Note: Descriptions are shown in the official language in which they were submitted.
- 2 137 44 3
The invention relateR to novel thienothiazine
derivatives, a process for their preparation and their
use .
US 4,180,662 discloses thienothiazine derivatives
which are unsubstituted on the pyridine nucleus. These
compounds display good cyclooxygenase inhibition.
Furthermore, Chemical Abstracts, Volume 11, No. 19,
166771 n and Chemical Abstracts, Volume 115, No. 3,
21532 h disclose thienothiazine derivatives which are
unsubstituted or substituted by a hydroxyl group on the
pyridine nucleus. These compounds also display a good
cyclooxygenase inhibition.
It has now been possible to find novel thieno-
thiazine derivatives which are substituted on the
pyridine nucleus and which, while substantially ret~;n;ng
the cyclooxygenase inhibition, bring about a signifi-
cantly increased inhibition of 5-lipoxygenase.
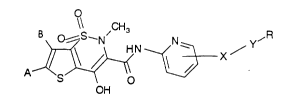
The invention accordingly relates to thieno-
thiazine derivatives of the formula I
o
A~N __~ X /
OH (I)
in which X is a single bond or a 5-12-membered mono- or
polycyclic, optionally partially hydrogenated aryl or
heteroaryl radical which can optionally be substituted by
halogen, lower alkyl or lower alkoxy,
Y is a single bond or a heteroatom,
R is a mono- or polycyclic, optionally partially hyd-
rogenated 5-12-membered aryl or heteroaryl radical which
can optionally be substituted one or more times by
halogen, lower alkyl or lower alkoxy,
A is halogen
and
B is hydrogen or halogen.
Preferred compound~ are those in which the
21374~3
-- 2
substituent is linked at position 6 of the pyridine.
X in the formula (I) is a single bond or a 5-12-
me_bered, mono- or polycyclic, optionally partially
hydrogenated aryl or heteroaryl radical. Examples of such
radicals would be 5-12-m~mhered, mono- or polycyclic,
optionally partially hydrogenated aromatic or hetero-
aromatic radicals, for example a phenyl, a thienyl, a
furyl, a pyrrolyl, a pyrimidyl, a pyranyl, a thiadi-
azinyl, an azepinyl radical, a quinolinyl radical and the
like.
These radicals can optionally be substituted one or more
times by halogen, for example F, Cl or I or by lower
alkyl or lower alkoxy.
Lower alkyl means a straight-chain or branched alkyl
radical with 1-4 C atoms, for example a methyl, an ethyl,
a propyl, an i-propyl radical, a butyl radical, an
i-butyl radical or a t-butyl radical.
Lower alkoxy means a straight-chain or branched alkoxy
radical with 1-4 C atoms, for example methoxy, ethoxy,
propoxy, i-propoxy, n-butoxy, i-butoxy or t-butoxy.
Preferred compounds are those in which X is a single bond
or a 5-7-membered aryl or heteroaryl radical, for example
a phenyl radical or a furyl radical.
Y in formula (I) is a single bond or a hetero-
atom, for example O, N or S.
Preferred compounds are those in which Y is a single bondor O.
R is a mono- or polycyclic, optionally partially
hydrogenated, 5-12-me_bered aryl or heteroaryl radical.
These are 5-12-membered, optionally partially hydrogen-
ated aromatic or heteroaromatic radicals, for example a
phenyl, a thienyl, a furyl, a pyrrolyl, a pyrimidyl, a
pyranyl, a thiadiazinyl, an azepinyl radical or a benzo-
furyl, a quinolinyl or a benzothienyl radical.
These radicals can optionally be substituted one or more
times by halogen, for example F, Cl, Br or by lower alkyl
or lower alkoxy.
Preferred compounds are those in which R is an
optionally mono- or polyhalogenated phenyl, a benzofuryl
213~4~3
-- 3
or a quinolinyl radical.
A can be halogen, for example Cl, F, Br.
B is hydrogen or halogen, for example Cl, F or
Br.
The invention furthermore relates to a process
for the preparation of compounds of the formula (I),
which is characterized in that a compound of the formula
(II)
_~OR2
OH (Il)
in which R2 is a straight-chain or brAnche~ alkyl group
with 1-4 C atoms, and A and B have the abovementioned
me~ning, is reacted with a compound of the formula (III)
-X ~ `R
(111)
in which X, Y and R have the abovementioned me~n;ng, and
the compounds of the formula (I) which are obtained in
this way are converted where appropriate into their
pharmaceutically utilizable salts.
The compounds of the formulae II and III are
known from the literature or can be prepared in analogy
thereto by conventional methods which are familiar to the
skilled worker.
The process according to the invention can
preferably be carried out in one of the following ways,
by
a) dissolving a compound of the formula II in an inert
~olvent such as diethyl ether, dioxane, toluene,
benzene or the like, and adding at a temperature from
-20C to 100C one equivalent of a strong base, such a~
2137~43
-- 4
butyllithium or LDA, in an inert solvent, for example
n-hexane, where appropriate under inert gas, adding to
this salt solution 1-10 equivalents of a compound of the
formula III, adding at least 1 equivalent of the strong
base and stirring at -20C to 100C for between 0.5 and
60 hours, preferably 1-48 hours, or
b) dissolving the compounds of the formula II and of the
formula III in an inert high-boiling solvent, such as
toluene, xylene, pyridine, quinoline, dimethylformamide,
dimethyl sulphoxide or hexamethylphosphoric triamide, and
heating this mixture at 100C to 200C for 1-30 hours.
The compounds of the formula I obtained in this way can,
where appropriate, easily be purified, for example by
recrystallization.
The compounds of the general formula I are acidic
compounds and can be converted in a conventional way with
inorganic or organic bases into their pharmaceutically
utilizable salts.
The salt formation can be carried out, for example, by
dissolving the compounds of the formula I in a suitable
solvent, for example water, a lower aliphatic alcohol,
for example methanol, ethanol and the like, in tetra-
hydrofuran, dioxane, benzene, diethyl ether, dimethyl-
formamide or dimethyl sulphoxide, A~;ng an equivalent
amount of the required base and, after the salt formation
is complete, distilling the solvent out under vacuum.
It is possible where appropriate for the salts to be
purified further after their isolation, for example by
recrystallization.
Examples of pharmaceutically suitable salts are
metal salts, in particular alkali metal salts and alka-
line earth metal salts, such as sodium, magnesium,
potassium or calcium salts. Other phAr~-ceutically
utilizable salts are easily crystallizable Ammo~ium
salts derived from A-mmonia or organic amines, such as,
for example, mono-, di- or tri-(lower alkyl, cycloalkyl
or hydroxyalkyl)amines, lower alkylenediamines,
(hydroxy-lower-alkyl)- or (aryl-lower-alkyl)-lower-
alkyl~mmonium bases, for example methylamine,
21374~3
-- 5
diethylamine, triethylamine, dicyclohexylamine,
triethanolamine, ethylenediamine, tris(hydroxymethyl)-
aminomethane, benzyltrimethylammonium hydroxide and the
like.
The compounds of the formula I according to the
invention and their salts have oral activity and surpris-
ingly show a significantly greater inhibition of 5-lip-
oxygenase compared with the compounds unsubstituted on
the pyridine nucleus, such as, for example, the 6-chloro-
4-hydroxy-2-methyl-N-(2-pyridyl)-2H-thieno[2,3-e]-1,2-
thiazine-3-carboxamide 1,1-dioxide ("Lornoxicam") dis-
closed in US 4,180,662, while the cyclooxygenase inhibi-
tion is substantially retained.
They are therefore particularly suitable for the
treatment of disorders partly caused by the natural
product of 5-lipoxygenase, namely leukotriene B4, such
as, for example, inflammations and pain associated with
allergic asthma, arthritis, skin allergy, etc.
By reason of these ph~r~-cological properties,
the novel compounds can be used, alone or mixed with
other active substances, in the form of conventional
pharmaceutical preparations as medicines for the treat-
ment of disorders which can be cured or alleviated by
inhibition of 5-lipoxygenase.
The invention furthermore relates to medicines
which are used, for example, in the form of pharma-
ceutical products which contain the compounds of the
formula (I) according to the invention or their phArm~-
ceutically utilizable salts mixed with a ph~rm~ceutical~
organic or inorganic vehicle suitable for oral, enteral,
parenteral or topical admini~tration, for example water,
gelatin, gum arabic, lactose, starch, magnesium stearate,
talc, vegetable oils, polyalkylene glycols, petrolatum
and the like.
The pharmaceutical products can be in solid form,
for example as tablets, film-coated tablets, sugar-coated
tablets, suppositories, capsules, microcapsules, or in
liquid form, for example as solutions, solutions for
injection, suspensions or emulsions, or in compositions
~1~7~43
with delayed release of the active substance.
They can be, where appropriate, sterilized and/or
they contain ancillary substances such as preservatives,
stabilizers or emulsifiers, salts to alter the osmotic
pressure or buffers.
In particular, phAr~-ceutical products may
contain the compounds according to the invention in
co_bination with other therapeutically valuable sub-
stances. The compounds according to the invention can be
formulated with the latter together with the above-
mentioned ancillary substances and/or vehicles to give
co_bination products.
The novel compounds may be present in the compo-
sitions according to the invention in an amount of
4-200 mg per tablet, the r~m~;n~er being a pharma-
ceutically acceptable b--l k; ng agent.
A suitable dose for the administration of the
compounds is about 4-200 mg/kg per day, but other doses
are also suitable, depending on the condition of the
patient to be treated. The novel compounds can be ~m; n; _
stered in several doses and by the oral route.
Example 1
N-[6-(2-Benzo[b]thienyl)-2-pyridinyl]-6-chloro-4-hydroxy-
2-methyl-2H-thieno[2,3-e][1,2]thiazine-3-carboxamide
1,1-dioxide
900 mg (3.98 mmol) of 6-(2-benzo[b]thienyl)-2-
pyri~;n~m;ne and 1236 mg (3.99 mmol) of methyl 6-chloro-
4-hydroxy-2-methyl-2H-thieno[2,3-e][1,2]thiazine-3-
carboxylate 1,1-dioxide are heated to boiling in 25 ml of
ab~. xylene for 10 hours. The solution is cooled and the
precipitate is filtered and digested several times with
acetone. This crude product is recrystallized from
toluene/active charcoal.
Yield: 1.37 g of yellow crystals (68% of theory)
Melting point: 257-261C (decomposition, toluene)
Thin-layer chromatogram: CH2Cl2:MeOH 40:1 RF = 0-70
H-NMR: (trifluoroacetic acid-dl)
~(ppm): 8.03 (m, lH, Py-H4); 7.69 (8, lH, Tz-H5); 7.40-
7.65 (m, 3H, Th-H4 + Th-H7 + Py-H5); 7.00-7.25 (m, 3H,
2137443
Th-H5 + Th-H6 + Py-H3); 6.95 (8, lH, Th-H3); 2.70 (æ, 3H,
CH3)
3C-NMR: (trifluoroacetic acid-dl)
~(ppm): 173.58 (8, CO); 162.38 (6, Py-C2); 151.55 (8, Tz-
C4); 146.73 (8, Th-C7a*); 146.26 (8, Tz-C4a); 144.95 (8,
Th-C2*); 143.02 (d, Py-C4); 142.13 (8, Tz-C7a); 135.26
(6, Tz-C6); 134.63 (8, Th-C3a); 131.89 (d, Th-C4); 130.90
(8, Py-C6); 129.80 (d, Th-C5*); 129.33 (d, Th-C6*);
126.52 (d, Tz-C7); 126.04 (d, Th-C7*); 122.91 (8, Tz-C3);
122.35 (d, Py-C3); 117.56 (d, Py-C5); 43.21 (q, CH3)
The starting material can be prepared as follows
2-Benzo[b]thienyltributylstAnn~ne
31 ml (77.5 mmol) of n-butyllithium are added to
8.498 g (63.3 mmol) of benzo[b]thiophene in 100 ml of
abs. THF at -20C, the mixture is stirred for one hour
and then cooled to -70C, and 20.756 g (63.8 mmol) of
tributyltin chloride in 20 ml of abs. THF are added.
The solution is allowed to warm to room temperature, and
the solvent is stripped off. The residue is partitioned
between 400 ml of water and 100 ml of ether, the aqueous
phase is extracted with 5xlO0 ml of ether, the combined
organic phases are washed with water and dried, and the
solvent i8 stripped off.
The resulting crude product is subjected to flash
chromatography (300 g of silica gel 60, PE).
Yield: 22.55 g of colourless liquid (84% of theory)
Thin-layer chromatogram: PE:EA 1:1 RF = 0-90
n-Hx RF = 0 45
PE RF = 0-50
lH-NMR: (CDCl3)
~(ppm): 7.90 (d, lH, Th-H4); 7.85 (d, lH, Th-H7); 7.40
(8, lH, Th-H3); 7.30 (m, lH, Th-H5); 7.25 (m, lH,
Th-H6); 1.65 (m, 6H, ~-CH3); 1.40 (m, 6H, X-CH2);
1.20 (t, 6H, ~-CH2); 0.95 (t, 9H, ~-CH3)
13C_NMR: (CDCl3)
~(ppm): 144.32 (8, Th-C2*); 141.04 (8, Th-C7a*); 139.70
(8, Th-C3a*); 132.03 (d, Th-C3); 123.64 (d, Th-
C5*); 123.26 (d, Th-C4*); 122.68 (d, Th-C6*);
121.78 (d, Th-C7*); 29.05 (t, x-C*); 27.33 (t-~-
- 2137443
C*); 13.70 (q, ~-C); 10.84 (t, ~-C*)
2-(2-Benzo~b]thienyl)-6-bromopyridine
20.02 g (47.3 mmol) of2-benzotb]thienyltributyl-
st~nn~ne, 11.23 g (47.4 mmol) of 2,6-dib ~ ~yLidine,
4.79 g (47.3 mmol) of triethylamine, 0.496 g (1.89 mmol)
of triphenylphosphine and 0.213 g (0.95 mmol) of
palladium diacetate are stirred in 300 ml of abs. DMF at
105C for one hour. The reaction mixture is poured into
600 ml of hydrochloric acid (1.6 N) and the precipitate
is filtered off. The crude product i8 recrystallized from
toluene/active charcoal and used without further separa-
tion in the next stage.
Yield: 4.94 g of colourless crystals (36% of
theory)
15 Melting point: 163-168C (toluene)
Thin-layer chromatogragram: PE:EA 40:3 RF = 0 35
H-NMR: (DMSO-d6)
~(ppm): 8.20 (8, lH, Th-H3); 8.10 (d, lH, Py-H3); 8.00
(m, lH, Th-H4); 7.90 (m, lH, Th-H7); 7.85 (dd,
lH, Py-H4); 7.60 (d, lH, Py-H5); 7.40 (m, lH,
Th-H5); 7.40 (m, lH, Th-H6)
3C-NMR: (DMSO-d6)
~(ppm): 152.87 (8, Th-C7a); 142.27 (8, Py-C6); 140.87 (8,
Th-C3a); 140.16 (d, Py-C4); 139.96 (8, Py-C2); 126.96 (d,
25 Py-C3); 125.57 (d, Th-C4); 124.78 (d, Th-C5); 124.45 (d,
Th-C6); 123.05 (d, Th-C7); 122.60 (d, Py-C5); 118.90 (d,
Th-C3)
6-(2-Benzo[b]thienyl)-2-pyri~; n~m; n e
3.47 g (12.0 mmol) of 2-(2-benzo[b]thienyl)-6-
30 bromopyridine and 2.63 g (67.4 mmol) of sodamide are
stirred in 200 ml of ammonia in an autoclave at 0C for
one hour.
After the autoclave has been cooled to -50C, the mixture
is quenched with 3.73 g (69.7 mmol) of Ammo~;um chloride,
and the ~o~;a is slowly evaporated. The residue is
partitioned between water and EA and extracted 8x with a
total of 1100 ml of EA, the organic phase is washed 2x
with water, dried, concentrated to crystallization and
cooled, and the crystals of the by-product from the
2137~43
g
previous stage are filtered off.
The mother liquor is completely evaporated, the residue
is digested 2x with 5 ml of HCl-saturated methanol each
time and evaporated, and digested 2x with pure methanol
and evaporated, the hydrochloride is digested with 20 ml
of cold benzene and filtered off, and the amine is
liberated again by partitioning between EA and saturated
sodium bicarbonate solution.
Yield: 1.30 g of pale brown crystals (48% of
theory)
Melting point: 225-231C (hydrochloride)
Thin-layer chromatogram: PE:EA 40:3 RF = 0.15
PE:EA 1:1 RF = 0-70
1H-NMR: as hydrochloride
~(ppm): 8.50 (8, lH, Th-H3); 8.05 (m, lH, Py-H4*); 7.90
(m, 2H, Th-H4* + Th-H7*), 7.45 (m, 2H, Th-H5* + Th-H6*);
7.10 (d, lH, Py-H3); 6.95 (d, lH, Py-H5)
ExamPle 2
N-[6-(4-Biphenyl)-2-pyridinyl]-6-chloro-4-hydroxy-2-
methyl-2H-thieno[2,3-e][1,2]thiazine-3-carbox~ide 1,1-
dioxide
609 mg (2.47 mmol) of 6-(4-biphenyl)-2-pyridin-
amine and 767 mg (2.48 mmol) of methyl 6-chloro-4-
hydroxy-2-methyl-2H-thieno[2,3-e][1,2]thiazine-3-carboxy-
late l,1-dioxide are heated to boiling in 15 ml of xylene
for 6 hours. The crude product is filtered off after
cooling and recrystallized from DMF/active charcoal.
Yield: 1.07 g of pale yellow crystals (83% of
theory)
Melting point: 242-245C (decomposition, DMF)
Thin-layer chromatogram: CH2Cl2:MeOH 40:1 RF = 0-50
CH2Cl2:MeOH 40:3 RF = 0 90
H-NMR: (trifluoroacetic acid-dl)
~(ppm): 8.05 (dd, lH, Py-H4); 7.60 (m, 4H, Bz-H2 + Bz-
H3); 7.50 (8, lH, Tz-H7); 7.25 (m, 3H, Py-H3* + Ph-H2);
7.05 (m, 3H, Ph-H4 + Ph-H3); 7.00 (d, lH, Py-H5); 2.75
(8, 3H, CH3)
3C-NMR (trifluoroacetic acid-dl)
~(ppm): 173.56 (8, CO); 162.44 (s, Py-C2); 152.61 (8,
2137443
- 10 -
Py-C6); 151.73 (d, Py-C4); 150.28 (8, Tz-C4); 146.51 (8,
Tz-C4a); 142.45 (8, Bz-Cl); 141.81 (8, Tz-C7a); 135.46
(8, Tz-C6); 132.65 (d, Bz-C3 + Bz-C5); 132.34 (d, Bz-C2
+ Bz-C6); 131.54 (d, Bz-C4); 130.53 (d, Ph-C3 + Ph-C5);
130.44 (d, Ph-C2 + Ph-C6); 126.48 (d, Tz-C7); 122.77 (d,
Py-C3); 117.44 (d, Py-C5); 112.66 (8, Tz-C3); 43.21 (q,
CH3)
The starting material can be prepared as follows
4-Biphenylboronic acid
35.0 ml (87.5 mmol) of n-butyllithium are added
to 18.034 g (77.4 mmol) of 4-bromobiphenyl in 200 ml of
THF at -70C. After the solution has been cooled to
-100C, 16.50 g (159 mmol) of trimethyl borate (diluted
with 10 ml of THF) are added dropwise, and the mixture is
allowed to warm to room temperature.
500 ml of 1 N hydrochloric acid are added to the
reaction solution which is then extracted with 3x120 ml
of diethyl ether, the organic phase i~ washed 2x with
water, dried and filtered, and the solvent is stripped
off. The crude product is crystallized with DIPE and
filtered off.
Yield: 11.30 g of colourless crystals (74% of
theory)
Melting point: 216-220C (DIPE)
Thin-layer chromatogram: PE:EA 1:1 RF = 0-50
H-NMR: (acetone-d6)
~(ppm): 7.95 (d, 2H, Bz-H2); 7.70 (d, 2H, Ph-H2*); 7.65
(d, 2H, Bz-H3*); 7.40 (d, lH, Ph-H4*); 7.35 (dd,
2H, Ph-H3*)
13C-NMR: (acetone-d6)
~(ppm): 141.16 (8, Ph-Cl*); 140.44 (8, Bz-C4*); 134.76
(d, Bz-C2* + Bz-C6*); 134.11 (d, Bz-C3* + Bz-
C5*); 128.88 (d, Ph-C3* + Ph-C5*); 126.67 (d, Ph-
C2* + Ph-C6*); 125.80 (d, Ph-C4*)
2-(4-Biphenyl)-6-bromopyridine
7.99 g (40.3 mmol) of 4-biphenylboronic acid,
9.67 g (40.8 mmol) of 2,6-dibromopyridine, 8.19 g
(80.9 mmol) of triethylamine, 0.470 g (1.79 mmol) of
triphenylphosphine, 0.184 g (0.82 mmol) of palladium
2137~3
11
diacetate and 9.36 g (40.4 mmol) of silver oxide are
stirred in 120 ml of abs. DMF at 105C for 2 hours.
The cooled Bolution i6 filtered through Hyflo and washed
with EA, and the solvent is stripped off. The residue is
crystallized from acetonitrile/active charcoal.
Yield: 4.37 g of pale beige crystals (35% of
theory)
Melting point: 154-159C (acetonitrile)
Thin-layer chromatogram: CHCl3:Ac 7:3 RF = 0 95
PE:EA 40:1 RF = 0-30
Bz:Et2O 1:1 RF = 0 90
H-NMR: (benzene-d6)
~(ppm): 8.05 (d, 2H, Bz-H3); 7.50 (d, 2H, Ph-H2); 7.40
(d, 2H, Bz-H2*); 7.10-7.30 (m, 3H, Py-H3 + Ph-
H3); 7.05 (d, lH, Ph-H4); 6.90 (d, lH, Py-H5*);
6.70 (m, lH, Py-H4*)
3C-NMR: (trifluoroacetic acid-dl)
~(ppm): 159.29 (8, Py-C2); 149.96 (8, Py-C6); 148.17 (d,
Py-C4*); 141.49 (s, Ph-Cl);
137.13 (s, Bz-Cl); 132.56 (d, Py-C3*); 131.96 (d,
Bz-C3 + Bz-C5); 131.85 (d, Ph-C3 + Ph-C5);
131.562 (8, Bz-C4*); 131.46 (8, Ph-C4*); 130.57
(d, Bz-C2 + Bz-C6): 129.73 (d, Ph-C2 + Ph-C6);
127.39 (d, Py-C5*)
6-(4-Biphenyl)-2-pyri~;nAm;ne
2.65 g (8.54 mmol) of 4-biphenyl-2-bromopyridine
and 1.82 g (46.7 mmol) of sodamide in 200 ml of ammonia
are stirred in an autoclave at 0C for 60 minutes. Excess
amide is eliminated with 2.49 g (46.6 mmol) of ammonium
chloride, and the ammonia is slowly evaporated. The
residue is partitioned between water and EA and extracted
7x with a total of 700 ml of EA, the organic pha~e is
washed 2x with water and dried over sodium sulphate, and
the solvent is stripped off.
The crude product is digested 2x with 20 ml of HCl-
saturated methanol each time and evaporated. The residue
is digested with 40 ml of hot benzene, filtered off and
washed with hot benzene. The pure amine is liberated by
partitioning between methylene chloride and saturated
2137443
sodium bicarbonate solution, the organic phase is washed
2x with water and dried, and the solvent is stripped off.
Yield: 1.07 g of beige crystals (51% of theory)
Melting point: 240-243C (benzene)
Thin-layer chromatogram: Bz:Et2O 1:1 RF = 0-50
HNMR: (CDCl3)
~(ppm): 8.05 (d, 2H, Bz-H3); 7.70 (d, 2H, Bz-H2); 7.65
(dd, 2H, Ph-H2); 7.35 - 7.55 (m, 4H, Ph-H4 + Ph-
H3 + Py-H4); 7.15 (d, lH, Py-H3); 6.45 (d, lH,
Py-H5)
3C-NMR: (CDC13)
~(ppm): 158.27 (8, Py-C6); 155.57 (8, Py-C2); 141.20 (8,
Ph-C1); 140.65 (8, Bz-C1);
138.52 (8, Bz-C4); 138.31 (d, Py-C4); 128.72 (d,
Bz-C3 + Bz-C5); 127.33 (8, Ph-C4); 127.17 (d, Ph-
C2 + Ph-C6); 127.15 (d, Ph-C3 + Ph-C5);
126.99 (d, Bz-C2 + Bz-C6); 110.77 (d, Py-C3);
107.11 (d, Py-C5)
Example 3
6-Chloro-N-{6-[3-(4-fluoroph~noYy)phenyl]-2-pyridinyl}-4-
hydroxy-2-methyl-2H-thieno[2,3-e]-1,2-thiazine-3-carbox-
amide 1,1-dioxide
A mixture of 1.00 g (3.57 mmol) of 6-(3-(4-
fluorophenoxy)phenyl)-2-pyridinamine and 1.11 g
(3.57 mmol) of methyl 6-chloro-4-hydroxy-2-methyl-2H-
thieno[2,3-e]-1,2-thiazine-3-carboxylate 1,1-dioxide in
29 ml of absolute xylene is heated to boiling for
22 hours.
The crude product which precipitates on cooling is
filtered off and recrystallized from acetonitrile/active
charcoal.
Yield: 1.19 g of yellow crystals (60% of theory)
TLC: Solvent CH2Cl2:MeOH = 40:1; 0.4
M.p.: 166-169C (acetonitrile)
1H-NMR: (DMSO-d6)
d(ppm): 8.14-7.70 (m, 6H, ThH, BzH2,6, PyH3,4,5);
7.52 (dd, lH, BzH5); 7.30-7.00 (m, 5H, BzH4,
FBzH2,3,5,6); 2.95 (8, 3H, N-C_ 3)
13C_NMR (DMSO-d6)
21~7~43
- 13 -
d(ppm): 166.3, 158.2 (d), 157.5, 155.5, 152.8,
152.6, 152.5, 150.1, 140.1, 139.1, 137.4, 136.3,
134.9, 130.4, 123.1, 121.7, 120.4 (d), 119.1, 116.7,
116.5 (d), 115.0, 110.3, 39.4
The starting material can be prepared as follows:
Tributyl-[3-(4-fluorophenoYy)phenyl]stAnnAne
21.3 ml (53.3 mmol) of a 2.5 M solution of
n-butyllithium in n-hexane are added dropwise to 14.23 g
(53.3 mmol) of 1-bromo-3-(4-fluorophenoxy)benzene in
150 ml of absolute tetrahydrofuran at -70C, the mixture
is stirred for one hour, and 17.35 g (53.3 mmol) of
tributyltin chloride are added at -70C.
After 30 minutes, the mixture i8 warmed to room tempera-
ture, the solvent is stripped off, partitioning between
250 ml of water and 200 ml of diethyl ether is carried
out, and one more extraction is carried out with 200 ml
of diethyl ether. The organic phase is dried over Na2SO4/
active charcoal and filtered, and the solvent is stripped
off.
Yield: 25.54 g of yellowish oil (90% of theory)
TLC: Solvent CCl4; 0.65
H-NMR: (CDC13)
d(ppm): 7.29 (ddd, lH, BzH5); 7.19 (ddd, lH, BzH6);
7.13-6.95 (m, 5H, BzH2, FBzH2,3,5,6); 6.87 (ddd, lH,
BzH4); 1.60-1.40 (m, 6H, BuH2); 1.40-1.25 (m, 6H,
BuH3); 1.11-0.96 (m, 6H, BuH1); 0.87 (t, 9H, BuH4)
3C-NNR: (CDC13)
d(ppm): 158.4 (d), 156.9, 153.1 (d), 144.1, 131.1
(t), 129.0 (t), 126.0 (t), 120.1 (d), 117.8, 116.0
(d), 29.0 (t), 27.2 (t), 13.5, 9.5 (t)
2-Bromo-6-t3-(4-fluorophenoxy)phenyl]pyridine
23.67 g (44.3 mmol) of tributyl[3-(4-fluoro-
phenoxy)phenyl]stAnnAne in 12 ml of abs. DMF are added to
10.94 g (44.3 mmol) of 2,6-dibromopyridine, 149 mg
(664 ~mol) of palladium(II) acetate and 349 mg
(1.33 mmol) of triphenylphosphine in 235 ml of abs. DMF
at 80C under a nitrogen atmosphere, and the mixture is
stirred for 10 hours. After the solvent has been stripped
off, the residue is partitioned between 400 ml of water
2137~43
- 14 -
and 200 ml of diethyl ether and the aqueous phase is
extracted twice more with 200 ml of diethyl ether each
time. The combined organic phases are washed three times
with 100 ml of water each time, dried over Na2SO4/active
charcoal and filtered, and the solvent is stripped off.
The crude product is subjected to a chromatographic
separation (400 g of silica gel 60, PE:Bz=3:2).
Yield: 9.00 g of colourless crystals 59% of theory
TLC: Solvent CCl4:Et2O = 10:1; 0.65
M.p.: 80-81C (SC)
-NMR: (CDCl3)
d(ppm): 7.72 (ddd, lH, BzH6), 7.66-7.60 (m, 2H,
PyH5, BzH2), 7.57 (dd, lH, PyH4), 7.41 (dd, lH,
BzH5), 7.41 (dd, lH, PyH3), 7.10-6.97 (m, 5H, BzH4,
FBzH2,3,5,6)
3C-NMR: (CDC13)
d(ppm): 158.7 (d), 158.0, 157.6, 152.7 (d), 142.0,
139.5, 138.9, 130.0, 126.6, 121.7, 120.3 (d), 119.1
(d), 116.9, 116.5, 116.0
6-[3-(4-Fluorophenoxy)phenyl]-2-pyri~; nAm; n~
7.00 g (20.3 mmol) of 2-bromo-6-[3-(4-fluoro-
phenoxy)phenyl]pyridine and 4.76 g (122.1 mmol) of
sodamide in 400 ml of liquid Ammo~;a are stirred at 0C
for 30 minutes, and 6.52 g (121 mmol) of Am~o~;um
chloride are added.
The AmmOn; a is evaporated off and the residue is par-
titioned between 250 ml of water and 100 ml of diethyl
ether, the aqueous phase is extracted once more with
100 ml of diethyl ether, the combined organic phases are
dried over Na2SO4/active charcoal and filtered, and the
solvent is 6tripped off.
The crude product i8 subjected to a flash chromatography
(134 g of silica gel 60, Bz:Et2O=3:1)
Yield: 1.63 g of brown oil (29% of theory)
TLC: Solvent Bz:Et2O = 1:1; 0.55
H-NMR: (CDCl3)
d(ppm): 7.67 (ddd, lH, BzH6); 7.59 (dd, lH, BzH2);
7.46 (dd, lH, BzH5); 7.38 (dd, lH, PyH4); 7.08-6.93
(m, 6H, BzH4, PyH5, FBzH2,3,5,6); 6.44 (d, lH, PyH3)
2137493
- 15 -
3C-NMR: (CDCl3)
d(ppm): 158.6 (d), 158.2, 157.8, 155.0, 152.9,
141.6, 138.3, 129.8, 121.6, 120.3 (d), 118.4, 116.8,
116.2 (d), 110.8, 107.4
ExamPle 4
6-Chloro-N-{6-[5-(4-fluoroph~noYy)-2-furyl]-2-pyridinyl}
4-hydroxy-2-methyl-2H-thieno[2,3-e~-1,2-thiazine-3-
carboxamide 1,1-dioxide
114 mg (422 ~mol) of 6-[5-(4-fluorophenoxy)-2-furyl]-2-
pyridinamine and 131 mg (422 ~mol) of methyl 6-chloro-4-
hydroxy-2-methyl-2H-thieno 2,3-e]-1,2-thiazine-3-c~hnYy_
late are heated to boiling in 3.4 ml of xylene for
20 hours. The solvent is stripped off and the residue is
recrystallized from acetonitrile/active charcoal.
Yield: 191 mg of yellow crystals (83% of theory)
TLC: Solvent CH2Cl2:MeOR = 40:1; 0.65
M.p.: 186-195C
H-NMR: (acetone-d6)
d(ppm): 8.05-7.83 (m, 2H, PyH3,4); 7.57 (8, lH, ThH);
7.45 (dd, lH, PyH5); 7.33-7.15 (m, 4H, BzH2,3,5,6); 7.07
(d, lH, FuH3); 5.80 (d, lH, FuH4); 3.10 (8, 3H, CH3)
13C-NMR: (acetone-d6)
d(ppm): 169.1, 169.3 (d); 159.8; 157.0; 154.3; 152.2 (d);
149.0; 147.0; 141.4; 140.3; 139.4; 125.0; 121.3 (d);
118.5 (d); 116.1; 114.8; 113.2; 112.8; 92.9; 41.4
The starting material can be prepared as follows:
2-Bromo-6-(2-furyl)pyridine
30.0 g (268.12 mmol) of 2-furanboronic acid,
42.35 g (178.75 mmol) of 2,6-dibromopyridine, 54.26 g
(536.24 mmol) of triethylamine, 2.8 g (10.73 mmol) of
triphenylphosphine and 1.2 g (5.36 mmol) of palladium
diacetate are suspended in 800 ml of N,N-dimethylform-
amide and stirred at 100C under a nitrogen atmosphere
for three hours.
After the solvent has been stripped off, the residue is
partitioned between 400 ml of dichloromethane and 200 ml
of 10% strength aqueous ammonia. The aqueous phase is
back-extracted once with 200 ml of dichloromethane, the
collected organic phases are washed with 200 ml of water,
213744~
- 16 -
dried with sodium sulphate and filtered, and the solvent
is stripped off.
For purification, the crude product is chromatographed on
1000 g of silica gel 60 with benzene and distilled under
high vacuum.
Yield: 31.0 g of colourless liquid (78% of theory)
TLC: PE:EA=2:1; 0.35
B.p.: 80-85C/0.1 mbar
nD20: 1.6468
lH-NMR: (CDCl3)
d(ppm): 7.54 (m, 3H, Py-H3, Py-H4, Fu-H5); 7.28 (d,
lH, Py-H5, 3JH H=8Hz); 7.10 (d, lH, Fu-H3, 3JH H=
4Hz); 6.51 (dd, lH, Fu-H4, 3JH H3=4HZ~ 3JH H5=2Hz)
13C-NMR: (CDCl3)
d(ppm): 151.9 (8, Py-C6); 149.9 (8, Fu-C2); 143.6(d,
Fu-C5); 141.7 (8, Py-C2); 138.6 (d, Py-C4); 125.7
(d, Py-C3); 116.8 (d, Py-C5); 112.0 (d, Fu-C4);
109.9 (d, Fu-C3)
6-(2-Furyl)-2-pyridinamine
20.0 g (89.26 mmol) of 2-bromo-6-(2-furyl)pyr-
idine and 13.8 g (357.05 mmol) of sodamide are dissolved
in 1700 ml of ammonia and stirred at -33C for 15 minutes
and then quenched with 18.8 g (357.05 mmol) of ammonium
chloride, and the ~m~o~;a is allowed to evaporate. The
residue is partitioned between 400 ml of diethyl ether
and 400 ml of water, and the aqueous phase is extracted
once more with 200 ml of diethyl ether. The collected
organic phases are dried over sodium sulphate and fil-
tered, and the solvent is stripped off.
The crude product is purified by coll~n chromatography on
400 g of silica gel 60 with CH2Cl2:Et2O = 2:1.
Yield: 8.3 g of pale yellow crystals (58% of theory)
TLC: CH2Cl2:Et2O = 2:1; 0.35
M.p.: 66-68C (chromatographed)
lH-NMR: (CDCl3)
~(ppm): 7.49 (d, lH, Fu-H5, 3JH H=2Hz); 7.45 (dd, lH,
Py-H4, 3JH H3=3JH Hs=8HZ); 7 07 (d, lH, Py-H5,
3JH H=8Hz); 6.92 (d, lH, Fu-H3, 3JH H=4HZ); 6.49 (dd,
lH, Fu-H4, 3JH H3=4HZ, 3JH H5=2Hz); 6.37 (d, lH,
2137~43
- 17 -
Py-H3, 3JH H=8Hz); 4.56 (8, 2H, Py-NH2)
3C-NMR: (CDC13)
~(ppm): 158.1 (8, Py-C2); 153.6 (8, Py-C6); 147.5
(8, Fu-C2); 142.8 (d, Fu-C5); 138.2 (d, Py-C4);
111.7 (d, Fu-C4); 108.9 (d, Py-C5); 107.9 (d, Fu-
C3); 107.2 (d, Py-C3)
1,1-Dimethylethyl N-(l,l-dimethylethoxycarbonyl)-N-[6-(2-
furyl)-2-pyridinyl]carbamate
6.0 g (37.46 mmol) of 6-(2-furyl)-2-pyridinamine
are heated to boiling with 16.35 g (74.92 mmol) of
bis(l,1-dimethylethyl) dicarbonate and 10.0 g
(74.92 mmol) of potassium carbonate in 200 ml of abs.
dioxane with a catalytic amount of N,N-dimethyl-4-pyrid-
; n~; ne for 90 minutes.
After the solution ha~ cooled, the potassium carbonate i8
filtered off and the sol~ent is stripped off in ~acuo.
The residue is taken up in 100 ml of 2 N aqueous hydro-
chloric acid and extracted three times with 100 ml of
diethyl ether. The collected organic phases are washed
once with 100 ml of 2 N aqueous sodium hydroxide solution
and twice with water, dried over sodium sulphate and
filtered, and the solvent is stripped off in ~acuo.
The residue is purified by digestion with diisopropyl
ether.
Yield: 11.5 g of beige crystals (85% of theory)
TLC: PE:EA = 4:1; 0.5
M.p.: 99-101C (diisopropyl ether)
H-NMR: (CDC13)
~(ppm): 7.73 (dd, lH, Py-H4, 3JH H3=3JH H5=8Hz); 7.57
(d, lH, Py-H3, 3JH H=8Hz); 7.49 (d, lH, Fu-H5,
3JH H=2Hz); 7.11 (d, lH, Py-H5, 3JH H=8Hz); 7.00 (d,
lH, Fu-H3, 3JH H=4HZ); 6.50 (dd, lH, Fu-H4,
3JH H3=4HZ~ 3JH H5=2Hz); 1.45 (8, 18H, -CH3)
l3C-NMR: (CDC13)
~(ppm): 152.9 (8, Py-C2); 151.9 (8, C=0); 151.1 (8,
Py-C6); 148.2 (8, Fu-C2); 143.1 (d, Fu-C5); 138.1
(d, Py-C4); 118.9 (d, Py-C3); 116.1 (d, Fu-C3);
111.9 (d, Fu-C4); 108.9 (d, Py-C5); 82.7 (8, C-CH3;
27.7 (q, C-CH3)
~ - 18 - 2137~43
1,1-Dimethylethyl N-t6-(2-furyl)-2-pyridinyl]carbamate
5.75 ml (31.91 mmol) of 30% strength sodium
methanolate in abs. methanol are added to 11.5 g
(31.91 mmol) of 1,1-dimethyl ethyl N-(1,1-dimethylethoxy-
carbonyl)-N-[6-(2-furyl)-2-pyridinyl]carbamate in 100 ml
of abs. methanol, and the mixture is stirred at room
temperature for 60 minutes. Then a further 2.9 ml
(16.09 mmol) of 30% 6trength sodium methanolate in abs.
methanol are added, the mixture is stirred for a further
30 minutes, and the solvent is stripped off in vacuo.
The residue is taken up in 100 ml of 2 N aqueous hydro-
chloric acid and extracted three times with 100 ml of
diethyl ether. The collected organic phases are washed
twice with 100 ml of water and dried over sodium sul-
phate, and the solvent is stripped off in vacuo.
The residue is purified by column chromatographyon 400 g of silica gel 60 with benzene.
Yield: 7.3 g of colourless foam (88% of theory)
TLC: PE:EA = 4:1; 0.6
H-NMR:(CDC13)
~(ppm): 7.81 (d, lH, Py-H3, 3JH H=8Hz); 7.66 (dd, lH,
Py-H4, JH,H3= JH~Hs=8Hz); 7.48 (d, lH, Fu-H5,
3JH H=2Hz); 7.32 (d, lH, Py-H5, 3JH H=8Hz); 7.30 (8,
lH, Py-NH); 6.93 (d, lH, Fu-H3, 3JH H=4HZ); 6.50 (dd,
lH, Fu-H4, 3JH H3=4HZ~ 3JH H5=2Hz); 1.49 (8, 9H, -CH3)
3C-NMR: (CDCl3)
~(ppm): 153.2 (8, Py-C2); 152.4 (8, C=0); 151.6 (8,
Py-C6); 147.5 (8, Fu-C2); 143.1 (d, Fu-C5); 138.7
(d, Py-C4); 113.2 (d, Py-C3); 111.9 (d, Fu-C3);
110.5 (d, Fu-C4); 108.5 (d, Py-C5); 80.8 (8, C-CH3);
28.1 (q, C-CH3)
1,1-Dimethylethyl N-[6-(5-bromo-2-furyl)-2-pyridinyl]-
carbamate
7.0 g (26.89 mmol) of l,1-dimethylethyl N-[6-(2-
furyl)-2-pyridinyl]carbamate are heated to boiling with
4.8 g (26.89 mmol) of N-bromosuccinimide in 50 ml of abs.
tetrachloromethane for two hours. After cooling, the
reaction mixture is filtered and the solvent is removed
from the filtrate. The residue is recrystallized from
2137443
- 19 -
methanol/active charcoal.
Yield: 7.5 g of colourless crystals (82% of theory)
TLC: PE:EA = 4:1: 0.6
M.p.: 115-117C (MeOH)
lH-NMR: (CDC13)
~(ppm): 7.81 (d, lH, Py-H3, 3JH H=8Hz); 7.67 (dd, lH,
Py-H4, 3JH H3=3JH H5=8Hz); 7.48 (8, lH, Py-NH); 7.30
(d, lH, Py-H5, 3JH H=8Hz); 6.88 (dd, lH, Fu-H4,
JH,H3=4HZ, JH,H5=2Hz); 6.39 (d, lH, Fu-H3,
3JH H=4HZ); 1.49 (8, 9H, -CH3)
3C-NMR: (CDC13)
~(ppm): 154.9 (8, Py-C2); 152.2 (8, C=O); 151.7 (8,
Py-C6); 146.3 (8, Fu-C5); 138.7 (d, Py-C4); 122.7
(8, Fu-C2); 113.6 (d, Py-C3); 112.8 (d, Fu-C4);
110.8* (d, Py-C5); 110.7* (d, Fu-C3); 80.7 (8, -
CH3); 28.0 (q, C-H3)
6-(5-Bromo-2-furyl)-2-(dimethylethoxycarbonylamino)-
pyridine l-oxide
7.5 g (22.11 mmol) of l,l-dimethylethyl N-[6-(5-
bromo-2-furyl)-2-pyridinyl]carbamate are stirred with
9.02 g (28.75 mmol) of 55% strength 3-chloroperbenzoic
acid in 150 ml of abs. trichloromethane at room tempera-
ture for 30 minutes. The solution is washed twice with
100 ml of a saturated aqueous sodium bicarbonate solution
each time and twice with 50 ml of water each time. The
organic phase is dried with sodium sulphate and filtered,
and the solvent is stripped off in vacuo.
The residue is recrystallized from acetone/active char-
coal.
Yield: 3.7 g of colourless crystals (48% of theory)
TLC: PE:EA = 4:1; 0.55
M.p.: 120-122C (acetone)
H-NMR: (CDC13)
~(ppm): 9.45 (8, lH, Py-NH); 8.00 (d, lH, Py-H3,
3JH H=8Hz); 7.91 (d, lH, Fu-H4, 3JH H3=4HZ); 7.50 (d,
lH, Py-H5, 3JH H=8Hz); 7.31 (dd, lH, Py-H4,
3JH H3=3JH H5=8Hz); 6-52 (d, lH, Fu-H3, 3JH H=4HZ);
1.53 (8, 9H, -CH3)
3C-NMR: (CDC13)
2137~ 13
- 20 -
~(ppm): 151.4 (8, C=O); 147.2 (s, Py-C2); 144.7 (8,
Py-C6); 137.0 (8, Fu-C5); 126.5 (d, Py-C4); 124.5
(8, Fu-C2); 118.6 (d, Fu-C4); 114.2 (d, Fu-C3);
113.5 (d, Py-C3); 109.6 (d, Py-C5); 81.9 (8, C-CH3);
28.0 (q, C-CH3)
6-[5-(4-Fluorophenoxy)-2-furyl]-2-pyri~;n~m;ne 1-oxide
2.37 g (6.66 mmol) of 6-(5-bromo-2-furyl)-2-
(dimethylethoxycarbonylamino)pyridine 1-oxide and 2.95 g
(22.0 mmol) of sodium 4-fluorophenolate are stirred in
23 ml of absolute dimethyl sulphoxide at 100C for
13 hour~ ;ng after 8 and 11 hours in each case 0.90 g
(6.66 mmol) of ~odium 4-fluorophenolate.
The reaction mixture i8 diluted with 500 ml of water, the
product i~ extracted with 4x200 ml of diethyl ether and
the combined organic phases are washed with 2xlO0 ml of
2 N sodium hydroxide solution and 100 ml of water. The
extract is dried over Na2SO4/active charcoal and fil-
tered, and the new solvent is stripped off. The crude
product is recrystallized from acetonitrile/active
charcoal.
Yield: 0.42 g of pale beige crystals (22% of theory)
TLC: Solvent Bz:Et2O = 1:1; 0.2
M.p.: 195-201C (acetonitrile, decomposition)
lH-NMR: (CDCl3)
~(ppm): 7.67 (dd, lH, PyH4); 7.58 (d, lH, FuH3); 7.24-
7.02 (m, 6H, PyH3,5, BzH2,3,5,6); 5.63 (d, lH, FuH4)
3C-NMR: (CDC13)
~(ppm): 161.0; 159.9 (d); 156.2; 150.3 (d); 137.2; 132.8;
130.5; 120.6; 120.1; 116.7; 109.7; 107.0; 90.0
6-[5-(4-Fluorophenoxy)-2-furyl]-2-pyri~;n~m;ne
226 mg (789 ~mol) of 6-[5-(4-fluorophenoxy)-2-
furyl]-2-pyri~;n~m;ne 1-oxide are vigorously stirred with
226 mg (4.05 mmol) of an iron powder in 4.5 ml of
analytical grade glacial acetic acid under a nitrogen
atmosphere at 80C for two hours, adding a further 100 mg
(1.80 mmol) of iron powder after one hour.
After the solvent has been stripped off, the residue is
partitioned between 20 ml of diethyl ether and 20 ml of
dilute hydrochloric acid. The aqueous phase is further
21~7~43
- 21 -
extracted with 3x15 ml of diethyl ether, and the combined
organic phases are washed with 2x20 ml of 1 N hydro-
chloric acid and 20 ml of saturated sodium bicarbonate
solution. The organic phase is dried over Na2SO4 and
filtered, the solvent is stripped off, and the crude
product i8 subjected to flash chromatography on 11 g of
silica gel 60 with benzene:ether = 1:1.
Yield: 178 mg of colourless crystals (83% of theory)
TLC: Solvent Et2O: MeOH = 50.1; 0.6
Solvent Bz:Et2O = 1:1; 0.3
M.p.: 98-100C (SC from benzene/diethyl ether)
H-NMR: (CDC13)
~(ppm): 7.41 (dd, lH, PyH4); 7.15-6.96 (m, 4H,
BzH2,3,5,6); 6.94 (dd, lH, PyH5); 6.90 (d, lH, FuH3);
6.33 (dd, lH, PyH3); 5.60 (d, lH, FuH4); 4.59 (broad 8,
2H, NH2)
3C-NMR: (CDCl3)
~(ppm): 159.2 (d); 158.2, 157.0; 152.3 (d); 147.3; 146.1;
138.2; 118.8 (d); 116.3 (d); 109.4; 108.2; 106.9; 90.8
ExamPle 5
N-[6-(2-Benzo[b]furyl-2-pyridinyl]-6,7-dichloro-4-
hydroxy-2-methyl-2H-thieno[2,3-e][1,2]thiazine-3-carb-
oxamide 1,1-dioxide
428 mg (2.03 mmol) of 6-(2-benzo[b]furyl)-2-
pyri~;n~m;ne and 700 mg (3.99 mmol) of methyl 6,7-di-
chloro-4-hydroxy-2-methyl-2H-thieno[2,3-e][1,2]thiazine-
3-carboxylate l,l-dioxide are heated to boiling in 7 ml
of abs. xylene for 9 hours. The solution is cooled and
diluted with 25 ml of diethyl ether, and the precipitate
is filtered off and digested several times with cold
diethyl ether. This crude product is recrystallized from
chloroform/active charcoal.
Yield: 734 mg of yellow crystals (57% of theory)
M.p.: 225-227C
Thin-layer chromatogram: PE:EtOH=2:1 RF = 0.25
H-NMR: (DMSO)
~(ppm): 8.09-7.88 (m, 2H, Py-H5, Bzfu-H7); 7.74-7.53 (m,
4H, Bzfu-H3,4,Py-H3,4); 7.43-7.21 (m, 2H, Bzfu-H5,6);
3.02 (8, 3H, CH3)
2137~43
- 22 -
Example A:
The formation of prostaglandin D2 by neutrophil6
was used as a measure of the cyclooxygenase activity, and
the formation of leukotriene B4 as a measure of the
5-lipoxygenase activity.
Male Sprague-Dawley rats (250-300 g) received 1 mg of
la~mbda-carrageenan (dissolved in 0.5 ml of distilled
water) by intraperitoneal injection.
After 16 hours, the rats were sacrificed by exposure to
diethyl ether. 15 ml of ice-cold Hanks balanced salt
solution (HBSS) were injected i.p., the neutrophils were
harvested by aspiration (10 ml) and centrifuged (5 min,
100 g, 4C), the supernatant solution was decanted, and
the cells were resuspended in HBSS at 4C to a
concentration of 5X106 cells/ml.
400 ~1 of cell suspension (2x106 cells), 0.5 ~l of com-
pound dissolved in DMS0 and 49.5 ~1 of HBSS were incu-
bated at 37C for 5 min. Then 50 ~1 of A23187 (2 ~mol/l
final concentration) were added and subsequently incu-
bated again at 37C for 5 min.
The reaction was stopped by centrifugation at 10,000 gfor 3 8, and the supernatant liquid was transferred into
precooled plastic tubes and left in an icebath for a
~-~imllm of 1 h before starting the radioimmllnoassay
measurement.
PGD2 and LTB4 were measured after dilution with HBSS
using commercial RIA kits.
The comparison substance used was 6-chloro-4-hydroxy-2-
methyl-N-(2-pyridyl)-2H-thieno[2,3-e]-1,2-thiazine-3-
carboxamide l,l-dioxide ("Lornoxicam", compound A)
IC50 (~mol/l)
Compound PGD2 LTB4
A 0.02 ~10
1 0.049 2
2 3.5 1.4
3 0.45 1.7
4 1.7 1.3
0.036 1.3