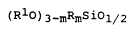Note: Descriptions are shown in the official language in which they were submitted.
214198~
Docket: WA 9343-S
Paper No. 1
PROCE~ FOR TH~: PREPARA'rIOII OF ORGAl~OPOLY~ILOXAI~I~
CONTAIl!II~IG ORGAIIYLOXY GROUP~3
Field of Invention
The invention relates to a process for the preparation of organopoly-
siloxanes containing organyloxy groups by reaction of hydroxysiloxanes with
0 organyloxysilanes, and the use thereof in compositions which can be cross-
linked at room temperatures.
Back~round of Invention
Processes for the preparation of organopolysiloxanes contz~inin~
organyloxy groups are already known. For ex~ple, U.S. 5,196,497 (Bayer
AG, issued on March 23, 1993) and the corresponding EP 468 239 A2
describe the reaction of a,~-dihydroxypoly(diorganosiloxanes) with alkoxy-
silanes in the presence of alkali metal hydroxides, which leads, by elimin5~-
tion of alcohol, to the desired polysiloxanes blocked by end groups. The
strong bases have a high equilibrating activity and, if the reaction time is
too long or the temperatures relatively high often lead to an undesirably
high content of monoalkoxy end groups which are not capable of crosslink-
ing. Deactivation of the catalyst with strong acids, such as, chlorosilanes or
phosphoric acid, must therefore be careried out at precisely the right time.
Since the reaction may occur literally within a minute, the time between
addition of the catalyst and deactivation of the catalyst of ~veral minutes,
customary during factory production, can result in a product which does
not meet the specification. Furthermore, the amount of deactivating reagent
should be precisely matched stoichiometrically to the amount of catalyst
employed, in order to quarantee the storage stability of the end product. In
practice, an excess of deactivating reagent will therefore often have to be
employed. Since these are strong acids having an equilibrating activity, this
excess must be removed from the product again.
U.S. 5,055,502 (Phone-Poulenc Chemie; issued on October 8, 1991)
describes a process in which zinc chelate complexes effect the blocking of
the ends of OH polymers with alkoxysilanes at relatively high temperatures.
2141985
DE 3428840 Al (General Electric Co.; published on February 21, 1985) and
the col~esponding GB 2144758 A disclose aluminum chelate complexes
which are employed as catalysts for alkoxy blocking of the ends of organo-
polysiloxanes containing OH groups. In U.S. 5,166,296 (General Electric
Co.; issued on November 24, 1992) and the corresponding EP 520 718 A2,
the preparation of polysiloxanes blocked by alkoxy end groups from aL~coxy-
nes and polysilox~qnes having terminal OH groups is car~ied out in the
presence of catalytic amounts of ammonium salts of carboxylic acids.
Sllmm~t~r of Invention
0 The present invention relates to a process for the preparation oforganopolysiloxanes which contain at least one unit of the formula
(RlO)3 mRmSiOl/2 (I)
in which
R is identical or different and is a hydrogen atom or monovalent,
optionally substituted hydrocarbon radical,
Rl is identical or different and is a monovalent, optionally substituted
hydrocarbon radical having 1 to 8 carbon atoms and
m is 0, 1 or 2,
which comprises, in a 1st step, reacting org~nosilicon compounds (1) which
20 contain at least one Si-bonded hydroxyl group with at least one silane (2) of the formula
(RlO)4 mSiRm (II)
and/or partial hydrolysates thereof, in which R, R' and m have the above
mentioned meaning, in the presence of a fluoride salt (3), and, optionally, in
25 a 2nd step, when the reaction has ended, adding component (4) which can
bond fluoride ions.
The term organopolysiloxanes in the context of the present invention
is also intended to include oligomeric siloxanes.
The radical R is preferably monovalent, optionally substituted hydro-
30 carbon radicals having 1 to 13 carbon atoms, where the methyl, vinyl and3-(N-cyclohexylamino)propyl radical are more preferred.
Examples of the radical R are allcyl radicals, such as the methyl,
ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert-butyl, n-pentyl,
2141985
-
iso-pentyl, neo-pentyl and tert-pentyl radical, hexyl r~ic~l~, such as the
n-hexyl radical, heptyl radicals, such as the n-heptyl radical, octyl radicals,
such as the n-octyl radical, and iso-octyl radicals, such as the 2,2,4-tri-
methylpentyl radical, nonyl radicals, such as the n-nonyl radical, decyl
radicals, such as the n-decyl radical, and dodecyl radicals, such as the
n-dodecyl radical; aLt~enyl radicals, such as the vinyl and the allyl radical;
cycloaLtcyl radicals, such as cyclopentyl, cyclohexyl and cycloheptyl radicals
and methylcyclohexyl radicals; aryl radicals, such as the phenyl and the
naphthyl radical; aL~aryl radicals, such as o-, m- and p-tolyl radicals, xylyl
radicals and ethylphenyl radicals; and araL~cyl radicals, such as the benzyl
radical and the a- and ~-phenylethyl radical.
Examples of substituted hydrocarbon radicals are haloaLtcyl radicals,
such as the 3,3,3-trifluoro-n-propyl radical, 2,2,2,2',2',2'-hexafluoroiso-
propyl radical and the heptafluoroisopropyl radical; haloaryl r~-lic~l~, such
as the o-, m- and p-chlorophenyl radical; the 3-thio-1-propyl radical;
acyloxyaL~cyl radicals, such as the acetoxyethyl radical and
(meth)acryloylo~y~ropyl radical; and A
CH2-CH-CH2-0-CH2-, HSCH2-,
H2NCH2-, 4,5-dihydroimi~1~701-l-yl-CH2-, imi~l~701-l-yl-CH2-,
pyrrolidinyl-CH2-, piperidyl-CH2-, N-morpholinyl-CH2-,
pipera_inyl-CH2-, cyclohexyl-NH-CH2-,H2N-CH2CH2-NH-CH2-,
H2C=C(CH3)C00-CH2-, 2-cyanoethyl-, 3-cyanopropyl-,
CH2-CH-CH2-0-(CH2)3-, HS(CH2)3-, H2N(CH2)3-, 4,5-dihydroimidazol-1-yl-
(CH2~3-, imida_ol-l-ly-(CH2~3-, pyrrolidinyl-(CH2~3-. piperidyl-(CH2~3,
N-morpholinyl-(CH2~3-, piperazinyl-(CH2~3-, cyclohexyl-NH-(CH2~3-,
H2N-CH2-CH2-NH-(CH2)3- and H2C=C(CH3)C00-(CH2)3- radical.
Examples of the radical Rl are the examples of optionally substituted
hydrocarbon radicals having 1 to 8 carbon atoms mentioned for R.
The radical Rl is preferably a methyl, ethyl, n-propyl, isopr< ~yl, pro-
pen-2-yl, n-butyl, sec-butyl or iso-butyl radical, methyl and ethyl radicals
being more preferred.
21419B~
The organosilicon compounds (1) cont~ining at least one Si-bonded
hydroxyl group which are employed in the process according to the inven-
tion are preferably those chosen from the group conci~ting of organopolysi-
loxanes having at least one Si-bonded hydro~yl group, and organosilanes
having a hydroxyl group.
The organosilicon compound (1) cont~ining at least one Si-bonded
hydroxyl group which is employed in the process according to the invention
can be any of the hydroxysiloxanes and monohydroxysilanes known to date.
The hydroxysiloxanes employed according to the invention can of course
contain other units containing Si-bonded hydroxyl groups, such as
(HO)2 ~R3~SiO2/2 and HOSiO3/2 units, in addition to units of the formula
(HO)3 tR3,SiOl/2, in which R3 has one of the me~nin~ given for R1 t is 0, 1 or
2 and sis0 or 1.
Ex~mples of the organosilicon compound (1) employed according to
the invention are a,Q)-dihydroxydiorganopolysiloxanes, such as
HOMe2Si(OSiMe2)l to looooOH and
HOMe2Si(OSiMe2)0 to lOOOO(OSiMeVi)o to looooOH, where this siloxane cont~ s at
least two silicon atoms, a-monohydroxydiorganopolysiloxanes and monohy-
droxysilanes, such as Me3Si(OSiMe2)0 ~ looooOH, HMe2Si(OSiMe2)~ ooooOH,
(H2C=CH)Me2Si(OSiMe2)0 to looooOH and
(H2C=CHCH2)Me2Si(OSiMe2)0 to looooOH, where Me is the methyl radical and
Vi is the vinyl radical, and branched hydroxy-functional organopolysilox-
anes and hydroxy-functional organopolysiloxane resins, such as described
in EP 540 039 Al (Dow Corning Japan Ltd.), column 5, lines 37 to 40 and
column 6, line 25, the olg~lyl r~-lic~ preferably being methyl r~tlic~
Fur~er examples are organosilicon compounds of the above mentioned type
which contain hydroxyl groups and, in addition to methyl groups, also con-
tain phenyl groups, vinyl groups, l-thio-3-propyl groups or 3,3,3-trifluoro-
propyl groups.
The hydroxysiloxanes (1) employed according to the invention have a
viscosity at 25~C of preferably 1 to 106 mm2/s, more preferably 10 to 5 x 105
mm2/s.
214198~
The organosilicon compounds (1) employed according to the invention
are more preferably a,~-dihydroxyldiorganopolysiloxanes.
The organosilicon compounds which contain hydroxyl groups and are
employed according to the invention can be one type of such organosilicon
5 compounds or a mixture of at least two different types of organosilicon com-
pounds.
The organosilicon compounds which contain hydroxyl groups and are
employed according to the invention are commercially available products or
can be prepared by processes customary in silicone chemistry.
Examples of the silanes (2) employed according to the invention are
Si(OCH3)4, Si(OCH2CH3)4, H3CSi(OCH3)3, CH3Si(OCH2CH3)3,
H2C=CH-Si(OCH3)3, H2C=CH-Si(OCH2CH3)3, C6Hs-Si(OCH3)3, (H3C)2Si(OCH3)2,
HSi(OCH2CH3)3, F3CCH2CH2Si(OCH3)3, H2C=CH(CH2)4-Si(OCH3)3,
N_C-CH2CH2-Si(OR')3, N-C-CH2CH2CH2-Si(OR')3, and XCH2CH2CH2Si(ORI)3
where X is ~O
CH2-CH-CH2-O-, HS-, H2N-, 4,5-dihydroimi~1~7ol-l-yl,
imidazol-l-yl, pyrrolidinyl-, piperidyl-, N-morpholinyl-, piperazinyl-, cyclo-
hexyl-NH-, H2N-CH2CH2-NH-, or H2C=C(CH3)COO- radical and Rl has the
above mentioned me~ning. Some of these silanes also react with OH-func-
tional organosilicon compounds even in the absence of catalysts. In such
cases, reaction times can be shortened and/or reaction temperatures low-
ered by the process according to the invention, which can bring advantages
during further proces~ing of the products.
The silanes (2) employed according to the invention are preferably
Si(OCH3)4, Si(OCH2CH3)4, H3CSi(OCH3)3, CH3Si(OCH2CH3)3,
H2C=CH-Si(OCH3)3, H2C=CH-Si(OCH2CH3)3, N=C-CH2CH2Si(OCH2CH3)3,
4,5-dihydroimi-1~7.ol- l-yl-CH2CH2CH2SitOCH2CH3)3,
(~2-CH-CH2-O-CH2CH2CH2Si(OCH3)3,
H2C=C(CH3)COOCH2CH2CH2-Si(OCH3)3,
cyclohexyl-NH-CH2CH2CH2-Si(OCH3)3,
H2N-CH2CH2-NH-CH2CH2CH2-Si(OCH3)3,
2141985
HS-CH2CH2CH2-Si(OCH3)3 and N-morpholinyl-CH2CH2CH2-Si(OCH3)3, where
H3CSi(OCH3)3, CH3Si(OCH2CH3)3, H2C=CH-Si(OCH3)3,
H2C=CH-Si(OCH2CH3)3, N-C-CH2CH2Si(OCH2CH3)3,
4,5-dihydroimiti~7~0l-l-yl-CH2CH2CH2Si(OCH2CH3)3 and
cyclohexyl-NH-CH2CH2CH2-Si(OCH3)3 are more preferred.
The silanes (2) employed according to the invention can be a single
type or a mixture of at least two different types of such silanes or partial
hydrolysates thereof.
If partial hydrolysates of the silanes (2) are employed in the process
0 according to the invention, these are preferably those which are liquid at
room te~ ature.
The silanes (2) employed according to the invention or partial hydro-
lysates thereof are commercially available products or can be prepared by
processes customary in silicone chemistry.
The silane (2) and/or partial hydrolysate thereof is advantageously
employed in the process according to the invention in a stoichiometric
excess with respect to Si-bonded hydroxyl groups. The silane (2) and/or
partial hydrolysate thereof is preferably employed in amounts of 1.0l to 20
mole per mole of Si-bonded hydroxyl groups of the compound (1), more
preferably 1.01 to 10 mole per mole of Si-bonded hydroxyl groups of the
compound (1). Higher excesses can be favorable if the preparation of com-
positions based on the ol~.yloxysiloxanes prepared according to the in-
vention which can be crosslinked by moisture at room temperature and in
which the excess organyloxysilane serves as the cros~1inking agent is
desired. Under certain circumstances, a further metering operation thus
becomes superfluous, which can have an advantageous effect in particular
during continuous preparation of RTV- 1 compositions.
The fluoride salt (3) employed in the process according to the inven-
tion is preferably one chosen from the group consisting of ammonium fluo-
rides of the formula
[R24N]F (III)
in which R2 can be identical or different and has one of the me~nings {pven
f~r R, adducts thereof with carbonyl compounds, such as ~-ketocarboxylic
214198~
acid esters of 1,3-diketones, (alkali) metal fluoAdes, such as potassium
fluoride, cesium fluoride, zinc fluoride, dibutyltin fluoride and copper fluo-
ride, and organic or inorganic ammonium hydrogen fluorides, phosphonium
fluorides, phosphonium hydrogen fluorides, tetratluoroborates, hexafluoro-
silicates and fluorophosphates.
The radical R2 is preferably the methyl, ethyl, n-butyl, n-propyl, iso-
propyl or the benzyl radical, where the methyl, n-butyl and benzyl radical
are more preferred.
The fluoride salt (3) employed in the process according to the inven-
tion is more preferably ammonium fluoride of the formula (III).
Examples of the fluoride salt (3) are l(H3C(CH2)3)4N]F (called TBAF
below), l(H3C)4NlF, lC6H5CH2-N(CH3)3]F and [H3CNH3]F and adducts thereof
with carbonyl compounds, where acetylacetone, methyl acetoacetate,
2-ethylhexyl acetoacetate and isopropyl acetoacetate are preferred and
acetylacetone and ethyl acetoacetate are more preferred as the carbonyl
compound.
Fluoride salts are commercially available products or can be prepared
by processes customary in organic chemistry. Reference may be made for
example to, Clark, J.H., Miller, J.M. in J. Chem. Soc., Perkin Trans.I, 1977,
1743- 1745.
The fluoride salts (3) employed according to the invention can be a
single type or a mixture of at least two different types of such fluoride salts.The fluoride salt (3) can be employed in the process according to the
invention as a mixture with organic solvents and/or organosilicon com-
pounds or in a form fixed to support materials, such as silicic acid, ion
exch~nger resin, titanium dioxide or aluminum oxide. Processes for the
preparation of fluoride salt bonded to a support material are described, for
example, in Gambacorta, ~, Turchetta S., Botta, M., Synth. Commun.,
1989, 19 (13-14), 2441-2448; Li, C., Lu, Y., Huang, W., He, B., Synth.
Commun., 1991, 21(12-13), 1315-1320.
All the known organic solvents which have no interfering effect on the
reaction procedure can be employed as solvents; the solvents are preferably
21~198~
_
organic solvents, which can easily be removed from the end product by
~olation. Examples of such solvents are diethyl ether, dibutyl ether,
tetrahydrofuran, dioxane, hexane, toluene, xylenes, chlorobenzene,
1,3-pentanedione, acetone, methyl t-butyl ketone, methyl ethyl ketone,
5 1,2-dimethoxyethane, acetonitrile, ethyl acetate, methyl acetate, butyl ace-
tate, N,N-dimethylformamide, N,N-dimethylacetamide, dimethyl sulfoxide,
methanol, ethanol, n-propanol, 2-propanol, n-butanol, 2-butanol and
isobutanol and mixtures of these solvents.
The fluoride salt (3) can also be employed in the process according to
lO the invention as a mixture with organosilicon compounds, such as silanes
or oligomeric or polymeric siloxanes.
In the ple~alation of mixtures which can be crosslinked by moisture
at room temperature after the preparation according to the invention of the
organyloxysiloxanes in particular it is advantageous to dissolve the fluoride
l5 salt (3) in the organyloxysilanes to be reacted, if api)ro~,iate with the addi-
tion of the corresponding free compound RlOH, where Rl has the above
mentioned meaning, or in another liquid constituent, such as OH-cont~ining
polysiloxane or a poly(diorganosiloxane) blocked by end groups, which is
often employed as plasticizer, such as (H3C)3SiO-[Si(CH3)20]70-Si(CH3)3, an
20 oligomeric siloxane, such as (H3C)3SiOSi(CH3)3, or a cyclosilox~ne, such as
Si(CH3)20]4.
Both the adducts with carbonyl compounds and the ammonium
fluorides adsorbed onto support materials often have the advantage that
they are less hygroscopic and therefore have a better storage stability than
25 the pure ammonium fluorides.
The fluoride salt (3) is employed in the process according to the
invention in amounts of preferably 0.1 to 1000 ppm (parts by weight per
million parts by weight), more preferably 1 to 100 ppm, in each case calcu-
lated as elemental fluorine and based on the total weight of organosilicon
30 compound (1). The amount of fluoride salt (3) to be employed depends in
particular on the reactivity of the individual reaction partners and on the
presence of constituents which accelerate or retard the reaction, such as
compounds having acid or basic radicals or fluoride-bonding constituents.
2141985
The conditions under which the process according to the invention
can be catried out primarily depend on the reactivity of the olg~-yloxysilane
(2) employed and on the nature and concentration of the fluoride salt ~3).
The process according to the invention is carried out at temperatures
of preferably 20~ to 100~C under a pressure of preferably 900 to 1100 hPa.
However, it can also be carried out at higher or lower temperatures and un-
der higher or lower pressures.
In most cases, the process according to the invention can be carried
out at room temperature. However, it may be advantageous, for example, if
a lower viscosity of the reaction mixture is required for technical reasons, to
carry out the reaction at elevated temperature; in this case acceleration of
the reaction is in general to be expected under otherwise the same condi-
tions.
The end of the reaction according to the invention can be detected by
lS measuring the SiOH content in the reaction mixture by means of IR spec-
troscopy, 29Si-NMR or lH-NMR spectroscopy or by a cros~linking test to
detect residual SiOH functions in polysiloxanes, such as by the cros~linking
test according to EP 468 239 A2 cited above, or by addition of aluminum
tri-sec-butylate; an immediate increase in viscosity, under certain circum-
stances up to gelling, indicates residual SiOH groups and therefore incom-
plete co.lversion.
When the reaction according to the invention has ended, the fluoride
salt (3) is preferably deactivated by addition of component (4), which can
bond fluoride ions, the aim being to suppress further unwanted reactions
and to ensure that the organopolysiloxanes which contain organyloxy
groups and are prepared according to the invention do not change during
storage.
Examples of component (4) are aluminum compounds and com-
plexes, such as aluminum alcoholates, pyrogenically produced or precipi-
tated silicic acid, calcium-cont~ining fillers, which are suitable for deactiva-tion of component (3) because of the high tendency towards formation of
calcium fluoride, such as calcium carbonate, calcium silicate, calcium
phosphate and chalks whose surface has been treated with carboxylic acids
214198~
such as 2-ethylhexanoic acid (so-called coated chaL~s), and mixtures
thereof.
Aluminum compounds or complexes are preferably employed as
component (4) in the process according to the invention.
Examples of compounds and complexes of aluminum are aluminum
carboxylates, aluminum thiolates, aluminum sulfonates, aluminum phos-
phonates, aluminum amides, aluminum sil(ox)anolates, aluminum halides,
aluminum alcohol~tes and aluminum alcoholates in which one or more
a~coxy radicals can be replaced by ~-dicarbonyl chelating ligands, for ex-
0 ample Al[OCH2CH3l3, Al[OCH(CH3)(C2Hs)]3, Al[OCH(CH3)2]3,
AllH3C-C(O)CHC~O)-CH3]3, AllOCH(CH3)2]2lH3CC(O)CHCOOCH2CH3], alumi-
num complexes according to formula (4) of DE 34 28 840 Al cited above,
such as aluminum di(methoxy)ethylacetoacetonate, aluminum methoxy-
di(ethylacetoacetonate), aluminum di(isopropoxy)acetylacetonate, aluminum
isopropoxy-di(acetylacetonate), aluminum isopropoxy-di(ethylacetoace-
tonate), aluminum bis(trimethylsiloxy)ethylacetoacetonate, aluminum
bis(dimethoxymethylsiloxy)ethylacetoacetonate, aluminum bis(dimethoxy-
methylsiloxy)acetylacetonate, aluminum tri(ethylacetoacetonate), aluminum
bis(dimethylamino)ethylacetoacetonate, aluminum 1,3-propanedioxyethyl-
acetoacetonate and aluminum di(isopropoxy)(methylsalicylate), and reaction
products of aluminum alcoholates and organyloxysilanes of the formula III),
such as di-sec-butoxyaluminoxytriethoxysilane and the reaction product of
aluminum di(isopropoxy)-ethylacetoacetonate and tetraethoxysilane.
An aluminum alcoholate is more preferably employed as component
(4) in the process according to the invention.
Component (4) employed according to the invention can be a single
type or a mixture of at least two different types of such components (4).
The aluminum compounds and complexes employed as component
(4) are commercially available products or can be prepared by processes
customary in chemistry.
The aluminum compound or complex (4) can be employed in the
process according to the invention as a mixture with organic solvents
and/or organosilicon compounds, which is preferred.
2141985
Solvents and organosilicon compounds which can be employed are
the same as those which were described above in connection with the fluo-
ride salt (3), the aluminum compound or complex ~4) preferably being
employed as a mixture with tetrahydrofuran and/or polydiorganosiloxanes,
such as (H3C)3SiO[Si(CH3~2l70-Si(CH3)3, (H3C)3SiOSi(CH3)3 and ¦Si(CH3)2O]4.
At least a stoichiometric equivalent of aluminum in the form of the
aluminum compound or complex ~4) with respect to the fluoride is prefera-
bly added in the deactivation step acco~ding to the invention. The alumi-
num compound or complex (4) is more preferably employed in amounts of
1.05 to 3 mole of aluminum per mole of fluoride of component (3).
The process according to the invention can be car~ied out continu-
ously or discontinuously.
The elimin~tion of the organopolysiloxanes according to the invention
cont~inin~ organyloxy groups after the reaction according to the invention or
after the deactivation step according to the invention can be carried out by
any desired and known methods. For example, after the deactivation step
according to the invention, the excess organyloxysilane (2), the compound
RlOH liberated as a cleavage product, where R' has the above mentioned
me~ning, and other possible cleavage products and solvents can be removed
by thorough heating and/or by reducing the pressure.
The organopolysiloxanes which contain organyloxy groups and are
prepared according to the invention can be employed for all purposes for
which organopolysiloxanes having organyloxy groups have also been
employed such as, for co~hngs to improve the water-repellent properties of
substrate surfaces, as an adhesion promoter additive, as a primer, for
adhesives, for tex~le coatings, for plasticizers (which can be crosslinked in ifthe siloxane is blocked by organyloxy at only one end) and as a base poly-
mer in organopolysiloxane compositions which can be cros~linked by mois-
ture, in particular RTV-l compositions.
Organopolysiloxane compositions which can be cross1inked by mois-
ture and processes for their preparation are generally known. They essen-
tially comprise base polymer, vll1c~ni7~hon catalysts, cros~linking agents
. 2141985
and, optionally, plasticizers (in general silicone oils which are blocked with
non-reactive end groups), fillers, adhesion promoters and stabilizers.
For certain intended uses of the organopolyci10~c~nes which contain
organyloxy groups and are prepared according to the invention, in particu-
5 lar for their use in compositions which crosslink by means of moisture, thereaction composition obt~ined according to the invention can be employed
without ~limin~ti~n of the organopolysiloxane which contains organyl
groups. In this case, an excess of the silane (2) employed in the process
according to the invention can serve as the cros~1inking agent. If pyrogenic
0 silicic acid is employed as a constituent, the amount of aluminum com-
pound can be greatly reduced proportionally, or its use can be dispensed
with entirely, because of the high adsorptive bonding of fluoride ions onto
the silicic acid surface.
It is essential, for the stability of the compositions which can be
5 crosc1ink~d by means of moisture, only that complete reaction of the
hydroxyl groups of the organosilicon compound (l) with the organyloxy-
silane (2) has taken place before addition of the pyrogenic silicic acid. This
applies to calcium-cont~ining fillers or additives, which are suitable for the
deactivation because of the high tendency toward the formation of calcium
20 fluoride.
If use of the polysiloxanes prepared by the process according to the
invention in organopolysiloxane compositions which cure by means of
moisture is intended, the process according to the invention can also be
carried out as a one-pot process or continuously in the mixin~ unit envis-
25 aged for preparation of the compositions which cros~link by means ofmoisture. In the latter case, the fluoride salts (3) and the deactivating rea-
gents (4) can be combined with the reaction medium in static mixer systems
with the aid of metering pumps.
The process according to the invention has the advantage that
30 organopolysiloxanes cont~ining organyloxy groups can be prepared in a
simple manner and selectively with a high rate of reaction.
2141985
The fluoride component (3) employed according to the invention has
the advantage that it has a highly accelerating action on the reaction accord-
ing to the invention and has only a moderate equilibrating activity.
If component (4) is added, there is a further advantage in that by the
S deactivation step with aluminum alcoholates carried out according to theinvention, storage-stable end products are accessible without an after-
treatment step, even if the deactivating aluminum compound is employed in
a small stoichiometric excess.
In the examples described below, all parts and percentage data relate
0 to the weight, unless stated otherwise. Furthermore, all the viscosity data
relate to a temperature of 25~C. Unless stated otherwise, the following
examples were carried out under a pressure of the surrounding atmosphere
at about 1000 hPa and at room te~l~elature at about 20~C, or a tempera-
ture which is established when the reactants are brought together at room
temperature without additional heating or cooling.
TBAF represents tetra-n-butylammonium fluoride
THF represents tetrahydrofuran
E~ample 1
A Preparation of the aluminum component
A mixture of 27.6 g of water and 230 g of THF is added to a solu-
tion of 210 g of aluminum di(isopropoxy)-acetoacetic ester chelate
(=Al[O-CH(CH3)2]2[H3C-C(O)CHC(O)OC2Hsl) and 319 g of tetraethoxy-
silane in 766 g of THF at room temperature in the course of 30 min-
utes. The mixture was then heated under reflux for one hour.
Thereafter, all the volatile constituents were stripped off at room tem-
perature under 3 hPa. After filtration, 344 g of a clear oily liquid, the
aluminum content of which was 3.9% by weight, were obtained.
A mixture of 2000 g of a polydimethylsiloxane having OH end
groups and a viscosity of 1000 mm2ts with 145 g of methyl-
trimethoxysilane was prepared in a planetary mixer. 4.3 ml of 1.1 M
solution of TBAF in THF were stirred into this mixture (=0.0047 mole
of F; 45 ppm of F, based on the weight of hydroxysiloxane). After 25
minutes, 7.74 g of a solution of 3.87 g of the aluminum component
2141985
'_
described under A) in 3.87 g of mel~lylL,i~ethoxysilane were added
(=0.0056 mole of A1). After the components had been mixed thor-
oughly for 5 minutes, a 29Si-NMR spectrum and a gel permeation
chromatogram of the reaction mixture were recorded. It was found
that all the OH end groups had been replaced by H3CSi(OCH3)2-O-
end groups. Gel permeation chromatography showed a molecular
size distribution (excluding the excess methyltrimethoxysilane) which
corresponded to that of the OH group-cont~ining polymer employed.
E~ample 2
1.3 ml of 1.1 M solution of TBAF in THF were added to a mixture of
150 g of polydimethylsiloxane having OH end groups and a viscosity
of 70 mm2/s and 48.96 g of methyltrimethoxysilane (0.0014 mole of
F, 181 ppm of F, based on the weight of hydroxysiloxane). After 20
minutes, the catalyst was deactivated by addition of 2.6 ml of a 50%
strength solution of the aluminum component described in Example
1 under A) in methhyltrimethoxysilane ~0.0019 mole of Al). The
volatile constituents were then distilled off up to 80~C/ 12 hPa. 155 g
of a clear colorless oil remained as the residue, the average formula of
which was obtained from the 29Si-NMR spectrum:
MeSi(OMe)2-(SiMe20)44-Si(OMe)2Me.
Comparison Example 1
0.55 ml of a 10% strength solution of aluminum tri-secbutylate in
THF was added to a mixture of 100 g of a polydimethylsiloxane hav-
ing OH end groups and a viscosity of 1000 mm2/s and 14.8 g of
methyltrimethoxysilane, after which the formation of gelatinous
regions occurred suddenly, which is to be interpreted as an indication
of incomplete saturation of the Si-OH groups of the polydimethylsi-
loxane having OH end groups. In addition to me~ imethoxysilane
and the dimethylsiloxy units of the OH-polymer, only HO-Si(CH3)2-O-
and no H3CSi(OCH3)2-O- end groups were detectable in the 29Si-NMR
spectrum.
2141985
~:xample 3
A mixture of 2000 g of a polydimethy~ ox~ne having OH end
groups and a viscosity of 1000 mm2/s with 145 g of mell~yllli-
methoxysilane was prepared in a planetary mixer. 4.3 ml of a 1.1 M
s solution of TBAF in THF were stirred into this mixture (= 0.0047 mole
of F; 45 ppm of F, based on the weight of hydroxysiloxane). After
storage at 25~C for two days, products of polymer degradation reac-
tions (equilibration) were detected from the 29Si-NMR spectrum: the
content of monomethoxy end groups, which are not capable of
0 crosslinking, was 20 mole %, based on all the end groups (80 mole %
of H3CSi(OCH3)2 end groups); the chain lengthening content of
Si~CH3)0CH3-groups incorporated, was the same size.
Dimethyldimethoxysilane was also detectable.
E~ample ~
5 B Preparation of catalyst solution F
150 ml of 4% strength hydrofluoric acid were added to 195 ml of a
40% strength aqueous solution of tetra-n-butyl-ammonium
hydroxide. The pH of the solution was 7. After addition of 60 g of
2,5-pentanedione, all the volatile constituents were distilled off on a
rotary evaporator at 40~C/ 1 hPa. 5.5 g of the solid residue were dis-
solved in 30 ml of methyltrimethoxysilane. A clear red-brown solu-
tion having a nuoride content of 0.014 g/ml was obtained.
0.1 ml of the catalyst solution F described above under (B) was
added to a mixture of 100 g of polydimethylsiloxane having OH end
groups and a viscosity of 1000 mm2/s and 7.4 g of methyltrimethoxy-
silane (=0.000074 mole of F; 14 ppm of F, based on the weight of
hydroxysiloxane~ and the mixture was stirred for 20 minutes. The
catalyst was deactivated by addition of 0.55 ml of a 10% strength
solution of aluminum tri-sec-butylate in THF (0.000223 mole of Al).
As a cros~linkin~ test showed (addition of aluminum tri-sec-butylate
to small samples taken from the reaction mixture after certain inter-
vals of time), all the SiOH groups had reacted in the desired sense
after only 15 minutes (no further gelling with aluminum tri-sec-
214198S
butylate). It was to be seen from the 29Si-NMR spectrum that aU the
OH end groups had been converted into H3CSi(OCH3)2- end groups.
E~ample 5
The procedure described in Example 4 was repeated with the modi-
fication that, instead of 0.1 ml, 0.2 ml of catalyst solution F
l= 0.000147 mole of F; 28 ppm of F, based on the weight of hydroxy-
siloxane) was added. From the crosslinking test for residual SiOH, it
was found that the reaction had already ended after 10 minutes.
Nevertheless, deactivation with the aluminum component was carried
0 out only after 20 minutes. The 29Si-NMR spectrum was identical to that from Example 4.
Example 6
The procedure described in Example 4 was repeated, with the
modification that inete~l of 0.1 ml, 0.3 ml of catalyst solution F
(= 0.00022 mole of F; 42 ppm of F, based on the weight of hydroxysi-
loxane) was added. It was found from the croselinking test for resid-
ual SiOH that the reaction had already ended after 5 minutes. Never-
theless, deactivation with the aluminum component was carried out
only after 20 minutes. The 29Si-NMR spectrum was identical to that
from Example 4.
The 29Si-NMR spectrum of a sample which had been subjected to
storage under heat in a closed polyethylene bottle in a drying cabinet
at 80~C for 7 days, showed no change compared with the starting
spectrum.
E~ample 7
The procedure described in Example 5 was repeated, with the
modification that instead of 0.2 ml of catalyst solution F as described
in Example 4 under B), 0.2 ml of a 1.1 M TBAF solution in THF
(0.00022 mole of F) was added. After 20 minutes, deactivation was
carried out with 1.1 ml of a 10% strength solution of aluminum tri-
sec-butylate in THF ~0.00045 mole of Al). The 29Si-NMR spectrum
was identical to that of Example 5.
16
2141985
The 29Si-NMR spectrum of a sample which had been subjected to
storage under heat in a closed polyethylene bottle in a drying cabinet
at 80~C for 7 days showed no change compared with the initial spec-
trum.
E~ample 8
The procedure described in Example 7 was repeated, with the
modification that after 20 minutes the deactivation was carried out
with 2.2 ml in~te:~tl of 1.1 ml of a 10% strength solution of aluminum
tri-sec-butylate (0.00089 mole of Al). The 29Si-NMR spectrum was
identical to that of Example 5.
The29Si-NMR spectrum of a sample which had been subjected to
storage under heat in a closed PE bottle in a drying cabinet at 80~C
for 7 days showed no change compared with the starting spectrum.
Compllrison E~mple 2
A mixture of 0.2 ml of a 1.1 M solution of TBAF in THF (0.00022
mole of F) and 0.6 ml of a 10% strength solution of aluminum tri-sec-
butylate in THF (0.00024 mole of Al) was added to a mixture of 100 g
of a polydimethylsiloxane having OH end groups and a viscosity of
1000 mm2/s and 10 g of methyltrimethoxysilane. It was found from
the 29Si-NMR spectrum of the reaction mixture that no reaction had
taken place.
E~ample 9
0.2 ml of a 1.0 M solution of TBAF in THF was added to a mixture
of 100 g of a polydimethylsiloxane having OH end groups and a vis-
cosity of 1000 mm2/s and 12.7 g of 3-glycido~y~ropyltrimethoxy-
silane (H2C(O)CHCH20(CH2)3-Si(OCH3)3 (0.0002 mole of F, 38 ppm of
F, based on the weight of hydroxysiloxane). After 45 minutes, deacti-
vation was carried out with 0.55 ml of a 10% strength solution of
aluminum tri-sec-butylate in THF (0.00022 mole of Al). It was found
in the 29Si-NMR spectrum of the mixture that all the SiOH functions
had been converted into Si-OSi(OCH3)2-(CH2)3-OCH2-CH(O)CH2. The
excess silane employed could be removed by thorough heating at
1 10~C/0. 1 hPa on a thin film evaporator.
2141985
Example 10
0.2 ml of a 1.0 M solution of TBAF in THF was added to a mixture
of 100 g of a polydimethyl~ilox~ne having OH end groups and a vis-
cosity of 1000 mm2/s and 13.4 g of 3-methacryloylo~y~royyl~i-
methoxysilane (H2C=C(CH3)COO(CH2)3-Si(OCH3)3) (0.0002 mole of F,
38 ppm of F, based on the weight of hydroxysiloxane). After 25 min-
utes, deactivation was carried out with 0.55 ml of a 10% strength
solution of aluminum tri-sec-butylate in THF (0.00022 mole of Al).
It was found in the 29Si-NMR spectrum of the mixture that all the
0 SiOH functions had been converted into
Si-OSi(OCH3)2-(CH2)3-OOC(CH3)C=CH2. The excess silane employed
could be removed by thorough heating at 110~C/0.1 hPa on a thin
film evaporator.
Example 11
0.2 ml of 1.1 M solution of TBAF in THF was added to a mixture of
100 g of a polydimethylsiloxane having OH end groups and a viscos-
ity of 1000 mm2/s and 14.2 g of 3-(N-cyclohexylamine)plo~yl~
methoxysilane (cyclohexyl-HN-(CH2)3-Si(OCH3)3) (0.00022 mole of F>
42 ppm of F, based on the weight of hydroxysiloxane). After 15
minutes> deactivation was carried out with 0.22 g of the aluminum
component described in Example 1 under A) (0.0003 mole of A1). It
was found in the 29Si-NMR spectrum of the mixture that all the SiOH
functions had been converted into Si-OSi(OCH3)2-(CH2)3-NH~cyclo-
hexyl).
Example 12
0.2 ml of a 1.0 M solution of TBAF in THF was added to a mixture
of 100 g of a polydimethylsiloxane having OH end groups and a vis-
cosity of 1000 mm2/s and 8.9 g of triethoxysilane (0.0002 mole of F>
38 ppm of F> based on the weight of hydroxy~ilo~c~ne). After 15 min-
utes> deactivation was carried out with 0.55 ml of a 10% strength
solution of aluminum tri-sec-butylate in THF (0.00022 mole of Al). It
was found in the 29Si-NMR spectrum of the mixture that all the SiOH
functions had been converted into Si-OSiH(OCH2CH3)2.
_ 214198S
Example 13
0.2 ml of a 1.0 M solution of TBAF in THF was added to a mixture
of 100 g of a polydimethylsiloxane having OH end groups and a vis-
cosity of 1000 mm2/s and 6.5 g of dimethyldimethoxysilane
(= 0.0002 mole of F, 38 ppm of F, based on the weight of hydroxysi-
loxane). After 15 minutes, deactivation was carried out with 0.55 ml
of a 10% strength solution of aluminum tri-sec-butylate in THF
(0.00022 mole of A1). It was found in the 29Si-NMR spectrum of the
mixture that all the SiOH functions had been converted into
Si-OSi(CH3)20CH3.
Example 14
1 ml of a 1.0 M solution of TBAF in THF was added to a mixture of
50 g of a branched polydimethylsiloxane having OH end groups and
the average composition [HOSi(CH3)201/2l4-[Si(CH3)20l;,2¦SiO2ll 2
(prepared by gentle hydrolysis of a reaction product, prepared in the
presence of PNC12, SiClq and a polydimethylsiloxane having OH end
groups and a viscosity of 5 Pas) and 35.4 g of methyltrimethoxysilane
(= 0.001 mole of F, 380 ppm of F, based on the weight of hydroxysi-
loxane). After 10 minutes, deactivation was carried out with 2.7 ml
of a 10% strength solution of aluminum tri-sec-butylate in THF
(0.001 mole of Al). It was found in the 29Si-NMR spectrum of the
mixture that all the SiOH functions had been converted into
Si-OSi(CH3)2CH3.
Example 15
0.2 ml of a 1.0 M solution of TBAF in THF was added to a mixture
of 100 g of a polydimethylsiloxane having OH end groups and a vis-
cosity of 1000 mm2/s and 7.4 g of methyltnmetho~silane at 75~C
(= 0.0002 mole of F, 42 ppm of F, based on the weight of hydroxysi-
lo~ane). After 4 minutes, cros~linkin~ test on SiOH (aluminum
sec-butylate) indicated complete co~ sion. Deactivation wa subse-
quently carried out with 0.55 ml of a 10% strength solution of alumi-
num tri-sec-butylate in THF (0.00022 mole of Al). It was found in the
19
214198S
29Si-NMR spectrum of the mixture that all the SiOH functions had
been converted into Si-OSi(OCH3)2CH3.
Comparison E~cample 3
2 g of a solution of 2 g of NaOH in 47.5 of tetraethoxysilane and
0.5 g of ethanol (= 0.002 mole of NaOH) were added to a mixture of
163 g of a polydimethylsiloxane having OH end groups and a viscos-
ity of 1000 mm2/s and 24.5 g of tetraethoxysilane. After 15 minutes,
the base was neutralized with 0.2 g of dimethyldichlorosilane
(= 0.0031 mole of Cl). All of the volatile components were subse-
quently stripped off at 50~C/2 hPa. It was found in the 29Si-NMR
spectrum of the mixture, recorded after three days, that all the SiOH
functions had been converted into Si-OSi(OCH2CH3)3.
Comparison E~ample 4
Comparison Example 3 was repeated, with the difference that the
volatile constituents were not distilled off after the neutr~li7~tic~n. It
was found in the 29Si-NMR spectrum of the mixture recorded after
three days that the desired triethoxysilyl end groups were present
only in traces, and instead Si-OSi(CH3)2(0CH2CH3) functions which
were not capable of crosslinking were chiefly detectable.
Example 16
A mixture of 90.9 g of melhylL,illlethoxysilane and 2.7 ml of 1.1 M
solution of TBAF in THF was added to 1000 g of a polydimethylsilox-
ane having OH end groups and a viscosity of 80 Pas (0.003 mole of F,
56 ppm of F, based on the weight of hydroxysiloxane) in a planetary
mixer. The mixture was stirred at room temperature for 25 minutes
before deactivation was carried out with 24.3 g of a 10% strength
solution of aluminum tri-sec-butylate in a polytdimethylsiloxane)
blocked by trimethylsilyl end groups (= 0.01 mole of A1) which had a
viscosity of 100 mm2/s. 524 g of this polydimethylsiloxane having
trimethylsilyl end groups, 72.7 g of hexamethyl~ 7~ne, 254.4 g of
a hydrophobic, pyrogenic silicic acid having a specific surface area of
120 m2/g and 4.91 g of dibutyltin diacetate were then mixed in suc-
cession. Half of the paste obtained was cured in air in a layer thick-
2141985
ness of 2 mm at room temperature for 14 days. An elastic vulcani-
zate which gave the following mechanical values was obtained:
Tear strength (DIN 53504): 0.9 N/mm2
Elongation at break (DIN 53504): 340%
Tensile stress at 100% elongation (DIN 53504): 0.2 N/mm2
Tear propagation resistance (ASTM D 624 B-91): 4.3 N/mm2
Hardness (Shore A) (DIN 53505): 17
To investig~te the storage stability, the other half of the paste was
protected from access of atmospheric humidity in polyethylene car-
tridges. After storage at 50~C for 3 weeks, the paste showed no
crosslinking phenomena when spread out, but then cured to an
elastomer under the influence of atmospheric moisture.
Comparison Example 5 - (analogous to Example 1 of EP 468239 A2 cited
above)
0.9 g of a solution of 2 g of NaOH in 47.5 g of melhyll,.~ethoxy-
silane and 0.5 g of methanol (= 0.0009 mole of NaOH) was added to a
mixture of 145 g of a polydimethylsiloxane having OH end groups
and a viscosity of 1000 mm2/s (about 0.0178 mole of OH) and 10 g of
methyltrimethoxysilane (0.0735 mole). After 5 minutes, the base was
neutralized with 0.73 g of a solution of 5 g of dimethyldichlorosilane
in 45 g of hexamethyldisiloxane (= 0.0011 mole of Cl). The mixture
was then heated thoroughly at 140~Ct25 hPa for 2 hours. 132 g of a
cloudy oil having a viscosity of 979 mm2/s remained as the residue.
The following average formula was obtained from the 29Si-NMR spec-
trum of the product:
[MeSi(OMe)2Ol/2)2[SiMe2O]220. Blocking of the SiOH end groups was
complete.
Compari~on E~ample 6
The procedure described in Comparison Example 5 was repeated,
with the modification that the base was neutralized only after 10
minutes. 138 g of a cloudy oil having a viscosity of 427 mm2/s were
obtained as the end product. The following average formula was
21~1985
obtained from the 29Si-NMR spectrum of the product:
[MeOSiMe2O~/2]2[Me(MeO)SiO][MeSiO3/2]05[SiMe2O]l,O. Although the
blocking of SiOH end groups was complete, rearrangements to an
extent such that the desired MeSi(OMe)2 end group was present only
in traces had already taken place by lengthening the reaction time by
5 minutes compared with Comparison Example 5.
E~ample 17
C Preparation of catalyst solution F1
60 ml of 25% strength sulfuric acid were added to a solution of
17.4 g of potassium fluoride in 30 ml of completely demineralized
water. After 30 minutes neutralization was carried out with 180 ml of
an approximately 40% strength aqueous solution of tetra-n-butyl-
ammonium hydroxide. The mixture was then extracted with 200 ml
of THF. The extract was concentrated to dryness on a rotary evapora-
lS tor and the residue was taken up on 300 ml of methyltrimethoxy-
silane. Volatile constituents were then stripped off at 25~C/ 10 hPa.
The mixture was filtered. 192 g of a colorless, clear liquid having a
fluoride content of 0.6 mole/l were obtained.
The advantage of this procedure lies in the fact that the hygroscopic
tetrabutyl~mminium fluoride is practically dried with methyltri-
methoxysilane. In the presence of the fluoride the residual moisture
led to hydrolysis or condensation of the methyltrimethoxysilane.
Liquid oligomers of methyltrimethoxysilane and an insoluble precipi-
tate of methylsilicic acid, which can be removed by simple filtration,
2s are formed.
0.2 ml of catalyst solution F1 described above under ~C) was added
to a mixture of 100 g of a polydimethylsiloxane having OH end
groups and a viscosity of 1000 mm2/s and 7.4 g of melhyll,imethoxy-
silane (= 23 ppm of F, based on the weight of hydroxysiloxane) and
the mixture was stirred for 20 minutes. The catalyst was deactivated
by addition of 0.7 ml of a 10% strength solution of aluminum tri-sec-
butylate in hexamethyldisiloxane. As a crosslinkin~ test showed
(addition of aluminum tri-sec-butylate to small samples of the reac-
214I9~5
tion mi~cture taken after certain intervals of time), all the SiOH groups
had reacted in the desired sense after only 10 minutes (no further
gelling with aluminum tri-sec-butylateJ. It was to be seen from the
29Si-NMR spectrum that all the OH end groups had been converted
into H3CSi(OCH3)2- end groups.
Example 18
The procedure described in Example 17 was repeated, with the
modification that 0.2 ml of catalyst solution F1 prepared in Example
17 under (C) (= 23 ppm of F, based on the weight of hydroxysiloxane)
which had been stored at a temperature of 70~C in a polyethylene
bottle for a period of 7 days was employed. As a cros~linkin~ test
showed ~addition of aluminum tri-sec-butylate to small samples
taken from the reaction mixture after certain intervals of time), all the
SiOH groups had reacted in the desired sense after only 10 minutes
~no further gelling with aluminum tri-sec-butylate). It was to be seen
from the 29Si-NMR spectrum that all the OH end groups had been
converted into H3CSi(OCH)2- end groups.
Example 19
0.2 rnl of a 1.0 M solution of TBAF in THF was added to a mixture
of 100 g of a polydimethy1~ilox~ne having OH end groups and a vis-
cosity of 1000 mm2/s and 11.7 g of 2-cyanoethyltriethoxysilane
(0.0002 mole of F, 38 ppm of F, based on the weight of hydroxysilox-
ane). As a cros~linkin~ test showed (addition of aluminum tri-sec-
butylate to small samples taken from the reaction mixture after cer-
tain intervals of time), all the SiOH groups had reacted in the
desired sense after only 10 minutes (no further gelling with alumi-
num tri-sec-butylate). After this period of time, deactivation was
carried out with 0.65 ml of a 10% sll~legth solution of aluminum tri-
sec-butylate in hexamethyldisiloxane (0.00026 mole of Al). It was
found in the 29Si-NMR spectrum of the mixture that all the SiOH
functions had been converted into Si-O-Si(OCH2CH3)2-(CH2)2-C~N and
the ratio of end groups/dimethylsiloxy units had not changed com-
pared with the starting value. The 29Si-NMR spectrum of a sample
which had been subjected to storage under heat at 70~C in a dosed
polyethylene bottle for 7 days showed no formation of monoethoxy
end groups and/or branchings.
Example 20
0.1 ml of catalyst solution Fl described in Example 17 under C)
was added to a mixture of 100 g of a polydimethylsiloxane having OH
end groups and a viscosity of 1000 mm2/s and 15.0 g of N-1~3-tri-
ethoxysilyl)-propyl]-4,5-dihydroim~ 4le (commercially obtainable
undcr the name"Dynasilan~ IMEO" from Hule AG, Marl) ~0.0006 mole
of F, 11 ppm of F, based on the weight of hydroxyeil- Y~n~). Since no
further gelling on samples taken occurred in the croselil.lnng test ac-
cording to Example 19 with aluminum tri-sec-butylate after 20 min-
utes, deactivation was carried out after this period of time with 0.16
ml of a 10% strength solution of aluminum tri-sec-butylate in hex-
amethyldisiloxane (0.000065 mole of A1). It was found in the 29Si-
NMR spectrum of the mixture that all the SiOH functions had been
converted into Si-OSi(OCH2CH3)2-(CH2)3-N-dihydroimidazole and the
ratio of end groups/dimethylsiloxy units had not changed compared
with the starting value.
Example 21
0.2 ml of a 1.0 M solution of TBAF in THF was added to a mixture
of 100 g of a polydimethylsiloxane having OH end groups and a vis-
cosity of 1000 mm2/s and 0.9 g of N-l(3-triethoxysilyl)-propyll-4,5-
dihydro-imidazole (commercially obtainable under the name
~Dyn~ n IMEO~ from Huls AG, Marl) and 7.6 g of vinyltriethoxy-
silane (0.0002 mole of F, 38 ppm of F, based on the weight of hy-
droxysiloxane). The end point of the reaction was determined by the
crosslinkin~ test described in Example 19. Since no further gelling
on samples taken occurred after 20 minutes, deactivation was carried
out after this time with 0.65 ml of a 10% strength solution of alumi-
num tri-sec-butylate in hexamethyldisiloxane (0.00026 mole of Al). It
was found in the 29Si-NMR spectrum of the mixture that practically
all the SiOH functions had been converted into
24
.,
~,
2141985
Si-OSi(OCH2CH3)2-CH=CH2 and the ratio of end groups/dimethyl-
siloxy units had not changed compared with the starting value.
E~ample 22
0.2 ml of a 1.0 M solution of TBAF in THF was added to a mixture
s of 100 g of a polydimethylsiloxane having OH end groups and a vis-
cosity of 1000 mm2/s, 7.15 g of cyanoethyltriethoxysilane and 7.6 g
of vi~ iethoxysilane (0.0002 mole of F, 38 ppm of F, based on the
weight of hydroxysiloxane). The end point of the reaction was deter-
mined by the crosslinkin~ test described in Example 19. Since no
further gelling on samples taken occurred after 10 minutes, deactiva-
tion was carried out after this time with 0.65 ml of a 10% strength
solution of aluminum tri-sec-butylate in hexamethyl~ ilo~ne
(0.00026 mole of Al). It was found in the 29Si-NMR spectrum of the
mixture that 93.3% of all the SiOH functions had been converted into
Si-OSi(OCH2CH3)2-CH2CH2-CN and 7.7% of all the SiOH functions
had been COn~ led into Si-OSi(OCH2CH3)2-CH=CH2, and the ratio of
end groups/dimethylsilo~y units had not changed compared with the
starting value. The 29Si-NMR spectrum of a sample which had been
subjected to storage under heat at 70~C in a closed polyethylene
bottle for 7 days showed no formation of monoethoxy end groups
and/or branchings.