Note: Descriptions are shown in the official language in which they were submitted.
r`W0 93/21187 2 1 3 ~ 3 ~ ~ PCr/U~93/02~41
7 DI~ 3TI~TEI)-Nl ~Yh- 4--0~0--3H~
PYRROLOL3,2-d~PY~I~IDINE AND P~R~ E~TICAL ~8E~
C~Ol~POSITION8 CONTAINING q~
The present inventlon relates to derivatives of 4-oxo-
3H,5H-pyrrolo[3,2~d]pyrimidine. In particular, it rela~es to 4-
oxo-3H,5H-pyrrolo[3,2-dJpyrimidinederi~ativessubstitutedatthe
7-position.
Purine nucleoside phosphorylase (PNP) catalyzes the
phosphorolysis of purine nuoleosides in a reversible reaction.
Individuals who are deficient in PNP exhibit impaired T-c~ll
development, resulting in lowered cell-mediated immunity, but
normal ~-cell de~elopment, resulting in normal humoral immunity.
Accordingly, specific inhibitors of PNP that selectively inhibit
T-cell development without damaging humoral immunity could be
potentially effective against disorders in which activated T-
cel-s are pathogenic.
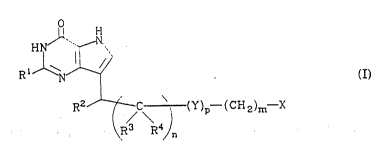
Accordingly, the present invention is a compound of the
formula
HN
R~
R2~ / C ~ ')p-(CH~)m-X
\ R3 F~ ,/n
i'
wherein R1 is H, NH~, or OCH~, R2 is an optionally substituted
cyclic group optionally containing one or more heteroatoms, R3
and R4 are independently H or C14 alkyl, m is 0-4, n is 0-6, p is
0-~, X is CN, CSNH2, PO~OH)2, COOH, SOzMH2~ NH2, OH, CN~N~2,
tetrazole, triazole or CoR5 wher~ Rs is C~ 4 alkyl, CF3, NH2~ or
~, . ' ~ !
; OC~,4 allcyl, and Y is O or NH. The compound of the present
3 0 in~rention is u5eful as a PNP inhi~itor . Also coIltempla~ed
. according to the present invention a~e a pharmac:eutical
composition for the selective suppression of mammalian T-cell
immunity comprising an pharmaceutic:ally effective amount of the t
:: compound of the pre5ent invent~ on and a pharmaceutirally
acceptable carrier or diluent and a method for ~he s lective
93/21187 ~ 3 3 3 il~ PCT/US93/02~
suppression of mammalian T-cell immunity without diminished
effect on humoral immunity comprising administering to a subject
a pharmaceutically effective amount of the compound of the
present invention.
The optionally substituted cyclic group (he~einafter
r~ferred to as cyclo) recited for the above formula includes
aromatic, heteroaromatic, alicyclic, and heteroalicyclic groups
prefera~ly containing five to nine atoms. Preferred optional
substituents include halogen, hydroxy, alkoxy, alkyl, and
trifluoromethyl. Exemplary su~stituents include chloro, fluoro,
methoxy, ethoxy, propoxy, butoxy~ methyl, ethyl, propyl, and
butyl. Preferred heteroatoms include oxygen, nitrogen, and
sulfur, which can be present in combination in the same group.
The preferred axomatic and heteroaromatic groups are phenyl, 2-
or 3-thienyl, 2- or 3-furanyl, 2-, 3-, or 4-pyridinyl, 2- or 3-
pyrrolyl, 2-, 4-, or 5-thiazolyl, 2-pyrazinyl, 3- or
4-pyridazinyl, and 3-, 4-, or S-pyrazolyl. The preferred
alicyclic and heteroalicyclic groups are 1- or 2-adamantyll
cyclohexyl, cycloheptyl, 2- or 3-tetrahydrofuranyl, 2- or
3-tetrahydrothienyl, 2- or 3-tetrahydropyranyl, 2-, 3-, or
4-piperidinyl, 3- or 4-pyrazolidinyl, 2-, 4-, or 5-thiazolidinyl,
2- or 3-piperazinyl, 2- or 3-morpholinyl, or 3- or
4-hexahydropyridazinyl. Examples include compounds wherein R1 is
NHz or H, R2 is phenyl, 3-chlorophenyl, or 3,4-dicholorophenyl,
and (C~3R4)n-(Y)p-(CH2)~-X is CH2C~2CN; CH2CH2CQOH; CH2CH2CH2OH;
C}I2CH2CH2CN; CH2CH2CHzCOOH C~I2C~I2CH2CX20H, or sub~tituents where an
oxygen atom replaces one or more of the methylene groups.
The present invention contemplates pharmaceutical
: compositions suitable for enteral, such as oral or rectal,
transdermal and parentera} administration to m~mmals including
man~ which are useful to inhibit purine nucleoslde phosphorylase
acti~ity and for the treatment of disorders responsive thereto,
comprising an effectiYe amount of a pharmacologically ac~i~e
compound of the invention, alone or in com~ination, with one or
more pharmaceutically acc~ptable carriers~
Preferred phaxmaceutical compositions are tablets and
gelatin capsules compr_sing the active ingredient together with
-2-
~ wo g~/2ll87 2 ~ 3 3 3 'i ~ PCT/US93/0~
a) diluents, e.g., lactose, dextrose, sucrose, mannitol,
sorbitol, cellulose and/or glycine, b) lubricants, e.g., silica, :
talcum/ stearic acid, its magnesium or calcium salt and/or
polyethyleneglycol; for tablets also c) binders, e.g., magnesium
aluminum silicate, starch pas~e, gelatin, tr~gacanth,
methylcellulose, sodium carbox~me~hylcellulose and/or
polyvinylpyrrolidone; if desired d) disintegrants, e.g., .
starches, agar, alginic a~id or its sodium salt, or efferYescent
mixtures; and/or e) abs~rbents, colorants, flavors and
sweeteners. Injectable compositions are preferably aqueous
isotonic solutions or suspensions, and suppositories are
ad~antageously prepared from fatty emulsions or suspensions.
Said compositions may be sterilized and/or contain adjuvants,
such as preser~ing, stabilizing, wetting or emulsifyin~ agents/
solution promoters, salts for regulating the osmotic pressure
and/or buffers. In addition,they may also contain ot~er
t~erapeutically valuable substances. Said compositions are
prepared according to conventional mixing, granulating or coating
methods, respectively, and contain about 0.1 to 7~%, pref2rahly
about 1 to 50%, of the active ingredient.
Suitable formulations for transdermal application in~lude
an effective amount of a compound of the invention with a
carrier. Advantageous carriers include absorbable
pharmacologically acceptable solvents to as~ist passage through
~he skin of the host. Characteristically, transdermal devices
are in the form of a bandage comprising a backing member, a
reservoir containing the compound optionally with carriers,
optionally a rate controlling barrier to deliver the compound to
the skin of the host at a controlled and predetermined rate over
a prolonged period of time, and means to secure the device to the
skin.
nother aspect o~ the present invention provides a method
of making a 2amino compoun~ (R1 = NX2) of the presen~ invention
and int~irmediates thereof. The first step of the met~od involves
reacting an~ optionally substituted cyclic aldehyde with
cyanaacetic acid at a molar ratio of about 1/1 to 1/5 in the
presence of ammonium ~acetate at about reflux temperature for
-3-
~ hO93~21187 ~13 3 3 ~ ~ PCT/US93/~2~1
about 10 hours to ~ days to make a 3-cyclo~substituted
pentanedinitrile as an intermediate~3 In the second step, the 3-
cyclo-pentanedinitrile is reacted with an alkyl formate such as
ethyl formate and a strong base such as the metal-containing
bases sodium hydride or sodium alkoxide, e.g., sodium me~hoxide,
at a molar ratio of about 1-2/3-6/1 3 and at a temperature of
about 20-65~C for about 10 hours to B days to make a 3-cyclo-
2-formylpentanedinitrile as a further intermediate. The next
step involves reacting the 3-cyclo-2~formylpentanedinitrile with
a glycine alkyl ester hydrochloride and sodium or ammonium
acetate at a molar ratlo of about 1-2/1.5-4~1~5-~ and at a
temperature of about 20-60-C for about 10-48 hours to make methyl
N-~(3-cyclo-2,4-~icyano)-2-butenyl]glycine as an intermediate.
In the subse~uent step, the~methyl N-[(3-cyclo-2,4~dicyano)-2-
15 butenyl ]glycine is reacted with an alkyl chloroformate such asethyl chloroformate and l,S-diazabicyclo[ 4.~.O]non-5-ene (DBN)
or 1,8-diazabicyclo~S.4.0]undec-7-ene (DBU) at a molar ratio of
about 1-2/1.5-5/1.5-4 and at a temperature of about 0-50-C for
abou~ 10 hours to 10 days to make methyl 3-amino-4-(2-cyano-l-
cyclo-ethyl)-1-ethyl-lH-pyrrole-1,2-dicarboxylate as an
intexmediate. The next step involves reacting the methyl 3-
~ amino-4-(2-cyano-l~-cyclo-ethyl)-1-ethyl-lH-pyrrole-1,2-
r ~ ~ dicarboxylate with a ba5e such as sodium car~onate at a molar
ratio o~ about 2~1 to 1~5 and at about room temperature for abou~
}0-48 hours to make methyl 3-amino-4-(2-cyano-1-cyclo-ethyl~-
~; ; lH-pyrrole-2-car~oxylate as an interm3ediate. In the next step,
the methyl 3-amino-4-(2-cyano~1-cyclo-ethyl)-lH-pyrrole-2-
`; carboxylate is reacted with ~enzoylisothiocyanate at a molar
ratic of about 2/1 to~ 1/2 and at about room temperature for about
3 0 3 0 minutes to 3 hours to make N-benzoyl-N'-~4-(2-cyano 1-cyclo-
ethyl~2-methoxy~arbonyl-lH-pYrrol-3-yl~thio~urea as an
intermediate. The next step~ reacts the N-benzoyl-N'-~4-(2-
cyano-l-cyclo33333ethyl~ 2-methoxycarbonyl-lH-pyrrol-4-3-yl]thiourea
With an alkyl halide such as methyl iodide at a molar ratio of
about 1/1 to 1/6 an~ at a temperature of about 0-30OC for a~out
10 min~tes to 10~hours to make N-~enzoyl-N -~ 2-cyano-1-cyclo-
ethyl)-2-methoxycarbonyl-}H-pyrrol-3 yl]S-m2thylthiourea as an
4-
`:; :
~ :
WO 93/211~7 213 3 3 ~ ~ PCI/US93/02841
intermediate . In the following step, the N-benzoyl-N ' - [ 4 - ( 2 -
cyano- l-cyclo-ethyl ) -2 -methoxycarbonyl-l~-pyrrol-3 -yl ] -S-
meth~ylthiourea ( about 1-2 mol ) is reacted with methanolic or
ethanolic ammonia at a ratio of a}:~out l/l to l/20 and at a
temperature of about 20-130'C for about 16~60 hour~ to make a
mixture of a 2-amino compound of t~e present invention 3-cyclo-
3 ~ amino-4 -oxo-3H~5H-pyrrolo [ 3, 2 -d]pyrimidin-7-
yl]propanenitrile and a 3-cyclo-3-~2-methylmercapto-4-oxo-3H~5H-
pyrrolo [ 3, 2 ~ ~ pyrimidin-7 -yl ] propanenitril e as an intermediate
in making another compound of the present invention.
In a further aspect of the present inYention there is
provided a method of making a 2-methoxy compound (R1 = OCH3) and
intermediates thereof. The intermediate 3-cyclo~3-[2-
methylmercapto-4-oxo-3H,5H-pyrrolo~3,2-d~pyrimidin-7-
yl]propanenitrile is reacted with an oxidi~ing agent such as
permanganate or hydrogen peroxide at a molar ratio of about 1/1
to 1/10 a~d at a temperature of about 25-120-C for about 3-48
hours to make 3~cyclo-3-~2-methylsulfonyl-4-oxo-3H,5H-
pyrrolo[3,2-d]pyrimidin-7-yl]propanenitrile as an intermediate.
In the next step, the 3-cyolo-3-[2-methylsul~onyl-4-oxo-3H~5H-
pyrrolo[3,2-d]pyrimidin-7-~lJpropanenitrile is reacted with a
sodium alkoxide such as sodium methoxide at 2 molar ratio of
about 1/1 to 1/10 and at a temperature of about 25-100^C for
about 1-48 hours to make a 2-methoxy compound of the present
invention 3-cyclo-3-[2-methoxy-4-oxo-3H,5H-pyrrolo[3,2-
d]pyrimidin-7-yl]propanenitrile.
In a further aspect of the present invention there is
provided a method of making a compound of the present invention
wherein Rl is hy~rogen. The methyl 3 a~ino-4-(2-cyano~l-cyclo-
3 0 ethyl ) -lH~-pyrrole~2 -carboxylate intermediate described su~ra is
reacted with dlmethylformamide dime~hyl acetal at a molar ratio
of about 1/1 to 1/4 and at a temperature of about 25-lOODC for
about 1-10 days to make methyl 4-(2 cyano-l-cyclo-ethyl)-3-[N
(dimethylaminomethylene)amino~ pyrrole-2-car~oxylate as an
intermedi~te. The next step involves reacting the methyl 4--~2-
cyano-l-cyclo-ethyl)-3-~N-(dimethylaminomethylene~amino)-lH-
pyrrole-2-carboxylate with methanolic or ethanolic ammonia at a
~`i` 213~3~
0g3/2~187 PCT/US93/~2~1
molar ratio of about 1/1 to 1/20 and at a temperature of about
20-130C for about 10-68 hours to make the compound of the
present invention 3-cyclo-3-~4-oxo-3H,5H-pyrrolo[3,2-d~pyrimidin-
7-yl}propanenitrile~
As will be apparent to the skilled artisan, variations of
the aforesaid procedures are useful in making the variety of
compounds of th~ present in~ention without departing frcm 'he
spirit thereof. For example, compounds having different values
for "n" and "~" in accordance with the present invention are
obtained by either stepping up or stepping down the series by.the
necessary number of carbon atoms in accoxdance with known
procedures. Also, reactions involving some intermediates require
protection of nitrogen or oxygen atoms on the intermediates using
known procedures.
The present invention provides a method of inhibiting purine
nucleoside phosphorylase activity in mammals and treating
diseases and conditions responsive thereto, e.g., autoimmune
disorders, rejection of transplantation, or psoriasis, which
comprises administaring to a mammal in need thereof an effective
amount of a compound of the in~ention or of a pharmaceutical
~ composition comprising a said compound in combination with one
.~ or more pharmaceutically acceptable carriers.
A ~urther aspect of the invention relates to a method of
~: inhibiting the phosphorolysis and metabolic breakdown of
2S anti~iral or antitumor purine nucleosides ~n mammals which
compris~s administering in conjunction therewith to a mammal in
need thereof, either separately or in combination therewith, an
:~ effecti~e purine nucleoside phosphorylase inhi~iting amount of
~: a compound of the invention or of a said compound in combination
with one or more pharmaceutically acc~ptable carriers. M~re
particularly,j such relates to a ~ethod o~ inhibiting Ithe
phosphorolysis and metabolic breakdown of purine nucleosides
known in the art, e.~, of 2' deoxyguanosine, 2',3'-
~id~oxyinosine, 2',3'-dideoxyguano~ine or 2',3'-dideoxyadenosine.
Furthermore, the in~ent1on thus relates to a method of
~ potentiating the antiviral or antitumor eff~ct of 2' or 3'
;~ monodeoxypurine nucleosides or of2',3' dideoxypurine nucleosides
': :
~ 6-
WO93/211~7 Z1 3 3 ~ ~ ~ PCT/U593/02~1
in mammals which comprises administering in conjunction ~herewith
to a mammal in need thereof, either separately or in combina~ion
with a said nucleoside, an effective purine nucleosidP
. phosphorylase inhibiting amount of a compound of the invention
preferably in combination wit~ one or more pharmaceutically
acceptable carriers. More particularly, such relates to a method
of enhancing or potentiating the effect of 2',3'-dideoxypurine
nucleosides known in the art, e.g., of 2',3'~dideoxyinosine,
2',3'~dideoxyguanosine or 2'-3'-dideoxyadenosine for the
treatment of retrovirus infections, eOg., HIV-retrovirus
infections (ac~uired immunode~iciency syn~rome, AI~S). 2',3'-
Dideox~purine nucleosides are kn~wn in the art as inhibitors of
HIV retrovirus infectivity and to be metaboli~ally degraded by
PNP, e.g., as described in Biochem~cal Pharmacoloqy 22, 3797
(1987). Such are administered at a pharmaceutically acceptable
dose which is effective in i~hibiting HIV-retro~irus infections.
Preferably the lowest possible effective dose is used.
The pharmaceutically acceptable ef~ective dosage of active
compound ~f the invention to be administered is dependent on the
species of warm-blooded animal (mammal), the b~dy weight, age and
individual condition, and on ~he fo~m of administration.
The pharmaceutical composition may be aral, parenteral~
suppository or ~ther form which delivers the compound of the
present invention into the bloodstream of a mammal to be treated.
An oral ~orm has from about 1 to about 150 mg of the compound of
the pr~sent invention for an adult (50 to 70 kg) which is mixed
together with pharmaceutically acceptable diluents such as
lactose. In a typ~cal capsule, 25 mg of the compound of the
present invention is mixed together with 192 m~ lactose, 80 mg
modified 5tarch and 3 mg magnesium stearate. Injectable forms
of the compound are also contemplated for administratio~. ~
'The present invention is also useful with other therapeutic
: agents. A daily dosage of the compound of the present invention
for a human weighing 50 to 70 kg of l-~0 mg/kg inhibits meta~olic
3S d~struction of certain anticancer agents such as ~-2'-deoxy-6-
thio~uanosine and anti~iral agents such as 2'j3'-dideoxyinosine,
an anti-AIDS drug. These t~pes of agents are known ~o be
`~ ~093~21187 ~ 1 3 3 3 ~ ~ PCT/US93/02~]
susceptible to cleava~e. Upon cleavage, the agents lose
ef~ectiveness~ The compounds of the present invention are
capable of reducing such cle~vage. This protection, therefore,
enhances the efficacy of other chemotherapeutic agents.
In order to more full~ describe the present invention`~the
following non-limiting examples are provided. In the examples
all parts and percentages are by weight unless indicated
otherwise. Proportions of so.Lvent mixtures used as
chromatographic eluents are by vol~me.
XA~PLE 1
~3 cl ~
cl ,
t:~ The a~ove intermediate compound is prepared in this Example
~!''' by the modi~ication o~ the procedure of Schiemenz, G. P.;
;~ Engelhard, H~ (Chem. Ber., l962, 95, 195).
t~ O A mixture of cyanoacetic acid t25.38 g, 298.38 mmol), 2,3,5-
.. t~ichlorobenzaldehyde (25.0 g, 119.35 mmol), ammonium acetate
,~,; (500 mg), toluene (120 mL), and pyridine (65 ml) is heated at
,.~ reflux for 16 h in a flask fitted with Dean Stark trap and
. condens~r. The solvents are evaporated in vacuo, residue is
~.,, 5 extracted with ~HCl~, which is washed with H20~ ~ried ~Na2SO~),
.~ and e~aporated to give the crude product, which is purified by
silica gel column chromatography using hexaneEtOAc mixture as
:;~' ~he eluent. Yield 23.69 g (73%); mp 90-91 C.
~A~PL~ 2
~CN~
C~ ~ ~HO
; ii
Cl
The a~ove intermediate compound is prepared in this Example.
To a stirred mixture of NaH (l.S6 g, 65005 mmol) and ethyl
?
,r'~ :`
':
,.~,j
r~ , . WO 93~2l~X7 2 ~ 3 3 ~ ~ J PCT/U593/02~1
." formate (14.~8 g, 199.51 mmol) in THF (100 ml) is added
substituted pentanedinitrile of Example 1 (10.17 g, 37.17 mmol)
at r~om temperature under a nitrogen atmosphere, and the
~ resulting reaction mixture is stirred for 24 h. Volatile matter
,.~ 5 is evaporated ln vacuo at room temperature. Water (50 ml) is
~ added to t~e residue at 0-5 C, and the solution is adjusted to
: pH 5-6 by 20% conc. HCl (_~v). The heavy oil is extracted into
ethyl acetate, washed with H20 (1 x 100 ml) and dried (~gSO4).
. The ethyl acetate layer is evaporated to give a red-brown oil
~ 0 g) that is used in the next step without further
. purification.
""
;.', ~AMP~ 3
~ j O~e
The above intermediate compound is prepared in this Example.
2Q Glycine met~yl ester hydrochloride (8.17 gl 65.06 mmol~ and
sodium acetate (5.33 g, 65.06 mmol) are added to a solution of
the crude formyl compound of Example 2 (11.0 g) in a mixture o~
; MeOH (80 m~) and H20 (20 ml), and the resulting solution is
.~ stirred at room temperatur~ for 22 h. A~ter evaporation of
solvent at room temperature, the residue is extracted with ethyl
acetate. The washed (H20) and dried (MgS04) organic layer is
evaporated to gi~e an oil. Flash column chromatography (silica
.. gel) using CHCl3 as eluent gave the pure desired enamine as a
mixture of ~rtra~s isomers which is recrystallized from MeOH,
;, 30 yield 10.41 g (75%), mp 142-143 C~
~AMPLE 4
!.'¦
Cl E~70C 0
.,¦ Cl ~ ~NH~
Cl ~ N
~, _g_
, ,~
2~333~G
';~ VO93/XllX7 PCT/US93/02~1
The above intermediate compound is prepared in this Example.
solution of enamine of Example 3 (10.0 g, 26.84 mmol) in dry
C~2Cl2 (100 ml) is cooled to 0 ~C and treated with 1,5
diazabicyclo[4.3.0~non-5-ene (10.53 g, 84.79 mmol) under a
nitrogen atmosphere followed by ethyl chloroformate (6.90 g,
63~57 mmol). The solution is stirred at 0 ~C for 1 h and then
at room temperature for 48 h. Volatiles are evaporated in vacuo
to give a viscaus dark gum which is purified by flash column
chromatography over silica gel using CHC13 as the eluent. All
the fractions containing the desired ~-protected pyrrole are
pooled and evaporated to give a foamy light pale yellow material
which is stirred in MeOH (100 ml) to give ~he crystalline
material which is recrystallized from CHCl3-MeOH, yield 8.92 g
(74.7%), mp 160-16~ C.
~MP~ 5
~ o M e
;20 Cl'``~ `NH2
Cl C~
The above intermediate compound is prepared in this Example.
A suspension of N-protected pyrrole of Example 4 (8.6 g, 19.34
2S .mmol) in MeOH (300 ml) is treated with NazCO3 (5.12 g, 48.34
mmoI) and the reaction mixture is stirred at room temperature for
17 h with separation of the deblocked pyrrole during the f irst
hour~ Solid sodium carbonate is removed by filtration and washed
well with MeOH. The filtrate is reduced to a ~olume of -25 ml
and kept in a refrigerator for 1 h to give 5.23 g of crystalline
prqduct. Fur~her concentration of the mother liquor Igave an
additional 0.14 ~ o~ pure product; total yield 6.45 g (89.5 ~),
mp 178-1~1 C.
"
: :,
~ `. ~og3/~l~$7 2~ ~ 3~ PCT/~S~3~2~1
PL~_6
C~ ~ o
Cl'" ,' ~ NH-ICl-NHC~Ph .
Cl CN S
The above inte~nediate compound is prepared in this Example.
To a suspension of pyrrole of Example ~ (5~83 g, 15.64 mmol) in
dichloromethane (lO0 ml) is added benzoylisothiocyanate (2.88 g,
17.64 mmol) at room temperature un~er nitrogen. The reaction
mixture is stirred for 30 min with the separation of the desired
t~ioureido compound. Additional benzoyl isothlocyanate (0.5 ml)
is added to it and again stirred for 30 min~ T~e solvent is
ev~porated to d~yness, and the light yellow residue is triturated
with methanol. The white crystalline material is isolated by
filtration and recrystallized from a chlorofo~m-ether mixture to
give the required thioureido compound as an analytically pure
sample, yield 7.71 g (92%), mp 210-211 DC.
_-S~MPL~ 7
ClH 0
' i ' \~ /i
Cl~ .~ `N=C-NHCOPh
Cl ~CN SMe
The above intermediate compound is prepared in this Example,
A solution of thioureido campour~d of Ex~mple 6 ( 6 . 75 g, 12 . 6
mmol ) arld 1, 5-diaæabicyclo [ 4 . 3 . O ] non-5-ene ~1. 7 ~ g, 14 . 2 0 ~ol )
in dry CHzC12 (200 ml) is cooled to o oc and treated with methyl
iodi`de (So20 g; 36.65 ~ol~ . The reaction mix~ure is istirred at
O C f or 10 min and then f or 1 h at room temperature . The
solvent is evaporat~d at room temperature, and the residue is
~ extracted with CHC13, washed with H20 (2 x 50 ml), dried (Na2S~4~
: 35 and evaporated to gi~e a glas5y foam (6095 g) which is used in
the next step without purification.
:
:
O 93/21~87 ~ ~ 3 ~ 3 ~ ~ PCr/US93/~)2~41
PLE ~
.~
O O
I, H 1I H
HN~--~> H I ~\1\
H 2 N~ M e S~\N--~/
~ /CN Cl \~CN
Cl Cl
A B
The above compo~nds A and B are prepared in this Example.
The compound A is a compound of the present in~ention and the
compound B is an intermediate. A solu~ion of the methylthio
intermediate of Example 7 (6.90 gl 12.~4 mol) ln MeOH (200 ml)
is saturated at o D C with ammonia and hPated at 100 ~C for 20 h
in a glass-lined stainless steel bo~h. The reaction mixture is
brought to room kemperature and the solvent is evaporated at room
temperature~ Purification of the crude mixtur~ by flash column
chromatoyraphy oYer silica gel using CHC13 as eluent gave 8B (l.l
g, 21%), mp 290-291 C then CHCl3-MeOH (95:5) gave pure ~ (2.76
gj 57.~), mp 284-285 C.
: :
P~E 9
The compound of the present invention of Example 8 is tested
for~ enzyme inhibition activity. A purine nucleoside:
~` phosphoxylas~ (~NP) enzyme a~say is performed in whlch the PNP
.30 activity (IC50) for the compound (8A) is Eound, which i5
det~mine~ radiochemicall.y by:measuring the formation ~f [1~.C]-
~:~ hyp~xanthine from ~14C~-in~sine (see Biomedic ne, 19~0, 3~, 39)
usi~g cal~ spleen as the enzyme source. At 1 mM phosphate the
IC5~ is 0. 64 ~M and at 50 mM pho5phate the IC50 is 10 ~M. ~ ;
.~ 3~:
,, ~
: ~ ~ : : :
` wog3/2ll~7 ~3.~ PCr/US93/0284~
E~PLE: l o
, ~
S H~
H2Nl\~
J~ CN
C1
lC
Following the procedure set forth in Examples 1-8, 3 - ( 3-
15chlorophenyl ) -3 - ( 2 -amino-4-oxo-3H, 5H-pyrrclo i' 3, 2 -d ] pyrimidin-
7-yl ) propanenitrile ( IC) is prepared using 3- ( 3-chlorophenyl ) -
pentanedinltrile as the starting material, yield 54 . 5%, mp 157-
158 ~ C.
2 OE:~PLB 11
Following the procedure set forth in Examples 1-8, the ~;
~o}lowing compounds are also prepared ( 1-9 ) .
O ,,.
;: 2 5H N~--
H~N'~,T~,
3-Aryl 3-i~2-amino -4-oxo-3H,5E~-pyrrolc: [3,2-d]-
pyrimidin 7-yl) propanenitrile
Where Ar is each of the following. (1,~ phenyl, 2,3
dich~orophenyl, 3-methylphenyl, and 3-methoxyphenyl, (2) thienyl
an~ 3 ~ ~, ( 3 ) furarlyl ( 2 - and 3 - ), ( 4 ) pyridinyl ( 2 - , 3 - , and .
~: 35 4~ 5) pyrrolyl (2~ and 3-), (6) thiazolyl (2-, 4-, and 5-),
~ . . .,
(7~ 2-pyrazinyl, (8) pyridazinyl (3- and 4-), and (9) pyrazolyl. ¦-
- 1 3 - .
:
;`: WO93/2~187 ~ 3 3 1 ~ PCT/US93/02~1
Eæ~MPLE 1~
Following th procedure set forth in Examples 1-8, the
,
following compounds 10-14 and 21 are prepared starting from the
appropriately substituted pentanedinitrile. Compounds 15-20, and
22 are prepared from the corresponding unsaturated Ar analogues
in Example }1. In this procedure, the nitrile group of the
unsaturated analogue is first converted to an amide group by
acid- or base-catalyzed hydrolysis, then the unsat1~rated Ar group
is converted to the saturated R2 group by Xnown catalytic
hydrogenation, followed by reconverting the amidei bac~ to the
nitrile by known dehydration procedures.
O
H
H 2 N ~/~
~;: 15R ~ ~`--~
3-(Substituted)-3-(2-amino-4-oxo-3H,5H-pyrrolo[3,2-d]- :
~`` pyrimidin-7-yl)propanenitrile
Where R2 is each of; 10) 1-adamantyl, 11) 2~-adamantyl, 12)
20cycloh~xyl, 13) cycloheptyl, 14) cyclopenkyl, 15
.
tetrahydro~uranyl, 16) tetrahydrothienyl, 17) tetrahydropyranyl,
18) pyrazolidinyl, ~19) thiazolidinyl, 20) piperazinyl, 21)
mo~pholinyl, and 22) hexahydropyridazinyl. ~:
:: :
~ 25
,. :
. ~
. ~ :
~ 14- ~
WO93121187 ~1 ~ 3 3 ~ 5 PCT/US93/02~1
E~n~E 13
.
~. O
S
The abo~e compound, 3-~2-amino-4~oxo-3H,SH-pyrrolo~3,2-
d]pyrimidin-7-yl)-3-phenylpropanenitrile, is prepared i~ this
Example. A solution of the compound A obtained in ExamplP 8 ~2.0
g, 5.22 mmol) in warm ethanol ~250 ml) and dimethylformamide
(DMF) (lS0 ml) is hydrogenated over 30% Pd/C catalyct (1.0 g~ in
the presence of triethylamine ~2.64 g, 5.O equivalent~ at
atmospheric pressure. After 5 h, the reaction is complete, and
the catalyst is filtered off u~der N2 pressure. The solid
obtained by evaporation o~ the filtrate is triturated and
sonicated w'th H20 and dried, yield 1.28 g (88%), mp 168-170 C.
20~MP~ 14
The compound prepared in Example 13 is tested for enzyme
inhibition activity as in Example 9. At 1 ~M phospha~e the IC5
,
is 0.023 ~M and at 50 mM phosphate the IC50 is 4.7 ~M.
I 1` ~ , ~
~
,;
-15-
`:, :
; :: :
: ~ : :
'.`~093/~1187 2 1 3 3 3 ~ ~ P~T/US93/02~
E~MFL~ ~s
o
. ~ H
~ ~ H
The above compoundl 3-(2-amino-4-o~o-3H r 5H-pyrrolo[3,2-
dJpyrimidin-7-yl)-3-phenylpropanoic acid, is prepared in thiis
example~ A solution of the compound obtainPd in Example 13
(0.200 g, 0.72 mmol) in 6N HCl (3,.~ ml) lS heated at reflux for
18 h. The solvent is evaporated ln vacuo and the residue is
triturated with H20 (6 ml), adjusted to pH ~lO by conc. ammonium
hydroxidei. Insoluble material is collected by filtration and the
filtrate is readjusted to pH ~6.8. Whiite material which is
pxecipitated out i5 colle ted, washed with wate~ and dried, yield
O.l9 g (89~), mp 290 C dec.
:32U~P~
: 20 The compound prepared in Example lS is tested for enzyme
:~ inhiibition activity as in Exampl~ 9. At 1 ~M phosphate the IC5
iig 0.012 ~ and`at 5Q mM phosphate the ICso is O.l9 ~M.
' ~
~ ,
., :
~ `W~93/~lY~7 2 1 3 3 3 '1 S PCT/U~93/0~l
,, E~PL~ 17
H ~ ..
"~ ~,J~ NH2
The above compound, 3-(2-amino-4-oxo 3H,5H-pyrrolo[3,2-
d~pyrimidin-7-yl)-3-phenylpropanamide, is prepared in this
example. A solution of the compound obtained in Example 13
(0.200 g, 0.72 mmol) in conc. HzS04 (0.5 ml) is stirred at room
temperature for 20 h and then poured onto crushed ice (5.0 g) and
adjust~d to pH ~6.8 by conc. NH40H. Th preoipitat~d solid is
collected, washed with H20 and dried, yield 0,180 g, mp 199-20
D C dec.
~P~ l ~
The compound prepared in Example 17 is tested for enzyme
inhibition activity as in Example 9. At 1 mM phosphate the IC50
is O.20 ~M and at 50 mM phosphate the IC50 is 6.6 ~M.
.
~ 17-
:
-;
. NO 93/21187 213 3 3 ~ ~ P~/US~3/0~2841
E~MPL}: l9
~2N ~ o
OMe
.
'
The above compound, 3-(2-amino-4-oxo-3~,5H-pyrrolo~3,2-
:: d}pyrimidin-7-ylj-3-phenylpropanoic acid, methyl ester, ~is
prepared in this example.~ Thionyl chloride (O.2 g, 0.l17 mmol)
is added to stirred mathanol (4.0 ml) a~ 0 C. The ~ompound
obtained in Example lS (0.~ g, 0.67 mmol) is added and the
mixture is stirred at~ro~m temperature ~or 18 h. The solvent is
5:~ removed on a water aspirator (30 C) and vacuum pump (lyophiIize)
to give a semisolid mass which iS purified on a silica gel column ~ : j
using~CHCl3-MeOH as the eluent, yield 0.1 g. :;~
20: ~ 7rJ~ 20
;: ,
The compound: prepared i~:Example 19 is tested ~or enzyme
inhibition acti~ity . S ignlf icant activity (ICso) is ~ound.
" W093~211~7 2 1 3 3 3 '~ J~ PCT/US93fO2~]
E~PL~
ll H
~ ~ OH
~ Amino-4-oxo~3H,SH-pyrrolo[3,2-d~p~rimidin-7-yl)-3-
cyclohexylpropanoic acld is prepared in this example. A solution
of the compound obtained in Example 15 ~83 mg, O.28 mmol) in
trifluoroacPtic acid ~TFA) (15 ml) is hydrogenated with PtO2 (83
mg) at 60 lb/in2 for 24 h. The catalyst is filtered off throu~h
15 a Celite bedt and t~e filtrate is evaporated at 25 C. The
residue is susp~nded in H20 (8 ml), and adjusted to pH 8.5 by
conc. N~OH and filtered through a Whatman filter paper to remove
brown colored impurities. The colorless filtrate is adjusted tG
pH -6.8, and t~e precipitated compound is filtered, washed with
~0 HzO, and dxied, yield 65 mg (77%), mp >300 C.
~ PLE 2 2
The compound prepared in Example 2l is tested for enzyme
inhibition activity as in Example 9. At l mM phosphat'e the'I~5
25 is 0.097 ~M and at 50 mM phosphate the IC~o is l.O ~M.
:
'
-19-
. ~093~21187 2 ~ 3 3 d ~ ~ PCT/US~3/02~1
~MP~ 23
A compound of the present invention is prepared wherein X
is PO(OH)2~ The nitrile group of the compound of Example 13 is
converted to the corresponding amide by treatment with sulfuric
S acid. Using a Hoffman degradation reaction, the amide is
converted to the corresponding amine, which is then converted to
the corresponding pyridinium salt using a pyrillium salt.
Canversion of the salt to the corresponding halide is
accomplished using sodium ~romide, which is then converted to the
phosphonic ester using triethyl phosphite. Hydrolysis of the
ester using trimethylsilylbromide yields the correspondinq
phosphonic acid wherein "n" is l and "m" is O.
`:
M~L~ 2
This Example makes a compound of the present invention ~y~
: stepping up the number of car~on atoms ~rom "m" is 0 to "m" is:
;~ l. The nitrile yroup oP the compound o~ Example 13 is reduced
to;~the corresponding aldehyde, which is then converted to the
corresponding alcohol. Using phosphorous tribromide the alcohol ;:
is converted to ~he corresponding alkyl bromide, which is the~n
: conYerted to the nitrile compound of the present invention
wher~in m is 1 using~p:otassium cyanide.
In this example a compound of the present invention LS~
prepared whereln "p"~ is 1 and "Y" is oxygen. The alcohol
prepared as~an intermedlate in the previous example is converted
to ~he corresponding :~diethy1 phosphonomethyl ether using~:
~ `;WO93~211~7 ~ 1 3 3 3 ~ ~ PCT/VSg3/02~1 ~
:C
diethy1chloromethyl phosphonate. Removal of ~he ethyl groups of
the ester is accomplished using trimethylsilylbromide to gi~e the
phosp~onic acid.
S~S~P~ 26
In this example a comp~und of the pre ent .invention is made
wherein "Y" is NH and "X" is SO~NH2. The nitrile group of the
compound of Example 13 is reduced to the amine using standard
catalytic hydrogenation wit~ palladium in acidic media (usually
l00.0l N to l N HCl), which is then converted to the sulfamide
uslng sulphamoyl chloride.
~MPL~ 27
: In this example a compound of the present invention is
15preparad wherein "X" is COOH and "Y" is NH by reacting the methyl
amine intermediate prepared in the previous example with
chloroacetic acid.
.
~SPIIE 2 8
;~ 20In this example a compound of the present invention is
prepared wherein "Xl' is PO(OH)z and "Y" is NH by reacting the
methyl amine intermediate prepared in Example 27 with ~.
diethylchloromethyl phosphonate, and reacti~g the resulting
p~od~uct with trimethylsiiylbromide.
~;~ 25~ :
~In this example a compound of the present invention is
:~:prepared wherein l'X7' is 5O2NH2 and "Y" is oxygen by reacting the
-21-
~ .
NO 93t21~i7 2 1 3 3 3 ~ ~ ~CrlUS93/0~84}
alcohol intermediate prepared in Example 24 with sulphamoyl
chloride.
i ' ,
P I~ 3 0 r ~
In this example a compnund of t~e present invention is
prepared wherein R1 is H, R2 is phenyl, R3 and R4 are hydrogen, m
is 0, n is 1, p is 0, a~d X is CN~ A modifiGation of the
proceduxe disclosed in Mu-Ill Lim, et al., J. Or~. Chem., VOlr
44, No. 22, 3826 (1979) is used. A mixture of the compound of
Example S and dimethylformamide dimethyl acetal is reacted at
room temperature ~or two days and then evaporated to dryness in
vacuo. The residue is crystallized to~ give the Ipure N-
(dimethylaminojmethylene deri~ative, which is cyclized with
saturated methanolic a~monia tQ give the desired end product.
~ 15
~ B~P1 31
~ -,
."~ In this example a compound of thç present in~ention is
~` prepared wherein R1 is 0C~3, R2 is phenyl, R3 and R4 are hydrogen;
m is 0, n is 1, p is 0, and X is CN. Usin~ the compound B of
; 20 Example 8, the S-methyl group is oxidized to methylsulfone, whlch
hen is converted to the final methoxy compound ~y treatment with :
sodium methoxid`e in methanol. ~ :
:
F~l~ 3 ~
; ~25 In this example a ~-compound of the pre~ent invention lS
`~ preparéd wherein X is tetrazole. The compound~of Exa~ple 13~is
~ : treated with= lithium:azide in the:presence of ammonium chloride
; : : :
. .~
2 2--
; ~
t; wo g3,21 ,87 2 ~ 3 3 ~ 4 ~ PCT/US93~2~1
as a catalyst in dimethylformamide ( DMF) at lO0 degrees C to give
t~e desired tetrazole.
.,.
In this example a compound of the present invention is
prepared wherein X is triazole. The campound of Example l9 is
treated with hydrazine hydrate to give the corresponding
hydrazide, which is then treated with imino ether to give the
desired triazole.
X~PL~_3~
The compound prepared in Example lO is tested $or en2yme
inhibition acti~ity as in Example 9. A~ 1 mM phosphate the ICso
is 0. 012 ~M and at 50 mM phosphate the ICso is 2 . 0 ~.
E~AMPL~ 35
In this example an amidine compound of the present invention
is prepared, i.e., wherein X in the recited generic formula is
CNHNH2. The compound A from Example 8 is reacted with sodium
methoxide in methanol at room temperature ~or abou~ 2 days to
~:~ give a methyl-imidate intermediate. The intermediate is then
reacted with ammonia in methanol to ~iYe the amidine product.
:, ~
-23-
` . WO g3/211~7 ~ ~ 3 3 ~ PCT/~S93/02~1
E~NPL~ 36-42
The followlng table gives the formula~ for the compounds
made in Examples 36-42 and the IC50 (nM) values obtained for
these compounds.
S ~ ~ H
--~R
CHR~
Table
No. _ Rl_ R2 - . R~ __ IC~ a
Ex~ 36 H2N 3-Chlorophenyl C~2Co2H 7
Ex. 37 H2N 3-Chlorophenyl CH2co2H (S)5-9
Exc 38 H2N 3-Chlorophenyl CH2co2H (R~160
Ex. 39 SMe 3-Chlorophenyl C~2C~ --
Ex. 40 H 3 Chlorophenyl CH2CN . 10
Ex~ 41 H2N 3-Chlorophenyl CH2CH20H 25
Ex. 42 H2N 3-Chlorophenyl CH2CO~Me 85
2,6-diamino~3,5-dihydro-7-(2-thienylmethyl)-4H-pyrollo-
[3~2-d]pyrimidine-4-one (available from ~arner-Lambert) 160
~a~PL~ 36
HN
~ H 2N
0!1
~ Cl
'~`' W093/~i87 2 1 3 ~ 3 ~ ù PCT/US93/02~1
The compound prepared in Example lO is hydrolyzed to the
cQrxesponding acid of the abo~e formula in this example. A
f ' solution of 3-~3-chlorophenyl)-3-(2-amino-4-oxo-3H,5H-
¦ pyrrolo(3,2-d~pyrimidin-~-yl)prop~nenitrile (2.0 g; 63.75 mmol)
in 6N HCl (60 ml) is heated at reflux for 8 h. T~e solvent is
evaporated in vacuo and the residue is dissol~ed in H2O (18 ml).
The resulting solution is adjusted to pH ~lO by conc. ammonium
hydroxide and any i.n~oluble material is removed by filtration.
The filtrate is then readj-~sted to pH ~6.8. The white
10 precipitated material was collected, washed with H2O, and dried
to yield 1.8 g of desired compound, m.p. 295-96-C dec, as a dl
racemic mixture.
~AMPLE 37
H~N
i ~N~<~
~ 20 C1
.
The above ~ompound, 3-(2-amino-3H,5H 4-oxo-
py~rolo[3,2-d]pyrimidin-7-yl1-3-(3-chlorophenyl-N-
:. (phenylethyl;)propanamide, is prepared in this example. A
solution of diphenylphosphoryl a~ide (O.72 g, 2.6 mmol) in D~F
(lO ml) is added dropwise during lO min to a mechanically-
stirred, cold (-5 to O'C) solution of the compound obtained in
~xample 36 t0.7~0 g; 2.4 mmol) and (R)d-~+)-~-methylbenzylamine
-25-
~::
. 093/21187 w~ 3 3 ~ ~ 5 PCT/US93/02~1
(0.32 g, 2.6 mmol) in D~F (lO0 ml). A solution of N-
methylmorpholine (0.48 g, 4.7~ mmol) in DMF (5 ml) is then added
dropwise during 5-lO min, and the solution is kept near O C for
5 h. It is then allowed to warm to room temperatur-~ and is
stirred overnight (18 h). A second portion of diphenylphosphonyl
axide (0~36 g), (R)d~ a~mQthylbenzylamine (0.16 g) and N-
methylmorpholine (O.24 g) is added at 0C and the reaction
mixture is stirred for 2 days. The solvent is removed in vacuo
and the residue is dissolved in an 8: 2 mixture of acetonitrile
and ammonium hydroxide (lM). The crude produc-t is adsorbed on
silica gel and dried in vacuo to remove the last traces of
solvent. Flash column chromatographic purification using
acetonitrile and lM ammonium hydroxide (95:5) gives the pure
desired material as a mixture of diastereomers (yield 0.630 g).
~: 15 These isomers are separated ~y repeated flash calumn
chxomatography on silica gel using acetonitrile and lM ammonium
~: hydroxide (98:2) as the eluent to yield 0~18 g of S,R-isomer
(Compound A), m.p. 170~75C dec. and 0.120 g of R,R-isome~
(Compound B), m.p. 155-60-C dec.
~NN~ ~NNz
~N--C--HzCJ~ N2C~
` ' Cl
.
-2~-
~ ` WO93/2~1~7 2 1 3 J 3 ~ s PCT/~S93/0~1
o
~I 2N
S
S-i~omer
I The above compound labeled "S-isomer," (S)-3-(2-amino-4-
ox o-3H,5H-pyrroloC3,2-d]py rimidin-7-yl) -3-(3 -
chlorophenyl)propanoic acid, is prepared in this example. A
10solution of the compound A (S,R~isomer) (0.170 g), obtained
above, in 6N HCl (30 ml) is heated at reflux for 6 h and then
le~t at room temperature for 6 h. The sol~ent is evaporated in
vacuo and the residue is dissolved in H20 (5 ml). T~e resulting
solution is adjusted to pH ~10 by conc. ammonium hydroxide~and
15any ins~luble material is removed hy flltration. The filtrate
~:; is then readjusted to pH -6.8 by ammonium hydroxide~ The white
precipitated material is collected, washed with H20, and dried to
yield 0.0~0 g o~ the crude material which was purified by flash
column chromatoyraphy usin~ a 98:2 mixture of acetonitrile~and
20ammonium hydroxide (1 M). Yield 48 mg, m~p. ~ 285~C dec.
: A purine nucleoside phosphorylase (PNP) enzyme assay is
performed in which the inhibitory activity (ICso) of the S-
; isomer compound is dete ~ ined by measuring the formation of
4C~-hypoxanthi~e from [14C]-inosine ~see Bigmedic~ne, 1980, 33~
253~) using calf spleen PNP in the presence and absence of
inhi~itor. At 50 mM phosphate the IC50 is 0.031 ~M and at 1 mM
~`~phosphate, it is 0.0059 ~.
: :
27-
` :~
:
` :~
~::
3 '~ ~
~093/~1187 PCT/US93/~2~1
E~l~ 38
o
HN
_ H2N ~
H
ao~c~2c
i R-i~om~r Cl
The procedure described in Example 37 is repeated to prepare
10 theabove compound, (R)-3-~2-amino~4~oxo-3H,5H-
pyrrolo~3,2-d]pyrimidin 7-yl)-3-(3-chlorophenyl)propanoic acid
from Compound B (R,R-isomer), obtained in Example 37. Yield 40%~
m.p. > 280 C dec. The compound prepared in Example 38 is tested
for enzyme inhibiti~n activity as in Example 37. At 50 mM
15 ph~sphate the IC50 is 0.900 ~M and at 1 mM phosphate the IC~o is
0.160 ~M. Thus the S-isomer (Example 383 .is ca. 30X as potent
as the R-isomer in the inhibition of PNP~ X ray crystalloyraphic
analysis of the enzyme-inhibitor complex ~ormed from a soak of
a crystal of the enzyme in a solution containing the unresolved
racemic mlxture (Example 36) showed that the S-isomer excluslvely
bound to the active site of the enzyme.
~3~Ph~ 3 9
The procedures of Examples 1 8 are followed, except that the
starting material used is the 3-chlorophenyl deriYative rather
than the 2,3,5-trichlorophenyl derivative used in the pre~iou5
Examples. The SMe derivative as shown in the Table is obtaine~.
-28-
.
~ 093/21~87 2 1 ~ 3 3 ~1 5 PCT/US93/0~l
~PLE 4~
The compound from Example 39 (1 g) in ethanol~(loO ml) is
suspended in in 30% palladium on carbon (1 g) and subjected to
reflux for a few minutes. Hydrazine hydrate (0.3 ml)-~s added
with stirrlng an the mixture refluxed for two days~ Additional
hydrazine hydrate (O.3 ml) and palladium on carbon (O.5 g) are
added and the mixture refluxed for an additional four days. The
catalyst is remo~ed by filtration, and the filtrate reduced to
25 ml and filtered vn Whatman filter paper and evaporated to give
the final product.
An altern~tive way of making the final product begins by
using the present Examples 1-5 except t~at the 3-chlorophenyl
derivative i5 used as the starting material. The resulting
material, 3-amino-4-~(3-chlorophenyl)methyl]methylester-1-H-
pyrolle-2-carboxylic acid, ~5 g) is disolved in dimethylformamide
dimethylacetal (50 ml) under ar~on and heated for 24 hours at 60
70 C. After e~aporation to dryness, the material is disol~ed
in dichloromethane (50 ml), filtered and diluted with patroleum
et~er until cloudy, triturated to induce crystallization, and
slowly diluted with and additional 40 ml of patroleum ether.
This mono-chloro intermediate is collected, washed with patroleum
eth~r and dried~ Yield 5 g (88%)~ mp 122-124 C. The resulting
: intermedi~te is heat~d in methanolic ammonia at 95-100 ~C for 24
hour~ in a s~ainles5 steel bomb, evaporated to a yellowish solid
crude product. The yellowish solid crude product (3 ~) in 175 ml
ho~ methano~ yiel~s a final pro~uct of 2.2 g (8B% yield~.
-29-
21 333~
, .
W093/2~1~7 PCT/US93/~2~1
E~Ph~ 41
T~e compound of Example 10 (6.80 g) in 6N HCl (400 ml) is
re~luxed for 10 h, cooled overnight, and evaporated under reduced
¦ pressure. The residue is added to methanol and evaporated and
then added to toluene, which results in a white foam in nearly
quantitative yield. A solution of the dried white foam in
an~ydrous methanol (400 ml) is cooled below O C in an ice salt
bath under dry conditionc. Thienyl chloride tlO.31 g) is added
510wly dropwisa, and the solution allowed to come to ambient
lQ temperature and stand overnight. The solvent is evaporated in
vacuo, ~resh methanol and toluene are added and then evaporated
to aid in the removal of acid vapors. A suspension of the solid
in cold water (200 ml) is neutralized in lN NaOH and the solid
is collected by filtration, washed ~ith cold water, and dried in
vacuo o~er P205 at llO-C. Yield 6.08 g (81% from the material of
Example 10)~ Thi5 product is of sufficient purity for use in.the
n~ixt step, but may recrystallize in methanol using Soxhlet
apparatus to fine white crystals having a m.p. o~ 302-303C
(dècompose~. An amount of 6g of the product from the pre~ious
paraqraph with 100 mg dry a~monium sulfate in
h~xamethyldisilazane (400 ml) is refluxed for 8 h under dry
condi~ions. The resulting clear solu~ion is evaporated in ~acuo
to a viscous gum that is further dried over P20~, which is used
in the next Example without further treatment.
~5
,~M~ 42
Under nitroqen, a s~lution of the product from the preivious
paragraph in anhydrous THF or ether (200 ml) is treated dropwise
: -3~-
'
; wo g3/211g7 213 3 3 ~1 a PCT/US93~02~1
with a 1 molar solution of lithium aluminum hydride (26 ml) in
T~F. After 1 h at room temperature, excess hydride is des~royed
by dropwise addition of ethyl acetate (50 ml), and the solvent
evaporated in vacuo. The residue is suspended in cold water (200
ml), adjusted to a pH of 1 with HCl, stirred for 15 min, adjusted
to a pH 7 with dilute sodium hydroxide, and filtered r The
resulting filter cake is wa~hed with cold water, dried, and
washed with ethyl to remove TMS by-products. Silica gel (50 g)
is added to a hot solution of the resulting crude solid (~ 8 g)
in a large volume of methanol, and tAe resulting slur~y is
evaporated to dryness. The resulting material is layered
carefully onto a silica gel column that is eluted with a
chloroform~methanol mixture (85/15) to give the desired alcohol
final product. Yield 4.65 g (84%). Two recrystallizations from
ethanol/water gives a white crystalline materiai. Yield 3036 g
(61~), m.p. 2S5-277~C (decompose).
-31-