Note: Descriptions are shown in the official language in which they were submitted.
~OECHST AKTIENGESELLSCHAFT HOE 93/F 133 Dr.Bl/PP
Description 2 1 2 3 2 ~ 2
Novel derivatives of 3-fluorophenol, proce~ses for
preparing them, and the use of these compound
The pre~ent invention relates to novel derivatives of
3-fluorophenol, to proce~ses for preparing them, and to
the use of these compounds.
.. . .
Derivatives of 3-fluorophenol are useful intermediates in
the preparation of plant protection agents, pharmaceu-
ticals and industrial products, such as, for example,
liquid crystals. Owing to the general importance, and
numerous uses, of this substance class, it represents a
rewarding object to prepare novel compounds from this
group of substances in order not only to supplement the
spectrum of their posslble applications, but also to
15 enxich and extend it by giving nuances to material i-;~
properties.
: .
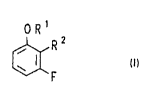
This object is achieved by compounds of the formula (I)
ORl
~R 2
in which
is hydrogen, (C1-C8~-alkyl or CH2-phenyl, where the
alkyl radical or the phenyl group can be substituted
by one to three ( Cl-C4 ) -alkoxy groups, fluorine,
chlorine or bromine atoms~ nitrc groups, cyano
groups, trifluoromethyl grcups or (C1 C4 ) -alkoxy-
! carbonyl groups, and
¦25 RZ is -CW, -CON~2, -NH2, -NCO or -N~CooR3, where R3 has
Ithe m~anings of R1 other than that of hydrogen.
¦Cyclic, straLght-chain or singly or multiply branched
f ~ 2 1 2 3 2 ~ 2
- 2 -
alkyl radicals are generally suitable as alkyl radicals.
Examples of ~uch radicals are the methyl, ethyl,
n-propyl, i-propyl, n-butyl, i-butyl, n-pentyl, i~pentyl,
n-hexyl, i-hexyl, n~heptyl, i-heptyl, n-octyl, i-octyl,
2-chloroethyl, 2-bromoethyl, 2-methoxyethyl, 2-ethoxy-
ethyl, 3-methoxypropyl, 3-ethoxypropyl, 4-methoxybutyl,
~- 3-chloropropyl, cyclopropyl, cyclobutyl, cyclopentyl,
cyclohsxyl and methylcyalohexyl radicals.
In addition to unsubstituted phenyl, phenyl radicals
which are substituted one to three times ars generally
suitable as phenyl radicals.
.1
Those which may be mentioned here by way of example are
2-methylphenyl~ 4-methylphenyl, 2-methoxyphenyl, 4-
methoxyphenyl, 2-fluorophenyl, 3-fluorophenyl, 4-fluoro-
phenyl, 4-trifluoromethylphenyl, 2,5-dichlorophenyl,
2,4-dichlorophenyl, 3-nitrophenyl, 4-cyanophenyl, 3,5-
dimethoxyphenyl, 2-nitrophenyl and2,4,5-trichlorophenyl.
is, in particular, hydrogen, a linear or branched
l(Cl~C~)-alkyl group or a CH2-phenyl group.
j20 A certain importance i~ attached to the phenols of the
,formula I (~l = H) in which R2 iB -CONH2 or NH2, in par-
ticular 2-amino-3-fluorophenol and 2-hydroxy-6-fluoro-
benzamide.
The compounds of the foxmula I in which Rl is CH2-phenyl
and R2 is -CN, -CONH2, NH2 or NHCooR3, in particular 2
1 benzyloxy-6-fluorobenzamide, 2~benzyloxy-6-1uoroaniline,
j 2-benzyloxy-6-fluoro-N-benzyloxycarbonylaniline and ~-
benzyloxy-6-fluorobenzonitrile, are also valuable inter-
mediates.
.,~
The compound~; of the formula I in which R' i6 (C,-Ca)-
alkyl and R2 is -CN or -CON~2, in paxticular 2-ethoxy-6-
fluorobenzonitrile and 2-ethoxy-6-fluorobenzamide, are of
interest.
,
`I
~ 2~23242
~ ,~
- 3 -
The present invention additionally relates to processes
~, for preparing the compound3 according to the in~ention.
. Thus, compounds of the formula I, in which R1 is
hydrogen, ~Cl-C8)-alkyl or CH2-phenyl, where the alkyl
radical or the phenyl group can be sub3tituted by one to
-~ three (C,-C4)-alkoxy group , fluorine, chlorine or bromine
atom~i, nitro groups, cyano groups, trifluoromethyl group3
! or (C1-C4)-alkoxycarbonyl groups, and R2 i8 -NH2 or
~ -NHCooR3~ can be prepared by reacting compounds of the
;i 10 formula (II)
~ .
~ C ~ N H 2
., .
in which X i5 OR', in a~ueous-alkaline medium, with
i chlorine, bromine, sodium hypochlorite or sodium hypo-
I bromite in the presence of alcohols and at temperatures
i of from -15C to ~80~C, in the ~en~ie of a ~ofmann
j 15 degradation. : -
In many ca6es, it ha3 proved to be of value to employ, as
the alcoh~l component, compounds of the formula ~oR3,
where R3 is ( C1_CB ) -a1kY1 or CH2-phenyll where the alkyl
radical or the phenyl group can be substituted by one to ~ . :
three (C1-C4)-alkoxy groups, fluorine, chlorine or bromine
~: atoms, nitro groups, cyano groups, trifluoromethyl groups :: -
;1: or (C1-C~]-alkoxycarbonyl groups.
~ The compound o the formula (I')
,~
~N H C O O R ~ ~:
:~ resulting fro:m the reaction can be isolated. However, it ~ :
25 i8 also possi.ble to cleave the carbamate group and/or,
for X = OR1, the group OR1 (when R1 i8 not hydrogen)
' ~' .
i
~1 :
` ` 2123242
. ~
-- 4 --
~ hydrogenolytically or hydrolytically without undertaking
'~! any intermediate isolation. It is likewise po6sible to
~elect the reaction condition~ such that the cleavage
already takes placP to a large extent at the same time a~
the rearrangementa
s
The compounds of the formula II are prepared by convert-
ing the corresponding nitriles into the benzamides by
methods which are generally known in the literature (J.
March, Advanced Organic Chemistry [1985], 788).
In this context, preference is given to the reaction of
2,6-di~luorobenzonitrile in aqueous~alkaline medium with
hydrogen peroxide (JP-OS 60-132 942), where appropriate
in correspondan~e with the pre~ent invention, in the
presence of from about 1 to about 20 mol, preferably from
about 0.1 to 10 mol, particularly preferably between
about 1.05 mol and 5 mol, of alcohol per 1 mol of 2,6-
difluorobenzonitrile. In this context, alkan-(C,-C8)-ols
of any structure, which can additionally be substituted
^~ by alkoxy(Cl-C4) groups, fluorine, chlorine or bromine
atoms, nitro groups, cyano groups, trifluoromethyl groups
or alkoxylCl-C4)-carbonyl gr~ups, or phenylmethanols,
where the phenyl radical can be substituted by alkyl(Cl-
C4 ) groups, alkoxy~Cl-C4~ groups, fluorine, chlorine or
bromine atoms, nitro groups, cyano groupY, trifluoro-
methyl groups or alkoxy(CI-C4)-carbonyl groups, can be
used as Rl-OH alcohols; primary alcohol~ are preferred,
`with methanol, ethanol and benzyl alcohol being particu-
larly preferred.
.~
2,6-Difluorobenzamide can be isolated; however, it is
l 30 also possi~le to obtain the compounds of the formula II
having X = ORl immediately in a one-pot process. Thi~ can
be achieved, in particular, by raising the reaction
il temperature.
The work is carried out at temperatures of between about
0 and about 90C, preferably of between about 20 and
;'j
'1
!
~ 2123242
s
about 70C.
If 2,6-difluorobenzamide, which has been isolated as an
intermediate, i~ employed for preparing the compounds ~f
the formula II having X - ORl, the work i~ preferably
carried out, in accordance with the invention, in excess
alcohol, as described above, and reaction takes place, in
the temperature range which has likewise already been
defined, with about 1 mol to about 1.8 mol, particularly
preferably with about 1.1 mol to about 1.5 mol, of alkali
metal or alkaline earth metal compounds having an
alkaline effect. Ex~mples of such compounds having an
1 alkaline effect are hydroxides, carbonates, hydrogen
carbonates, phosphatesl hydrogen phoæphates, dihydrogen
phosphates, oxides, or similar compounds, or mixtures ~
15 thereof; sodium or potassium hydroxide or carbona$e are -1
1 preferred. The reaction times amoun to from about 1 to
'1 about 16 h, with the reaction product generally being
¦ isolated, in the event that isolation is ~esired, by
diluting the reaction mixture with water and filtering
off (extraction, crystallization, chromatography).
~ Alternatively, it is possible to react 2,6-difluorobenzo-
`1 nitrile, as explained below, or 2,6-difluorobenzamide
i with the above-describ2d quantities and types of ~ ~
~ compounds having an allcaline ef~ect and al~ohols in ~ -
¦ 25 dipolar, aprotic solventsr Acetone, tetrahydrofuran
(THF), acetonitrile, 1,2-dimethoxyethane (DMAc), N,N-
dimethylformamide (DMF), N-methylpyrrolidone (NMP),
dimethyl ~ulfoxide (DMS0), dimethyl ~ulfone, diphenyl
sulfoxide, diphenyl sulfone, tetramethylurea, tetra-n-
l 30 butylurea or 1,3-dimethylimidazolidin-2-one (DMI), or
3 mixtures thereof, can be used as dipolar, aprotic
solvents. Such solvents are u~ed in ~uantities of about
li 50 to 500 ~ by mass, based on 2,6-difluorobenzamide or
J 2,6-difluoroblenzonitrile, preferably in quantities of
I 35 between about 80 and about 250 % by mass. The use of
!l these solvent:s makes it possible to ~eparate off by
~ filtration the alkali metal or alkaline earth metal
~ .
21232~2
- 6 -
fluoride which i~ formed in the reaction; the product i8
' crystallized from the mother liquor by adding water and
the solvent is recovered by di~tillation. In this variant
of the process, the work is carried out at reaction
5 temperatures of between about 30 and about 150C,
preferably of between about 40 and about lOO~C. A~
l compared with aqueous variants of the process, this
! variant has the advantage of higher yields.
The preparation of 2-(primary-alkoxy)-6-fluorobenzamides
is preferred, with that of 2-methoxy-6-fluorobenzamide,
2-ethoxy-6-fluorobenzamide and 2-benzyloxy-6-fluoro-
benzamide being particularly preferred.
In the sase of 2-benzyloxy-6-fluorobenzamide, 2-hydroxy-
6-fluorobenzamide can be obtained by hydrogenolytic
elimination OI the benzyloxy group.
i
Compounds of the formula I (R' ~ hydrogen) having R2 = CN
i can be obtained in an analoyous mannerc For this purposs,
2,6-difluorobenzonitrile is reacted with R30H alcohols in
aqueous-alkaline or dipolar, aprotic medium in the
presence of alkali~.
In this context, alkan-(CI-C8)-ols of any ~tructure, which
can additionally be substituted by alkoxy(C1-C4~ groups,
fluorine, chlorine or bromine atoms, nitro groups, cyano
group~, trifluoromethyl gxoups or alkoxy-(Cl-C~)-carbonyl
groups, or phenylmethanol~, where the phenyl radical can
be 0ub~tituted by alkyl( C,-C4 ) groups, alkoxy(Cl-C4)
groups, fluorine, chlorine or ~romine atom~, nitro
roups, cyano groups, trifluoromethyl group~ or alkoxy-
~C,-C~)-carbonyl groups, can be u~ed a~ Rl-OH alcohols;
! 30 primary alcohols are preferred, with methanol, ethanol
and benzyl alcohol being particularly preferred.
~ It i8 al~o pos~ible to obtain compounds of the formula I
¦ (Rl ~ hydrogen) having R2 - NH2 in a one-pot process by
¦ reacting either 2,6-difluorobenzonitrile or 2,6-difluoro-
,, :
~123242
- 7 -
benzamide in aqueous-alkaline medium with alcohols to
form compounds of the formula II having X = OR1 ~R1 ~
hydrogen), and then reacting the latter~ after inter-
~mediate isolation, but preferably without any
'!5 intermediate isolation~ in a one-pot proces3, with
chlorine, bromine, sodium hypochlorite or sodium hypo-
bromite, at temperatures of from -15C to ~80C and in
~the presence of alcohols, in the sense of a Hofmann
ddegradation, and ~ubsequently, where appropriate,
`i,10 cleaving ~arbamates which have been formed hydrolytically
~or hydrogenolytically.
j':!In this context, (C,-C8)-alkanols or phenylmethanols,
`Alwhere the alkyl radical or the phenyl radical can be
`'substituted by one to three (Cl-C4)-alkoxy groups,
fluorine, chlorine or bromine atoms, nitro groups, cyano
groups, trifluoromethyl groups or (Cl-C4)-alkoxycarbonyl
groups, in particular primary alcohols, can also be
employed as alcohols.
jIn many cases, the use of methanol, ethanol or benzyl
alcohol has proved to be advantageous.
These alcohols are employed in mixtures with aqueous
solutions having an alkaline effect, a~ described above,
ipreferably sodium or potassium hydroxide solutions. The
compounds having an alkaline effect are used, dissolved
or su~pended in water, in quantities, based on the amide
to be dsgraded, of between about 1 mol and about 30 mol,
preferably of between about 3 mol and about 15 mol,
particularly preferably of between about 5 mol and about
10 mol. While the concentrations of the aqueous solutions
depend on the amide employed, the concentrations are
typically between about 1 mol/l and about 20 mol/l,
~;~preferably between about 3 mol/l and about 10 mol/1. In
¦practice, the quantity of the aqueou~ suspension having
an alkaline effect i~ chosen such that it is still
possible to stir the reaction mixture.
.1
`` 2~23242
- 8 -
The procedure can be carried out using hypohalite solu-
tions (bleaching liquors), which is completely equivalent
to metering elemental halogen into solutions having an
I alkaline effect. An indication of the quantity of halogen
5 employed is suPficient to describe the reaction ,~ondi-
tions since, in situ, hypohalite solution~ are formed
when halogen makes contact with the aqueous solutions
having an alkaline effe,ct which are employed. Chlorine or
~ bromine are used, therefore, in quantities of between
i 10 about 1 mol and about 5 mol, preferably of from about
1.01 mol to about 2 mol, particularly preferably of
between about 1.02 mol and about 1.2 mol, in each case
based on 1 mol of amide to be degraded. Chlorine is
preferably used since it is more readily available
15 industrially. The halogen can be added dropwise (bromine)
or passed in in gaseous form (chlorine). In thi~ regard,
suitable metering times on a laboratory ~cale are,
~ depending on the reaction temperature, between about
1 0.5 h and about 8 h, preferably from about 1 h to about
20 4 h. Owing to the reaction being exothermic, the metering
times on an industrial scale mu~t be adapted to the
cooling capacities available.
The reaction in the ~en~e of the Hofmann degradation is
carried out at temperatures of between about -15C and
about 100C, preferably of between about 0C and about
60C, particularly preferably of between about 10C and
about 40~C.
::
If bleachin~ liquor~ e. aqueous hypohalite solutions,
are metered in, which generally has operational advan-
tage~ as ~ompared with the use of elemental halogen,
~ solutions are then used having a content of active
1 chlorine of from about 30 to about 250 ~ per kg of
solution, preferably of between about 100 and about 160 g
of active ch~orine per kg of solution, or from about 60
to about 550 g o~ active bromine per kg of solution,preferably oP between about 200 and about 350 g per kg of
solution. These solutions may be obtained by metering the
I 2123242
, . .~
, ~ 9
corresponding quantities of chlorine or bromine into
aqueous solutions having an alkaline effect.
~.~
In the variant according to the invention, the primary
reaction product of the reaction in the ~en3e of a
i 5 Hofmann degradation is pressmably the alkyl carbamate of
the desired aniline, ~ince the isocyanate ari~ing as an
intermediate i8 probably captured by the alcoholates,
i nucleophilic under the reaction conditions~
The anilines can be obtained from the carbamates by
10 processes known in principle from the literature
(T.W. Greene, P.G.M. Wuts, Protective Group~ in Organic
¦ Chemistry (1991), 317-348). As a rule, liberation of the
amino group is achieved by heating the reaction mixture
according to the invention, once any excesses of chlorine
15 or bromine have been destroyed. Optionally sub~tituted
, benzyl carbamates can be hydroly~ed under alkaline
3 conditions in less than 10 h at from 80 to '00C, whereas
¦ simple alkyl carbamates require reaction times of up to
96 h at the same temperatures. ~he use of optionally
20 substituted benzyl alcohol is therefore particularly
~ preferred since the resulting benzyl carbamates can be
I cleaved without any great difficulty, and therefore cost-
effectively, by hydrogenation (T.W. Greene et al., loc.
cit., 335-341). The hydrogenolytic cleavage is preferably
25 carried out in the presence of transition metal
catalyst~, in particular platinum, nickel or palladium
cataly~ts. In general, the reaction products can ~e
~ i~olated simply by phase separation, resulting in
I mixtures with the alcohols employed. The phase separation
30 can be improved or induced by adding additional solvents,
such a~ t for example, toluene or xylene.
. .
The resulting solutions of the free amine~ can, provided
they contain compounds which posse s benzyl group~, be
hydrogenated by methods which are generally known from
35 the literature (T.W. Greene et al., loc~ cit., 47-68,
156-160, 335-341). Hydrogenation u~ing hydrogen gas in
,:! .
- 1o_2~23242
the presence of transition metal catalysts, preferably
palladium catalysts, particularly preferably palladium on
active charcoal, is preferred. An alternative option is
to use the so-called transfer-hydrogenation method for
~l 5 synthesizing the novel intermediates (T~W. Greene et al.,
-lj loc. cit., 156-160). Besides palladium, nickel or
,! platinum, in particular, are suitable for employment as
transition metals. The hydrogenations proceed smoothly
under a hydrogen pressure of between about 1~1 bax and
about 100 bar at temperatures of between about 10 and
about 80C in lower aliphatic alcohols or simple aromatic
or aliphatic hydrocarbons, such as, for example, hexane,
methylcyclohexane, toluene, xylene, methanol, butanol,
isopropanol or ethanol as solvent, or mixtures thereof.
The precious metal catalysts are employed in quantikies
of between about 0.05 and about 3, preferably of from
i about 0.3 to about 1, % by mass (calculated as pure
transition metal). The concentration of the end product
(in particular 2-amino-3-fluorophenol) in the hydrogena-
`1 20 tion mother liquor is typically between about lD and
j about 500, preferably between about 100 and about 300,
'37 g/l. If the unsubstituted benzyl compounds are employed
, under these circumstances, the free hydroxyl groups and
!~7 toluene are obtained during the hydrogenation. Toluene is
~3 25 therefore preferred as the ~iolvent ~or the hydrogenation.
j The end product, 2-amino-3-fluorophenol, can be i~iolated
by concentrating the solutions, from which, where appro-
priate, the catalyst has been filtered off while the
3 solutions were hot, and subsequently filtering them. In
30 this context, it is advi~able to carry out the procedure
`i in the presence of oxidation-preventing additivPs, such
l I as hydrazine or hydrazinium Ralts, or 2,6-di-tert-butyl-
~7 4-methylphenol, since the end product exhibits a high
`~l degree of lability towards atmospheric oxygen, particu-
j 35 larly on heating. Other phenol ether groupings, such as
the methyl ethers or ethyl ethers which can prefferably be
prepared according to the invention, may be cleaved by
methods known from the literature (T.W~ Greene, loc.
cit., 14.68, 145-161) before or after hydrogenating any
~` ff,;~
21 23242
: .
benzyl groups which may be present.
In the process according to the invention, the use of
benzyl alcohol is particularly preferred in those
reaction steps which proceed in the presence of alcohols.
6-Fluorosalicylamide can be prepared in high yield by
cleaving the phenyl ether grouping in 2-alkoxy-6-fluoro-
benzamides by the methods which have already been
described. The compound can be reacted, likewise in the
,described manner and optionally in the presence of
110 alcohols, with halogens in alkaline iolutions in the
sense of a Hofmann degradation. Under these circum-
~stances, the reaction proceeds as far as 2-amiino-3-
¦fluorophenol, as described above, provided that it
involves formation of the carbamates. Otherwise, 6-
fluorobenzoxazolone i6 obtained as an intermediate and
has to be hydrolyzed in acid solution to yield the
product, resulting in lower yields being obtained.
The compounds o~ the formula I, in particular compounds
in which Rl ii hydrogen f methyl, ethyl or CH2 phenyl, and
R2 is -CN, -CON~2, -NHCooR3 or NH2, with R3 being methyl,
ethyl or CH2-phenyl, may be used for preparing liquid-
crystalline compounds, plant protection agents and
pharmaceuticals.
The following examples illustrate the invention without
limiting it.
1~
~ Example 1
! !
¦ 2-Amino-3-fluorophenol
180 g o~ benzyl alcohol and 269 g (6.725 mol) of sodium
hydroxide are introduced into 450 g of water. 220 g
1 30 (0.9 mol) of 2-benzyloxy-6-fluorobenzamide are added at
20C, and chlorine i9 passed in (15 l/h) at 40C while
stirring thoroughly. The reaction is monitored by gas
2123242
- 12 -
chromatography and terminated after 2.5 h when conversiion
is complete. The aqueou~ mother liquor is heated at 95C
for 3 h once any excess of chlorine has been destroyed
~ (sodium sulfite).
:,
5 At this point, 2-benzyloxy-6-fluoroaniline can be
iEfolatsd as a pale brown-colored viscous oil by extract-
ing with MTBE, drying over magnPff~ium sulfate and removing
the ~olvent (see Example 3 a~ well); however, this ~tep
is omitted here.
'I
'J
' 10 Procedure a
,
The organic phase is separated from ths aqueous phase,
taken up in 200 ml of methanol, and then stirred
~- vigorously (15 h~ together with 5 g of Pd/C (5 % Pd, 50
f moist) under an H2 atmosphere (slight exaess pressure)
15 until compounds po~se~sing benzyloxy groupings can no
longer be detected. The catalyst i6 filtered off and than
washed with methanol. Most of the methanol is distilled
~ off under an inert qas, and 300 g of toluene are added.
3 After cooling (0C), 60.9 g tO.48 mol, 53 %) of 2-amino~
20 3-fluorophenol, which i~ colored pale-brown to dark-gray
after drying, can be obtained (content (GC): 100 %). A
I further 24.2 g (0.19 mol, 21 %) of product are contained
J in the black mother liquor, as is dstermi~ed by quanti-
tative ga~ chromatography. The mother liquor i8 reU8ed
25 for further batches.
; .
3 Procedure b
~ I After some minutes without stirring, the phases can be
i separated after adding 200 g of toluene, and 300 g of
;l methanol are added to the organic phase which is then
~l 30 ~tirred vigorously (20 h~ together with 5 g of Pd/C ~5 %
¦ Pd, 50 % moist) under an H~ atmosphere ~slight excess
pressure). GC analysi~ reveals solvent and, apart from
minor quantities of ~yproducts, 2-amino-3--fluorophenol,
as the main component. The catalyst i5 filtered off and
.1
1!
' 21232i~2
- 13 -
then washed with methanol. ~o~t of the methanol is
~ distilled off from the $iltrate under an inert gas, and
; 300 g of toluene are added. After cooling (0C), and
-$~ drying, 65.3 g (0.51 mol, 57 %) of 2-iamino-3-fluoro-
~ 5 phenol, which is colored pale-brown, can be obtained
;~ (content (GC): 100 %). A further 28.2 g (0.22 mol, 25 %)
of product are contained in the mo~her liquor/ as was
determined by quantitative gas chromatography The mother
~ liquor is reused for further batches.
¦ 10 2-Amino-3-fluorophenol:
M.p. 124-126C; 126.3C (DSC)
:~ B.p. 250.7C (DSC)
Solubility in water ~ 15 g/l
Bulk density approximately 0.2 g/cm3
,; : '.
1H-NMR [DMSO-d6/TMS, ppm]:
= 6.39 (C) (ddd, lH, JBC = 8.28 HZ~ JCD = 6-36 H~
JAC = 7.83 HZ, Ar-HS)
6.50 ~B) (ddd, lH, J8c = 8-28 Hz, Ji3D = 8-30 Hz,
J~ = 1.48 Hz, Ar-H4)
6.52 (A) (ddd, lH, JAC = 7.83 ~Z~ JAD = 2-30 HZ,
J~ = 1.48 Hz, Ar-H6)
.: ' ',''.
'9F-NMR [DMSO-d6/CFCl3, ppm]: :
= -133.94 (D) (ddd, lF, JaD = 6.36 ~Z, JAD = 2-30 Hz,
JCD = 636 H Z, Ar-F3 )
13C-NMR [DMSO~d6/T~S, ppm]
= lQ6.1 (J = 19.1 Hz, Ar-C2), 110.5 (Ar-C6), 11504
(J = 9O4 Hz, Ar-C1), 124.5 (J = 14.8 Hz, Ar-C4) 146.1
(J = 7.9 Hz, Ar-C5), 151.5 (J = -234.2 Hz, Ar-C3)
~' .
MS: m/z (%) = 51 ~14), 52 (16), 57 ~2), 63 (3), 70 ~6),
71 ~7)/ 78 ~7), 79 ~13), ~0 ~5), 81 (4),
82 (8), 98 (57), 99 (5), 126 ~11), 127
(100, ~), 128 (7)
: ~
-~ 21232~2
- 14 -
Example 2
A. Preparation of 2-benzyloxy-6-fluorobenzamide in
j aqueous medium
J (1) 261.2 g (1.66 mol) of 2~6-difluorobenzamide are
su~pended in 232.3 g (2.15 mol) of benzyl alcohol/ and
the su~pension is then heated to 55C and 233.3 g
(2.08 mol) of 50 ~ potassium hydroxide ~olution are ad~ed
dropwise within the space of 2 h. At the end c>f this
time, the temperature i~ raisecl to 60C and the mixture
is stirred for a further 3 h.
i
¦ (2) The entire reaction mixture i~ allowed to flow,
¦ while 6tirring~ i~to 1900 g of water, and the precipi-
tated solid i8 filtered off with suction. Washing takes
place 3 times with 200 ml of water on each occasion, and
the product i8 dried at 70C in vacuo. 297.7 g (1.21 mol,
73 %) of 2-benzyloxy-6-fluorobenzamide are obtained from
381 g of moist product as a slightly yellowish powder
(purity (GC) > 98 %).
B. Preparation in dipolar, aprotic ~olvents
165.~ g ~1.2 mol) of potassium carbonate are suspended in
230 g of N,N-dimethylacetclmide (DM~c), and 113.4 g
(1.05 mol) of benæyl alcohol are added to thi~ suspen-
sion. After the addition of 157.1 g (1 mol) of 2,6-
difluorobenzc~mide, the mixture is heated at 130C for
~ 25 24 h, and the fine salt which has precipitated is then
I filtered off and washed with 250 g of DMAc in several
'portions (184.5 g, dry). The organic phase (658.6 g) is
added dropwiæe, while stirring, into 1100 g of water,
resulting in the reaction product crystallizing out.
Filtration takes place, and the filter cake i8 washed
three times with 150 g of water on each occasion. 405 g
of moist product are obtained as is, after drying,
170.7 g (0.696 mol, 70 %) of 2-benzyloxy-6-fluorobenza-
mide a~ a powder which is colored yellow-beige.
;-~ 2~232~2
- 15 -
2-Benzyloxy-6-fluorobenzamide:
N.p. 142.6C (DSC)
H-NMR [DMSO-d6/~MS, ppm]:
8 = 5.15 (I) (s, 2H, Ar-CH2)
~l 5 6-81 (G) (ddd~ J~G = 8-2 Elz, JPG = 0-5 ~Z~
JGI~ = 8 9 HZ ~ HS
~J 6.95 (F) (d(dd), 1~, J~ ~- 8-5 ~Z~ JFG - 0-5 HZ~
JF~ = 0 - 5 HZ ~ Ar-~3)
7.31 (~) (tm, 2~, Ar(benzyl)~H3s)
7.33 (E) (ddd, lHI J~ = 8-5 HZt J~G ~ 8-~ ~Z~
J~K = -6-9 ~Z, Ar-H4)
J 7.38 (D) (tm, 1~, Ar(benzyl)-~4)
7.45 (C) (dm, 2~, Ar(benzyl)-H26)
7.49 (B) (8(br), 1~, Ar-NH2Ci~/tr~n~)
7.80 (A) (s(br), 1~, ~r-N~2 i
9F-NMR [DMSO-d6/CFCl3, ppm~:
= -116-40 (K) (ddd~ 1F~ J~:R = -6~9 HZ~ JF~ = 0-5 HZ~
JG~ = 8 - 9 HZ ~ Ar_F6 )
~S: m/z (%) = 63 (5), 65 ~17), 91 (100), 92 (8), 98(3),
110 (6), 1~3 (4), 138 (3), 139 (5), 155
(1), 199 (1), 200 (1), 2~8 (20), 229 (3),
~ 245 (8.1, M+), 246 (1)
3 Example 3
: 2-Benzyloxy-6-fluoroaniline
120 g of methanoll 70 g of water, 30 g (0.75 mol) of
60dium hydroxide and 24.5 g (0.1 mol) of 2-benzyloxy-6~
fluorobenzamide are initially introduced and heated to
40C. Chlorine is passed in (4 l/h), whereupon, after a
short time, the colorless suspension assumes a slightly
brownish color and the heating can be removed since the
temperature i.s then maintained by the exothermic nature
of the reaction. A~ter 25 min, the reaction i complete,
as can be demonstrated by GC. A clear solution is
2 1 2 3 2 4 2
- 16 -
: obtained in place of the initial su~pension. The methanol
is distilled off (50C) under a weak vacuum, and the
resulting suspension of 2-benzyloxy-6-fluoro-N-carbo-
~l methoxyaniline is heated at 100C for 48 h. After
3 5 cooling, 50 g of toluene are added, the pha~es are
~eparated, and 2 g of MgS04 and 1 g of active ~harcoal
are added to the organic pha~e, which is stirred for ~ome
hours. After filtration and removal of the solvent on a
rotary evaporator, 19.8 g (91 mmol, 91 ~ of 2-benzyloxy-
10 6-fluoroaniline are obtained as3 a brownish, clear oil,
which exhibits a purity (GC. > 95 %) which is excellent
! for subsequent reactions.
2-Benzyloxy-6-fluoroaniline:
H-NMR [DMS0-d6/TMS, ppm]:
a = 4.57 (H~ (s(br~, 2H, Ar-NH2~
5.13 ~G) (s, 2H, Ar(benzyl)-CH2)
~ 6.51 (F) (ddd, lH, J~ = 8-35 HZ~ JDF = 8-15 Hz~
I JIF = 6.35 Hæ, Ar-H4)
¦ 6-68 (E) (ddd, lH, JBF = 8-35 HZI JD~ = 1.30 ~Z~
¦ 20 JEI = 10.6 Hz, Ar-Hs)
6-77 (D) (ddd~ JED = 1-30 HZI JDF = 8.15 Hz,
JDI 1 . 2 0 Hz, Ar-H3)
7.21 (C) (tm, lH, Ar(benzyl)-H4)
7.39 (C) (tm, 2H, Ar(benzyl)-H3s)
1 25 7.48 (B) (dm, 2H, Ar(benzyl)-~26)
9F-NMR rDMS0-d6/CFCl3, ppm]:
133.77 (I) (ddd, lF, J~;I = 10-6 HZI 3FI = ~;-35 ~Z~
JDI 1 . 2 0 ~ Z ~ Ar-F )
MS: m/z (%) = 51 (9), 63 (4), 65 (16), 91 (100), 92
(8), 98 ~16), 126 (10), 138 (0.5), 217
(17.6, M~), 218 (2.6)
2-Ben~yloxy-6-fluoro-N-carbomethoxyaniline can be iso-
lated if the procedure indicated above i~ followed but
the 48-hour long hydroly~is of the intermediate i~
omitted and the latter i~ isolated and purified in
J2~23
- 17 -
accordance with customary methods (in particular
filtration and recrystallization)~ -
MS: m/z (%) = 51 (3.2), 59 ~1.7), 63 (3.0), 65 tl4.0),
70 (3.2), 83 (1.6), 89 (2.5), 91 (100),
92 (9.7), 112 (1.2), 152 (1.6), 153
(1.3), 216 (1-6), 243 (800~, 244 tl.3),
275 (15.0, M+), 276 (2.6)
..
2-Benzyloxy~6-fluorophenyl isocyanate ari6es as an
intermediate in the synthe~is elnd can be isolated if ~o
desired.
MS: m/z (%) = 51 (4.2), 63 (3.1), 65 (15.1), 76 (5.3),
89 (3.2), 91 (100), 92 ~8.2), 9~ (2.6),
108 (1.1), 152 (8.9), 124 (3.7), 152
(1-6), 243 (15.9, M')
Example 4
2-Ethoxy-6-fluorobenzamide
69.6 g (~.5 mol) of 2,6-difluorobenzonitrile and 237 g
(2 mol) of 6-normal sodium hydroxide solution ~re
¦ initially introduced in 270 ml of ethanol. 221 g
(1.95 mol) of 30 % hydrogen peroxide solution are added
~ dropwise to this mixture within the ~pace of 30 min,
¦ during which the temperature, which was 20~C at the
beginning of the addition, rise~ to 50C and is then
maintained at this value. After 5 h, the mixture is
cooled and ths precipit~ted white solid i8 filtered off
I ! with suction (1902 g, m.p. 187.8C)~ The latter i5
- recrystallized from aqueous ethanol, yielding 16.5 g
(90 mmol, 18 ~) of pure, colorle~s 2-ethoxy-6-fluoro-
benzamide. lB.6 g (0.118 mol, 24 %) of 2,6-difluoro-
benzamide can additionally be isolated from the mother
liquor by distilling off 62 g of ethanol, and a further
17.5 g of this compound can be obtained in impure form
(purity about 60 ~) by extracting the mother liquor with
2123~2
- - 18 -
~ dichloromethane and removing the solvent.
-~, 2-Ethoxy-6-fluorobenzamide:
~. M.p. 195.5C (DSC)
.,
.~' l9F-NMR [DMSO-d6/CFCl3, ppm]:
r 5 ~ = -115.98 ~D) (ddd, lF, JAD = 6.65 HZ~ JCD = 9-07 E~Z,
J9D = 0-70 EIZ, Ar_F6)
,, .,
MS (EI~ 70 eV): m/z (%) = 40 (3), 44 (8), 57 (~), 63
. ~5), 74 (2), 82 (9), 81 (17), 9~ (4), 110 -
(48), 111 (4), 123 (34), 124 ~3), 138
, 10 (10~), 139 (32), 140 (4), 151 ~16), 166
(39) ~ 168 137) ~ 169 (3), 183 (4.5, M~)
H-NMR [Acetone-d6/TMS, ppm]:
8 = 1 . 3 6 ~ t, 3H, -CH2-CH3)
4 .1 2 ( qu , 2 H , -CH2 -C~3 )
6- 7 (s(br), lH, Ax-NH2i~/tr3n~)
6.71 (C) (ddd, lH, JAC = 8-4 HZr JBC = 0-78 HZ~
JCD = 9 07 ~Z, Ar-~4 )
6.84 (B) (ddd, 1H, J~ 0078 HZ, J~ = 8.4 ~Z,
J~D = 0-70 ~Z, Ar-HS)
6.9 ( 8 ( br ), lH , Ar-NH2Ci~/tranD )
7.31 (A) (ddd, lH, J~ = 6.65 Hz, J~ = 8.4 Hz,
JAC = 8 4 Hz, Ar-~13 )
.~ .
IR (KBr, cm~l): 3375, 3190, 2990 (W), 2985 (w), 2940
(W), 1650, 1570, 1~90, 1460, 1400, 1390,
1270, 1240, 1115, 1060, 1000, 810, 785, :
~, 755, 700, 645, 620, 530, 420
Example 5
'~ .. . ' '
I 2-~enzyloxy-6-fluorobenzonitrile
, .
100 g of benzyl alcohol and 55.6 ~ t0.4 mO1) Of 2,6-
difluorobenzonitrile are initially introduced and heated
to 40C. 49.2 g (0.44 mol) of 50 ~ potassium hydroxide ~ :
' .
',
~j .
~! 2 ~L 2 3 2 ~L 2
.~ - 19 - .
",j
solution are then added dropwise, with stirring, in such
a way that the temperature ca~ be maintained (cooling).
';
After 4 h, the mixture i~ diluted with 200 ml of water,
the precipitated product is iltered off with suction,
and 50 g of toluene are added to the mother liquor in a
separating funnel. The yield of moist product ie 62.0 g,
and, after drying, 56.2 g (0.247 mol, 62 ~) of 2-benzyl-
oxy-6-fluorobenzonitrile (purity (GC) 99.3 %) remain in
the form of a slightly yellowish solid. 31.5 g of 92.4 %
pure material (29.1 g, 0.128 mol, 32 %) are obtained ~rom
the organic phase by removing the toluene in vacuo (up to
190C). After dissolving the crude products in
ethanol/water and crystallizing without filtration, the
yield of pure 2-benzyloxy-6-fluorobenzonitrile i 81.2 g
(0.36 mol, 90 %); the product is obtained in the form of
colorless shiny platelets.
,.: .
¦ 2-Benzyloxy-6-fluorobenzonitrile
~ M.p. 71.9C (DSC)
3 MS: m/z (%) = 50 (3), 51 (6), 57 (3), 63 (7), 65 (27),
89 (5), 91 (100), ~2 (15), 108 (7), 120
(1), 136 (1), 170 (1), 227 (M~, 13.7), ~28
(2.4)
Example 6
6-Fluorosalicylamide
': ~,
24.3 g (0.1 mol) of 2-benzyloxy-6-fluorobenzamide are
stirred vigorously (lS h), at 20C and under an H2 atmo~-
phere (slight excess pressure), in 150 g of toluene
together with 5 g of Pd/C (5 %, 50 % moist). After that,
only the desired ~alicylamide can be detected in the
~ 30 mixture. The catalyst ie filtered off and then washed
3 with 100 ml of methanol in several portion~ in order to
dissolve off undisfiolved product from the solid. The
filtrate is concentrated (about 150 g) under a weak
232~2
~' '' .
- 20 -
vacuum, and then cooled down to 0C, whereupon colorless
crystals of 2-hydroxy-6-fluorobenzamide precipitate out.
~l ~hese latter are filtered off with ~uction and washed
with cold toluene and hexane. After drying, 14.2 g
(91.5 mmol, 92 %) of colorlesFi 6-fluorosalicylamide are
obtained.
2-Hydroxy-6-fluorobenzamide (6--fluoro~alicylamide)
M.p. 144-146.5C
,'
MS: m/z (%) - 44 (14), 57 (20), 63 (19), 71 (5), 81
(12), 82 (24), ~i3 (25), 98 (5), 110
(100), 111 (11), 113 (8)/ 138 (~0~, 139
i~i (19), 141 (19), 155 (74.4, M~) t 156 (~.43,
157 (11.5)
Example 7
~i :
2-Amino-3-fluorophenol via 2-hydroxy-6-fluorobenzamide
(6-fluorosalicylamide) -~
15.5 g (0.1 mol) of 6-fluorosalicylamide are introduced
into 100 g of 30 ~ ~iodium hydroxide solution, and ~
chlorine ~ passed (4 l/h) into the resulting clear ~-
solution. After 30 min, the chlorine str~am is shut off,
and excess chlorine is destroyed (sodium sulfite). The p~
of the æolution is adjus~ed to 6 with sulfuric acid and -~
the precipitated 3-fluorobenzoxazolo~e is filtered off
~; with suction at 0C. The moist product (16.6 g) i heated ~at 130C for 3 h in 80 g of 70 % sulfuric acid, and, ~ -
after cooling, the pH of the mixture i8 once again
adju~ted to 6, and filtration with suction takes place at
0C. 7.3 g (57 mmol, 57 %) of dark-gray 2-amino-3-fluoro-
phenol are obtained with a purity of 98 % (GC); the black
mother liquor i5 discarded.
.............. .................................................................... ...
2-Hydroxy-6-fluoro N-carbomethoxyaniline, inter alia, i8
,~ formed if a little methanol is added to the reaction
mixture.
,i
~1232~2
:. f . -: '
~ 21 ~
'~ MS ~ Z (%) = 51 (19.7) ~ 59 (18.8) ~ 70 (15.4) ~ 71
(lloO) ~ 97 (13~ 98 (69~2) r 109 (6-3) ~
126 (100) 1 127 (8~4) ~ 140 (5~9) ~ 153
(32~4) ~ 154 (5~3) ~ 185 (71~ 186
(6~4)
Example 8
' ;~
2-Amino-3-fluorophenol in a one-pot proce~s starting from
2,6-difluoroben amide
.
The procedure described in Example 2A. (1) iB carried
10 out. After that, a further 600 g of water, 280 g (7 mol)
of sodium hydroxide and 185 g (1.71 mol) of benzyl
¦ alcohol are added at 10C, and chlorine i~ passed in
15-18 l/h~ at 40C for 3 h. The phase~ are separated,
and 500 ml of methanol and 7 g of Pd/C (5 %, 50 ~ moist)
are added to the organic phase and hydrogenation is
carried out as described in Exampl~ lb. After working up ~ ~ :
I in analogy to Example lb, 123.5 g (0.97 mol, 59 %) of 2~
i~ amino-3-fluorophenol are obtained a~ pale-gray shiny
~ platelets.
3~ 20 Example 9
~ .
2-Amino-3-fluorophenol in a one-pot proces~ starting from
2,6-difluorobenzonitrile
69.6 g (0.5 mol) of 2,6~difluorobenzonitrile and 237 g
(1.2 mol) of 6-noxmal sodi~m hydroxide solution are
~ ~ 25 initially introduced in 200 ml of benzyl alcohol. 221 g
! ( 1. 95 mol) of 30 ~ hydrogen peroxide solution are added
~ drQpwise to this mixture within the space of 30 min,
¦ during which the temperature, which was 20C at the
¦ beginning of the addition, rises to 50C ~nd i8 then
maintained at this value. After 5 h (complete conversion
to 2-benzyloxy~6-fluorobenzamide can be detected by means
of GC), the mixture is cooled down and ~upplemented with
200 g o~ wate!r and 60 g ( 1~5 mol) of ~odium hydroxideO
2~23242
- 22 -
Chlorine is passed in (8 10 l/h) at 30~C for 2 h, with
the reaction being monitored by gas chromatography and
terminated when the amide has disappeared. The phases are
separated, and 180 g of methanol are added to the organic
5 phase, and this is followed by further working up as
3 indicated in Example 8. 32.4 g (0.255 mol~ 51 %) of 2-
amino-3-fluorophenol are obtained as a brown-black
powder.
.
.,,,,~ ~ ,.
:~ .
' ~, .
;:~
1. '.:.
~,