Note: Descriptions are shown in the official language in which they were submitted.
WO 93/21518 prL~/US93/037
NITRIC OXIDE SENSOR
RELATED APPLICATION
This is a continuation of U.S. Application Serial No. 07/871,463, entitled "Nitric
Oxide Sensor,U filed April 21, 1992, by Tadeusz Malinski, incorporated herein by5 reference.
FIELD OF THE INVENTION
The present invention generally relates to sensors and sensing techniques
which can selectively and quantitatively deteet NO in solution in both biological and
chemical media. More specifically, the present invention relates to NO sensors which
10 utilize conductive catalytic materials deposited on microfibers or other supports to
monitor the presence or release of NO using amperometric, voltammetric or
coulometric methods.
BACKGROUND OF THE INVENTION
Nitric oxide (NO) has recently been shown to be a key bioregulatory molecule
1~ in a number of physiological processes. For example, NO plays a major role in the
biological activity of endothelium derived relaxing fac~or (EDRF), abnormalities in which
are associated with acute hypertension, diabetes, ischaemia and atherosclerosis. NO
is also considered a retrograde messenger in the central nervous system, appears to
be involved in the regulation of macrophage cytotoxic activity and platelet aggregation
20 inhibition, and has been implicated in endotoxic shock and genetic mutations. In
ad~ition, a number of drugs and other xenobiotics are metabolized to produce NO as
either the eflector molecule or as a harmful m~tabolite. See e.g. Furchgott, P(.F. et al.,
Natvre 288:37~376 (19803; Palmer, P~.M. et al., Nature 327:~24-526 (1987); Furchgott,
R.F., "Mechanism of Vasodilation", IV;401414 (ed. Vanhoutte, P.M.~ (Raven, NY)
25 (1988); Ignaro, LJ. et al., PNAS (USA) 84:926~9269 (1987); Wei, E.P. et al., Clr. Res.
57:781-~87 (1985); Piper, G.M. et al., J. ArT1. J. Pt~ysiolO 24:48254833 (1988);Vanbethuysen, K.M. et al., J. Clin. Invest. 79:26~-274 (1987~; Freiman, P.C. et al., Circ.
Res. 58:783-789 (1986); and Schuman, E.M. et al., Science 254:1503 (1991).
Several different methods have been employed in the past to measure NO
30 concentration in aqueous solution. For example, analysis of the ultimate aerobic
oxidation products of NO, i.e. nitrite/nitrate ~NO2-/NO3-), has been used as a measure
of NO. Monitoring of UV-vis spectral changes resulting from the conversion of
oxyhemoglobin to met-hemoglobin has also been used as an indicator of NO
concentration. These methods, however, provide only an indirect and thus less
35 accurate measurement of NO. NO has also been measured in biological systems
WO 93/21518 213 3 9 9 ~ PCr/US93/03701~
using a Thermal Energy Analyzer (TEA), in which NO reacts with ozone to produce a
characteristic chemilumineseent response. Downes, M.J. et al., Analyst 101:742-748
(1978). This approach, however, requires a lengthy regeneration time, the isolation
of NO from solution, and cannot be miniaturized for in situ monitoring of N0 release.
5 Mass spectrometry has also been attempted, but has problems similar to the TEA approach. ~
Recently, a modified oxygen electrode for thç detection of nitric oxide has alsobeen reported. Shibuki, K., Neurosa Res. 9:6~76 (1990). This electrode, however, has
a relatively large diameter (0.25 mm), a slow response time and a narrow
10 concentration range (1-3 ~.M). Although this method is advantageous in that it
discriminates against the N02- produced in the outer solution of the electrode, it is not
selective for NO in the presence of any NO2- produced in the electrode inner solution.
There is also some question of the validity of this technique owing to the small current
observed and the lack of standards done in less than ~lM concentration.
Although the above-described methods can be used to measure NC3 in
biological or chemical media, they are not sufficiently sensitive nor specific to provide
a direct and accu!ate quantitative measurement of NO, particularly at low NO
concentra~ions. Furthermore, none of the methods or sensors employed to date canrapidly and selectively measure NO release by the cell In SitU in the presence of
20 oxygen and/sr N02-. Development of this methodology is crucial in order to evaluate
endogenous NO release, distribution and reactivity on molecular ievel in biological
systems.
Thus, there exists a need for a sensitive and selective sensor for direct
quantitive measurements of N0. An optimal sensor for monitoring NO release should
25 be sturdy and capable of sufficient miniaturiza~ion for in Situ measurement in a single
cell. The senscr should also be sensitive enough to produce an adequate signal to
be observable at the low levels of N0 secreted in biological environments. Due to the
variation in the amount of NO secreted by different types of cells (e.g. from
nanomoles/106 cells in maorophages to picomoles in endothelial cells), the signal
30 produced by the sensor should also change linearly over a wide range of
concentrations. See Marletta, M.A., Trends B~ochem. Sci. 14:488492 (1989). The
short half-life of NO in biological systems, on the order of about 3 - 50 seconds, also
mandates a fast response time. See Moncada, S. et al., Pharmacol. Rev. 43:10~142(1991). The NO sensor and method of the present invention exhibit these desirable
35 characteristics.
' WO 93/21518 21 ~ 3 9 9 '¦ PCr/US93/03701
- 3 -
SUMMARY OF THE INVENTION
The NO sensor and method of the present invention provide a direct and
accurate measurement of NO in biological and chemical media. A sensor of the
present invention generally comprises an electrode having a catalytic material capable
5 of catalyzing oxidation of NO coated with a cationic exchanger. The sensor provides
a direct measurernent of NO through the redox reaction of NO - NO+ ~ e~ and is
selective for NO through the discrimination of the cationic exchanger against nitrite
(NO2~. Although the sensor can be fabricated on any scale, it can be miniaturized
to provide a microsensor which can accurately measure NO in situ at the cell level.
In one preferred embodiment of the invention, the amount of NO released from
a single cell can be selectively measured in situ by a microsensor with a response
time better than about 10 msec. The rnicrosensor comprises a thermally-sharpenedconductive carbon fiber with a tip diameter of about 0.5-0.7 ~m covered with several
layers of polymeric porphyrin capable of catalyzing NO oxidaiton with a cationicexchanger deposited thereon. Using either a two ~r three electrode arrangement, the
~nicrosensor can be operated in either the amperometric, voltammetric or coulometric
mode. The microsensor is characterized by a linear response up to abou~ 300 ~M
and a detection limit oS about 10 nM NO concentration, which allows detection of NO
rsl~ase at the levels present in a single biological cell. Th8 sensor also discrimina~es
against N02-, the most problematic interferant with current NO sensing techniques.
In other embodiments of the present invention, larger scale NO sensors are
used to measure NO concentrations in chemical media, cell culture, extracellular fluids
and tissue, rather than in single cells. For example, a carbon electrods Witll a larger
tip diameter, platinum mesh or a tin indium oxide layered plate is coated with aconductive catalytic polymeric pvrphyrin and a cationic exchanger. A linear response
and low detection limits similar to tl e NO microsensor are observed.
Other features and advantages of the present invsntion will become apparent
from the following description and appended claims, taken in conjunction with the
accompanying drawings.
BRIEF DESCRIPTION OF THE DRAWINGS
Figures lA and B depict preferred monomeric porphyrin structures used in
sensors of the present invention.
Figurc 2 is a dmerential pulse voltammogram of NO at various concentrations.
Figure 3 is a graph showing nitric oxide response (nA) of NO solutions
3~ measured by a sensor of the invention.
WO 93/21518 2 1 3 3 9 9 4 PCI/US93/03701 .
Figure 4A is a microscopic photograph of a carbon fiber microsensor of th
present invention.
Fi~ure 4B is an electron scanning micrograph of the portion of the microsensor
covered with a coat of isolating wax-resin mixture.
Figure 4C is an electron scanning micrograph of the thermally-sharpened tip
of the microsensor covered with conductive polymeric porphyrin.
Figure 5 is a scan showing the grou~h patterns~or poly-TMHPPNi, deposited
from 5 x 1 o4M TMHPPNi, 0.1 M NaOH solution by continuous scan cyclic voltammetry
on a carbon fiber microelectrode.
Figures 6A, B, C and D are voltammograms showing the response of the
rnicrosensor in the differential puise voltametric mode.
Figures 7A, B, C and D are scans showing the response of the microsensor
in the amperometric mode.
Figure 8A - C are schematic overviews of macrosensors of the present
invention used in cell culture.
Figure 9 shows the response of a platinum nlesh macrosensor to NO release
by a cell culture grown directly on the sensor surface.
DETAIL~D DESCRIPTION OF THE PREFERRED EME~ODIMENTS
The basic strategy used in the design of a pref~rred embodiment of the NO
20 sensor is based on catalytic oxidation of NO which uses a specHic potential unique
to NO - NO+ ~ e~. The normal oxidation potential for NO is about 1.0 V vs SCE ona standard platinum electrode, which potential can be lowered with various materials
capable of catalytically oxidizing NO. The current or charge generated thereby is high
enough ts be used as an analytical signal in microsystem.
In accordance with the principles of the pr~sent inv~ntion, a working electrode
of a sensor of the present comprise a conductive solid support with a catalytic surface
for NO oxidation. A catalytic surface on a conductive support can be provided using
several approaches. For example a conductive catalytic material capable of catalyzing
NO oxidation can be layered on a conductive solid support; the conductive catalytic
material can be layered on a conductive material coated on a conductive or
nonconductive base material; or the conductive catalytic material can itself comprise
the conductive support. The third approach can be accomplished by fashioning theelectrode directly from the conductive catalytic material or by incorporating or doping
a catalyst into the support material. A working electrode of a sensor of the described
- 35 embodiment of this invention preferably comprises a solid conductive support coated
~`1 WO 93/21518 21 339 9 4 PCI/US93/03701
with one or more layers of a conductive material capable of catalyzing oxidation of
NO, hereinafter referred to as catalytic material.
It will be appreciated that several types of catalytic materials can be used in
a sensor of the present invention, as long as tha catalytic material exhibits electronic,
5 ionic or redox conductivity or semiconductivity, collectively referred to herein as
conductivity. Such materials include, but are not limited to, polymeric porphyrins and
polypthalocyanines. The above-mentioned rnaterials can contain central metals,
preferably transition or amphoteric metals. Polymers which can also be used but
require doping include, tor example, polyvinylmetaliocenes (e.g. ferrocene),
10 polyacetylene doped with different metal redox centers and polypyrroline doped with
different redox centers such as, e.g. methyl viologen.
Preferred catalytic conductive materials for a sensor of the present invention
are polymeric metalloporphyrins, which are organic p-type semiconductors with
relatively high conductivity and which can be successfully deposited on a supporting
15 conductive material. Polymeric metalloporphyrins have been shown to have highcatalytic effect tor the electrochemical oxidation of several small organic and inorganic
molecules. Bennett, J.E. et al., Chem. Materials 3:49~495 (1991). Polymeric
porphyrins polymerized and copolymerized from monomeric porphyrins N,N'~i(~p-
phenylene-10,1 5,2~tri(3-methoxy-4-hydroxyphenylkorphyrin;1,1 0,-phenantroline4,7-
~0 diamine, and 5-p-(pyrole-1 -yl) phenylene-10,1 5,20-tri-(3-methoxy-4-
hydroxyphenyl)porphyrin with Fe, Mn, Co and Ni as central metals are more preferred
given their high catalytic effect for selective electrochemical oxidation of NO. Even
more preferred compounds include tetrakis(3-methoxy-4-hydroxyphenyl) porphyrin
(TMHPP) and meso-5'-O-p-phenylene-2',3'-O-isopropylidene uridine-~ri(n-methyl-4
25 pyridinium)po7phyrin (PUP), shown in Figures 1A and B.
In order to discrimina~e against NO2-, the porphyrinic catalysts used in the
present invention are also preterably covered with a thin layer of a cationic exchanger
to prevent anion diffusion to the catalytic surfac~. Suitable cationic exchangers
include AQ55D available from Kodak and Nafion. Nafion, which is used in the Specific
30 Examples, is a negatively charged cationic exchange polymer which prevents diffusion
of anions 5ike N02- to the electroactive surface of the polymeric porphyrin, but is
highly permeable to NO.
The thin layer ot polymeric porphyrin film can be electrochemically deposited,
as described in detail below, on any solid conductive support. As previously
35 discussed, a conductive support can comprise a material that in itself is conductive
W O 93/21518 PC~r/US93/03701 '~
213399 ~ 6-
or a conductive or nonconductive base material coated with a conductive material.
Conductive materials which do not need to be coated with additional conductive
materials are preferred. It will also be appreciated that, although the catalytic
component of the invention is preferably layered on a conductive support, the
5 conductive catalytic material can also compris~ the conductive s~pport. Conductive
support materials particularly suitable for sm~r scale sensors of the invention include
carbon fibers, and gold or platinum wire. ~ùe to their mechanical properties as well
as the possibility for controlled miniaturization, carbon f~bers are preferable support
materials for microsensors in single cell applications. See e.g. Malinski, T. et al. Anat.
10 Chem. Acta. 249:35-41 ~1991); Bailey, F. et al., Anlal~ Chem. 63:395-3g8 (1991).
It will be appreciated that the dimension of the sensor of the invention can be
varied to produce virtually any size sensor, including microsensors with a tip diameter
of about 1 ~m or less and macrosensors, including fibers with a larger tip diameter
(e.g. about 1 - 10 mm) and metallic mesh and conductive layered plates. Thus, while
15 Specific Examples Il-V describe the production and use of a microsensor for use in
small environments such as single cells ar syr:apses, the same techniques can beapplied to a larger support, such as described in Specific Examples I and IV, toproduce convenient macrosensors for tissue, cell culture or chemical media studies.
In measuring N0, a two or preferably thres electrode system can be employed.
20 The workin~ electrode, comprising the coated carbon fiber, with mesh or plate, is
connected to a conductive lead wire (e.g. copper) with conductive (~.9. silver) epoxy,
with the lead wir~ connecting to the voltammetric analyzer, potenffostat or coulometric
measuring instrument. The auxiliary or counterelectrode generaliy comprises a
chemically inere conductivs material such as a nobet metal (e.g. platinum wire), carbon
25 or tin indium oxide which is also connected to the measuring instrument with a lead
wire. In a three electrode system, a reference electrode, such as a standard calomel
electrode (SC~3, is also employed and connected to the measuring instrument witha third conductive lead wire.
In use, the working electrode, with the other electrode(s) in proximity, is placed
30 into the analytic solution. It will be appreciated that by "analytic solution" is meant any
aqueous or nonaqueous solution in which N0 is to be detected or measured~ The
term thus includes both chemical and biological media, including tissue fluids and
extracellular and cellular fluids. It will also be appreciated that the sensor of the
invention can be used quantitatively to detect the presence of N0 and also
35 quantitatively to measure the levels o~ N0 present in the analytic solution. To detect
~j WO 93/21518 2 1 3 3 ~ 9 ~ PCT/US93/03701
- 7 -
or measure NO release in a singie biological cell, a microsensor of appropriate
dimension can be either inserted into or placed close to the cell membrane.
The cell membrane surface concentration of NO is influenced by the following
factors: release of NO due to the action of bradykinin~ adsorption and chemisorption
5 o~ NO on the surface of the cell membrane, oxidation by 2 and organic molecules,
and diffusion into other cells and to the bulk solution. The decay in NO response
following the addition of standard amounts of oxygen was studied. Decreases of only
22% and 35% were observed a~ter 4 and 10 min respectiveiy, tollowing the addition
of 100 ~M 2 to a 20 ~lM NO solution in the absence of biological material. These
10 measurements in the presence of 2 indicate that Hs role in the oxidation of NO may
have been overestimated, and that NO oxidation is due mainly to the biological
material as was previously suggested by Monocada. See Moncada, S. et ai.,
.Pharmacol. Rev. 43:109-142 (1991).
SPECIFIC E)CANIPLE I
A carbon macroelectrode covered with cont1uctive porphyrin polymer was
prepared as ~ollows. A glassy carbon electrode ~GC~) (diameter about 2 mm) was
coated wrth conductive polymeric porphyrins by cyclic voltammetry or controlled
potential oxidation (4 min3 at 0.7 V vs SCE of the monomeric porphyrin in 0.1 M NaOH
solution (5 ml). The auxiliary electrod~ was a platinum (Pt) rod and the re~erence
20 electrode was a standard calomel electrode (SCE). The porphyrin-coated (about 0.8 -
1.5 nmlcm2) electrode was removed from the solution and stored in 0.1 M base. The
porphyrins used were Ni2+~ Co2~, or Fe3+ TMHPP or PUP as shown in Figures 1A
- and E3. The porphyrin-coated electrodes were then further coated with 4 ~l of 5%
- Nafion solution.
2~ A stock solution of saturated nitric oxide was prepared anaerobically in pH =
7.4 (0.1 M) phosphate buffer. This stock solution was then added in the correct
volume to obtain the desired final concentration of NO (10, 20 and 40 ~M). The
slectrochemical celi had a Pt rod counter electrode and SCE re~erence electrode and
the working electrode was the glassy carbon electrode coated with polymeric
30 porphyrin film and Nafion, as described above. It will, also be appreciated that a two
electrode arrangement, i.e. the working and auxiliary electrode, can be utilized.
Measurements were performed in 5 ml phosphate buffer (pH = 7.4, 0.1 M) which
served as the supporting electrolyte. All solutions were degassed prior to use and
kept under nHrogen. A base line scan was taken using linear sweep voltammetry
35 (range = 0 to +0.9 V vs SCE) or differential pulse voltammetry (range = +0.4 to 0.9
WO 93/21~18 213 ~ 9 9 ~ PCl`/US93/03701 ?e~
V vs SCE). Aliquots of the N0 stock solution were introduced to the cell via a gas-
tight syringe. The final dilution was taken as the final N0 concentration. A ~ypical N0
response by differential pulse voltammetry is shown in Figure 2 (Epa = 0.7 V vs SCE),
which shows the results using a GCE/Ni(ll)TMPP Nafion-coated (4 ~L) electrode. As
5 shown in Figure 3, the electrode gave a linear response (as did all three electrodes)
in the range of lNO] - 1 = 100 llM (0.7 V vs SCE).
SPECIFIC EXAMPLE l!
Carbon rnicrofiber conductive supports for th:e microsensor were produced by
threading an individual carbon fiber (7 ~m) through the pulled end of a capillary tube
10 with approximately 1 cm left protruding. Non-conductive epoxy was put at the
glass/fiber interface. When the epoxy that was drawn into the tip of the capillary dried,
the carbon fiber was sealed in place; The carbon flber was sharpened following
standard procedure using a microburner. See Bailey, F. et al., Anal. Chem. 63:395-
398 (1991). The sharpened fiber was immersed in melted wax-resin (5:1 ) at controlled
15 temperature 10r 5 - t5 sec. After cooling to ro~m temperature, the fiber was
sharpened again. During burning, the flame temperc~ture and the distance of the flber
from the centcr o~ the tlame need to be carefully con~rolled. While the diameter of the
sharpened lip is smaller, the tip length is larger, with the overall effect ot the resulting
electrode being a slim ~ylinder with a small diameter rather than a short taper. This
20 geometry aids in implantation and increases the active surface area. Scanningeléctron microscopy of the f~ber produced shows that the wax is burned approximately
to the top of the sharpened tip. The area of the tip, controllably fabricated with
appropriate dimensions, is the only part of the carbon fiber where electrochemical
processes can occur. A typical length of the electrochemically active tip is between
25 4 6 ~m. For the sensor to be implanted into a cell, this length must be smaller than
ths thickness of the cell. The unsharpened end of the carbon fiber was attached to
a copper wire lead with silver epoxy.
Referring now to the Figures, Figure 4A is a microscopic photograph of a
complete N0 microsensor of the present invention. Figure 4~ is an electron scanning
30 ~micrograph illustrating the part of the microsensor covered with the coat of isolating
wax-resin mixture. Figure 4C is an electron scanning micrograph of the thermally-
sharpened tip of the microsensor covered with conductive polymeric porphyrin andNafion as described below.
3 WO 93/21518 21 3 3 9 9 ~ Pcr/us93~o37ol
SPECIFIC E)CAMPLE lll
The growth patterns for poly-TMHPPNi were examined. Poly-TMHPPNi was
deposited from a solution of 0.1 M NaOH containing 5 x 104 M monomeric tetrakis(3-
methoxy-4-hydroxy-phenyl)porphyrin (TMHPP), with Ni as a central metal (TMHPPNi)by continuous scan cyclic voltammetry from 0.0 to 101 V, on a carbon fiber
microelectrode (16 ~m2 surface area), as generally described in 16. As shown in
Figure 5, peaks la and Ic correspond to the oxidation of Ni(ll) to Ni(lll) and teduction
of Ni(lll) to Ni(ll), respectively, in the film. The Ni(ll)/Ni(lll) redox couple observed at
0.5 V allows porphyrin surface coverage, (r), to be monitored (optimal r = 0.7-1.2
nmol cm2). Surface coverage is calculated ~rom the charge transferred under process
la (r = 0.8 nM/cm2). The surface coverage depends upon the initiall concentration
of TMHPPNi, electrolysis time and potential.
Following deposit on the fiber, the porphyrin film was conditioned by 5 - 10
scans from 0.4 to 0.9 V. At this stage, the electrode should be stored in 0.1 M NaOH.
1~ Sensor fabrication was completed by dipping in the Nafion solution ~5h) for 15 - 20
sec and lefl to dry (5 min) and stored in pH 7.4 buffer. Since the Ni(ll)/Ni(lll) reacffon
requires dfflusion of OH- to neutralize a charge generated in the poly-TMHPPNi and
OH- cannot diffuse through Nafion, the later absence of the Ni(ll)/l~li(lll) voltammetric
peaks in 0.1 M NaOH demonstrated the integrity of the Na~lon film coverage.
SPECIFIC EXAMPLE IV
NO monitoring was done by dfflerential pulse voltammetry using a classic three
electrode system: the sensor as the working electrode, a saturated calomel electrode
(SCE) reference electrode and a platinum (PE) wire auxiiiary electrode. The pulse
amplitude was 40 mV and the phospha~e buffer solution was pH 7.4. Differerltial pulse
25 voltammograms were obtained for oxidation of NO on poly-TMHPPNi without Na~lon
(depicted as A in Figure 6) and with Nafion (depieted as C in Figure 6~ and for 1 I~M
NO in the presence o~ 20 ~M N02- on poly-TMHPPNi without Nafion (depicted as B
in Figure 6) and with Nafion (depicted as D in Figure 6).
DPV of NO on poly-TMHPPNi without Nafion showed a peak at 0.63 V in buffer
30 pH 7.4 (see ~igure 6A). DPV of a solution of 1 ~ M NO and 20 ~.M N02- showed a
single peak at 0.80 V (see Figure 6B). The peak current was thus three times higher
than that observed at 0.63 V for NO alone. This indicated that the oxidation of NO2-
and NO occur at a similar potential, but that the current increase is not proportional
to the concentration of NO2-. The NO peak current with the Nafion-coated sensor was
3~ observed at 0.64 V (see Figure 6C). Although the observed current is lower, Nafion
W O 93/21518 Pc~r/uss3/o37ol ~ ~
213399~
coverage provides high selectivity against NO2-. Only a 1% increase in current and
no change of potential was observed for oxidation of 1 ~.M NO in the presence o~ 20
~M N02- (see Figure 6D). Thus the porphyrinic microsensor was selective for NO and
insensitive for NO2- up to a ratio of at least 1:20.
A linear relationship was observed between current and NO concentration up
to 300 ~M (r = 0.994; slope = 2.05 nA/IlM; n = 21). The response time (time tor the
signal increase ~rom 10% to 75%) in the amperometric mode was less than 10
milliseconds. The detection limit calculated at a signal/noise ration = 3 was 20 nM
~or DPV and 10 nM for the amperometric method. Since, in a volume equivalent to
that of an average singie cell (1 o~l2 L)~ about 1 o~2 attomoles (1 o~20 moles~ of NO can
be detected, the detection limit of the sensor is 24 orders of magnitude iower than
the estimated amount of NO released per single cell (1-200 attomol/cell)12~13.
SPECIFlt: EXAMPLE V
Amperometric detection of NO by the microsensor under various biological
conditions was also studied. Ring segments from porcine aorta (about 2-3 mm wide)
and porcine aorta endothelial cell culture were prepared according to previouslydescribed procedures.17 Using a computer-controlled micropositioner (0.2 mm X-Y-Z
resolution), ~he microsensor could be implanted into a single cell, or placed on the
surface o~the cell membrane, or kep~ at a controlled distance~rom the cell membrane.
Alternating cuJrent was measured in three electrode systems, as described
abovs, at constant potential of 0.75 V modulated with 4û mV pulse in time intervals
of 0.5 sec. The background, shown in Figure 7A, was measured in cell cu~ure
mediurn at 37C (DMEM-Dulbecco's Modrfled Eagie Medium, 100 mg/L D-glucose, 2
mM glutamine, 110 mg/Lsodium pyruvate, ~ 5% controlled process serum replacement25 TYPE 1). No change of the background was observed aiter the addition ~ 50 nM of
bradykinin to 5 ml of cell culture medium. As shown in Figure 7B, 2 nm of NO were
injected by microsyrlnge into the cell culture medium, a S mm distance from the
microsensor. As shown in Figure 7C, one microsensor was placed on the surface of~he single endothelial cell in the aortic ring, and another was implanted into the
30 smooth muscle cell. 2 nm of bradykinin was injected into the medium near the
endothelial cell. After 3 ~ 0.5 sec (n = 7), NO release was detected and a steady
increase of surface concentration to a plateau at 450 + 40 nM was observed after 200
sec (see Figure 7C). After 16 min, the surface concentration of NO decreased to zero.
No signfficant difference in NO surface concentration was found for the endothelial cell
35 trom cell culture (430 ~ 40 nM, n = 7). NO was detected in a single smooth muscle
d` 1 WO 93/21518 213 3 9 9 4 PCr/US93/û3701
cell within 6.0 ~ 0.5 s0c (n = 7) after injection of bradykinin and a maximum
concentration (130 ~ 10 nM, n = 7) was observed after 90 sec (see Figure 7D). The
observed current indicates that the initial concentration around the sensor is 230 nM
and decreases to 40 nM after 17 sec due mainly to depletion of NO by dfflusion and
5 also reaction with 2
SPECIFIC E)(ANIPLE Vl
Sensors utilizing a layer of tin indium oxide on a glass plate base used as
either a counterelectrode or as the conductive layer of a working electrode were also
- constructed. Figure 8A illustrates the use of a layer of indium oxide (14) as a
10 counterelectrode (10), (10), whereas Figure 8B illustrates its use as a conductive l~yer
of the working electrode of a macrosensor (12). BCH1 n-yocytes (16) were grown
under standard culture conditions at 2 x 107 celllcm2 on a glass plate (18) (Figures
8A and C) or on a plate layered with catalytic polymeric iron porphyrin with Nafion
coated thereon (20) (Figure 8B). As shown in Figures 8A and B the tin indium oxide
15 semiconductor layer in both cases was attaehed to the measuring instrument by a
copper wire lead (22) with silver epoxy (24).
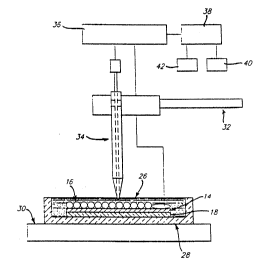
The schematic of Figure 8C depicts the set up for NO measurements of the
cell culture in Figure 8A. Celis were grown on a tin indium oxide (14) layered glass
plate ~183 placed in a Petri dish (20) with standard culture media (26). A microsensor
20 working electrode (34) constructed as described in previous Examples was then used
to measure NO release in sit~. The culture was micr3scopically monitored (30 -
inverted microscope) and the working electrode positioned with a micromanipulator
(32). As shown in Figure 8C, microsensor was atta~hed to a measuring instrument
such as a voltammetric analyzer (36) with the results fed ~o a computer (38)
25 connected to a plot~er (40) and printer (42) for result readout. NO response results
observed were on the order of those in the previously described Specific Examples.
Similar results were also obtained in cell cultures grown on fine platinum mesh and
are shown in Figure 9.
The foregoing discussion discloses and describes merely exemplary
30 embodiments of the present invention. One skiJled in the art will readily recognize
from such discussion, and from the accompanying drawings and claims, that various
changes, modifications and variations can be made therein without departing from the
spirit and scope of the invention as described herein and defined in the following
claims.
All publications clted herein are incorporated by reference.