Note: Descriptions are shown in the official language in which they were submitted.
W093/23563 PCT/EP93/01204
Loop Structures
This invention relates to loop structures at the
3'-terminal of a single strand of DNA.
It is known in molecular biology that single
stranded nucleic acids are known to fold back on
themselves and form loop structures and short double ;:.
stranded ~egments held together by hydrogen bonding.
Such secondary structures can affect the
susceptibility of the nucleic acid to interaction with
enzymes, e.g. polymera~es, hydrolases and such like. In
~, . .
some instances the formation of loop structur s by
single stranded nucleic acid will compe~e with the
hybridisation of a probe or a primer ko a target
sequence of the nucleic acid. In the ca~e where a
primer is being used, e.g. for sequencing D~A, the
formation of loops at or near the seque~ce at which the
primer hybridiæes will lower the efficiency of
hybridisation and thereby effect the clarity of
sequencing results.
The present invention is based on the concept of
usi~g PCR to introduce loop structures which provide a
primer at the 3'-terminal of a DNA strand of interest.
:DNA molecules are often present in samples in s~all
~uantities and in order to amplify such DNA, the
polymerase chain reaction (PCRj method has been
developed. In this technique a pair of polymerisation
primers specific to known se~uences of the target DNA
are selected, one hybridising at or near the 5' end of
one of the strands and the other at or near the 5' end
- of the complementary strand such that in the presence of
a polymerase, each primer produces a DNA sequence
extending the full length of the target DNA ~emplate.
If the DNA so produced is then subjected to strand
separation, typically by melting at a temperature of
W093/2356~ j6~ ~ PCT/EP93/01204
about 90C, the newly formed single stranded DNA
sequences will hybridise to excess primer present in the
mixture, usually after reducing the temperature to the
range suitable for annealing, whereupon in the presence
of the polymerase, further DNA strands are synthesised,
this time extending only between the termini of the two
primers. The polymerase is preferably capable of
surviving the hig~ temperature used in the strand ~`
separation step, a suitable thermophilic polymerase, ~:
namely Taq, having recently become available. If an
excess of the two primers and of nucleotides needed for
DNA synthesis is maintained in the medium, it is
possible to operate a repeated cyclic process in which
.the separate strands are synthesised, separated,
15 annealed to primer and new strands synthesised, ~erely :
by raising and lowering the temperature between the
optimal temperature~ for each of the above ~tages. In
this way, it is found that amplificakion o~ the original
target DNA can be exponentia1 and million-fold increases
20 of concentration can be effected in a relati~ely short ::
tîme. Such amplified DNA may then be sequenced or
otherwise investigated. `-
The invention provides a method of introducing a
3'-terminal loop structure onto a target sequence of one
strand of double stranded DNA, said target sequence
having a region A at the 3'-terminùs thereof and there
: being optionally a DNA region B which extends 3' from
region A, whereby sai,d double-stranded DNA is subjected
to polymerase chain reaction (PCR) amplification using a ~:
first primer hybridising to the 3'-terminus of the
sequence complementary to the target sequence, which
first primer is immobilised or provided with means for
attachment to:a solid support, and a second primer
having a 3'-terminal sequence which hybridises to at
least a portion of A and/or B of the target sequence
while having at its 5'-end a sequence substantially
identical to A, said amplification producing double-
W093/23563 7 ~ ~ J ~ ~ PCT/EPg3/01204
-- 3
stranded target DNA having at the 3~-end of the target
sequence, in the following order, the region A, a region
capable of forming a loop and a se~uence A'
complementary to sequence A, whereafter the amplified
double-stranded DNA is subjected in immobilised form to
strand separation whereby the non-immobilised target
strand is liberated and region A' is permitted or caused
to hybridise to region A, thereby forming said loop. ~`
It will be understood that the region capable of
10 forming a loop comprises all or part of region B (if ;.
present) and/or a sequence complementary to an optional
loop-forming linker sequence in the second primer
.~. between region A~ and A. If the second primer
~ybridises to region A on the target sequence it will be
highly desirable that a lvop-forming linker seguence is
present in the primer in order to allow substantially
*ull hybridisation of A' to A in loop formation. If the
second primer hybridises to a part of regi~n B remote
from region A on the target sequence, the the portion of
B between region A and the region of hybridisation will
~ form the loop together with the region of hybridisation
: and any loop-forming linker sequence (if present) in the
primer.
Although the second primer has a sequence A'-B (if
: 25 present) and/or a loop-forming linker sequence-A and
there is the possibility of the primer looping backing
: on itself (rather than acting as a primer), this
possibility of "self priming" is avoided by selecting a
higher annealing temperature than that at which the
primer preferentially anneals to itself.
It will be appreciated that both the immobilised
strand and the non-immobilised strand will be capable of
forming loop structures. However, only the immobilised
strand will have the sequence A' in the riqht
orientation to serve as a primer for chain extension.
Further, it will be noted that the convention of raading
sequences in a 5' to 3' direction has been followed.
W093~2356~ 6 4 - PCT/EP93/01204
Thus the sequence of the primer substantially identical ~-
to A is substantially identical reading in a 5' to 3' :~
direction. ~-
An advantage of this invention is that a primer is
incorporated into the 3' end of a strand of DNA and the
primer can then be used, for example, in sequencing the
strand or other procedures. It is clear that each
immobilised template will have a built-in primer and
that the primer will be kept in relatively close ~.
lO proximity to the region where it hybridises by virtue of :`
the linker sequence. Thus, even if conditions are
~aried such that strand separation occurs, the primer,
: being joined to the template, will readily re-hybridise.
If desired, it is possible to form an restriction
endonuclease (RE) site close to the loop so that it may
be removed, e.g. to allow strand separation during .
sequencing. This arrangement will be desirable, for
example when the sequencing is by the Sanger method
(e.g. S~nger F. et al 51977) PNAS (USA~ 74- 5463-5467)
and lengths of newly formed dideoxynucleotide terminated
strands need to be separated from target DNA which has
served as a template. Suitable RE sites and their mode
of incorporation into a primer will be known to the
skilled worker and are taught by the literature, for
example Molecular Cloning: a laboratory manual by T.
Maniatis et al. For example, a linker sequence in the
second primer may contain complementary palindromic
sequences ad7acent A and A' respectively so that on loop
formation these adjacent regions hybridise to ~orm an RE
site in the double stranded portion which site allows
for removal of the loop. Alternatively, region A A' may
be chosen such that it contains an RE site.
Advantageously the present invention can be
combined with the invention taught in our co-pending
application of even date entitled "Chemical Method~'
(Agents ref.: 75.S7465).
our co-pending case is b~sed on the concept of
W093/23563 `;~ PCT/EP93/012U4
amplifying and then immobilising the DNA of interest
followed by a p~l~merase reaction carried out on four
aliquots of the immobilised DNA in single stranded form.
Each aliquot uses the same specific extension primer and
5 a different dideoxynucleotide but no deoxynucleotides so -~
that only the dideoxynucleotide complementary to the :::
base in the target position is incorporated; the target
position being directly adjacent to the 3' end of the
specific extension primer hybridising to the DNA. Put
another way, the target position on the immobilised
strand is immediat~ly 5' of where the extension primer
hy~ridises to the DNA. Chain extension using normal
. deoxynucleotides is then effected using the specific
primer so that the dideoxy-blocked DNA will remain
I5- unreacted while the un-blocked DNA will form double
stranded DNA. Various methods may then be used to
distinguish double stranded DNA from non-extended, i.e.
substantial~y single stranded DNA, and ~hus enable the
base in the target position to be iden ~.fied.
Preferably, the DNA of interest is amplified by PCR.
:A primer is normally added to each of the aliquots
: for the dideoxy and extension reactions. However, using
the present invention, the target DNA may be provided
with a loop-attached primer which, as indicated above is
stable to variations in conditions and substantially
reduces or elimina ~s losses in passing from the dideoxy
reaction to chain extension.
Specifically: the invention taught in QUr co-pending
: application may be modified in accordance with the
present invention so that sample DNA is provided with a
loop-attached 3' primer which hybridises to the
immobilised DNA immediately adjacent to the target
position; each of four aliquots of the immobilised
single stranded DNA is then subjected to a polymerase
reaction in the presence of a dideoxynucleotide, each
aliguot using a ~ifferent dideoxynucleotide whereby only
the dideox~nucleotide compleme~tary to the base in the
W O 93/23563 ? ~ 6 PC~r/EP93/01204
~ - 6 -
target position becomes incorporated; the four aliquots
are then subjected to extension in the pres2nce of all
four deoxynucleotides, whereby in each al iquot the DNA -
which has not reacted with the dideoxynucleotide is
5 extended to form double stranded DNA while the dideoxy-
blocked DNA remains as non-extended stranded DNA;
followed by identification of the double stranded and/or
non-extended DNA to indicate which dideoxynucleotide was
incorporated and hence which base was present in the
target position.
It is desirable that the effectiveness of the PCR
is assessed, e . g . to determine whether or not su f f icient
DNA has been formed to give clear results with a
relatively low level of background. Various tests are
15 known in the art but we prefer to use the solid phase
approach we described earlier for detection of
immobilized amplified nucleic acids, designated DIANA
(PCT/EP90/00454), which has been used for example in its
preferred embodiment in the colorimetric det~ction of in
v tro amplified DNA. The assay is based on the use of a
biotin~lated or otherwise functionalised PCR primer,
which is used to capture in vitro amplified material on,
f or example ~ streptavidin-coated magnetic beads. The
other PCR primer contains a "handle", such as a lac
operator sequence, allowing colorimetric detection of
the captured ~NA using a LacI repressor-~-galactosidase
: fusion protein. (Wahlberg, J., Lundeberg, J., Hultman,
T. and Uhlen, M. (1990) "General colorimetric method
for.DNA diagnostics aIlowing direct solid-phase genomic
se~uencing of the positive samples." Proc~ Natl. Acad.
Sci U.S.A. 87, 6569-6573). The preferred form of the
qualitative DIANA assay combines the advantages of the
: PC~ method with the high specif icity and stability of
the biotin-streptavidin system and the simplicity of a
colorimetric detection based on ~-galactosidase. The
strong interaction between biotin and str~ptavidin
(Kd~10-15 M-1) accentuates the efficiency of the system.
WO 93/23563 ~ r~ PCT/EP93/01204
~ 7 --
The magnetic beads as solid support ensure that no
centrifugations, filtrations or precipitations are
needed (T. Hultman, S. Stahl, E. Hornes and M. Uhlén
Nucl. Acids Res. 17 ~ 4937 (1989) ) ~
A number of proteins are known which bind to
specific DNA sequences and are often involved in genetic
processes such as switching operons on and off. One
such protein is the lac repressor ~acI which reacts with
the lac operator (lacOP) to inhibit transcription.
Thus, if the recognition site is the DNA sequence lacOP,
the label can be attached via the protein LacI. It is
particularly convenient to devise a fusion protein of a
DNA binding protein such as LacI with a further protein
which can be subsequently used for detection for example
15 using methods based on colour fluorescence or ..
chemiluminescence. Examples of such proteins are
galactosidase, alkaline phosphatase and peroxidase.
It is preferred to use as a label a LacI repressor-
~-galactosidase fusion protein which recognises a 21
base pair lac operator se~uence introduced at the end of
the amplified DNA, by one of the primers, preferably the
immobilised primer. The fusio~ protein ~ill bind to the
lac OP sequence of the DNA and the addition of ONPG
~ortho-nitropheny~-~-D~galactoside) will lead to a
colour formation which can be assessed
spectrophotometrically. Use of this fusion protein and
ONPG allows for a fast simple colorimetric assay which
does not have the safety problems associated with using
radiolabels. IPTG (n-isopropyl ~-D-
thiogalactopyranoside~, for example, can be added torelease the fusion protein from the DNA.
The specificity of the process is greatly increased
by including a first-stage PCR amplification step. By
such preliminary amplification, the concentration of
target DNA is greatly increased with respect to other
DNA which may be present in the sample and a second-
stage amplification with at least one primer specific to
W093~23563 -~ ~ 6 PCT/EP93/01204
~ 8 -
a different sequence of the target DNA, as described in
PCT/EP 90/00454, significantly enhances the signal due
to the target DNA relative to the 'background noise'.
Two-stage PCR (using nested primers), as described
in our co-pending application PCT/EP90/00454, may be
used to enhance the signal.to noise ratio and thereby
increase the sensitivity of the method according to the
invention~
Immobilisation of the amplified DNA may either take
place as part of the PCR amplification itself, as where
one or more primers are attached to a support, or
alternatively one or more of the primers may carry a
~: functional group permitting subse~uent immobilisation,
eg. a biotin or thiol group. Immobilisation by the S'
end of the primer allows the strand of DNA emanating
from that primer to be attached to a solid support and
have its 3' end r~mote from the support and available
for subsequent chain extension by polymerase.
The solid support may conveniently take ~he form of
microtitre wells, w~ich are advantageously in the
conventional ~ x 12 fonmat, or dipsticks which may be
made of polystyrene activated to bind the primer DNA (K
Almer, Doctoral Theses, Royal Institute of Technology,
Stockholm, Sweden, 1988). The support may also comprise
particles, ~ibres or capillaries made, for example, of
agarose, cellulose, alginate, Teflon or polystyrene.
The support may also comprise magnetic particles eg the
superparamagnetic beads produced by Dynal AS (0510,
Norway).
The solid support may carry functional groups such
as hydroxyl, carboxyl, aldehyde or amino groups, or
other moieties such as avidin or strepta~idin, for the
attachment of primers. These m~y in general be provided
by treating the support to provide a surface coating of
a polymer carrying one of such f~nctional groups, e.g.
polyurethane together with a polyglycol to provide
hydroxyl groups, or a cellulose derivative to provide
W093/23563 ~ PCT/EP93/01204
hydroxyl groups, a polymer or copolymer of acrylic acid
or methacrylic acid to provide carboxyl groups or an
aminoalkylated polymer to provide amino groups. US
Patent No. 4654267 describes the introduction of many
such surface coatings.
Any suitable polymerase may be used, although it is
preferred to use a thermophilic enzyme such as Taq
polymerase to permit the repeated temperature cycling
without having to add further polymerase, e.g. Klenow
lO ragment, in each cycle. `
The target DNA may be cDNA synthesised from RNA in
the sample and the method of the invention is thus
` applicable to diagnosis on the basis of characteristic
RNA. Such preliminary synthesis can be carried out by a
preliminary treatment with a reverse transcripta~e,
conveniently in the same system of buffers and bases to
be used in the subsequent PCR steps. Since the PCR
procedure requires heating to effect strand separation,
the reverse transcriptase will be inactivated in the
first PCR cycle. When mRNA is the sample nucleic acid,
it may be advantageous to submit the initial sample,
e.g. a serum sample, to treatment with an immobilised
polydT oligonucleotide in order to retrieve all mRNA via
th~ terminal polyA sequences thereof. Alternatively, a
specific oligonucleotide sequence ma~ be used to
retrieve the RNA via a specific RNA sequence. The `
oligonucleotide can then serve as a primer for cDNA
synthesis, as described in International Patent
Application PCT/89EP/00304.
PCR has been discussed above as a preferred method ~.
of initially amplifying target ~NA although the skilled
person will appreciate that other methods may be used
instead of in combination with PCR. A recent :.
developmènt in amplification techniques which does not ~:.
require temperature cycling or use of a thermostable
polymerase is Self Sustained Sequence Replication (3SR).
3SR is modelled on retroviral replication and may be
W093/23563 ,~ Q ~ PCT/EPg3t~1204
used for amplification (see for example Gingeras, T.R.
et al PNAS (USA) 87:1~74-1878 and Gingeras, T.R. et al
PCR Methods and Applications VOL. 1, PP 25-33).
Advantageously, the primers are sufficiently large
5 to provide appropriate levels of hybridisation, yet ~:
still reasonably short in order to avoid unnecessary
chemical synthesis. It will be clear to persons skilled
in the art that the size of the primers and the
stability of hybridisation will be dependent to some
degree on the ratio of A-T to C-G base pairings t since
more hydrogen bonding is available in a C-~ pairing.
Also, the skilled person will consider khe degree of
homology between the extension primer to other parts of
the amplified sequence and choose the degree of
stringency accordingly. Guidance for such routine
çxperimentation can be found in the literature, for
example, ~olecular Cloning: a laboratory manual by
Sambrook, J,, Fritsch~ E.F., and Maniatis, T (1989).
The polymerase reaction in the presence of dideoxy
nucleotides is carried out using a polymerase which will
: incorporate dideoxynucleotides, e.g. T7 polymerase,
Klenow or Sequenase Ver. 2.0 (USB U.S.A.~. However, it
is known that many polymerases have a proof-reading or
error checking ability and that 3' ends available for
chain extens.ion are sometimes digested by one or more
: nucleotides. If such digestion occurs in the method
according to the invention the level of background noise
increases. In order to avoid this problem it is
desirable to add to each aliquot fluoride ions or
nu~leotide monophospha~es which suppress 3' digestion by
polymerase.
Identification of the double stranded and/or non-
extended ~NA is possible via a variety of means. With
regard to the double stranded DNA, conventional
techniques such as radiolabel incorporation during chain
extension are possible but it is preferred to use the
lac operator sequence which is preferably incorporated
W0~3/23563 ~ 5 PCT/EP93/01204
into the DNA during amplification, as discussed above.
Full chain extension creates the double stranded DNA
se~uence which is bound by the lac I repressor-
~galactosidase fusion protein. Bound fusion protein can
then be identified colorimetrically as discussed above
and this identifies the three aliquots which have been
extended, thereby identifyi~g the di~eoxy base which was
added in the remaxning aliquot~
With regard to the non-extended DNA, where
extension of the loop-attached 3' primer was blocked by
a dideoxynucleotide~ again a number of means for
identification are possible and will be readily apparent
to the skilled person. Preferably, a probe which
hybridises downstream of the 3' end of the loop~attached
primer is used, i.e. the probe hybridises to the
immobilised strand between 5' end of immobilisation and
the 3' loop struct~re. The probe is suitab~y labelled
or provided with means for attaching a label. Such a
probe will bind to the single strand D~A but will not
bind to the double ~tranded DNA.
If desiredt both double and single stranded DNA can
be identified and this provides additional checking for ~;
the accuracy of the results. It will usually be
desirable to run a control with no dideoxynucleotides .
and a 'zero control' containing a mixture of all four
dideoxynucleotides.
Another means of identification is that disclosed
in our co-pending application of even date tAgents ref.:
75.57799) which relates to detection of pyrophosphate
released during chain extension. When each nucleotide
is incorporated a pyrophosphate group is split off the
nucleotide triphosphate and the remaining nucleotide
monophosphate is incorporated at the end of the growing
nucleic acid chain. In those aliquots which have not
35 incorporated a chain terminating dideoxynucleotide there -
is extensive pyrophosphate release during chain
extension. This release of pyrophosphate can be
WO 93/23~63 ~ r~ 6 - 12 - PCT/EP93/01204
measured using luciferin and luciferase which emit light
in substantially direct proportion to the amount of
pyrophosphate present.
In many diagnostic applications, for example
genetic testing ~or carriers of inherited disease, the
sample will contain heterozygous material, that is half
the ~NA will have one nucleotide at the target position
and the other half wil~ have another nucleotide. Thus
of the four aliquots used in the method of the
invention, two will show a positive signal and two will
show half the positive signal. It will be seen
there~ore that it is desirable to quantitatively
determine the amount of label detected in each sample.
In the case of a homozygous sample it will be clear that
there will be three negatives and one positive signal of
the four aliquots.
The invention will now be described by way of a
non-limiting example with reference to the drawings in
which:
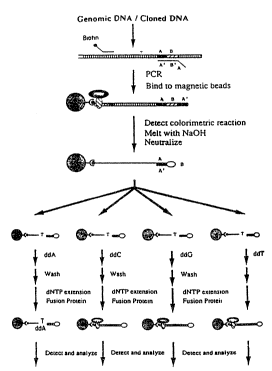
: 20 Fig.l shows a protocol for identifying a base in a
single target position using the method according to the
: invention;
. ~
~igs.2 and 3 show oligonucleotide primers used in
Example l; and
:25 ~ Fig.4 is a graph showing the results obtained in the
Example.
.~
MATERIALS AND METHODS
,
Bacterial strains and enzymes~ Escherichîa coli RRI ~Ml5
(Ruther, U(l982~, Nucl. Acids Res., lO 5765-5772) was used
as bacterial host. The plasmid vec~or used was pRIT 28
(Hultman, T., Stahl, S., Moks, T. and Uhlén, M. ~1988)
~i'Approaches to solid phase DNA sequencing", Nucleosides and
: 35 Nucleotides 7 629-638~. Restriction endonucleases, DNA
polymerase I (Klenow Fragment), T7 DNA Polymerase, CIP and
T4 polynucleotide Kinase were obtained from Pharmacia,
W093/23563 2~ 3 ;i ~ ~ g PC~/EP93/01204
- 13 -
Sweden. The Taa DNA polymerase used was purchased from
Perkin-Elmer, Ca., U.S.A. (AmpliTaq).
Example 1
Synthesis of oligonucleotides. 5 oligonucleotide
primers ~See figures~ RIT 321, RIT 322, RIT 331, RIT ~33
and RIT 338, complementary to regions encoding a part of
the active site of the HIV reverse transcriptase gene
(RT) (bases 625 to 1165 Myers, G., Korber, B.,
Berkovsky, J.A. Smith, R.F. and Pavlakis, G.N. Human
Retroviruses and AIDS l991 (Los Alamos National
Laboratory, New Mexico l991)), were synthesized by
phosphoramidite chemistry on an automated DNA synthesis
apparatus (Gene Assembler Plus, KABI-Pharmacia, Sweden)
as described by the manufacturer. RIT322 was
biotinylated by using a biotin phophoramidite
(Clonetech, Ca, U~S.A.). Purification was performed on
a pepRPC 5/5 re~ersed phase colum~ (KABI Pharmacia,
Sweden3.
.'
PCR cloning
The HIV RT fragment was cloned by amplification
from a clinical sample obtained from a patient with HIV-
l (Swedish Bacteriol~gy Laboratory, SBL, Stockholm~
Sweden) using 5 pmol each of the oligonucleotides RIT331 ;~
and RIT333 (figure 3~ both containing "handles" in order `~
to introduce an upstream Bam HI and a downstream Eco RI
recognition sites. The PCR reaction mix contained 200
~M dNTPs, 20 mM Tris-HCl (pH 8.7), 2 mM MgCl2, 0.1% Tween ~-
20 and 0.5 units AmpliTaq resulting in a final volume of -~
50 ~l. The temperature profile was set up by a
denaturation step at 95C for 0.5 min. followed by a
primer annealing step at 55C for 0.5 min. and a final
extension step at 72C for 2 mins. These steps were
repeated 30 times using a Gene Amp PCR System, PE 9600
SlJE~iTlTVTE S~E~T
W093/23563 7 ~ ;fi~ 14 - PCT/EP93/01204
(Perkin Elmer, Ca., U~S.A.). The PCR amplified HIV RT
fragment and the pRIT 2~ vector were both restricted
with Bam ~1 and Eco Rl, cut out and purified from
agarose and then iigated for 1 nour in room temperature.
The construction was transformed into competent
RRIQM15cells and spread on TBAB (Sambrook, J. et al loc.
Cit. ) plates containing IPTG(n-iso,propyl-~-D-
thiogalactopyranoside) X-gal (5-bromo-4-chloro-3-
indolyl-~-D-galactoside) and ampicillin allowing
blue/white selection ~Langley, E.K. et al (1975~ PNAS
~USA) 72, 1254-1257). Fi~e white colonies containing
the plasmid with a correct insert was confirmed by solid
, phase sequencing (Hultman, T. et al ~1991) Biotechniques
10, 84-93). one of those clones was designated pRIT-RT
and choosen for further studies. This clone is stor~d at
the Department of Biochemistry, Royal Institute of
Technology, Stockholm, Sweden.
Template preparation for DIANA detected Mini Sequencing
A colony harbouring pRIT28-RT was transferred to a .:
: vial and 1~,7sed at 99~C for 5 min. in 10~1 20 mM Tris-HCl ,~-
(pH 8.7). 1 ~1 lysate was subsequently transferred to a
PCR mixture of 5 pmol RIT135 and RIT322 (biotinylated),
0.25 pmol RIT321, 200 ~M dNTPs, 20 mM Tris-HCl (pH 8.7),
2 mM MgCl2, 0.1% Tween 20 and 0.5 units AmpliTaq to a'
f:inal volume of 50 ~1. It will be noted that primer
RIT322 compris~es a 5' Biotin, for subsequent attachment
to a streptavidin coated solid support, and the 21 bases
which define the lac Op recognition sequence.
Amplification was performed as above and the resulting
PCR product was subsequently immobilized (Hultman, T. e5
al ~1989) Nuc. Acids Res. 17, 4937-4946) on prewashed
streptavidin coated paramagnetic beads, (Lea, T. et al
(1988) J. Mol. Recognit 1, 9-18) Dynabeads M280-
Streptav din (Dynal AS, Norway~, prewashed with binding
solution according to manufacturer. After
Sa,el~3TlT~TE SHEET
W093/23563 ~ 3a~ PCT/EP93/01204
immobilization, the beads were rinsed with 50 ~l
bindiny-washing solution and assayed for bound DNA. The
beads with the immobilized DNA were mixed with 50 ~l o~
the fusion protein, lacI-~-galactosidase (Dynal AS,
Norway), and incubated for 20 minutes. Excess of the
fusion protein was removed by washing the beads 4 times
with DIANA buffer (Dynal AS, Nsrway) and changing to new
tubes in the last step in order to avoid background due
to coating of the walls. l00 ~l of chro~ogenic
substrate, ortho-nitrophenyl-~-D-galactoside ~ONPG, l.25
mg/ml), was added and after 6 min. the reaction was .;
stopped by an addition of l00 ~l lM Na2CO3 and the
supernatant was analyzed in an EAR340AT ELISA plate ;
. reader (5~T-Labinstruments, Austria) by measuring the
absorbence at 405 nm. The strands were separated by
melting by incubation with 20 ~l 0.l M NaOH fQr 5 min. .-
generating single stranded immobilized DNA template,
which was once again washed with 50 ~l binding solution,
50 ~l l x TE.
Mini Sequencing reactions .
' ''''
Six separate extension reactions with respect to ~;
the appropr}ate dideoxy nucleotide were set up (one with.'i~;
25 only ddATP, one with only ddCTP, one with only ddGTP, ;:~
one with only ddTTP, one with all four ddNTPS present
and one without any of dd~TPs3 in a ~otal of lO ~
containing 2 ~l of the annealing mixture, 17 mM Tris-HCl
(pH7.5), 6 mM MgCl2, 1 mM DTT, 1 ~M of the appropriate
dideoxy nucleotide and 0.13 units of Sequenase ver. 2.
A schematic outline of the experiment is shown in figure
l. The dideoxy incorporation was performed at room
~emperature for 5 mins. and stopped by adding 20 ~l 0.5M
EDTA. Thereafter the beads were washed twice with 30 ~l
l0 mM Tris-HCl (pH 7.5). In the following extension
step 200 ~M dNTP concentration was used together with 25
mM Tris-HCl (pH 7.5), 12.5 mM MgCl2, 1 mM DDT and 0.13
S~35T3~5TE ~i~lEET
WOg3/23563 ~ 6 ~ ~ - 16 - PCT/EP93/01204
units Sequenase in a total of 10 ~1. In the aliquots
where a dideoxy nucleotide had not been incorporated,
the Sequenase leads to a chain extension and to full
double stranded DNA being attached to the beads. After
a 5 min. incubation in room temperature 20 ~1 0.5 M EDTA
was added and the beads were washed with 40 ~1 DIANA
buffer (Dynal AS. Norwa~) (O.l M Tris-HCl tpH 7.5), 0.15
M NaCl, O.1~ Tween 20, 1 mM MgC12 and 10 mM ~- :
mercaptoethanol).
Detection by DIANA
. .
The results were detected by DIANA ~Wahlberg, J., -:
. Lundeberg, J., Hultman, T. and Uhlén, M. (1990)
15 "General colorimetric method for DNA diagnostics ~:~
allowing direct solid-phase genomic sequencing of the
positive samples." Proc. Natl. Acad. Sci U.S.A. 87,
6569-6573). The beads with the immobilized DNA were
mixed with 50 ~1 of the fusion protein, lacI-~-
galactosidase (Dynal AS, Norway), and incubated for 20
minutes. Excess of the fusion protein was remov~d by
washing the beads 4 times with DIANA buf~er (Dynal AS,
Norway) and changing to new tubes in the last step in
order to avoid background due to coating of the walls.
100 ~1 of chromogenic substrate, ortho-nitrophenyl-~-D-
galactoside (ONPG, 1.25 mg/ml), was added and after 6
: : min. the reaction was stopped by an addition of 100 ~1
lM Na2CO3 and the supernatant was analyzed in an EAR340AT
ELISA plate reader (SLT-Labinstruments, Austria) by
measuring the absorbence at 405 nm. The results are
shown in figure 4. The assay show that a low signal is
obtained when all four dideoxynucleotides ~ddNTP) are
used as well as when only ddATP is used. Since the
complementary base next to the 3'-end of the sequencing
primer is a dideoxythymidine, the result demonstrates
that the assay can be used to detect a base sequence at
a speci~ic point.
S~E3~Tllr~T~ 5~EET