Note: Descriptions are shown in the official language in which they were submitted.
94/00607 PCT/CA93/00261
- 1 -
HYDROMETALLURGICAL COPPER EXTRACTION PROCESS
FIELD OF THE INVENTION
This invention relates to a process for the
extraction of copper from copper ore or concentrates, in
particular sulphide ores, in which the concentrate is
subjected to acid leaching to extract the copper
therefrom.
BACKGROUND OF THE INVENTION
Most copper ores or concentrates from which
copper is extracted on a commercial scale contain copper
in sulphide form, such as CuFeS2 (chalcopyrite), Cu5FeS4
(bornite) and Cu2S (chalcocite) (hereinafter referred to
as "sulphide ores"), as opposed to copper in oxide form
(hereinafter referred to as "oxide ores").
Extraction processes for copper may be broadly
classified into two categories, i.e. smelting and
leaching. In general, smelting processes are applied to
sulphide ores, whereas leaching is more often used with
oxide ores. It is noted that the preliminary step of
concentration of ores (by flotation) is usually applied to
sulphide ores, not oxide ores.
The reasons for the differing treatment for the
two types of ores are generally technical, and thus also
economical. Sulphide ores float well, producing
relatively high grade concentrates from low grade ores.
These sulphide concentrates are well suited to the proven
smelting process. Oxide ores on the other hand, do not
concentrate easily and therefore it is difficult to
provide a feed material that is sufficiently high grade
WO 94/00607 PCT/CA93/0026~
21~~38~ ~.
- 2 -
for smelting. In addition, oxide ores do not have any
natural fuel, comparable to the sulphur in sulphide
concentrates. Fortunately though, oxide ores do leach
easily in sulphuric acid solutions, and thus a sizeable
industry has been established based on heap leaching of
oxide ores, followed by solvent extraction and
electrowinning.
Despite all of the above, smelting of sulphide
ores has some serious drawbacks, mostly concerned with the
need to avoid air pollution due to the sulphur gases
emitted. In locations where there is no market or use for
sulphuric acid, it is very difficult to justify a smelter
on economic grounds, assuming it is not permitted to vent
the gases freely to the atmosphere. Copper mines which
produce sulphide concentrates in such locations usually
ship the concentrates to distant (offshore) smelters.
When smelting/refining/shipping charges are low, this may
be economic, but there are times when the available world
smelting capacity for custom concentrates is limited and,
consequently, smelting charges rise sharply according to
the law of the marketplace. At such times, concentrate
producers. may be placed at a severe disadvantage.
Therefore there is a need for an economical and
technically sound leaching process that can treat sulphide
concentrates at the mine site, particularly concentrates
based on chalcopyrite, as this is the most widely
distributed copper mineral worldwide.
One of the problems which arises with the
leaching of copper from sulphide ore is that, while a high
acid concentration may be desirable during the leaching
stage, such a high concentration is undesirable during
the subsequent solvent extraction stage, due to
unfavourable equilibrium conditions which are created and
CA 02138381 2004-07-06
- 3 -
leading to higher equipment and working costs. This
requires neutralization in order to yield a pregnant liquor
suitable for solvent extraction, rendering the process
uneconomical, in particular where low grade ores are
involved.
It is accordingly an object of the present
invention to alleviate the above-mentioned difficulties.
SiJN~fARY OF THE INVENTION
According to the invention, there is provided a
process for the extraction of copper from a sulphide copper
ore or concentrate, comprising the steps of subjecting the
ore or concentrate to agitation leaching at an elevated
temperature and pressure to obtain a resulting acidic leach
liquor containing dissolved copper; and reducing the
acidity of the resulting acidic leach liquor by effecting
percolation leaching of a bed of low-grade copper ore or
granular rock with said resulting acidic leach liquor,
whereby the pH of the leach liquor is raised.
In one embodiment, the agitation leaching is
carried out in the presence of oxygen and water to obtain
an acidic leach liquor containing sulphate and dissolved
copper. In another embodiment, the agitation leaching
comprises a first leaching step at an elevated temperature
and pressure in the presence of oxygen and an acidic
chloride solution as lixiviant and a second leaching step
at atmospheric pressure with an acidic sulphate solution.
Also according to the invention, there is
provided a process for the extraction of copper from a
sulphide copper ore or concentrate, comprising the steps of
leaching the ore or concentrate a.n a first leaching
WO 94/00607 ~ ~ , PCT/CA93/00261~
- 4 -
step at an elevated temperature and pressure with an
acidic chloride solution to produce an insoluble basic
copper salt; leaching the basic copper salt produced by
the first leaching step in a second leaching step at
atmospheric pressure with an acidic sulphate solution to
dissolve the basic copper salt to produce a leach liquor
containing copper sulphate in solution; subjecting said
leach liquor from the second leaching step to a solvent
extraction process to produce a copper concentrate
solution and an acidic raffinate: and heap leaching a mass
of low-grade copper ore or granular rock by means of said
acidic raffinate to raise the pH of said raffinate.
The process may further comprise the step of
recycling the raffinate from the heap leaching step to the
first or the second leaching step. The insoluble basic
copper salt may comprise a mixture of basic copper
chloride and basic copper sulphate.
Further objects and advantages of the invention
will become apparent from the description of a preferred
embodiment of the invention below.
BRIEF DESCRIPTION OF DRAWINGS
Figure 1 is a flow diagram illustrating, by way
of example, a copper extraction process according to one
embodiment of the invention;
Figure 2 is a flow diagram illustrating a copper
extraction process according to another embodiment of the
invention; and
Figure 3 is a flow diagram illustrating a copper
extraction process according to yet another embodiment of
the invention.
94/00607 PCT/CA93/00261
- 5 -
DETAILED DESCRIPTION OF PREFERRED EMBODIMENT
In order to be amenable to treatment by the
process of Figure 1, the copper ore or concentrate, being
predominantly chalcopyrite in the present example, should
be in finely divided particulate form. It is preferable
that the starting material be at least 90% minus 325 mesh
standard Tyler screen. In preparing a copper ore or
concentrate for carrying out the extraction process, the
concentrate is therefore reground, if necessary, to obtain
a finely divided concentrate slurry with a moisture
content of about 20-30%.
The concentrate slurry is then subjected to
leaching with water in the presence of oxygen in an
autoclave 12. The leaching is carried out at an elevated
temperature, e.g. of about 180°C-220°C, preferably about
200°C, and an oxygen partial pressure of about 150-300
psig (approximately 1000-2000 kPa). Water is continuously
added during the pressure leaching process and a solids
content of about 10-20% is maintained. The residence time
in the autoclave 12 is about 0.5 to 2 hours under vigorous
agitation.
The pressure leaching process can be carried out
in one or more autoclaves. The autoclaves may be of the
conventional type having several internal compartments,
e.g., from two to eight, typically four, and provided with
agitators in the compartments. In the present example a
single autoclave 12 having four compartments 12.1, each
provided with an agitator 12.2, is shown. The ore is
introduced through feedline 12.3 and oxygen under pressure
is introduced through feedline 12.4. Water is introduced
through feedline 12.5.
WO 94/00607 ~ ~. , . PCT/CA93/0026~
- 6 -
As an example of a typical reaction taking place
in the autoclave 12, during the leaching process, the
following equation is given:
4CuFeS~ + 1702 + 4H20 -~ 4CuS04 + 4H2S04 + 2Fe203
Iron is precipitated as hematite, whereas copper
is dissolved and sulphuric acid is formed during the
leaching process. Additional acid may be added to the
autoclave 12, e.g. to dissolve the iron oxide, if desired.
The product slurry will typically contain in the solution
about 20-200 grams per litre Cu, 0.1-100 grams per litre
Fe and about 20-100 grams per litre H2S04. Depending on
the final acidity of the solution, any iron present will
precipitate out or remain in the solution.
The product slurry from the autoclave 12 is
flashed down to atmospheric pressure in a flash tank 13
operating at about 90°C-100°C.
The product slurry ,is then directed to a heap 14
for carrying out a heap leaching process. The heap 14 is
in a closed circuit operation with a solvent extraction
plant 16 in which the solvent extraction raffinate
(aqueous stream left over after the copper has been
extracted) is recycled to the top of the heap 14. The
solvent extraction plant 16 in turn is in a closed circuit
operation with an electrowinning plant 18.
The product slurry, which also includes all the
solids produced by the agitation leaching process, such as
iron solids and gangue minerals (silica, etc.) is first
filtered, as shown at 20 in Figure 1, and then the
filtrate or product liquor is mixed with the recycled
solvent extraction raffinate, prior to being applied to
the heap 14, in a ratio of about 1:50, i.e. one part
94/00607 . ~ PCT/CA93/00261
product slurry to about 50 parts raffinate by weight.
However, it is conceivable that a ratio in a broader range
of from about 1:50 to about 1:500 may be used.
The heap leaching process is carried out
primarily to neutralize and filter the product liquor
rather than for the purpose of leaching copper values from
the heap itself, which, in the present example, comprises
ordinary waste rock normally generated by a copper mine,
or low grade copper ore piled up in dumps as run-of-mine
waste. Such waste is uncrushed and typically contains
large boulders of up to one meter across, together with
considerable fine material. Copper values in most waste
ore from open pit mines in British Columbia, Canada, is
typically about 0.1~ Cu. However, copper values can be
higher in other countries. In general, the copper values
in the waste rock is typically less than 1 or 2~ of the
total mass of rock. The rest of the rock is primarily
gangue or silicates. Therefore, despite the fact that the
main purpose is not to recover copper from the heap 14
itself, some useful leaching can take place. Any leaching
which does take place is enhanced by the addition of the
product slurry to the raffinate because it increases the
acidity as well as the iron levels in the liquor being
sprayed onto the heap 14. The low grade ore in the heap
14 may be sulphide ores or oxide ores, or both.
By passing the product slurry through the heap
14, neutralization of the acid and removal of dissolved
iron in the leach liquor by the minerals in the waste rock
is achieved. It has been found that in such heap leaching
situations, an equilibrium condition appears to be
established between the leach liquor and the heap 14
regarding acid and iron content. Thus, irrespective of
the acid and iron content of the product slurry being
poured onto the heap 14, and irrespective of the make-up
WO 94/00607 . PCT/CA93/0026~
_ g _
of the waste heap 14, the acid and iron content of the
resultant leach liquor coming off the bottom of the heap
14 is relatively constant in composition at about pH
1.5-2.0 and about 1-2 grams per litre Fe.
A possible explanation for this phenomenon is
that minerals normally found in waste rock, such as
feldspars and clays, reach an equilibrium condition with
the solution when a large excess of rock over the
solution, e.g., of about 100:1, is present, so that the
rock does not become saturated. The feldspars and clays
slowly absorb or neutralize the acid from the leach liquor
and the degree of this reaction is dependent upon the
strength of the acid solution. The net result is that the
resultant pregnant leach liquor is always about the same
in acidity and iron content. Therefore, the heap 14 may
be composed of any rock which will serve this purpose and
need not necessarily contain any copper ore.
The above explanation is given solely for the
purpose of providing as much information as possible and
is believed to be correct. However, in the event that in
the future the explanation is found to be incorrect or
imprecise, the applicant does not wish to be bound
thereby.
The pregnant leach solution from the heap 14 is
passed to the solvent extraction plant 16 and the
electrowinning plant 18 to produce cathode copper. The
raffinate is recycled to the top of the heap 14 as
previously mentioned.
In the above example, the process was carried
out with a predominantly chalcopyrite ore, but the process
can also be carried out with other sulphide ores, such as
ores containing bornite.
94/00607 PCT/CA93/00261
j
- 9 -
Tests have been carried out to determine the
neutralization capacity of waste rock, both in finely
ground form as well as in crushed form, firstly using only
sulphuric acid and water and secondly using actual leach
liquor as the source of acid. Tests have also been
carried out to determine the effectiveness of the pressure
leach stage by subjecting finely ground samples of
concentrate to pressure leaching. These tests are
described in more detail below but first a description of
the samples of concentrate and waste rock used in the
tests is given.
A. Samples of Concentrate used for Testwork
Samples of Cu concentrate for pressure leaching
tests were obtained from two Cu mines in Canada, i.e.
Gibraltar Mine at Williams Lake, B.C. (referred to as
Mine "A") and Highland Valley Copper Mine near Kamloops,
B.C. (referred to as Mine "B°').
The Gibraltar concentrate is considered a fairly
typical Cu concentrate, in terms of grade and mineralogy
(chalcopyrite), whereas the Highland Valley concentrate is
a high grade concentrate, due to the occurrence of
bornite, a secondary Cu mineral with high Cu content in
the ore.
B. Samples of Waste Rock used for Testwork
Samples of waste rock were obtained from the
same two mines for use in neutralization tests. These
were actually low grade Cu ore samples. The
neutralization process involves the reaction of the
various silicate minerals in the waste rock. Therefore,'
it is useful to have a description of the mineralogy of
the waste rock samples. These two mines are both porphyry
WO 94/00607 ~ ~ ~ ~' ~ ~ ' PCT/CA93/0026~
- 10 -
Cu mines, in which the host rocks are a complex series of
aluminum silicates, with substantial amounts of lime,
soda, potash, magnesia, iron oxides, etc. Assaying
produced the following results (per cent by weight):
Sample Mine Cu Fe S Principal Cu Minerals
1 p 0.33 2.40 0.96 Chalcopyrite
2 B 0.24 0.68 0.20 Bornite, Chalcopyrite
Whole Rock Analysis
am le - - - - - -Ca0 K_20 P_205 Si02 A1~03 M_g0_ Na20 Fe203 Ti02 Mn0 Total
1 3.00 1.44 0.11 66.21 14.37 1.94 2.52 5.55 0.42 0.09 98.56X
2 2.74 1.29 0.10 71.43 14.73 0.49 4.13 1.40 0.26 0.05 99.37%
Mineralocxy of Sample 1
The host rock of Sample 1 is described in the
literature (See: "Gibraltar - Regional Metamorphism,
Mineralization, Iiydrothermal Alteration, and Structural
Development", by A.D. Drummond et al, in Porphyry Deposits
of the Canadian Cordillera, Part B --Porphyry Copper and
Copper-Molybdenum Deposits of the Calc-Alkalic Suite--
Paper 19, CIM Special Volume #15, published by Canadian
Institute of Mining and Metallurgy) as having the
following general composition:
- Duartz Si02 z5-suz
- Plagioclase a mixture of albite-epidote-zoisite-muscovite
typically: NaAlSi308, CaAl2Si08, H2KAl3(Si04)3 50-55X
- Chlorite typically: H4Mg3Si209 10-20%
In determining the actual mineral assemblage of
the rock, specimens of the rock were prepared as thin
94/00607 . PCT/CA93/00261
- 11 -
sections, each incorporating two or three fragments of the
predominantly macroscopically distinguishable rock types
from a bag of rock chips. The rock fragments were found
to consist mainly of quartz diorite, showing moderate to
strong pervasive sericitization and epidotization, and a
mineralized quartz-sericite rock, probably representing a
sheared and intensely altered form of quartz diorite.
Estimated composition
Quartz 31
Plagioclase 49
Sericite 8
Epidote 5
Chlorite 5
Carbonate 1
Sphene trace
Pyrite 1
Chalcopyrite 0.2
Fe oxides trace
The estimated composition was established by
point counting of the various components in a series of
random fields.
The predominant constituents were found to be
quartz and plagioclase. The latter showed mild to locally
intense sericitization, and was often more or less
strongly dusted with minute granules of epidote. These
same two components also occurred in coarser-grained form,
concentrated as pockets in aggregates of quartz and/or
feldspar. Minor carbonate was found to occur in like
manner.
Chlorite was the characteristic mafic
constituent.
WO 94/00607 ~ , PCT/CA93/0026~
- 12 --
Mineralogy of Sample 2
The host rock of Sample 2 is similarly described
in general terms in the literature (See: "Valley Copper",
by M. Osatenko and M.B. Jones, Paper 15, CIM Special
Volume #15, published by Canadian Institute of Mining and
Metallurgy).
- Quartz Si02 Sox
- Plagioclase see above 55X
- K-Feldspar KAlSi30$ 10%
- Biotite Mg-Fe mica
typically: HZK(Mg,Fe)3(AL,Fe)(Si04)3 5X
Specimens of this rock were prepared similarly
to Sample 1 above as thin sections. The selected
fragments were found to be mildly sericitized quartz
diorite (quartz-plagioclase rock with minor altered
mafics) .
Estimated composition
Quartz 37
Plagioclase 53
Sericite 6.5
Hornblende 0.3
Biotite trace
Chlorite 1
Carbonate 1
Chalcopyrite 0.2
Bornite trace
Fe oxides 1
Like Sample 1, it consisted predominantly of
quartz and plagioclase. The plagioclase differed from
that of Sample 1 in its lesser degree of pervasive
alteration, being essentially fresh but for a mild
argillic turbidity and a light dusting of minutely fine-
grained sericite. Epidote was notably absent. The
majority of the accessory sericite in this rock was found
to occur as meshwork clusters of relatively coarse flakes,
94/00607 ~ - PCT/CA93/00261
- 13 -
intergranular to quartz and/or feldspar. Mafics were
found to be notably sparse. They consisted of chlorite,
minor fresh hornblende and traces of biotite. Carbonate,
Fe oxides and Cu and Fe sulphides were found to be minor
accessories.
The results of the tests referred to above will
now be described in the Examples below.
Example 1: Neutralization capacity of waste rock
Samples from each mine were tested for their
(maximum) neutralization capacity by the following
procedure (Sample 1 was tested more than once):
The samples were first subjected to crushing to
about 1/2" size in a laboratory jaw crusher, and then wet
grinded in a small rod mill, to produce a slurry in which
the solids were typically 99~ minus 325 mesh. The slurry
was removed from the rod mill and then further diluted to
about 30~ solids with water and put into an agitated
vessel. Dilute sulphuric acid was then slowly added to
the slurry to reduce the pH of the slurry from its natural
value (near neutral) to pH 1.5. The pH was monitored at
intervals over the next few hours and acid added as
necessary to maintain the target pH 1.5. This procedure
lasted for 24 hours. The temperature was ambient (about
25 ° C) .
The total volume of acid added was measured and
at the conclusion of the 24 hour period, the slurry was
filtered to produce a residue and a filtrate. The residue
was dried and weighed and the filtrate volume measured.
Some solution analyses were also carried out on the
filtrate.
PCT/CA93/0026
WO 94/00607
- 14 -
The results are as follows:
Neutralization
Sample Test Vol Solids off Acid Addition Capacity of rock
No. No. Slurry Init. Final g/l Vol. Wt
1. grams 1. g. kg acid/ton rock
1 1 2.0 752 7.9 1.8 158 .143 23 30
1 2 2.0 778 - 1.5 158 .220 34.7 45
1 3 8.0 3165 8.3 1.5 158 .810 128 40
2 4 15.0 4754 - 1.5 158 1.867 295 62
Solution analyses indicated that about 15~ of
both the Cu and Fe contained in the rock was leached
during the tests.
The above results show that both types of rock
(after fine grinding) absorb substantial amounts of acid,
i.e. at pH 1.5, 40 kg acid and 62 kg acid per metric ton
rock respectively for samples 1 and 2. Since finely
ground rock probably reacts much faster than coarser
(crushed) rock, these results represent the upper limits
of neutralization capacity for the rock if it is used in
crushed form rather than in a finely-ground form. In
carrying out the method according to the invention, it is
preferable to use the waste rock in coarse sizing, for
economic reasons. Then the neutralization can be done on
heaps of crushed or run-of-mine rock, rather than in
agitated tanks, which require the extra expense of
grinding as well as agitation, filtering, etc. The test
procedure used here does not indicate the rate at which
the rock absorbs acid either, particularly the rate at
which crushed rock reacts.
To obtain data on crushed rock, further tests
were carried out as described below:
94/00607 ~ ~ _ PCT/CA93/00261
- 15 -
Example 2: Neutralization with crushed waste
rock. usinq~ synthetic acid.
A sample of rock from Mine A from the same lot
as described above was crushed to about 1/4" size and
placed in a small glass column having a 2-inch diameter
and 24 inches high. The column was placed upright and
fitted with layers of plastic screen at the bottom of the
column to hold the rock in place with plastic tubing
connections to the top and bottom of the column. The
total weight of rock placed in the column was 1865 grams
(wet weight) at 3.54 moisture, or about 1798 g
(approximately 1.8X10'3 metric ton) of dry rock.
The tubing leading from the bottom of the column
was connected to a small (product) reservoir, for
collection of liquor draining out of the column. A second
small (feed) reservoir was used to supply solution to the
top of the column, by means of a small peristaltic pump,
controlled by a timer.
The purpose of the test was to determine how
much acid could be absorbed or reacted by the column of
rock, by slowly percolating an acidic solution through the
column of rock, and at which rate. For this test, the
acid was supplied in two solutions, an initial solution
and then a supplemental acid as required.
The initial solution was derived from Test #1 in
Example 1 above, i.e. by agitation of a slurry of ground
rock in dilute acid at pH 1.5 for 24 hours. The solution
was separated from the solid fraction of the slurry as
described by filtration to yield 1.08 litres. In
addition, the filter cake was washed and the wash filtrate
35' (0.62 litres) added to the first filtrate to give a total
solution of 1.70 litres which was placed in the feed
reservoir at the start of the test.
PCT/CA93/0026
WO 94/00607
- 16 -
The supplemental acid was a (synthetic)
solution prepared by mixing pure (98~) chemical grade
H2S04 with water, to obtain a dilute solution of acid,
which was determined by filtration to contain 250 grams
per litre H2S04.
The test was started by slowly pumping the
initial solution from the feed reservoir onto the top of
the column and allowing it to percolate through the rock
at a rate of approximately 550 millilitres per day, which
corresponds to typical leaching rates used for heap
leaching, i.e. 0.005 gallons per ft2 per minute. The
solution coming off the bottom of the column was collected
in the product reservoir.
At suitable intervals (1-3 days typically) the
product solution was sampled, pH measured and acid added
to adjust the pH down to 1.5 - 1.7 range. This pH
adjustment was made using the supplemental acid solution
described. Then the (acidified) product solution was
emptied into the feed tank and the process continued.
Samples of the solution were assayed for dissolved Cu and
Fe. A record was kept of total acid additions. The test
was continued for 121 days.
Initially, the acid consumption was rapid,
averaging about 1.2 kg acid/ton rock per day, for the
first 20 days. It then decreased to about 0.1 kg acid/ton
rock per day after 60 days and remained roughly constant
at this rate until the end of the test.
,94/00607 ~ $ ~ ~ , _* PCT/CA93/00261
- 17 -
The results are as follows:
Days pH CCu7 CFe1 Acid Consumption XCu
ppm ppm gH2S04 RatioRate Leached
from rock
0 1.75 210 1189 - -
1 6.57 - - - -
1.67 790 2700 45 25 1.26 22
41 1.63 725 3305 56 31 0.28 21
62 1.68 610 2570 60 33 0.11 20
83 1.54 700 3700 66 37 0.16 24
15 100 1.52 750 3700 69 39 0.12 25
121 1.54 670 3500 T3 41 0.09 25
The "ratio" in the above table is the ratio of
20 the amount of acid consumed to the amount of rock present,
i.e. 1.8x10-3 ton rock. The "rate" refers to the rate of
acid consumption as the daily rate over the last entry in
the table, expressed as kg acid/ton rock per day.
The total acid consumption after 121 days was 41
kg acid per ton rock, which is similar to the values
(average 38 kg/acid per ton rock), obtained with finely
ground rock in 24 hours, as described in Example 1.
Therefore, the neutralization with crushed waste rock
appears to give similar final results, albeit at a much
slower rate, as would be expected.
The amount of Cu and Fe leached from the rock is
of interest also. Any Cu leaching from the waste rock
will augment the Cu obtained from leaching concentrate.
The results indicate that about 20~ of the Cu in the rock
leached very quickly, but after that Cu leaching
practically stops. It is known that the Chalcopyrite (the
' PCT/CA93/0026~
WO 94/0060 ~7
_ 18
predominant Cu mineral in this sample) leaches very slowly
under these conditions, which approximate heap leaching,
and even this low rate is dependent on bacterial action
(bioleaching) to some extent. No attempt was made in
these tests to introduce or cultivate bacteria, such as
thiobacillus ferrooxidans or thiobacillus thiooxidans,
which are well-known to improve leaching of Cu sulphide
minerals. The 20~ Cu that did leach may have been due to
some surface oxidation on the sulphide particles.
The amount of Fe leaching is of interest for the
following reasons: It is important that Fe in the
neutralized liquor, coming off the bottom of the column,
should stabilize at some reasonable value such as 1000 to
5000 ppm, rather than continue to build up as the solution
is recirculated. In actual practice, neutralized liquor
will go to solvent extraction where Cu is removed leaving
a raffinate stream containing all the Fe which will go
back onto the heap. Therefore, there is no removal
mechanism for Fe from the circuit other than the heap
itself, which should come to equilibrium with dissolved Fe
at about 1000 to 5000 ppm, depending on the pH. Some Fe
will leach from the minerals in the waste rock, but
eventually this dissolution rate will be balanced by other
chemical reactions such as hydrolysis which remove the Fe
from solution, thus leading to a stable equilibrium value.
The results in the above Example 2 indicate that indeed
the Fe has stabilized at about 3700 ppm in solution.
The ratio of Fe++ to Fe+++ in the solution was
monitored, along with the redox potential (Pt electrode
vs. Ag/AgCl reference electrode):
9.x/00607 PCT/CA93/00261
- 19 -
Days CFe++7 CFe+++~ Redox
ppm ppm mV
0 1000 189 427
20 1750 950 415
41 2000 1305 420
62 150 2420 520
83 - - 533
100 - - 536
121 60 3440 541
The results show that initially the oxidation
potential on the column was quite reducing, as evidenced
by the high Fe++ to Fe+++ ratio and low redox potential.
After about 60 days, the solution became more oxidizing,
probably as a result of surface oxidation of the reducing
minerals in the column of rock. There was no attempt to
introduce or restrict the access of air to the column, so
it is likely that some atmospheric oxygen gradually reacts
with the rock minerals over time. The small size of the
test equipment in this example, compared to a practical
application, i.e. many thousands or millions of tons of
rock on a heap, tends to give greater access to
atmospheric oxygen than would be realized in a practical
application. However, the same general principle is
likely to be found, i.e. initial reducing conditions,
slowly converting to more oxidizing over time. The
neutralization process is not dependent on any special
oxidation conditions and it is to be expected that it will
work satisfactorily on the larger scale of actual heaps as
well as on the micro scale of laboratory columns.
PCT/CA93/0026,~
WO 94/00607
- 20 -
Example 3~ Pressure leaching of Concentrate
Concentrate samples were assayed for Cu, Fe and S:
Cu Fe S_
Gibraltar 28.6 29.4 29.9 X
Highland Valley 41.4 22.2 28.0 X
Screen analyses of the concentrates as received
were also carried out:
-200 -325 Mesh
Gibraltar 86 39 X
Highland Valley 79 29 X
Both concentrates samples were subjected to
regrinding in a laboratory rod mill for 35 minutes, in
preparation for pressure leach testing. Screen analysis
of the reground concentrates indicated 99~ passing minus
325 mesh (44 micron).
Pressure leaching of the concentrates was done
in a two litre titanium autoclave equipped with an
agitator including two impellers, and a temperature
control system including internal cooling coils and
external heating system.
Leaching tests were done in batch mode. The
_ autoclave was charged with a fixed weight of concentrate
and lixiviant before the test and all slurry products
discharged completely from the vessel at the end of the
35' test. The lixiviant used was water with no addition of
94/00607 ~ PCt"/CA93/00261
- 21 -
acid. Oxygen used for the tests was high purity bottled
oxygen. No other reagents were used in the tests.
The procedure used for the tests is as follows:
The autoclave is charged with concentrate and water,
closed up and sealed. The agitator is turned on. The
autoclave is heated to about 180°C by an external heating
system. The autoclave is then pressurized with oxygen to
300 psig (approximately 2000 kPa). This marks the
starting time. The temperature which rapidly increases to
the reaction in the autoclave is controlled to remain in
the region of 200-210°C by the use of a cooling system.
During the test allowance is made for a small bleed of
vent gas from the 'autoclave, while maintaining a pressure
of 300 psig (approximately 2000 kPa) in the autoclave with
an external oxygen cylinder. The test is continued for 60
minutes while the temperature is maintained about 200°C.
At the end of 60 minutes the contents of the autoclave is
cooled to about 95°C. The autoclave is depressurized and
opened. All the slurry contents is then discharged using
wash water as required. The combined slurry and washings
is then filtered on a vacuum filter. The filter cake is
washed with hot water, keeping the wash filtrate separate.
The filter cake is then dried, weighed and analyzed. The
filtrate and wash filtrate are also analyzed.
35
WO 94/00607 ~ ~ PCT/CA93/0026
- 22 -
The results are as follows:
TEST No. 1 2
Feed Materials
Source of conc. Gibraltar Highland Valley
Concentrate wt (dry) 170 171 grams
Water volume 1.0 1.0 litres
Products
Filtrate -volume 1.34 0.91 litres
-CCu7 35.8 78.3 9/l
-IFeI 0.7 0.4 9/l
-CH2S047 40.0 30.6 g/l
Wash -volume 1.07 1.50 litres
-CCu7 1.4 2.0 9/l
Residue -weight 92.0 69.0 grams
-XCu 0.70 1.07 X
-XFe 43.2 43.2 X
Cu Extraction 98.7 99.0 X
Mass Balance on Cu
Cu in Feed material 48.6 70.6 grams
Cu in combined products50.1 75.0 grams
X in Recovery 103 106 X
In the above table, the "Cu extraction" referred
to is calculated on the basis of Cu in the residue as a
fraction of Cu in the feed concentrate. The "~ recovery"
referred to is defined as Cu in the combined products as
of Cu in feed materials.
The data indicate excellent Cu recovery to leach
liquor from both concentrates by the pressure leaching
94/00607 PCT/CA93/00261
- 23 -
process, with minimal solubilization of Fe. Most of the
Fe, though oxidized, appears to hydrolyze as Fe203, or
some hydrated form. The leach liquor contains excess acid
in both cases, indicating that a high proportion of the
sulphur in the concentrates is oxidized to the sulphate
form. The results generally support the chemical reaction
as given earlier for chalcopyrite in the concentrate:
4CuFeS2 + 1702 + 4H20 ~ 4CuS04 + 4H2S04 + 2Fez03
A corresponding reaction can be written for
bornite, Cu5FeS4, giving the same products but in
different ratios.
The solutions produced by these and similar
pressure leach tests were used as the source of acidic
leach liquor for the subsequent tests on neutralization
using waste rock.
Example 4: Neutralization of pressure leach liquor
with waste rock
The test described in Example 2 above was
repeated using actual pressure leach liquor as the source
of acid, rather than a synthetic acid solution.
Separate tests were carried out on each of the
two types of waste rock described, Gibraltar and Highland
Valley, using the corresponding pressure leach liquor, as
in Example 3.
The procedure used was essentially the same as
in Example 2, with the product liquor coming off the
bottom of the column being acidified to pH 1.5, and then
this acidified product solution was used as the feed
solution. Due to the Cu content of the leach liquor, this
resulted in a considerable buildup of Cu in the recycling
WO 94/00607 . ~ ~ ~ ~ ' PCT/CA93/00261~
- 24 -
solution, i.e. 14 grams per litre Cu for the Gibraltar
test, and 45 grams per litre Cu for the Highland Valley
case, as no attempt was made at this stage to remove Cu
from the product solution by solvent extraction.
The results are as follows:
~xamole 4A Gibraltar waste rock Column Test Ho. 1
Weight of Waste rock used in Column: 1824 grams
Composition of pressure leach liquor used for acidification:
Days 1 - 55: 52.5 g/l H2S04, 47.8 g/t Cu
Days 56 - 83: 40.0 g/t H2S04, 35.8 g/t Cu '
Days pH tCu) IFe7 Acid Consumction Volume of
ppm ppm 9H2S04 Ratio Rate leach liqr
Used (mL)
0 1.63 283 1953 6.28 3.44 - -
3 4.43 550 643 13.79 ~ 7.561.37 143
9 2.43 5230 2000 22.61 12.40 0.81 311
20 1.66 8500 2832 27.09 14.85 0.22 396
30 1.48 10125 3250 32.01 17.55 0.27 490
1.88 10188 4588 39.59 21.70 0.42 634
59 1.58 14500 6300 48.58 26.63 0.26 814
83 1.55 14000 6700 53.77 29.48 0.12 944
35
_ ~~.~888~.
~, 94/00607 PCT/CA93/00261
- 25 -
Example Hig hland wasterock Column Test No. 2
4B Vallev
Weight rock ed in 1864 grams
of Waste us Column:
Compositionofpressureleach usedfor
liquor acidification:
Days 1 - 30.6 g/l H2S04,78.3g/tCu
15:
Days 15- 28.5 g/t H2S04,67.9g/tCu
18:
Days 18- 25.8 g/l H2S04,55.0g/lCu
36:
Days 36- 25.3 g/l H2S04,50.0g/lCu
40:
Days 40- 26.8 g/l H2S04,53.7g/lCu
50:
-Days 50- 24.0 g/l H2S04,54.3g/lCu
68:
Days 68- 16.5 g/l H2S04,35.5g/lCu
71:
Days pH CCuI CFeI Acid Consumption Volume
of
ppm ppm gH2s04 RatioRate Leach liqr
Used (mLl
0 - - - 6.24 3.35
5 5.53 170 374 10.75 5.77 0.48 147
11 3.95 11480 6320 27.58 14.741.50 698
13 3.61 17180 855 27.58 14.74- 698
15 2.90 20550 1145 35.88 19.252.22 989
25 2.25 26375 3350 51.18 27.450.82 1582
36 1.73 32400 4000 60.89 32.670.47 1962
' 1.6231450 5550 63.70 34.170.37 2067
40 50 1.50 42000 5950 68.92 36.970.28 2262
64 1.59 41100 8000 70.05 37.580.04 2309
- 71 1.56 41750 8000 73.28 39.310.13 2505
WO 94/00607 ~ ~ ~ ~ ~ PCT/CA93/0026~
- 26 -
The results indicate that the absorption of
acid by Gibraltar rock from actual pressure leach liquor
is somewhat slower than for the corresponding test with
synthetic acid, i.e. 30 kg acid consumed/tonne rock
compared to 37 kg acid for the synthetic acid test, after
the same time (83 days).
Similarly the amount of acid consumed by the
Highland Valley rock is less after 71 days (39 kg
acid/tonne rock) than by the corresponding test with
ground rock (60 kg acid/tonne rock).
It appears therefore that the strong dissolved
salts, Cu and Fe sulphates, have some effect on the amount
of acid absorbed. It is expected that in the commercial
plant, using solvent extraction, to keep Cu levels down to
about 3-5 grams per litre Cu, will not suffer this effect
nearly as much.
After the conclusion of the two tests in this
Example, the rock was washed thoroughly with water to
remove any entrained liquor, and subjected to
mineralogical examination, to see what effect the acid
neutralization tests may have had on the waste rock.
Evidence was found of conversion of silicates,
i.e. feldspars to gypsum, in the fine fraction of the
rock. These conclusions are tentative and must be
confirmed by further investigation.
In situations where the concentrate contains
significant precious metal values, such as gold and
silver, the process of the invention includes a
filtration step or other liquid-solid separation means for
the flash tank slurry before application thereof to the
94/00607 $ PC'T/CA93/00261
- 27 -
heap 14. The solids obtained during the filtration step
can then be removed for further treatment in a refinery.
It is an advantage of the process according to
the invention that by combining the pressure leaching
step with a percolation leaching step involving low grade
ore or waste rock, the neutralization of the product
slurry is economically and effectively carried out without
the need for extraneous neutralizing agents. An
additional advantage is that further copper values are
simultaneously recovered during the neutralization process
in situations where low grade copper ore is used in the
percolation leaching step.
The result of the above is that low grade ore,
containing less than 0.25 copper and as low as 0.1~
copper can be successfully processed, whereas processing
of such ores with the conventional processes would be
uneconomical.
With reference now to Figure 2, a copper
extraction process according to another embodiment of the
invention is illustrated.
The stages of the process are shown in the flow
diagram of Figure 2. The stages comprise a pressure
leach stage 30, an atmospheric leach stage 32, first and
second solvent extraction stages 34 and 36, respectively,
a washing stage 38, first and second stripping stages 40
and 42, respectively, a neutralization stage 44 and an
electrowinning stage 46. After each of the leaching
stages 30 and 32, filtration is carried out as indicated
at 48 and 50, respectively, to separate the liquids and
solids.
WO 9.~f00607 ~ _ PCT/CA93/0026~
- 28 -
The process will now be described in greater
detail by way of a specific example in which a
chalcopyrite ore is treated.
Copper concentrate is first'ground in a ball
mill to reduce the size of the particles to about 95~
minus 325 mesh or smaller. Although satisfactory results
are obtainable without regrinding, it has been found that
there is a small but significant improvement with
regrinding.
The concentrate is leached at an elevated
pressure and temperature with a lixiviant containing about
3 grams per litre copper, 12 grams per litre chloride, 7
grams per litre sodium at a pH of about 3 to 5 (leaching
stage 30). About 80-90~ of the lixiviant is leach liquor
which is recycled after the leaching stage 30 and
filtration 48. The remaining 10-20~ comprises raffinate
from the subsequent solvent extraction step 36, to be
described below.
The temperature of the leach 30 is about 150°C
and the pressure about 200 psig (1380 kPa). This is
total pressure comprising oxygen pressure on top of the
steam pressure. The retention time is about 2.5 hours in
a batchwise process.
The solids content is maintained at about 11-
13~, i.e. 120-150 grams per litre solids as determined by
the heat balance. A higher percentage solids would
require some form of heat removal to prevent the
temperature from rising above the desired limit of 150°C.
Sodium chloride in solution is added as makeup
to maintain the chloride concentration of the lixiviant at
about 12 grams per litre.
94/00607 _ ~ ~ ~ PCT/CA93/00261
- 29 -
The slurry produced by the leach 30 is cooled to
below 100°C and then filtered 48 to separate the residue
from the leach liquor, which is recycled to the leaching
stage 30 as noted above.
The residue contains the copper originally
present in the concentrate as insoluble basic copper
chloride and basic copper sulphate together with all the
other solid elements, such as iron and sulphur.
There is a gain in the weight of the leach
residue, typically it has 30-40~ more weight than the feed
concentrate. The leach residue contains about 0.5-2~
chloride, as well as the copper, iron and sulphur, which
is due to the presence of the basic copper chloride and
the basic copper sulphate.
The iron in the chalcopyrite concentrate is
converted almost completely to hematite, while sulphur is
mostly converted to the elemental form with a fraction
being oxidized to sulphate.
The leach liquor produced by the leaching step
has much the same composition as the feed lixiviant
25 except that there is a drop in the chloride concentration
from about 12 grams per litre to about 7-10 grams per
litre, depending on the conditions, due to the formation
of the basic copper chloride. Some sodium is also trapped
in the residue.
The filter cake or leach residue is repulped in
raffinate from the subsequent solvent extraction process
36, which comprises an acidic sulphate solution containing
about 20-25 grams per litre HZS04 and a small amount of
copper (about 1-3 grams per litre). This second leaching
step 32 takes place at atmospheric pressure and a
WO 94/00607 PCT/CA93/00261~
2~~~3~~.
- 30 -
temperature of about 40°C for a retention time of about
60-120 minutes. The percentage solids is about 6~ or
about 70 grams per litre. The final acidity of the slurry
is about pH 1.5-2.0 or about 2-5 grams per litre HZS04.
During the atmospheric leach 32, the basic
copper salts dissolve almost completely with very little
of the iron going into solution. Typically, the leach
liquor produced after filtration 50 contains about 15-20
grams per litre copper with less than 1 grams per litre
iron and about 0.3-1.0 grams per litre chloride.
The percentage solids is kept low during the
atmospheric leach 32 because higher copper concentrations
cannot be treated satisfactorily by the subsequent solvent
extraction circuit.
The copper extraction has been found to be
about 97-98~ based on the original feed to the pressure
leach. Iron extraction to solution is less than about 5~.
The main constituents of the solid residue
after filtration 50 are hematite and elemental sulphur, as
well as any gold or silver which may have been present in
the original concentrate. The sulphur can be recovered by
screening or flotation to separate it from the hematite
into a high-grade sulphur concentrate, which can be
further treated for recovery of sulphur. The gold and
silver can be recovered by cyanidation after sulphur is
removed from the leach residue.
The copper leached from the atmospheric leach 32
is extracted by means of solvent extraction 36 to produce
a loaded copper electrolyte suitable for electrowinning.
. After the solvent stage 36, the loaded organic extractant
is subjected to one stage of wash 38 to remove chloride
94/00607 PC'T/CA93/00261
- 31 -
and two stages of stripping 40, 42. The high copper
concentration of about 15-20 grams per litre derived from
the atmospheric leach 32 provides significant advantages
over conventional solvent extraction/electro-
winning plants because much higher loading of the organic
is possible, thus reducing the size of the plant for a
given tonnage of copper. Stripping of the loaded organic
is effected by means of spent acid from the electrowinning
stage 46, to obtain a pure copper sulphate solution which
is then passed to the electrowinning stage 46.
During the pressure leach 30, some of the
sulphur present, which may be about 15-20~ of the sulphur
in the concentrate, is oxidized to sulphate. This is
removed by subjecting about one-third of the raffinate
from the atmospheric leaching step 32, after solvent
extraction 36, to neutralization 44 by heap leaching waste
rock or low grade ore with the raffinate. The remainder
of the raffinate is recycled to the pressure leach and
atmospheric leach steps 30 and 32, respectively.
The raffinate after solvent extraction 36
contains about 20-25 grams per litre acid and about 1-2
grams per litre copper and iron. During neutralization 44
the acid is reduced to about 2-5 grams per litre by
contact with the silicate minerals in the rock, whilst
copper passes through the heap leach. Iron will be
hydrolyzed on the rock only if it builds up beyond its
solubility limit, depending on the acidity. In addition,
any copper minerals in the waste rock or low grade ore
will be partly leached depending on the time of leaching,
the type of mineral, bacterial activity, iron
concentration, temperature, etc.
Solutions derived from the waste rock
neutralization step 44 are passed to the first solvent
WO 94/00607 PCT/CA93/0026~
- 32 -
extraction stage 34 to recover dissolved copper, typically
1-3 grams per litre using stripped organic. After solvent
extraction 34, the raffinate is treated at 52 to remove
soluble impurities derived from leaching, such as zinc and
magnesium, which are not removed by the neutralization
step. This is carried out by using lime at a pH 10-10.5.
The final effluent from this process is suitable for
disposal in a tailings pond. The partially loaded organic
from the first solvent extraction stage 34 is passed on to
the second solvent extraction stage 36.
The embodiment of Figure 2 has been described
using a chalcopyrite ore which does not contain any
significant amounts of pyrite (FeS2), resulting in little
acid production during the pressure leaching stage. When
treating ore containing significant amounts of pyrite,
more acid is formed due to the oxidation of the pyrite to
sulphate. In such cases a greater degree of
neutralization is required and a greater proportion than
one-third of the raffinate after the solvent extraction
step 36, or indeed all the raffinate, may be subjected to
neutralization 44 by heap leaching waste rock or low-grade
ore with the raffinate and recycling the raffinate to
either the pressure leach or atmospheric leach steps or to
both the pressure leach and atmospheric leach steps 30,
32.
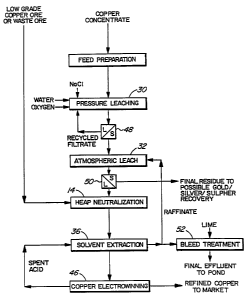
In more drastic situations, where large amounts
of acid is created, the neutralization step can be
introduced prior to the solvent extraction step 36, as
shown in the flow diagram of Figure 3. Thus, in such a
situation, the pressure leach and atmospheric leach steps
30 and 32 are followed by a heap leach step 14.
The solvent extraction 36 is operated in a
closed circuit with the atmospheric leach step 32, whereby
~~~8~~~
94/00607 PCT/CA93/00261
_ 33 _
the raffinate after solvent extraction is recycled to the
atmospheric leach step 32. Spent acid from the
electrowinning step 46 is recycled to the solvent
extraction step 36 to strip the copper from the loaded
extractant. A bleed from the solvent extraction raffinate
is subjected to lime treatment 52 to remove impurities and
disposed of in a tailings pond.
In situations of acid formation, water is also
introduced at the pressure leaching stage, as in the case
of the Figure 1 embodiment, to compensate for the water
consumed in the reaction taking place during the pressure
leaching.
While only preferred embodiments of the
invention have been described herein in detail, the
invention is not limited thereby and modifications can be
made within the scope of the attached claims.
25
35