Note: Descriptions are shown in the official language in which they were submitted.
W(~ 93/1 1 14X 2 1 2 2 4 7 0 Pcr/US92/10043
OLIGONUCI.EOTIDES ~AVING ~INO~YDRQCARBON
PllOSP~0~113 MOIETIES
Thi~ application is a continuation-in-part of
Application Serial No. 79~,804, filed November 25, l991.
This invention relates to oligonucleotides which bind
to RNA (such as mRNA), DNA, protein~, or peptides,
including, for example, oligonucleotides which inhibit mRNA
function. More particularly, this invention relates to
oligonucleotides in which one or more of the nucleotides
include an aminohydrocarbon phosphonate moiety.
Watson-Crick base pairing enakles an oligonucleoti~e to
act as an antisense complement to a target sequence of an
mRNA in order to block processing or effect translation
arrest and regulate selectively gene expre~sion. ~Cohen,
OliaodeoxYnucleotides, CRC Presæ, Boca Raton, Florida
(lg89)); Uhlmann, et al., Chem. Rev., Vol. 90, pgs. 543-584
(1990)). Oligonucleotides have also been utilized to
interfere with gene expression directly at the DNA level by
formation of triple-helical (triplex) structures in part
through Hoogsteen bondi~g interactions (Moffat, Science,
Vol. 252, pgs 1374-137~ (l991)). ~urthermore,
oligonucleotides have been shown to bind specifically to
protein~ (Oliphant, et al., Molec. Cell. Biol. Vol g, pgs.
2944-2949 (19B9)) and could thus be used to block
undesirable protein function.
SuasT~
WO93/1114X 2 122 ~7 0 -2- PCT/US92/10043
Natural oligonucleotides, which are negatively charged,
however, are poor candidates for therapeutic agents due to
their poor penetrability into the cell and their
susceptibility to degradation by nucleases. Thereore, it
is expected that relatively high concentrations of natural
oligonucleotides would be reguired in order to achieve a
therapeutic effect.
To overcome the above shoxtcomings, various strategies
have been devised. U.S. Patent No. 4,469,863, issued to
Miller, et al., discloses the manufac'ure of nonionic
nucleic acid alkyl and aryl phosphonates, and in particular
nonionic nucleic acid methyl phosphonates. U.S. Patent No.
4,757,055, also issued to Miller, et al., discloses a method
for selectively controlling unwanted expression of foreign
nucleic acid in an animal or in mammalian cells by binding
the nucleic acid with a nonionic oligonucleotide alkyl or ~
aryl phosphonate analogue. ~`
Oligonucleotides have also been synthesized in which
one non-bridging oxygen in each phosphodie ter moiety is
replaced by sulfur. Such analogues sometimes are referred
to as phosphorothioate (PS) analogues, or "all PS"
analogue~, (Stein, et al., Nucl. Acids Res., Vol. 16, pgs.
3209-3221 (1988)). Another approach has been to attach a
targeting mo~ety, such as cholesterol, which improves the
uptake of the oligonucleotide by a receptor-mediated
process. (Stein et al., Biochemistry, Vol. 30, pgs.
243g-2444 ( 1991 ) ) . ' '
Examples of oligonucleotides with positive charges have
been reported. Letsinger, et al. (JACS, Vol. 110, pgs.
4470-4471 ~1988)) describe cationic oligonucleotides in
which the backbone is modified by the attachment of diamino
compoundæ to give positively charged oligonucleotides with
phosphoramidate linkages. Phosphoramidate linkages,
however, are known to be somewhat labile, especially at
SUBSTITUTE SHEEr
W093/1114X 2 i 2 2 1 7 0 PCT/US92/10043
3--
acidic pH levels, and therefore the cationic group could be
lost under certain conditions. Conjugates with the
positively charged molecule polylysine have been described
by Lemaitre, et al., Proc Nat. Acad. Sci., Vol. 84, pgs.
648-652 (1987), and have been shown to be more active in
cell culture than unmodified oligonucleotides. Polylysine,
howe~er, is not a preferred molecule for conjugation due to
its relatively high toxicity.
Mononucleotides with aminomethyl phosphonate moieties
have been synthesized in order to study their susceptibility
to nucleotide degrading enzymes. Holy, et al. (Journal of
CarbohYdrates, Nucleosides and Nucleotides, Vol. l, pgs.
85-96 (1974)) disclose the synthesis of uridine-2'
(3'-aminomethyl) phosphonate and thymidine -3'-aminomethyl
pho~phonate by the reaction of th~ corresponding 5'-0-trityl
nucleo~ide with N-benzyloxycarbonyl-aminomethyl phosphonate.
Gulyaev, et al., (FEBS Letters, Vol. 22, pgs. 294-296
(1972)) disclose the formation of ribonucleoside
5'-aminomethyl phosphonates.
In accordance with an aspect of the present invention,
there is provided an oligonucleotide wherein at least one
nucleotide unit includes a phosphonate moiety having the
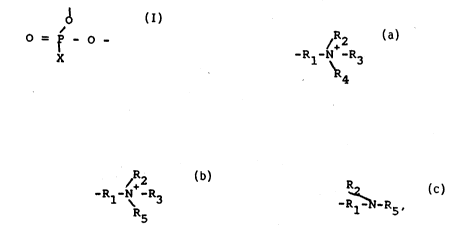
following structural formula:
O = P - O -
X,
wherein X is:
Rl-N-R3
R4.
SUBSTITUTE SHEEl-
WO93/11148 PCT/US92/10043
2 122 4~ 0 _4_
Rl is a hydrocarbon, preferably alkylene, phenylene, or
naphthylene, more preferably an alkyl group having from l to
15 carbon atoms, and most preferably l to 3 carbon atoms,
with methylene being preferred. Each of R2 R3, and R4 is
hydrogen or a hydrocarbon. Preferably, the hydrocarbon is
an alkyl group having from l to 15 carbon atoms, more
preferably from l to 3 carbon atoms, and most preferably a
methyl group. Each of R2, R3, and R4 may be the same or
different. Most preferably, each of R2, R3, and R4 is
hydrogen.
The term "oligonucleotide", as used herein, means that
the oligonucleotide may be a ribonucleotide or a
deoxyribonucleotide; i.e., the oligonucleotide may include
.- ribose or deoxyribose sugars. Alternatively, the
oligonucleotide may include other 5-carbon or 6-carbon
sugars, such as, for example, arabinose, xylose, glucose,
galactose, or deoxy derivatives thereof.
In general, the oligonucleotide has at least two
nucleotide units, preferably at least five, more preferably
from five to about 30 nucleotide units.
As hereinabove stated, at least one nucleotide unit of ;
the oligonucleotide includes a phosphonate moiety which is
an aminohydrocarbon phosphonate moiety, as hereinabove
described. An aminohydrocarbon phosphonate moiety may be
attached to one or more nucleotide units at the 3' end
and/or at the S' end of the oligonucleotide. In one
embodiment, an aminohydrocarbon phosphonate moiety may be
attached to alternating nucleotide units of the
oligonucleotide. In another embodiment, an aminohydrocarbon
phosphonate moiety may be attached to each nucleotide unit
of the oligonucleotide.
The oligonucleotides also include any natural or
unnatural, substituted or unsubstituted, purine or
pyrimidine base. Such purine and pyrimidine bases include,
SU~STITUTE SHEF~
wo 93/11148 2 I 2 2 ~ 7 0 ` P~/US92/10043
--5--
but are not limited to, natural purines and~ pyrimidines such
as adenine, cytosine, thymine, guanine, uracil, or other
purines and pyrimidines, such as isocytosine,
6-methyluracil, 4,6-dihydroxypyrimidine, hypoxanthine,
xanthine, 2, 6-diamino purine, azacytosine, 5-methyl
cytosine, and the like. -
In a most preferred embodiment, X is an aminomethyl
moiety. The synthesis of an oligonucleotide having such
aminomethyl pho~phonate moieties may be accomplished through
the synthesis of a monomer unit with a protected aminomethyl
group, followed by incorporation of one or more
such monomer units into an oli~onucleotide; or by synthesis
of an oligonucleotide followed by subsequent attachment of
the aminomethyl groups.
Monomer units which may be incorporated into an
oligonucleotide, may, in one embodiment, be prepared as
follows:
Aminomethyl phosphonic acid may be reacted with a
suitable reagent, ~uch as trifluoroacetic anhydride,
fluorenyloxycarbonylchloride, or phthalyl chloride to
protect the amino group, and to give one of the following :~
protected derivatives, (1), ~2), or (3):
o
~ ~ CI~Iz~ CO~
OH OA~
-- ~o~ Gg
=~--C h,'~ o - O ~
o~ 3
Alternatively, the phthalimide derivative (1) may be
prepared by reaction of chloromethyl phosphonic acid with
pht~alimide, or by demethylation of commercially available
TITI IT~ FFT
WO93/11148 PCT/US92/10043
212~ 4~ -6- : ~
dimethylphthalimidomethyl phosphonate using trimethylsilyl
bromide.
Hydroxymethyl phosphonic acid can also be used as a
starting material for the synthesis of aminomethyl
phosphonate derivatives. The reaction of hydroxymethyl
phosphonic acid with trifluoroacetic anhydride produces an
ester which can be converted into a pyridinium intermediate,
the reaction of which with ammonia produces aminomethyl
phosphonic acid.
Reaction of one of the protected derivatives (1), (2),
or (3) with a partially protected nucleoside, such as one
having the structural formula (4):
~c~3
C~o ~ C--~¢~
0
(~)
wherein B is a protected or unprotected purine or pyrimidine
base, in the presence of a condensing aqent such as
dicyclohexylcarbodiimide or triisopropylbenzene-sulfonyl
chloride would produce an ester having the following
structural formula (5):
~C~3
,~
C~C~C- 0~
~ )/
S~--C
o
wherein Q is the protected amino group.
SUBSTITUTE SHEET
WOg3/11148 21~2470 PCT/US92/10~3
Preferably, the protected amino group is selected from
the group consisting of:
~a~
Cb~ c~
cc) ~ C ~7 - o--C ~
The ester having the structural formula 5 can be used
as a monomer unit for oligonucleotide synthesis by coupling
to a protected mononucleotide or oligonucleotide attached to
a ~olid support. After the solid support~attached
oligonucleo~ide is synthesized, the material is treated with ;~
ammonia to cleave the protecting groups and generate~an
oligonucleotide having one or more aminomethyl phosphonate
moieties. Alternatively, the phthalimide protecting group
can be rémoved by treatment with hydrazine or a substituted
hydrazine to ~enerate the aminomethyl compound. By this
route, the aminomethyl modified units can be introduced at
any position in the oligonucleotide as desired.
Alternatively, a modified mononucleotide may be
prepared by reacting a partially protected nucleoside æuch
as hereinabove described with a protected aminomethyl
phosphite derlvative to form a nucleoside phosphonamidite.
The nucleo8ide phosphonamidite can then be used in place of
a nucleoside pho~phoramidite in a DNA synthesizer. At the
conclusion of the synthesis, the protecting groups can be
r~moved from the aminomethyl moieties by treatment with
ammonia or with amines such as ethylenediamine.
It is also contemplated that aminomethyl phoiphonate
moieties may be introduced into preformed oligonucleotides.
One appro~ch is to carry out a synthesis of an
oligonucleotide on a solid support using a DNA synthesizer,
except that the iodine oxidation step which is normally used
,~
9UBSTITUTE SHEET
.. :.
, ~
W093/1114X 2 1 2 2 4 7 0 PCT/US92/1~43
-8-
to oxidize the phosphite intermediate to a phosphate is
eliminated, and instead the oligonucleotide phosphite
attached to the solid support is reacted with
phthalimidomethyl bromide. Subseguent treatment with
ammonia removes the phthalimido protecting group to give the
aminomethyl oligonucleotide.
Alternatively, a methyl phosphonate oligonucleotide can
be prepared by using commercially available nucleoside
methyl phosphonamidites, and the methyl phosphonate
oligonucleotide is then treated with iodine in pyridine to
give a methyl pyridinium intermediate which can be converted
into an aminomethyl oligonucleotide by treatment with
ammonia.
~, In another embodiment, some oligonucleotides in-
accordance with the present invention may be prepared such
that the oligonucleotides may be isolated as pure
stereo~omers in either the--R- or S- form. Such
oligonucleotide~ include those with one aminohydrocarbon
phoæphonate moiety at, or adjacent to, either the
3'-terminus or the 5'-terminus; oligonucleotides having
aminohydrocarbon phosphonate moieties at both the 3'- and
5'-termini; oligonucleotides having aminohydrocarbon
phosphonate moieties at internal positions, provided that
the am~nohydrocarbon phosphonate moieties are not present on
adjacent nucleotide units; oligonucleotides in which
aminohydrocarbon phosphonate moieties alternate with natural
phosphodiester linkages throughout the entire sequence; and
oligonucleotides possessing a mixture of aminohydrocarbon
phosphonat~ and other modified backbone substituents, such
as phosphorothioates.
Such oligonucleotides may, in one embodiment, be
prepared by synthesizing protected aminohydrocarbon
phosphonate dinucleotides which are mixtures of R- and S-
isomers, followed by separation of the R- and S- isomers by
SUE~STITUTE SHEET
W093/l1148 - 2 1 2 2 ~ 7 0 PCTtUSg2~10043
conventional means, such as high pressure liquid
chromatography or sîlica gel column chromatography. The
pure isomers may then be attached to oligonucleotides by
conventional means to produce single isomer aminohydrocarbon
phosphonate oligonucleotides.
The administration of the oligonucleotides as pure
steroisomers in either the R- or S- form may fùrther improve
the binding capabilities of the oligonucleotide and/or
increase the resi~tance of the oligonucleotide to
deqradation by nucleases.
The oligonucleotides may include conjugate groups
attached to the 3' or 5' termini to improve further the
uptake of the oligonucieotide into the cell, the stability
of the oligonucleotide inside the cell, or both. Such
conjugates include, but are not limited to, polyethylene
glycol, polylysine, acridine, dodecanol, and cholesterol.
The oligonucleotideæ of the present invention may be
employed to bind to RNA seguences by Wat~on-Crick
hybridization, and thereby block RNA proce~sing or
translation. For example, the oligonucLeotides of the
present invention may be employed as "antisense" complements
to target sequences of mRNA in order tQ_e~fect translation
arrest and regulate selectively gene expression.
The oligonucleotides of the present invention may be
employed to bind double-stranded DNA to form triplexes, or
triple helices. Such triplexes inhibit the replication or
transcription of DNA, thereby disrupting DNA synthesis or
gene transcription, respectively. Such triplexes may also
protect DNA binding sites from the action of enzymes such as
DNA methylases.
The RNA or DNA of interest, to which the
oligonucleotide binds, may be present in a prokaryotic or -
eukaryotic cell, a virus, a normal cell, or a neoplastic
cell. The sequences may be bacterial sequences, plasmid
Sl~3ST~TUTE SHEET
WO 93/1 1 14X 2 1 2 2 ~ ~ ~ PCT/US92/10043~
--10--
seguences, viral sequences, chromosomal sequences,
mitochondrial sequences, or plastid sequences. The
sequences may include open reading frames for coding
proteins, mRNA, ribosomal RNA, snRNA, hnRNA, introns, or
untranslated 5'- and 3'-sequences flanking open readin~
frames. The target sequence may therefore be involved in
inhibiting production of a particular protein, enhancing the
expression of a particular gene by inhibiting the expression
of a repressor, or the sequences may be involved in reducing
the proliferation of viruses or neoplastic cells.
The oligonucleotides may be uced in vitro or in vivo
for m~diEying the phenotype of cells, or for limiting the
proliferation of pathogens such as viruses, bacteria,
protists, MYcoplasma species, Chlam~dia or the like,-or for
inducing morbidity in neoplastic cells or specific classes
of normal cells. Thus, the oligonucleotides may be
administered to a host subject to or in a diseased state, to
inhibit the transcription andjor expression of the native
genes of a target cell. Therefore, the oliqonucleotides may
be used for protection from a variety of pathogens in a
host, such as, for example, enterotoxigenic bacteria,
Pneumococci, Nei~seria organisms, Giardia organisms,
Entamoebas, neopla~tic cells, such as carcinoma cells,
sarcoma cells, and lymphoma cells; specific B-cells;
8pecific T-cells, such as helper cells, suppressor cells,
cytotoxic T-lymphocytes (CTL), natural killer (NK) cells,
etc.
The oligonucleotides may be selected so as to be
capable of interfering with transcription product maturation
or production of proteins by any of the mechanisms involved
with the binding of the subject composition to its target
sequence. These mechansims may include interference with
proces~ing, inhibition of transport acro~s the nuclear
membrane, cleavage by endonucleases, or the like.
SUE~STITUTE SHEET
W093/1114X 2 1 2 ~, ~ 7 0 PCT/US92/10043
The oligonucleotides may be complementary to such
sequences as sequences expressing growth factors,
lymphokines, immunoglobulins, T-cell receptor sites, MHC
antigens, DNA or RNA polymerases, antibiotic resistance,
multiple drug resi~tance (mdr), genes involved with
metabolic processes, in the formation of amino acids,
nucleic acids, or the like, DHFR, etc. as well as introns or
flanking s~quences a~sociated with the open reading frames.
The following table is illustrative of some additional
applications of the subject compositions.
Area of ADDlication Specific AvPlicati-on Taraets
Inectious Diseases: -
Antivirals, Human AIDS, Herpes, CMV
Antivirals, Animal Chicken Infectious Bronchitis
Pig Transmissible
Gastroenteritis Virus
Antibacterial~ Human Drug Resistance Plasmids,
E. coli
Antiparasitic Agents Malaria
Sleeping Sickness
(Trypanosomes)
Cancer
Direct Anti-Tumor Oncogenes and their products
A~ents
Ad~unctive Therapy Drug Resistant Tumors-Genes
and Products
Auto Immune Diseases
T-cell receptors Rheumatoid ~rthritis
Type I Diabetes
Systemic Lupus
SUBSTITUTE SHEET
W093/1114X 2 1 2 2 ~ ~ O PCT/US92/10043
-12-
Multiple sclerosis
''
Organ Transplants Kidney-OTK3 cells cause
GVHD
The oligonucleotides of the present invention may be
employed for binding to target molecules, ~uch as, for
example, proteins including, but not limited to, ligands,
receptors, and/or enzymes, whereby such oligonucleotides
inhibit or stimulate the activity of the target molecules.
The above techniques in which the oligonucleotides may -
be employed are also applicable to the inhibition of viral
repl-ication, as well as to the interference with the
expression of genes which may contribute to cancer
development.
The oli~onucleotides of the present invention are
administered in an effective binding amount to an RNA, a
DNA, a protein, or a peptide~ Preferably, the
oligonucleotides are ad'ministered to a host, such as a human
or non-human animal host, so as to obtain a concentration of
oligonucleotide in the blood of from about O.l to about 100
~mole/l. It is also contemplated, however, that the
oligonucleotides may be administered in vitro or ex vivo as
well as in vivo.
The oligonucleotides may be administered in conjunction
w~th an acceptable pharmaceutical carrier as a
pharmaceutical composition. Such pharmaceutical
compositions may contain suitable excipients and auxiliaries
which facilitate processing of the active compounds into
preparations which can be used pharmaceutically. Such
oligonucleotides may be administered by intramuscular,
intraperitoneal, intraveneous or subdermal injection in a
suitable solution. The preparations, particularly those
which can be administered orally and which can be used for
SU9STITUTE SHE T
WO93/1114X 2 ~ 2 ,~ ~17 a PCT/US92/10043
-l3-
the preferred type of administration, such as tablets,
dragees and capsules, and preparations which can be
admini~tered rectally, such as suppositories, as well as
suitable solutions f~r administration parenterally or
orally, and compositions which can be administered bucally
or sublingually, including inclusion compounds, contain from
about O.l to 99 percent by weight of active ingredients,
together with the excipient. It is also contemplated that
the oligonucleotides may be administered topically.
The pharmaceutical preparations of the present
invention are manufactured in a manner which is itQelf well
known in the art. For example, the pharmaceutical
preparations may be made by means of conventional mixing,
granulating, draqee-making, dissolving or lyophilizing
processes. The proce~s to be used will depend ultimately on
the physical propertie~ of the active ingredient used.
Suitable excipients are, in particular, fillers such as
sugar, for example, lactose or sucrose, mannitol or
~orbitol, cellulose preparations and/or calcium phosphates,
for example, tricalcium phosphate or calcium hydrogen
pho~phate, as well as binders such as starch or paste,
u~ing, for example, maize starch, wheat starch, rice starch,
potato starch, gelatin, gum traqacanth, methyl cellulose,
hydroxypropylmethylcellulose, sodium
carboxypropylmethylcellulose, sodium carboxymethylcellulo~e,
and/or polyvinyl pyrrolidone. If desired, disintegrating
agents may be added, such as the above-mentioned starches as
well as carboxymethyl-starch, cross-linked polyvinyl
pyrrol$done, agar, or alginic acid or a saIt thereof, such
as ~odium alginate. Auxiliaries are flow-regulating agents
and lubricants, such as, for example, silica, talc, stearic
acid or salts thereof, such as magnesium stearate or calcium
stearate, and/or polyethylene glycol. Dragee cores may be
provided with suitable coatings which, if desired, may be
SU~STITUTE SHER
WO93~11148 212 2 4 7 0 - PCT/US92/10043
-14-
resistant to gastric juices. For this purpose, concentrated
sugar solutions may be used, which may optionally contain
gum arabic, talc, polyvinylpyrrolidone, polyethylene glycol
and/or titanium dioxide, lacquer ~olutions and suitable
organic solvents or solvent mixtures. In order to produce
coatings resistant to gastric juices, ~olutions of suitable
cellulose preparations such as acetylcellulose phthalate or
hydroxypropylmethylcellulose phthalate, are used. Dyestuffs
and pigments may be added to the tablets of dragee coatings,
for example, for identification or in order to characterize
different combinations of active compound doses.
Other pharmaceutical preparations which can be used
orally include push-fit capsules made of gelatin, as well as
soft, 8ealed capsules made of gelatin and a plasticizer such
as glycerol or sorbitol. The push-fit capsules can contain
the oligonucleotide in the form of granules which may be
mixed with fillers such as ~actose, binders such as
starches, and/or lubricants such as talc or magnesium
stearate and, optionally, stabilizers. In soft capsules,
the active compounds are preferably dissoIved or suspended
in suitable liquids, such as fatty oils, liquid paraffin, or
liguid polyethylene glycols. In addition, stabilizers may
be added.
Possible pharmaceutical preparations which can be used
rectally include, for example, suppositories, which consist
of a combination of the active compounds with a suppository
base. Suitable suppository bases are, for example, natural
or synthetic triglycerides, paraffin hydrocarbons,
polyethylene glycols, or higher alkanols. In addition, it
is also posible to use gelatin rectal capsules which consist
of a combination of the active compounds with a base.
Possible base materials include, for example, liquid
triglycerides, polyethylene glycols, or paraffin
hydrocarbons.
SUBSTlTUT~ ~:~FFT
WO93/1114X 2 1 2 . '1 ~ O PCT/US92/10043
-l5-
Suitable formulations for parenteral administration
include aqueous solutions of the active compounds in
water-soluble or water-dispersible form. In addition,
suspen~ions of the active compounds as appropriate oil
injection suspensions may be administered. Suitable
lipophilic ~olvents or vehicles include fatty oils, for
example, sesame oil, or synthetic fatty acid esters, for
example, ethyl oleate or triglycerides. A~ueous injection
suspensions may contain substances which increase the
viscosity of the suspension including, for example, sodium
carboxymethyl cellulose, sorbitol and/or dextran.
Optionally, the suspenQion may also contain stabilizers.
Additionally, the compounds of the present invention
may al~o be administered encapsulated in liposomes,-wherein
the active ingredient is contained either di6persed or
variously present in corpuscles consisting of aqueous
concentric layers adherent to lipidic layers. The active
ingredient, depending upon its solubility, may be present
both in the aqueous layer, in the lipidic layer, or in what
i8 generally termed a liposomic suspension. The hydrophobic
layer, generally but not exclusively, comprises
phospholipids ~uch as lecithin and sphingomycelin, steroids
such as cholesterol, surfactants such as dicetylphosphate,
stearylamine, or phosphatidic acid, andjor other materials
of a hydrophobic nature. The diameters of the liposomes
generally range from about 15 nm to about 5 microns.
It is also contemplated that oligonucleotides having
aminoalkyl phosphonate moieties may be used as diagnostic
probes. Thus, in accordance with another aspect of the
present invention, there is provided an oligonucleotide
wherein at least one of the nucleotide units of the
oligonucleotide includes a phosphonate moiety having the
following structural formula:
SUBSTITUTE SHEEl~
WO93~11148 2 1 2 2 ~ 7 ~ PCT/US92/1~043
-16-
O = ~- o -
X, wherein X is:
,R2 ~R2
-Rl-N -R3, or -Rl-N-R5,
R5
wherein Rl, R2, and R3 are as hereinabove described, and R5
i 8 a detectable marker.
Detectable markers which may be employed include, but
are not limited to, colorimetric markers, fluorescent
markers, enzyme markers, luminescent markers, radioactive
markers, or ligand recognition reporter groups. Specific
examples of detectable markers which may be em~loye~
include, but are not limited to, biotin and derivatives
thereof (such as, for example, e-aminocaproyl biotin, and
biotin amidocaproyl hydrazlde), fluorescein (including
derivatives such as fluorescein amine), rhodamine, alkaline
phosphatase, horseradish peroxidase, and 2, 4-dinitrophenyl
markers. Such oligonucleotides which include a de~ectable
marker may be used as DNA or RNA probes. The probes may be
used a~ diagnostics as known in the art.
The invention will now be described with respect to the
fo~lowing example~; however, the scope of the preRent
invention i8 not intended to be limited thereby.
Exam~le 1
Production of the pyridinium salt of
Dhthalimidomethvl Dhos~ gnic ~
To 2.0g (7.42 mmole) of dimethylphthalimidomethyl
phosphonate, dried by coevaporation of pyridine and
dissolved in 40 ml of dry pyridine, was added dropwise 2.45
ml (2.5 equivalents) of trimethylsilyl bromide under
nitrogen. After 2.5 hours, the reaction mixture was
SUB5TITUTE SHEET
:
2 1 ~1 ~47 0 ``
W093/1114X PCT/US92/10043
-17-
filtered through a sintered glass funnel and the eluant was
treated with H20. The resulting mixture was concentrated
under high vacuum and the residue remaining was dissolved in
methylene chloride. Upon addition of ethyl a~etate, the
desired product was precipitated out. The precipitate was
collected, washed with ethyl acetate, and dried over P205 to
yield 1.2g of pure material.
ExamPle 2
PreDaration of the Triethylaminonium Salt
of Phthalimidomethyl PhosDhonate
.- Dimethyl phthalimidomethyl phosphonate (2.0 ~, 1.4
mmole) was di~solved in chloroform (15 ml), and
bromotrimethyl~ilane (2 ml, 15 mmol) wa~ added dropwise to `
the solution. After 2 hrs. the reaction mixture was
concentrated under reduced pre~sure, and the residue was
dissolved in chloroform (8 ml) followed by dropwisé addition
of triethylamine (20 ml) with cooling in ice bath. After
stirring at room temperature for 2 hrs. the mixtuxe was
filtered and concentrated to dryness. The residue was
dissolved in methanol (10 ml) and then added dropwise to
anhydrous diethyl ether (4 ml). The precipitate was
filtered, washed with ether and dried over P205 to yield 2.1
g (65X) of pure pthalimidomethyl pho~phonate,
triethylammonium salt.
ExamDle 3
PreDaration of 5'-dimethoxvtritYl-thvmidine-3'
-DhthalimidomethYl PhosPhonate
SVBSTtTUTE SHEET
WO93/1114X 2 1 2 2 4 7 0 PCT/US92/10043
-18-
The triethylammonium salt of
phthalimidomethylphosphonate (1.7 g, 5 mmol) was dried by
coevaporation with pyridine (3 x l0 ml), dissolved in dry
pyridine (40 ml) and treated with
triisopropylbenzenesulfonyl chloride (3.0 g, 9.9 mmol)
followed by a solution of 5'-0-dimethoxytritylthymidine (2.0
g., 3.67 mmol) in dry pyridine (40 ml) which was previously
dried by coevaporation with pyridine. The resulting mixture
was stirred at room temperature overnight under a dry
nitrogen atmosphere and the solvent was removed under
reduced pre~ure. The re~idue was purified by silica gel
column chromatography u~ing CH2Cl2/MeOH~Et3N (30:l:0.3, 2.8
L followed by 30:2:0.3, l.l L) as solvent. The appropriate
fractions were colIected and combined to yield 1.8 ~ (57%)
of a pure compoOu~d having the following structure:
o~ . 6
o~
- o
(wherein Bl is thymine) as a white foam.
ExamDle 4
SYnthesis of an aminomethYl d nucleotide
A commercially available nucleoside attached to a
controlled pore gla~s (CPG) support, and having the
following structural formula:
D~r- O ~o~l 2
~_ O
~h/f~- CP~
SuE3sTlTuT~
W093/11148 2 1 2 2 ~ 7 0 PCT/US92~10043
--1 9--
(wherein B2 is a protected or unprotected purine or
pyrimidine base) was treated with 3% dichloroacetic acid to
remove the dimethoxytrityl (DMT) protectinq group, and then
reacted with the phthalimidomethyl nucleoside phosphonate
(6) of Example 3 in the presence of
trii~opropyl-3-nitro-1,2,4-triazole as coupling agent and
1-methylimidazole as catalyst in dry acetonitrile for 15
minutes to give a phthalimidomethyl dinucleotide having the
following structural ~ormula: ~
O
p_ o ~!?
O
C~
The protected dinucleotide was then treated with
dichloroacetic acid to remove the dimethoxytrityl protecting
group, and then treated with ammonium hydroxide at 55C to
remove the phthalimido group and cleave the dinucleotide
from the solid CPG ~upport to gi~e an aminomethyl dimer
having the following ~tructural formula 8, wherein each of
Bl and B2 i~ an unprotected purine or pyrimidine base.
t~o
0'~
0~
Example 5
SYnthesis of a 3' aminomethYl end
SUBSTITUTE. SHEEl'
- W093/1114X 212 2 4 7 O PCT/US92/10043
~ -20-
The protected dinucleotide (7) is prepared as described
in Example 4. The protected dinucleotide is then loaded
into a l~mole size column, installed on an Applied
Biosystems DNA synthesizer (Model #394), and synthesis of a
modified oligonucleotide is performed using standard
phosphoramidite chemistry. Deprotection is carried out with
28~ aqueous ammonium hydroxide at 55C and then freeze dried
in vacuo. The crude oligonucleotide is con~erted into its
sodium salt form by passage of an aqueous solution through a
cation exchange resin (Na ) using water as an eluant, and is
purified by Sephadex G-25 column chromatography using water
a~ an eluant to give the aminomethyl 3' end-capped
oligonucleotide.
Example 6
Preparation of 5-'0-dimethoxYtritvl-thYmidYl-3'-
Dhthalimidomethvl-DhosPhonvl-5'-thvmidine, mixed isomers
5'-0-dimethoxytritylthymidine-3'-phthalimidomethylpho phona-
te (Example 3, 2.0 g, 2.3 mmol) was dried by coevaporation
with pyridine (3 x 15 ml), redissolved in dry pyridine (80
ml) and treated with
1-(2,4,6)-trimethylbenzenesulfonyl-nitrotriazolide (0.75 g,
2.5 mmol), for 15 min. at room temp. Thymidine (0.6 g, 2.3
mmol) was dried by pyridine coevaporation in the same way,
di~ olved in pyridine (15 ml) and added to the solution of
5'-0-dimethyoxytritylthymidine-3'-phthalimidomethylphosphon-
~te. The reaction mixture was stirred at room temperature
under a dry nltrogen atmosphere for 2-3 hrs., then diluted
with aqueous ~odium bicarbonate (5%, 300 ml) and extracted
with ethyl acetate (3 x 200 ml). The organic layers were
combined, dried over anhydrous magnesium sulfate and
concentrated under reduced pressure to give the mixed
isomers of 5'-0-dimethoxytrityl-
SUBSTITUTE SHEEl'
.
W093/11148 ~1 2 2 ~ 7 ~ PCT/US92/10043
-21-
thymidyl-3'-phthalimidomethyl-phosphonyl-5'-thymidine, 1.5 g
(65%).
Example 7
SeParation of i~omers of S'0-dimethox~tritvlthvmidvl-3'-
Dhthalimidomethvlpho~phonyl-5'-thvmidine by HPLC
~ The mixture of isomers of
5'0-dimethoxytritylthymidyl-3'-
phthalimidomethylphosphonyl-S'-thymidine from a 200 mg scale
reaction was dissolved in triethylammonium acetate (0.1 M,
TEAA)/ acetonitrile (60/40, 1.5 ml) and injected into a
reversed phase C4 column, Radial Pak Cartridge (Waters RCM
25 x 100 mm). The column was eluted with a linear gradient
of TEAA/acetonitrile in which the concentration of
~-' acetonitrile increased from 35-80%. The individual~isomers
were eluted at 31-35 and 39-41 minutes respectively. This
separation procedure was repeated six times and the
appropriate fractions were pooled, extracted with ethyl
acetate (3 x 50 ml), evaporated and dried in vacuo over
P205. This procedure yielded 80 mg of a faster isomer and
110 mg of a slower isomer, total yield 83%. Analysis of the
composites by analyti~al HPLC using a rever~ed phase C*
column (Radial Pak cartridge, 8x 100 mmh 15 um, 300 A)
indicated that pure isomers were obtained in each case.
ExamPle 8
SeDaration of 5'0-dimethoxvtritvlthvmidvl-3'-Dthalimido-
methviDhocDbonvl -5'-thymi~ne~ bv ~ilica column
chromato~raphY
The residue from a 1.72 g preparation of mixed isomers
of
5'-0-dimethoxytritylthymidyl-3'phthalimidomethylphosphonyl--
5'-thymidine was purified by column chromatography on silica
gel (lOOg) using CH2Cl~/CH30H/Et3N (30:1:0.3) as the
solvent.
SUE3~;TIT(JT~ FT
W093/1l148 212 2 4 7 0 PCT/US92/10043..
^ -22-
Fractions 120-126 contained the faster eluting isomer,
fractions 127-157 contained a mixture of both isomers, and
fractions 158-170 contained the slower eluting isomer. The
appropriate fractions were collected, evaporated to dryness
and dried in vacuo over P205 to give 0.17g of the faster :
eluting isomer, 0.2 g of the slower eluting isomer, and 0.7
g of a mixture of isomers.
ExamDle 9
S~nthe~is of isomers of S'-0-dimethoxYtritylth~midYl-3'
DhthalimidomethvlDhosphonyl-5'-thymidine -3'-cYanoethYl-N,
N-diisoproDYlaminoDhosPhoramidite.
A sample of the faster iso~er of
5'-0-dimethoxytritylthymidyl
-3'phthalimidomethylphosphonyl-5'-thymidine (O.g2 g,~0.42
mmole) was i8 dried by coevaporation with pyridine,
dissolved in dry acetonitrile (10 ml) under nitrogen, and
treated with stirring with cyanoethoxy-
(N,N,N',N'-tetra-isopropylamino)- phosphine (0.33 ml, 1.05
mmol), tetrazole (30 mg), and diisopropylamine (0.08 ml,
0.58 mmol). After 50 minutes at room temperature the
mixture was partitioned between 5% aqueous ~odium
bicarbonate and acetonitrile (S0 ml of each). The organic
layer was washed with water (2 x S0 ml) and concentrated in
vacuo to a gum. The crude product were purified by column
chromatography on silica gel (40g) using CH2C12/MeOH/Et3N
(100:2:1). The appropriate fractions were combined and
evaporated to yield 0.36 g (71%) of the faster isomer of
5'-0-dimethoxytritylthy-
midyl-3'-phthalimidomethylphosphonyl-S'-thymidine-3'-cyanoe-
thyl-N,N-diisopropylaminophosphoramidite.
An~identical procedure was followed to produce a
phosphoramidite.from the slower isomer.
Exam~le 10
Procedures for oliqonucleotide s~nthesis and deProtection
SUE3STITUTE SHEFT
WO93/1114X 212 ~ ~ 7 0 PCT/USg2/10043
-23-
a) 5'-End capped oligonucleotide
A 12 base, thymine-containing oligonucleotide is
prepared on a 1 umole scale using an Applied Biosystems
Model 394 DNA ynthesizer, with phosphoramidites and other
reagents as supplied by the manufacturer. After nine
coupling cycles with the commercially available monomer
5'dimethoxytritylthymidine-3'-N,N-
diisopropylamino-cyanoethoxyphosphoramidite, the final cycle
employs a 0.1 M solution of either the faster or slower
isomer of the phthalimidomethyl dinucleotide phosphoramidite
of Example 9. Upon completion of the synthesis, the
modified oligomer i8 treated with concentrated ammonia for
20 min, partially concentrated under a stream of nitrogen,
lyophilized to dryness and purified as described ~elow.
This procedure produces a twelve base oligonucleotide with a
Yingle isomer aminomethyl phosphonate moiety at the
5'terminus.
b) Synthesis of a tridecanucleotide with an alternating
single i80~er uoinomethyl phosphonate/phosphodie~ter
backbone.
A thymine-containing tridecanucleotide with an
alternating, single isomer aminomethyl
phosphonate/phosphodiester backbone i~ prepared on a 1 umole
scale u~ing an Applied Biosystem Model 394 DNA synthesizer,
using a standard phosphoramidite cycle with either the
fa~ter or slower i~omer of Example 9 as the phosphoramidite.
Coupling time~ of 2 min. per cycle are used. Upon
completion of the synthesis, the modified oligomer is
treated with concentrated ammonia for 20 min.~ partially
concentrated under a stream of nitrogen, lyaphilized to
dryness and purified as described below.
This procedure produces a thirteen base oligonucleotide
with single i~omer aminomethyl phosphonate moieties
alternating with pbosphodiesters throughout the sequence.
SUE~STITUTE SHEET
W093/1l148 212 2 4 7 0 PCT/US92/10043
-24-
c) 3',5'-Aminomethyl phosphonate end capped
oligonucleotide
A 12 base thymine-containing oligonucleotide is
prepared on a 1 umole scale using an Applied Biosystems
Model 394 DNA synthesizer. The initial cycle employs a 0.1
M solution of either the faster or slower isomer of
phthalimidomethyl phosphonate dinucleotide phosphoramidite
(Example 9) which is coupled to the solid support to which a
thymidine residue is attached. After nine subsequent
coupling cycles with the commercial available monomer
5'-dimethoxytritylthymidine-3'-N,N-diisopropylamino-cyanoet-
hoxyphosphoramidite, the final cycle again employs a 0.1 M
~olution of either the fa~ter or slower isomer of
phthalimidomethyl dinucleotide phosphoramidite of Example 9.
Upon completion of the synthesis, the modified oligomer is
treated with concentrated ammonia for 20 min, partially
concentrated under a stream of nitrogen, lyophilized to
dryne~s and purified as described below.
d) General procedure for oligonucleotide purification by
E~PLC '
The oligonucleotide possessing a 5'-0-dimethoxytrityl
~roup was purified by reverse phase HPLC (C4 Radial Pak
Cartridge, 100 x 25 mm, l5u, 300A). After detritylation
with 0.1 M acetic acid the product was again purified by
reverse phase HPLC (C4 column) using a linear gradient of
O.1 M TEAA/aceton~trile, with the concentration of
acetonitrile being varied from 5 to 70%. Deprotection was
carried out using ethanol/ethylenediamine (1:1) at room
temperature for 45 minutes to give the desired aminomethyl
backbone modified oligonucleotide.
ExamPle 11
SYnthesis of a biotinYlated 3' aminomethYl
oliaonucleotide
S(IBSTITUTE SHE~T
W043/l1148 21 2 2 ~. 7 0 PCT/US92/10043
-25-
.
The 3'-aminomethyl end capped oligonucleotide of
Example 5 i~ placed in aqueous sodium bicarbonate buffer, pH
8. This solution i8 then treated with a solution of bi~tin
N-hydroxysuccinimide ester (50 equivalents) in
dimethylsulfoxide for 18 hours at room temperature. The
resulting solution i~ passed through a Sephadex G25 column
to remove the bio~in and other ~mall molecules and the
fractions containing the olîgonucleotide are concentrated
and purified by high performance liquid chromatography using
a C18 rever~ed pha~e silica column. The appropriate
fractions are collected and evaporated to drynes~ to give
the biotinylated 3'-aminomethyl end capped oligonucleotide.
Advantages of the present invention include improved
olubility o the positively charged oligonucleotides in
agueou~ ~olution~ as compared with nonionic
oligonucleotides, improved uptake into the cell a~ compared
with natural oligonucleotides which are negatively charged
and are poorly taken up by the cell, and re~istance to
degradation by nucleases a~ compared with natural
oligonucleotides which are readily degraded by cellular
enzymes. By virtue of their positively-charged regions, the
oligonucleotide~ of the present invention are ta~en up by
the cell more readily and are less readily degraded because
of their modified backbone~. In the ca~e of
oligonucleotides having aminomethyl phosphonate moieties,
the cationic groups are ~maller and therefore les~ likely to
disrupt base pairing than previously synthesized cationic
oligonucleotides. Also, the carbon-phosphorus bonds are
more ~table than nitrogen-phosphorus bonds of other cationic
oligonucleotides, and thus the oligonucleotides of the
present invention are less likely to lo~e the cationic group
by chemical or enzymatic hydrolysis.
~ minomethyl oligonucleotides bearing detectable markers
such as reporter groups have the advantage that the reporter
C~ I~'rtT~ ITr ~J~_ .
WO93~11148 - PCT/US92/10043
2122~70 -26-
groups are on the outside of the duplex produced by
hybridization to its target DNA or RNA and are therefore
more accessible towards detection, and also do not interfere
with the hybridization sites on the bases.
It is to be understood, however, that the scope of the
preqent invention is not to be limited to the specific
embodiments described above. The invention may be practiced
other than as particularly described and ~till be within the
scope of the accompanying claims.
! :UBSTITUTE~ S~EET