Note: Descriptions are shown in the official language in which they were submitted.
CA 02136350 2001-09-10
75154-6
1
Xylanases From Trichoderma Reesei and
Methods for their Production
Field of the Invention
The present invention is related to enzyme preparations with unique
enzyme profiles. Methods for the production of such enzyme preparations
by genetically engineering members of the species Trichoderma are
disclosed. These preparations contain high levels of xylanase enzymes and
are especially useful in the pulp and paper industries.
Background of the Invention
r- Cellulose is a linear polysaccharide of glucose residues connected by
0-1,4 linkages. In nature, cellulose is usually associated with lignin
together with hemicelluloses such as xylans and glucomannans. In the
pulp and paper industry, in chemical pulping (cooking) of the wood, the
major part of the lignin is extracted to get acceptable cellulose pulp
product. However, the resulting pulp is brown, mainly because of the
small portion of the lignin still remaining in the pulp after cooking. This
residual lignin is traditionally removed in a multi-stage bleaching procedure
using typically a combination of chlorine chemicals and extraction stages.
Peroxide, oxygen and ozone are also used when the use of the chlorine
chemicals is wanted to be reduced or avoided totally.
Hemicellulases can be used in enzyme-aided bleaching of pulps to
decrease chemical dosage in subsequent bleaching or to increase brightness
of the pulp (Kantelinen et al., International Pulp Bleaching Conference,
WO 93/24621 PCT/F193/00221
2136350
2
Tappi Proceedings, 1-5 (1988); Viikari et al., Paper and Timber 7:384-389
(1991); and Kantelinen et al.,"Enzymes in bleaching of kraft pulp,"
Dissertation for the degree of Doctor of Technology, Technical Research
Centre of Finland, VTT Publications 114, Espoo, 1992). Naturally, in this
use, the hemicellulose should be free of cellulases, which would harm the
cellulose fibers.
The use of hemicellulose hydrolyzing enzymes in different bleaching
sequences is known from WO 89/08738, EP 383,999, WO 91/02791, EP
395,792, EP 386,888 and WO 91/05908.
Other industrial applications for hemicellulolytic enzymes are in the
production of thermomechanical pulps, where the aim of the use of
hemicellulolytic enzymes is decreased energy consumption.
Hemicellulolytic enzymes can be used to improve drainage of recycled
pulp or in the production of dissolving pulps (Viikari et al.,
"Hemicellulases for Industrial Applications," In: Bioconversion of Forest
and Agricultural Wastes, Saddler, J., ed., CAB International, USA (1993)).
The use of hemicellulolytic enzymes for improved water removal
from mechanical pulp is known from EP 262,040, EP 334,739 and EP
351,655 and DE 4,000,558).
When the hydrolysis of biomass to liquid fuels or chemicals is
considered, the conversion of both cellulose and hemicellulose is essential
to obtain a high yield (Viikari et al., "Hemicellulases for Industrial
Applications," In: Bioconversion of Forest and Agricultural Wastes,
Saddler, J., ed., CAB International, USA (1993)).
Also, in the feed industry, there is a need to use a suitable
combination of enzyme activities to degrade the high 0-glucan and
hemicellulose containing substrate.
To make the use of hemicellulolytic enzymes an economically realistic
possibility in different applications, the production costs of the
enzyme must be lowered. This means that the production levels must be
high and no expensive purification steps can be included. The most
SUBSTITUTE SHEET
"-*'VO 93/24621 2136350 PCT/F193/00221
3
economical way would be to choose the production strains and conditions
in such a way that the culture supematant would be rich in required
activities and the side activities would be only minor components.
Consequently, there is a clear demand for enzyme preparations,
which contain unique enzyme profiles, taylor-made, that is, designed
spesifically for the purposes of the industry in which they are to be used,
and which can be obtained in a cost-effective manner, such as, for
example, directly from the culture medium of the microorganism which
has been modified so that it produces the desired enzymes, but not
appreciable quantities of undesired enzymes.
Xylans are complex heteropolymers mainly consisting of xylose and
arabinose. Xylans have a backbone consisting of P-1,4-linked
xylopyranose units, which may be substituted with acetyl residues and
residues of arabinose and niethyl glucuronic acid (Timell, T.E., et al.,
Wood Sci. Technol. 1:45-70 (1967)). Xylans are, after cellulose, the
second most abundant carbohydrate fraction of plant biomass. A number of
enzymes are needed for complete hydrolysis of xylan, the most important
ones belonging t o-rhe group of endo-j3-xylanases (Biely, P., Trends
Biotechno13:286-290 (1985); Dekker, R.F.H., in Hignehi, T., ed.,
Biosynthesis and biodegradation of wood components (Academic Press
Inc., Orlando), pp. 505-533 (1985); Woodward, J., Top Enzyme Ferment.
Biotechnol. 8:9-30 (1984)).
Various microorganisms are capable of degrading xylans, and
xylanases have been found in both prokaryotes and eukaryotes (Dekker,
RF.H., Richards, G.N., Adv. Carbohydrate Chem. Biochem. 32:277-352
(1976)). Xylanolytic micro-organisms often produce multiple xylanases to
attack the different bonds in these molecules. In a recent review,
Bastawde (Bastawde, K.B., World J. Microbiol. Biotechnol. 8:353-368
(1992)) presented a compilation of the current knowledge about the
properties and modes of action of microbial endoxylanases. There are
several reports on the molecular cloning of these enzymes from bacteria
SUBSTITUTE SHEET
CA 02136350 2005-10-06
75154-6
4
(e.g. Ghangas, G. S. et al., J. Bacteriol. 171:2963-2969
(1989); Lin, L.-L., Thomson, J. A., Mol. Gen. Genet.
228:55-61 (1991); Shareck, F. et al., Gene 107:75-82 (1991);
Scheirlinck, T. et al., Appl Microbiol Biotechnol.
33:534-541 (1990); Whitehead, T. R., Lee, D. A., Curr.
Microbiol. 23:15-19 (1991)) but few from fungi (Boucher, F.
et al., Nucleic Acids Res. 16:9874 (1988); Ito, K. et al.,
Biosci. Biotec. Biochem. 56:906-912 (1992); Maat, J. et al.,
in Visser, J. et al., eds., Xylans and Xylanases (Elsevier
Science, Amsterdam), pp. 349-360 (1992); van den Broeck, H.
et al., "Cloning and expression of xylanase genes from
fungal origin", EP 463,706 Al (1992)).
Trichoderma reesei is an efficient producer of
cellulolytic and xylanolytic enzymes (Suominen, P. et al.,
in Kuwahara, M., Shimada, M., eds., Biotechnology in Pulp
and Paper Industry (Uni Publishers Co., Ltd., Tokyo),
pp. 439-445 (1992); Tenkanen, M. et al., Enzyme Microb.
Technol. 14:566-574 (1992)).
Trichoderma reesei also produces all the enzymes
needed for complete hydrolysis of native substituted xylans
(Poutanen, K., et al., J. Biotechnol. 6:49-60 (1987)).
Multiple endo-R-1,4-xylanases have been purified from
culture filtrates of Trichoderma (Baker, C. J., et al.,
Phytopathology 67:1250-1258 (1977); Hromova, M., et al.,
Arch. Microbiol. 144:307-311 (1986); John and Schmidt,
Methods Enzymol. 160A:662-671 (1988); Lappalainen, A.,
Biotechnol. Appl. Biochem. 8:437-448 (1986); Sinner and
Dietrichs, Holzforschung 29:207-214 (1975); Tan, L. U. L,
et al., Enzyme Microb. Technol. 7:425-430 (1985); Wood and
McCrae, Carbohydr. Res. 148:321-330 (1986)). Two specific
CA 02136350 2005-10-06
75154-6
4a
endoxylanases of T. reesei with isoelectric points at pH 5.5
(endoxylanase I) and pH 9.0 (endoxylanase II) have been
characterized (Tenkanen, M., et al., Enzyme Microb.
Technol. 14:566-574 (1992)).
There is a need for efficient microbial and
especially fungal xylanase producers, for example, for
producing enzyme mixtures for enzyme-aided pulp bleaching
(Kantelinen, A., et al., "Hemicellulases and Their Potential
CA 02136350 2002-05-22
75154-6
Role in Bleaching," Int. Pulp Bleaching Conference, Tappi Proceedings 1-
9 (1988); Lahtinen, T., et al., "Using Selective Trichoderma Enzyme
Preparations in Kraft Pulp Bleaching," in Biotechnology in the Pulp and
Paper Industry, Kuwahara and Shimada, eds., Uni Publishers Co., Ltd.,
5 Tokyo, Japan, pp. 129-137 (1992); Viikari, L., et al., "Bleaching with
Enzymes," Biotechnology in the Pulp and Paper Industry, Proc. 3rd Int.
Conf., Stockholm, pp. 67-69 (1986); Viikari, L., et al., Proc. 4th Int.
Symp. Wood and Pulping Chemistry, Paris 1:151-154 (1987)).
Summary of the Invention
Recognizing the importance of producing, in an economical way,
higher amounts of hemicellulolytic enzymes, and producing these enzymes
as an ideal combination with other wood or plant material degrading
enzymes, the inventors have investigated the use of recombinant DNA
techniques in the design of hosts which would be useful as a large-scale
source of recombinantly produced enzymes of interest to the applications
of hemicellulolytic enzymes.
These studies have resulted in the development of fungal hosts
which express large amounts of desirable enzymes and preferably large
amounts of hemicellulolytic enzymes.
These studies have also resulted in the developmeiit of fungal hosts
which not only express large amounts of desirable enzymes, preferably
hemicellulolydc enzymes, but also are deficient in at least one enzymatic
component of the cellulase degradation system.
The invention relates to fungal hosts belonging to
the genera Trichoderma which are capable of expressing high levels of
xylanase activity, or, fungal hosts which, in addition, are partially or
completely deficient in cellulase activity.
This invention also relates to fungal hosts which
produce high levels of xylanolytic enzymes and certain cellulolytic
CA 02136350 2008-03-25
75154-6
6
enzymes, preferably endoglucanases, but which, in addition,
are partially or completely deficient in other cellulase
activity, and preferably deficient in cellobiohydrolases.
This invention also relates to enzyme compositions
which are enriched in such hemicellulase activity or which
in addition are partially or completely deficient in certain
cellulolytic activities.
This invention also relates to isolation and
cloning of xylanase genes from Trichoderma.
This invention further relates to new hosts
(plant, yeast, fungal or bacterial) which contain and
express such Trichoderma xylanases.
According to one aspect of the present invention,
there is provided an isolated nucleic acid molecule
comprising a nucleic acid sequence encoding the amino acid
sequence of T. reesei pI 5.5 xylanase (SEQ ID No. 4) or
another nucleic acid molecule encoding a protein having
endo-beta-l,4-xylanase activity and which hybridizes to
the complement of SEQ ID NO. 3 at 60 to 68 C in the presence
of 6 x SSC + 0.1% SDS and washing at 60 to 68 C in the
presence of 6 x SSC + 0.1% SDS.
According to another aspect of the present
invention, there is provided an isolated nucleic acid
molecule, comprising a nucleic acid sequence encoding the
amino acid sequence of T. reesei pI 5.5 xylanase set forth
in SEQ ID No. 4.
CA 02136350 2008-03-25
75154-6
6a
According to still another aspect, the invention
provides an isolated nucleic acid molecule comprising a
nucleic acid sequence encoding the amino acid sequence of
T. reesei pI 9 xylanase set forth in SEQ ID NO. 2.
According to still another aspect, the invention
provides an isolated polypeptide encoded by the nucleic acid
as described herein.
According to still another aspect of the present
invention, there is provided a method for the production of
an enzyme preparation enriched in xylanase activity, said
method comprising: (1) transforming a host cell with the
recombinant vector comprising an isolated nucleic acid
molecule as described herein; (2) culturing said host cell
in culture medium under conditions wherein a protein having
xylanase activity expressed by said recombinant construct is
synthesised from said transformed host cell of step (1); (3)
and collecting said culture medium.
According to yet another aspect of the present
invention, there is provided a recombinant vector comprising
an isolated nucleic molecule described herein. In certain
embodiments, the nucleic acid molecule in the vector is
operably linked to a T. reesei promoter selected from the
group consisting of the xlnl promoter, the xln2 promoter,
the cbhl promoter, the cbh2 promoter, the egll promoter, and
the e912 promoter.
According to a further aspect of the present
invention, there is provided a recombinant host cell
transformed with the recombinant vector described herein.
According to yet a further aspect of the present
invention, there is provided a culture medium obtained after
culture of a host cell as described herein.
CA 02136350 2008-03-25
75154-6
6b
According to still a further aspect of the present
invention, there is provided an enzyme preparation having
endo-beta-1,4-xylanase activity or pI 9 xylanase activity,
obtained by purifying, drying, concentrating or immobilizing
the culture medium as described herein.
According to another aspect of the present
invention, there is provided a use of the culture medium or
the enzyme preparation described herein in the pulp and
paper or the feed industries.
Brief Description of the Drawings
Figure 1 shows the general strategy for deleting a
gene.
Figure 2 shows the restriction map of the 5.7 kb
(KpnI) insert of pALK475. The location of xln2 gene is
marked with an arrow. The inserts in the subclones pALK573,
pALK574, pALK570 and pALK476 are shown by separate lines.
The SmaI site at the 5' end of the fragment is derived from
the polylinker of pUC19.
Figure 3 shows the nucleotide sequence of the
T. reesei xln2 gene [SEQ ID No.:1:]. The coding regions are
expressed in upper case letters [SEQ ID No.:2:1. Peptide
sequences obtained from purified protein are indicated by an
underline; sequence indicated by a double underline was used
for preparing the PCR primer. The TATA box is indicated by
dotting. Putative signal cleavage site is marked with an
arrow (T) and putative sites for N-glycosylation with a
triangle (V). The N-terminal of the mature protein is shown
with an arrow (Y_). The two glutamic acids proposed to be
involved with an active site are boxed.
WO 93/24621 PCT/F193/00221
~136350
7
Figure 4 shows plasmid pALK476, a plasmid for targeting the xln2
gene to the cbhl locus. The xln2 gene is under the control of its own
promoter.
Figure 5 shows the pALK476 fragment used in the transfonnations.
The 1.9 kb cbhl S'-flanking region (Scal-EcoRI) is from 2.2 kb upstream
of the cbhl-coding region, the 1.8 kb 3'-region (BamHI-EcoRI) is from
1.4 kb downstream of the end of the cbhl-coding region. The amdS gene
was from p3SR2 (3.1 kb SpeI-XbaI fragment) and the xln2 gene was from
pALK475 (5.0 kb SmaI fragment, see Figure 2). The promoter area of the
xln2 gene in the fragment is 2.3 kb in size.
Figure 6 shows the relative xylanase production levels of the
transformants of Figure 5 calculated relative to that of T. reesei
VTT-D-79125. The columns show the mean values and the range of
production levels among the transformants of each host strain. One flask
of each transformant was grown. The numbers of transformants tested
were (CBHI+'"): T. reesei VTT-D-79125 19/38, ALK02221 16/26 and
ALK02721 22/31.
Figure 7 shows the construction of pALK174 (xln2 gene is fused to
the cbhl promoter). Only the relevant restriction sites are shown.
Figure 8 shows the relative increase in production of xylanase
activity in pALK174 transformants.
Figure 9 shows the restriction map of the 2.3 kb EcoRI insert of
pALK572. The location of xlnl gene is marked with an arrow.
Figure 10 shows the nucleotide sequence of the T. ressef xlnl gene
[SEQ ID No.:3:]. The coding regions are indicated by upper case letters
[SEQ ID No.:4:]. Peptide sequences obtained from purified protein are
shown underlined; the sequence with a double underline was used for
preparing the PCR primer. The TATA box is indicated by dotting.
Putative signal cleavage site is marked with an arrow (T). The N-terminal
of the mature protein is shown with an arrow (-). The two glutamic acids
proposed to be involved with an active site are boxed.
SUBSTITUTE SHEET
2136350
.W
WO 93/24621 PCT/F193/00271
8
Figure 11 shows the construction of plasmid pALK807 (xlnl gene is
fused to the cbhl promoter). Only the relevant restriction sites are shown.
Figure 12 shows an FPLC analysis of the CBHI negative
transformant VTT-D-87312; Figure 12A shows its comparison to the
untransformed host.
Figure 13 shows a diagram of the plasmid pALK99.
Figure 14 shows a diagram of the replacement of the chromosomal
cbh2 gene with the argB gene.
Figure 15 shows the construction of the plasmid pALK412.
Figure 16 shows a diagram of plasmid pPLE3.
Figure 17 shows the construction of a Acbhl acbh2 strain; trpC-
mutant of VTT-D-79125 (UV).
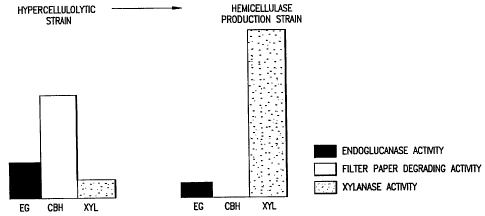
Figure 18 shows the enzyme profiles of the hypercellulolytic mutant
strain of T reesei VTT-D-79125 (UV) and the genetically engineered
derivative of that strain that lacks cellobiohydrolase.
Figure 19 shows modified enzyme profiles of the genetically
engineered T. reesei strains #1, #2 and #3.
Detailed Description of the Invention
I. Definitions
In the description that follows, a number of terms used in
recombinant DNA (rDNA) technology are extensively utilized. In order to
provide a clear and consistent understanding of the specification and
claims, including the scope to be given such terms, the following
definitions are provided.
Cellulase. Cellulase is a collective term which encompasses
enzymes which catalyze reactions which participate in the degradation of
insoluble cellulose to soluble carbohydrate. The term "cellulase" is known
in the art to refer to such a group of enzymes. For efficient hydrolysis of
SUBSTITUTE SHEET
WO 93/24621 2143 C 350 PCT/F793/00221
U
9
cellulose to glucose, at least three cellula.se enzymes (three types of
cellulase enzyme activity) are needed: randomly cleaving endoglucanases
(1,4,-P-D-glucan glucanohydrolase, EC 3.2.1.4) which usually attack
substituted soluble substrates and show no activity to crystalline cellulose;
cellobiohydrolase (1,4-A-D-glucan cellobiohydrolase, EC 3.2.1.91) which is
capable of degrading crystalline cellulose but has no activity towards
derivatized cellulose and P-glucosidase ((3-D-glucoside glycohydrolase, EC
3.2.1.21) which degrades cellobiose and cello-oligosaccharides to yield
glucose. Each of the three main types of enzymes listed above occurs in
multiple forms. For example, two immunologically distinctive
cellobiohydrolases, CBH I and CBH II are known. In addition, 5-8
electrophoretically distinct endoglucanases are known. Synergistic action
between some of these enzymes has been demonstrated. Cellulase activity
is synonymous with cellulolytic activity.
The biosynthesis of cellulases is provoked or induced by cellulose,
cellobiose, sophorose and lactose, and repressed by glucose or other readily
utilizable carbon sources.
By a Trichoderma host which is "substantially incapable" of
synthesizing one or more cellulase enzymes is meant a Trichoderma host in
which the activity of one or more of the cellulase enzymes is depressed,
deficient, or absent when compared to the wild-type (untransformed)
Trichoderma.
Hemicellulolytic enzymes (hemicellulases). For the enzymatic
degradation and modification of hemicelluloses, several different enzymes
are needed, each of which are termed "hemicellulase." The two main
glycanases depolymerizing the hemicellulose backbone are
endo-1,4-(3-D-xylanase and endo-1,4,(3-D-mannanase. Small
oligosaccharides are further hydrolyzed by 1,4-(3-D-xylosidase, 1,4-(3-D-
mannosidase and 1,4-(3-D-glucosidase. The side groups are split off by
a-L-arabinosidase, a-D-glucuronidase and a-D-galactosidase. Esterified
side groups are liberated by various esterases.
SUBSTITUTE SHEET
WO 93/24621 2136350 P(.T/F193/00271
,...
The defnzition of hemicellulolytic enzymes is taken from Viikari
et al., "Hemicellulases for Industrial Applications," in Bioconversion of
Forest and Agricultural Wastes (1992).
Enqme preparation. By "enzyme preparation" is meant a
5 composition containing enzymes which have been extracted (either
partially or completely purified) from fungi or the medium used to grow
such fungi. Therefore, the term "enzyme preparation" is meant to include
a composition comprising medium previously used to culture such fungi
and any enzymes which the fungi have secreted into such medium during
10 the culture.
Culture medium. By culture medium is meant a medium previously
used to culture a fungi ("spent" culture medium), such culture medium
containing enzymes which the fungi have secreted into the medium during
the culture. The culture medium is usable as such or as partially or
completely purified, concentrated, dryed or immobilized. If desired, the
expressed endoglucanase protein may be further purified in accordance
with conventional conditiosn, such as extraction, precipitation,
chromatography, affmity chromatography, electrophoresis, or the like.
Bio-bleaching. By "bio-bleaching" is meant the extraction of
lignin from cellulose pulp after the action of hemicellulose degrading
enzymes with or without lignin degrading enzymes. Removal of the lignin
may be restricted by hemicelluloses either physically (through
reprecipitation onto the fiber surface during cooking) or chemically
(through lignin-carbohydrate complexes). The hemicellulase activity
partially degrades the hemicellulose, which enhances the extractability of
lignins by conventional bleaching chemicals (like chlorine, chlorine
dioxide, peroxide, etc.) (Viikari et al., "Bleaching with Enzymes" in
Biotechnology in the Pulp and Paper Industry, Proc. 3rd Int. Conf.,
Stoclholm, pp. 67-69 (1986); Viikari et al., "Applications of Enzymes in
Bleaching" in Proc. 4th Int. Symp. Wood and Pulping Chemistry, Paris,
Vol. 1, pp. 151-154 (1987); Kantelinen et al., "Hemicellulases and their
SUBSTITUTE SHEET
WO 93/24621 2136350 PC'T/F193/00221
11
Potential Role in Bleaching" in International Pulp Bleachrng Conference,
Tappi Proceedings, pp. 1-9 (1988)). The advantage of this improved
, bleachability is a lower consumption of bleaching chemicals and lower
environmental loads or higher final brightness values. Hemicellulolytic
enzymes improve bleachability by making the lignin removal with
conventional bleaching chemicals easier.
Gene. A DNA sequence containing a template for a RNA
polymerase. RNA that codes for a protein is termed messenger RNA
(mRNA) and, in eukaryotes, is transcribed by RNA polymerase II.
However, it is also known to construct a gene containing a RNA
polymerase II template wherein a RNA sequence is transcribed which has a
sequence complementary to that of a specific mRNA but is not normally
translated. Such a gene construct is herein termed an "antisense RNA
gene" and such a RNA transcript is termed an "antisense RNA." Antisense
RNAs are not normally translatable due to the presence of translational
stop codons in the antisense RNA sequence.
A"complementary DNA" or "cDNA" gene includes recombinant
genes synthesized by reverse transcription of mRNA and from which
intervening sequences (introns) have been removed.
By an enzyme homologous to a Trichoderma host of the invention is
meant that an untransformed Trichoderma of the same species as the host
species naturally produces some amount of the native protein; by a gene
homologous to a Trichoderma host of the invention is meant a gene found
in the genome of an untransformed Trichoderma of the same species as the
host species.
By an enzyme heterologous to a Trichoderma host of the invention
is meant that an untransformed Trichoderma of the same species as the
host species does not naturally produce some amount of the native protein;
by a gene heterologous to a Trichoderma host of the invention is meant a
gene not found in the genome of an untransformed Trichoderma of the
same species as the host species.
SUBSTITUTE SHEET
Z136350
:i..
WO 93/24621 PCT/F193/00221
12
HybridiZation. By hybridization are meant conditions, under which
all Trichoderma xylanase genes hybridize to the nucleic acid sequence
encoding the amino acid sequence of T. reesei p1 5.5 xylanase or to the
nucleic acid sequence encoding the amino acid sequence of T. reesei pI 9
xylanase. These conditions are characterized by hybridization preferably in
60 to 68 C in the presence of 6 x SSC + 0.1% SDS and washing in in 60
to 68 C in the presence of 6 x SSC + 0.1% SDS.
Cloning vehicle. A plasmid or phage DNA or other DNA sequence
(such as a linear DNA) which provides an appropriate nucleic acid
environment for the transfer of a gene of interest into a host cell. The
cloning vehicles of the invention may be designed to replicate
autonomously in prokaryotic and eukaryotic hosts. In Trichoderma, the
cloning vehicles generally do not autonomously replicate and instead,
merely provide a vehicle for the transport of the gene of interest into the
Trichoderma host for subsequent insertion into the Trichoderma genome.
The cloning vehicle may be further characterized by one or a small number
of endonuclease recognition sites at which such DNA sequences may be
cut in a determinable fashion without loss of an essential biological
function of the vehicle, and into which DNA may be spliced in order to
bring about replication and cloning of such DNA. The cloning vehicle
may further contain a marker suitable for use in the identification of cells
transformed with the cloning vehicle. Markers, for example, are
tetracycline resistance or ampicilltin resistance. The word "vector" is
sometimes used for "cloning vehicle." Alternatively, such markers may be
provided on a cloning vehicle which is separate from that supplying the
gene of interest.
Expression vehicle. A vehicle or vector similar to a cloning vehicle
but which is capable of expressing a gene of interest which has been
cloned into it, after transformation into a desired host. In a preferred
embodiment, such expression vehicle provides for an enhanced expression
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
13
of a gene of interest which has been cloned into it, after transformation
into a desired host.
In a preferred embodiment, the gene of interest which is provided to
a fungal host as part of a cloning or expression vehicle integrates into the
fungal chromosome. Sequences which derive from the cloning vehicle or
expression vehicle may also be integrated with the gene of interest during
the integration process.
The gene of interest may preferably be placed under the control of
(i.e., operably linked to) certain control sequences such as promoter
sequences provided by the vector (which integrate with the gene of
interest). If desired, such control sequences may be provided by the fungal
host's chromosome as a result of the locus of insertion.
Expression control sequences on an expression vector will vary
depending on whether the vector is designed to express a certain gene in a
prokaryotic or eukaryotic host (for example, a shuttle vector may provide a
gene for selection in bacterial hosts) and may additionally contain
transcriptional elements such as, enhancer elements, termination sequences,
and/or translational initiation and termination sites.
II. Genetic Engineering of the Trichoderma Hosts
The process for genetically engineering the hosts of the invention is
facilitated through the cloning of genetic sequences which are capable of
encoding a desired enzymic activity and through the expression of such
genetic sequences. As used herein, the term "genetic sequences" is
intended to refer to a nucleic acid molecule (preferably DNA). Genetic
sequences which are capable of encoding a desired enzyme are derived
from a variety of sources. These sources include genomic DNA, cDNA,
synthetic DNA, and combinations thereof.
The mesophilic imperfect fungus Trichoderma reesei (formerly
T. viride) is classified as a member of Fungi imperfecti. Fungi imperfecti
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
...
14
is a catch-all category of fungi which have no sexual reproduction or
obvious affinities with sexually reproducing genera, such as the highly
characteristic Aspergillus. Although Trichoderma has been reported to
possess a poorly defmed sexual stage being an imperfect state of the
perfect ascomycete species Hypocrea, the genera Aspergillus and
Trichoderma are clearly to be considered taxonomically very different.
The improved enzyme preparations according to this invention are
produced by the fungus Trichoderma which has been modified by
recombinant DNA techniques. The Trichoderma hosts of the invention are
modified so as to be able to produce high levels of enzymes, preferably
hemicellulases. In addition, these hosts may be modified so as to be
totally deficient in at least one cellulase enzyme (whose activity is
undesirable during pulp and paper processing). Thus, although the
remaining cellulase activities may be unaffected, the Trichoderma hosts of
the invention are partially or completely deficient in the necessary
complement of enzymes which will fully degrade cellulose to glucose, and,
as a result, such degradation is greatly lowered or completely blocked.
Ila. Transformation of Trichoderma with a New Genetic Construct
In one embodiment, the Trichoderma hosts of the invention which
may be partially or completely deficient in at least one cellulase activity
are transformed with a genetic construct capable of expressing at least one
desired pulp and paper processing enzyme which is homologous to
Trichoderma, so as to provide for increased amounts of this enzyme in the
Trichoderma host. Examples of desired pulp and paper processing
homologous enzymes include, for example, hemicellulases and pectin-
degrading enzymes. Trichoderma is inherently capable of producing a
variety of hemicellulases including endoxylanases, mannanases, 0-
xylosidase, a-arabinosidase, a-glucuronidase, and acetyl esterase, the
activity of any of which may be a desired enzyme in the enzyme
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
2136350
preparations of the invention. Also, native Trichoderma produces minor
amounts of pectin degrading enzymes like polygalacturonase which may be
classified as a desired enzyme in the enzyme preparations of the invention.
Further, any other Trichoderma enzyme which oxidizes cellulose may be
5 utilized in the enzyme preparations of the invention and may be a desired
enzyme.
Comparison with xylanolytic enzymes produced by Trichoderma
reesei QM 9414, Aspergillus awamori VTT-D-75028, Fusarium
oxysporum VTT-D-80134, Bacillus subtilis ATCC 12711 and
10 Streptomyces olivochromogenes ATCC 21713 has shown that the highest
xylanase activity was produced by T. reesei. Therefore, under conditions
where it is desired to retain xylanase activity, T. reesei is an advantageous
host. (Poutanen, K., "Characterization of Xylanolytic Enzymes for
Potential Applications" in Disssertation for the degree of Doctor of
15 Technology, Technical Research Centre of Finland, Publications 47).
Further, although the above ireparations from the different microbial
origins differed with respect to (3-xylosidase activity and side-group
cleaving activities, the T. reesei culture filtrate contained all the side-
group
cleaving activities assayed (acetyl esterase, a-glucuronidase and a-
arabinosidase) whereas those from F. oxysporum and S. olivochromogenes
only contained esterase. Thus Trichoderma is also advantageous as a host
because it naturally produces a wide spectrum of xylanolytic enzymes the
proportions of which can be manipulated by genetic engineering for
different applications to provide enzyme preparations tailored for those
purposes.
According to this invention, the genetic constructs which encode
homologous enzymes which are desirable for pulp and paper processing
purposes may be introduced into the genome of Trichoderma and enhanced
expression can also be achieved by using strong promoters such as cbhl
and, if desired, additional or modified regulatory regions, such as, for
example, enhancer sequences. Preferably, such regulatory sequences are
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/0027'
,....
16
homologous to Trichoderma. A regulatory region, and especially a
promoter, may be modified to contain only those sequence elements
needed for expression and/or to retain a region which is responsible for
high expression levels. Enhancer sequences may be introduced
concurrently with the gene of interest as a separate DNA element but
operably-linked to such gene of interest, for example, as a DNA sequence
which is colinear with that providing the gene of interest (for example, in a
5' or 3' non-translating sequence, or in an intron).
In a highly preferred embodiment, the homologous gene introduced
to the genome of Trichoderma is a gene encoding a homologous
hemicellulase, preferably xylanase. The two main xylanases produced by
T. reesei have been purified (Tenkanen, M., Enzyme Microb. Technol.
14:566-574 (1992)). The enzymes have isoelectric points of 5.5 (XYLI)
and 9.0 (XYLII) and their molecular masses are 19 and 20 kDa,
respectively.
The xylanase I enzyme from Trichoderma reesei has optimal activity
in the more acid pH region, whereas xylanase II has its pH optimum in the
near-neutral pH range. Because of the different properties of the two
xylanases, either one or a mixture of these enzymes can be used in
different applications (WO 92/03541 and Viikari et al., "Important
Properties of Xylanases for Use in Pulp and Paper industry" In:
Biotechnology in Pulp and Paper Industry, Kuwahara, M. and Shimada,
M., eds., Proc. 5th Int. Conf. Biotechnology in Pulp and Industry, Uni
Publishers Co., Ltd., Tokyo, 1992, pp. 101-106).
According to the invention, it is possible by using genetic
engineering methods to enrich the desired xylanolytic activity to the culture
medium and use this culture medium in a desired application. For
exarnple, in some bleaching sequences and bleaching conditions it may be
advantageous to use xylanase I (more acid pH region), in some other
bleaching sequences and bleaching conditions xylanase II may be
preferably (alkaline or near neutral pH range).
SUBST6TUTE SHEET
WO 93/24621 PC.'I'/FI93/00221
17
In some applications, although one cellulolytic activity may be
eliminated, reduced, inactivated, or repressed, it may be desirable to
introduce a gene encoding a different cellulolytic enzyme into the host
cells so as to enhance one specific cellulolytic activity. In addition to
hemicellulolytic activity, for example, in feed industry, an enzyme
preparation comprising an elevated amount of endoglucanase may be used.
Thus, in those preparations in which such hydrolysis is desired, a host
which expresses elevated levels of endoglucanases, in addition to xylanases
or other hemicellulases may be used.
In another embodiment, the Trichoderma host which already
expresses a homologous form of an enzyme is transformed with a genetic
construct encoding a heterologous form of the same enzyme. In a further
embodiment, a Trichoderma host which does not express a certain enzyme
is transformed with one or more genetic constructs encoding enzyme(s)
heterologous to Trichoderma.
According to this invention increased amounts of a heterologous
enzyme whose activity is desired for pulp and paper processing purposes
are achieved by introducing the gene producing such heterologous desired
enzyme into a specific locus and/or introducing the gene in multicopies
into the genome of Trichoderma as described above.
In a preferred embodiment, the gene encoding a desired enzyme is
inserted into the cbhl locus such that it is operably linked to the strong
cbhl promoter. As described below, enhanced production is achieved by
using strong promoters such as cbhl. Increased amounts of the desired
heterologous enzyme are also achieved when Trichoderma's cellulase
producing capacity is lowered in general, even if the heterologous gene is
not inserted into the cbhl locus.
A gene encoding a desired enzyme, either homologous or
heterologous, such as a hemicellulose hydrolyzing enzyme, can be
integrated into the genome of Trichoderma by inserting the gene into a
general expression vector, for example, pAMH110, which is described in
SUBSTITUTE SHEET
WO 93/24621 P(.'I'/FI93/00221
e3 J !~ ~,
18
the patent application EP 244,234. pAMH 110 is derived from pUC 19
(Yanish-Perron et al., Gene 33:103-119 (1985)) and includes the promoter
and terminator of the cbhl gene and a stuffer fragment between the
promoter and terminator sequences which can be removed by digestion
with SacII and Ndel. After the ends are made blunt, any DNA, cDNA or
chromosomal DNA can be inserted between the promoter and terminator.
The desired gene can be inserted to the cbhl expression cassette in the
plasmid pAMH110 between the cbhl promoter and terminator.
Transcriptional regulatory elements of other genes may be used
where it is desired not to use the cbhl elements. For example a vector
construction comprising the 3-phosphoglycerate kinase gene (pgk)
transcriptional regulatory regions may be used as 3-phosphoglycerate
kinase, a key enzyme for ATP generation by glycolysis, is expressed in the
presence of glucose under which conditions the synthesis of cellulases is
repressed.
While the inventors do not intend to be bound by any particular
theory, the effectiveness of the expression of the desired gene seems to be
dependent both on the number of copies of the desired gene integrated to
the genome of Trichoderma and on the location of integration of the gene
in the genome. In a preferred embodiment, the integration of a desired
gene is directed into a specific locus. The use of a linear DNA helps in
directing the integration into a homologous locus. In a highly preferred
embodiment, the integration of a desired gene is directed into the
Trichoderma cbhl locus.
The DNA constructions prepared according to this invention can be
used to transform any Trichoderma strain. Such strains include, for
example, T. reesei strains QM9414 (ATCC 26921), RUT-C-30 (ATCC
56765), and highly productive mutants like VTT-D-79125, which is a
descendant of QM9414 (Nevalainen 1985, Technical Research Centre of
Finland Publications 26, (1985), Espoo, Finland). The transformation of
SUBSTITUTE SHEET
WO 93/24621 P(,'I'/FI93/0022]
2136350
19
Trichoderma may be performed by any technique known in the art and
especially by the technique taught in EP 244,234.
The Trichoderma host cells may be cultivated and the desired
enzymes produced by cultivating the host strain having the desired
properties under any conditions which allow expressing of the desired
enzymes. For example, a Trichoderma host strain having the desired
properties may be cultivated in a liquid cultivation medium, which may
comprise, for example, 6% Solka Floc cellulose, 3% distiller's spent grain,
0.5% KH2PO4, 0.5% (NH4)ZSO4, 0.1% struktol. The cellulase production by
Trichoderma strains is sensitive to glucose repression and require an
inducer such as, for example, cellulose, lactose or sophorose (Allen et al.,
Biotechnology and Bioengineering 33:650-656 (1989)). The pH in
Trichoderma cultivation should be kept at approximately pH 5 by the
addition of phosphoric acid or ammonia and the temperature may be kept
at 30 C during the cultivation. However, the temperature should be
adjusted according to the strain and according to the enzyme preparation to
be produced (Merivuori et al., Biotechnology Lett. 12:117-120 (1990)).
IIb DNA Cloning
Vector systems may be used in the method of producing
Trichoderma hosts for the production of the enzyme preparations of the
invention. One element provided by such vector construction may encode
the sequence of at least one homologous gene the activity of which it is
desired to eliminate, reduce, inactivate, delete or repress. Such vector
construction (a) may further provide a separate vector construction (b)
which encodes at least one desired gene to be integrated to the genome of
Trichoderma and (c) a selectable marker coupled to (a) or (b).
Alternatively, a separate vector may be used.
The cloned DNA which is used in the hosts of the invention may or
may not include naturally occurring introns. Moreover, such genomic
SUBSTITUTE SHEET
WO 93/24621 2136350 PCT/F193/00221
.....
DNA may be obtained in association with the native 5' promoter region of
the DNA genetic sequences and/or with the 3' transcriptional termination
region if such sequences are capable of functioning in Trichoderma.
Further, such genomic DNA may be obtained in association with the
5 genetic sequences which encode the 5' non-translated region of the mRNA
and/or with the genetic sequences which encode the 3' non-translated
region. To the extent that the Trichoderma host cell can recognize the
transcriptional and/or translational regulatory signals associated with the
expression of the mRNA and protein, then the 5' and/or 3' non-transcribed
10 regions of the native gene, and/or, the 5' and/or 3' non-translated regions
of the mRNA, may be retained and employed for transcriptional and trans-
lational regulation. Genomic DNA can be extracted by means well known
in the art (for example, see Guide to Molecular Cloning Techniques, S.L.
Berger et al., eds., Academic Press (1987)). Alternatively, mRNA can be
15 isolated from any cell which produces or expresses the desired protein, and
used to produce cDNA by means well known in the art (for example, see
Guide to Molecular Cloning Techniques, S.L. Berger et al., eds., Academic
Press (1987)). Preferably, the mRNA preparation used will be enriched in
mRNA coding for a desired protein, either naturally, by isolation from a
20 cells which are producing large amounts of the protein, or in vitro, by
techniques commonly used to enrich mRNA preparations for specific
sequences, such as sucrose gradient centrifugation, or both.
For cloning into a vector, such suitable DNA preparations (either
genomic DNA or cDNA) are randomly sheared or enzymatically cleaved,
respectively, and ligated into appropriate vectors to form a recombinant
gene (either genomic or cDNA) library.
A DNA sequence encoding a desired protein may be inserted into a
DNA vector in accordance with conventional techniques, including blunt-
ending or staggered-ending termini for ligation, restriction enzyme
digestion to provide appropriate termini, filling in of cohesive ends as
appropriate, alkaline phosphatase treatment to avoid undesirable joining,
SUBSTITUTE SHEET
WO 93/24621 PC1/F193/00221
2136350
21
and ligation with appropriate ligases. Techniques for such manipulations
are disclosed by Maniatis, T., et al., supra, and are well known in the art.
Libraries containing clones encoding a desired protein may be
screened and a clone to the desired protein identified by any means which
specifically selects for that protein's DNA such as, for example, a) by
hybridization with an appropriate nucleic acid probe(s) containing a
sequence specific for the DNA of this protein, or b) by hybridization-
selected translational analysis in which native mRNA which hybridizes to
the clone in question is translated in vitro and the translation products are
further characterized, or, c) if the cloned genetic sequences are themselves
capable of expressing mRNA, by immunoprecipitation of a translated
protein product produced by the host containing the clone.
Oligonucleotide probes specific for the proteins desired in this
invention which can be used to identify clones to such protein can be
designed from knowledge of the amino acid sequence of the protein.
Because the genetic code is degenerate, more than one codon may be
used to encode a particular amino acid (Watson, J.D., In: Molecular
Biology of the Gene, 3rd Ed., W.A. Benjamin, Inc., Menlo Park, CA
(1977), pp. 356-357). The peptide fragments are analyzed to identify
sequences of amino acids which may be encoded by oligonucleotides
having the lowest degree of degeneracy. This is preferably accomplished
by identifying sequences that contain amino acids which are encoded by
only a single codon.
Although occasionally an amino acid sequence may be encoded by
only a single oligonucleotide sequence, frequently the amino acid sequence
may be encoded by any of a set of similar oligonucleotides. Importantly,
whereas all of the members of this set contain oligonucleotide sequences
which are capable of encoding the same peptide fragment and, thus, poten-
tially contain the same oligonucleotide sequence as the gene which encodes
the peptide fragment, only one member of the set contains the nucleotide
sequence that is identical to the exon coding sequence of the gene.
SUSSTiTUTE SHEET
WO 93/24621 36350 PCT/F193/0027'
22
Because this member is present within the set, and is capable of
hybridizing to DNA even in the presence of the other members of the set,
it is possible to employ the unfractionated set of oligonucleotides in the
same manner in which one would employ a single oligonucleotide to clone
the gene that encodes the peptide.
Using the genetic code (Watson, J.D., In: Molecular Biology of the
Gene, 3rd Ed., W.A. Benjamin, Inc., Menlo Park, CA (1977)), one or more
different oligonucleotides can be identified from the amino acid sequence,
each of which would be capable of encoding the protein. The probability
that a particular oligonucleotide will, in fact, constitute the actual
protein's
sequence can be estimated by considering abnormal base pairing
relationships and the frequency with which a particular codon is actually
used (to encode a particular amino acid) in eukaryotic cells. Such "codon
usage rules" are disclosed by Lathe, R., et al., J. Molec. Biol. 183:1-12
(1985). Using the "codon usage rules" of Lathe, a single oligonucleotide
sequence, or a set of oligonucleotide sequences, that contain a theoretical
"most probable" nucleotide sequence capable of encoding the protein's
sequence is identified.
The suitable oligonucleotide, or set of oligonucleotides, which is
capable of encoding a fragment of the protein's gene (or which is
complementary to such an oligonucleotide, or set of oligonucleotides) may
be synthesized by means well known in the art (see, for example, Synthesis
and Application of DNA and RNA, S.A. Narang, ed., 1987, Academic
Press, San Diego, CA) and employed as a probe to identify and isolate the
desired cloned gene by techniques known in the art. Techniques of nucleic
acid hybridization and clone identification are disclosed by Maniatis, T., et
al. (In: Molecular Cloning, A Laboratory Manual, Cold Spring Harbor
Laboratories, Cold Spring Harbor, NY (1982)), and by Hames, B.D., et al.
(In: Nucleic Acid Hybridization, A Practical Approach, IRL Press,
Washington, DC (1985)).
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
2136350
23
Cloning techniques using oligonucleotides and PCR are also well known in
the art ("PCR Protocols" in A Guide to Methods and Application, Innis
et al., eds., Academic Press, San Diego (1990)). Those members of the
above-described gene library which are found to be capable of such
hybridization are then analyzed to determine the extent and nature of the
desired genomic coding sequences which they contain.
To facilitate the detection of the desired DNA encoding sequence,
the above -described DNA probe is labeled with a detectable group. Such
detectable group can be any material having a detectable physical or
chemical property. Such materials have been well-developed in the field
of nucleic acid hybridization and in general most any label useful in such
methods can be applied to the present invention. Particularly useful are
nonradioactive labels such as digoxigenin-nucleotides (dUTP) or radio-
active labels, such as 3zP, sH, 14C, 35S, tzsl, or the like. Any radioactive
label may be employed which provides for an adequate signal and has a
sufficient half-life. The oligonucleotide may be radioactively or
nonradioactively labeled by several methods known in the art and
commercial kits for these purposes are available.
Alternatively, polynucleotides are also useful as nucleic acid
hybridization probes when labeled with a non-radioactive marker such as
biotin, digoxigenin, an enzyme or a fluorescent group. See, for example,
Leary, J.J., et al., Proc. Natl. Acad Sci. USA 80:4045 (1983); Renz, M., et
al., Nucl. Acids Res. 12:3435 (1984); and Renz, M., EMBO J. 6:817
(1983).
Thus, in summary, the actual identification of protein's sequence (or
a partial sequence of the protein) permits the identification of a theoretical
"most probable" DNA sequence, or a set of such sequences, capable of
encoding such a peptide. By constructing an oligonucleotide comple-
mentary to this theoretical sequence (or by constructing a set of
oligonucleotides complementary to the set of "most probable" oligo-
nucleotides), one obtains a DNA molecule (or set of DNA molecules),
SUBSTITUTE SHEET
WO 93/24621 2136350 PC.'I'/FI93/0022'
24
capable of functioning as a probe(s) for the identification and isolation of
clones containing the protein's gene.
In an alternative way of cloning a gene, a library is prepared using
an expression vector, by cloning DNA or, more preferably cDNA prepared
from a cell capable of expressing a desired protein, into an expression
vector. The library is then screened for members which express the
protein, for example, by screening the library with antibodies to the
protein.
The above discussed methods are, therefore, capable of identifying
genetic sequences which are capable of encoding a desired protein or
fragments of this protein. In order to further characterize such genetic
sequences, and, in order to produce the recombinant protein, it is desirable
to express the proteins which these sequences encode. Such expression
identifies those clones which express proteins possessing characteristics of
the desired protein. Such characteristics may include the ability to
specifically bind antibodies directed against the protein, the ability to
elicit
the production of antibody which are capable of binding the protein, and
the ability to provide a protein specific function to a recipient cell, among
others.
The cloned protein encoding sequences, obtained through the
methods described above, and preferably in a double-stranded form, may
be operably linked to sequences controlling transcriptional expression in an
expression vector, and introduced into a Trichoderma host cell to produce
recombinant protein or a functional derivative thereof. Depending upon
which strand of the protein encoding sequence is operably linked to the
sequences controlling transcriptional expression, it is also possible to
express an antisense RNA or a functional derivative thereof.
A nucleic acid molecule, such as DNA, is said to be "capable of
expressing" a polypeptide if it contains expression control sequences which
contain transcriptional regulatory information and such sequences are
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
"operably linked" to the nucleotide sequence which encodes the
polypeptide.
An operable linkage is a linkage in which a sequence is connected to
a regulatory sequence (or sequences) in such a way as to place expression
5 of the sequence under the influence or control of the regulatory sequence.
Two DNA sequences (such as a protein encoding sequence and a promoter
region sequence linked to the 5' end of the encoding sequence) are said to
be operably linked if induction of promoter function results in the
transcription of the protein encoding sequence mRNA and if the nature of
10 the linkage between the two DNA sequences does not (1) result in the
introduction of a frame-shift mutation, (2) interfere with the ability of the
expression regulatory sequences to direct the expression of the mRNA,
antisense RNA, or protein, or (3) interfere with the ability of the template
to be transcribed by the promoter region sequence. Thus, a promoter
15 region would be operably linked to a DNA sequence if the promoter were
capable of effecting transcription of that DNA sequence.
The precise nature of the regulatory regions needed for gene
expression may vary between species or cell types, but shall in general
include, as necessary, 5' non-transcribing and 5' non-translating (non-
20 coding) sequences involved with initiation of transcription and translation
respectively.
Expression of the protein in the Trichoderma hosts requires the use
of regulatory regions functional in such hosts. A wide variety of
transcriptional and translational regulatory sequences can be employed,
25 since Trichoderma generally recognize eukaryotic host transcriptional
controls, such as, for example, those of other filamentous fungi. In
eukaryotes, where transcription is not linked to translation, such , -itrol
regions may or may not provide an initiator methionine (AUG) Ion,
depending on whether the cloned sequence contains such a methionine.
Such regions will, in general, include a promoter region sufficient to direct
the initiation of RNA synthesis in the host cell.
SUBSTITUTE SHEET
WO 93/24621 PCT/FI93/0027'
213~`~5~
26
As is widely known, translation of eukaryotic mRNA is initiated at
the codon which encodes the first methionine. For this reason, it is
preferable to ensure that the linkage between a eukaryotic promoter and a
DNA sequence which encodes the protein, or a functional derivative
thereof, does not contain any intervening codons which are capable of
encoding a methionine. The presence of such codons results either in a
formation of a fusion protein (if the AUG codon is in the same reading
frame as the protein encoding DNA sequence) or a frame-shift mutation
(if the AUG codon is not in the same reading frame as the protein
encoding sequence).
Ilc Regulatory Regions
In a preferred embodiment, a desired protein is secreted into the
surrounding medium due to the presence of a homologous Trichoderma
secretion signal sequence. If a desired protein does not possess its own
signal sequence, or if such signal sequence does not function well in
Trichoderma, then the protein's coding sequence may be operably linked to
a signal sequence homologous or heterologous to Trichoderma. The
desired coding sequence may be linked to any signal sequence which will
allow secretion of the protein from a Trichoderma host, for example, the
signal sequence of the Trichoderma cellobiohydrolase I protein. Such
signal sequences may be designed with or without specific protease sites
such that the signal peptide sequence is amenable to subsequent removal.
Transcriptional initiation regulatory signals can be selected which
allow for repression or activation, so that expression of the operably linked
genes can be modulated. Of interest are regulatory signals which are
temperature-sensitive so that by varying the temperature, expression can
be repressed or initiated, or are subject to chemical regulation, e.g.,
substrate or metabolite regulation. Also of interest are constructs wherein
both (a) a desired protein's mRNA and (b) antisense RNA directed to a
SUBSTITUTE SHEET
w0 93/24621 PCT/F193/00221
27
cellulase enzyme are provided in a transcribable forms such that expression
of the desired protein's mRNA is accompanied by antisense RNA
repression of the expression of one of the host's cellulase enzymes.
Translational signals are not necessary when it is desired to express
antisense RNA sequences.
If desired, the non-transcribed and/or non-translated regions 3' to the
sequence coding for a protein can be obtained by the above-described
cloning methods. The 3'-non-transcribed region may be retained for its
transcriptional termination regulatory sequence elements; the 3-non-
translated region may be retained for its translational termination
regulatory sequence elements, or for those elements which direct
polyadenylation in eukaryotic cells.
The vectors of the invention may further comprise other operably
linked regulatory elements such as enhancer sequences.
IId Detecting Stable Transformants of Trichoderma
In a preferred embodiment, genetically stable transformants of
Trichoderma are constructed whereby a desired protein's DNA is integrated
into the host chromosome. The coding sequence for the desired protein
may be from any source. Such integration may occur de novo within the
cell or, in a most preferred embodiment, be assisted by transformation with
a vector which functionally inserts itself into the host chromosome, for
example, DNA elements which promote integration of DNA sequences in
chromosomes.
Cells which have stably integrated the introduced DNA into their
chromosomes are selected by also introducing one or more markers which
allow for selection of host cells which contain the expression vector in the
chromosome, for example the marker may provide biocide resistance, e.g.,
resistance to antibiotics, or heavy metals, such as copper, or the like. The
selectable marker gene can either be directly linked to the DNA gene
SUSSTITUTE SHEET
WO 93/24621 PC.'1'/F193/0022'
64. ,~~63.,o
28
sequences to be expressed, or introduced into the same cell by co-
transfection.
Factors of importance in selecting a particular plasmid or viral
vector include: the ease with which recipient cells that contain the vector
may be recognized and selected from those recipient cells which do not
contain the vector; the number of copies of the vector which are desired in
a particular host; and whether it is desirable to be able to "shuttle" the
vector between host cells of different species.
Once the vector or DNA sequence containing the construct(s) is
prepared for expression, the DNA construct(s) is introduced into an
appropriate host cell by any of a variety of suitable means, including
transformation as described above. After the introduction of the vector,
recipient cells are grown in a selective medium, which selects for the
growth of transformed cells. Expression of the cloned gene sequence(s)
results in the production of the desired protein, or in the production of a
fragment of this protein. This expression can take place in a continuous
manner in the transformed cells, or in a controlled manner.
The DNA encoding sequences, obtained through the methods above,
will provide sequences which by definition, encode a desired protein and
which may then be used to obtain a desired protein's antisense RNA
genetic sequences as the antisense RNA sequence will be that sequence
found on the opposite strand of the strand transcribing the peptide core's
mRNA. The antisense DNA strand may also be operably linked to a
promoter in an expression vector such that transformation with this vector
results in a host capable of expression of an antisense RNA in the
transformed cell. Antisense RNA and its expression may be used to
interact with an endogenous DNA or RNA in a manner which inhibits or
represses transcription or translation of the gene in a highly specific
manner. Use of antisense RNA probes to block gene expression is
discussed in Lichtenstein, C., Nature 333:801-802 (1988).
SUBSTITUTE SHEET
w0 93/24621 2~ 36350 PCT/F193/00221
.,,,.
29
Trichoderma is an especially useful and practical host for the
synthesis of the enzyme preparations of the invention because Trichoderma
is capable of secreting protein at large amounts, for example,
concentrations as much as 40 g/L culture fluid have been reported; the
homologous Trichoderma cbhl promoter provides a very convenient
promoter for expression of genes-of-interest because it is a strong, single
copy promoter which normally directs the synthesis of up to 60% of the
secreted protein from the Trichoderma host; the transformation system is
highly versatile and can be adapted for any gene of interest; the
Trichoderma host provides an "animal cell type" high mannose
glycosylation pattern; and culture of Trichoderma is supported by previous
extensive experience in industrial scale fermentation techniques.
M. Trichoderma Hosts Deficient in At Least One Cellulase En2yme
According to this invention, it is also possible to enrich Trichoderma
hosts for an enzyme whose activity is desirable for pulp and paper
processing purposes by inactivating or eliminating at least one cellulase
enzyme. In one embodiment, the cbhl gene is merely mutated. Since the
majority of the secreted proteins of Trichoderma may be the cellulase
activity encoded by the gene cbhl, (the cellobiohydrolase, CBHI, protein),
by constructing Trichoderma hosts in which the cbhl gene is mutated to an
inactive form, the relative percent of the remaining proteins secreted by
Trichoderma in the culture medium may be increased. In another
embodiment, a desired gene is inserted preferably into the cbhl locus such
that expression of the desired gene is operably linked to the strong cbhl
promoter. In a highly preferred embodiment, a casette comprising a
desired gene already operably linked to the homologous cbhl promoter is
inserted into the cbhl locus.
In the hosts of the invention, any one, some, or all of the cellulolytic
enzymes may be eliminated, reduced, inactivated, or repressed by methods
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/0027'
known in the art so as to result in the host's partial or complete inability
to
degrade cellulose to glucose. Undesired cellulolytic enzyme activities can
be eliminated, reduced, inactivated, or repressed by several methods, e.g.,
by inactivating the gene(s) encoding such enzyme (for example, by
5 introducing a frame-shift mutation to the gene), by deleting the entire
whole gene or large segments of the gene, by replacing the gene with
another DNA via homologous recombination, by compensation of the gene
region, by additional integration, by double crossing-over, and by
transforming the host cell with a genetic construct capable of expressing an
10 antisense RNA directed against the coding sequence for that gene, etc.
For example, inactivation of genes coding for cellulolytic activities
may be performed as described in European Patent Applications EP
137,280 and EP 244,234.
Trichoderma fungi produce large amounts of identical,
15 predominantly haploid uninucleate conidia which constitute excellent
material for various mutagenic treatments. However, even a haploid
mutated nucleus can produce a heterokaryotic colony (mycelium) if a
mutation becomes initially fixed only in one of the two strands of the
DNA double helix (mosaicism). The amount of mosaic mutants depends
20 on both the mutagen and dose used. In fungi forming haploid uninucleate
conidia, the problem of heterokaryotic mycelium can be handled by
allowing conidiation and by reisolation of colonies originating from single
separate conidia. This cycle can be repeated several times.
Examples of chemical mutagens useful for mutengenizing the
25 Trichoderma hosts of the invention include alkylating agents, such as, for
example, N-methyl-N'-nitro-N-nitrosoguanidine (NTG),
ethylmethanesulphonate (EMS) and diethylsulphate (DES). Hydroxylamine
and chemicals deaminating DNA bases such as nitrous acid are also useful.
Ionizing radiation (y- and X-rays) as well as ultraviolet irradiation (UV)
30 are examples of physical mutagens useful in Trichoderma strain
mutagenesis.
SUBSTITUTE SHEET
CA 02136350 2001-09-10
75154-6
31
The use of solid media permits rapid screening of thousands of
colonies arising from mutagenized conidia for the presence or absence of
specific enzymes and allows quantitative estimation of the amount of
enzyme produced. Several types of solid media for detection of
enzymes, for example, extracellular amylolytic enzymes, pectinase,
protease, chitinase, 0-galactosidase and cellulase, lipase, urease, RNAase
and DNAase are known in the art.
Many fungi form large diffuse colonies when grown on solid media.
Addition of chemical agents restrictive to colony growth may therefore be
desired to allow development of more than one (up to 100) colony per one
plate. Among agents used for the purpose are rose bengal, oxgall and
TM
phosphon D, Triton X-100 and saponin. With some fungi, replica plating
technique analogous to that developed for bacteria can, iri certain cases, be
used to test the properties of fungal colonies on different growth media.
Screening on plates is usually followed by cultivation of the selected
colonies in shake flasks in a liquid production medium for measurement of
enzyme activity. The best isolates showing enhanced enzyme production
in shake flask scale may be in a second round of mutagen treatment if
desired.
Homologous genes which it is desirable to inactivate or delete
according to this invention include, for example, the cellulase genes cbhl,
cbh2, egll, egl2 (formerly eg13; Saloheimo et al., Gene 63:11-21 (1988))
(which encode the proteins cellobiohydrolase I, cellobiohydrolase II,
endoglucanase I and endoglucanase II) or combinations of these genes.
Eliminating the activity of any of these genes will result in a host which is
partially or completely deficient in its ability to degrade cellulose to
glucose. Such elimination of cellulolytic activity may be achieved at the
genomic level, by eliminating the gene or modifying it into a form which
is incapable of expression. Such elimination may also be achieved at the
translational level, by hybridizing the mRNA which encodes the protein to
WO 93/24621 PCT/FI93/002''
32
an antisense RNA to a degree which prevents the translation of the
hybridized RNA.
In a preferred embodiment, a cellulase activity is selectively
inactivated so that some, but not all of the cellulase components are
inactivated. For example, if it is desired to maintain the host's ability to
hydrolyze R-glucan, then the endoglucanase genes would not be
inactivated.
The inactivation of, e.g., one of the cellulase genes can be based on
transformation of Trichoderma reesei with a plasmid carrying a defected
gene as described in patent application EP 244,234. Homologous
recombination of the plasmid at the chromosomal cellulase gene locus
causes insertional inactivation of the endogenous T. reesei cellulase gene.
The plasmid used for transformation contains only part of the cellulase
coding region and produces inactive protein. No 5' flanking sequences are
included. A frameshift mutation can also be introduced to the truncated
coding region. A selection marker, (for example amdS (acetamidase) or
argB (ornithine carbamoyl transferase)) or a marker for screening (for
example, lacZ) can be coupled to the plasmid used for the transformation
or the transformation can be done as a cotransformation, which means that
the selectable marker and the defected gene are on different plasmids (EP
244,234). Inactivation of a gene with homologous recombination may be
done with a circular DNA, which integrates in a colinear manner into the
Trichoderma chromosomal DNA.
The deletion of an undesired gene can be done by using a strategy
the principle of which is described in Figure 1. The recipient strain is
transformed with a linear DNA fragment containing a selectable marker
gene (like trpC, argB or amdS) and/or a foreign desired gene of interest
which is to be expressed, flanked by the 5' and 3' flanking regions of the
gene to be deleted. Homologous recombination at the A locus will thus
lead to replacement of the A gene with the selection marker and/or desired
gene B. If the 5' region in the transforming fragment is taken upstream
SUBSTITUTE SHEET
WO 93/24621 213 6 ~ ~ ~_, PCT/FI93/00221
33
from the promoter area, the promoter will also be removed in the resulting
replacement strain. Gene A can be any Trichoderma gene, preferably a
cellulase gene, the flanking regions of which can be cloned/isolated.
Moreover the linear DNA fragment can be ligated to form a circular
plasmid or in addition the circular form may contain DNA needed for
replication in bacteria (e.g., in E. coli). The linear DNA fragments used in
deletion of undesired genes can be constructed for example from pUC 19
plasmids (Yanish-Perron et al., Gene 33:103-119 (1985)).
This method is described in more detail in the Example 1 B which
describes the deletion of cbh2 gene from the genome of Trichoderma by
said method.
Clones of the cellulase enzymes have been described which may be
used to design mutant sequences for inactivation of homologous sequences
in the hosts of the invention. Any mutant sequence which results in the
inactivation of the enzyme's activity may be used. For example, the gene
for the native cellobiohydrolase CBH I sequence has been cloned by
Shoemaker et al. (Shoemaker, S., et al., BiolTechnology 1:691-696 (1983))
and Teeri et al. (Teeri, T., et al., Bio/Technology 1:696-699 (1983)) and
the entire nucleotide sequence of the gene is known (Shoemaker, S., et al.,
Bio/Technology 1:691-696 (1983)). From T. reesei, the gene for the
major endoglucanase (EG I) has also been cloned and characterized
(Penttilii, M., et al., Gene 45:253-263 (1986); EP 137,280; Van Arstel,
J.N.V., et al., BiolTechnology 5:60-64). Other isolated cellulase genes are
cbh2 (Patent Application WO 85/04672; Chen, C.M., et al.,
BiolTechnology 5:274-278 (1987)) and eg12 (originally eg13 (Saloheimo,
M., et al., Gene 63:11-21 (1988)).
IV. The Engyme Preparation
According to the invention, there is provided a method for producing
high levels of enzymes, preferabaly hemicellulases, desirable for pulp and
SUBSTITUTE SHEET
WO 93/24621 2 11~~~~ O PCT/F193/0027'
,.~
34
paper processing. There is also provided a method for producing an
enzyme preparation partially or completely deficient in cellulolytic activity
(that is, in the ability to completely degrade cellulose to glucose) and
enriched in enzymes desirable for pulp and paper processing, preferably
hemicellulases. By "deficient in cellulolytic activity" is meant a reduced,
lowered, depressed, or repressed capacity to degrade cellulose to glucose.
Such preparations may be obtained directly from the hosts of the invention.
Further, if desired activities are present in more than one recombinant host,
such preparations may be isolated from the appropriate hosts and combined
prior to use in the method of the invention.
It is envisioned that enzyme preparation which are enriched or
partially or completely deficient in specific enzymatic activities will be
provided so as to satisfy the requirements of a specific utility in various
applications in the pulp and paper industry and in fodder production.
Enzyme activities may be added or deleted as described above to provide,
remove or retain or lower a desired activity. For example, if the intended
application is improvement of the strength of the mechanical mass of the
pulp, then the enzyme preparation of the invention may provide enzymes
which enhance or facilitate the ability of cellulose fibers to bind together.
In a similar manner, in the application of pulp milling, the enzyme
preparation of the invention may provide enzymes which enhance or
facilitate such swelling.
To obtain the enzyme preparations of the invention, the recombinant
hosts described above having the desired properties (that is, hosts
substantially incapable of expressing one or more cellulase enzymes and
capable of expressing the desired enzymes) are cultivated under suitable
conditions, the desired enzymes are secreted from the Trichoderma hosts
into the culture medium, and the enzyme preparation is recovered from
said culture medium by methods known in the art.
The enzyme preparation can be produced by cultivating the
Trichoderma strain in a fermentor having the desired properties for
SUBSTITUTE SHEET
WO 93/24621 2136350 PCT/F193/00221
~.~ 35
example in a liquid cultivation medium, which may comprise for example
6% Solka Floc cellulose (BW40, James River Corporation, Hackensack,
NJ), 3% distiller's spent grain, 0.5% KH2PO4, 0.5% (NH4)2SO4, and 0.1%
struktol as an antifoaming agent (struktol SB 2023, Schill & Seilacher,
Hamburg, FRG). The cellulase production of Trichoderma strains is
sensitive to glucose repression and require an inducer (cellulose, lactose or
sophorose) (Allen et al., Biotechnology and Bioengineering 33:650-656
(1989)). The pH should preferably be kept at approximately pH 5 by the
addition of phosphoric acid or ammonia and the temperature at 30 C
during the cultivation. However, the temperature may be adjusted
according to the strain and according to the enzyme preparation to be
produced (Merivuori et al., Biotechnology Letters 12(2):117-120 (1990)).
The enzyme preparation is recovered from the culture medium by
using methods well known in the art. However, because the hosts of the
invention may be partially or completely deficient in cellulase activity, it
is
an advantage of the invention that the enzyme preparations of the invention
may be utilized directly from the culture medium with no further
purification. If desired, such preparations may be lyophilized or the
enzymatic activity otherwise concentrated and/or stabilized for storage.
The enzyme preparations of the invention are very economical to provide
and use because (1) the enzymes may be used in a crude form; isolation of
a specific enzyme from the culture fluid is unnecessary and (2) because the
enzymes are secreted into the culture medium, only the culture medium
need be recovered to obtain the desired enzyme preparation; there is no
need to extract an enzyme from the Trichoderma hosts.
If desired, an expressed protein may be further purified in
accordance with conventional conditions, such as extraction, precipitation,
chromatography, affinity chromatography, electrophoresis, or the like.
The Trichoderma and enzyme preparations of the invention have
further application in fodder production. For example, fodder treated with
SUBSTITUTE SHEET
WO 93/24621 PCT/FI93/00221
36
the enzyme preparations of the invention would be of great food benefit to
farm animals because it would be easier for them to digest.
The invention is described in more detail in the following examples,
These examples show only a few concrete applications of the invention. It
is self evident for one skilled in the art to create several similar
applications. Hence the examples should not be interpreted to narrow the
scope of the invention only to clarify the use of the invention.
Examples
Materials and Methods
Transformation of T. reesei
Transformation of T. reesei and selection of AmdS+ and ArgB+
transformants were carried out as described by Penttila et al., Gene 61:155-
164 (1987).
Phleomycin resistant transformants were screened as described by
Durand et al. in: Biochemistry and Genetics of Cellulose Degradation, p.
135-151, 1987, J.-P. Aubert, P. Beguin and J. Millet (eds.), Academic
Press, New York.
In cotransformation with p3SR2 and pAMH111, equal molar
amounts of plasmid DNA (5-10 g) were used. In transformations
conferring phleomycin resistance, the relative amounts of plasmid DNA
used were 1:1 or 2:1 for pAMHl l l and pAN8-1 respectively. When
cotransformation was carried out using p3SR2 and pMS4, the plasmid
pMS4 was added in 3-4 times molar excess. Transformants were purified
through conidia; that is, the conidial suspension was plated again on the
selective medium so that every colony started from a single conidia.
In transformations with a linear DNA fragment, the amount of DNA
used varied from 2 to 5 g. The selection marker (amdS (acetamidase) or
SUBSTITUTE SHEET
WO 93/24621 2136350 POW193/00221
37
argB (ornithine carbamoyl transferase, OTCase, E.C. 2.1.3.3)or trpC
(tryptophane)) was within the transforming fragment.
Isolation and Analysis of DNA
Plasmid DNA from E. coli was isolated using standard methods
(Maniatis et al. 1982, Molecular Cloning: A Laboratory Manual, Cold
Spring Harbor Laboratory, Cold Spring Harbor, N.Y.). Chromosomal
DNA was isolated from T. reesei using the method of Raeder and Broda,
Lett. Appl. Microbiot :l.47-20 (1985)). Southern and Northern
hybridizations were performed by standard techniques (Maniatis et al.
supra, 1982). Western blotting was carried out according to Maniatis et
al., supra, 1982).
Liquid Cultivation Media and Conditions for Trichoderma
All Trichoderma liquid cultures were started from conidiospores
grown on potato dextrose agar as described by Bailey and Nevalainen,
Enzyme Microb. Technol. 3:153-157 (1981)). Liquid cultivations in shake
flasks were performed according to Bailey and Nevalainen, Enzyme
Microb. Technol. 3:153-157 (1981), except that Finnfloc was replaced with
Solca Floc cellulose. Medium used in fermenter cultivations contained 6%
Solka Floc cellulose, 3% distiller's spent grain, 0.5% KH2PO41 0.5%
(NH4)2SO4 and 0.1% struktol. The pH was kept between 4.0 and 4.8 by
addition of phosphoric acid or ammonia. Fermentations were carried out at
C. Maximum yield of enzymes was obtained in 5 days in laboratory
fermentations and in 4 days in 100 liter fermenter scale.
SUBSTITUTE SHEET
CA 02136350 2001-09-10
75154-6
38
EnUme Assays
All assays for enzyme activity were carried out from culture
supernatant fractions after removing the mycelia by centrifugation for 20
min at 3000 rpm. Endoglucanase activity using hydroxyethylcellulose as
substrate (IEC, mittelviskos, Fluka AG 54290, pract. grade) was measured
as described in Bailey and Nevalainen, Enzyme Microb. Technol. 3:153-
157 (1981) and Commission on Biotechnology, International Union of Pure
and Applied Chemistry; Measurement of Cellulase Activities, Biochemical
Engineering Research Centre, Indian Institute of Technology, Delhi, New
Delhi - 10016 (1984)) and xylanase activity using birch xylan (Roth No.
7500) as substrate, were measured as described by Bailey et al., J. Bact.
23:257-270 (1992). The TCA precipitated proteins were assayed with the
method of Lowry et al., J. Biol. Chem. 193: 2-65-275 (1951) using
bovine serum albumin as standard. Cellobiohydrolase activity against filter
paper (filter paper unit, FPU) was measured as described in Commission
on Biotechnology, International Union of Pure and Applied Chemistry,
Measurement of Cellulase Activities, Biochemical Engineering Research
Centre, Indian Institute of Technology, Delhi, New Delhi -10016 (1984)).
ELISA Assay for Endoglucanase I
Endoglucanase I protein concentration in the culture supennatant
fractions was determined by a double antibody sandwich ELISA. The
assays were performed in 96-well flat bottomed microtiter plates at 37 C
(except were noted). Each step was terminated by washing 3 times with
TM
phosphate buffered saline pH 7.2 containing 0.05% Tween 20 and 0.02%
sodium azide (PBS/Tween).
The plates were coated with mouse monoclonal antibodies directed
against endoglucanase I (anti-EGI antibody EI-2) overnight at 4 C.
Unoccupied sites on the plastic surface were blocked with 1% BSA in
WO 93/24621 213t7 350 PCT/F193/00221
39
PBS/Tween for 1 hr. Appropriate dilutions of culture supematant fractions
and purified endoglucanase I were then added and incubated for 2 hrs
followed by an incubation with rabbit polyclonal antibodies against
endoglucanase I for 2 hrs. Bound rabbit antibodies were detected by
incubation with swine polyclonal antibodies against rabbit IgG conjugated
to alkaline phosphatase (Orion Diagnostica, Espoo, Finland) for 2 hrs. In
an end step p-nitrophenylphosphate (1 mg/nil) was added and the reaction
stopped after 30 min at room temperature with 2 N NaOH. The developed
yellow color was measured photometrically at 405 nm. The concentration
of endoglucanase I in culture supematant fractions was then calculated by
comparing their 0D405 values with a standard dilution curve prepared using
purified endolgucanase I and performed at the same time on the same
plate.
Fractionation of the Culture Supernatant Fraction by Chromatofocusing
The chromatographic system consisted of a Pharmacia FPLC
apparatus equipped with a Mono P HR 5/20 column for chromatofocusing.
The resin was stabilized in 25 mM Bistris-HCl buffer, pH 6.5. The crude
enzyme mixture produced by T. reesef in shake flask culture was diluted
with the same buffer to 1 mg/ml protein content. 500 l enzyme samples
were injected into the column and eluted with Pharmalyte/Polybuffer
(Pharmacia, 1 ml PharmalyteR 2.5 - 5 and 5 ml PolybufferT' PB 74 in a
total 100 ml, adjusted pH to 3.0 with HCl) forming a pH gradient from 6.5
to 3Ø The flow rate was 30 ml/h. Column effluents were collected in
600 l fractions and the pH and EGI activity were assayed.
SUBSTITUTE SHEET
CA 02136350 2001-09-10
75154-6
Example
Cloning, Sequencing and Enhanced Expression of the Trichoderma
reesei Endoxylanase II (pl 9) Gene, xln2
5 In the results presented in this example, the Trichoderma reesei xln2
gene coding for the pI 9.0 endoxylanase was isolated from the wild-type
strain QM6a. The gene contains one intron of 108 nucleotides and codes
for a protein of 223 amino acids in which two putative N-glycosylation
target sites were found. Three different T. reesei strains were transformed
10 by targeting a construct composed of the xln2 gene with its own promoter
to the endogenous cbhl locus. Highest overall production levels for
xylanase were obtained using the T. reesei ALK02721, a genetically
engineered strain, as a host. Integration into the cbhl locus was not
required for enhanced expression under xln2 promoter.
15 Materials and Methods
Organisms, plasmids and growth conditions. Plasmids were
propagated in Escherichia coli strain XL1-Blue (Bullock, W.O., et al.,
BiolTechniques 5:376-378 (1987)) or in E. coli INV 1 aF' (Invitrogen, San
Diego, CA, USA). The recipient organisms for the x1n2 gene were the
20 high cellulase-producing mutants T. reesei VTT-D-79125 (Bailey and
Nevalainen, Enzyme Microb. Technol. 3:153-157 (1981)), ALK02721 and
ALK02221. T. reesei ALK02221 is a low protease mutant of the strain
VTT-D-79125. T. reesei ALK02721 is a trpC minus (trpC ') UV mutant
of VTT-D-79125, in which the cbh2 locus has been replaced with the trpC
25 gene of Aspergillus nidulans (Yelton, M.M., et al., Proc. Natl. Acad. Sci.
USA 81:1470-1474 (1984)). The strain also carries several other
integrated copies of the trpC gene.
TM
The plasmids used in this study included pBluescript (Stratagene,
San Diego, CA, USA), pCR1000 (Invitrogen) and pUC19 (Yanish-Perron,
WO 93/24621 2136350 PCT/F193/00221
41
C., et al., Gene 33:103-119 (1985)). The selectable marker, amdS, came
from the plasmid p3SR2 (Kelly and Hynes, EMBO J. 4:475-479 (1985))
kindly donated by Dr. M. Hynes. The cbhl flanking regions were isolated
from the T. reesei strain ALK02466 (Harkki, A., et al., Enzyme Microb.
Technol. 13:227-233 (1991)), using plasmid rescue.
E. coli cultures were grown at 37 C overnight in L-broth (Maniatis,
T., et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor
Laboratory, Cold Spring Harbor, NY (1982)) supplemented with ampicillin
(50 g/ml) as needed. PD (Potato Dextrose Broth, Difco, Detroit, USA)
agar slants were used for growing the Trichoderma strains. For xylanase
production, the T. reesei strains were grown for seven days in shaker flasks
(30 C, 250 rpm) in a cellulase- and xylanase-inducing medium (pH 5.5)
containing of 4% whey, 1.5% complex nitrogen source, 1.5% KH2PO4 and
0.5% (NH4)2SO3.
Peptide digestion and amino acid sequencing. Purified xylanase II
(Tenkanen, M., et al., Enzynie Microb. Technol. 14:566-574 (1992)) from
T. reesei (VTT-D-79125) was digested with 2% (w/w) trypsin (TPCK-
treated, Sigma Chemical Company, St. Louis, MO, USA) in 1% (w/v)
ammonium bicarbonate at 37 C for 2.5 h. After addition of 3% (w/w)
trypsin, incubation was continued overnight (13 h). The peptides were
separated using high-performance liquid chromatography (HPLC) with a
Rexchrom Prep-5/300 ODS reverse-phase column. Elution was
performed at the rate of 1 ml/min in a linear solvent gradient running from
5% (v/v) acetonitrile (ACN) containing 0.1% trifluoroacetic acid (TFA) to
60% (v/v) ACN containing 0.06% TFA in 60 min at 24 C. Absorbance at
218 nm was measured. Prior to amino-terminal sequencing, 10 g of
xylanase II was treated with 1 U of pyroglutamate aminopeptidase
(Boehringer Mannheim, Mannheim, Germany) at 37 C overnight in
100 mM potassium phosphate buffer, pH 8.0, containing 10 mM EDTA
and 5mM DTT. Amino-terminals of the protein and the peptides were
sequenced by degrading them in a gas-pulsed-liquid-phase sequencer
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
21
42
(Kallckinen and Tilgmann, J. Protein Chem. 7:242-243 (1988)). The
released PTH-amino acids were analyzed on-line using narrow-bore
reverse-phase HPLC.
DNA manipulation and transformations. Standard DNA methods
described by Maniatis, T., et al., Molecular Cloning: A Laboratory
Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1982)
were used for the DNA constructions. Plasmid and cosmid DNAs were
isolated using Qiagen columns (Diagen GmbH, Diisseldorf, Germany)
according to the manufacturer's instructions.
E. coli was transformed according to Hanahan, D., J. Mol. Biol.
166:557-580 (1983), and T. reesei strains by the method of Penttild, M.,
et al., Gene 61:155-164 (1987) with the following modifications: the
protoplasts were treated by heat shock (Berges and Barreau, J. Gen.
Microbiol. 135:601-604 (1989)) before transformation, 25% PEG6. was
replaced with 60% PEGWO and the transformation was performed at room
temperature instead of that on ice. The transformants were purified
through conidia on selective acetamide-CsCl plates (Penttild, M., et al.,
Gene 61:155-164 (1987) before transferring them to PD slants.
RNA isolation, T. reesei. VTT-D-79125 was cultured in a
fermentor in xylanase-inducing medium for three days. The mycelia were
harvested, frozen and ground into a fine powder under liquid nitrogen.
Total RNA and, subsequently, mRNA were isolated using Proteinase K
digestion, phenol extractions and oligo dT-cellulose purification, as
described by Bartels and Thompson, Nucleic Acid Res. 11:2961-2978
(1983). The mRNA obtained was size-fractionated using DMSO-sucrose
gradient centrifugation (Boedtker, H., et al., Biochemistry 15:4765-4770
(1976)).
Cloning of xylanase H cDNA. The first strand of cDNA was
prepared from 1 g of mRNA by using the cDNA synthesis kit
(Boehringer Mannheim), replacing the oligo dT-primer with a hybrid
dTWadapter primer (5' GAC-TCG-AGA-ATT-CAT-CGA-dT17 3')
SUBSTtTUTE SHEET
WO 93/24621 2136350 PCr/F193/00221
43
[SEQ ID No.:5:]. This cDNA was used as a template for polymerase
chain reaction (PCR) amplification (Frohman, M.A., "Race: Rapid
Amplification of cDNA Ends," in PCR Protocols, Innis, M.A., et al., eds.,
Academic Press Inc., San Diego, CA, pp. 28-38 (1990)) directed by a
gene specific primer (sense 5' GG(A/C/G)-TGG-CA(A/G)-CCN-GGN-
ACN-AA 3') [SEQ ID No.:6:] deduced from a peptide sequence and by
the adapter primer (5' GAC-TCG-AGA-ATT-CAT-CGA 3') [SEQ ID
No.:7:]. The 100 l PCR reaction mixture contained 5 l of 1:25 diluted
cDNA, 100 pmol of each primer, 5 M dNTP, 1 x PCR buffer and 1.5
units of Taq DNA polymerase (Boehringer Mannheim). Amplification in a
programmable thermal controller (M.J. Research Inc.) comprised 30 cycles
at 95 C for 1 min, at 55 C for 1 min and at 72 C for 2 min. After the last
cycle, the elongation period was extended to 10 min. The PCR fragments
obtained were cloned using a TA Cloning kit (Invitrogen) according to the
manufacturer's instructions and verified by sequencing.
Isolation of the x1n2 gene. A genomic cosmid library (Suominen et
al., MGG, In press) of T. reesei QM6a (Mandels and Reese, J. Bacteriol.
73:269-278 (1957)) was screened by hybridization with a xylanase II PCR
probe according to the supplier's instructions (Boehringer Mannheim,
Germany).
Nucleotide sequencing. The templates for nucleotide sequencing
were generated by unidirectional deletions according to the manufacturer's
instructions for the pBluescript Exo/Mung DNA sequencing system
(Stratagene). DNA was sequenced in both directions by using ABI
(Applied Biosystems, Foster City, USA) kits based on fluorescence-
labelled T7 and T3 primers and a Taq cycle sequencing method according
to the supplier's instructions. Sequencing reactions were analysed on an
ABI 373A Sequencer.
Enzyme and protein assays. Enzymes were assayed from the
culture supernatants after removing the mycelia. Xylanase activity was
measured, using birch xylan (Roth 7500) as substrate, by the method
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
44
described by Bailey, M.J., et al., J. Biotechnol. 23:257-270 (1992).
Production of cellobiohydrolase I(CBHI) protein was detected by Western
blot or dot blot methods using the CBHI specific monoclonal antibodies
CI-89 or CI-261 (Aho, S., et aL, Eur. J. Biochem.. 200:643-649 (1991)).
Results
Peptide sequences
Several tryptic peptides from the purified endoxylanase II were
obtained and directly sequenced (see Figure 3 for peptide sequences).
However, no amino-terminal sequence for the native protein was detected,
suggesting that it was blocked. After treatment with pyroglutamate
aminopeptidase, an amino-terminal sequence of (Q)-X-Ile-Gln-Pro-Gly-
Thr-Gly-Tyr-Asn [SEQ ID No.:8:] was obtained. The first amino acid (X)
of the pyroglutamate aminopeptidase treated sample could not be
deterinined without ambiguity because of confounding peaks in the HPLC
chromatogram.
Cloning of T. reesei xln2 cDNA
The accumulation of xylanase II specific mRNA in T. reesei cultures
was determined by Northern hybridization using a nucleotide oligomer
deduced from the peptide (marked with a double underline in Figure 3)
sequence as a probe. The results indicated that the xylanase II nmRNA was
most abundant in mycelia grown for three days and that the size of the
xylanase II specific mRNA was about 0.7 kb (data not shown).
The xylanase II specific oligomer (see above) primer and an
unspecific dT-adapter primer were used to amplify the xylanase sequence
from the first strand of cDNA. The amino acid sequence deduced from the
nucleotide sequence of the subclone pALK564, containing the longest of
the fragments synthesized in a PCR reaction, contained several of the
SUBSTITUTE SHEET
WO 93/24621 2136350 PCT/F193/00221
xylanase II peptide sequences obtained by direct sequencing. This
confumed that the PCR clone coded for xylanase II.
Isolation and nucleotide sequence of the xln2 gene of T. reesei
Four clones were isolated from a genomic cosmid library of T. reesei
5 through hybridization with the insert of pALK564 (insert= PCR-
synthesized xln2 cDNA). A subclone, pALK475, containing a hybridizing
5.7 kb KpnI fragment from the cosmid clone in the pUC19 vector was
analysed by restriction mapping (Figure 2). The hybridizing 2.3 kb HindIII,
1.5 kb HindIIl/XhoI and 1 kb Xhol fragments from pALK475 were
10 subcloned and partially sequenced to reveal the xln2 gene.
The nucleotide and deduced amino acid sequences of the xln2 gene
are shown in Figure 3. Sequence resembling the TATA box was found in
the DNA at a distance of -140 nucleotides from the ATG, (Figure 3). The
primary translation start site (ATG), flanked by the highly conserved
15 consensus sequence 5' CA C/A A/C ATG 3' found in filamentous fungi
and other eukaryotes (Ballance, J.D., "Transformation systems for
filamentous fungi and an overview of fungal gene structure," in Molecular
Industrial Mycology: Systems and Applications for Filamentous Fungi,
Leon and Berka, eds., Marcel Dekker Inc., New York, pp. 1-29 (1991)), is
20 99 nucleotides upstream of the codon for the determined N-terminal amino
acid of xylanase II. The xln2 gene codes for a protein of 223 amino acids.
In this protein, the N-terminal, obtained by direct peptide sequencing, is
preceded by 33 amino acids. This preprosequence contains a putative
signal peptidase cleavage site (von Heijne, G., Nucl. Acid Res. 14:4683-
25 4690 (1986)) between Alal9 and A1a20. Comparison of the genomic
sequence with the cDNA sequences revealed one intron of 108 nucleotides.
This indicates that the mature xylanase II protein consists of 190 amino
acids and has a calculated molecular weight of 20.8 kDa. This is in good
agreement with the 20 kDa obtained for purified xylanase II (Tenkanen,
30 M., et al., Enzyme Microb. Technol. 14:566-574 (1992)). Sequence
SUBSTITUTE SHEET
2136350
WO 93/24621 PGT/F193/00221
46
analysis also revealed two putative targets (Asn-X-Ser/Thr where X is not
Pro; Gavel and von Heijne, Protein Engineering 3:433-442 (1990)) for N-
glycosylation (Asn38 and Asn6l of the mature protein).
Construction of pAL%476
Construction of the plasmid pALK476 is shown in Figure 4. This
plasmid is useful for targetting the x1n2 gene to the cbhl locus.
Expression of the x1n2 gene is under the control of its native promoter.
The 5.0 kb SmaI fragment from the plasmid pALK475 (Figure 2),
containing the x1n2 gene and promoter (2.3 kb of the gene's upstream
area), was ligated to Spel site (filled in with Klenow) of the plasmid
pALK425.
Enhanced expression of xln2 in T. reesei
To enhance xylanase expression in T. reesei, three strains, VTT-D-
79125, ALK02221 and ALK02721, differing in their enzyme production
profiles, were transformed with the EcoRI fragment from the plasmid
pALK476 (Figure 4). The expression cassette containing the xln2 gene
with its own promoter (Figure 5) was targeted to the cbhl locus, using the
cbhl flanking regions.
Transformants, 57 from T. reesei VTT-D-79125, 42 from
ALK02221 and 53 from ALK02721 transformations, were purified and
grown in shake flask cultures in xylanase-inducing conditions. Xylanase
production was measured as birch xylan degrading activity in the culture
supernatants. The transformants were tested for CBHI protein production to
determine the targeting frequencies to the cbhl locus and to distinguish
between transformants in which the x1n2 expression cassette was located at
the cbhl locus and those in which it was integrated elsewhere.
Replacement of the cbhl locus by the x1n2 expression cassette results in a
SUBSTITUTE SHEET
WO 93/24621 2436350 P(.'I'/F193/00221
47
CBHI negative (CBHI ) phenotype which was detected in Western blots
and dot blots by using a CBHI specific monoclonal antibody. Targeting
efficiency to the cbhl locus, determined as the CBHI- phenotype was high
in each case: 67%, 62% and 58% in transformants of VTT-D-79125,
ALK02221 and ALK02721, respectively.
Figure 6 shows the relative xylanase activities of the transformants
grouped according to their CBHI' phenotype. Increase in the xln2 copy
number resulted in increased production of xylanase activity in both CBHI-
and CBHI+ transformants of each strain. The best transformants yielded
about twofold (T. reesei ALK02221 and ALK02721) to 4.5-fold (T. reesei
VTT-D-79125) xylanase activity compared with the respective host strains.
The highest activities (nkat/ml) were obtained by using T. reesei
ALK02721 as a host. The best transformants of T. reesei ALK02221
produced less than half the activity obtained with the best transformants of
the two other strains. There was little difference in xylanase activity
between the CBHI+ and CBHI- phenotype in the transformants of T. reesei
ALK02221 and ALK02721 (Figure 6). Among the T. reesei VTT-D-
79125 transformants, those with the CBHI+ phenotype were on average
better producers of xylanase.
To evaluate the effect of the genetic background on the expression
of the xln2 gene in the three hosts, the relative increase in xylanase
activity
in the CBHI- transformants was calculated. The CBHI- transformants were
used to eliminate any effects of differences in the site of integration on
gene expression. Within each host strain, most of the CBHI- transformants
(42-86%, Table 1) exhibited similar levels of increase in xylanase activity.
Most of the transformants excluded from this comparison produced higher
xylanase activities. From this we conclude that it is likely that the
transformants shown in Table 1 have one extra copy of xln2 at the cbhl
locus, in addition to the natural xln2. The average xylanase activity of
these transformants and the average increase compared with the host strain
are shown in Table 1. The average increase ranged, depending on the host
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
48
strain, from about 600 nkat/ml (ALK02221) to about 2700 nkat/ml
(ALK02721).
Table 1
Relative Increase in Xylanase Production
in a Chosen Set of CBHI- transformants
Average Activity Average Increase in
Strain (nkat/ml) activity (nkat/ml)
VTT-D-79125 3700 (+/- 20%)
transformants (50%) 5100 (+/- 20%) 1400
ALK02221 3800 (+/-10%)
transfornnants (85 %) 4400 (+/- 15%) 600
ALK02721 10200(+/- 15%)
transforniants (42%) 12900(+/- 10%) 2700
* The proportion (%) of CBHI' transformants apparently having an extra
copy of xln2 is shown in the parentheses.
Discussion
There are several reports on molecular cloning of bacterial xylanases
(e.g., Ghangas, G.S., et al., J. Bacteriol. 171:2963-2969 (1989); Lin and
Thomson, Mol. Gen. Genet. 228:55-61 (1991); Shareck, F., et al., Gene
107:75-82 (1991); Whitehead and Lee, Curr. Microbiol. 23:15-19 (1991)).
Recently, reports on the cloning of xylanases of filamentous fungi have
also been published, including those of Aspergillus tubigensis (van dert
Broeck, N., et al., "Cloning and Expression of Xylanase Genes from
Fungal Origin," EP 0 463 706 Al (1992)), A. niger var. awamori (Maat,
J., et al., "Xylanases and Their Application in Bakery," in Xylans and
Xylanases, Visser, J., et al., eds., Elsevier Science, Amsterdam, pp. 349-
360 (1992)), A. kawachii (Ito, K., et al., Biosci. Biotechnol. Biochem.
56:906-912 (1992)) and T. reesei (Suominen, P., et al., "Genetic
Engineering of Trichoderma reesei to Produce Suitable Enzyme
Combinations for Applications in the Pulp and Paper Industry," in
SUB8TITUTE SHEEIr
WO 93/24621 2136350 P(.'I'/F193/00221
~-
49
Biotechnology in Pulp and Paper Industry, Kuwahara and Shimada, eds.,
Uni Publishers Co., Ltd., Tokyo, Japan, pp. 439-445 (1992); T6rr6nen, A.,
et al., BiolTechnology 10:1461-1465 (1992)). The genes for T. reesei
xylanase II (pI 9) cloned of the wild-type strain QM6a by us (see also
Suominen, P., et al., "Genetic Engineering of Trichoderma reesei to
Produce Suitable Enzyme Combinations for Applications in the Pulp and
Paper Industry," in Biotechnology in Pulp and Paper Industry, Kuwahara
and Shimada, eds., Uni Publishers Co., Ltd., Tokyo, Japan, pp. 439-445
(1992)) and of the mutant strain RutC-30 by TarrSnen, A., et al.,
Bio/Technology 10:1461-1465 (1992) were not completely identical. The
main differences were between the sequences of pre-propeptides thus
suggesting differences in the signal processing.
According to our results, the x1n2 gene of T. reesei contains one
long intron of 108 bp. The introns in filamentous fungi are usually
smaller, around 50 bp in length, but, as shown by Vanhanen et al., Curr.
Genet. 12:181-186 (1989) for the gene of 3-phosphoglycerate kinase in
T. reesei, they can also be considerably longer. The fairly long pre-
prosequence (33 amino acids) of the x1n2 gene contains one primary
cleavage site for the signal peptidase. This is compatible with a signal
sequence of 19 amino acids followed by a propeptide of 14 amino acids,
which may be post-translationally cleaved from the mature protein.
T6rr6nen, A., et al., Bio/Technology 10:1461-1465 (1992) suggested that
the signal pre-propeptide of the xylanase II (pI 9) of T. reesei RutC-30
consists of 32 amino acids and has two putative signal peptidase cleavage
sites very close to the translation start site (5 and 11 amino acids,
respectively). This would result in rather short signal sequences. Two-
step protein processing, similar to what is proposed here for the
xylanase II, has been shown to occur with A. niger glucoamylase (Innis,
M.A., et al., Science 228:21-26 (1985)) and suggested to occur with
T. reesei cellobiohydrolase II (Teeri, T., et al., Gene 51:43-52 (1987)).
Overall structure similar to T reesei xln2 is found in the A. niger xylanase
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
gene (van den Broeck, N., et al., "Cloning and Expression of Xylanase
Genes from Fungal Origin," EP 0 463 706 Al (1992)) but not, for
instance, in the A. kawachii xylanase A gene, which contains nine introns
and codes for a larger (32.7 kDa) protein (Ito, K., et al., Biosci.
5 Biotechnol. Biochem. 56:906-912 (1992)).
Production of xylanase could be increased in three T. reesei strains
by increasing the copy number of the x1n2 gene. Of these strains, T. reesei
VTT-D-79125 is a high cellulase-producing mutant and ALK02721 was
used as a host because of its pre-existent high xylanase production. We
10 also wanted to produce xylanase in the low-protease T. reesei ALK02221,
both to obtain a xylanase product with a low protease background and to
increase the relative amount of xylanase in the culture medium.
The highest increase in xylanase activity ranged from about twofold
to over fourfold compared with the respective host strain. Xylanase
15 activities of the transformants of the low-protease strain ALK02221 and
those of the genetically engineered host ALK02721 were independent of
whether the x1n2 gene was integrated into the cbhl locus or elsewhere in
the genome (CBHI' phenotype). For T. reesei VTT-D-79125
transformants, integration of the x1n2 gene elsewhere (CBHI+ phenotype)
20 than into the cbhl locus seemed to result in higher enzyme yield. These
observations are somewhat contradictory to those of Harkki, A., et al.,
Enzyme Microb. Technol. 13:227-23 3(1991), who suggested that for
optimal expression the expression cassette would require its integration
into the cbhl locus. It should be noted, however, that in the present study
25 _the xln2 promoter was used for expression instead of the cbhl promoter as
done by Harkki, A., et al., Enzyme Microb. Technol. 13:227-233 (1991)
with egll cDNA. This indicates that the cbhl locus may not be the best
environment for expression under the xln2 promoter.
The genetic background seemed to have a significant effect on xln2
30 expression, despite the fact that the strains T. reesei ALK02221 and
ALK02721 originate from VTT-D-79125. The increase of xylanase
SUBSTITUTE SHEET
WO 93/24621 ~13 6 35 0:-? PCT/F193/00221
51
production was highest in the transformants of the strain T. reesei
ALK02721 (2700 nkat/ml) which also had the highest xylanase activity.
The expression of one additional copy of xln2 varied over fourfold
depending on the host (Table 1).
Exaynple 2
Increased production of XYLII: x1n2 gene fused to the cbhl promoter
Construction of p.ALK174 and transformation of three
Trichoderma reesei strains
Construction of pALK174 is shown in Figure 7. First, a 674 bp
PCR fragment containing an exact fusion of the cbhl promoter to the xln2
signal sequence (and xln2 gene to the internal XhoI site) was synthesizeu
Oby using plasmid pALK475 (Figure 2) as a template. The
olig-,nucleotides used were as shown below: 5'-primer contained the end
of the cbhl promoter and the sequence of the beginning of the putative
xln2 signal sequence, 3'-primer had sequence from ihe x1n2 gene including
the gene's internal Xhol site (see the pALK476 map, Figure 5 or the x1n2
sequence).
S'-primer (39-mer) [SEQ ID No.:11:J:
5'- C AAC CGC GGA CTG CGC ATC ATG GTC TCC TTC ACC TCC
CT
SacII beginning of the putative
end of the cbhl promoter x1n2 signal sequence
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
~136370
52
3'-primer (26-mer) [SEQ ID No.:12:J:
5'-GGG AGC CGC TCG AGC GGT GGT TGC GG
Xhol
x1n2 sequence
The 100 l PCR reaction contained in PCR buffer (10 mM Tris pH 8.3, 50
mM KCI, 1.5 mM MgC12, 0.01% gelatin, w/v) 50 pmol of each primer, 10
ng of the template DNA, 0.2 mM dNTP and 2U of Taq-polymerase
(Boehringer Mannheim). The reaction conditions were: denaturation 1
min. at 95 C, annealing 1 minute at 60 C, extension 2 minutes at 72 C for
30 cycles, fmal extension was 9 minutes.
The PCR fragment was purified by using the Mermaid kit (BIO
101 Inc., La Jolla, California, USA). After treating it with the T4 DNA
polymerase, it was cut with SacII for fusion to the cbhl promoter.
The plasmid pAMH110, containing the cbhl promoter, was cut with
NdeI (filled in by Klenow) and SacII. The PCR fragment described above
was ligated to the digested pAMH110 (pALK174X). The fusions and the
sequence synthesized by PCR were ensured by sequencing. The plasmid
pALK174 was constructed by replacing the x1n2 promoter in pALK476 by
the cbhl promoter: the 2.9 kb KpnI XhoI fragment from the plasmid
pALK174X, containing the cbhl promoter fused to the xln2 gene and x1n2
sequence to the internal XhoI, was ligated to the isolated vector containing
fragment of pALK476, after cutting pALK476 with Kpnl and Xhol.
The plasmid pALK174 contains, in addition to the amdS and cbhl
3'-areas as in pALK476 (Figure 4), the x1n2 gene, fused to the cbhl
promoter and 1.9 kb terminator (3'-area) of the x1n2 gene.
The 9.7 kb EcoRl fragment (expression cassette free from the vector
sequences) was used to transform three Trich'oderma strains, VTT-D-
79125, ALK02221 and ALK02721. The strains, transformation method
and methods for purification, analyzing and growing the transformants
were as described above and in Example 6.
SUBSTITUTE SHEET
WO 93/24621 2136350 PCT/F193/00221
õ~..
53
2.2 Production of xylanase by the pAL%174 transformants
VTT-D-79125, ALK02221 and ALK02721 transformants, 39, 41
and 50, respectively, were purified and grown to measure the xylanase
activities produced. The targeting frequencies to the cbhl locus were,
measured by using dot blot method and a CBHI specific monoclonal
antibody (as in Example 4) 82, 46 and 84%, respectively. The production
of xylanase activity, shown as relative activity compared to the activity
produced by VTT-D-79125, of the CBHI+ and CBHI- transformants is
shown in the Figure 8. The columns show the mean values and the range
of production levels among the transformants of each host strain. One
flask of each transformant was grown. The xylanase activity was
determined from the culture supernatants as described in Example 3
(cloning of xlnl ).
High production of xylanase was obtained in transformants of all the
three Trichoderma strains. Tlie best VTT-D-79125 and ALK02221
transformants produced about 10 times the amount of xylanase activity
compared to their host strains. The best ALK02721 transformants
produced about the same amount of xylanase activity as the best VTT-D-
79125 and ALKO2221 transformants and the increase compared to the host
was over three fold. In average, the CBHI- transformants were better
producers than those with the CBH+ phenotype. Naturally, in the CBHI-
transformants the cellulolytic activity has decreased because of the deletion
of the cbhl gene.
The production levels of xylanase, when pALK174 construction was
used, were higher compared to those obtained by using pALK476 (xln2
under the control of its own promoter, see Example 1).
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/0022'
54
Example 3
Structure of the Endoxylanase I, xlnl, Gene of Trichoderma reesei
In this example, the cloning of the T. reesei gene xlnl which codes
for the low pl (5.5) endoxylanase I is presented.
Materials and methods
Bacterial strains and plasmids
The plasmids used in this study were pBluecscript (Stratagene, San
Diego, CA, USA) and pCR1000 (Invitrogen, San Diego, CA, USA). The
plasmids were propagated in Escherichia coli strain XLI-Blue (Bullock,
W.O. et al., Bfo/Techniques 5:376-378 (1987)) or in E. coli INV1aF
(Invitrogen). E. coli cultures were grown at 37 C overnight in L-broth
(Maniatis, T. et al., Molecular Cloning: A Laboratory Manual (Cold
Spring Harbor Laboratory, Cold Spring Harbor, New York) (1982))
supplemented with ampicillin (50 g/ml) as needed.
Peptide digestion and amino acid sequencing
Purified xylanase I (Tenkanen, M. et al., Enzyme Microb.
Technol. 14:566-574 (1992)) from T. reesei (VTT-D-79125; Baily, M.J.,
Nevalainen, K.H.M., Enzyme Microb. Technol. 3:153-157 (1981)) was
digested with 2% (w/w) trypsin (TPCK-treated, Sigma Chemical
Company, St. Louis, MO, USA) in 1% (w/v) ammonium bicarbonate at
37 C for 2.5 h. After addition of 3% (w/w) trypsin to the sample,
incubation was continued over night (13 h). The peptides were separated
using high-performance liquid chromatography (HPLC) with a Rexchrom
Prep-5/300 ODS reverse-phase column. Elution was performed at the rate
of 1 ml/min in a linear solvent gradient running from 5% (v/v) acetonitrile
(ACN) containing 0.1 % trifluoroacetic acid (TFA) to 60% (v/v) ACN
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
2136j5o
containing 0.06% TFA in 60 min at 24 C. Absorbance at 218 nm was
measured. Amino terminals of the protein and the peptides were
sequenced by degrading them in a gas-pulsed-liquid-phase sequencer
(Kalkkinen, N., and Tilgmann, C., J. Prot. Chem. 7:242-243 (1988)). The
5 released PTH-amino acids were analysed on-line using narrow-bore
reverse-phase HPLC.
DNA manipulation and transformations
Standard DNA methods described by Maniatis et al. (Maniatis, T. et al.,
Molecular Cloning: A Laboratory Manual (Cold Spring Harbor
10 Laboratory, Cold Spring Harbor, New York) (1982)) were used for the
DNA constructions. Plasmid and cosmid DNAs were isolated using
Qiagen columns (Diagen GmbH, Diisseldorf, Germany) according to the
manufacturer's instructions. E. coli was transformed according to Hanahan
(Hanahan, D., J. Mol. Biol. 166:557-580 (1983)).
15 RNA isolation
For RNA isolation, T. reesei (VTT-D-79125) was grown in a fermentor in
a xylanase-inducing medium at 30 C for three days (as described hereinfor
the isolation of the xln2 gene). The mycelia were ground into a fine
powder under liquid nitrogen. Total RNA and mRNA were isolated as
20 described by Bartels and Thompson (Bartels, D., and Thompson, RD.,
Nucleic Acid Res. 11:2961-2978 (1983)) and size-fractionated using
DMSO-sucrose gradient centrifugation (Boedtker, H. et al., Biochemistry
15:4765-4770 (1976)).
Cloning of xylanase I cDNA
25 The synthesis of the first strand of cDNA and the subsequent PCR
amplifications were performed as described hereir. for x1n2 except that here
an oligomer (5' AA(T/C)-TA(T/C)-GA(T/C)-CA(G/A)-AA(T/C)-TA(T/C)-
GA 3') [SEQ ID No.:9:] deduced from the N-terminal sequence
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
56
of the xylanase I protein was used as the gene-specific primer. The 0.6 kb
PCR fragment obtained was subcloned into the pCR1000 vector and
sequenced.
Isolation of xlnl gene
A T. reesei (QM6a, Mandels, M., Reese, E.T., J. Bacteriol. 73:269-280
(1957)) genomic cosmid library was screened with a digoxigenin-labelled
PCR fragment of xylanase I cDNA (see Example 1) according to the
supplier's instructions (Boehringer Mannheim, Mannheim, Germany). One
positive cosmid clone was obtained. A restriction map of the xlnl area of
the cosmid was prepared, and a 2.3 kb EcoR1 fragment was subcloned into
pBluescript (pALK572) for sequencing.
Nucleotide sequencing
The templates for nucleotide sequencing were generated by unidirectional
deletions according to the manufacturer's instructions for the pBluescript
Exo/Mung DNA sequencing system (Stratagene). DNA was sequenced in
both directions by using ABI (Applied Biosystems, Foster City, USA) kits
based on fluoresence-labelled T7 and T3 primers and a Taq cycle
sequencing method according to the supplier's instructions. Sequencing
reactions were analysed on an ABI 373A Sequencer.
Results
Cloning of T. reesei xlnl cDNA
The amino-terminal sequence of purified xylanase I was found to be Ala-
Ser-Ile-Asn-Tyr-Asp-Gln-Asn-Tyr-Gln-Thr-Gly-Gly-Gln- V al- Ser-Tyr-(Ser)-
Pro-(Ser)-Asn-Thr-Gly-Phe-Ser [SEQ ID No.:10:]. Five
tryptic peptides were also obtained and directly sequenced (see Figure 10
for peptide sequences).
SUBSTITUTE SHEET
WO 93/24621 2136350 PCT/F193/00221
57
Northern hybridization using the oligomer deduced from the xylanase I N-
terminal peptide sequence (see above) as a probe indicated that xylanase I
mRNA was approximately 0.7 kb in size.
PCR amplification of the first strand of cDNA resulted in a fragment of
0.7 kb. The nucleotide sequence of this DNA when translated into a
protein contained all the xylanase I peptide sequences, including the
N-terminal (see Figure 10).
Isolation and nucleotide sequence of the T. reesei xlnl gene
One positive clone was isolated from a genomic cosmid library of T.
reesei. The gene for xylanase I(xlnl ) was found in a 2.3 kb EcoRI
fragment of the cosmid (Figure 9).
The nucleotide sequence of the xlnl gene was determined and is presented
in Figure 10 together with the deduced amino acid sequence. A TATA
box was found approximately 90 nt upstream of the putative translation
start site. Of the three different putative translation initiation sites
preceding the N-terminus of the mature protein, only one was in an
environment resembling the highly conserved consensus sequence 5' CA-
C/A-A/C-ATG 3' for the translation start site among filamentous fungi
(Ballance, J.D., in Leone, S.A., Berka, RM., eds., Molecular Industrial
Mycology. Systems and applications for filamentous fungi (Marcel Dekker,
Inc., New York), pp. 1-29 (1991)). Thus the gene codes for a protein of
229 amino acids. In this protein, the N-terminal (obtained by direct
peptide sequencing, see above) is preceded by a 51 amino acid long signal
propeptide containing a primary signal sequence cleavage site (von Heijne,
Nucleic Acids Res. 14:4683-4690 (1986)) between the amino acids Ala19
and Met2O (Figure 10). Comparison of the genomic sequence with the
cDNA sequence revealed one intron of 62 bp. On the basis of these data,
it can be concluded that the mature xylanase I is a protein of 178 amino
acids with a calculated molecular weight of 19.1 kDa. This is in good
agreement with the 19 kDa obtained for purified xylanase I from T. reesei
SUBSTITUTE SHEET
WO 93/24621 PCT/F793/00221
2136350 58
(Tenkanen, M. et al., Enzyme Microb. Technol. 14:566-574 (1992)).
Unlike the gene of xylanase II (see herein), there is no sequence (Asn-X-
Ser/Thr where X is not Pro; Gavel, Y., von Heijne, G., Protein
Engineering 3:433-442 (1990)) for N-glycosylation.
Comparison of T. reesei xylanase I with other xylanases
Both of the proposed catalytic glutamic acids of xylanases (Katsube, Y.
et al., Proc. 2nd Int. Conference Protein Engineering (Japan Scientific
Societies Press, Tokyo), pp. 91-96 (1990); Wakarchuk, W. et al., in Visser
J. et al., eds., Xylans and Xylanases (Elsevier Science, Amsterdam), pp.
439-442 (1991)) were found in xylanase I by sequence alignment,
corresponding to Glu75 and Glu164 of the mature enzyme (Figure 10).
Table 2 presents a sequence alignment comparison showing the percent
identity between different xylanases. The upper right half of the matrix
shows the results of an alignment of complete mature sequences, and the
lower left half of the sequences between the proposed active site glutamic
acids of xylanases.
SUBSTITUTE SHEET
WO 93/24621 Z1363j U PCT/F193/00221
59
Table 2
Identity % of sequence of alignments between different xylanases
expressed as a matrix. Upper right half: mature enzyme sequence alignments.
Lower left half: sequence alignments between the two proposed active site
glutamic acids.
TI, T. reesei xylanase I; TII, T. reesei xylanase II; TV, T. viride
(Yaguchi, M. et al., in Visser, J. et al., eds., Xylans and Xylanases
(Elsevier
Science, Amsterdam), pp. 149-154 (1992a)); TH, T. harzianum (Yaguchi, M.
et al., in Visser, J. et al., eds., Xylans and Xylanases (Elsevier Science,
Amsterdam), pp. 435-438 (1992b)); AT, A. tubigensis (van den Broeck, H.
et al., "Cloning and expression of xylanase genes from fungal origin," EP 0
463 706 Al (1992)); SC, Schizophyllum commune (Oku, T. et al., Canadian
Fed. Biol. Soc. Annu. Meet. (Quebec City), Abst. 676 (1988)); AK, A.
kawachii (Ito, K. et al., Biosci. Biotec. Biochem. 56:906-912 (1992), the
first
190 amino acids were aligned); BC, Bacillus circulans (Yang, R. C. A. et al.,
Nucleic Acids Res. 16:7187 (1988)); BP, P. pumilus (Fukasaki, E. et al.,
FEBSLett. 171:197-201 (1984)); BS, B. subtilis (Paice, M.G. et al., Arch.
Microbiol 144:201-206 (1986)); CA, Clostridium acetobutylicum (Zappe H.
et al., Nucleic Acids Res. 18:2179 (1990)); SS, Streptonryces sp. 36a
(Nagashima, M. et al., Trends Actinomycetologia:91-96 (1989)).
TI TII TV TH AT SC AK BC BP BS CA SS
TI 100 52 52 51 47 42 16 49 40 49 40 43
TII 60 100 98 94 43 54 17 52 48 52 46 52
TV 60 99 100 94 43 54 25 52 49 52 46 51
TH 57 98 97 100 43 56 19 52 48 52 44 51
AT 62 57 57 58 100 38 17 40 34 40 32 33
SC 52 51 51 52 50 100 15 52 42 52 44 50
AK 20 21 20 21 23 21 100 14 18 14 20 19
BC 53 54 54 56 53 54 16 100 48 100 45 58
BP 52 57 57 58 47 48 17 59 100 48 71 49
BS 53 54 54 56 53 54 14 99 60 100 45 58
CA 52 51 52 52 42 47 19 57 73 58 100 46
SS 50 53 53 53 46 49 18 64 56 64 52 100
It can be seen that T. reeset xylanase II (pI 9.0) is almost identical with
the T. viride and T. harzianum small, high pI xylanases (Yaguchi, M.
et al., in Visser, J. et al., eds., Xylans and Xylanases (Elsevier Science,
SUBSTlTUTE SHEET
WO 93/24621 2 1 3 635 ~ PCT/FI93/00221 60
Amsterdam), pp. 149-154 (1992); Yaguchi, M. et al., in Visser, J. et aL,
eds., Xylans and Xylanases (Elsevier Science, Amsterdam), pp. 435-438
(1992)). Alignment of the region between the two active site glutamic
acids resulted in most cases in a higher identity than when complete mature
sequences where aligned.
Discussion
T. reesei produces at least two different xylanases, xylanase I with a pI of
5.5 and xylanase II with a pI of 9, which are small proteins of 19 kDa and
20 kDa, respectively (Tenkanen, M. et al., Enzyme Microb. Technol.
14:566-574 (1992)). The corresponding genes xlnl and xln2 have been
cloned and described herein. In overall structure the xlnl and xln2 genes
are very similar; they are about the same size and both contain only one
intron and a long pre-propeptide. However, these two xylanases only
show a 54 % identity at the DNA level and a 52 % identity at the amino
acid level. Thus, they are clearly different not only in primary structure
but also in enzymatic properties such as tolerance of different pH values or
temperatures and kinetic parameters as shown by Tenkanen et al.
(Tenkanen, M. et al., Enzyme Microb. Technol. 14:566-574 (1992)).
Based on hydrophobic cluster analysis, xylanases have been divided into
two subfamilies, F and G (Henrissat, B. et aL, Gene 81:83-95 (1989)).
Subfamily F comprises the high-MW xylanases whereas the low-MW
xylanases belong to subfamily G. Henrissat (Henrissat B., in Visser, J.
et al., eds., Xylans and Xylanases, Proc. Int. Symp. Wageningen (Elsevier,
Amsterdam), pp. 97-110 (1991)) suggests that the xylanases of these two
subfamilies undergo different folding. The high identity of T. reesei
xylanases I and II to all other xylanases compared (Table 2), except the A.
kawachii xylase A, suggests that they too, like the other low-MW
xylanases, belong to subfamily G. In the case of the A. kawachii xylanase
A (Ito, K. et al., Biosci. Biotec. Biochem. 56:906-912 (1992)), an identity
SUBSTITUTE SHEET
WO 93/24621 2136350 PCr/F193/00221
61
of only about 20% to the T. reesei xylanases was found. This xylanase has
a higher MW of 32.7 kDa, and it has been proposed to belong to subfamily
F (Ito, K. et al., Biosci. Biotec. Biochem. 56:906-912 (1992)).
Example 4
Increased production of XYLI by Trichoderma transformed with the
construction containing the xlnl gene fused to the cbhl promoter
(pAL%807)
Construction of the plasmid pAL%807 and transformation of the
Trichoderma strain ALK02221
The plasmid pALK807 was constructed by using the same strategy as in
construction pALKl74 (Figure 11). The 98 bp PCR fragment containing
the fusion of the cbhl promoter to the putative xlnl signal sequence and
xlnl sequence to the internal XhoI site (see the pALK572 map in Figure 2
or the xlnl sequence) was made by PCR using the following
oligonucleotides:
5'-primer (39-mer) [SEQ ID No.:13:1:
5'- C AAC CGC GGA CTG CGC ATC ATG GTT GCC TTT TCC AGC CT
SaciI beginning of the putative
end of the cbhl promoter xlnl signal sequence
3' primer (30-mer) [SEQ ID No.:14:]:
5'-CAG GCT CGA GGC CTG TGG GCA TCG CCA GAG
XhoI
x1nl. sequence
SUBSTITUTE SHEET
WO 93/24621 PCf/F193/00221
2136350
62
Plasmid pALK572 containing the xlnl gene was used as a template for the
PCR reaction. The reaction and purification of the product were done like
in constructing pALK174. To obtain pALK805, the purified and SacII-
XhoI digested PCR fragment was ligated to the SacII-Xhol cut plasmid
pALK486 containing the cbhl promoter. To construct pALK806, the xlnl
sequence downstream from the XhoI site was isolated from the plasmid
pALK572 by PstI (filled by T4 DNA polymerase) -XhoI digestion and the
fragment was ligated to the BamHI (filled in with Klenow) - XhoI digested
pALK805. pALK805 was obtained by ligating the EcoRI-SpeI fragment
(filled in by Klenow) from pALK424, containing the amdS gene and the
chbl 3'-flanking region (as in pALK174), into the EcoRV cut pALK806.
T. reesei strain ALK02221 was transformed with the 8.2 kb NotI
fragment from the plasmid pALK807.
Production of xylanase by the pALK807 transformants,
Total of 51 pALK807 transformants were purified, analyzed and
grown as in Example 2. Targeting efficiency to the cbhl locus was 35%.
The xylanase activity was measured as in Example 2, but at the pH 4.3
which is at the optimum pH range for the XYLI activity (Tenkanen et al.,
Enzyme Microb. Technol. 14:566-574 (1992)). The results, as an increase
in xylanase activity produced compared to that of the host strain
ALK02221 are shown in the Table 3. Thirty best xylanase producers
obtained are included. One bottle of each transformant was grown.
The best transformants produced over 10 fold the amount of
xylanase activity compared to the parent. Differing from the XYLII
(pALK174) transformants, the best xylanase producers were CBHI+.
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
2136350
63
Table 3
x
transformant CBHI+/- compared to
number (dot blot) ALK02221
(control) (+)
ALK02221
20 (+) 11.8
5 (+) 11.7
18 (+) 10.4
4 (+) 9.8
33 (+) 9.5
32 (-) 9.2
21 (+) 8.9
3 (+) 8.9
46 (+) 8.8
40 (+) 8.2
1 (+) 8.2
43 (+) 8.0
10 (+) 8.0
19 (+) 8.0
8 (+) 7.9
51 (+) 7.8
39 (+) 7.6
12 (-) 7.5
7 (-) 7.2
36 (+) 6.9
41 (-) 6.8
(+) 6.8
49 (-) 6.6
29 (-) 6.6
30 24 (+) 6.3
50 (-) 5.9
14 (-) 5.9
26 (+) 5.8
15 (-) 5.8
35 52 (+) 5.1
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
Z136350
64
Example 5
A. Inactivation of the Major Cellulase cbhl Gene
The cbhl gene which encodes the major cellulase in T. reesei was
inactivated by homologous recombination with plasmid pMS4 containing a
0.8 kb internal fragment of the cbhl cDNA bearing a frame shift mutation.
The pMS4 plasmid was prepared on the following way: the plasmid
pTTcOl (Teeri et al., Anal. Biochem. 164:60-67 (1987); Penttila et al.,
Gene 63:103-112 (1988)), which contains the full length cDNA clone of the
cbhl gene in the pUC8 vector (Vieira and Messing, Gene 19:259-268
(1982)), was digested with Bgll cutting in the signal sequence (Shoemaker
et al., BiolTechnology 1:691-696 (1983)) and with BgIII. The resulting
0.8 kb DNA fragment bearing the 5' region of the cbhl cDNA was made
blunt-ended with S, nuclease and was ligated to an EcoRl cut, blunt-
ended pUC18 vector (Yanish-Perron et al., Gene 33:103-119 (1985)).
The clone obtained was cut in the middle of the cbhl fragment with
EcoRI. The EcoRI generated termini were then filled in and back-ligated.
The resulting plasmid pMS4 thus contains a frameshift mutation in the
middle of the truncated cbhl cDNA fragment.
T. reesei VTT-D-79125 (Bailey and Nevalainen, Enzyme Microb.
Technol. 3:153-157 (1981)) was cotransformed with pMS4 and p3SR2.
p3SR2 carries a 5 kb DNA fragment containing the A. nidulans amdS gene
cloned into pBR322 (Kelly and Hynes, EMBO J. 4:475-479 (1985)).
Transformants were selected on the basis of the AmdS+ phenotype after
which they were purified from conidia. About 600 clones from 200
independent transformants were then grown on microtiter plates and their
cellulase phenotype was tested by the Ouchterlony immunodiffusion
(Ouchterlony, Progr. Allergy 5:1-78 (1958)) using undiluted growth medium
and the CBHI specific sheep antiserum.
A number of strains produced no detectable CBHI. The CBHI
negative character of one of these strains VTT-D-87312 was confirmed by
SUBSTaTUTE aHEET
WO 93/24621 213635t1 PCr/FI93/00221
V
analyzing the growth medium in SDS-PAGE and in FPLC, in which no
peak corresponding CBHI was seen (compare Figure 12 and Figure 12A).
The amount of total secreted protein of the CBHI negative strain was about
half of that secreted by the T. reesei VTT-D-79125. The filter paper
5 degrading activity, FPU (Mandels et al.,"Measurement of Saccharifying
Cellulase," in: Biotechnol. Bioeng. Symp. no. 6., p. 21-33, Gaden, E.L.,
Mandels, M.H., Reese, E.T., and Spano, L.A. (eds.), John Wiley and Sons,
New York, 1976) activity detected in the culture supematant fraction of the
strain VTT-D-87312 was significantly reduced and was about 20% of
10 normal. The lack of the major cellobiohydrolase which normally
represents about 60% of the total secreted protein did not notably change
the growth properties of the strain.
B. Deletion of the cbh2 Gen ti Its Promoter
The cbh2 gene of Trichoderma replaced with the Aspergillus argB
15 gene as described below. Plasmid pAI.K99 was constructed to be the
source of the transforming fragment (Figure 13). Plasmid pALK99 was
constructed in the following way. The PvuH fragment (containing the
multilinker) of the plasmid pUC19 was replaced by a new synthetic
multilinker fragment containing recognition sites for the following
20 restriction enzymes: XhoI-StuI-SmaI XbaI-PvuII-SalI XhoI. The new
plasmid was called pALK96. This plasmid was cut with Xbal and PvuH
and a 2.1kb XbaI-PvuII fragment from the 3' region of the cbh2 gene (see
Figure 14) was ligated into it. The resulting plasmid was cut with PvuII
and HincII and ligated with the 3.4 kb PvuII-fragment from the 5' area of
25 the cbh2-gene (see Figure 14). Both the 3' and 5' fragments were
originally from the X clone cbh2lambdal (Teeri et al., Gene 51:43-52
(1987)). The resulting plasmid was called pALK98. The Aspergillus
nidulans argB gene (2.6kb Sali fragment) was then ligated between the 3'
and 51 regions of the cbh2 gene into the unique PvuII site of plasmid
SUBSTiTUTE SHEET
WO 93/24621 PCf/F793/00227
....,
66
pALK98. The resulting plasmid was called pALK99. Thus, the
transforming fragment which is isolated from pALK99 as a XhoI fragment
contains the Aspergillus argB gene as a 2.6 kb Sall fragment (Berse et al.,
Gene 25:109-117 (1983)) between 3.4kb (PvuII-PvuII fragment) of the 5'
flanking region and 2.1kb (PvuII-Xbal fragment) of the 3' flanking region
of the cbh2 gene (see Figure 14). T. reesei VTT-D-87305 ArgB- mutant
strain (Penttila et al., 1987, Gene 61:155-164) was transformed with this
fragment using selection for arginine prototropy. ArgB+ transformants were
then screened for CBHII- phenotype by Western blotting using monoclonal
antibody against CBHII. Replacement of the cbh2 locus by the
transforming fragment in the CBHII-transformants was then confirmed by
Southern blots. Strain ALKO 2564 is an example of this kind of
"replacement" strain and thus does not contain the cbh2 gene any more.
With the method described above, the cbh2 gene of Trichoderma
was replaced with the Aspergillus trpC gene.
The Aspergillus nidulans trpC gene (4.2 kb Xhol fragment blunt-
ended with Klenow enzyme) was ligated between the 3'and 5'regions of the
cbh2 gene into the unique PvulI site of the plasmid pALK98. The resulting
plasmid was called pALK402. The transforming fragment which is isolated
from pALK402 as XhoI fragment contains A. nidulans trpC gene as a 4.2
kb XhoI fragment (Yelton et al., PNAS, 91:1470-1474 (1984) between the
3.4 kb of the 5'flanking region and 2.1 kb of the 3'flanking region of the
cbh2 gene. T. reesei ALK02319 trpC' mutant strain was transformed with
this fragment using selection for tryptophane prototrophy. TrpC+
transformants were screened for CBHII- phenotype by Western blotting
using monoclonal antibody against CBHII. Replacement of the cbh2 locus
by the transforming fragment in the CBHII- transformants was confirmed by
Southern blots. Strain ALK02721 is an example of this kind of
"replacement" strain and thus does not contain the cbh2 gene any more.
SUBSTITUTE SHEET
WO 93/24621 2136350 PC,'I'/F193/00221
67
Example 6
Construction of a CBHI Negative Trichoderma Strain
Producing Elevated Amounts of EGI
The CBHI negative strain VTT-D-87312 described in Example 5A
was transformed with the plasmid pAMH 111 to enhance EGI expression
in a CBHI negative background. The plasmid pAMH 111 was constructed
using the general expression vector pAMH 110 (both of these plasmids are
described in EP 244,234). pAMH 110 was built from pUC19 (Yanish
Perron et al., Gene 33:103-119 (1985)). First the single Ndel site of
pUC19 was destroyed by filling in the recessed ends with Kienow
polymerase, and then the plasmid was digested with EcoRI and PstI and
ligated to cbhl promoter and terminator fragments to make an expression
cassette. The promoter fragment was a 2.6 kb EcoRI-PstI fragment from
the plasmid pAMH 102 (Harkki et al., BiolTechnology 7:596-603 (1989)).
The terminator was a 0.75 kb AvalI fragment contained in a PstI fragment
which also included an adaptor with the TAA stop codon in all three
reading frames. pAMH 110 was then digested with SacII and Nde1 to
remove a piece of DNA between the cbhl promoter and terminator, and
the digested ends were made blunt-ended with S, nuclease and Klenow
polymerase. The egll cDNA to be expressed was taken from the plasmid
pTTc11 ((Teeri et al., Anal. Biochem. 164:60-67 (1987); Penttilli et al.,
Yeast 3:175 -185 (1987); as a 1.6 kb EcoRI-BamHI fragment, made
blunt-ended with Klenow polymerase, and ligated into the expression
cassette to give plasmid pAMH 111. Transformation was carried out as a
cotransformation with pAMH111 and the plasmid pAN8-l (Mattern et. al.,
"Transformations of Aspergillus oryzae," In: Abstracts of the 19th Lunteren
Lectures of Molecular Genetics of Yeasts and Filamentous Fungi and its
Impact on Biotechnology, Lunteren, the Netherlands, p.34, (1987)) carrying
the phleomycin resistance gene of Steptoallotheicus hindustanus under the
SUBSTOTUTE SHEET
WO 93/24621 PCT/F193/00221
68
A. nidulans gpd promoter. Another marker must be used if, as in this
example, the strain VTT-D-87312 was already AmdS+. Transformants
were purified and tested for endoglucanase production in shake flasks
cultures. In about 20 % of the transformants, the level of
hydroxyethylcellulose (HEC) hydrolyzing activity was higher than in the
recipient strain. The amount of EGI protein (Table 4) in the shake culture
supernatant fraction was analyzed from three transformants showing high
HEC activity. Southern blot analysis of these transformants showed that in
the best endoglucanase producing clone (ALKO 2466) the expression
cassette containing the egll cDNA between the cbhl promoter and
terminator sequences was integrated in the chromosomal cbhl locus
through the terminator sequences on the insert. The amount of secreted
EGI protein in this transformant strain (ALKO 2466) was increased about
four fold over that of the control (Table 4).
Table 4
Characterization of EGI production in T. reesei VTT-D-87312 (CBHI
negative transformant of VTT-D-79125) and in ALKO 2493, ALKO 2466 and
ALKO 2494 which arise from VTT-D-87312 transformed with the plasmid
pAMH111. TT-D-79125 is the untransformed high-cellulase producing T.
reesei mutant strain. All strains were grown in shake flasks as described in
the
Materials and Methods. The amount of EGI protein and total secreted protein
were measured after 7 days cultivation as described in the Materials and
Methods.
EG1 protein Total secreted protein % EGI
% of the total
(mg/ml) (mg/ml) secreted protein
VTT-D-87312 0.35 4.5 7.7
ALKO 2493 1.25 5.2 24.0
ALKO 2466 1.90 5.8 32.8
ALKO 2494 1.40 5.9 23.7
VTT-D-79125 0.75 10.6 7.1
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
2136350
69
Example 7
Simultaneous Inactivation of the cbhl Gene and
Multiplication of the egll Copy Number
The cbhl gene of T. reesei VTT-D-79125 was replaced with the
Trichoderma egll cDNA and amdS gene. The egll cDNA was ligated
between the promoter and terminator of cbhl gene. Plasmid pALK412
was constructed to be the source of the transforming fragment. The
plasmid pALK412 was prepared as in Figure 15.
The plasmid p3SR2 which contains the Aspergillus nidulans amdS gene
cloned into pBR322 (Kelly and Hynes, EMBO J. 4:475-479 (1985)) was
digested with Sphl and with XbaI. The resulting 3.2 kb DNA fragment
bearing the whole amdS gene was ligated to the Sphl and Xbal cut pUC 19
vector (Yanisch-Perron et al., Gene 33:103-119 (1985)). The resulting
plasmid was called pALK410.
A DNA fragment containing 1.65 kb of the 3' region of the cbhl gene
starting from the ScaI site in the coding region was isolated as a ScaI-
BamHI fragment and blunt-ended with Klenow-enzyme. This fragment
was ligated to the Xbal site (blunt-ended with Klenow enzyme) of the
plasmid pALK410. In this case the 3' fragment was isolated from the
plasmid pTTl l.
Plasmid pTTI l(Teeri et al., BiolTech 1:696-699 (1983)) contains 1.8 kb
fragment of the cbhl region, 3' from the BamHI site in the coding region,
cloned into the BamHI site of pBR322. The gene can also be isolated
from other sources, for example, from ak clone 44A (Teeri et al.,
BiolTech 1:696-699).
The plasmid obtained was pALK41 1. It was digested with Scal and with
Sphl. The 5.8 kb fragment was ligated to ScaI and Sphl cut plasmid
pPLE3 (Nevalainen, et al., In: Molecular Industrial Mycology: Systems and
Applications for Filamentous Fungi, Leong et al., eds., pp. 129-148
(1990)) which contains egll cDNA between the promoter and terminator
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
2136150
regions of cbhl gene cloned into pUC18 (Figure 16). The promoter and
terminator regions are from the expression vector pAMH 110 (Nevalainen
et al., In: Molecular Industrial Mycology: Systems and Applications for
Filamentous Fungi, Leong et al., eds., pp. 129-148 (1990)).
5 The resulting plasmid pALK412 was cut with EcoRI to remove the
bacterial DNA. The 9.3 kb pALK412F fragment was also backligated to
form plasmid pALK412L.
T. reesei VTT-D-79125 (Bailey and Nevalainen, Enzyme Microb.
Technol. 3:153-157 (1981)) was transformed with plasmid pALK412, with
10 pALK412F linear fragment, with backligated pALK412L and with
pALK412F and pALK412L at the same time with a molar ratio of 5:1
respectively. Transformants were selected on the basis of the amdS+
phenotype and purified from conidia on selective medium containing
acetamide as a sole nitrogen source.
15 Purified transformants were grown on microtiter plates and were
screened for CBHI- phenotype by Western blotting using polyclonal
antibody against CBHI protein. About one third of the pALK412F
transformants produced no detectable CBHI. There was one CBHI"
transformant among forty strains that had been transformed with the
20 plasmid pALK412.
CBHI' transformants were tested for endoglucanase production in
shake flasks cultures. In all of these transformants the level of
hydroxyethylcellulose (HEC) hydrolyzing activity was higher than in the
recipient strain. The best transformants secreted 4-5 times the
25 endoglucanase activity of the recipient strain.
Southern blot analysis of the CBHI' transformants showed that their
cbhl locus was replaced by the vector fragment. The chromosomal DNA
of the transformants was digested with Xhol and hybridized with the 0.5 kb
fragment of the cbhl coding region probe.
30 The best endoglucanase producing strains had more than one copy of
the vector fragment which carries the gene of interest inserted into the
SUBSTITUTE SHEET
WO 93/24621 ~ ~ ~ ~ ~ ~ Q PCT/FI93/00221
71
Trichoderma genome.
Example 8
Construction of a cellobiohydrolase negative T. reesei strain
The hypercellulolytic mutant VTT-D-79125 (Bailey, M.J. et al., Enzyme
Microb. Technol. 3:153-157 (1982)) is a good cellulase producer, and also,
has a markedly better viability and increased capability to produce secreted
protein as a result of the mutagenesis program that created it. Thus, we
wanted to make use of the high protein production capacity of this mutant
to produce xylanases free of cellobiohydrolases to be used in pulp
bleaching.
An UV-induced tryptophan auxotrophic mutant of VTT-D-79125 was
transformed with a linear fi-< -ient of DNA (Figure 17), on which the cbh2
flanking regions were used to target the transforming fragment into the
cbh2 locus. Purified TrpC+ transF rmants were grown on inicrotiter plates
and were screened for CBHII" phenotype by Western blotting using
monoclonal antibody against CBHII. Replacement of the cbh2 locus by
the transforming fragment was confirmed by Southern blotting. One such
cbh2 negative transformant was transformed with a second DNA fragment.
This time cbhl flanking regions were used in order to replace the wild type
cbhl locus with the amdS marker for acetamidase. Purified amdS+
transformants were grown on microtiter plates and screened for CBHI'
phenotype by Western blotting using polyclonal antibody against CBHI
protein. The replacement was again confirmed by Southern blotting. The
resulting strain has neither the cbhl nor the cbh2 gene and thus is unable
to produce cellobiohydrolases in any conditions.
Figure 18 illustrates the enzyme production profiles of the hypercellulolytic
strain and its cellobiohydrolase negative derivative. Production of
endoglucanases is lowered by choosing suitable fermentation conditions as
well known in the art. With the new strain xylanase can be produced as
SUBSTITUTE SHEET
WO 93/24621 PCT/F193/00221
2136350
72
an enzyme composition secreted from the host cell, such enzyme
composition not containing cellobiohydrolases, and thus lacking activity
against crystalline cellulose. The very small amount of endoglucanase
activity still present does not have any harmful effects on the pulp
properties when this kind of preparation is used in enzyme aided pulp
bleaching.
Example 9
Construction of Strains Producing
Different Combinations of Cellulases
Using similar gene replacement strategy as in the previous strain
constructions (Figure 17), sets of T. reesei strains have been constructed
that produce different combinations of cellulases (Table 5). In the first set,
strains A-D, one cellulase is eliminated at a time. Strains A-D synthesize
everything the parent strain does except for CBHI (strain A), CBHII (strain
B), EGI (strain C) or EGII (strain D). The second set, strains E-J,
provides mutants missing all pairs of the four cellulases. Strain E lacks
CBHI and CBHII. Strain F lacks CBHI and EGI. Strain G lacks CBHI
and EGII. Strain H lacks CBHII and EGI. Strain I lacks CBHII and
EGII. Strain J lacks EGI and EGII.
When genes for both EGI and EGII (strain J, Table 5) are deleted, the
activity against hydroxyethylcellulose drops to less than 10% of the
activity produced by the parent hypercellulolytic strain, VTT-D-79125.
Lack of CBHI or CBHII proteins (strain E, Table 5) can be assayed by
examining for activity against filter paper as known in the art. When both
CBHI and CBHII are eliminated, no measurable activity is produced. One
of skill in the art would recognize that further modifications, such as to
eliminate three or more activities may also be constructed by the same
strategy.
SUBSTITUTE SHEET
WO 93/24621 2136350 PCT/F193/00221
1 .
73
Table 5
Genetically Engineered Strains
to Produce Novel Cellulase Mixtures
Strain CBHI CBHII EGI EGII
type
A - + + +
B + - + +
C + + - +
D + + + -
E - - + +
F - + - +
G - + + -
H + - - +
I + - + -
J + + - -
K + - - -
L - - + -
Figure 19 illustrate the powerfulness of gene technology to produce the
compositions of the invention. Genetic engineering has been successfully
used to construct derivatives of the hypercellulolytic mutant to produce
different cellulase-xylanase mixtures. In strain 3, the cbhl gene coding
sequence is replaced with the egll gene coding sequence, which in strain 3
is expressed under the cbhl promoter. Thus, the proportion of
endoglucanases produced and secreted by these hosts is increased.
Strain 1 produces xylanases as the main activity, and in different
production conditions, as known in the art, endoglucanases without any
cellobiohydrolases can be produced. In the enzyme mixture produced by
strain 3, the proportion of endoglucanases is increased, and strain 2
produces cellobiohydrolases essentially free of endoglucanases.
SUBSTITUTE SHEET
WO 93/24621 2136s;? r~ 50 PCT/F193/0027'
74
Example 10
Use of the Enzyme Preparations in Biobleaching
Oxygen-delignified softwood kraft mill pulp (kappa 16.1, brightness 35.4%
ISO) was used. The pulp pH was adjusted with sulphuric acid to 6. The
pulp samples of 250 g dry matter were treated with enzyme A3273
("enzyme A") or A2799 ("enzyme B") so that the xylanase dosage was 100
nkat/g pulp dry matter, at pH 6 for 55 C for 2 hours and washed. Then
they were bleached using bleaching sequence D. (EO) DED. Reference
pulp was bleached without enzyme treatment.
Following standard methods were used: brightness (ISO 2470), kappa
number (ISO 302), viscosity (ISO 5351/1) and pc (post-coloring) number
(TAPPI 260 pm-81). Pulps were beaten with a PFI mill (ISO 5264/2),
handsheets were made according ISO 5269/1 and the strength properties
were tested (ISO 5270).
The enzyme treated pulps were bleached using 25% lower C1O2 dosage in
the first bleaching stage than in the reference pulp. Enzyme pretreated
pulp achieved the full brightness with about 15% less aCl in total (Table
6). This is about the same as achieved in earlier experiments using
conventionally cooked pulps (kappa number 31-32) without 02-
delignification and bleaching sequences containing elementary chlorine
(Lahtinen, T., et al., In: Biotechnology in Pulp and Paper Industry,
Kuwahara, M., Shimada, M. (eds.), Uni Publishers Co., Tokyo, Japan, pp.
129-137 (1992)).
The strength properties (viscosity and tear index) correlated with the
cellulolytic side activities: with low side activities (enzymes A), strength
properties similar to the reference were obtained. Optical properties were
also comparable to reference, with the exception of the pc-number (the
measure of brightness reversion) which was even better in enzyme treated
pulps.
SUBSTi i'fdTE SHEE'f
WO 93/24621 2136350 PCT/F193/00221
Table 6. ECF bleaching experiments
Enzymes Ref
A B
Enzyme sta, :
Dose, nkat/g 100 100 -
Temp., C 55 55 -
5 pH 6.0 6.0 -
time, hours 2 2 -
DO stage
consumption of C102, act.
Cl kg/t 24.2 24.1 31.9
10 EO stage
Brightn, % 62.5 62.4 63.1
Kappa 4.6 4.6 4.2
D 1 stage
Consumption of C10Z,
15 act. Cl kg/t 17 17 17
Brightn, % 82.9 82.5 83.5
D2 stage
Consumption of C102,
act. Cl kg/t 6.5 6.3 6.0
20 Final
Brightn., % 90.2 89.5 89.7
pc number 0.45 0.54 0.87
Viscosity 830 810 840
Total
25 Consumption of C102,
act. Cl kg/t 47.7 47.4 54.9
Consumption of C1021
act. Cl kg/t, at ISO 90' 46.9 49.4 56.1
Reduction 16% 12% --
30 Pulp properties after refming
(at Tensile Index 70 Nm/g)
PFI revs
Tear, mNm2/g 1123 945 871
Light scatt. coeff. m2/kg 13.8 2.2 14.2
35 Light abs. coeff. m2/kg 20.7 ". ,6 21.6
0.06 J8 0.06
Fiber length
mm 2.00 1.99 1.97
< 0.20 tnm, % 3.23 3.22 3.39
1: assuming that 4 kg aCl increases brightness by 1 ISO unit
SUBSTOTUTE SHEET
CA 02136350 2001-09-10
75154-6
76
Bleaching Conditions:
Stage DO (EO) D 1 E D2
Consistency, % 3 10 9 9 9
Temperature, C 55 75 70 70 70
Time, min. 60 60 180 60 180
End pH 2.9-3.2 10.8 3.7-4.0 10.8 3.5-3.8
C102 dose, kg/t 24.2 - 17 - 8
active Cl
OZ pressure, bar - 2 - - -
NaOH dose, kg/t - 17 2.5 9 -
Having now fully described the invention, it will be understood by
those with skill in the art that the scope may be performed within a wide
and equivalent range of conditions, parameters and the like, without
- affecting the spirit or scope of the invention or any embodiment thereof.
2136350
The Swedish Patent O{fice PCT/ Fl 9 3 / 0 0 2 2 1
PCT International Application
02 -08- 1993
77
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT:
(A) NAME: OY ALKO AB et al.
(B) STREET: Salmisaarenranta 7 H
(C) CITY: Helsinki
(D) COUNTRY: Finland
(E) POSTAL CODE: 00180
(ii) TITLE OF INVENTION: Novel Enzyme Preparations and Methods
for Their Production
(iii) NUMBER OF SEQUENCES: 14
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Release #1.0, Version #1.25
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER: PCT/F193/00221
(B) FILING DATE: 24-MAY-1993
(C) CLASSIFICATION:
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 07/524,308
(B) FILING DATE: 16-MAY-1990
(2) INFORMATION FOR SEQ ID NO:1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 1015 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ix) FEATURE:
(A) NAME/KEY: CDS
(B) LOCATION: join(176..448, 557..952)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:1:
AAGCTTGATG AGGCCAAATT ATCCGTCAAC TGTCTTATAA AGGAGCCCAT GCCAAACCCC 60
CCCTAAAGAC TCAAGAAGCC AAACCTGAAC AACCCCAGCA CCTGAACAGT CATACAACCC 120
CTCCAAGCCC AAAAGACACA ACAACTCCTA CTAGCTGAAG CAAGAAGACA TCAAC ATG 178
Met
1
GTC TCC TTC ACC TCC CTC CTC GCC GGC GTC GCC GCC ATC TCG GGC GTC 226
Val Ser Phe Thr Ser Leu Leu Ala Gly Val Ala Ala Ile Ser Gly Val
10 15
TTG GCC GCT CCC GCC GCC GAG GTC GAA TCC GTG GCT GTG GAG AAG CGC 274
Leu Ala Ala Pro Ala Ala Glu Val Glu Ser Val Ala Val Glu Lys Arg
20 25 30
CAG ACG ATT CAG CCC GGC ACG GGC TAC AAC AAC GGC TAC TTC TAC TCG 322
Gln Thr Ile Gln Pro Gly Thr Gly Tyr Asn Asn Gly Tyr Phe Tyr Ser
35 40 45
SUBSTITlJTi E SHEET
rl.l/ rl y-) / UU127
The Swedish Pa'.ent Office 02 -08- 1993
pCT Interna-ionat Application 2136350
.,. _ 78
TAC TGG AAC GAT GGC CAC GGC GGC GTG ACG TAC ACC AAT GGT CCC GGC 370
Tyr Trp Asn Asp Gly His Gly Gly Val Thr Tyr Thr Asn Gly Pro Gly
50 55 60 65
GGG CAG TTC TCC GTC AAC TGG TCC AAC TCG GGC AAC TTT GTC GGC GGC 418
Gly Gin Phe Ser Val Asn Trp Ser Asn Ser Gly Asn Phe Val Gly Gly
70 75 80
AAG GGA TGG CAG CCC GGG ACC AAG AAC AAG TAAGACTACC TACTCTTACC 468
Lys Gly Trp Gln Pro Gly Thr Lys Asn Lys
85 90
CCCTTTGACC AACACAGCAC AACACAATAC AACACATGTG ACTACCAATC ATGGAATCGG 528
ATCTAACAGC TGTGTTTTAA AAAAAAGG GTC ATC AAC TTC TCG GGA AGC TAC 580
Val Ile Asn Phe Ser Gly Ser Tyr
AAC CCC AAC GGC AAC AGC TAC CTC TCC GTG TAC GGC TGG TCC CGC AAC 628
Asn Pro Asn Gly Asn Ser Tyr Leu Ser Val Tyr Gly Trp Ser Arg Asn
100 105 110 115
CCC CTG ATC GAG TAC TAC ATC GTC GAG AAC TTT GGC ACC TAC AAC CCG 676
Pro Leu Ile Glu Tyr Tyr Ile Val Glu Asn Phe Gly Thr Tyr Asn Pro
120 125 130
TCC ACG GGC GCC ACC AAG CTG GGC GAG GTC ACC TCC GAC GGC AGC GTC 724
Ser Thr Gly Ala Thr Lys Leu Gly Glu Val Thr Ser Asp Gly Ser Val
135 140 145
TAC GAC ATT TAC CGC ACG CAG CGC GTC AAC CAG CCG TCC ATC ATC GGC 772
Tyr Asp Ile Tyr Arg Thr Gln Arg Val Asn Gln Pro Ser Ile Ile Gly
150 155 160
ACC GCC ACC TTT TAC CAG TAC TGG TCC GTC CGC CGC AAC CAC CGC TCG 820
Thr Ala Thr Phe Tyr Gln Tyr Trp Ser Val Arg Arg Asn His Arg Ser
165 170 175
AGC GGC TCC GTC AAC ACG GCG AAC CAC TTC AAC GCG TGG GCT CAG CAA 868
Ser Gly Ser Val Asn Thr Ala Asn His Phe Asn Ala Trp Ala Gln Gln
180 185 190 195
GGC CTG ACG CTC GGG ACG ATG GAT TAC CAG ATT GTT GCC GTG GAG GGT 916
Gly Leu Thr Leu Gly Thr Met Asp Tyr Gln Ile Val Ala Val Glu Gly
200 205 210
TAC TTT AGC TCT GGC TCT GCT TCC ATC ACC GTC AGC TAAAGGGGGC 962
Tyr Phe Ser Ser Gly Ser Ala Ser Ile Thr Val Ser
215 220
TCTTCTTTTG TGATGTGTGA AXAAAAAAAA AAGGATGGTG GATAAAAGGG GGT 1015
(2) INFORMATION FOR SEQ ID NO:2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 223 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: protein
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:2:
Met Val Ser Phe Thr Ser Leu Leu Ala Gly Val Ala Ala Ile Ser Gly
1 5 10 15
np.nnr*~e.~-r- ~+iv~rT
13635 0
The Swedish Pa vnt O`:`ice PCT/ FI 9 3/ 0 C 2 2 1
PCT Interna-ional AaplicGtion 0 2-08- 1993
79
Val Leu Ala Ala Pro Ala Ala Glu Val Glu Ser Val Ala Val Glu Lys
20 25 30
Arg Gln Thr Ile Gln Pro Gly Thr Gly Tyr Asn Asn Gly Tyr Phe Tyr
35 40 45
Ser Tyr Trp Asn Asp Gly His Gly Gly Val Thr Tyr Thr Asn Gly Pro
50 55 60
Giy Gly Gln Phe Ser Val Asn Trp Ser Asn Ser Gly Asn Phe Val Gly
65 70 75 80
Gly Lys Gly Trp Gln Pro Gly Thr Lys Asn Lys Val Ile Asn Phe Ser
85 90 95
Gly Ser Tyr Asn Pro Asn Gly Asn Ser Tyr Leu Ser Val Tyr Gly Trp
100 105 110
Ser Arg Asn Pro Leu Ile Glu Tyr Tyr Ile Val Glu Asn Phe Gly Thr
115 120 125
Tyr Asn Pro Ser Thr Gly Ala Thr Lys Leu Gly Glu Val Thr Ser Asp
130 135 140
Gly Ser Val Tyr Asp Ile Tyr Arg Thr Gln Arg Val Asn Gln Pro Ser
145 150 155 160
Ile Ile Gly Thr Ala Thr Phe Tyr Gln Tyr Trp Ser Val Arg Arg Asn
165 170 175
His Arg Ser Ser Gly Ser Val Asn Thr Ala Asn His Phe Asn Ala Trp
180 185 190
Ala Gln Gln Gly Leu Thr Leu Gly Thr Met Asp Tyr Gln Ile Val Ala
195 200 205
Val Glu Gly Tyr Phe Ser Ser Gly Ser Ala Ser Ile Thr Val Ser
210 215 220
(2) INFORMATION FOR SEQ ID NO:3:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 941 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ix) FEATURE:
(A) NAME/KEY:*CDS
(B) LOCATION: join(102..392, 455..850)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:3:
GAATTCTGCA TATATAAAGC CATGGAAGAA GACGTAAAAC TGAGACAGCA AGCTCAACTG 60
CATAGTATCG ACTTCAAGGA AAACACGCAC AAATAATCAT C ATG GTT GCC TTT 113
Met Val Ala Phe
1
TCC AGC CTC ATC TGC GCT CTC ACC AGC ATC GCC AGT ACT CTG GCG ATG 161
Ser Ser Leu Ile Cys Ala Leu Thr Ser Ile Ala Ser Thr Leu Ala Met
10 15 20
CCC ACA GGC CTC GAG CCT GAG AGC AGT GTC AAC GTC ACA GAG CGT GGC 209
Pro Thr Gly Leu Glu Pro Glu Ser Ser Val Asn Val Thr Glu Arg Gly
25 30 35
cs RQcTITIITF SHEET
2136350
The Swedish Patent Office r~ ~~ r I y 3/ 0 0 2 2 1
PCT International AFplication 02_0$_ 1993
ATG TAC GAC TTT GTT CTT GGA GCT CAC AAT GAT CAT CGC CGT CGT GCT 257
Met Tyr Asp Phe Val Leu Gly Ala His Asn Asp His Arg Arg Arg Ala
40 45 50
AGC ATC AAC TAC GAC CAA AAC TAC CAA ACT GGC GGA CAA GTC AGC TAT 305
Ser Ile Asn Tyr Asp Gln Asn Tyr Gln Thr Gly Gly Gln Val Ser Tyr
55 60 65
TCG CCT TCC AAC ACT GGC TTC TCA GTG AAC TGG AAC ACT CAA GAT GAC 353
Ser Pro Ser Asn Thr Gly Phe Ser Val Asn Trp Asn Thr Gln Asp Asp
70 75 80
TTT GTT GTG GGC GTT GGT TGG ACG ACT GGA TCT TCT GCG TCGGAGGATT 402
Phe Val Val Gly Val Gly Trp Thr Thr Gly Ser Ser Ala
90 95
CTCATCATTC TGCACTTTGA AAGCATCTTC TGACCAACAA GCTTCTCTTA GT CCC 457
Pro
ATC AAC TTT GGC GGC TCT TTT AGT GTC AAC AGC GGA ACT GGC CTG CTT 505
Ile Asn Phe Gly Gly Ser Phe Ser Val Asn Ser Gly Thr Gly Leu Leu
100 105 110
TCC GTC TAT GGC TGG AGC ACC AAC CCA CTG GTT GAG TAC TAC ATC ATG 553
Ser Val Tyr Gly Trp Ser Thr Asn Pro Leu Val Glu Tyr Tyr Ile Met
115 120 125 130
GAG GAC AAC CAC AAC TAC CCA GCA CAG GGT ACC GTC AAG GGA ACC GTC 601
Glu Asp Asn His Asn Tyr Pro Ala Gln Gly Thr Val Lys Gly Thr Val
135 140 145
ACC AGC GAC GGA GCC ACT TAC ACC ATC TGG GAG AAT ACC CGT GTC AAC 649
Thr Ser Asp Gly Ala Thr Tyr Thr Ile Trp Glu Asn Thr Arg Val Asn
150 155 160
GAG CCT TCC ATC CAG GGC ACA GCG ACC TTC AAC CAG TAC ATT TCC GTG 697
Glu Pro Ser Ile Gln Gly Thr Ala Thr Phe Asn Gln Tyr Ile Ser Val
165 170 175
CGG AAC TCG CCC AGG ACC AGC GGA ACT GTT ACT GTG CAG AAC CAC TTC 745
Arg Asn Ser Pro Arg Thr Ser Gly Thr Val Thr Val Gln Asn His Phe
180 185 190
AAT GCT TGG GCC TCG CTT GGC CTG CAC CTT GGG CAG ATG AAC TAC CAG 793
Asn Ala Trp Ala Ser Leu Gly Leu His Leu Gly Gln Met Asn Tyr Gln
195 200 205 210
GTT GTC GCT GTC GAA GGC TGG GGT GGT AGT GGT TCT GCC TCA CAG AGT 841
Val Val Ala Val Glu G1y,Trp Gly Gly Ser Gly Ser Ala Ser Gln Ser
215 220 225
GTC AGC AAC TAGGTTCTGT TGATGTTGAC TTGGAGTGGA TGAGGGGTTT 890
Val Ser Asn
GAGCTGGTAT GTAGTATTGG GGTGGTTAGT GAGTTAACTT GACAGACTGC A 941
(2) INFORMATION FOR SEQ ID NO:4:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 229 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
e=. ~~~~v"rvrTC cUCr-T
2136350f PCT/ FI 93/00221
The Svredish Pa.ent O.fice 02 -U8-
PCT Interna-ional Application ~~
81
(ii) MOLECULE TYPE: protein
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:4:
Met Val Ala Phe Ser Ser Leu Ile Cys Ala Leu Thr Ser Ile Ala Ser
1 5 10 15
Thr Leu Ala Met Pro Thr Gly Leu Glu Pro Glu Ser Ser Val Asn Val
20 25 30
Thr Glu Arg Gly Met Tyr Asp Phe Val Leu Gly Ala His Asn Asp His
35 40 45
Arg Arg Arg Ala Ser Ile Asn Tyr Asp Gln Asn Tyr Gln Thr Gly Gly
50 55 60
Gln Val Ser Tyr Ser Pro Ser Asn Thr Gly Phe Ser Val Asn Trp Asn
65 70 75 80
Thr Gln Asp Asp Phe Val Val Gly Val Gly Trp Thr Thr Gly Ser Ser
85 90 95
Ala Pro Ile Asn Phe Gly Gly Ser Phe Ser Val Asn Ser Gly Thr Gly
100 105 110
Leu Leu Ser Val Tyr Gly Trp Ser Thr Asn Pro Leu Val Glu Tyr Tyr
115 120 125
Ile Met Glu Asp Asn His Asn Tyr Pro Ala Gln Gly Thr Val Lys Gly
130 135 140
Thr Val Thr Ser Asp Gly Ala Thr Tyr Thr Ile Trp Glu Asn Thr Arg
145 150 155 160
Val Asn Glu Pro Ser Ile Gln Gly Thr Ala Thr Phe Asn Gln Tyr Ile
165 170 175
Ser Val Arg Asn Ser Pro Arg Thr Ser Gly Thr Val Thr Val Gln Asn
180 185 190
His Phe Asn Ala Trp Ala Ser Leu Gly Leu His Leu Gly Gln Met Asn
195 200 205
Tyr Gln Val Val Ala Val Glu Gly Trp Gly Gly Ser Giy Ser Ala Ser
210 215 220
Gln Ser Val Ser Asn
225
(2) INFORMATION FOR SEQ ID NO:5:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 18 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:5:
GACTCGAGAA TTCATCGA 18
(2) INFORMATION FOR SEQ ID NO:6:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
QOJCCT
2136350
The Swedi:. ~h Patent O{llice PCT/ FI 9 3/ C022 1 PCT {nternational
Application 02 -08- 1993
82
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:6:
GGVTGGCARC CNGGNACNAA 20
(2) INFORMATION FOR SEQ ID NO:7:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 18 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:7:
GACTCGAGAA TTCATCGA 18
(2) INFORMATION FOR SEQ ID NO:8:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 10 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ix) FEATURE:
(A) NAME/KEY: Peptide
(B) LOCATION: 1
(D) OTHER INFORMATION: /label= Peptide
/note= "The amino acid at position 2 labelled Xaa is
unknown."
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:8:
Gln Xaa Ile Gln Pro Gly Thr Gly Tyr Asn
1 5 10
(2) INFORMATION FOR SEQ ID NO:9:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:9:
AAYTAYGAYC ARAAYTAYGA 20
(2) INFORMATION FOR SEQ ID NO:10:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 25 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
SUBSTITUTE SHEET
2136350
The Swedish Patent Office PCT/F193/00221
PCf Intemational Applicatian
10-08-1993
83
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:10:
Ala Ser Ile Asn Tyr Asp Gin Asn Tyr Gln Thr Gly Gly Gln Val Ser
1 5 10 15
Tyr Ser Pro Ser Asn Thr Gly Phe Ser
20 25
(2) INFORMATION FOR SEQ ID NO:11:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 39 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:11:
CAACCGCGGA CTGCGCATCA TGGTCTCCTT CACCTCCCT 39
(2) INFORMATION FOR SEQ ID NO:12:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 26 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:12:
GGGAGCCGCT CGAGCGGTGG TTGCGG 26
(2) INFORMATION FOR SEQ ID NO:13:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 39 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:13:
CAACCGCGGA CTGCGCATCA TGGTTGCCTT TTCCAGCCT 30
(2) INFORMATION FOR SEQ ID NO:14:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 30 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:14:
CAGGCTCGAG GCCTGTGGGC ATCGCCAGAG 30
"rBELT