~ATION
USEFVL THE~ OR
The present invention relates ~o analytical methods. Ln a particular
aspect, the present invention relates to methods and apparatus ulseful for perforl2~ing
a rapid assay of gold or other elemental conccntrations, especially low concentra~ons
of gold in rock and soil samples. In another aspect, the present invention relates to
S analytical methods and apparatus that are both rapid and porta~le, so as to be useful
in field applications.
Background of the Invention ~ -
Most of the gold deposits being discovered today are made up of gold
10 grains that are invisible by normal microscopy. These gold particles are often smaller
than one micr~n in size. B~cause ~e gold is not detectable by any readily available,
portable m~s, prospecting presently depends almost completely upon reliaUe
analytical techniques which can only be G~rried out in large analy~cal laboratwies.
Such facili~es are, by necessi~r (due to the nature of ~eir equipment), large,
15 st~onary facilides. E~cplora~on cr~ws must, therefore, label and ship geologic
samples to these stationary facilities for analysis, and must ~en wait a week or longer
until analytical results ~e obtained. This method of exploling for valuable n~ineral
content is laborious, slowt ine~ficien~, and expensive. A gold analysis method (and
apparatus usef~l therefor) which could be carried out at or near the exploration site,
~0 and which could provide lapid and reliable analyses of very small quan~es of &~ld
in rock matrices, would cons~tute a major improvement in the field of gold
exploration.
At the current price of gold, rock bodies containing concen~ations as
low as one part per million of gold are economically minable However, during t}le
2~ exploration process subeconomic amounts of gold may act as a tracer to lead the
geologist toward an economic deposit. Typically, gold values as low as ~ parts per
billion may constitute significant hints that other, higher values might be found
WO 93/07472 PCI ~US92/Q8363
nearl)y. Therefore, an analytical technique that is sensitive in the range of parts per
billion levels of gold would be useful for this pwpose.
Most gold analyses presently perfonned in commercial laboratories
utilize the fire assay technique. Fire assay laboratories are large, labor-intensive
S facilities which consume large quantities of power and water, and frequently emit
hazardous lead fumes. Another drawback to the fire assay method of analysis is the
fact that cross contamination can readily occur between samples.
At the present time, the only commercially v;able modern analytical
technique for the analysis of gold is neutron activation. The requirement for access
to a nuclear research reactor makes neutron activation even less readily available than
fire assaying. Also, neutron activation i~herently requires an eight day "cool down"
period for short-lived radioisotopes to decay before counting the gold. Thus, inaddition to being available on1y where a nuclear reactor is available, it is impossible
for neutron activa~on to satisfy the need in the field for a rapid analytical technique.
Summarv of th~ Inver~oll
In accordance witb the present invention, an assay method is provided
which is capable of measuring gold concent~ations over a broad range (i.e., from a
few parts per billion up to appro~cimately 30 thousand parts per billion or higher).
The invention assay method comp~ises solubilizing the gold content of
the sample, if necessary, then gene~ating a phot~res~onsive complex with the gold,
e.g., a ~odamine B-gold c~mplex. The gold concentration in the sarnple is then
determined by analysis of ~e gold complex using optical me~ns.
In accordarce with anothes as~ect of the present invention, a portable
25 fluorometer instrumentation apparatus is provided that perfonns a rapid analysis of
rocks, soil and other samples to determine the concen~a~on of gold therein.
Advantageously, the apparatus is easily transportable to those locations where ~e
rock or soil samples are found. The instrumentation apparatus is easy to operate, and
provides an accurate indication of the gold concentration within the samples in a
30 relatively shon time, e.g., in less than about three hours. Further, the
Wo 93/07472 PCI /US92/08363
` 21191~
instrumentation apparatus is capable of measuring gold concen~ations ~ver a broad
range from a few parts per billion (~pb) to app~oximately 30 thousand ppb.
In accordance with another aspect of the invention, there is provided
a system for determining the concentration of gold in a field sample, such as a rock
5 or soil sample.
One embodiment of the present invention may thus be characterized as
a method for determining the concentration of go1d in so1utions containing the same.
Such method comprises carrying out the following steps: (a) contaetirlg the gold-
containing solution with: (i) an oxidizing agent, and (ii) at least one crown ether
10 polymer; wherein the contacting is ~Tied out in acidic media under condidons
suitable to convert substantially all of tt e gold ions in solution into their highest
oxidation state, and for a time sufficient to allow capture of substantially all of the
gold ions in the solution by the crown ether polymer; ~b) sepa~ting the gold~rown
ether complex from the remaining components of ~he solution; (c) recovering the gold
15 ions from the gold~rown ether comples~; (d) contacting the metal containing soluffon
pr~pared as described in step (c) with label means, wherein ~e label means is capable
of binding to the gold in its highest oxidadon state, and wherein the label means is
capable of ready analysis; wherein the contacting is ca~ried out in acidic media under
condi~ons sufficient to allow substantially all of the gold in the solution to become
20 bound to the label means, and then sepa~ating unbound label means from the solu~on;
and ~ereafter (e) measunng the amount of bound label means in the solution.
In a~rdance with another embodiment, the invention may be fur~er
cha~acte~ized as a method for detennining the concentra~on of gold in a matri~c.Such method comprises the steps: (a) contacting the matrix with an aqueous cyanid~
25 containing solution in ~e presence of an oxidizer in alkaline condition; (b) contacting
the solution obtained from step (a) with: (i) hydrochloric acid in the presence of an
ox;dizing agent, and (ii) at least nne crown ether polymer; wherein the contacting is
c:amed out under conditions suitable to convert su~stantially all of the gold ions in
solution into their highest oxidation state, and for a time sufficient to allow captu~e
30 of substantially all of the gold ions in the solution by the crown etherJ (c) sepa~a~ng
the gold-crown ether complex from the remaining components of the solution;
Wo 93/07472 Pcr/uS92/08363
2~
- 4 - .
(d) recovering ~he gold ions from the gold-crown ether complex; (e) contacting the
gold-containing solution prepared as described in step (d) with labe1 means, wherein
the label means is capable of binding to the gold in their highest oxidadon state, and
wherein the label means is capable of ready analysis; wherein the contacting is calTied ;
S out in acidic media under conditions sufficient to allow substanti~lly all of the gold
in the solution to becomc bound to the label means, and then separating unbound label
means from the soludon; and thereafter (f) measuring the amount of label means in
the soludon.
A furtha embodiment of the invention may be characterized as
10 instrumentation apparatus useful in the practice of the present invention. Such
instrumentation apparatus includes: (a) means for generating radiant energy within a
- first narrow band of wavelengths; (b) coupling means for coupling the radiant energy
to a prepared sample under investigation; and (c) detection means radiantly coupled
to the sample fot de~ang any transmissive light that passes through the sample and
lS falls within the first luw band of wavelengths, or any fluoresced light emitted from
the sample that falls within a second narrow band of wavelengths.
In operation of the instrumcntadon apparatus, the p~esence of the
t1ansmissive or fluoresced light within the first or second narrow band of
wavelengths, respectively, indicates the presence of a par~cular element, e.g. gold,
20 within the sample. The intensity or magnitude of the detected transn~issive or
fluoresced light provides a measu~e of the concent~ation of ~e particular element
within the sample. As ~equired or desired, ~ conversion chart or table is generated,
e.g., by measuring samples contsining known concentrations of ~e par~cular
element, that allows for the direct conveFsion of the measured fluoresced light to a
25 concen~ation ~ralue of the element within the sample. Hence, by merely measuring
the intensity or magnitude of the transmissive and/or fluoresced ligh~ within ~e first
and/or second narrow band of wavelengths, a convenient and quick measurement is
provided as to the concentration of the particular element witllin the sample.
Advantageously, in one embodiment of the instrumentation appa~atus,
30 a fiber optic bundle is used to couple radiation energy to and from the sample under
test avoids the problem of ~inner filter e~fect~ commonly found in prior art
.
Wo 93/07472 Pcr/Us92/~8363
21i~134
fluorometer devices. TSle inner filter ~ffect, when present, produces ambiguous
results in the signal output from ~e detector.
Yet another embodiment of the invention may be characterized as an
analysis system for determining the concenhation of gold in a field sample. SuchS system includes: (a) binding means for binding a portion of the field samp1e to a
suitable label means, such as Rhodamine B, thereby producing a ~iation complc~c
of the label means plus gold; (b) irradiating means for irradiating the label means to
which the sample is bound with radiation energy falling within a first narrow band
of wavelengths, ~is in~diating means including fiber optic means for direc~ng
10 radiation to and collecting radiation emitted from the samp~e ~omplex; and (c)
detecting means coupled to the fiber op~ means for deteeting radiation ernitted ~rom
the sample within a second narrow band of wavelengths.
In operation of the analysis system, the metal-label means complex
emits radiation energy wi~in the second narrow band of wavelengths in response to
15 irradia~on with radiation energy within ~e first nalTow band of waveleng~s only
when ~e field sample bound to ~e label means contains ~e par~cular metal being
assayed. Further, b~cause ~e amount or intensity of ~e radiation thus emitted isproportional to the amount of metal bound to ~e label means, the magnitude of ~ede~ted radiation provides a simple and quick measure of ~e metal concen~ation
20 within ~e field sample. Addi~ional alhancements of ~e system optionally include
processing means ~or automa~cally conYerting ~he detected ~adiation within ~he
speciff~d band to a me~sure of the gold concentlation within ~e field sample.
It is 2hus a feature ~f the invendon to provide portabie apparatus ~at
includes all the ins~mentation and other compoflents needed to carry out the m~thod
25 of ~e inven~on.
It is another featu~e of the invention to provide fluorometer
instrumentation apparatus for assaying a sample complex for the presence of gold.
The sample complex comprises a suitable ligand to which a portion of a soil or rock
sample believed to contain gold has been bound. An advantageous feature of such
30 instrumentation apparatus is that fluorescence and transmissive readings may be
obtained over a wide dynamic range. A related feature provides good proportionality
Wo 93~07472 Pcr/us92/08363
z~ 3 4 .~
between the fluorescence/ transmissive readings and the gold concentrations. Other
features of such apparatus include an overall accuracy of i 15%.
It is another feature of the invention to provide such
fluorometer/transmissive instrumenhtion apparatus that avoids the "inner filter
S effect."
lt is a further feature of ~e invention to provide
fluorometer/transmissive instrumentation appa~tus that is relatively insensitive to
variations in the intensi~y of the source radiation.
It is still another feature of the invention to combine process
10 methodology with a novel fluorometer/transmissive instrumentation apparatus
comprising a particular selection of che,lnicals, treatment conditions, instruments,
circuits and shielding configuration th~t effectively eliminates the e~fects of extraneous
sign~ls and background radia~on, thereby permitting noise repression and the reading
of very small signals. ~-
~rief Pescription of the D~a~nes
The above and o~er aspects, features and advantages of the p~esent
- invention will bc more apparent from the following more particular d~ption
~ereof, presented in conjunc~on with ~he following drawings and appendi~ wherein:
FIG. 1 is a block diagram illus~a~ng the basic operation of a first
embodiment of a fluorometer ins~mentation apparatus made in accordance with the
- present inven~don;
FIG. 2 is a more detailed block dia~am of the first embodiment of ~e
fluorometer instrumenhtion apparatus of ~he p~esent invention as generally illustrated
25 in FIG. l;
F~G. 3 is a graph illus~rating the transmissivi~ of the two filters used
within the apparatus of FIG. 2;
FIG. 4 is a re~resentative cali~ration graph used with cr by ~e
apparatus of FIG. 2 in order to eonven the measured intensi~ of ~he fluoresced light
30 to a measure of gold concentradon;
WO 93/07472 PCr/Uss~/08363
211~13~
- 7 -
FIG. S is a top view block diag~un illustrating the basic opera~don of
a combined fluorometer/transn~issive instrumentation appa~atus madc in accordance
with the preferred embodiment of the present invention;
FIG. 6 is a more detailed top view block diagram of the combined
S fluorometerttransmissive instrumentation apparatus of the pre~erred embodiment as
generally illustrated in FIG. S; and
FIG. 7A and 7B are a flow diagram showing the ste~s taken when
utilizing the preferred embodiment, as shown in FIG. 6, to measure gold
concent~ation in a sample.
Like components or elements are referred to with like reference
numerals throughout the various views o~ the drawings.
etailed Desc~iption of the lnven~Qn
The following description is of the best motle presently contemplated
15 for carrying out ~e invention. This descrip~on is not to be taken in a limiting sense,
but is made merely for the puIpose of describing the general principles of the
invention. The sco~ of the invention should be determined with rference to ~e
cla~ms.
The present inven~on provides a method, and instrumentadon appaIatus
20 for rapidly ~ng out the me~hod, for determining the concentration of a specihc
element, e.g., gold, in a matenal (field sample). The method and instrumenta~on
appa~tlls have been summarized above. Advantageously, the invention method can
be calTied out in both bateh and continuous modes. Where ~e mat~ to be
analyzed is in par~culate form such as an ore, rock, or the like, it is desirable ~o first
25 clush the partic~late material into a fine powder to improve the contacting of reagents
the components of ~e particulate material. The ore powder is then roasted
under an oxidizing atmosphere in the temperature Iange of 500 800 oC for up to one
hour to removevolatile elements, thereby minimizing the likelihood of false positives.
To ensure that substantially all of the gold in the par~culate material
30 is taken up into solution, the finely ground particulate is suspended in an aquoous
WO 93/~7472 PCr/US92/083~3
3 4 ~s
- 8 -
cyanide containing solution in the presence of an oxidizer e.g. calcium peroxide,
sodium peroxide, potassium persnaganate, bromine, chlorine and hydrogen pero~ide.
Cyanide compounds contemplated for use in this solubilization step
include sodium cyanide, potassium cyanide, and ~e like. Potassium cyanisde is
5 presently preferred. The quanti~ of cyanide employed can vary widely, so as toprovide a final cyanide concenhation in tbe me~al contaiSning aqueous solution which
typically ~sls in the range of about 0.001 M - 0.5 M. Currenely, final cyanide
concentrations in the range of about 0.1 M arè employed. In order to prevent theloss of cyanide by hydrolysis or reaction with CO2 and to neutralize the acidic
10 components in the ore, a base, e.g., potassium hydroxide, is added to the cyanide
solution.
Contac~ing of the optionally crushed metal-containing matri~ with
cyanide and oxidiær is ca~ied out under conditions suitable to allow solubilization
of substantially all the metal contained in ~e mat~
Once the gold is leached into ~e cyanide solution, it is treated wi~ an
o~idi~er (e.g., H20~) and hydr~ loric acid. The oxidizer oxidizes gold from +l to
~3 ~alence sta~e. The function of hydroclll~e acid is two-fold: one is to des~oycyanide, the o~er is to pruvide a chloride ligand to the Au3~ ions. After ~s step,
gold is ~ the form of AuCl4- which is appropria~e for the next interference rem~val
20 s~p by the use of crown ether polymer. AuCI4- comple~ mol~cules can be t~app~d
into the cavi~es of the crown ether polymers in the HCl coneenha~on of 0.1-6 M.
By selec~ng 0.6 M HCI concentra~on, ~o~ example, AuCI~- is adsorbed by ~he
polymer, letting most other metal ~pecies pas~ by.
A key element of this invention is the discovery that the crown ether
25 polymer ava!i able frosn all commercial sollrces must be pre~eated in order to obtain
consistent recovery of the gold eomplex, especially when the sample gold
concent~ation is below about 1000 ppb. The variation in gold recovery percentageis caused by adsorption sites of o~er ~an the gold complex dimension that wQuld
~ap Au complex molecules but not release them at a later stage. The pretreatment30 invention requires filling ~e crown ether polymer with some metal species (e.g., Ga,
As, Fe, Au) that fill the adsoIption sites, and then ex~acting most of the metal
WO 93/07472 211~ 1 ? 4 Pcr/us92/o8363
species out of ~e cavities leaving the "bad sites" that are filled. This treatment gives
about 40% improvement in consistency of recove~ing gold complex concentration in~e range of 100 ppb.
Any crown ether polymer capable of complexlng with the ~metal to be
5 assayed is contemplated for use in ~e practice of the presen~ invention. Exemplary
crown ether polymers include poly(dibenzo 18-crown-6) [poly(DB18C6)], and the
like. The amount of crown ether polymer employed is in the ~nge of about 2~100
mg/ml of metal-containing solution.
The crown ether polymer bound-metal material is ~en sepa~ated from
10 the remaining components of the solution. This sepa~tion can readily be carried out
by removal of the liquid from the particulate material using standard techniques such
as decantation, filtration, aspiration or the like. It is preferred, to insure removal of
substantially all e~t~eous species, to then wash the particulate material with 0.6 M
HCl.
It is preferred dlat the crown e~her polymer sepaFation be used in the
column form. This is because the time to perform ~he s~ tion is reduced
drama~ically when tlle column form is used ~rom wha~ would be ~equired if o~er
sepa~tion techniques, e.g., tumbling, were ernployed. For example, the crown ether
sepa~ation may take 2Ih hours when tumbling is used, but may only require 30
20 minutes when the eolumn form is used.
Once t~e gold~wn ether ~omplexes ha~re been separated from the
~emaining componetlts of the metal containing solution, the gold ions are recovered
~rom the eomplex. This is done, for example, by e~c~c~ng ~e ions ~rom the
comple~ wi~ a polar oxygen containing organic solvent such as alcohol, ketone, etc.
25 Acetone (a ketone) is preferred.
When the gold ions have been recovered apart from the crown ether,
~e organic extractant is driven off of the sample by gentle heatîng in the presence of
an oxidizer (e.g. H2O2) to prevent Au-reduc~on, and the material is contact~d wi~ a
label means. As employed herein, ~label means" refers ~o a chemical species which
30 is capable of binding to AuCl~-, and which is also capable of ready analysis.Exemplary label means include chromophores, metal-complexing agents which ar~
W093/07472 ~ - 10 - ,r
capable of fluorescing when excited with incident light of proper wavelength, and the
like.
Exemplary label means include l~hodamine B (i.e., N-(9~
carboxyphenyl)-6-(diethylamine)-3H-xanthen-3-ylidene)-N-ethyl ethanaminium
S chloride; also hlown as tetraethyl rhodamine~, Brilliant Green,
PQPP (i.e., 2-phenylbenz~8,9]-quinolizino~4,5,6,7-fed]phenanthridinylium cation),
and the like. Rhodamine B is the pre~ently preferred label means because it gives
very low levels of background fluorescence when gold concentrations are determined.
Substantial binding between AuCl4- and the dye is obtained by
10 vortexing the mixture for a few seconds at room ternperature. Following such
treatment, it is desirable t~ separate substantially all unbound label means from the
sample so as to reduce background noise,in the invention analytical method. Suchremoval can be carried out, for example, by extraction of the label means-metal
complex with an organic solvent (e.g. C~I6, ether, preferably diisopropyl ether) from
15 ~e aqueous media in which the comple~ is generated.
The above~escribed removal of unbound label means should be done
with care so that substantial quan~ties of unbound label means do not remain in ~e
sample to be analyzed.
Once sample prepara~on has been completed, it is desirable to analyæ
20 ~e sa~nple relatively promptly so that the opportuni~ for sample degradation is
minimized. A s~unp~e thus prepared is ready for analysis of ~e gold content
~erein. Metal analysis in ac~ordance wi~ ~e present invention is accomplished bydetern~ining the amount of label means incolporated by the sample. Where ~e label
means is capable of emit~ng fluoresoent ~adiation upon excit~tion, analysis can be
25 accomplished by e~ci~ng the label means at a s~eeified wavelength, then measuring
the intensity of the emissions at a specified wavelength di~ferent from the wavelength
used for excita~on. Various means for carrying out such analysis employs novel
instrumentation apparatus described hereinbelow. Alternatively, where the label
means is a chromophore, analysis can be accomplished by spectrophotometric means,
30 i.e., by measuring ~e amount of radia~on absorbed by the sample when radiation of
lmown intensity is passed through ~e sample.
Wo 93/07472 2 1 1 3 1 ~ 4 Pcr/US92/08363
. .
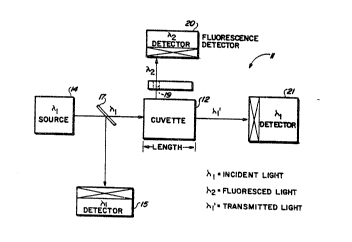
A first embodiment of the apparatus of the present invention facilitates
the practice of the above~escribed assay method by providing portable fluoromcter
instrumentadon a~paratus tha~ allows the assay method to be quicldy carried out in
the field at or near the locadon of the soil or rock samples being assayed for the
5 presence of gold. A block diagram illustrating the basic components of fluorometer
appa~atus 10 made in accordance with the invention is shown in FIG. 1. These basic
components include a source of radiant energy 14, a dichroic mirror 16, a fiber optic
bundle 18, and a detector 20. A second detector 15 may also be employed. The
- - dichroic mirror 16 is positioned so as to direct radiant energy generated by the source
10 14 into the flber optic bundle 18. The detector 20 is positioned relative to the
dichroic mirror 16 so as to receive radiant energy emitted from the fiber optic bundle
18. The detector 15 is positioned to receive radiant energy generated by the source
in order to detect variations in the intensity thereof that occur over time. Such
variations, if not corrected, could introduce an error in the measurements made by
15 the apparatus.
A sample comple~ 12, comprising a suitable label means, such as
Rhodamine B, to which a desired element, such as gold (when present) has been
bound, is prepared as descdbed above in connection with the method of ~e presentinvention. When ~e comple~t 12 contains gold, ~e gold-Rhodamine B complex
20 fluoresces in ~e spectra~ ~egion of 560 to 580 nanometers (nm) when e~ccited by
~adiant energy ~ight) in the spect~l region of 540 to 550 nm. When gold containing
comple~ 12 is a gold-PQPP complex, exalta~on in the range of about 300 nm~ leadsto fluorescence in the spectral region of about 460 nm. Advantageously, the amount
of emitted fluo~escence is a monotonically incr~sing function of the gold
25 concen~ation in ~e comple~.
To determine the concenbation of gold in a given sample comple~c 12,
the fluorometer instrumentation apparatus 10 operates as follows: The source of
radiant energy 14 generates radiation of a first wavelength ~" where ~, for the gold-
~hodamine B complex falls wi~in the ~ange of 540 to 550 nm (about 300 nm for
30 gold-PQPP). This radiation is directed into ~e fiber optic bundle 18 using ~edicWic mirror 16, and is also directcd into the detector 15. The sample compleJc,
` in response to the ~l radiation, fluoresces radiation of a second wavelength ~2. where
.
wog3/07472 9~ 12- PCr/USs2/08363
(for the gold-Rhodamine B complex) falls within the range of ~60 to 580 nm
(about 460 nm for gold-PQPP complex). llle ~2 radiation is directed from the sample
comple~ 12 through the fiber optic bundle 18, through ~e dichroic mirror 16 and to
the detector 20. The detector 20 is configured to detect the amount of radiadon of
S` wavelength A2. This detec~on thus provides a direct measure of ~e gold
concentration within the sample complex 12.
The output intensi~ of the source 14 will vary to some degree during
the measurement of a group of references and samples. In order to minimize the
error due to source intensity variation, the detector 15 allows the source intensity to
10 be monitored so that appropriate corrections in the determination of the gold concentration can be made, as described below.
Fiber optic bundles are wldely available, and can be constructed of a
variety of materials, including silica, glass, polymethyl methacrylate, polycarbonate,
p~lystyrene, and the like. Commercially available fiber optic bundles can be used as
15 received, without the need for any special treatment and/or preparation prior to use.
Typically, the only modification made is to rem~ve the cladding from the bundle from
that part of ~e bundle which will be directly e:cposed to sample.
1 he primary considerations in selecting a ~icular fiber optic bundle
are the stability of the probe material when exposed to the solvent system containing
20 the metal to be analyzed, and the degree, if any, to which the probe mate~ial and
sample components are prone to interact, there~y introducing interference into the
analysis. Where unstable signa~ is observed, it is advisable t~ consider the use of a
different fiber optic bundle, a different solvent system for the analytes, and/or a
diffe~ent label means to overcome the pFoblem.
Referring next to FIG. 2, a more detailed block diagIam of one
embodiment of fluorometer instrumenta~on apparatus made in accordance wi~ the
present invention is shown. (It is noted that like numerals are used to r~present like
parts in FIG. 2 and FIG. 1). As seen in FIG. 2, the apparatus includes the same
basic components shown in FIG. l, i.e., a source of radiant energy l4, a dichroic
mirror 16, a Sber optic bundle 18, a detector 20, and a second detector lS. As with
FIG. 1, the fiber op~dc bundle 18 directs radiation to and from the sample c~mple~
12 under test. As shown in ~G. 2, the source of radiant energy 14 comprises a
2 1 1 ~
Wo 93/07472 PCI/USg2/08363
- 13-
power supply 22 coupled to a broad band light source 24. The light generated by the
broad band light source 24 is directed through a suitable optical system 27 to a filter
30. The optical system 27 includes any suitable means for efficiently coupling ~e
radiant energy from the broad band light sour~e 24 to the filter 30. Ille filter 30 is
S a narrow band filter and filters out all of energy except that of the desir~d wavelength
It is to be understood that 1~ may comprise a narrow band of wavelengths as
well as a single wavelength, depending upon the bandwidth of the filter 30.)
As shown in FIG. 2, the op~cal system 27 includes a fiber optic
coupler 26 having one end optically coupled to the light source 24 and the other end
10 at the focal point of a lens 28. Radiant energy ~rom the light source 24 is thus
directed through the fiber optic coupler 26 to the lens, where it is directed to the filter
30.
After passing through the filter 30, only a narrow band of radiant
energy remains, e.g, of wavelength ~,. The ~, radiant energy is optically reflected
15 ~rom the dichroic mi~or 16 to an additiona~ lens 32 so as to be ~irected or focused
into the fiber optic bundle 18.
Still refernng to FIG. 2, fluoreseed light from the comple~t sample 12
is emitted from ~e fiber opdc bundle 18 and is focused through ~e lens 32 back to
the dichroic miITor 16. This fluoresced light passes through the mi~ror 16 to ~e20 detector 20. The detector 20 includes a narrow band filter 34 configur0d to pass only
radiation having a w~velength A2. (It is to be unclerstood that ~2 may comprise a
narrow band of waveleng~s, as well as a single wavelength.) That is, ~adiation or
light of wavelengths other than ~2 iS significantly attenuated by the filter 34. Hence,
if the radiation flu~res~d ~rom the comple~ sample 12 includes the desired element,
25 e.g., gold, it will fall wi~hin the wavelength band )~2~ and such radiation will pass
through the filter 34.
Afeer passing through the narrow band filter 34, the radiation is
directed or focused through a second lens 36 ~o a photodetector 38. llle
photodetector 38 operates in conventional manner and detects the amount of radiation
30 incident thereon. Thus, any radiation of wavelength ~2 that makes its way through
the filter 34 is detected by the pho~odetector 38. In response to such detection, the
photodetector 38 generates an electrical signal. The amplitude of this signal is
wo 93/07472 PCr/USs2/08363
~9 ~3 - 14 - ?
propor~onal to the intensity of the detected radiation. The electrical signal generated
by the pbotodetector is amplified by amplifier 40. The output signal of the amplifier
40 may then be monitored to determine how much, if any, radiation or light of
wavelength ~`2 was detected by the photodetector 38. A large output signal indicates
5 ~2 radiation of a high intensity, which in tum indicates a high concentration of the
particular element within the complex sample (assuming a uniform volume of the
sample). Similarly, a small output signal indicates ~2 hdiation of a low intensity,
which in turn indicates a low concentration of the particular element within a uniform
- volume of the sample complex. Advantageously, as explained more fully below in10 connection with FIG. 4, the output signal from the amplifier 40 may be calibrated to
provide a direct measure (indicated, e.g., in parts per billion, or ppb) of the
concentration of the element within the s~mple complex.
As seen in FIG. 2, the output signal from the amplifier 40 is measured
with a digital voltmeter (DVM) 42. Further, as desired, this output voltage may be
15 recorded and/or stored in a data logger 44, or oquivalent device. Moreover, for some
applications, it may be desirable to couple a suitable processor 46, such as a portable
personal computer (PC), to the output voltage of the amplifier 40. Such couplingmay be accomplished direc'dy f~om ~e output signal of the amplifier 40 if the
p~ocessor 46 includes intemal analog-t~digital (A/D) conversion means (for
20 converting the analog output signal from the amplifier 40 to a digital signal suitable
for use with the PC); or, alterna~vely, if the DVM 42 includes a digital output port,
as many commercially available DVM's do, the coupling may be made from the
- DVM 42 to ~e processor 46.
Tbe processor 46, when used, performs various processing functions
25 associated with the amplifier output signal. For e~ample, the processor may include
a ~look-up table" or equivalent (e.g., an equation) that is stored therein that allows
the measured output voltage of the amplifier 40 to be converted directly to a
concentration value of the desired element. Fur~her, the processor may include
various digital processing steps that analyze the output voltage data obtained from the
30 amplifier 40 over a specified period of time in order to funher enhance such data,
e.g., by Jemoving noise therefrom using conventional digital filtering techniques.
wo g3,0,4,2 2 1 1 9 1 3 I PCr/USs2/08363
Sdll referring to FIG. 2, it is seen that the detector lS is positioned to
sense the incident radiation of wavdength Al generated by the source 14. Such
radiation is directed through a suitable lens 29 to a photodetector 39. The
photodetector 39 operates in convendonal manner and detects the amount of ~adiadon
S incident thereon. Thus, any radiadon of wavelength ~I from the source 14 that makes
its way through the filter 30 is detected by the photodetector 39. Addidonal filters,
positioned in front of the lens 29, for filtering out- all radiation except that of
wavelength A, may also be used, as required. In response to detecdon of the A
radiadon, the photodetector 39 generates an electrical signal. The amplitude of this
10 signa1 is proportional to the intensity of the detected radiadon. The electrical signal
generated by the photodetector is arnplified by amplifier 41. The output signal of the
amplifier 41 is then monitored to det~rmine how much radiation or light of
wavelength ~I was detected by the photodetector 39. A large output signal indicates
~ radiadon of a high intensity. Similarly, a small output signal indicates ~1 ~diation
15 of a low intensity.
The output signal from the amplifier 41 is measured with a digital
voltmeter (DVM) 43. Further, if desired, this output voltagc may be recorded and/or
stored in a data logger 45, or equivalent device. Moreover, for most applications,
it is ade~uate to couple the output of the amplifier 41 direc~y to the processor 46,
20 such as a ponable pewnal computer (PC). Such output signal may ~en be used toprovide a reference signal that indicates vasiations that may have occurred in the
intensity of the ~, radiation.
ln opera~on, ~e detector 15 thus monitors ~e intensi~ of the ~1
source. When an ini~l sample is measured, the measured intensity thus represents25 a reference signal, SRC~. For subsequent measurements, ~e intensity of the ~
fluoresced ~ignal is another signal,~ Sp. Advantageously, *e signal Sp may be
corrected for variations ~at may have occured in the intensi~ of the ~1 source as
follows:
SCo~r = (Sl, SRd)lS~l
30 where SCO" represents the correction of the signal $, and SAI represents the current
~tensity measurement of the source 14 as made by the detector 15. Advantageously,
Wo 93/0747~ 4 Pcr/US92/0~363
g~ 16~ s
by storing tbe signal SRe~ in the processor 46, or equivalent device, the described
correction can be made by the processor for ~ach measurement made.
An additional embodiment of the detector 20 may include a plurali~r
of filter-pbotodetector-amplifier sets, each adapted to sense radiation of a particular
S wavelength. Tbe output signals from all of such sets may then be monitor~d, e.g.,
using the processor 46, to provide an overall assay report of the eontents of the
sample complex, including an indication of the concentrations of a plwality of
elements within the sample comple~.
As an altemative embodiment of the source of radiant energy 14, a
10 conventional laser may be used, e.g, a He-Ne laser that provides an output
wavelength of 543.5 nm. The narrow output of such He-Ne laser may be coupled
through any suitable means directly int~ the fiber optic bundle 18, with a smallportion thereof being coupled directly into the detector 15. Typically, such coupling
will utilize at least the dichroic ~r 16 and the lens 32, thereby allowing the laser
15 to be positioned off al~is from the fluoresced radiant energy traveling out from ~e
complex sample 12.
FIG. 3 is a ~ph illustrating ~e t~nsmission pr~perties through the
two filte~s 30 and 34 used wi~in the apparatus of FIG. 2. These t~sn~ission
pro~erties are selected for the detec~on of gold as a gold-Rhodamine B complex. lt
20 is to be understood, of course9 that similar proper~es m~y be selected for ~edetection of other gold complexes. A first ~ or band 70 is centered approximately
at 545 nm. This peak or band 70 re~resents the desired transmission prope~es of
the filter 30 coupled to ~e bro~d band energy source 24. A second peak or band 72
is centered appro~imately at 580 nm. This peak or band 72 thus re~esents the
25 desired transmission p~pe~es of the filter 34 placgd prior to the photodetector 38.
If a He-Ne laser is us~d in plaee of the broad band energy ~urce 24, the wavelength
of the He-Ne laser is 543.5 nm, whieh is roughly centered in the desired peak orband 70.
If other complexes are being detected, the location of the band 70 and
30 the band 72 would be selected a~cordingly. For e~ample, if a given comple~ were
to fluoresce radiation having a wavelength of 590 nm in response to being irradiated
WO 93/07472 2 1 I 9 1 3 4 PCI/VS92/û8363
- 17-
with radiadon having a wavelength of 520 nm, then the first band 70 would be
centered at 520 nm, and the second band 72 would be centered at 590 nm.
Refening ne~t to FIG. 4, there is shown a re~rOEntative calibration
graph used with or by the apparatus of FIG. 2 in order to conven the measured
5 intensity of the fluoresced light to a measure of gold concentration. It is to be noted
that equivalent calibration techniques, such as using a look-up table, or an ~quation,
may also be used to convert measured intensity to gold concentration. The graph of
FIG. 4, or equivalent table or equation, is generated by measuring the fluorescent
signal amplitude for a series of samples of uniform volume containing hlown
10 concentrations of gold. That whieh is shown in FIG. 4 is only a po~ion of ~e
overall calibration curve that can be obtained. In general, good calibration data is
obtained using the fluorometer apparatus o~the invention over a wide dynamic range,
e.g., of from 10 ppb to 3,000 ppb. Moreover, with a system response time on the
order of one second, the fluorometer apparatus is able to measure coneentra~on
15 ~ifferences on the order of 1 ppb. The accu~acy of the combined chemical and
~ptical method ~rom the onginal samples is approximately i 15%.
Advantageously, ~e use of the fiber op~c bundle 18 avoids a
mea~urement p~oblem, hlown as the "inner filter effect,~ common in spectro
fluorometers of the prior art. This problem resul~s m the amount of emitted
20 fluor~scent light reaching a peak l~vel at low metal concentrations, and ~ereafter
decreasing with increasing metal concentration. ~his "inner filter ef~ect" ~us results
in a double-valued oulput and almbiguous readings ~ver the range of metal
ooncentration of interest. See, ~r example, Yuan et al., I'Calculadon for
Fluorcscence Modula~on by Abso~bing Species and lts Application to Measurements
25 Using Optical Fibers9" naly~ hemi~y, Vol. 59, No. 19, 2391-94 (October 1,
1987).
With reference to FIG. 2, it is noted that the light source 24 may be
realiz~d with a Xenon Arc (ILC No. 131), an incandescent lamp, such as the Gilway
Technical I~mp No. L7394, or a Helium-Neon laser (No. LSGR~lSOM, obtained
30 from Panicle Measuring Systems (PMS)). As indicated below, the H~Ne is
pseferred, but the other sources may also be used. The light source 24 is positioned
at the focal point of the lens 28, which may be an aspheric lens No. 06 3097,
Wo 93/07472 ~ PCr/USg2/08~63
3 18
available from Spindler & Hoyer. Altematively, the light may be t~nsmitted t~ the
focal point of the lens 28 by the fiber optic bundle 26. If used, the fiber opdc bundle
26 may be realized using 1000 micron diameter fibers available from, c.g., Ensign-
Biclcford.
S The collimated (focused) beam from the light source 28 is transmlt~
through the filter 30, which may be obtained from Omega Optical Inc., as Part No.
546BP10. The dichroic mi~or 16 may also ~e obtained from Omega Optical Inc. as
Part No. 440 DES P. The lens 32 focuses the light onto the end of the fiber op~cbundle 18. This lens 32 may be realized with an aspheric lens, No. 06 3097,
10 obtained from Spindler & Hoyer. The fiber optic bundle 18 is preferably comprised
of 1000 micron diameter fibers obtained from, e.g., Ensîgn-Bickford. The filter 34,
through which the fluoresced light is dire~ted, may further be obtained from Omega
OptiG~l Inc as Part No. 577BP10. The lens 36 may be ~e same as the lens 32. The
photodetector 38 may be any suitable photodetector, such as a photodiode detector,
15 of which numerous types are commercially available7 e.g.9 a silicon phot~-diode No
S2386, manufa~tured by Hamamatsu.
The electric~ signal from ~e photodiode detector is amplified by ~e
amplifier 40. A low noise, low drift opera~donal amplifier is preferable for dlis
pu~pose. A Burr-Brown No. OPA128LM amplifier is well suited for ~is pulpose.
20 The amplified outl)ut from the amplifier 40 is preferably printed and stored using ~e
data logger 44, which ~ata logger may be obtained i~rom Omega Engineering Inc asPart No. OM-550. The DVM 42 may be obtained ~rom any suitable manufacturer.
1 he components of the detec~or 15 may be the same as co~esponding
components of the detector 20.
The par~cular sel~ction of components described above in connection
with FIG. 2 advantageously permits the reading of exce~tionally low ~ralues of
fluoresced light by repressing noise. The preferred amplifier 40, for example, (Burr-
Brown No. OPA128LM) has ultra-low bias current, very high signal-to-noise ratio
and common-mode rejection, and thus allows ~xtremely small signals (electncal
30 currents) to be r~ceived from the photodetector 38, which small signals corre~ond
to small sample values. The amplifier 40 converts such small signals to a relatively
large ou~ut voltage.
WO 93/07472 2 1 1 9 1 3 ~ Pcr/us~2/o8363
` ~
,9
In addition to suppressing noise, it is important in the cons~ruction of
the fluorometer apparatus to select optical components ~at provide large amounts of
light energy and yet can func~on wi~in a very nalTow band without e~t~aneous
signals (which exh~neous signals would be seen as noise). The preferred filters 30
5 and 34, the photodetector 38, the lenses 28 and 36, as well as the H~Ne laser (if
used), as identified above, are all selected with these considerations in mind. For
example, by utilizing a He-Ne laser as the radiant energy source 14 in PIG. 2 (in lieu
of broadband light source 24) to generate a narrow band energy at 543.5 nm having
an output power level of approximately 1.5 milliwatts, the photodetector 38 may
10 respond to very low power incident radiation levels from test samples having
extremely low amounts of the metal of interest. For this reason, ~he use of a He-Ne
laser as the radiation source 14 is preferred over the use of a broad-band source, as
shown in FIG. 2.
In ~ew of the very small signals and the necessity of low
15 accompan~ng noise, the fluorometer appasatus of- ~e present invention fur~er
utilizes appropriate shielding in the packaging of ~e components. Such shielding is
necessasy to avoid radiative and electrical interference, e.g., background Iadia~don,
and is especially needed in paGlGIging ~e photodetector 38 and ~e amplifier 40.
Such shielding thus includes a special enclosure 39 in which the photodeteetor 38, the
20 operational amplifier 40, and related components (e.g., a source of elec~ical power
for these components) are housed. The erclosure 39 may be made from any suitablemetal, such as copper or aluminum, th~t prevents low level radia~ion from passing
therethrough and that is a good electrical conductor.
In addidon, in order to funher ~educe noise and extran~ous signals, ~e
25 size of tlle fiber optic bundle 18 should be selected for maximum delively of the ~l
and ~2 signals with minimum noise. In the prefe Ted embodiment, this t~quirementis met by utilizing fiber bundles having a diameter of abou~ 1000 microns.
Advantageously, all of the components of the apparatus 10 are
s~fficiently small and lightweight so that they may be e~sily transpotted, e.g., in a
30 ~iler, truck, van, by backpack, ot the like, thereby allowing the entire apparatus to
be portable and easily ~ansported to a field location where soil or rock samples are
to be assayed.
wo 93/0~472 Pcr/us92/08363
?.I!L~9~3 ~ 20 - ~`
A second embodiment of the apparatus of the present invention
facilitates the p~acdce of the abov~mentioned assay method by providing a combined
portablc fluorometcr/transmissivc ins~umentation apparatus that, like the first
embodiment, allows the assay method to be quic!~y ca~ied out in the field or ncar
S the location of the soil or rock samples being assayed for the presence of gold. The
second embodiment further improves upon the first embodiment by providing a
fluorometer/transmissive instrumentation apparatus that detects a wide range of gold
concentradons (e.g., from a few parts per billion (ppb) to about thiny-thousand ppb)
without the use of a fiber-optic bundle. By eliminating the need for the fiber-optic
10 bundle, various problems associated with positioning the fiber-optic bundle within the
sample complex are eliminated. By combining a fluorometer instrumentation
apparatus and a transmission measurement,apparatus, a much wider range of desired
element concentrations can be measured.
A block diag~m of a combined fluorometer/t~nsmissive
15 instrumentation apparatu~ 11, made in accordance with the second embodiment of
the invenaon is shown in FIG. 5. As seen in FIG. 5, the apparatus 11 indudes a
sourcc of Iadiant energy 14, a source detector 15, a florescence detector 20, and a
hansmission detector 21. The floresccnce detoctor 20 is positioned relative to the
sample 12 so as to reoeive radiant energy fluoresced from the source 14 by the
20 samplé 12. The source detector 15 is positioned relative to a suitable dichroic n~irsor
17 so as to receive a small portion of the radia~on emitted by ~e source 14. Thetransmission detector is positioned relative to the source 14 so as to receive radiant
energy t~ansmitted ~rou~ the sample 12.
It is noted ~at with o~tically dense solutions, for example, samples
25 with high pp~ of gold, ~e Linear ~ange of the fluo~ence signal is limited. Toincrease thisl range, a ligbt shield 13 with a narrow slit 19 is placed on the side of the
cuvette that faces the fluorescenee detector 20. This in effect, reduces the path length
of the incident light and allows the fluorescence intensity to be propor~onal to the
exciting light over an expanded range.
As with the first cmbodiment, a sample complex 12 comprising a
suit~ble label means to which gold (when present) has been bound, is prepared asdescribed above in connection with the method of the present invention. When the
~' ,
WO 93/07472 2 1 1 9 1 3 4 Pcr/US92/0~363
complex 12 contains gold, the gold~omplex fluoresces in the spec~al range at about
577 nm and ~ansmits r~diant energy at 543.4 nm in response to a ~adiant energy
source that emits radiation at 543.5 nm. At low gold concentrations ~e.g., below 2000
ppb), the intensity of the fluoresced radiant energy is a monotonically increasing
S function of the gold concen~ation in the complex whereas at hi~her gold
concentrations (e.g., above 2000 ppb), the intensi~ of the transmitted radlant energy
is a monotonically decreasing function of the gold concentration in the complex.To determine the gold concen~ation in tbe sample complex 12, the
combined fluorometer/transmissive instrumentation apparatus 11 operates as follows:
10 The source of radiant energy 14 generates radiation of a first wavelength A" where
~ for the gold-Rhodamine B complex is about 543.5 nm. This radiation is directedinto the sample 12. The sample complex,~in response to the ~I radiation, fluoresces
radiation of a second wavelength )~2, where ~2 for the gold-Rhodamine complex isabout 577 nm. The 3\2 radiation is detected by the detector 20. The detector 20 is
15 configured to detect ~e amount of radiation of w~velength A2. Additionally, the
sample complex transmits t}le 3~, ~adiation. The transmitted radiation has been
designated ~`1' in FIG. S so as to distinguish it fr~m the ineident radia~on A, and so
as to indicate ~at the t~ansmit~ed ~adia~on ~,' is of a lowe~ intensi~ ~han the incident
radiation ~,. However, the wavelengths of incident radiation ~1 and ~ansmitted
20 radiat~on ~,' are the same. The Al' ~adiation is detected by the detector 21. The ~1
radiation is detected by the detector 1~. The de~ector 21 is eonfigured to detect the
amount of radiation of wavelength ~,'. The combined detections Of ~\2 and ~,' thus
provide a di~ect measure of the gold concent~tion within the ~nple comple~ 12.
The de~ection of ~he incident radiation by the detec~or 15 allows a sui~able cor~ction
25 to be made to the measurments made by the detectors 20 and 21 so as to ~rrect for
variatinns in the intensity of ~e source 14.
Refe~ring next to FIG. 6, a more detailed block diagram of the s~cond
embodiment of the combined fluorometer/transmissive instrumentation apparatus 11is sho~m. (It is noted that like numerals are used to represent like parts in ~IG. 6,
30 FIG. 5" FIG. 2, and FIG. 1). As seen in FIG. 6, ~e appa~atus includes the basic
components shown in FIG. 5, i.e., a source of radiant energy 14, a calibra~on
detector 15~, a fluorescence detector 20, and a transmission deteetor 21. As further
WO 93/07472 ,~31~ PCr/USs2/08363
~ - 22 -
shown in FIG. 6, the source of radiant energy 14 comprises a power supply 22
coupled to a narrow band laser light source 25. lneident light generated by the laser
25 is directed through an dectro stop or solenoid-controlled shutter 29. The electro
shutter 29 is controlled by a solenoid 23 to be either open or closed. Operation of the
5 solenoid in response to a programmed personal computer (PC) or equivalent processor
46, or other control means, is e~cplained more fully below with respect to FIGS. 7A
and 7B. When the electro stop is open, the light generated by the laser is further
directed through neutral density (ND) filter 31 and aperture 33 into the sample
complex 12. The neutral density filter 31 reduces the intensity of the light generated
10 by the laser 25 to an appropriate level,; and the aperture 33 is used to confine the
light beam to the sample 12.
Also referring to FIG. 6, the fluoresced light or radiation from ~e
sample comple~ 12, generated in response to the incident light, passes to the
fluorescence detector 20. T~he detector 20 includes a light shield 13 with a narrow slit
15 19 as dcscribed abovc in COMeCbOII with FIG. 5. The detector 20 further includes
a first fluorescence band filter 35 oonfigured to pass only radiation hav~g a
waveleng~ ~2. In ~e preferred embodimcnt ~2 iS 577 nm. That is, ladiation or light
of wave1engths other than )~2 iS significan~y attenuatod by the filter 35. Hencc, if the
sample includes ~e desired element, c.g. gold, ~e radiation fluoresccd from ~e
20 complé~ sample 12 will fall within the wavelength band ?~29 and such radiation will
pass through the filter 35.
After pas~g thr~ugh ~e first band filter 35~ the radiation p~sses
through a second fluor~ce band filter 37. The second fluorescence band filter 37is also eonfigured to pass only radia~don having wavdength A2. Hence, the second25 fluo~escence band filter functions in- a manner similar to the f~rst fluorescence band
Slter so as to further attenuate light of wavelengths other than wavelengtb ~2 that
passes through the first fluorescence band filter 35.
After passing through the second fluorescence band filter 37, the
radiation is directed to a photodetoctor 38. The photodetector 38 operates in a
30 conventional manner and detects the amount of radiation incident thereon. Thus, any
radiation of wavelength ~ that makes its way through the first fluorescence bandfilter 35 and the second fluorescence band filter 37 is detected by the photodetector
wo g3,07472 2 1 1 g 1 3 ~ Pcr/US92/08363
- 23 -
38. In response to such detection, photodetector 38 generates an electrical signal
wherein the amplitude of the signal is proportional to the intensity of the detocted
radiation. The dectrical signal generatod by photodetector 38 is amplified by
amplifier 40. The output signal of the amplifier 40 may then be monitored to
S determine how much, if any, radiation or light of wavelength )~2 was detecSed by the
photodetector 38. A large output signal indicates A2 radiation of high întensity, which
in turn indicates a high concentration of the particular element within the complex
sample (assuming uniform volume of the sample). Similarly, a small output signalindicates radiation of low intensity, which in turn indicates a low concentration of the
10 particular element within a uniform volume of the sarnple complex. Advantageously,
as e~plained more fully below, the output signal may be calibrated to provide a direct
measure (indicated, e.g., in parts per billion, or ppb) of the concentration of the
element within the sample complex.
As seen in FIG. 6, the output signal from the arnplifier 40 may be
15 measured with a digital voltmeter ~DV~ 42. As c'esired, this output may be recordod
or stored in a~data logger 44, or oquivalent device.
Still eferring to FIG. 6, any light that passes through the sample 12,
refe~ed to as transmitted ligllt, passes from the sample to the transmission detector
21. The transmission detector 21 includes an aperture 43 to collimate the transmitted
20 light and direct it to a desired detection location. Fur~er, the aperture helps
minimize scattering intc the detector from outside sources. Light entenng through
the aperture 43 is directed ~rough a transmission band filter 45. The filter ~5 is
configured to pass only radiation having a wavelength of A,'. In ~he prefe~ed
embodiment, the filter 45 is centered at about 546 nm so as ~ readily pass
25 t~nsmitted radiation having a frequency of ~l (543.5 nm). l hat is, radiation or light
of wavelengths o~er tnan ~`1' is significantly attenuated by the filter 45. Hence, the
radiation fluoresced from the comple~ sample 12, which has wavelength A2, will be
significantly attenuated and therefore have minimal effect on the transmission
measurements made by the transmission detector 21. However, the radiation
- 30 ~ansmitted through the sample 12, which has wavelength ~,', will pass through the
filter 45. The light passing through the filter 45 is directed through an ND filter 47,
Pcr/uss2/~
which ND filter 47 reduces ~e intensity of the light transmitted therethrough by a
controlled amount.
After passing through the ND filter 47, the radiation is direc~d to a
photodetector 48. The photodetector operates in a conventional manner and detects
S the amount of ~adiation incident thereon. Thus, any radiation of wavelength ~`!' that
makes its way through the transmission filter 45 and the ND filter 47 is detected by
the photodetector 48. In res~onse to such detection, pbotodetector 48 generates an
electrical signal wherein the amplitude of the signal is proportional to the intensity
of the detected radiation. The electrical signal generated by photodetector 48 is
10 amplified by amplifier 50. The output signal of the amplifier 50 is then monitored to
determine how much, if any, radiation or light of wavelength ~,' is detected by ~e
photodetector 48. A decreasing signal indicates a high concentration of the particular
element within the complex sample (assuming uniform volume of the sample).
Similarly, a higher output signal indicates a lower concentration of the par~cular
15 element within a uniform volume of the sample complex. Advantageously, the output
signal may be calibrated to provide a direct measure (indicated, e.g., in parts per
billion, or ppb) of ~e concen~tion of the dement within ~e sample complex.
As soen in FIG. 6, the output signal from ~e amplifier 50 may be
- measured with a digital voltmeter (DVM) 52. As desired, ~is output may be recorded
20 or stored in a data logger 44, or equivalent device.
It is noted that all of the elements shown in FlG. 6, e.g., the
photodetectors, amplifiers, DVM's, data lo~gers, laser source, etc., may be realized
using the same types of de~rices and equipment as described previously in connection
wi~ FIG. 2.
In the second embodiment, a personal computer (PC) 46, or equivalent
processor, is coupled to the output of the amplifier 40 and the out~ut of the amplifier
50. Such coupling may be direct if the computer 46 includes internal analog-to~digîtal
(A/D) conversion means (for converting the analog output signal from the amplifier
40 and the amplifier 50 to a digital signal suitable for use with the PC 46); or~
30 alternatively, if the DVM's 42 and S2 include a digital output port, as many
commercialb available DVM's do, thc coupling may be made f~m the DVM's 42
and 52 to the processor 46.
.~ .
-
Wo 93/07472 P~US92/û8363
211313~
Further details associated with the ope~ation of the embodiment shownin Fig. 6 may be found in Applicants' c~pending U.S. patent app1ica~on Serial No.
07/769,531, filed October 1, 1991, incorporated herein by reference.
Advantage~usly, the signals receîved by ~e processor 46 from the
S amplifiers 40 and 50 are used to automatically Gllcula~e the concenh~ion of the
desired element, e.g. gold, present in ~e c~mplex sample. As in the first
embodiment, a calibration graph, such as that shown in FIG. 4, can be generated (or
mathematically expressed) by analyzing complex samples from hlown desired element
concentrations, e.g. hlown gold concen~ations. In accordance with the present
10 invention, a first calibration curve is generated based on the fluorescence dete~tor 20
out~ut, and a second calibration curve is generated based on the ~ansmission detector
21 output. (As used herein, the tenn ~calibration curve" refers to any means, whether
a mathematical equation, or equivalent, that relates one variable, i.e., the detector
output, to another vanable, i.e., the gold concentration.) The first ealibration curve
15 is generated, based on b~own complex sample concentrations, and may be
mathema~dcally expressed as:
R
C: = (l/ael) ~c In 1~] (1) :
K- (S - Sa~
where (:, is the gold concentration9 S is the fluorescence detector output, is an
intnnsic absorption constant for the matenal, ~1 is the effec~ve length of the cuvette,
and K, and S0 are constants. The constants are determined empincally using samples
25 containing hlown concen~a~ons of gold. Re~resen~ative values of such constants
al, = .00035, X-0.755, and So=0.0045 (volt~. Unfortunately, the ability of Eq.
(1) to accurately estimate the gold concen~ation is limite(l to a lower range ofconcentrations, e.g., about 0 ppb to 3,000 ppb.
If the range of concent~ations is higher than this range, then the second
30 calibration curve is used. The second calibration curve is also derived from h~own
comple~ sarnple conoentrations, and may be mathematically expressed as:
C = (l/at~) ~c ln (To ~1~ (2)
W093/07472 '~ 26- Pcr/uSs2/0~363
where C is the gold concentration, T is the transmissive detector ou~put, and 11~2
and To are constants. Again, the constants are determined empirically using comple~
samples containing Icnown concentra~ons of gold. Re~resentative values of such
constants are a~2=.0000751; and To=1.75 (volts). l~e ability of Eq. (2) to
S accurately approximate the second calibration curve is limited to a higher~range of
concentrations, e.g. 1,000 ppb to 25,000 p~b.
Hence, in operation, after the amplifier 40 output signal and ~e
amplifier 50 output signal are generated, the processor 46 calculates a first
concentration, based on the fluorescence formula, Eq. (1), and a second
10 c~ncentration, based on the transmission formula, Eq~ (2). Because the range of
accuracy for the fluorescence formula, equation (1) is limited to approximately 3000
ppb or less, equation (2) is used for concentrations in the range from 2000 ppb to
approximately 30,000 ppb. ln the overlap range between 2,000 to 3,000 ppb eitherequation may be used. In this way, this second embodiment provides a significant15 increase in the dynamic range over tbe first embodimen~.
Advantageously, use of the source detector 15 allows the signals
detected by ~e detectors 39 and 49 to be corrected for variations that occur in ~e
intensity of ~e inadent source 14. The mirror 17 functions as a beam splitter and
directs a small portion of ~e incident sadiation (e.g., approximately 4%) to ~e
20 detector 15. An initial reading of the source intensity is represented as the signal SRd;
a current reading of the source intensity is represented as a signal S,~,; a current
reading of the fluoresced radiation is represented as a s;gnal Sp; and a current reading
of the transmitted radiation is rcpresented as a signal ST. Both the fluo~escence and
t~ansmission signals Sp and ST may then be correcte~l by u~lizing the following
25 relat;onships:
SP(COSr) = (SP SR~)/SAI and
ST(Corr) = (ST SRCr)/S~I -
A more detailed description of the steps taken by the processor 46 incalculating an accurate gold concentration is presented below with respect to ~:IGS.
30 7A and 7B.
It is noted that the ~nsmission properties of the filters 35, 37 and 45
used with the apparatus of FIG. 6 are sel~cted for the detection of gold as a gold-
~ c - ~--
r ~ r ~ ~ ~ ~ r
r ~ r
r r ~ ~ ~ O ~ r ~ r ~ r
2 1 1 ~J 1 ~3 ~
- 27 -
Rhodamine B complex in the same manner as used in connection with the filters 30and 34 associated with the apparatus of FIG. 2. The filter 45 includes a first peak
or band centered at approximately 546 nm. The filters 35 and 37 include a secondpeak or band centered at approximately S77 nm. The laser radiant energy source 25
5 provides radiant energy of approximate wavelength 543.5 nm, which is roughly
centered in the peak or band of the filter 45.
Refernng next to FIGS. 7A and 7B, a flow diagram is shown that
illustrates the manual and programmed steps used to measure the concentration of the
desired element within the sample 12 using the tluorometer/transmissive
10 instrumentation apparatus of the second embodiment. Each main step of the method
showll in FIGS. 7A and 7B is depicted as ar'!block". It is submitted that those skilled
in the art can readily perform the steps shown in FIGS. 7A and 7B (if manual) orwrite an appropriate program or code for use by the processor 46 (if automatic).Referring then to FIG. 7A, bloclc 110 represents the preparation, as
15 described above in connection with the method of the present invention, of the sample
complex 12, including contacting the prepared matenal with a suitable label means
- to which gold, when present, has been bound. After preparing the desired element-
label means complex, e.g., gold-Rhodamine B complex, the solution is transferredinto a cuvette, or transparent container, through which the sample solution can be
20 irradiated. Further, during the reading of the initial reference sample, a`measurement
is made of the intensity of the radia~ion source 14 in order to obtain the signal~
as described a~ove.
Af~er applying power to the apparatus 11, the processor 46 monitors
- a switch 60 (FIG. 6). The switch 60 assumes an open position or a closed position
25 responsive to the opening or closing of a door to a test chamber that holds the cuvette
containing the sample 17 while the sample 12 is tested for the desired-element
concentration. Al~ernatively, other manual or automatic means for detecting whether
the cuvette has been put in the test chamber and/or the test chamber door has been
closed may be used, e.g., using optoelectronic sensvrs, load cells or by displaying a
30 message and requestnlg operator confirrnation that the cuvette is in place and the test
chamber door is closed.
wo 93/07472 Pcr/US92/08363
~91~3~ - 28 -
Thus, as soon as possible after applying power to the apparatus 11
(e.g., after a suitable warmup period), the switch 60 or the other detection means
detects whether the test chamber is empq or whether the test chamber door is opcn.
Responsive to the "open" or ~empty~ detection, tbe processor 46, under the control
S of a suitable control program, signals the operator to put a cuvette containing a
known concentration of desired element-label means complex into the test chamber.
Sucb signalling is acbieved by displaying a message on a CRT display tern~inal, or
by using other signalling means, e.g. Iights or audible alarms. Block 120 of FIG. 7A
represents the signalling of the operator to place the cuvette in the test chamber and
10 to close the test chamber door.
Upon detecting that the cuvette is in the test chamber, as re~resented
by block 130, tbe processor 46, responsive to the control program, activates solenoid
23 which opens electro shutter 29 (FIG. 6) thereby directing radiant energy into the
sample~ontaining cuvette as explained hcreinabove. After a sufficient delay to allow
15 transient responses to dissipatc, as ~presented by bloc~ 150, a measurement is made
of ~e intensity of thc n dia~on sou~ce 14 is made in order to obtain tlle signal SRCf
(block 152), as desc~iW above. Ne%t, the source intensity is cheebed to obtain ~e
signal S,, *lock lS4), the output of the fluorescenoe detector 20 is read to obtain the
fluorcscence signal Sp (block 160), the output of the transmission detector 21 is read
20 to obtain the transmissive signal ST (block 170), and such signals (which are read as
output voltages) ~re then coITected as required (block 172). (As men~oned above,the recordation of ~e output voltages may be achieved via analog-todigital
conversion means coupled to the PC 46 or by the digital ou~put ports of the DVM's
42 and 52.)
Afterrecording the ou~putvoltages generat~d in response to the hlown-
desired~lement concen~adon sample, the electro shutter 29 is closed. If sufficient
known samples have been measured, the processor 46 uses the volsages to
mathematically calculate values for the above mentioned constants, i.e., 1/~l, K, S0,
and To. This calculation is represented by block 190.
In ~e preferred embodiment, the steps indicated at blocks 120, 130,
140, 150, 152, 154 160, 170, 172 and 180 are repeated or iterated 12 times, thereby
achieving highly accurate approximated constants, as indicated at block 190. In this
, . . .
W O 93/07472 P(~r/US92/08363
, -
-29' 211~134
way the apparatus 11 is calibrated to measure unknown desired elemcnt
concentrations.
Next, as represented by blocl~ 200, the processor or PC 46 signals the
operator to put the cuvette containing the unknown concentration of desired element-
S label means in the test chamber. The operator then places the cuvette containing theunknown sample into the test chamber. After sensing that the cuvette is in place and
that thc test chamber door is c!osed, represented by block 210, that the cuvette is in
place and that the test chamber door is closed, the processor 46 (as controlled by the
control program) activates solenoid 23 which opens the electro stop 29, thereby
10 directing radiant energy into the cuvette contair~ing the sample 12. The opening of
the electro stop is represented by block 220. After a sufficient time delay to allow
transient responses to dissipate, as represcnted by block 230, the processor 46 reads
and stores the output voltage or signal SA1 from the source detector 15, the output
voltage or signal Sp from the fluoresoence detector 20, and the output voltage or
15 signal ST from the transmission de~ector 21, after which the electro stop is closed.
Such reading and storing is represented by blocks 232, 240 and 250 in FIG. 7B.
- After reading the output voltages of ~e source detector 15, the
fluorescence detector 20, and the transmission detector 21, the processor 46 corrects
such readings for any variations that`may have occurred in the intensity of the source
20 radiation using the aboverdescribed correction method. Such correction is r;epresented
by bl~ck 252. Next, the processor calculates the desired element ~oncen~ation using
the abov~mention~d formu~s, Eqs. ~1) and (2), or uses other means for
approximating the desired elemen~ concentra~don based on the amplifier output
voltages. That is, the processor 46 calculates a first concentration based on the
25 fluorescence detector output, and a second concentration based on the transmission
detector out~ut. The processor 46 then compares the first and second concentrations
with a reference value as mentioned above. The reference value is selected to be a
desired element concentratdon in the overlap range or in the range of accuracy for
botb the fluorescence forrnula and the t~ansmission formula. If the concentrations are
30 below the reference value, the first concentration, based on the fluorescence detector
output, is used or selected as an indication of the concentra~on. lf tbe concentrations
are above the reference va1ue, the second concentration, based on the tIansmission
WO93/07472 ~9~3~ PCI/US9~/08363
- 30 - ~
detector output, is used or selected as an indication of the concentration. l'heselection of the appropriate concentra~on is re~resented by bloc~ 260. In ~is way,
the more accurate concentration is used as an indication of the gold (or other metal)
concen~ation. The appropriate equation is then used to calculate the concentra~on
S (block 270). This selected concent~ation is then displayed by the prooessor 46 (block
280) ~nd/or suitably recorded by the processor 46.
Finally, the processor 46 signals the operator to indicate whether
additional unknown~esired-element~ncentration samples to be tested (block 280)
If the operator indicates that additional sarnples are to be tes~, the steps represented
10 by blocks 200, 210, 220, 230, 232, 240, 250, 252, 260, 270 and 275 are repeat~d
or i~erated. Such re~etition continues until the operator indicates that ~ere are no
more samples to be tested. In this way, multiple unhlown samples can be accurately
tested for desired element concentrations, and the most accurate detee~on means, i.e.
fluorescence detection or transmission detection, can be utiliæd in r~onse to a
15 programmable controlled processor 46.
Advantageously, all of the components of the preferred embodiment 11
are sufficiently small and light weight ~o that they may be easily transport~d, e.g.,
in a ~ailer, truck, van, by backpack or the like, ~ere~y allowing the en~re apparatus
to be portable and easily ~ oned to a field loca~on where soil or rock samples are
20 to be assayed. Fu~er, the second embodiment advantageously eliminates the need
for ~e fiber op~c bundle, thereby elimina~ng the problems associated with the use
of a fiber op~c bundle, e.g., cFacl~ng, while providing a larger overall range of
accuracy by using bodl a fluorescence de~tor 20 and a ~smission det~ctor 21.
Hence, by using the ~nd embodiment, the advantages of the first embodiment and
25 other advantages are maximiæd and the disadvant~es of the first embodiment are
minimized.
In one embodiment of the invention, a small and light weight device
is provided that is adapted for ~ransport in a back pack. Such small, portable unit
(hereafter the "back-pack unit~) is thus readily carried to a desired field location
30 where the sample analysis is to be performed. Advantageously, such back-pack unit
is battery powered, thereby allowing the analysis to be carried out at remote locations
where other power sources are not readily available.
WO 93/07472 2 1 1 3 1 3 4 Pcr/us92/o~363
~ 31 ~
In order to conse~ve the limited energy available from the battery of
the back-pack unit, a flashlamp is used as the source of radiant energy 14 (FIG. 5)
rather than a laser. The flashlamp provides a very short pulse or "flash" of int~nse
radiation, similar to the flash provided from a flash camera. Such pulse or flash of
S radiation is ~en optically filtered, as needed, in order to significantly attenuate all
wavelengths except the wavelength of interest, ~,. (It is noted, of course, that even
a laser may be pulsed.)
As des~ibed above in connec~on wi~ FIGS. 5 and 6, the back-pack
unit also employs three separate sensors or detectors. A first sensor detects the
10 intensity of the radiation from the source 14 ()~, detector 15). A second sensor
detects the intensity of the radiation that fluoresces from the sample (A2 de~ector 20).
A third sensor detects the intensity of the radia~on transmitted through the sample (A,
detector 21). Because the intensity of the source 14 from a typical flashlamp varies
a great deal from flash to flash, the use of ~e ~1 detector 15 to detect ~e Mtensity
15 of the ~ ladiation at ~e sour~e, thereby allowing the~eadings at the ~l transmissive
detector 21 and the ~2 fluoresced detector 20 to be corrected for such vanations as
described above, becomes extremely imp~rtant to the successful operation of ~he
back-pack unit.
A typical flashlamp that may be used widl ~e back-pack unit is the
20 XLS-S42 flashlamp, available from Xenon Corp. of Wobum, Massachusetts. The
weight of the back-pack unit is less than about four (43 kg (8 lbs.); and its
approximate volume, ~cluding battery, is no more ~an about 16000 cm3 (1000 ill3).
The flashlamp is powered using a conventional power supply that steps up ~e voltage
from the battery (e.g., 9 volts) to a suitable flashlamp operating voltage (e.g., 600
2$ volts). Advantageously, ~e sensors 15, 20 and 21 consume very little power, and
the PC (computer) 46, when used, may be of a laptop or notebook size, which
computers are already made for portability.
The invention will now be described in greater detail by reference t~
the following non-limiting examples.
Wo 93/07472 Pcr/US~2/08363
32
EXAMPLF~
The general procedure followed for each of the analyses describ~d
below is as follows: . `
Samples ar~ prepared carefully to obtain representative ore samples.
The ore samples are roasted under o~idizing atmos~here in ~e temperatur@~ range of
600 800OC for about one hour. Gold (Au) is leached out by tun~bling the ore in
potassium cyanide/potassium hydroxide solution in the presence of hydrogen
pero~ide. The cyanide extract is then acidified with hydrochloric acid in the presence
of hydrogen peroxide at about 90OC. The resulted Au(: 14- complex is removed from
10 interference ions ~y tumbling with pretreated poly(dibenzo 18-crown-6) beads in 0.6
M HCl. 1 he polymer beads ælectively trap AuCli complex leaving the interferenceions in HCl. After several washes, Au~14- is recovered by tumbliDg the AuCL- -
loaded beads with acetone, the acetone is further evaporated in the presence of H2O2
leaving AuCl4- in HCl. The AuCl; is labelled with Rhodamine B dye. The AuCl4-
15 dye complex is further ext~acted into diisopropylether, and thefluo~escence/transmittance of the complex is measured.
E~ample L:
To explore the range of the signal obtained with ~e invention appa~atus
20 and method when ~e amount of gold is increased, a range of gold concen~tions
were studied (09 1 2000S and 20000 ppb of gsld). The results in millivolts
response per sample, are set for~ below in Table 1.
WO 93/~7472 2 1 1 ~ 1 3 4 ~cr/US92/083~3
- 33 -
Gold con~n~b Millivolts
0 8
500 199
1~00 516
20~0 946
4000 1658
8000 2629
1 0 ~
The results of Table 1 are plotted in Figure 4, which reveals a smooth
non-linear response over a wide range of concentradons.
These results demonstrate that the invention method and apparatus are
useful over a wide concen~on range.
Exam~?le 2:
The invention technique was employed to est;mate ~e amount of gold
p~nt in two rock samples; these sample were designated samples "A" /and "13",
and were independently determined by fire assay analysis to contain about 1325 and
20 166~ ppb of gold, ~espec~vely. The signals o~tained for 0, 500, 1000, 2000, and
4000 ppb of gold were 0, 410, 770, 1430, and 2080 millivolts, respectively. Whenplotted, ~ese va1ues give a smooth non-linear response over the entire concen~a~on
range evaluated~
The fluorescent sigr,als for samples ~A~ and ~B" were 970 and 1120
25 MY (afte~ s~tra~n~ the ~values ~or ~ zero control sample), corresponding to about
}325 and about 1660 ppb of gold, respecdvely.
~am~le 3:
The fluorescent/transmissive apparatus described above in connection
30 with ~IGS. 5, 6, 7A and 7B was employed to measure the gold concentration of four
test samples, 1abele~ , "2~, "3~ and "4", having ppb gold concen~a~dons of 0,
r ~ ~ ~ r r
- ~ r 2 1 3 9 ~ 3 4
- 34 -
100, 200Q and 20000, respectively. The resul~s obtained are summarized below in
Table 2.
TABLE 2
S CALCULATED PPB OF TEST SAMPLES
FROM THEIR MEASURED READINGS
(Example 3)
CALIBRATION DATA
PPBFLUORE~SCENCE (VOLTS) TRANSMIl~ANCE ~OLTS)
0 0.00346 1.71 1
10Q 0.0341 ~ 1.678 --
2000 G.402 1.333
1~ 200~0 0.676 0.0618
CALCULATED CURVE CONSTANTS
a~l K ae2 S0 To
0.00Q493 0.636 7.41E-05 0.00346 1.711
GOLD CALCULATIONS OBTAINED USING ~QUATIS:)NS (1) A~D (2)
TEST
SAMPLE GOID(PPB) S(VOLTS) T~VOLTS~ CAL GOLD~PPB~
.
1 o 0.00363 1.7 0.54
2 loo 0.34s 1.681 lo
3 2000 0.401 1.337 lg~o
4 20~ 0.~86 0.06~2 19615
l'he invention method and apparatus are seen to rapidly provide
results which agree quite well w;th values obtained by traditional analy'dcal means.
S~ TITU~E SHE~T
... .