Note: Descriptions are shown in the official language in which they were submitted.
\~'0~4/~ 669 PCT/~'S93/0783~ ~
, ,
,:.. `. 1
2 i ~
.
. .
--1 .
1o TITLE OF T~E INVENTION --`
CATIONIC-2-~ETEROARYLPHENYL-CARBAPENEM ANTIBACTERIAL
AGENTS
BACKGR0UND QF T~E INVENTIQN
The present invention relates to
~:~ antibacterial agents of the carbapenem class, in
which the 2-position sidechain is characterized by a
heteroarylphenyl moiety, substituted by various
cationic and neutral substituents~ as described in
more detail below.
:~ ;'
~ 30 ~
:
- . '
: ~ -
: ~
.
wos~/~;669 PCT/~593/07830
2 l ~ 2 -
Thienamyci~ was an early carbapenem
antibacterial agent having a broad spectrum; it has
the following formula: ,`
HO
H H
o~N~ ~ 2
C~
I
0
Later, N-formimidoyl thienamycin was discovered; it
has the ~ormula:
,~ -
, ~ ~
'~
: HO
~ H H
1 . H
OH
Thelclationic 2-heteroarylphenyl-carbapenems of.the, , . j~
present invention are not characterized by a broad : .'
:30 antibaeterlal spe~ctrum such as~that of thienamy~in or ~ ~;
N-formimidoyl thienamycin. Rather, their spectrum of ` ~
a~:tivity is la~igely;limited to gram positive : - ~7
microorganisms, esp~ecially methicillin resistant :~ :
t~phylococ~us ~ ~ I(MRSA), methici:Llin
1 `;-`
/0~669 PCT/~S93/07830
2 :~ ~ 2 7 3 ~ !
resistant S~hvlococcus ~ ermidis ~MRSE), and
methicillin resistant coagulase negative
StaphvlQcoçci (MRCNS). The antibacterial compounds
o~ the present invention thus comprise an important
contribution to therapy of these difficult to control
pathogens. Moreover, there is an increasing need for
agents effective against such pathogens (MRSA/MRCNS)
which are at the same time safe, i.e., ~ree from
undesirable toxic side effects. No ~-lactam
antibacterial has yet been found which meets these
requirements. And, the current agent of choice,
vancomycin, a glycopeptide antibacterial, is
e~periencing an ever increasing amount of resistance
in the MRSA/MRCNS pathogens.
More recently, carbapenem antibacterial
agents have been described which have a 2-substituent
which is an aryl moiet~ optionally substituted by,
e.g., aminomethyl and substituted aminomethyl. These
agents are described in U.S. Patent Nos. 4,543,257
and 4,260,627 and have the formula:
E~2 H or CH~
R~ H NH
CQOH
~ 30 However, there is no description or
: suggestion of a cationic heteroarylphenyl ~,
~.
~ .
; :
! ~
WO 9~/~)5fi69 PC~/~,'S93/07830
~2 ~ '3 ~ 4 -
2-substituent such as characterizes the compounds of ~ `
the present invention, nor is there any suggestion of 1:.
the surprisingly be~ter anti-MRSA/MRCNS ac~ivity of
the compounds of the present invention.
U.S.P. 4,978,659 describes a particular
class of compounds of the formula: i
: :
R2 H R Ra
R1 ~ -N 3 R (1_
: 15 y ~b
~ '
: but this limited teaching in no way suggests the
totally different compounds of the present invention,
` nor their surprisingly hetter anti-MRSA/MRCNS
: 20 actlvity
~ S~M~ARY OF INV~NTIO~
:~ Thc presen~ in~ention provides novel
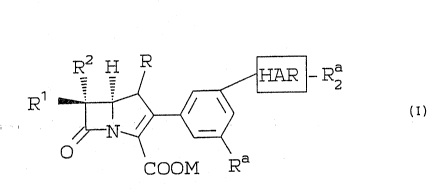
;`~ carbapenem compounds of the formula: :
~; 25 ~ :
R2 ~ H ~ R ~- R2 , i-
R~ L'
`~ C03M Ra~
,~
~ 94/0,6~'~ PCT/~'S93/07830
,.,~. I`` 2 7 a ~
. 5
wherein:
R is H or C~3;
Rl and R2 are independently H, C~3-, CH3CH2-,
(CH3)2CH-, HOC~2-, CH3C~(OH~-, (CH3)2C(OH)-,
FC~2C~(OH)-, F2C~CH(O~)-, F3CC~(OH)-,
CX3C~(F)-, CH3CF2-, or (CH3)2C(F)-;
is a 5- or 9-membered mono or bicyclic
~ heteroaryl ring system wherein 1 atom is O
or S, or an 8-membered bicyclic heteroaryl
ring system wherein 2 atoms are O and/or S;
Ra is each independently selected from the group
lS consisting of hydrogen and the radicals
set out below,~provided that one and
only one Ra is selected from Type I
substituents:
I.
a)
c~
-- Ap- N~;
~ere -~
A is (CH2)m Q-(CH2)n, where m is 0 to 6 and n is
1 to 6 and Q is a covalent bond, O, S, SO,
S2 NH~ -S02N~ S02-~ -CUN~ CO
-SO2N~Cl-C4 alkyl)-, -N(Cl-C4 alkyl)SO2-
'
.
:
~;~
~:
~: .
...... ...... . . ..
~'O9~/0~669 PCT/~S93/07830
.
2 1 1~ ~3 - 6 -
-CON(C~-C4 alkyl)-, -N(Cl-C4 alkyl)CO-,
-C~=CH-, -CO-, -OC(O)-, -C~O)O- or N(Cl-C
alkyl) and (CH2)m is attached to the phenyl
aromatic moiety;
is a 5- or 6-membered monocyclic heterocycle
or an 8-, 9- or 10-membered bicyclic
heterocycle, the heterocycle containing a
first nitrogen in an aromatic 5- or 6-membered
first ring, with attachment of the heterocycle
to A by way of said first nitrogen and said
first nitrogen is quaternary by virtue of the
attachment and ring bon~s, with the first ring
containing 0 or 1 of either O or S, with the
first ring containi~g 0 to 3 additional
nitrogen atoms, with the first ring optionally
fused to a 3- or 4 membered moiety to form the
~:~ optional second ring, with the moiety
containing at least one carbon atom, with the
moiety containing 0 or 1 of either O or S,
with the moiety containing 0 to 2 nitrogen
atoms, and with the moiety being saturated or
unsaturated and the second ring aromati~ or
non-aromatic;
c is Ra as defined under II belows hydrogen, or .
: -NRYRz ~where RY and RZ are defined in lI
below), but independently selected from Ra and
from each other-if more than one Rc is
prese~t, and is attached to a carbon ring atom
or a nitrogen heteroatom the valency of which
is not satisfied by the r~ing bonds;
~,~, wo9~J~)~h6s PC~/~S93/n7~30
~j ~.. , l' '
2 1 ~.~ 2 ~ ~ VJ 1~
- 7
` 1
p is O or l;
b)
.j ,.
~<R ~ 0 - 2
-A' ~ N~R~
,/ (o-~ )
~ere
10 ~ ~ is a 5- or 6-membered monocyclic heterocycle
or an 8--, 9- or 10-membered bicyclic
heterocycle, the heterocycle containing a
first nitrogen in an aromatic 5- or
6-membered first ring, with said first
nitrogen quaternary by virtue of a
substituent Rd in addition to the ring bonds
thereto, with said first nitrogen neutral in
the absence o~ a substituent Rd, with
~;~ attachment of the heterocycle to A' by way of
~ a carbon atom of a xing, with the first ring
containing O or 1 of either O or S, with the
fir~ ring containing O to 2 additional
nitrogen atoms, with the first ring
: optionally fused to a 3 or 4-membered ~oiety
to form the optional seeond ring, with the
moiety containing at least one carbon atom,
with the moiety containing O or 1 of either O
or S,; with,the moiety containing O to 2
nitroge~ atoms,-and with the moiety being
saturated or unsaturated and the second ring ~-.
aromatic or non-aromatic;
,.
~,
: ~ :
,~ ~
1,, ~ .
~v~st/0~669 PCT/~S93/07830
21~`%7`~
- 8 -
Rc is defined above;
~d is hydrogen, NH2, 0~ or Cl-C4 alkyl (where
the alkyl group is optionally
mono-substituted with Rq as defined under IIc
below~;
A' is (CH2)m-Q-(CH2)n, where m is 0 to 6 and n
is 0 to 6 and Q is defined above;
C)
-Ap-N+RY(P~w)o-l(Rz)~ where
RY and RZ are as defined under II below,
RY and RZ may further be together a C2-C4
alkylidene radical to form a ring (optionally
mono-substituted with R~ as defined below)
;nterrupted by N(O)Re or N~(Re)2 (where Re is
~ hydrogen, Cl-C4 alkyl, or C~ C4 alkyl
;~ mono-substituted with Rq as defined below),
Rw is hydrogen, Cl_4 alkyl, 0~, Na2, or absent in
which case the N+ is neutrals
RW, RY and RZ may ~urther together form a C5-C10
tertiary alkylidene radical which with N~
~: forms a bicyclic ring, where the terti~xy
alkylidene radical is optional~y :
mono-substituted wi~h Rq as defined below and
where the tertiary carbon of the tertiary ~.
alkylidene radical is optionall~ replaced
with nitrogen, N+Re (where Re is defined
above),:or N+-0
p is 0 or 1, and
A is as defined a~ove;
,
. :
. ~ :
'`' ~ ~ :
' :`~:
~ O~ 669 l~c~/~ss3/o7B3o t^ `
~`. . 1-
2 i '~ 2 ~
. _ 9 _ I
1-
d ~
S -A' p ~d
~ere
is a 5- or 6-membered monocyclic heterocycle
~N~ or an 8-, 9- or 10-membered bicyclic
heterocycle, the heterocycle containing a
~irst nitrogen in a first ring, with the first ring
saturated or unsaturated and non-aromatic, with the
f.irst nitrogen quaternary by vi.rtue of one or two
; substituents Rd in addition to the ring bonds
~: thereto, with the first nitrogen alternatively
neutral by virtue of zero or one substituents Rd in
: addition to the ring bonds thereto with attachment~ 25 of the heterocycle to Al by way of a carbon atom or
non-quaternary nitrogen atom of a ring, with the
first ring eontaining in addition to carbon and the
, flirFt~ nitrogen 0 to 1 of a memb;er selected ~rom the
group consisting~of the non-quaternary nitrogen of: 30 attachment, O, S, S(0), S(0)2 and NRe where Re is
defined above, with the ~irst ring optionally fused ~:
to a 2-, 3~ or 4-membered moiety:to form the
~ .
. ~ :
:
.~. .
' ~09~/0~66~ PCT/~IS93/07830
Z i 42 l O ~
.. -- 10 --
optional second ring, with the moiety
optionally containing in addition to carbon
I the non-quaternary nitrogen of attachment,
I and with the moiety saturated or unsaturated
S and the second ring non-aromatic;
Rd is defined above and where more than one Rd
is present on a nitrogen, at least one Rd is
hydrogen or Cl-C~ alkyl;
A is defined above; and
p is defined abo~e;
R~ is defined below;
II.
a) a trifluoromethyl group: -CF3;
:~ 15 b) a halogen atom: -Br, -Cl~ -F, or -I;
c) Cl-C4 alkoxy radical: -OCl_4 alkyl,
wherein the alkyl is optionally
-: mono-substituted by Rq, where
~r.'~
20 Rq is a member selected from the group consisting ; -~
~ of -0~ t -OCH3, -CN, -C(O)NH2, -OC(O)MH~, CH0, --
"`~ -OC(O)N(c~3)~, -S02MH2.. -S02N(CE3)2, -SOCH3,
`~ -S02C~3, -F, ~CF3, -COOMa ~where Ma is
: hydrogen, alkali metal, methyl or phenyl),
~: 25 tetrazolyl (where the point of attac~ment is
the carbon atom of the tetrazole ring and one :
of the~nitrogen atoms is mono-substituted by
~ Ma as~ defined above) an~ -SO3Mb (where Mb is
:~; hydrogen or an alkali metal);
d) a hydroxy group: -OH;
e) a carbonyloæy radical: -O(C=O)Rg,
where
, , ~
~'O9~/0~669 Pcr/~ss3/o783o ,~
.;, ~ :,
.J. ~ i
2 1 4;~
11 I
~.
Rs is Cl-C~ alkyl or phenyl, each of which is
optionally mono-substituted by Rq as defined
above or tri-suhstituted with -~; 5
~) a carbamoyloxy radical:
-O~C=O)N(RY)Rz~ where
RY and RZ are independently H, Cl_4 alkyl
(optionally mono-substituted by Rq as defined
above), together a 3- to 5-membered
alkylidene radical to form a ring (optionally
substituted with R~ as defined above) ~r
together a 2- to 4-membered alkylidene
radical 9 interrupted by -0-, -S-, -S(0)-,
-S(0)2- or -NRe-, to form a ring (where Re is
: lS hydrogen, Cl-C4 alkyl, and Cl-C4 alkyl
mono-substituted with Rq and the ring is
optionally mono~substituted with Rq as
defined above);
: g) a sulfur radical:
` 20 -S(O)n-RS where n - 0-2, and RS is defined
above;
h) a sulamoyl group:
-S02N(RY~RZ where RY and Ræ are as defined
above;
~ 25 i) azido: N3
: i) a formamido group: -N(Rt3-C(0)~,
where .
t I ~ or Cl-C4 alkyl, and the alkyl thereof is
optionally mono-substituted by Rq as defined
~ abo~e; : J
:~ ,
.
.
~ 9~/~)s~69 PCT/~Ss3/07~30
,. . .-
~1~2~ 0 ~
- 12 -
.,
k) a (Cl-C4 alkyl)carbonylamino radical:
-N(Rt)-C(O)Cl-C4 alkyl, where Rt is as
defined above, and the alkyl group is
also optionally mono-substituted by Rq
as defined abo~e;
~ 1) a ~Cl-C4 alkoxy) carbonylamino
3 radical: -N(Rt)-C(O)OCl-C4 alkyl,
where Rt is as defined above, and the
alkyl group is also optionally
mono-substituted by R~ as defined above;
m) a ureido group:
-N(Rt)-C(O)N(RY)Rz where Rt, RY and RZ
are as defined above;
. n) a sul~onamido group: -N(Rt)S02RS,
where Rs and Rt are as defined above;
o) a cyano group: -CN;
p) a formyl or acetalized formyl radica~:
_c(O)H or -C(0cH3)2~;
q) (Cl-C4 alkyl)carbonyl radical wherein
the carbonyl is aceta:Lized:
-C(OC~3)2C~-C4 alkyl, where the alkyl
is optionally mono-substituted by Rq as
defined aboYe;
r) carbonyl radical: ~C(O)RS, where Rs is
as defined above;
s) a hydroximinomethyl radical in which
the oxygen or carbon atom is optionally
substitutcd by a Cl-C4 alkyl ~roup:
-C(RY)=NORæ where RY and R2 are as
defined above, except they may not be
I joined to~ether to form a ring;
~ "
:.
' ~vo~/n~66s PC~/~S93/07X30
,
2 i ~ 3
- 13 -
t) a (Cl-C4 alkoxy)carbonyl radical:
-C(O)OCl_4 alkyl, where the alkyl is
optionally mono-substituted by Rq as
defined above;
5 u) a carbamoyl radical:
-C(0)N(RY)RZ where RY and RZ are as
defined above;
v) an N-hydroxycarbamoyl or N(Cl-C4
alkoxy)carbamoyl radical in which the
nitrogen atom may be additionally
substituted by a Cl-C4 alkyl group:
~(C=O)-N(ORY)RZ where RY and Ræ are as
defined above, except they may not be
joined together to form a ring;
w) a thiocarbamoyl group: -C~S)N(RY)(Rz)
where RY and RZ are as defined above;
x) carboxyl: -COOMb, where Mb is as
defined above;
y) thiocyanate: -SCN;
z3 trifluoromethylthio: -SCF3;
: aa) tetrazolyl, where the point of
attachment is the carbon atom of the
tetrazole ring and one of the nitrogen
atoms is mono-substituted by hydrogen,
~ 25 an alkali metal or a Cl-C4 alkyl
:~; optionally substituted by Rq as defined
above;
ab~ a~ ani~onic function selected ~rom the
group consisting of:
~ : f:
'~'` ` '
,`: :
~:
::
~VO 'J~/~)5669 PCI/I 59~/0 830
21~2708
- 14 -
phosphono [P=O(OMb)2}; alkylphosphono
{P=O~OMb)-[O(Cl-C4 alkyl)~}; alkylphos-
phinyl [P=O(OMb)-(Cl-C4 alkyl)];
phosphoramido CP=O(OMb)M(RY~RZ and
S P=O(OMb)NHRX]; sulfino (SO2Mb); sulfo
(SO3Mb); acylsulfonamides selected from
the structures CONMbSO2RX,
CONMbSO2N(RY)R~, SO2NMbCON(RY~RZ; and
S02NMbCN, where
lo Rx is phenyl or heteroaryl, where heteroaryl is a
monocyclic aromatic hydrocarbon group having
5 or 6 ring atomsj in which a carbon atom is
the point of attachment, in which one of the
carbon atoms has been replaced by a nitrogen
atom, in which one additional carbon atom is
optionally replaced by a heteroatom selected
from 0 or S in the case of a 5-membered ring,
; and in which from 1 to 2 additional carbon
atoms are optionally replaced by a nitrogen
heteroatom, and where the phenyl and
heteroaryl are optionally mono-substituted
by Rq, as de~ined above; ~b is as defined
above; and RY and RZ are as defined ab~ve;
~ ac) C5-C7 cycloalkyl group in which one of
: 25 the carbon atoms in the ring is
replaced by a heteroatom selected from
Q, Sl N~ or N(Cl~C4 alkyl) and in which
, ; l one additional carbon atom may:~e
replaced by NH or N(Cl-C4 alkyl), and
~ in which at least one carbon atom ~:;
ad~acent to each nitrogen heteroatom
~: .
'
~ ~09~/05669 PC~ S93/07830
"` 21-~2738 ` ~,`
... ~ . .
,,.~
15 -
has both of its attached h~drogen atoms
replaced by one o~ygen thus fo.rming a
carbonyl moiety and there are one or
two carbonyl moieties present in the
ring;
ad) C2-C4 alkenyl radical, optionally mono-
~,~ substituted by one of the substituents
a) to ac) above and phenyl which is
optionally substituted by Rq as defined
~ 10 above;
i~ ae) C2-C4 alkynyl radical, optionally mono-
,`3 substituted by one of the substituents
a) to ac) above;
af) Cl-C4 alkyl radical;
ag) Cl-C4 alkyl mono-substituted by one of
the substituents a) - ac) above;
ah) a 2-oxazolidinonyl moiety in which the
point of attachment is the nitrogen atom
of the oxazolidinone ring, the ring
~ oxygen atom is optionally replaced by a
heteroatom ~elected from -S- and NRt
(where Rt is as defined above) and one
of khe saturated carbon atoms of
the oxa~olidinone ring is optionally
mono-substituted by one of the
substituents a) to ag) abo~e; and
~ ,
M lisjselected from: i~ hydrogen~
ii) a pharmaceutically acceptable
estcrifying group or removable ~.
carboæyl protecting group;
'
:
~VO ~t/O~fi69 PCI/I.'S93/07830
2 1 ~ 2 rl 0 8
-- 16 --
~ iii) an alkali metal or other
¦ pharmaceutically acceptable
cation; or
iv) a negative charge which is
balanced by a positively
¦ charged group.
I
¦ The present invention also pro~ides novel
carbapenem intermediates of the formula:
~ O , ~_ ~?a
// N
COOM Ra
y
wherein:
; R is H or CH3;
Ra is defined above, with the proviso that R~
additionally includes OP' where P' is
defined below, that Ma and Mb of Rq both
include M and that the Type d) hydroxy
substituent additionally may be protected
hydroxy, Opl;
P' is a rsmovable protecting group for hydroxy;
M is a remo~able protecting group for carboxy; and
the Type I, Ra substituent is counterbalanced with ~`
the anionic form of Z where
: Z is methanesulfonyloxy, trifluoromethanesulfonyl- ri
.
';
t
~ 669 PCT/~iS93/07~30
` 21 Ll: 2 ~ ~ 3
- 17 - I
~ .
oxy, fluorosulfonyloxy, p-toluenesul-
fonyloxy, 2,4,6-triisopropylbenzene-
sulfonyloxy, p-bromobenzenesulfonyloxy,
p-nitrobenzenesulfonyloxy, bromo, or iodo.
Preferred intermediates have the formu~a:
r ~ `z
COOM Ra
lS
wherein:
R is ~ or CH3;
P' is a removable protecting group for hydroxy;
M is a removable protecting group for carboæy;
Ra is selected ~rom the group consisting of H, OP', `~--
Cl, Br, I, SC~3, CN, ~0, SOCH3, SO2CH3, :`
C02M , C~20P' or COM~2; and with the proviso
that the -C~2Z substituent is in the 2- or
3-position of the heteroaromatic ring,
X is O or S; and
;~ Z is as defined above.
ETAILED D~SCRIPTIQN OF T~E INVENTION
The manufacture of compounds of Formula I
3 may be carried out in a three-stage synthetic scheme ~`:
~:: followed by deprotection. The ob:jeetive of the first
synthetic s~age is to produee a base heteroarylphenyl t~''`
(hereinafter HAP) compound which mày be converted to
,~
09~/~669 PCT/~S93/07830
2`14`2708
- 18 ~
be the two-position substituent of the carbapenem of
Formula I. The objective of the second synthetic
stage is to attach the base HAP to the carbapenem.
Finally, the objective of the third synthetic stage
S is to substitute the HAP with the desired Ra. This
third synthetic stage may either be performed after
the first synthetic stage or after the second
~ synthetic stage according to the nature of the
¦ desired Ra.
Flow Sheet A demonstrates a suggested first
stage synthesis. Flow Sheets Bl and B~ demonstra~e a
second stage synthesis. The third stage synthesis
varies according to the selec~ed Ra.
~0 .
,
' .
~-- O ~ f/~): 66~ PC~/~ S93/07830
21~ 7û~ lf
-- 19 - I
f
FLOW S~IEET A ~ 1
Br
~13(0H)z + Br- ~-Rz
Ra A1 A2
.
Pd( PPh3 ) 4 Br
aqueous ~[3- ~2
Na2CO3,
T,a
To l ue ne, ~ A3
EtOH
Alt ernat ively,
2 0
: Eir
~X +E~t2B--~3-Rz
Ra
:~ ~ A4 A5
,
Pd~ PPh3) 4
A3
3 0 n- Bu4NBr
~ ~: KOH t
; ~ T~ : ~:
~-~ ~ere X=Br, I s
:: ~ ; :
9 ~o s~/ns66s PC r/~ ss3/n7x30
1;``~
21~?~7~ 20-
Flow Sheet A
Substituted bromophenylboronic acids Al ar~d
substituted heteroaryldiethylboranes A5 may be
prepared by conventional methods. Exposure of ei~her
of these boron compounds to aryl halides in the
presence of a catalytic amount o~ palladium catalyst
yields the desired synthons A3.
Some of these desired synthons A3 may be
prepared by the general synthetic routes published in
the literature.
Flow Sheet Bl
The second stage synthesis is to attach the
base HAP to the 2-position of the carbapenem. With
compatible Ra or suitable precursor substituents
therefor, HAP ~ may be added to azetidin 2-one Bl in
a Grignard reaction as shown in Flow Sheet B. ~1 is
subgeneric to the more general Bl*. Replacing Bl by
* (where M is as de~ined above under ii) produces a
~ hroader class of compounds analogous to B2, B3, and
B4.)
The Grignard reaction requires that ~ be
converted to a Grignard reagent by reaction with
magnesium and 1,2-dibromoethane in THF from 20C to
~S 60~C and subsequently contacting A3 as a Grignard
reagent with ~1 in T~F at from -70C to about 20C to
produce azetidin-2-one B2. Alternati~ely, A3 may be
! 1 rea~ted with t-butyllithium, n-butyllithium, or the
like in Et20 or THF at from ~78 to -50~C followed by
t~e addition of magnesium bromide to produce the same
Grignard reagent. Ri of Bl is in practice pyridin-2-
yl but may clearly be a variety of substituents
including aromatic and hetéroaromatic substituents.
~'Q94/0~669 PCT/~S93/078~0
2 1 -1 2 7 ~
,
- 2~ ~
!
Further, Rl might be, for example, phenyl,
pyrimidinyl or thiazolyl.
Azetidin-Z-one B2 is an intermediate that
may be ring closed to a carbapenem. It is on this
intermediate that Ra or precursor substituent such as
t-butyldimethylsilyloxy-methyl group should be
modified where such modification is incompatible with
the carbapenem nucleus. For example, a convenien~
reaction to remove the t-butyldimethylsilyl group
from a hydroxymethyl substituent of the HAP on
compound B2 is to expose compound B2 to a dilute
solution of sulfuric acid or hydrochloric acid in
methanol at 0C. If the t-butyldimethylsilyl group
were removed from carbapenem B3 under the same
conditions, a substantial portion of carbapenem would
be degraded and lost. Thus, modification of the ~ -
precursor substituent in this instance and
replacement with another precursor substituent or
even Ra is best performed hefore closing the
carbapenem. Of course it is possible to remove the
t-butyldimethylsilyl group from carbapenem ~ in
reduced yield by e~posin~ B3 to tetra-n-butylammonium
fluoride and acetic acid in T~F.
;~ Compound ~ may be ring closed to carbapenem
;~ 25 ~ by refluxing in xylene with p-hydroquinone for
about 1 to 2 hours. It is on this intermediate that
final elaboration of Ra from a precursor substituent,
e.~i hydro~ymethyl, may be accomplishçd. Removal of
the protecting groups then provides the final
compound Formula I. Such final elaboration and
deprotection is described in further detail be~ow.
~ t"'
'`` ;:
~"
0 9~ fi69 PC[/~S9~/07~33()
, , ~ .
`2'~`0'~`
-- 2
FL~:)W SEEET B 1
C02allyl
A3 ~ 2 R li 1
o N~PPh3 ~ o ~PPh3
CO2allyl \ CO2M / _
C02allyl
O N~PPh3 R
CO2allyl
~32 B2
CO2allyl
' ~3R2
C02allyl
133
Pd( PPh3 )
,~{j-R
~` 3 0 \~-- o R~
COOH CO(~tM
C~l~C~ ' E~20
O ~/0:~669 PCr/~ S93/0~1~30
. ~ r `
21~2~0~
- 23 -
~_ow Sheet B2
Flow Sheet B2 shows an alternative second j,~
stage synthesis, i.e. attachment of the base ~AP such
as iB5 to the 2-position of the carbapenem. This
synthesis involves a palladium catalyzed cro~s-
coupling reaction between a carbapenem triflate and a
suitably su~stituted arylstannane, a process which is
described in U.S. Ser. No. 650,111 filed February 4,
1991. In order to apply thîs synthesis, it is first
necessary to modify B5 to the trimethylstannylhetero-
arylphenyl B6. This is accomplished by reacting B5
with t-butyllithium in T~F at from -78 to -50C
followed by the addition of trimethyltin chloride.
Alternatively, iB6 may be prepared by simply heating
B5 with hexamethylditin in the preisence of
tetrakistriphenylphosphine palladium in toluene
solution. At this intermediate stage, it may be
desirable to remove certain protecting groups if
employed on a precursor substituent Ra. For
instance, a protecting group such as t-butyldimethyl-
silyl on a hydroxymethyl substituent may be removed
by exposure to tetra n-butylammonium fluoride in T~F
yielding a particular B6. If the t-butyldimethylsilyl
group were removed from carbapenem B7 under the same
conditions, a substantial portion of the carbapenem
would be degraded and lost. Thus, modi~ication of
the prccursor substituent in this instance and
r~pllacement~with another precursor substit~ent or
even an Ra is best performed before attachment to the ~ ;~
30 carbapenem. ~ ;
..'"
~,
:
: ~ ` :
. .
~'O 9~ 6fi'~ PCI/~ S9~s~078~0 ~.~
2 1 ~ 2 7 0 ~3
- 24 -
The steps for preparing the 2-oxocarbapenam
intermediate B8 are well known in the art and are ' r. . '
explained in ample detail by D.G. Melillo et al., ' ~i-
Tetrahedron Letters, 21 , 2783 (1980), T. Salzmann
S ~ . J Am. Chem. Soc., 102, 6161 (1980), and L.M. :~
Fuentes, I. Shinkai, and T.N. Salzmann, J. Am. C~
~, 108, h675 (1986). The syntheses are also
disclosed in U.S. Patents 4,269,772; 4,350,631;
4,383,946; and 4,414,155 all incorporated herein ~y
lo reference
Referring again to Flow Sheet B2, the
2-oxocarbapenam, B8, is reacted at -78OC to -50C
with a suitable trifluoromethanesulfonyl source, such
as trifluoromethanesulfonic anhydride, trifluoro- ~:
methanesulfonyl chloride and the like, in the
presence of an organic nitrogen base, such as
triethylamine, diisopropylamine and the like, in a
polar aprotic solvent, such as tetrahydrofuran or ;~
methylene chloride. Optio~ally, an organic nitrogen
base, such as triethylamine and the like, is then
added to the reaction solution ~ollowed immediately
by a silylati~g agent, such as trimethylsilyl
trifluoromethanesulfonate to provide intermediate
B9. An aprotic polar coordinating solvent, such as
DME, l-methyl 2-pyrrolidinone and the like, is
optionally added. This is followed by the addition
of a palladium compound, such as tris(dibenzylidene-
. ; ace~one)dipalladium-chloroform (Pdz(DBA)3~CHCl
palladium acetate and t~e like, optionally, a
30 suitably substituted phenylphosphine, such as ~.;
tris(4-methoxyphenyl)phosphine9 tris(2,4,6-trimethoxy
.
9~ 669 pcT/~s9~/o7~3n ~.
~27~`3
- 25
phenyl)phosphine and the like, and thè stannane ~6.
A halide source such as lithium chloride, zinc
chloride or ammonium chloride and the like, is added t
and the reaction solution is allowed to warm and is
stirred at a suitable temperature, such as 0 to 50C
for from a few minutes to 48 hours. The carbapenem
B7 is obtained by conventional isolation/purification
methodology known in the art.
Generally spea~ing, the milder conditions of
10 the synthesis shown in Flow Sheet B2 allow for a
wider range of functional group Ra to be present than
the synthesis illustrated in Flow Sheet Bl ~owever,
in certain cases, it is ad~antageous for the Ra
substituent(s~ of the stannane B6 to be introduced in
15 a protected or precursory form. Final e}aboration of
Ra from a precursor substituent, e.g. hydroxymethyl,
: may be accomplished on earbapenem intermediate B7. . `
Removal of hydroxyl and ester protecting groups then
provides the final compound, C5 of Formula I. Such
f.inal elaboration and deprotection is described in
detail below.
,.
3 73v.
~; :
~ ' ' '
.".
~'O 9~/0~66~ P~r/~ss3/o7~o !~
21'1~0g
- 26 -
FkOW S~IEET B2 ~
1. `
HO H H R
H ,g I ~) Br ~ 3~ R2
H CO2- p- NI3
E3~ 1
~ B5 F;'
~33S~ ~33Sn~3-Rz
E~9 CO2 - p- NE + R~ ~6
~3SiO H H R ~3-R2a
B7 ¦ CO2- p~
~ R3
C5( I~ . COOM
~ere
p-NE3 = -CH3 ~NO~ ,
-:
~'O 9 ~/05fi69 PCr/~S93/07~30
.: .,....................................................................... ,`,.,
21~270~ ~-
- 27 -
~1Q~_Sheet C
Azetidin-2-ones Bl and Bl* ~Flow Sheet Bl),
pyridyl-thioesters, are well known compounds in the
production of carbapenems. Di~erse synthetic schemes
useIul to make Bl and Bl* may be imagîned by the
ski.lled artisan. Particularly useful to ~he instant
inventors is a synthetic scheme set out further in
Flow Sheet C below in which the symbol R is.as
defined above. The steps for preparing intermediate
and Bl* are analogous to the procedures described,
for example, in U.S. Pat. Nos. 4,260,627 and :~-
4,543,257; L.D. Cama et al. Tetraked_Qn 39, 2531 :;
(1983); R.N. Guthikonda et al. J. Me~. Chem., 30, 871
~1987) hereby incorporated by reference, as discussed
below.
'~
r
:~ 5
f
"~'j,
" ` : :
~VO'~ 6~i9 PCT/-S93/n7~30 ~,,'
i . ~
; ` " ' '; `
2~ 42r7 0 ~ ~ 28 -
FLQW ~HEET C ~ ,
~ . . .
t - E~uMe S iO R
2 I H H I
C2 Me a~ NaOH/M~OH ~;
o I b. carbonyl
~, a diirr~Ldazole/
t - ~3uMe2SiO R TM~
I H H I ~
/~)\ CCz H c. 1 OHCCO
,~NH ii. SOC12
iii. Ph~P
.
t - BuMe2SiO R d. 6N HCl~M~30H
I H H
2 0 ~ ~TMS
H~NH
O I . i:
t-~3uMQ2SiO R ~
I H H ~
/~\C2 /\~TMS
H ~N
3 0 0// \~ PPh~ 3
; 2 \~'~\
: ~ :
d
; ~
.
~:
~0 4~/0;664
- P~/~ S93/07~30 i`~
`:, ` :
2 ~ 2 7 ~ ~
- 29 -
FLOW SHEET C ( CONT ' D
~ ,.
HO R
S ~ TMS
~NyP
C2 ~ _.. ,, ; ~
O2CO R ~
¦ H H ~
~ f\~TMS
1 S ~ ~ ~ PPh3 ~ ;
e ClCOz ~ /DMAP
~--2C R F. nE3u4NF
g. Pyr-SS-Pyr, / Ph3P
i ~j ~2CO ~ ` :R ~
~VO 9J./~fi9 PCr/~iS93/07830
(' :, .'
2 1 4 2 7 0 ~ !
-- 30 --
,
The general synthesis description depicted
above in the Flow Sheets shows a protected
l-hydro~yethy~ substitution on the 6-position of the
carbapenem. After final deprotection, a
l-hydroxyethyl substituent is obtalned, which is
preferred in most cases. ~owever, i~ has been found
that with certain 2-side-chain selections, the
ultimate balance of favorable properties in the
overall molecule may be enhanced hy selection of the ;
6-(1-fluoroethyl) moiety instead. Preparation of
6-fluoroalkyl compounds within the scope of the
present invention is carried out in a straightforward
manner using techniques well known in the art of
preparing carbapenem antibac~erial compounds See, ;~
lS e.g., J. G. deVries et al., ~erocvçles, ~ , 1915
(1985); BE 900 718 A (Sandoz) and Japanese Patent
Pub. No. 6-0163-882-A (Sanruku Ocean).
In the compounds of the present invention,
one of the Ra substituents must be of Typie I. As a
general matter, it is conjectured that anti-MRSA/MRCNS
activity results from the configuration of the overal:l ;
molecule uniquely conferred by the HAP nucleus. The
Type I substituent proYides still greater anti~
MRSA/MRCNS activity to the molecule. `
~ The Type II Ra substituents are distin-
guishable from Type I substituents chemically and
with respect to the biological properties which they
confer. In related compounds, it has been ~oun~ that
!` t~e Type II substituted compounds afford greater
30 water solubility and reduced potential for CNS s.ide ~.;
ef~ects. Substituents which tend to confer improved
?
''' ';
'~
~; :
~vo~/0~669 ~CT/-S93/~7830
;; 21~2~0 3 i ~
~ - 31 - I
water solubility on the overall compound have been ~ ¦
found useful, since they are contemplated to thereby
improve the transport o~ the compound involved.
Although a substantial number and range of Type II
substituents have been described herein, all of these
are contemplated to be a part of the present
invention based on the biologic.al performance o~
substituents related iIl terms of their medicinal
chemistry.
Since it is possible to combine, in the
compounds of the present invention, the required Type .
I substituents with the optional Type II
substituents, there can be obtained a combination of
desired attributes in the final overall molecule not
lS attainable with a single substituent, i.e., i~proved
~ anti-MRSAlMRCNS activity together with enhanced water ~.
: solubility.
Type X substituents employed in the
compounds of the present invention may have .
quaternary nitrogen groups, and these include both
cyclic and acyclic types, as is described under Type
I. As already pointed out above, it is required that
one, but no more than one, of the substituents Ra
~: must be a member selected from the group consisting
~25 of the definitions under Type I. It is optional that
one, or at most two, of the remaining substituents ~. .
may be a member selected ~rom the group consisting of ~ I
; definitions under T~ype II. For example, Ra;attached
to the phenyl group may-be Type I and Ra at a
3Q position on~the EAR moiety may be OL` Type II, while ~ .
the rema`ini~g~substituent~ are hydrogen.
~ ~ ,
~'09~ 66~ l~CT/~S93/078~0 ~.
.. . . ~ .
~ , ~ i, j.
21~27~8
- 32 -
In preferred compounds of ~ormula I, Rl is
hydrogen. More preferably, Rl is hydrogen and R2 is
(R)-CH3CH(OH)- or (R)-CE3CH(F)-. In the most
preferred case, Rl is hydrogen and R' is
(R)-CH3CH(O~)-. While R = H is usually preferred,
there are instances in which R = C~3 may provide
.improved chemical stability, water solubility, or
pharmacokinetic behavior. The substituent R = CH3
may be of either configuration, i.e., the ~ or
r3-stereoisomer. Additionally, in preferred
compounds, at least one Ra is other than hydrogen. ~
In the most preferred compounds, in total, up to two :
Ra substituents are other than hydrogen;
. :'~
. . .
~5
::
1' ~ ; .
~- ~
. ..
'~.
:
W(~ 9~ 669 P~/I~S93/07830
21 4 ~ 7 ~
- 33 -
Preferred Type I. a) substituents include:
~N ~ 0-2; ~N ~ R 0-2 ;
"
1 0 . -~ ~
A + ~ N R 0-2 ; + ~ , Y 0-2
: ' `
: 15
-A-N 0 t R 0-2 + ~N~RC
~ier~ t he ring ~ere t he ring ~ -
. . . ,~j.
cont ains t hree cont ains t ~
carbon atoms; carbon~ atoms;
~G ~ 2 ;
s ~ i ~
66() PC r/~ S93~078~
. ~-....... . .
2~42708
-- 34 --
X ~, ,.
\\~ C - A- N~
N 0-2 ; N 0-2
'.
.
A N/ ~N N~X~RC
~J ; ~A
+
RC
0 - 2
2 0
< ~RCo~ RCO-~ ;
: 2 5 A `
~ / :
1` ~ ' ' " I ` , .,
.
:~ ,
; ~ ` `
~:0 9~/0~669 PCT/I 'S93/07~30
21~2~0~
- 35 - ~ :
R o_1 X A N/ \N
N~ ' ~N ' :~`
/ + RCo_l :
l o A
N--I + ~X~
RCo 1 A_ `~
R O-1 ~X ~ N/ \ ~ `
X 5 N N )--N i~
RCo,
,. .. -` .
3 0
.
.
'
\~0 9-i/()~669 PCT/I~S93/07830
21~L27~ -
-- 36 --
R 0-2 ~R 0-2
R 0-2 RC0-2
~ ; X~3R o_~
~Co Z
:~ 20 A
Rc
: ~ -
R 0-2
: : ,
where X = 0, S ~ or NRC . For structures of Type I . :~
~, , a) ,~ Iwhere Rc is shown to have an inde~inite position,
it may be attached to any carbon of the ring.
~ -.
~n 9~ 69 PCT/~S93/07~30 ~
2 1 L~ 2 7 '~ g ` I ~ .
. - 37
Preferred Type I.b) substituents include~
-A~ --~3-RCo-2 -A~ ~Rc0-2
Rd Rd ~
,
-A~ ~\~RC0 2 -A _~ 3RCO ~ ~
Rd Rd ~ :
-
+hd -A _~-R 0-2;
where t he ring
cont ains t hree
~s ..
carbon atoms;
..
-A~ RC0-2 ; G ~'
~.1
~,
"~
\-O 9~ 669 PCr/~S9~/0783n
214~708
` 38 -
s
--A ~N~
Rd
1 s
--A ~RCo l; ,A '~ ?N + d ;~
~ 0-1 ~
.,
2. 5
'~,
; ' ~
o ' `~ ~
', ;~
: .
~O 9-1/(1:.669 PCT/I S9~07~?10 t ~
;;, . ~ ~
~ 1 4 ,~ D &
.~ -- 39 --
R o ~X~ R
RS R 0-
N--N ; Rd ~`N
R~ --A
2.0 A' ~X'
N~N ' ~) N
; + ~d _ A~ ~ R
A --~ ~N Rd N ~ , N- Rd
N N ; ~N
';
: ..
~: :
~VO 9~1/():`66() PCl /~ S93/07830
. ~ ~ . . . .
21~270~
-- 40 --
Rd
-A' ~ ~ - RCo z; -A ~ RC
RC0 2
Rd
\ RCo z and A ~ R 0-2
RC 1
0-2 Rd -
where X = O, S, or NRC and X' = O or S. For
2S structures of Type I. b), where Rc and/or A' are
: shown to have inde~inite positions, they are
independently attached to any carbon atom of the ring. ', ~;
3 0 : ~
~. ~
., ~ ''~'.
`.
--
, .
\~'09~ 566') PCT/~S93/07830 ~
~ 2 ~ 7 ~ ~ ~
- 41 -
Preferred Type I. c) substituents include~
1~ . `
-Ap-+N(CH3)3. ~Ap-+N(C~2cH3)3
-Ap-+N(CH3)2CH2Rq, -Ap--+N(CH2cH3)2cH2cH2R ~ .
10~AF~RqO, C/H~Rq Rqo
- ~ -N O-l -Ap N ~ ~ -Ap-N ~
CH2CH2Rq CH~ ~qo-t Rqo 1 ;
. ...
.
+ ~ N o~
-Ap-N ~ -Ap-N ~
Rqo-l R O-l ;
'.
, .
where W is O, S, NRe, N(O)Re, SO, S02 or N+(Re)2 an~
.W' is M~Re or NO.~ For structures of Type I.c), where ~ .
30 R~ is shown to have an indefinite position, it may be ';
attached to ~ny carbon atom of the r ing .
'.' :.,
' :~
! ~
~-'09~ 669 PCT/~S9~/07X~0
2 1 ~27 0`~ i i ` ` ``
- 42 -
Preferred Type I. d) substituents include:
d
~ N ~ ;
Rd
Rqo
Rd ` ::
N
-A~ p ~
Rqo
/d Rqo
2 0 ~d
--A' --N N a nd
P \_~,/ \Rd
~q
L~ O_ 1
Rd
: - A' p ~
For structures of~ Type I.d), where Rq and/or A'p is
30 shown to have an ind~efinite position, it may be s
attached to any carbon atom of the ring.
~ , . .
~ 669 PCT/~S93/07X30
:,',~'. ~;.,
2 ~ ll 2 ~
43 - I
~' '
The Rc substituents herein are intended to ~ '
represent suitable further substituents on the Type {
I. a) or b) substituents for ~AP~ As seen above,
these Type I. a) or b) substituents are monocyclic or
bicyclic aromatic groups containing heteroatoms.
Given this class of primary sub~tituent, further
suitable substituents may be readily discovered in
the penem and carbapenem art. For example, suitable ;
substituents for Type I. a) or b) substituents are
generally taught in U.S.Patent No. 4,729,993 assigned
to Merck and Co. or in U.S.Patent 4,746,736 assigned
to ~ristol-Myers Co. These patents are hereby .:'
incorporated by reference.
Broadly, Rc may be the same or different and
15 may be selected on an independent basis from the
group as defined above. While a single such
substitution is preferred, there is occasion to use
up to two such substituen~s on an Ra, e.g., where it
is desired to enhance the effect of a particula~
20 substituent group by employing multiple
substituents. The particular choice of Rc will
depend upon the situation. For instance, a specific
may le~d particular stability to a nitrogen
cation At other times it may be desired to employ a - ;
25 substituent knoFwn to enhance antibacterial activity
of the overall molecule against a particular
bacterium, for example, while also employing a
substituent known to improve some other property such
as water solubility or the duration of action of the
30 overall molecule.
The scope of Rc herein includes tFwo specific s
Types of further substi~uent attached to the Type I.
',
~ 0 9~ h69 PCr/~'S93/0783~) i ~,.
;,. .`
21~~
a) or b) substituent. A first Type of Rc are those
attached to a ring carbon and a second Type of Rc are
those attached to a neutral ring nitrogen. Persons
skilled in the art will readily recognize that a wide
range of organic substituents are suitably used as
RC. Persons skilled in the art will also recognize
that some substituents including the -NRYRZ
substituents, useful for one purpose of RC, i.e.
carbon substitution, are not equally useful in the
other, i.e. nitrogen substitution.
Preferred Rc attached to ring carbon atoms
are -NH2, -SCH3, -soc~3, -C~20~, -(CH2)2OH, -OC~3,
-COOMb, -CH2COOMb, -CH2CH2COOMb, -CH2SOCH3, -C~2SC~3,
CM, -SG3Mb, -C~2S03Mb, -C~2C~2SO3Mb, -Br, -Cl, -F,
~ CH3, CR2CH3, CH2CON~2 and CH2CON(Cl-C4 alkyl)
where Mb is defined above. Preferred~RC attached to
neutral ring nitrogen atoms are -CH20H, -(C~2)20~,
-C~2COOMb, -C~2CH2COOMb, -CE2SOC~3, -CE2SC~3, CN,
_CH2SO3Mb, -C~2CH2SO3Mb, -~3~ C~2C~3~ C~CON~2 a
C~2CON(Cl-C4alkyl) where Mb is defined above.
It is preferred that each Type I. a) or b~
substituent have no more than two Rc substituents
which are other than hydrogen. Thus, the formula
shown above for Type I. a) substituents has up to two
Rc substituents with the remainder of course being
hydrogen. Further, the formula for the Type I.-b)
substituent also allows up to two Rc In accordance
with these $ormulae, the previously listed more ! !~
specific structures should be interpreted to ha~e no
more than two Rc for each monocyclic or bicyclic
group. Similarly for Type I. c) or d) subs~ituents i~
it is preferred that any monocylic or bicyclic group
have no more than a single R~ substituent.
~0 91/1)~669 PCT/~rS93/07830
2 i ~2705~ i :
~ 45
The scope of Rd includes a single type of
further substituent attached as a Type I. b3 or d)
subst.ituent. The Rd substituents are attached to a < :;
cationic nitrogen which may or may not be aromatic. :
Preferred Rd attached to cationic nitrogen atoms are
hydrogen, -CH3, CH2CH3, -CH2CH2C~3~ -C~2cOoM -
-C~2S03Mb, -~I2 and a(-), where Mb is defined above.
The formulas depicting Type Ib, Ic, and Id
substituents show positively charged states for those
substituents. It is understood that certain of those
substituents, which are cationic by virtue of having
a protonating hydrogen atom attached to the nitrogen,
may also exist or be produced under certain
conditions as a neutral substituent by ~irtue of the
absence of such a hydrogen atom (i.e., in Type Ib,
when the~e is no Rd; in Type Ic, when there is no RW; :;
and in Type Id, when there is zero to one Rd,
depending on Type of heterocycle). Whether such a
Type Ib, Ic, or Id substituent will be predominately
cationic or neutral in a given physical state will be
governed by principles of acid-base chemist~y, which
are well ~nown to those skilled in the art. ~or
example, the particular ratio of neutral form to
cationic form will depend upon the basicity of the
amine and acidity of a solution. When such a
substituent is in a prot~nated quaternized stat~, the
compound exists as a zwitterion which is internally
balanced as to charge or as an ammoni~m salt which is
externally balanced. In illustration, if there is no
30 Rd on a Type Ib substituent, it is understood that ~. `
such a substituent is neutral (there is DO positive
\~09~t~669 PCT/~:S93/07830 tr
` .,,: !`: .
2 1 ~ 2 ~ 0 '~ - 46 - `
charge on the nitrogen). A compound containing such
a substituent is typically produced in this form as a
salt, wherein M is an alkali metal, and may exist in
solution in its neutral form. However, depending
upon conditions, a compound containing a neutral Type
Ib substituent may be in equilibrium with, and may
also be represented by a formula showing, the
corresponding compound containing the quat~rnized
protonated substituent where Rd is present and is a
hydrogen atom. Furthermore the same compound may
exist with the Type Ib substituent in a completely
protonated quaternized form, for instance in an
aqueous solution in the presence of a stoichiometric
amount of a strong mineral acid. It is intended
herein that both the protonated (cationic) and the
unprotonated (neutral) forms of Type Ib, Ic and Id
substituents of the type just described are within
the scope of the present invention.
Suitable A spacer moieties include ~CH2-,
C~2CE~2~ H2C~E2CH2- ~ -cH2c~2c~2c~2- ~ -0CH2CH2-
-SOCH2-, -S02C~2-, -SC~I2(:~H2,-, -SOCH2C~I2-,
-SOZC~2c~2-. -N~CH2C~2-, -N(C~3)C~2c~2 ,
-C~2N(CH3)CH2C~2-, -COM~C~2C~2-, -S02N~C~2C~2 ~
-COCH2-, -C~=CHCH2- and -CH?OCH~CH2-. Preferably,
where Q is 0, S, N~ or N(Cl_4alkyl), then n is 2-6.
Suitable A' are listed for A above. Further
A' may suitably be -0-, -S-, -NH-, -S02-, -S02N~-,
-C,ONH- ~ -CH=CH-, -CH2S-, -CH2NH-, -CON~CH2- or ~ ~
S02N~C~2- . ~ .
; 30
'.`,.
''. ;'
' ~,
,,~",
:,
;-
~ o~ 669 rcr/~ss3/o,s30
2 i~2~
- 47 ~ I
The Type I. cationic substituents are
generally added to ~AP following attachment of HA* to
the carbapenem. Conveniently, the ~AP side-chain
should be synthesized with a precursor substituent
which may be elaborated into the desired cationic
substituent. The identity of the precursor
substituent will vary according to thè particular Ra
desired. For example, one such precursor substituent
is -A-OH, such as hydroxymethyl.
The hydroxymethyl precursor substitue~t may
be elaborated into cationic substituents of Type I.a)
by converting the hydroxyl into an active leaving
group such as an iodide (giving -A-I~ followed by
reaction with a desired nitrogen containing aromatic
compound. More particularly, two alternative
procedures may be utilized to produce a leaving group
on the moiety -A- and subsequently to replace such a
leaving group with cationic substituents of the type
just described.
For a first procedure, the hydrox~l group of
-A-OH may be con~erted to a methanesulfonate group by
treating with methanesulfonyl chloride in the
presence of triethylamine. A suitable solvent ,
e.g., dichloromethane, is employed and the reaction
is carried out at reduced temperatures. In turn, the
methanesulfonate intermediate may be converted to the
reacti~e iodide derivative by treatment with sodium
iodide in a suitable solvent, e.g., acetone, at
reduced or ambient temperatures. Alternatively, the
30 hydroxyl group may be directly converted into the ~.
iodide group by common methods known to the art. For
...
:`'
~-094/1)~669 pcTt~ss3/o7~3n
.
. - 48 -
example, treatment of the hydroxyl group with methyl
tripheno~yphosphonium iodide in a suitable solvent, .
such as dimethylformamide, at reduced or ambient
temperatures, directly provides the desired iodide.
S Once the iodide has been formed, the introduction of
the cationic substituent is accomplished simply by
treating the iodide with the desired nitrogen
containing compound, e.g. a heteroaromatic compound
such as pyridine. The reaction wil~ proceed in a
suitable solvent, such as acetonitrile, at or about
room temperature. This displacement reaction may
also be facilitated by the.addition of excess silver
trifluoromethanesulfonate to the reaction mixture, in
which case reduced temperatures are often desirable.
For a second procedure, the hydroxyl group
of -A-O~ may be converted into the reactive
trifluoromethanesulfonate (triflate) group. ~owever,
such an activating group cannot be isolated by . ~
conventional techniques but may be for~ed and used in ~.
20 ~, Thus, treatment of the hydroxyl group with ~.
trifluoromethanesulfonic (triflic) anhydride in the
presence of a hindered, non-nucleophilic base such as
2,6-lutidine, 2,4,6-collidine, or
2,6-di-tert-butyl-4-methylpyridine in a suitable :~
solvent, such as dichloromethane, at reduced
temperatures provides for the generation of the
triflate activating group. Introduction of the ~,
cationic group is then accomplished by reacting the
above triflate in .situ with the desired nitrogen
containing compound at reduced temperature. In
~: certain cases it is possible and desirable to use the
.
,:
. ~ :
~-~9~ 66'~ P~T/~IS93/07~30
21~08 , :
- 49 -
., j .
reacting nitrogen containing compound as the base for ~ I .
the formation of the triflate activating group. In
this case treatment of the hydroxyl group with
triflic anhydride in the presence of at least two
e~uivalents of the reacting nitrogen compound under
the conditions described above provides the cationic
substituent. . .
The above are representative of suitable ::
leaving groups: alkylsulfonyloxy, substituted
alkylsulfonyloxy, arylsulfonyloxy, substituted
arylsulfonyloxy, fluorosulfonyloxy and halogen. The
cGmmon sulfonate leaving groups are:
methanesulfonylo~y, trifluoromethanesulfonyloxy, .;
fluorosulfonyloxy~ p-toluenesulfonyloxy,
2,4,6-tri-isopropylbenzenesulfonyloxy, p-bromo-
benzenesulfonyloxy and p-nitrobenzenesulfonyloxy.
The preferred halo leaving groups are bromo and
iodo. These alkyl and arylsulfonate leaving groups
may be prepared using an analogous route to the one ~.
described above using the sulfonyl chloride or the
sulfonic anhydride.
Where the cationic substitution has a
substituent RC, the most ~acile method of providing
such a substituent is to employ as the reactant in
the preparation methods described above a nitrogen
containing compound which already has the desired
substituent. Such substituted compounds are readily
available starting materia~s or may be prepared in a
d;
.,:
'`
, ~
~v094/0~669 PCT/~S93/07830
~'. . , '-'
2 1 ~'10 ~ - 50 -
straight-forward manner using known literature
methods.
The Type I.b) cationic substituents are
prepared by quaternization of an aromatic ring
nitrogen of a neutral precursor substituent on either
of the HAP rings. Examples of neutral precursor
substituents are -CONHC~2~ pyridyl),
-CONHC~2-(4-pyridyl) or -S02CH2-(4-pyridyl).
Quaternization is accomplished by reacting the
nitrogen compound in an inert organic solvent
(e.g.CH2C12) at about 0C to room ~emperature with an
alkylating agent Rd-Y where Rd is given above and Y
is a leaving group such as iodide, bromide, mesylate
(methanesulfionate), tosylate (p-toluenesulfonate) or
triflate. Alternati~ely, the aromatic ring nitrogen
may be quaternized by reaction with an oxidizing
agent such as 3-chloroperbenzoic acid (giving the
N-oxide~ or an aminating reagent such as
0-(2,4,6-triisopropylbenzenesulfonyl)hydro~ylamine
(giving the N-amino derivative) in a suitable solYent
(e.g. dichloromethane or CH3CN) at about room
temperature. In addition, the neutral precursor
substituent may be rendered cationic through
protonation of the basic aromatic ring nitrogen.
This may be accomplished by treatment of the neutral
precursor with a suitable inorganic or organic acid,
e.g. hydrochloric acid, phosphoric acid, hydrobromic
acid, acetic acid or benzoic acid. Protonation may
!' I fu~lther be açcomplished by a carboxylic acid function
elsewhere in the molecule, including the C-3 carboxyl
on the carbapenem. The neutral precursor substituent
may be already attached to HAP at the time of its
. .
: ,:
\V~94/05~6~ PCT/~'S93/07830
;~ 2l~2~a~
- 51 - ,
I .
connection to the carbapenem, or it may be elaborated
from a simpler precursor after connection to the
carbapenem. An example of a precursor substituent
for elaboration is -A'-0~ such as hydroxymethyl. In
one suggested synthesis, the hydroxyl may be
converted to a reactive leaving group such as iodo as
described above. The iodide is then reacted in a
nucleophilic displacement reaction with a nitrogen
containing aromatic compound which has a nucleophilic
side-chain substituent such as CH~SH or C~2NH2. In
this displacement reaction, it is the side-cnain
substituent that is the reacting nucleophile and not
the aromatic ring nitrogen. Suitable substrates for
this reaction include 2-(mercaptomethyl~pyridine,
2-amlnopyridine, 2-(aminomethyl)pyridine or
4-(mercaptomethyl)pyridine. The reaction is
carried-out in an inert organic solvent, e.g.
methylene chloride, at from about 0C to room
temperature in the presence of a non-nucleophilic
base such as triethylamine or diisopropylethylamine.
Quaternization or protonation of the aromatic ring ;-
nitrogen as described above then gives the Type X.b~
cationic substituent. A second suggested synthe~is
of a Type I.b) cationic substituent starting from a
precursor ~A'-O~ (e.g. hydroxymethyl) consists of
oxidation of the alcohol functionallity to an
aldeh~de follow~d by Wittig-type olefination with an
appropriate nitrogen-containing aromatic substituted
reagent, and finally quaternization. The oxidation
may be co~veniently accomplished by a Swern oxidation
employing oxalyl chloride-dimethylsulfoxide followed ,-
by triethylamine. The reaction is conducted in ~` -
.1 . .
`: ' ' ' .'
~'09~ 6S9 PCT/~S43/07830
` 2 1~
- 52 -
methylene chloride as a solvent at from -70C to
0C. The Wittig reaction is carried-out by reacting
the aldehyde with the desired Wittig reagent in a -~
polar solvent such as acetonitrile or dimethyl-
sulfoxide at about room temperature. Suitable
Wittig reagents include: pyridylmethylene-
triphenylphosphorane, quinolylmethylenetriphenyl-
phosphorane, and thiaæolylmethylenetriphenyl- ~;
phosphorane. Quaternization or protonation as
described above then completes the synthesis of the
Type I.b) cationic substituent. Depending on the ~.
particular Ra of Type I.b) that is desired, many
other synthesis schemes may be employed, as would be
apparent to an organic chemist skilled in the art.
The Type I.c) cationic substituents may be
prepared in an analogous manner to that described for
I.a) substituents except that the nitrogen containing
compound employed in the displacement reaction ls an `.
aliphatic amine (i.e. NRYRZRw). However, in cases
where the amino group is directly bonded to HAP (i.e.
ApN+RYRZRw where p-~), the amine is most -.
conveniently attached to ~AP prior to its
incorporation into the carbapenem system. If such an
amine is primary or secondary, it may require
pxotection with a suitable amine protecting group
during the steps employed~to attach ~AP to the ;
carbapenem. Tertiary ami~es require no pro~ection
and may be quaternized or protonated as described for
$he!Type I.b~, catibnic substituents. ~ I /
The Type I.d)~~cationic substituents are ;
` 3 prepared by quaternizàtion or protonation of a
non-aromatic ring nitrogen of an appropriate neutral
:;
YO ~ 66') ~ S93/~)7P.30
~ 2l~27a~ ~ -1`''
precursor substituent on HAP. Quaternization or
protonation is accomplished as described above for
the Type I.b) substituents. As with the Type I.b)
substituents, the neutral precursor may already be ~-
attached to HAP at the time of its connection to the
carbapenem, or the neutral precursor may be
elaborated from a simpler precursor substituent on
~AP after its connection to the carbapenem. Examples
of neutral precursor substituents are
-CONH(3-quinuclidinyl),
-CONH[4-(N-methylpiperidinyl)],
-SO~CH2CH2t2-(N-methylpyrrolidinyl)],
-S02N~ (4-methylpiperazinyl)] and
-C~2~1-(4-methylpiperazinyl)J. Elaboration of -the
neutral precursor substit.uent from a simpler
substituent such as hydroxymethyl may be accomplished
in an analogous manner to that described previously
for the Type I.b) substituents by employing
appropriate reagents to introduce the Type I.d)
non-aromatic ring nitrogen moiety which is
subsequently to be quaternized or pro~onated.
It should be clear that for any of the Type
I.a) to I.d) substituents, the substituent may be
suitably ~ormed on ~AP prior to addition to the
carbapcnem. Thus, the substituent may be formed on
a~d reacted with B9 to ~orm the protected
carbapenem B7. For e~ample, 2-hydro~ymethyl-5-(3'- J
trimethylstannylphenyl)thiophene, i.e. B6, may be
substituted by reaction with triflic anhydride and
N-methylimidazole in a suitable solvent, such as, ~.
dichloromethane under nitrogen at -78C to room
temperature to form a Type I.a) substituted HAP, i.e.
i
.
.
~'O9~ 5669 PCT/US93/~7830
- 1`'
. . : .;: ; ~ .
..... '' ~
` 21~`10~ - 54 -
B6. This substituted HAP may be reacted with B9
employing conditions otherwise described herein and
specifically using an ammonium chloride source. 1`~"
In the compounds of the present invention,
the Ra substituents can be selected based on the `~
biological properties which they confer. In related ::
compounds, it has been found that the neutral or ~`
anionic substituted compounds afford greater water
solubility and reduced potential for CNS side
effects. Substituents which tend to confer improved
water solubility on the overall compound have been :
found useful, since they are contemplated to thereby :
improve the transport of the compound involved.
Although a substantial number and range of ;
5 substituents have been:described herein, all of these ~ 1-
are contemplated to be a part of the present
invention based on the biological performance of ~
substituents related in terms of their medicinal ~ -.
chemistry. 1`
: ~ is a 5-, 8-j or 9-membered mono- or
bicyclic aromatic ring system wherein ~ -
up to two carbon atoms are replaced by 0 or S.
HAR can be represented by
25` :
(where X is~ o or s)~ or
or ~
30~ ~ (Whete ~ i3 phrnyl~ne or a bivalent : ~ }`~
: 5-nen~er2d aronatlc ring wher~in ~
~ Feplacrd by o o- s~
w094/0~669 PCl/~S93/07830
- 55 -
':
Thus, this aryl structure may be the radical
of a S-membered furan or thiophene, of an 8-membered
~urofuran, thieno.furan, or thienothiophene, or of a
9-membered benzofuran or benzothiophene. The carbon
atom at the point of attachment, however, cannot be
replaced by a heteroatom.
The Ra ~ubstituents are on the carbon atoms
of the aryl ring system but not on the one at the
point of attachment. It is preferred that Ra =
when it is a to the point of attachment.
In preferred compounds of Formula I, R~ is
hydrogen. More prefera~ly, Rl is hydrogen and R2 is
(R)-CH3CH(OH)- or (R)-CH3C~(F)-. In the most
preferred case, Rl is H and R2 is (R)-CE3CE(OH).
While R = H is usually preferred, there are instances
lS in which R = C~3 may provide improved chemical
stability, water solubility, or pharmacokinetic
behavior. The substituent R = CH3 may be of either
con~iguration, i.e., the ~ or ~-stereoisomer.
Additionally, in preferred compounds, at least one Ra
in the meta-position of the HAP moiety from the point
of attachment to the other aromatic ring is other
than hydrogen. In the most pre~erred compounds, in
total, up to two Ra substituents are other than
hydrogen.
Among preferred Ra substituents are Cl-C
alkyl mono-substituted with hydroxy, such as,
hydroxymethyl; formyl; carbamoyl, such as, -CONE2;
hiydroxyiminomethyl, ~uch as, -C~=NOH; cyano; or
halogen such as chloro, bromo, and iodo.
~
-
.
:: "
.
,
~094/()~669 PCT/~S93/1)7830 ~;i
2 1i~27 0 8 - 56 -- :
Flow Sheet D
In regard to this preferred substitution,
the hydroxymethyl group may be obtained in the Ra
position of the phenyl portion of EAP as shown in
Flow Sheet D, in which A3 is obtained as given in
Flow Sheet A. Selective metallation of A3 and
formylation with N,N-dimethylformamide provides
synthon Dl . Reduction of ~l with sodium borohydride
in methanol yields the preferred substituent which is
protected as its silylether in the next step to give
D~i. The latter reagent is then incorporated into -
Flow Sheet Bl as A3. The preferred hydroxymethyl
group may also be obtained in the appropriate Ra
positions of the heteroaryI portion of HAP. Thus, by
a judicious choice of starting materials as exhibited `;
in Flo~- Sheet A, the desired substitution pattern is ~`
rea~ily available.
: ,.:.
;~ ..
: 20
1' '.'
. ,-
' ,.
~: : 25 ::
:.
~ .:
: ~ :
i" '
:~ 30 ,
~ . ~ : ,, .
, . ~,
WO ~ 669 PCr/~S93/07830 ~``;.
2 1 ~ 3
- 57 --
FLOW S~EET D
Br Br
Ra 1 ) BuL~ Ra
~/ 2 ) DMF ~=/ 2
Br O~C
A3 ( phenyl R~= Br ) D1
Br
NaSH" ~-R2 33CSi(1~22)Cl~
HO
D2
Br
~-~2
~3CSiO
I D3
'.
~-
~ ' ,
\~0~ 669 PCT/~S93/07830
: ^~
21~2:~'3 58 -
The preferred formyl substitution on the HAP
moiety may be obtained from the hydroxymethyl
substitution of B3 or isomeric B3~'~ described in Flow i-
Sheet Bl by a Swern oxidation. For example, isomeric
~* is oxidized in methylene chloride at from -70C
to room temperature employing oxalyl
chloride-dlmethyl sulfoxide as the active agent.
Obviously, the position of the resultant formyl
substitution will depend upon the position o~ the
hydroxymethyl substitution in isomeric B3*.
The preferred -C~=NOH substitution on the
HAP moiety may be conveniently obtained ~rom the
formyl substitution just described. This is
accomplished simply by exposing the formyl
substituted compound to hydroxylamine in an
appropriate sol~ent at room temperature.
The preferred cyano substitution on the HAP
moiety may be obtained from the -C~=MO~ substitution
JUSt described. The -C~=NO~ substituted compou~d is
dehydrated with triflic anhydride and triethylamine
in a solvent at -70~C.
.
The preferred carbamoyl substitution,
CONH2, may be obtained from B2 or 'iisomeric" ~* by
oxidizing hydroxymethyl with Jones reagent to the
corresponding carboxylic~acid substitution as (,
described above. This carboxylic acid is converted
t`o -CONH2 by seque~tially contacting with
l-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide
hyd~rochloride; l-hyldroxy-bènzotriazole, and a~monia
in~an organic solvent at~room temperature.
Substituted amides may of course be obtained by
replac~ing ammoni~a with the correspondlng substituted
,
~v~9.l~5669 PCT/~S93/07830
t
~'
_ 59 _ 21~270~ `
amine. In contrast to the carbo~ylic acid
substitution, this carbamoyl substituent requires no
protection from the conditions of carbapenem
cyclizatlon. Deprotection following cyclization is
carried out with palladium catalyzed deallylation in
a solution containing potassium or sodium 2-ethyl-
hexanoate as described in McCombie and Jeffrey, 1- -
Q~g. Chçm., 47, 2505 ~1~83). Deprotection in such a
solution yields the desired potassium or sodium salt.
In addition to or including the above,
1 suitable Ra of Type II include: ~:
-oc~3
-OCH2CH20~ ocH2co2c~3,
-F -CF3
-Br -Cl . :.
-OH -I
-OCON~2 -OCOCH3
-SOCH3 -SCH3
-SC~[2CEr20H S02CE3
-S2N~2 -SOC~2C~2OE
-N~C~O -SO2N(C~3)2
-NRCO~CH3 -N~COC~3 -.
-CN -NHS02C~3
-COCH3 -CHO
-CH=NOH -COCH2OE
-CH=NOCH~CO2CH3 -CH=NOCH3
-S2CH2CH2~I -CH=NOCMe2CON~2 ,, ,
-CH=NOCMe2C02Me -C2cH2c~2H
CONH2 -CO~C~3
-CoN(~3)2 -C:ON:E~C~2CN i,.
-CON~CH2COMg2 -CONECH2CO~CH3 ~:
-CON~OH -CON~C~3
.-.
'~
.
\-09~/()s6~9 PCT/~IS93/07830 ~ ~
2i427~
- 60 -
, . .
-tetrazolyl -C02CH3 ;
-SCF3 -CON~S02NH2
-CONHS02N~2 -S02NHCN
-S02CF3 -CH=CHCN
-502NHCON~2 -cH=c:EIco2cH3
-CH=CHCON~2 -C_C-CN
-C-C-CONH2 -C~2N3 and
-CH20~I -C~2I
-C~2c02cH3 ~.
. . ..
In the preparatlon methods described above,
the carboxyl group at the 3-position and the hydroxyl
group at the 8-position of the carbapenem remain
blocked by protecting groups until the final product
is prepared. Suitable hydroxyl protecting groups,
P', are silyl groups s~ch as trialkylsilyl, : `
aryl(alkyl)alkoxysilyl, alkoxydiarylsilyl and :: ~ r;
diarylalkylsilyl and carbonate groups such as
alkyloxycarbonyl, substituted alkyloxycarbonyl,
benzyloxycarbonyl, substituted ben2yloxycarbonyl,
allyloxycarbonyl and substi~uted allyloæycasbonyl.
The p~referred protecting groups, in addition to or
including those shown in the schemes, are
~-butylmethoæyphenylsiIyl, t-butoxydiphenylsilyl,
trimethylsilyl, triethylsilyl, t-butyldimethylsilyl,
o-nitrobenzyloxycarbonyl, p-nitrobenzyloxycarbonyl t
benzyloxycarbonyl, t-butyloxycarbonyl, 2,2,2
trichloroethyloxycarbonyl and allyloxycarbonyl. ,
Suitable carbo~yl protecting groups, M, in addition i ~ I`` ~ `
to or including those shown in the schemes are : i ;
described herein below. ; ` ~ ~
; ~: ' ~, r'
~ ~ : '' '`
:: ~ : : :
: : .
:,
U~9~ 669 pcT/us93/n7~3o
2~ a~
- 61 -
Deblocking may be carried out in a
conventional manner, with care being taken to avoid a -
procedure which is so harsh as to disrupt other
portions o~ the final product molecule. For
compounds prepared according to Flow Sheet Bl,
deprotection may be carried out in a palladium
catalyzed reaction in a solution containing potassium
2-ethylhexanoate and 2-ethylhexanoic acid or,
alternatively, another suitable nucleophile such as
pyrrolidine. Alternatively, for those prepared via
Flow Sheet B2, deprotection is conducted
sequentially. Thus, compound B7 is exposed initially
to aqueous acidic conditions, acetic acid or dilute
HCl or the like, in an organic solvent such as
tetrahydrofuran at 0C to ambient temperature for
from a few minutes to several hours. The resulting
desilylated carbapenem may be isolated by
conventional techniques, but is more conveniently
taken into the final deprotection process. Thus,
addition of an inorganic base such as NaHC03 or K~C03
; and a catalyst, such as, 10% Pd/C or 5a~ Rh/A1~03
~ollowed by hydrogenation provides for the removal of
the p-nitroben2yl protecting group and the formation
o~ the final compound of For~ula I.
The overall molecule must be electronically
balanced. Since a quaternary nitrogen is present in
the compounds of the pr~sent invention, a balancing
anion must also, in that case, be present. This is
usua~ly accomplished~by allowing COOM to be COO~.
~owever 9 where M is, e.g., a pharmaceutically
3~ accopeable ester, a counterlon (anion) Z~ must be i~
~ , ~
;~
~09-~/05669 PCT/~S93/07830
2 ~ ~ 2 r~ 0 8 - 62 - ~
provided, or alternatively, an anionic substituent
might be utilized. A counterion must also be
provided or additional anionic substituent utilized -
where there is more than one quaternary nitrogen.
Further, it is within the scope of this invention to
utilize an anionlc substituent where the quaternary
nitrogen is already balanced by COOM i~ C00~. In that
case, it will be understood that it is necessary to
provide a counterion (cation) for the anionie
substituent. However, it is well within the skill of
a medicinal chemist, to whom there .is available many
suitable anionic and cationic counterions, to make
such choices.
With reference to the above definitions,
"alkyl" means a straight or branched chain aliphatic
hydrocarbon radical.
The term "guaternary nitrogen" as used `;
herein refers to a tetravalent cationic nitrogen atom ,``;
including the cationic nitrogen atom in a -
tetra-alkylammonium group (e.g., tetramethylammonium,
N-methylpyridinium), the cationic nitrogen atom in a ~ i`
protonated ammonium species (e.g., trimethyl-
hydroammonium, N hydropyridinium), the cationic
nitrogen atom in an amine N-oxide ~e.g.,
N-methylmorpholine-N-oxide, pyridine-N-oxide), and
the cationic nitrogen atom in an N-amino-ammonium
group (e.g., N-aminopyridinium).
The term "heteroatom" means N, S, or 0, ~-
selected on an independent basis.
.
~
, .
, ,~
,
~ 94/()~669 PCT/~S93/07830 ~:
:~` 1``
2~2~0~ ` ' j
- 63 ~ i
The term "heteroaryl" has been defined
herein, in relation to the ~ group~ to have a
specific and limited meaning t being only monocyclic.
It is required that the monocyclic heteroaryl have at
least one nitrogen atom, and optionally at most only
S one additional oxygen or sul~ur heteroatom may be
present. Heteroaryl~ of this t~pe are pyrrole and
pyridine (1 N); and oxazole, thiazole or oxazine ~1 N
~ 1 0 or 1 S). While additional nitrogen atoms may
be present together with the first nitrogen and
oxygen or sulfur, giving, e.g., a thiadiazole (2N's
lS), the preferred heteroaryls are those where only
nitrogen heteroatoms are present when there is more
than one. Typical of these are pyrazole, imidazole,
pyrimidine and pyrazine (2 N's) and triazine (3 N's).
lS The heteroaryl group of Rx is always
optionally mono-substituted by Rq, defined above, and
substitution can be on one of the carbon atoms or one
of the heteroatoms, although in the latter case
certain substitutent choices may not be appropriate.
Listed in Table I are specific compounds of
the instant invention. In the table, R2 substituents
containing a chiral center (i.e., -CH(F)CH3 and
-C~(O~)C~3) have the (R) configuration, and the Ra :
column refers to the substituent on the phenyl ring.
, . i . ~
's.
,
.
` : .
~VO ~i05669 PC~/US43/07830
2142 1 ~ - 64 - '
TABLE I ~¦
R2
COO~ Ra ','`.
I
1 o -~ , .
HAR_Ra ! . ,'~
No. R R2 Ra 2 ~
. .,
~: 1 S 1 H - CH( OH) CH~ H _~, N~NCH3
2 o - CH( OEI) CH3 Cl ~ N~ ~NCE~3
, . .
3 H -CH(OH~CH3 3r ~3 N~NCH3
: :
: ~ 4 H -CH(OH)CH3 I ~ N~ CH3 :
H -CH(OH)CH3 S~3 ~ 'N;~NCH3
1, "
~VO 91/()~669 PCr/~!S93/07830 j~
2 ~ ~ 2 ~
- 6 5 -
TABLE I (C0NT~ )
,
HAR- R~
No. R R2 Ra 2
6 H - CH~ OH~ CH3 S~ O) ~ ~N~NCH3
S
7 H -CH(O~CH3 S2~ ~N~NCH3 `
lS B H -CH(O~CH3 F ~N~NCH3
~ ~ N~"NCH3
9 H - CH( OEI) C~3 H S
~0 ;'
~N~"NCH3
H -CH(OH)CH3 H S
:
:~5 ' ~ ''
_~\N~NCH3 . 7 '
11 H -CH(OH)CH3 F S
1' ' I ' , : ~ ! , ~ '
1 2 H - CH( OH~ CH, F ~ ~,N~;~NCH~
' :~
,, ,, ,. ,,, " . .......... . . , ~- . -
~ 1 ) 9 ~ 6 6 9 I C~ / ~ S 9 3 / 0 7 8 3 0
S~. ~
. 2 1 0 ~ - 6 6
TABL~ I (CONT. )
No. R R2 R~ R- R2
~ ~.
13 H - C~¢ OH~ CH3 ~ N~,NCH3
1 0 14 H - CH~ OH) CH3 13r ~N~NCH3
1 5 15 H - CH( OH) C~I3 I ~N~NCH3
1 6 H - CH( OH) CH3 I ~,N~NCH3
17 H - CH~ OH~ CH3 Cl ~NCH3
13 H -CHtOH~CH3 Cl ~ ;~H3
.. . .
, 1 9 H -C~(OH)CH3 CH2-N~NCH3 ~3
\~'O 9~ 66'~ PCI/~S93/0783() ~.
2~2~0~ ~
-- 6 7 -- . 1
' i
TABLE I (CONT. )
~.
No. R R2 R~H~- R2
.. . .. ~
2 O H - CE1( OH) C H3 - C Hz- N~NCH3 ~3
Z1 H -CH~ OH~CH3 -CH2-N~NCH3 ~--CHO
CHO
22 H -CH(OEl~CH3 -CH2-N~,NCH3
23 H -CH(OH)CH3 -CH!-N~,NCH3 ~HO
2 0 Z 4 H - Cl~ OH) CH3 - CHO ~NCH3
',
~ICH3
2 5 2 5 H - CE~( OH) CH3 - CN
2 6 H - CE~( OH) CH3 -ICINH2 ~CH3 `
1 I i O
: ~
1/05669 PCl /~'S93/Q7830
2~ 0S - 68 -
TABLE I (CONT. )
No. R R2 Rn H~- R2
27 H -CH~OH)CH3 ~HO ~ ,NCH3
28 H -CH~OH)CH3 -CN 1~NCH3
29 H -CH~OH)CH3 - IClNHz , 5~NCH3
H -CH~OH)CH3 11 --~--~N~lz
31 H - CH( OH) CH3 H ,~ ~,+N~
32 H -CH~OH)CH3 H l~N~SCH3
I , i , `: .
33 H -CH~OH)CH3 -SCH3 ~NCH3
w 0 94/05669 PC-r/US93/07830
2 ~ ~ ~ rl ~ ~3
.:
- 69 -
1 .:
TABLE I (CONT.)
. .
t; .
No. R R2 Ra H~- R2
- - _
34 H -CH(OH)CH3 -SCH3 ~\~
H -CH(OH)CH3 -SCH3 ~ilCH3
36 H -CH~OH)CH3 -SCH3 ~\~\NCH
~7 H -CH(OH)C~3 -SCH3 ~/=\ : :
20~ ,;
o :
38 H -CH(OH)CH3 -SCH3 ~,~ H ` ~
~:, .
~H2
39 H -CH~OH)CH3 H
' ! : " i: :
~ ~ 40 H -CH(OH)CH3 F ~, ~`.~? ' ;~;
`- :`~ ' : ;
,
. ~ .: `
~'O 9~ 669 PCI/~'S93/()7~0
:` ` `' l ~ .,`
21~2 10 8 70
~ ~
TAB?LE 1 ( CONT ~
No. R _ R Ra HAR- R ? ` ` "
41 H -CH~OH)CH3 Cl
NH2 . .
42 H --CH~OH)CH3 ~3r /~ ~?
43 H -CH~OH)CH3 I
S NHz . ~ I
44 H - CH( OH) C~13 - S CH3 Q ~ `
1~/ NH2
H -CH~OH)CH3 -S~CH3 ~
M~2
46 H -CH(OH~CH3 -ScH3
3 0 2
47 CH3 -C~(OH)CH3 ~ ~NCH3 : :~
`:
~'0 ~ Sfih9 rc~ s~3/n~830
```` 2~12~8 1
- 71
T BLE l (CONT.)
No. R ~? Ra H~R- R ~,
48 H -CH(FjCH3 H ~N~NCH3
,~\ ~H3 ;
lS 49 H - CH~ F) CH3 H S :
H -CH(F)CH3 H\~5~ ~/ 3
~ /~\ ' `
51 CH3 - CHC OH) CH3 H~ N~NCH3 ~ `
52 CH3 -CH(OH)CH3 H ~N~NCH3 3
j ~
~30 ~
,' '-' ,''~`,.
''' ,.'
:
~-'O 9~ .66~ PCr/~S93/07~30
4~1~ - 72 -
TA~3I.E I ~CONT.
No. R R2 ~a H~R-Ra
- _ 2
5 3 H - C H( OH) C H~ C N ,~N~N( C H2 ) 2 OH
,~ ( CH2) 20
54 H-CH(OH~CH3 CN S `
: , .,
1/ ~ ~'NCH2CN
H-CH{O.H)CH3 CN S \=/ ;
!
56 H-CH(OH)CH3 CN ~tJ
57 HCH~OH)CH3 CN ~5~` N~N--CONH
';",',
s~
~`
\~'O 9~ 669 PCr/~!S93/0783()
```` 2~ 7a~i !``~`
-- 7 3
TABLE I ( CON~2.
~ ``
!
;
No. R R~ R~ HAR- R~ .
58 H CH( OH)CH3 CN ~~li jN--~H
,.
59 H CH(OH)CH3 CN --~N~CONHz
~N~N--OH `
H CH( OH~CH3 CN --~S~
~ `~
61 H CH( OH) CH3 CN --~F ~ON~2
S ` ~ ;` '
62 H CH(OH)CH3 CN ~ ~ Z
2 S
-
.`,.
3 0
j.~ . . ~
' ~ ' '`"'~
66(~ PCr/~'Ss3/07s3n
r~ O I ~ 7 4
The carbapenem compounds of the present
invention are useful per se and in their
pharmaceutically acceptable salt and ester forms in . ~
the treatment of bacterial infections in animal and ~ ~`
human subjects. The term "pharmaceutically
acceptable ester or salt" refers to those salt and
ester forms of the compounds of the present invention
which would be apparent to the pharmaceutical ~`~
chemist, i.e~, those which are non-toxic and which ~
would favorably affect the pharmacokinetic properties ~ ;
of said compounds, their palatability, absorption, :~
distribution, metabolism and e~cretion. Other
factoræ, more practical in nature, which are also
important in the selection, are cost of the raw
materials, ease o~ crystallization, yield, stability, ~-
hygroscopicity, and flowability of the resulting bulk
drug. Conveniently, pharmaceutical compositions may ;:.
be prepared from the active ingredients in
combination with pharmaceutically acceptable
carriers. Thus, the present invention is also
concerned with pharmaceutical compositions and
methods of treating bacterial infections utilizing as ~;
an active ingredient the novel carbapenem compound~ ~-
of the present invention.
The pharmaceutically acceptable salts ~`
referred to above may take the form -COOM. The M may
be an alkali metal cation such as sodium or
potassium. Other pharmaceutically acceptable cation~
~orl M may be calcium, magnesium, ~inc, ammonium, or
alkylammonium catio~s such as tetramethylammonium,
3 tetrabutylammonium, choline, triethylhydroammonium,
meglumine, triethanolhydroammonium, etc.
:`,
~iO')~/056611 P(~/~S93/07830 ~i
`` 21~270~
- 7 5 -
... .
The pharmaceutically acceptable salts ~,
referred to abo~e may also include non-toxic acid
addition salts. Thus, the Formula I compounds can be
used in the form of salts ~erived ~rom inorganic or
organic acids. Inc~uded among such salts are the
S following: acetate, adipate, alginate, aspartate,
benzoate, benzenesulfonate, bisulfate, butyrate,
citrate, camphorate, camphorsulfonate,
cyclopentaIlepropionate, digluconate, dodecylsulfate,
ethanesulfonate, fumarate, glucoheptanoate,
glycerophosphate, hemisulfate, heptanoate, hexanoate,
hydrochloride, hydrobromide, hydroiodide,
2-hydroxyethanesulfonate, lactate, maleate,
methanesulfonate, 2-naphthalene-sulfonate,
nicotinate, oxalate, pamoate, pectinate, pcrsul~ate,
3-phenylpropionate, picrate, pivalate, propionate,
succinate, tartrate, thiocyanate, tosylate, and
undecanoate.
The pharmaceutical acceptable esters of the
novel carbapenem compounds of the present i~vention
20 are such as would be readily apparent to a medicinal :`
chemist, and include, for example, those describe~ in
detail in U.S. Pat. No. 4,309,438, Column 9, line 61
to Column 12, line 51, which is incorporated here:in
by reference~ Included within such pharmaceutically
acceptable esteræ are tho3e which are hydrolyzed
under physiological conditions, such as
pi~aloyloxymethyl, acetoxymethyl, phthalidyl, indanyl
and,methoxymejthy~, and those described in detail in I :~
U.S. Pat. No. 4,479,947, which is incorporated herein
by reference.
. - .
:
'
.,,
. . .~ .
E; ~`` ``
~v~ /0~66s PCT/~S93/07830 ~ ~
2 1 ~ 2 7 ~ ~
- 76 - I :
The novel carbapenem compoun~s of the ~`
present invention may take the form COOM, where M is
a readily removable carboxyl protecting group. Such ` :
conventional blocking groups consist of known ester ;;
groups which are used to protectively block the ;~
carboxyl group during the synthesis procedures
described above. These conventional blocking groups
are readily removable, i.e., they can be removed, if ~:
desired, by procedures which will not cause cleavage
or other disruption of the remaining portions of the
molecule. SUCh procedures include chemical and
enzymatic hydrolysis, treatment with chemical
reducing or oxidizing agents under mild conditions,
treatment with a transition metal catalyst and a
nucleophile, and catalytic hydrogenation. Broadly,
such ester protecting groups include alkyl,
substituted alkyl, benzyl, substituted benzyl, aryl, :~
substituted aryl, allyl, substituted allyl and
triorganosilyl. Examples of speciic such ester ~
protecting groups include benzhydryl, p-nitrobenzyl, ~:
2-naphthylmethyl, allyl, 2-chloroallyl, benzyl,
t-butyl, 2,2,2-trichloroethyl, t-butyldimethylsilyl, `
t-butyldiphenylsilyl, trimethylsilyl,
2-~tri.methyl)silylethyl, phenacyl, p-methoxybenzyl,
acetonyl, o-nitrobenzyl and 4-pyridylmethyl. `~
The compounds of the present inventîon are
valuable antibacterial agents active against various
Gram-positive and to a lesser extent Gram-negative
bacteria and accordingly find utility/in human and
veterinary medicine. The antibacterials o~ the
invention are not limited to utility as medicaments;
.,:
.,
': '
`
~V~} 9~ .fi69 PCT/~'S93/n7830 ~
" 2~27û~
- 77 -
they may be used in all manner of industry, for
example: additives to animal feed, preservation of
food, disinfectants, and in other industxial systems ,~
where cont.rol of bacterial growth is desired~ For
example, they may be employed in aqueous compositions
in concentrations ranging ~rom 0.1 to 100 parts of
antibiotic per million parts of solution in order to
destroy or inhibit the growth of harmful bacteria on
medical and dental equipment and as bactericides in
industrial applications, for example in waterbased
paints and in the white water of paper mills to
inhibit the growth of harmful bacteria.
The compounds of this invention may be used
in any o~ a variety of pharmaceutical preparations. -
They may be employed in capsule, powder form, in
liquid solution, or in suspension. They may be
administered by a variety of means; those of
principal interest include: topically or
parenterally by injection (intravenously or
intramuscularly).
Compositions for injection, a preferred
route of deli~ery, may be prepared in unit dosage ;`~
form in ampules, or in multidose containers. The
compositions may take such forms as suspensions,
solutions, or emulsions in oily or aqueous vehicles, ~`~
S and may contain ormulatory a,gents. Alternatively,
the active ingredient may be in powder form for ~ .
reconstitution, at the time of delivery, with a
3uiitable vehicle,~such as sterile water. Topical
applications may be formulated in hydrophobic or `~
hydrophilic bases as ointments, creams, lotions, i
paints, or powders~
' .
i
.,:
: : , .,
-, .
~09J/()566~ PCI/~593/(17830
`` ' `` .
. . .
~ l ~f~7 ~ 78 - ;~
The dosage to be administered depends to a ~ -
large extent upon the condition and size of the
subject being treated as welI as the route and , ~:
frequency of administration, the parenteral route by
injection being preferred for generalized
in~ectiors. Such matters, however, are left to the
routine discretion of the therapist according to
principles of treatment well known in the anti-
bacterial art. Another factor influencing the
precise dosage regimen, apart from the nature of the
infection and peculiar identity of the individual
being treated, is the molecular weight of the chosen
species of this invention.
The compositions for hu~nan delivery per unit
dosage, whether liquid or solid, may contain from
0.1% to 99% of active material, the preferred range
being from about 10-60%. The composition will ;
generally contain from about 15 mg to about 1500 mg
of the active ingredient; however, in general, it is
preferable to employ a dosage amount in the range of
2 from about 250 mg to 1000 mg. In parenteral ;~ administration, the unit dosage is usually the pure
compound I in sterile water solu~ion or in the form
of a soluble powder intended for solution. ~;;
The pre~erred method of administration of
the Formula I antibacterial compounds is parenteral
by i.~. infusion, i.v. bolus, or i.m. injection. t .
For adults, 5-50 mg of Formula I ,
an~ibacterial compounds per kg of bod~y weight given
2, 3, or 4 times per day is preferred. Preferred
3 dosage is 250 mg to ~000 mg of the Formula I ~; ;
antibacterial given two (b.i.d.) three ~t.i.d.) or
`~
~. .
\VO~ 66') rcTl~ss3/o7s3o ~ ~
2 ~ ~ 2 ~ ~ o
- 79 - `;
four (q.i.d.) times per day. More specifically, for
mild infections a dose of 250 mg t.i.d. or q.i.d. is ~ !
recommended. For moderate infections against highly ~;
susceptible gra~ positive organisms a dose of 500 mg
t.i.d. or q.i.d. is recommended. For severe,
life-threatening infections against organisms at the
upper limits of sensitivity to the antibiotic, a dose
of 1000 mg t.i.d. or q.i.d. is recommended.
For children, a dose of 5-25 mg/kg of body
weight given 2, 3, or 4 times per day is preferred; a
a dose of lO mg/kg t.i.d. or q.i.d. is usually
recommended.
Antibacterial compounds of Formula I are of
the broad class known as carbapenems or l-carbade-
thiapenems. Naturally occuring carbapenems are
susceptible to attack by a renal enzyme known as :~
dehydropeptidase (D~P). This attack or degradation `~
may reduce the efficacy o~ the carbapenem
antibacterial agent. The compounds of the present
in~ention, on the other hand, are significantly less
2 subject to such attack, and therefore may not require
the use of a DRP inhibitor. However, such use is
optional and contemplated to be part of the present
invention. Inhibitoss of D~P and their use with
carbapenem antibacterial agents are disclosed in the
~5 prior art ~see European Patent Applications No.
79102616.4 filed July 24, 1979 (Patent No. ~ 007 ' .
614); and No. 82107174.3, filed Au~,ust ~, 1982
(Publication No. 0 072 014)].
The compounds of the present in~cntion may,
30 whese DHP inhibition is desired or necessary, be ~ -
combined or used with the appropriate DHP inhibitor ~
. .. .
. ~ .
. .~
; .. '
, .
,,,
~-09~Jn;S6') pcT/~s~3/n7s
æ ~ 'i~) - 80 -
as described in the aforesaid patents and published
application. Thus, to the extent that the cited ~ , :
European patent applications 1.) define the procedure
for determining DHP susceptibility of the present
carbapenems and 2.) disclose suitable inhibitors,
combination compositions and methods of treatment,
they are incorporated herein by reference. A
preferred weight ratio of Formula I compound: D~P
inhibitor in the combination compositions is about
1:1. A preferred DHP inhibitor is 7-(L-2-amino-2- ::
carbo~yethylthio)-2-(2,2-dimethylcyclopropaneearbox-
amide)-2-heptenoic acid or a useful salt thereof.
The invention is further de~ined by ~-;
reference to the following examples, which are
intended to be illustrative and not limiting.
All temperatureæ are in degrees Celsius.
~ARI'ING MAT~RI~L SYNT~ESES
Br
~(OH)~ ~
~-BROMOF~ENXLBO~QNIC ACID:
N-Butyllithium (2.5M; 44 mL; 0.11 M~ was added
dropwise over 15 mins. to a vigorously stirred
soIution of m-dibromobenzene (25g; 0.106 M) in 500 mL ~:
30 of anhydrous ether at -78 under nitrogen. After ~ ~
~-'.
'~,',
'.': `'
~94/(~669 ` PCT/~S93/0783
2 i Ll ~ l O
- 81 -
,~ I
stirring 10 mins. more, a solution of
triisopropylborate (25.3 mL; O.llM) in anhydrous ~ I
ether (200 mL) was added over 20 mins. The coolin~ ~
bath was then removed, and the stirring solution was i `;
allowed to warm to R.T. over ~2 hrs. A small amount
of solid separated. After stirring 15 mins. more at
R.T., 150 mL o~ ice cold 8% aqueous hydrochloric acid
was cautiously added, and the stirring was continued
~or 15 mins. The organic phase was separated, washed
with 2 x 100 mL of saturated sodium chloride
solution, and dried over anhydrous magnesium
sulfate. Solvent removal gave ~~OG of crude product
as a semi-solid, which was shaken well with 150 mL of `~
hexane. The solid was filtered and washed with 2 x
25 m~ of hexane. The resulting silky solid (mp
178-9C after softening at ~160C) (6.5 g) was used
as 3-bromophenylboronic acid with a small amount of
contamination. The hexa~e filtrate was concentrated ~;~
and the residue was stirred well with 150 mL of ;~
petroleum ether (30-60). The resulting solid was
~iltered and washed with 2 x 25 mL of petroleum
ether. This resulting solid (4.4 g) melting at
178.3-179C was the desired 3-bromophenylboronic acid.
NMR: 7.38-7.46; 7.70-7.80; 8.1-8.18; 8.31 (aromatic
~I ' s ) ..
''.
, -
, .
. ~ , I , ~. ...
`~ --
~ ~,...
i. -. .
`~ .,
: .
........
"
~ ~,
\~0 '~ fi69 PCI`/I_'S93/07830 ~`
I f``
21~270~') 82~
!
.
Br ~ I
s (\~
2-(3 1-BRQ~QP~E~YL)THIOP~EN~
To a stirred solution of m-bromoaniline (34.4 g;
0.2M) in thiophene (200 mL) was added isoamylnitrite
(46.86 g; 0.4 M) dropwise over a period of 30 mins. at ~;
0C. The resulting mi~ture was cautiously warmed to
R.T. and heated to reflux for 16 hours. The reaction `
mixture was cooled, diluted with 400 mL of ether,
washed with 3 x 100 mL of satd. sodium chloride ; `
soluti~n, and dried over anhydrous magne~ium sulfate.
Sol~ent and e~cess thiophene were~removed. A solution
of the residue in 200 mL of ether was filtered through
50 G silica gel bed. Solvent was removed, and the ~
residue was distilled to give 34% of 2-(3' bromo- :
phenyl) thiophene as a yellow liquid boiling at ~:
130-2/~0.2 mm. This liquid solidified on standing
in the refrigerator.
MMR: 7.06-7.78
.~
.
2~ 4'-BRO~OP~ENYL)THIOPHENE:
Similarly, 2-(4'bromophenyl)thiophene was
prepared ~rom 4-bromo aniline in 29% yield as a
:
W09~/0~669 P~ S93/07830
-~ 2lA2~0~i
- 83 -
yel.Low oil boiling at 146-8/-0.5 mm.
NMR: (C6D6): 6.68-6.92(thiophene H's~ 7.03-7.20 ~ .
(p-phe~yl H's).
2-P~ENYLTEIQ~HENE:
The above method was used to prepare
2-phenylthiophene from aniline in 11% yield as
colorless liquid boiling at 110~113/~3 mm. . :-
''''. ~
Br ;-;
S ~.
,,.
- :,
~3'-B~OMOP~ENYL)T~IOPHE~E~
2 The thiophene was prepared according to G.
Martelli Q~ . Chem. Soc (B)., 901, (1968).
, .j
, ~
~ ;30
~VOs~ 66s ` PcT/~ss3/o783o
- 84 ~
, ~ ;
E~r ~`
Br
2-(3'~5'-DIBROMO~EENYL~T~I~PHENE:
3,5-dibromoaniline was treated as described
in the procedure for 2-(3~-bromophenyl)thiophene to
give 2-~3',5'-dibromophenyl)thiophene in 55% yield as
a yellow oil, which solidified as a glassy solid.
MMR: 7.04-7.68 (aromatic ~'s) ;;
E3r
C HO
2-FQRM~L-5-(3'-BROMOP~ENYL)TEIOPHENE:
Phosphorus oxychloride (1.15 mL; 15.4 mM)
was added slowly to stirring dimethylformamide (0.95
mL; 12.2 mM) at -10 under nitrogen. The resulting
mixture was stirred for 1~ mlns. ~ romOrhe~ly}~
thiophene (2.12 g; 9 mM) was then added. The reaction
mixture was then warmed slowIy to 110 over a period ~;
o~ 1 hr. cooled and poured into ice, and cautiously
neutralized with sodium carbonate. Extraction~with j :
~` ethyl acetate and drying the or~ganic phase with
anhydrous magnesium sulfate provided upon
conccntration 2.16 g of the desired aldehyde as an
oily solid.
NMR: 7.26-7.84 ~aromatic ~s~; 9.92(-C(O)E; S)
,
~ , .
.
' ~ ,:
\V09~/0~669 PCT/~S93/07830
214~
,
`; . , .
- 85 -
i ~;
Br ; . :
S
2-(HYDROXYMETHYL)-~-3l-BROMOPHENYL)THIGPHENE:
Sodium borohydride (400 mg; 10 mM) was added
portionwise over 5 min. to a stirred suspension of :~
the above crude aldeh~de (2.16 gj in 100 mL of ,~
methanol at 0C. The resulting clear solution was
stirred 30 mins. Solvent was then removed in ~S~Q
at R.T. The residue was taken up in 50 mL of ethyl
acetate, washed with 3 x 20 mL of sat'd. sodium
1 ~;
:~ chloride solution, and dried over anhyd. magnesium
~: sul~te. Solvent removal followed b~ silica gel
chromatography with methylene chloride gave 1.355 g .;
of desired alcohol as an amorphous solid.
~: NMR: 1.83 (OH; t; J-6~3); 4.82~CH2; d; J-6~3);
6.96-7.75 (aromatic H's)
/ ~ \ ~ OSi
: 2-(t~ TYLDIMET~YLSXLYLO~M~T~YL)-5-(3~-BROMOP~NYL)-
IOPHENE i~
~30 ~ To a stirred ~olution~of 2-~hydroxymethyl)-5~
: (3'-bromophenyl)thiophene (1.08 G; 4 mM) and :~ t
t~-~tb ~ U i~ ~ ml ~ t~ e
~O~ 669 l~CT/~93/078~0
2 1 ~ 2 7 0 ~ - 86 - 1~
chloride at R.T. was added t-butyldimethylchloro-
silane (1.5 g; 10 mM). This mixture was stirred ~ ~
overnight, diluted with 30 ml of ethyl acetate, '~ :
washed with 2 x 15 ml of sat'd. sodium chloride ~.
solution, and dried over anhyd. magnesium sulfate. ~:
Solvent was removed to give a residue, which was
puri~ied on silica gel with ether:petroleum ether
(1:20) as solvent mixture. Eluate was distilled to :~
give 0.9~ g of 2-(t-butyldimethylsilylo~ymethyl)-5-
(3'bromophenyl~thiophene as colorless liquid boiling
at 167-170/~0.2 mm.
~R: 0.17 & O 95 (silyl methyls); 4.88 (s, C~2);
6.88-7.75 (aromatic ~1s)
Br
~S
2-r3'-~ROMO-5'-M~TH~LT~IO~P~ENYLlT~IOP~ENE:
2,5M n-Butyllithium (1.5 mL; 3.75 mM) was
added dropwise to a solution of 2-(3',5'-dibromo-
pheny~)thiophene (1.06 g; 3.33 mM) in anhydrous
tetrahydrofuran (7 mL) at -78 under nitrogen. The
reaction mixture was then stirred 10 min. a~d a
solution of dimethyldisulfide(0.9 mL; 10 mM) in 3 mL
of;anhydrous tetr:ahydrofuran was added. ~he
re~ulting mixture was stirred overnight at R.T. after
which 5 mL o~ sat'd. ammonium chloride and 20 mL of
'.
'.
,
.. .........................
wo s4/n~6~s Pcr/uss3/07~30 ~ ,
21~270~ , i
- 87 -
ethyl acetate were added. The organic phase was
separated, washed with 2 x 10 mL of sat'd. sodiu~
ch~or.ide solution, and dried over anhyd. magnesium ~.
sulfate. Solvent removal, and purification on silica t ;~
gel using hexane as solvent gave a liquid, which was
distilled to give 52r/o of ~-~(3 '-bromo)-(5'-methyl-
thio)]phenylthiophene as a colorless oil boiling at
~150-152/~ 0~2 mm. (oil bath temp. 180)
NMR 1~52(SCH3; s); 7~00~7~50 (aromatic ~'s)
,. . .
Br ;
Br
2-BROMO-5- r 3'-BROMOPHENYLlTHIOPHENE:
A solution of bromine (8 g; 50 mM) in ~0 mL
o~ glacial acetic acid was added dropwise to a
vigorously stirred solution of 2-(3'-bromophenyl)-
thiophene (12 g; 50 mM) in 80 mL of glacial acetic
acid. The resulting mixture was heated to reflu~ 5 1~
hrs, cooled and poured onto ice. A solid separated ~`
which was filtered and washed with ice water, and
puri~ied on silica gel using heæane as solvent to
give 68~/o of 2-bromo-5-r3'-bromophenyl]thiophene as an ~ ;
amorphous solid.
!' I ' N~R 7.00-7.68;(aromatic H's) ; I I .
, ,
~ ~
s . ..
. .
: ~ ,
WO 94/05669 PCr/US93/07830
1 `"
21~'.1 0'~
-- 88 --
.
3 - ( 3 1 -BROMOP~ENYL ) - 5 -BROMOTHI OPHENE:
Br Br
S \~
~r
To a,solution of 3-(3'-bromophenyl)thiophene,
(G. Martelli e~ al., J. Chem. Soc., (B), 901, 1968)
(712 mg, 3 mmol) in acetic acid (6.2 ml) with '
stirring under N2, a solution of Br2 (154 ~1, 3 mmol) '
in acetic acid (4.8 ml) was added dropwise. The
15 resultant orange-red solution was heated for 5 hours `,
at 100C. After cooling, the reaction mi~ture was ' ~:
poured into ice water with stirring. A non-
~ilterable milky precipitate was extracted 2X with
Et20. The combined Et20 layers were carefully
extracted 3X with Na~C03 solution a~d then 2X with
brine. Ater drying (MgS04), ~iltering and
concentrating, the residue was chromatographed on a ,'
column of Bakers Si Gel (60-200 mesh) packed, applied
and eluted with hexane. Those fractions containing
the slightly less polar product were combined and
concentrated in ~cuo ~763 mg). Preparative TLC of
663 mg of this material on 7-1000 ~ Si Gel GF plates
(eluting with hexane and extracting with CE~C12)
~'' provided a purer sample o~ the desired 5~bromo isomer
(416 mg~ (i.e., les~ of the uniesired Z-bromo iso=er
.
W094~05669 PCT~US93/07830
2 ~ ~ i2 7 ~
- 89 -
,. : :
was present). Approximately 162 mg of this material
was further purified by preparative TLC on 4-1000~ ~ ¦
Si Gel GF (elutin~ and extracting as above) to give
3-(3'-bromophenyl)-5-bromothiophene pure enough for t
further reaction (196 mg).
MS: m/z 316/318/320 (MI).
lH NMR (300 MHz, CDC13)): ~ 7.01 (d, J-6~z, ~4 of `~.
2-Br compound~; 7.23-7.70 ~series of m's, phenyl and
thiophene protons of the minor 2-Br and the desired
5-Br isomers). ...
, ;,
13r ~3r \ E3r 1 .
~ ~ ~3 ,:
, .
,.: ....
3-(3'-BROMOP~ENYL2-5-T~IOPHENE CARBOXALDE~XD~:. .~`
;l ,.
To a solution of the brominated thiophene
~:` (mainly the correct 5-bromo isomer; 196 mg, 0.62 - .` ~:
mmol) in ether (2.7 ml) at -78C under N2, 1.6M BuLi .`
in hexane (388 ~1, 0.62 mmol) was added dropwise.
~5 A~ter 15 mln. at -78, DME (63 ~1, 0.81 mmol) was
:~: added, and the reaction mixture was stirred overnight ~ j"'!".,
at ambient tempera~ur.e. The reaction was partitioned i .
between EA and brine. After phase separation, the
:~l orgànie layer was agai~ extracted with brine, dried, ~-
~iltered and conce~trated to provide crude formylated
.-
~ ;: ~ .'
w09~/0~669 PCT/US93/07830 ~'
~.~
J 1 O g
~ 90 ~ .
.
product ~151 mg). Preparative TLC on 3-lOOO ~ Si Gel
GF plates (eluting with 20% Et20/he~ane and
e~tracting with CH2C12) provided a major band
containing a mixture (80 mg) of the desired 5-formyl
isomer contaminated with a small amount of the
~-formyl isomer.
MS: m/z 266/268 (MI.)
IR(CH2Cl2): 1670 (formyl) cm~l
~H NMR (300 MHz, CDC13): ~ 7.22 (low amplitude d,
J=S~z, H4 o~ minor amount of 2-formyl); 7.29-8.00
(series of m's phenyl & thiophene protons); 9.88 (d,
J=lHz, long range splitting minor amount of
2-formyl); 9.98 (d, J=l~z, C~O of S-formyl, allylic
splitting).
Br :
~ \ 3 4
~
3- r (3'-BRO~OPEENYL)- ~-HYpROXYMETKYL)lT~IOPHENE:
To a solution of the formylated thiophene
(79 mg, 0.3 mmol) stirred in MeOH ~3 ml) at 0, NaBH4 s
(13.6 mg, 0.34 mmol) was added, and stirring was ~ r
continued for 40 min. Upon concentration to an oil
30 under a N2 stream, the residue was partitioned .
W09~/0~669 PCT/~IS93/07830
~,. .
2 i~2~0~ ~ `
- 91 -
between EA and brine, the organic lay~r was again
~ashed with brine, dried, filtered and concentrated
in vacuo to give the crude product ~77 mg).
Preparative TLC on 2-lOOO ~ Si Gel GF plates (eluting
with C~2C12 and extracting the major W band with 10%
MeOH/CH2Cl2) provided the purified 5-hydroxymethyl ~:
compound (70 mg, 88% yield). A small amount of faster
running material (5 mg, 6% yield) proved to be the
undesired 2-hydroxymethylthiophene compound havi.ng
the widely split H4-doublet (J=4.5 Hz). The desired
1 product contained none of this impurity.
MS: m/z 268/250(MI) ;
~(C~C12) 3600(0H) cm~l
lH NMR (300M~z, C~C13): i~ 1.84 (t, J=6Hz, OE); `;
4.85(dd, J=O.S (allylic coupling to ~4) and 6Hz; ;~
C~20~); 7.24, 7.42, 750 and 7.70 (4 sets of m1s;
phenyl and thiophene H's).
NMR data for the less polar 2-hydroxymethylthiophene l~
(5 mg above): i;
1~ ~MR (300 mHz, CDCl3): ~ 1.81 (t, J=6 Hz, OH); 4.82
(d, J=6Hz, C~20H); 4.86 (br d, J=6 Hz, C~20H of small l`
amount of 5-isomer); 7.08 (d, J=5 Hz, ~4); 7.25~7.71 :`
(series of multiplets, phenyl and thiophene protons).
,,.
t ., .
3~ ; .
? ~
~, . .
~ '
:,
, = .
,,
~vos4/0~66~ PCT/US93tO7830
21 4
Br
~ ..
S H2 QS i ~
3-(3'-~ROMOPHENYL)-5~ BUTYLDIMET~YLSILYLOXYMET~YL)
THIOPHENE: _
, ~
To a solution of the 5~hydroxymethyl
thiophene (62 mg, 0.23 mmo~) in C~2C12 (1.1 ml) at `
0C with stirring under N2, TB~SiCl (183 mg, 0.55
mmol) and Et3N (80 ~1, 0.59 mmol) were added. The
cooling bath was remove~, and the reaction mixture
was stirred overnight at ambient temperature.
Wor~-up of an aliquot showed incomplete conversion.
Therefore, D~F (20 ~1) in CH2C12 (l ml) was added,
and stirring was resumed for a few hours*. ~rine
containing 1 M K2EP04 (1 ml) and additional CH2Cl2
wexe added to the reaction mixture with stirring.
Af~er phase separation, the aquPous layer was again
extracted with C~2Cl2, and the combined organic
layers were washed with brine, dried, filtered and
concentra~ed ~a vacuo to give the crude product (97
mg). Preparative TLC on 2-1000 ~ Si Gel GF plates
(eluting with 20% Et20/hexane and extracting with . .;
C~2C12) provided the purified 5-si~yloxymethyl , ,~
thiophene (79 mg, 90Z yield).
*In later runs, the DMF was introduced initially
~i.e., starting material (~20 mg~; C~2C12 (10.5 ml);
' .'"'~
: : ~ ' `',
;
` ~.,
W094/05669 PCT/US93/07830
; ~ ~, . 1'
- 93 _ 21~?,7~
TBDMSiCl (820 mg); Et3N (788 ~1); DMF (830 ~1)] and
o~ernight reaction provided complete silylation.
MS: m/z 325/327 (MI - t-butyl); 251/253 (MI - ¦ ~
OTBDMSi). ' ~:
H NMR ~300M~z, CDC13): ~ 0.13 (s, Si(C~3~); 0.94 ;
(s, t-butyl-Si); 4.8g (s, CH2OTBDMSi); 7.16-7.70
(series of m's, phenyl and thienyl ~'s). .-
Br
~3
2-(3~-BROMOPHENYL)FURAN:
BP: 98-105/0.1 mm . ~;~
Reference: E. L. Plummer, J. Agric. Food Chem.,
~1. 718-721 (1983).
. .
~ o+i/
2~ BUTYL~lMET~YLSILYL~XYMETHYL)FURAN: ::
To a stirring mi~ture of 2-furan methanol
(33 g, ~0.335M) and triethylamine (47 mL, ~0.335 M)
in anhydrous methylene chloride (200 mL) under l `;
nitrogen was;added t-butyldimethylchlorosilane
portionwise at room temperature. 20 mL of :.
t
.. . .
`.:
. :,
w094/05669 PCT/~S93/07830 i~i~
2l~ ~ ~
~ 94 -
N,N-dimethylformamide was added. The resulting
mixt~re was stirred 3 hrs. After dilution with 400
mL of ekher, the reaction mi.xture was washed with 3 x . ~
100 mL of ice-water, 100 mL of saturated sodium -:
chloride solution, and dried o~er anhydxous magnesium
S sulfate. Solvent removal gave a crude product, which
was distilled to afford 39.8 g of the desired silyl
ether as a colorless liquid boiling at 76-7/~0.5 mm.
Br
~ o+i~
2-(3'-BROMOPHENYL)-5-(~-BUTY~__METHYLSILYLOXYMET~YL)-
1 F~
To a stirred solution of m-bromoaniline
~6.88 g; 0.04 M) in 42.2 g (0.2M) of 2-(t-butyl-
dimethylsilyloxymethyl)furan at 0 was added
isoamylnitrite (10.75 mL: 0.08M) dropwise over a
period of 0.5 hr. The resulting mixture was then
heated 16 hours at 50C. The reaction mixture was - :
cooled and diluted with 150 mL of ether and washed
with 2 x 100 mL of ice cold water. The organic phase ;
was dried over anhydrous magnesium sulfate, and the
residue was distilled after filtering through 50G of
silica gel bed, to give 26% of the desired
2-(3'-bromophenyl)-5-(t-butyldimethylsilyloxymethyl)-
! I ~furan as a colorless liquid boiling at 163-7/~0.5 mm.
'' ~
'`.
' :'
W~9~JOSfi69 PCT/~S93~07830 k-~
. ;,. . ,, ' .
_ 95 _ 2i~2'7~ ~
STEP Al: GENERAL SYNT~ESI~__F ARYLKETONES:
, `
O O ., .:'.
~CO ~ ~CO R
// ~ H H I ~ Ar-~r ~ '
~ N ~ M~ ~ N
O ~ Ph3 0/ ~ Ph3
Icoo = Icoo = ''i,
,~
MET~OD l:
. Aryl bromide (l mM) was added to a stirred ~:
suspension of magnesium chips (1.25 mM) in 2mL of
anhydrous tetrahydrofuran under nitrogen at R.T. 8 ~L
f 1,2-dibromoethane was then added. The resulting
: mixture was stirred 3 hours, when most of the metal
was digested. The resulting dark yellow solution was :
used as 0.5 M solution of the aryl Grignard reagent. l'~
This Grignard reagent solution was added
20 dropwise to a stirred solution of (3S, 4R)~
~(allyloxy)carbonyl~(triphenylphosphoranylidene~-
methyl}-3~ (allyloxy)carbonyloxy]ethyl]-4-
~2'-pyrid~lthio)carbonyl]methyl]azetidin-2-one,
(~0.5 mM) in 2 mL of anhydrous tetrahydrofuran at 0 : ~;
25 under nitrogen. The reaction mixture was stirred 15
mins at 0. Satd ammonium chloride solution (5mL? and
10 mL of ethyl acetate were added~ The organic layer :
was separated, and washed with 2 x 5 mL of satd;
sodium chl orî~de ;solution and dried over anhyd
30 magnesium sulfate. Solvent removal followed by ~.
silica gel chromatography using mixtures (1:1 to 2~ c
of ethyl acetate:hexane as eluant ga~e the desired
ylid arylketone as a pale yellow fQam.
.
, ~
, .
~.
. .
. - . .
:,
....
WO g4/056~9 ~Cr/US93/07830 ~
2l~C~rln~ ' ; ','
-- 96 --
~r~-oD 2:
Ar - Br n- BULi r ~ E~r 2 [
ETHER THF
O O
[ ~r~ ~o
o / `FPPh 3 o / `FPPh3
1 0 Icoo = CQO
1 5
To a stirred solution of 3 mM aryl bromide in
anhydrous ether (12 mL) at -78 under nitrogen was
added n-butyllithium (2.5 molar solution; 1.32 mL;
3.3 mM) dropwise. The resulting mixture was stirred
O.S hr. A solution of magnesium bromide, freshly
prepared by stirring 6.6 mM of magnesium ~urnings in
~4 mL of anhydrous tetrahydrofuran with 6 mM of 1,2-
dibro~oeth~ne for about 1 hr under nitrogen at ambient :~-
temperature, was then added dropwise to the above
stirring lithium salt at -78. The resulting mixture `
was stirred 15 mins at -78, and 30 mins at 0. The
thus obtained turbid solution was used as a 0.0833
molar~solution of ~he required aryl magnesium bromide.
30`
', ;'
, :'
1;
w094/0~669 PCT/US93/07830
2 ~ 7 ~
- 97 -
:.
This solution of the Grignard reagent was ~ ;
added slowly to a stirred solution of 1.4 mM of (3S,
4R)-l-[[allyloxy)carbonyl](triphenylphosphoranyli-
dene)methyl]-3-~(lR)-l-~(allyloxy)carbonyloxy]ethyl]-
2-~[~21-pyridylthio)carbonyl]methyl]azetidin-2-one,
in 5 mL of anhydrous tetrahydrofuran at 0 under
nitrogen. The reaction mixture was stirred l5 mins.
at 0, and satd. ammonium chloride (l5 mL) and 30 mL -`
of ethyl acetate were added. The organic layer was
se.parated, washed with 2 x 15 mL of sat'd. sodium `~
chloride solution, and dried over anhydrous magnesium .`~
sulfate. Solvent removal and purification on silica ;`
gel using a (l:l to 2:l) mixture of ethyl
acetate:hexane gave the desired aryl ketone, as a
light yellow foam. `~`
.~ .
STEP A~: DESILYLATION OF T~E YLIDE SILYLET~ER KETONE
TO YLI~ ALCO~OL KF.TON~:
A solution of the arylketone (from Step Al)
(~20 mg) was dissolved in 3.5 mL of an ice-cold .
mixture of 2% H~S04 in methanol. After stirring the
mixture 75 mins at 0, it was diluted with 5 mL of
ethyl acetate and washed with 3x 5 mL of 10% sodium
bicarbonate solution followed by 5 mL of saturated
sodium chloride solution and dried over anhyd.
magnesium sulfate. Sol~ent removal afforded l63 mg ~;
of almost pure alcohol as white foam. ~`
"~
~ ~
~.~ ,. .
,.
WO 94/1~56~9 P~/US93/~)7830
2~ 98-
';
STE.P Bl: GENERAL PROCEDURE FQR CYCLIZATION :
O O
Il 11
rOCO R r) -
S /~
CO~
A solution of the ylid ketone (0.~5 mM) in
2 mL of p-xylene containing a tiny crystal of
hydroquinone was heated 45 mins. to 3 hours
(depending on the nature of R) at 130C under
nitrogen. The solution was cooled, applied in a
suitable solvent to a silica gel column packed with
he~ane and then eluted first with hexane and then
with 4:1 to 2:3 mixtures of hexane:ethyl acetate to
give the desired carbapenem analogs.
STEP B2: GENERAL PROCEDURE FOR QUATERNIZATION:
,1','
To a solution of 120 mg (0,2 mm~ of the
carbapenem carbinol from Step B2 in 3 mL of methylene
chloride at 0 under nitrogen were added 0.5 mm of
amine, and O.25 ~m o~ trifluoromethane sulfonic
anhydride. After stirring 15 mins at 0, the
reaction mixture was diluted with 10 ~L of methylene
ch~oride and washed with 5 mL of ice water. The
organic phase was dried over anhydrous magnesium
sulfate. Solvent remo~al afforded 118 mg of the
desired quaternary salt as yellow foOEm. ~ :
: ~ .
W094/05~69 PCT/~JS93/07830
~: 2 ~ ~ i~ 7 0 ~ `
- 99
STEP C: GE.NE,RAL PROCED~RE FOR DEALLYLATION:
O
rOCO HO :
/~
o~ O / ~
COO - COOM x
~0 ;~'`'`
To a stirred solution of the carbapenem (0.2 ~`
mM) in 3 mL o~ a 1:1 mixture of methylene chloride:
ether in a centrifuge tube at 0~ under nitrogen were `
added 2-ethylhexanoic acid (0.2 mM~, triphenyl-
phosphine (0.05 m~), tetra~is-(triphenylphosphine)-
palladium (0.04 mM), and O.2 mM of sodium or potassium ;~
2-ethylhexanoate. This mixture was stirred 2 hrs when
a solid precipitated out. After diluting with 10 mL
of ether, the mixture was centrifuged and the super-
natant liguid was decanted. The remaining solid was
stirred with 2 mL of ethyl acetate and centrifuged.
The resulting solid was dissolved in 1 mL, of water and
applied to a 1000 ~ reverse phase silica gel plate.
Elution with mixtures of acetonitrile:water or EtO~:
water gave an ultra~iolet active area, which was
scraped and stirred with 5 mL of 4:1
acetonitrile:water mixture. The solid was filtered ~ ;
andlwashed with 3~ 2 mL of a 4:1 acetonitrile:w~ter
mixture. The f iltrate was washed with 4x 10 mL of ~ ;~
hexane, concentrated to 1 mL in v~cuo at R.T. and
lyophiliæed to give the sodium or potassi~m salt of
the carbapenem as a white to cream~, fluf~y mass.
~.,
.
wo94~ 6~s PCT/~S93/0,830 ~;~
;. . ~.:
?,~411~ - lol~ -
In the following examples: -
the IR data are in cm~l;
the UV data are in nanometers for ~.a~
water; and `
the NMR spectra are measured in CDCl3
solvent unless otherwise specified.
EXAMPLEI , '
STEP A
STEP AI: PREP~RATION OF YLIDE KETONE
STEP A2: DESILYLATION OF SILYL ET~E~ ;
':
STEP Al: I
: .
sr
~r-~r=
~Si~
` i:`
Conditions: l) MgjT~F; 3 hr./R.T. ~:
2) 0; 15 min; THF; pyridylthioester
~ield: 67% .
.STE~ A2
Conditions: C~30H/H2S04; 0; 1.25 hrs i~
Yield: 81% ~:
' I ~
S~EP B . ~ :
STEP ~1: CYCLIZATION OF YLIDE KETONE TO CARBAPENEM
STEP ~2: QUATERNIZATION OF CARBINOL
,
W094/~5669 PCT/US93/07~30 ~
.'`,: , i
- 101- ?,~
Conditions ~ i :
Bl: Xylene; 130; 1.5 hrs.
Yield: 83~L . ~;
~2:
~N\>
~ NC H3; :
triflic anhydride; CH2C12; 0; 15 min. -
Spectra:
IR: 1775; 1745; 1720
NMR: ~6. 3.42-3.50; dd, J = 3 ~ 8 Hz
H5: 4.24-4.36; ddd; J - 3, 9 & 10 Hz `~
NC~3: 3.96 (s); M+CH2: 5.62 (s);
N+-CHN: 9.44 (s)
s~æ_~: DEALLYLATION
Conditions: ~Ph3; Pd(PPh3)4
\ ~ ,
CO2K ~,~
.
CO2H ~ i
~.:
C~2C12; 0; 4 hrs
Yield: 67% . ~`
, .
Wo~4/os66s PCT/US91/07830
`21~270~
- lQ2 -
Spectr~:
W : 2~2
E ext 5074 `
EX~MPLE 3
H ~ -CH~
co- Br
i.
~RTING.nATERI~h SYNT~ESIS
Br
9 ~, :H~osi t ~ ~
Br :~:
25 2-~t-~UTYLDIiMETHYLSILYLOXYMET~YL)-5-(3',5'-DIBROMO- ~;~
E~E~, ; ' `'
Conditions: l) POCl3, DiMF; 0C; l0 min
3~ 2) 2-(3',5'-dibromophenyl)thiophene; :
ll0-120; l.5 hours
~ ' ' ~: '' ;';
,'`,.
.
~ .
w094/05669 PCT/VS93/0783~ ~ `
;~` 2~ 7~
`:
. - 103
Yield: 40% -
Spectra:
IR: 1670(CHO) cm~~
1~ NMR (300 M~z, CDC13); ~ 7.4-7.76 ~phenyl & ::
thiophene H~s); 9.92 (C~O). -
R~DUCTIQN
Conditions: 1) NaBH~/MeO~ ~-
2) 0; 1 hour
o Yield: 99%
Spectra:
MS m/z: 346, 348, 350 (MI). `:
1~ NMR (300 M~z, CDC13): ~ 1.82 (t, 0~); 4.84 (d, ~:
C~20~); 6.88 & 7.18 ~thiophene protons); 7.55-7.64
(phenyl protons)
~ .
~IkyLATIQN
Conditions: 1) TBDMSiCl; Et3N; DME/CH2C12 ;
2) R.T.; few hours; 10; o.n. :~
Yield: 95~L :~
Spectxa:
MS m/z: 460, 462, 464 (MI); 403, 405, 407 (MI-t-butyl) ;;.
1~ NMR (300 M~z, CDC13); ~ 0.12 (s, Si(C~3)2); 0.94 ~:
(s, t-butyl); 4.86 (s, CX20); 6.87-7.62 (phenyl & j-`.
thi.ophene protons)
~A~BINOL PREPARATIO~ ~
. i i . ~.
3 ~~ 02 ~ H
. WO 94~05669 PCr/~JS93/07i~30
.`, . ~:
I
2 1 llt~
. - 104 -
STEP Al: PREPARATION OF YLIDE KETONE
STEP A2: DESILYLATION OF SI~YL ETEER TO CARBINOL
Br
Ar-Br = ~ Si' ~
~r :
Conditions:
Al: 1) Mg/THF; 3 hrs.; R.T.
2) 0; 15 min; pyridylthioester
Yield: 23% of ylide ketone
Spectra;
IR: 1740; 1690; 1645; 1620
A2: CH30~; H2S4
Yield: 72% of carbinol
Spectra: :~
IR: 3100(0~); 1740; 1685; 1620
~0 . . .:
. .,`~;
Conditions: Xylene; 130; 3 hrs.
~ield 81%
Spectra:
IR: 3500(0~); 1740; 1720 :~
NMR: 0~ 6-2.06; t; J = 6
C~20: ~.80-4.86; d; J = 6 ~æ
~6: 3.40-3.49; dd; J =~3 ~ 8 ~z
5: 4.24-4.38; ddd; J = 3, 8.5 & 9.5 ~z
~; ~
:: . '' '.
, ~
w~94/osc69 P~T/US93/07830 ;~
~ 7 ~3
-- 105 --
Q~ATERNIZATION OF CARBINOL
C~nditions: 1) 2.5 eq. l-methylimidazole/C~2C12
2) 1.1 eq tri1ic anhydride
3) 0~; 45 min.
Yield: 85% ::
Spectra:
lH NMR (300 MHz, CDCl3): ~ 1.48 (d~ CH3); 3.49 (dd,
E6); 3.96 (s, N-CH3); 5.58 (thiophene-C~-); 9.26 (s~ ;:
N=CH-N).
DEALLYLATION
Conditions: PPH3; Pd(Ph3)
CO2K CO2H i"'~:'
CH2C12/EtOAc; 2 hours :-:
Yield: 31%
Spectra: -
W(H20): ~max = 295 m~
1~ NMR (300 MEz, D20):(no inte~nal standard - DOH at :::4.80); ~ 1. 24 ~d, C~3C~OE-); 3.37 (dd, ~6); 3.80 (s,
N-C~3); 5.50 (s, thiophene-CH2-); 8.78 (s, N=CH-N).
t
EXAMPLE 4
~,.
3 HO ~ ~ ~ N ~ N-CH3 ~:
O - N
~2 ~
.
~.~
W09~1JnS669 PCr/US93/07830 ~
21~ ~0~
-- 106 --
5~RIAL SYNTHESI S
s ~ -H2 I t
~
2-(t-BUT~LDIMET~IYLSIL~LOX~MET~YL)-5-(3 ', S ' -DIIODO-
10 P~EN~L ) THI OPHENE _ _
I
~ NO~
':
I
13.8 g (0.2M) of sodium nitr;te was added
p~rtionwise slowly to 96 mL of conc. sulfuric acid at
0C. The resulting thick mixture was stirred at 0C
~or 10 minutes. A suspension of 68.25 g (0.175M) of
finely powdered 4-nitro-2)6-diiodoaniline in 175 mL
of glacial acetic acid was added cautiously
portionwise to the above mixture at 0C. After the i
addition, the mi~ture was stirred 30 mins. The~
~esulti~g slurry was then added slowly to a
~igorously stirred suspension of 4 g of cuprous oxide ~:
in 420 mL of absolute ethanol over a pèriod of 25
mins. Vigorous effe:vescence was observed~during ~ 3
'''` ,';
,'''
'.
w094/056~9 PCT/US93/07~3U
2 ~ a ~
- 107 -
this addition. The resulting mixture was stirred 20 ~ :~
mins. at room temperature and then heated to reflux
for 30 minutes. After cooling, this reaction mixture
was poured onto a large amount of ice. The solid
S which separated was filtered, and washed with water.
This solid was dissolved in a minimum amount of .
chloroform, dried over anhyd. magnesium sulfate, and
the solvent was removed to give 62 g of
3~5-diiodnitrobenzene as a yellow solid.
,~
~ ~ 2 ~
I
3.5-DIIO~OANILINE:
A mixture of 61.5 g (Q.164M~ of
3,5-diiodonitrobenzene and 111.1 g (0.4924M) of
stannous chloride in 900 mL of ethanol was heated to
re1ux l.S hrs under nitrogen. The reaction mixture
was cooled and most of the solvent was removed
va~uo The residue was partitioned between ethyl
acetate and excess ice-cold 5N sodium hydroxlde and
ice. The organic phase was separated, washed with : --
sat'd sodium chloride solution, and dried o~er
anhyd. magnesium sul~ate. Solvent remoYal afforded ;
a crude oil which was purified on silica gel using '
8:1:1 mixture of hexane:methylene chloride ethyl
acetate to gi~e 35 g of 3,5~diiodoaniline as a light
tan colored solid.
W~:) 94/05669 PCI /US93/07830
,: - . .`
~ ~ 4 2 1 0 ~
- 1 0 8 ~ .
DIAZOTIZATION & CONDENS.~N
Conditions: l) Dliodoaniline;thiophene as solvent &
reactant
2) isoamylnitrite
3) 600; 2 hours
Yield: 237/o
Spectra`:
MS m/z: 411.71 (MI)
1~ NM~ (400 MHz, CDC13): ~ 7.06-7.35 (m's; thiophene ~
lo protons~; 7.~8-7.94 (m's, phenyl protons). ~:
FO~MYLATION ;
Conditio~s: l) POC13; DME; oo; 10 min
2) 2-~3',5'-diiodophenyl)thiophene from
abo~e; 110; 3 hours
Yield: 77Z (used immediately)
R~DU~ION
Conditions: l~ NaBH4lMeO~
2) 0; 1.5 hours . :.
Yield: 69%
Spectra:
1~ NMR (400 Mhz, CDCl3): ~ 1.79 (t, OE); 4.8 (d,
: C~20H); 6.95 & 7.13 (2d's, thiophene protons);
7.80-7.~4 (phenyl protons)
SIL~LATION :
Condition: 1) TBDMSiCl; Et3N; DMF/C~2C12
2) R.T.; few hours; 10; o.n. ! ;.:
30 ~ield: 97/~
: Spectra: ~.
N~R (400 ~z, CDC13): ~ 0.12 (s, 0.12); 0.94 (s, ,,
~butyl~; 4.84 (s, C~20Si); 6.87 & 7.12 (2d's~ ~:
thiophene protons); 7.85-7.91 ~phenyl proto~s). , ~
-
,~"
~ . :
WO~ 5~69 PCT/USs3/07$30
- lOg -
I
PREPARATION OF YLIDE KETONE
Ar-I= Si t
Conditions~ BuLi/T~F; -78 to -20 (5 min)
2) MgBr2/THF; -20; 5 min
3) Pyridylthioester; 0; 4 hours
15 Yield: 25%
Spectra:
MS m/z: 1028 (M~l); 262 (Ph3R).
1~ NMR (400 MHz, CDC13): ~ 0.15 (s, Si(CH3)2); 0.95
(s, ~-butyl); 1.18 (d, CE3~; 4.86 (C~O); 5 8-5.98
20 (m, C~2=C~--CH2-). ;
~SILYATION OF KETONE ~LIDE TO CARBINOL
Conditions: l)aq. ~Cl/MeOH
2)0; 1.25 hours ~:.
~ields: 90%
Spectra: . ' -~
MS m/z: 914 (M~l); 262 (P~3P).
H!NMR (400 M~æ, CDC13): ~ 1.16 (d, CH3); 4.78 (dd, ~ .
~6); 5.8-6.0 (m, CH2-CH-CH2-) ~.
~ :
~Ç~IZATION QF CA~BINOL ~LIDE ~O CARBAP~NEM ~ :~
Conditions: Benzene; 80; o.n.
~ield: 8~C/o
~vos4t~66~ PCT~US93/07830
! ~ ~
0 8
- 110 - ;
Spectra:
MS m/z: 636 (MI+l); 618 (MI-OH)
IR: 1787 ~-lactam C=O); 1750 & 1725 (C=O~s) cm~l ` `
lH NMiR (400 MHz, CDC13); ~ 1~50 (d, CH3~; 1.81 (t,
0~); 3 44 (dd, H6); 4.31 (m, H5); 6.98 & 7.16 ~2d's.
thiophene protons); 7.50-7.87 (phenyl protons).
QUATERNI ZATI ON OF CARBINOL
Conditions: l) 2.4 eq. 1-methylimidazole/CH2C12
lo 2) 1.2 eq. triflic anhydride
3) 0; 45 min
Yield: 84% (used immediately) :
PEALLYLATION
Conditions: PPh3; Pd(PPh3)4
,',.:
ca2~
C02H . - ~
1:1 CH2C12:EtOAc; 2 hours
25 ~ield gz
Spectra: ' ~;
W (H20): f~max _ 296 m~
1~ NMR ~400 MHz, 2:1 D20:C~3CN) (no lnternal standard
- DOH at 4.80); ~ 1.47 (d, C~j3C~OR-) 3.63 (dd, H6); ~`~
4.05 (s, N-CH3); 5.75 (s, thiophene CH2N~
: 7.45 8.05 (thiophene, N-C~=C~-N, & phenyl protons); i` :
8.98 (partially eæchanged N=CH-N).
f
:
, -
~.
WO9~/05669 PCr/US93/07830 ~
.. -........................................................................... . . .
2 1 ~ 2 7 ~
:`
1 1 1 -
EXAMPLE S 5
S ; ~ , S
CO2il n = 0, 1, 2
()n
~TA~TI~NG ~ ~IATERIAL SYNTHESI S
Br
~ ~
~S CH2o i t
S C ~
2~ B~TYLDIMET~YLSILY~O~YMETH~L)-5-(3'-BROMO-5'
M~TEyLT~Iop~NyL)THIop~Er~ _
Conditions: l) 2-(~-butyldimethylsilyloxymethyl)~
5-~3',5'-dibromophenyl)thiophene; ~ -
1.1 eq. BuLi/THF, -78; 5 min
2) excess (CH3S)2; -78 to r.t. (o.n.) , ;:
~ield: 71/~ ''
,~
...
,, ~.,
W094/05669 PCT/USg3/07830 ~ ;
- ,: . .
~ 1~2 10 ~ _ 112 -
Spectra:
MS m/z: 428, 430(MI); 371, 373~MI-t-butyl); 297,
299(MI-TBDiMSiO)
1~ NMR (200 M~z, CDC13): ~ 0.13 (s, Si(C~3)2); 0.94
(s,t-butyl); 2.50 (s, SCH3); 4.89 (s, CH20);
6.88-7.47 (thiophene ~ phenyl protons).
~PARATION OF YLIDE KETQNE
Br
Ar-Br= ~H20 li t :
SCH3 :-
Conditions: 1) t-BuLi/T~F; -78 to -20 (5 min)
2) MgBr2/THF; -20; 5 min ~.
3) Pyridylthioester; 0; 5 hours
: Yield: 41%
Spectra: :.
MS m/æ: 949(MI~2); 262(Ph3P).
~ NMR (400 MHz, CDC13): ~ 0.14 (s, Si(CH3)2); 0.94 ~:
: (s,~butyl); 1.16 (d, CH3); 2.55 (s, SCH3); 5.76-5.98
~ ~5 (m~ C~2=C~-CH2-).
: ~ , .
~` DESILXI.ATION TO ~ARBINOL YLIDE KETONE
; Conditions: f 1 ) ! aq. ~Cl/MeO~ ~
2) Oc; lh ~ ;
Yield: 86%
Spectra~
MS m/z: 555(MI-Ph3PO); 262(Ph~P). ~ ;^.
(300 M~z, CDC13): ~ 1.15 (d, C~3); 2.54 (s,
SC~3); 5.74-6.0 (m, C~2 - C~-CH2-)-
,,
WO 94/05669 PCI /IJS93/07830 ~ ~
;`'`'`~ 1'. '
2 ~ ~ 2 ~
-- :L13 -- ~
,.
CYCLIZATION TO METHYLT~IIOCAR:BAPENE~I CARBINOL _ ~
1.:
Conditions: Benzene; 800; o.n.
Yield: 83%
S Spectra: ; :
MS m/z: 555(MI); 385 (MI-J3-lactam cleavage).
IR: 1787 (J3-lactam C=O); 1750 & 1725 ~other C=O's)
1~ NMR (300 MHz, CDC13): ~ 1.49 (d, ClI3); 1.80 (t,
OH); 2.50 (s, SMe); 3.43 (dd, H6); 4.30 (m, H5);
5.76-6.0 (m, C~2=CH-CH2-); 6.97-7.40 (phenyl & ~ ~
thiophene protons). :
.
OXIDA~It;)N TO SULFOXIDE AND SULFONE
i
15 Conditions: 1) excess ~-ClPBA; aq. Nc~CO3/CH2Cl2
2) oo; 1 hour
3) aq. Na2S203; 0; 2 hrs.
Yield: 52% Sulfoxide
32% Sulfone :;
20 Spectra (for sulfoxide):
l~I NIIR (400 MEIz, CDC13): ~ 1.48 (d, CH3); 1.86 (t,
OH); 2.76 (C~I3-S(0)-); 3.45 (dd, E6); 4.32 (m~ ~5);
4.84 (d, C~;20H); 7.0-7.78 (thiophene & phenyl
2.5 protons).
7 '' ;.
Spectra ~for sulfone):
~I N~ (400 ~z, CDC13) ~ 1.49 (d, ÇE3); ~.85 (t,
OE); 3.09 (s, C~3-S02-); 3.47 (d, ~I6); 4.34 (m, ~5);
4.85 (d, C~3;20~); 7.01-8.06 (thiophene & phenyl ~ :
protons). ;
:~'
w094/05669 PCT/US93/078N ~
Z ~ ~
- - 114 -
OUATERNIZATION OF ~ARBINOL
Conditions:* 1) 2.4-2.6 eq. 1-methylimidazole/CH2Cl2
2) 1.2-1.3 eq. triflic anhydride
3) oo; 45 min
*identical for C~3S(O)n
where n = O, 1, 2
Yield: methylthio ~97%)
methylsulfinyl (82%);
methylsulfonyl (84%).
Spectra:
lH NMR (400 MHz, CDC13) of methylthio: ~ 1.48 (d,
CH3); 2.50 (s, SCH3); 3.45 (dd, ~6); 3.97 (CE3N);
l~ 6.15-7.35 (thiophene, phenyl and two imidazole ~
~protons); 9.38 (N=CU-N). ~:
Methylsulfinyl & methylsulfonyl qu~ternaries used
immediately.
DEALLYLATION OF METHYLTHIO, METHYLSULFINYL, AND
: ~3~ FQNY~ CARBAPE~S _ _
; , ,
~: Conditions: 1) PPh3; Pd(PPh3)4 : ;i
. .
~ 25 ;~ .
C C r~ "
v 2 1~ : 2
.,
.~
,...
:~ :
:~ :
W09~/05669 PCT/US~3/~783~ ~
2 ~ dc ~ ~'7 ~
- 115 - !
i
Solvent TTime Yield
methylthio 2:1 CH2C12 ~t2~ 0 2h 16%
methylsulfi~yl 1:1 CH2C12:DMF r.t. 2h 19%
methylsulfonyl 1:1 CH2Cl~:D~ r.t. 2h 26%
Spectra:
H NMR (400 M~z, 2:1 D20:CD3CN) of methylthio: ~ 1.53
(d, C~3CHO-); 2.76 (s, SCH3); 3.67 ~dd, H6); 4.10 ~N-
C~3); 5.81 (thiophene -CH2-+N); 7.47-7.78 (thiophene,
} phenyl, & two imidazole protons).
H NMR (400 M~z, 2:1 D20: CD3CN) of methylsulfinyl:
1.58 (d, CH3CHOH-); 3.16 (s, SC~3); 3.75 ~dd, E~);
4.16 (s, CE3N); 5.89 (thiophene-CH2-~N); 7-60-8-13
(thiophene, phenyl, and two imidazole protons); 9.10
(s, N=CE-N). :~
1~ NMR (400 M~z, 2:1 D20:CD3CN) of methylsulfonyl:
1.55 (d, C~3CHOH-); 3.51 (s, CH3SO2); 3.73 (dd, H6); ~.
4.14 (s, CH3N); 5.87 (thiophene-CH2-+N); 7.59-8.25
(thiophene, phenyl, & two imidazole protons); 9.08 ` ::
(+N=C~-N). :-
~.
E~AMPLE 9
~-C~3 '~
3 0 _ ~ .
CO2 '
' ~'
L .,',~
WO 94~05669 PC~/US93/07830 ~.-
, ,:`'~ ":,
2 1 ~ z rl o ~ ;
" -- 1~6 --
.
STARTlN_ATERIA~ SYNTEESI ~ ~ ;
!
S
.
2-(3~-BROMOP~ENYL)-4-(~-BUTLYDIMETHYLSILYLOXYMET~YL)
I~IOP~ENE . _
~ROMINATION ,
Conditions: 3-thiophenecarboxylic acid (Aldrieh)
was selectively brominated to 5-bromo- .`
3-thiophenecarboxylic acid using the
method of Campaigne et al., J.A.C.S.,
76, ~445 (1954). -
~ield: 53%
: 1~ NMR (400 MHz, CDC13): ~ 7.51 (d, J= 1.5 ~z); 8.11 :
: ~dl J- ~.5 ~z).
2S
~EDUCTION TO 2-~R~Mo-4-T~LQ~NEM~rEA~QL ~ :
Conditions: 1) excess BH3-Me~S/T~F ' '`
2) O to r.t.; o.n.
3) MeO~; 0; slow addition then 15 min.
` 30 Yi ld 82%
~ . , ,. !,
.f"~
. ~ ' '.`'`:
~ ` '''"'
: : !,.,
W~4/0~669 PCT/US93/07~30 ~ :
~ ~ 2 ',~ ~ 2 ~
: - 117 -
Spectra:
MS m/z MI (192, 194).
1~ NMR ~400 MHz, C~C13): ~ 1.68 (t, 0~); 4.61 (br.d,
C~2OE); 7.05 (br. d); 7.11(m), when decoupled at ;.
C~OH- 1.67 (s, OH); 7.04 & 7.11 (sh d's, J= 1.5 Hz,
thiophene protons).
~ONDENSATIO~ WI~ 3-BRQMOPHENYLBQRQNI~_ACID
Conditions: 1) 2-bromo-4-thiophenemethanol/toluene
2) (Ph3P)4Pd
3) 3-bromophenylboronic acid/EtoH
4) aq. Na2CO3
5) 80; 5-24 hours
15 Yield: 43%
Spectra:
MS m/z: 268, 270 (MI); slight impurity (344, 346 -
dicondensat~on product)
1~ N~ (400 M~z, CDC13): ~ 1.67 ~t, 0~); 4.69 (d,
2n C~2OH); 7.20-7.74 (thiophene & phenyl protons).
SIL~ATION
Conditions: 1) TBDMSiCl; Et3N; DME/C~2C12
2) 0 to r.t.; few hours
Yield: 82%
Spectra:
MS mlz: 382, 384(MI); 325, 327(MI-t-butyl); 251, 253
(MI- OSiMe2t-butyl).
1~ NMR (400 MHz, CDC13): ~ O.12 (s, Si(C~3)2), 0.96 ~.
(s, ~-butyl); 4.72 (s, CH2O-); 7.13-7.73 (thiophe~e &
., phenyl proto~s).
i~ '
,,
~i.j
, ~ .
wos4/o5~69 P~T/U~93/07830 ~ ~
2 1 4 2 7 118 -
PREPARATION OF YLIDE KETONE ~ .
.
s ~3r
Ar-Br= ~ I t
Conditions: 1) Mg; BrCH2CH2Br/T~F; rflx; 2 hours
2) Pyridylthioester; 0; 3 hours
Yield: 45%
Spectra:
MS m/z: 908tMI ~ 7(Li)]; 262 (Ph3P).
1~ NMR (400 M~æ, CDC13): ~ 0.13 (s, Si(CE3)2); 0.95
(~, t-butyl); 1.17 (d, CH3); 5.78-6.06 (m,
C:~12=C~ C~2- )
D~SXLYLATION OF YLID~ KETONE CARBINOL
Conditions: 1) aq. HCl/MeOH
: 2) 0; 1 hour
Yield: 94%
~ 25 Spectra:
:~ MS m/z: 794~MI + 7(Li)]; 262 (Ph3P).
1~ NMR ~400 M~æ, CDC13): ~ 1.16 ~d, CH3); 2.80 (dd,
H6); 4.71 ~d, C~208); 5.77-5.89 (m! CE~=C~-CH2~
~ . . .
:~ 30
~, :
, ~ .
~, ~
: ~ -
`~
` W0~4/~669 PCT/US93/07830
2 1 ~ ~ ~ 0 3
` . ` .
i
- 119 -
Qy~ATERNIZATION Q~ CARBINOL
Conditions: 1) 2.5 eq. 1-methylimidazole/C~2C12
2) 1.~6 eq triflic anhydride ;:
3) 0; 45 min i:.
Yield: 86% (crude~; no data; used immediately.
DEALLYLATION
Conditions: PPh3; Pd(PPh3)4
CO2K CO2H ''.
.:
1:1:1 CH2C12:EtOAc:DME; r.t.; 2 hours
,
20 Yield 16%
Spectra: :
W (~2): ~ max - 290 m~ `
1~ NMR (400 ~H~, 2:1 D2O:CD3CN): (no internal
standard - DOH at 4.80); ~ 1.52 (d, CH3C~O~-); 3.64
(dd, ~6); 4.08 (C~3N); 5.58 (s, thiophene-C~2);
7.55-7.91 (thiophene, phenyl, & two imidazole
protons); 8.99 (s, N=CE-N). :~
' ~
~ ~:
:
`
W094/05669 PCr/US93/07830
~ ~ - ~^` .,
214~7~)~
-- 120 --
'-:
~EXAMPLE 1 0
~EP A~: `
/~2C H H ~=
o~ /= PPh3 ~OS i
C2 ~'
.,
Yield: 53%
Spectra:
MS: m/z 901(MI); 262 ~Ph3P). .
IR(C~2CL2): 1740 ~carbonyls); 1620 ~ylid) cm~
: lH NMR ~300 M~z, C~C13): selected absorbances ~ O.14
(s, Si~C~3)2); 0.94 ~s, t-butyl Si); 1.16 ~d, J=6 ~z, :
CE3C~OSi-); 2.79 (dd, ~6); 4.90 ~s, CH20Si~
5.75-6.00 (m, two -CH2C~C~2); 7.11-~.21 (all
aromatic protons,)
;, .......... ................................................................. ....... .. .~.
S~P A2:
~; '~
~,
/ ~ Z '~
O 1- PPh3 ~ ~ H
: Yl~ld. 8~%
" , , ` "
:: ~ ` : ,.~`
W094/0~6~ PCT/US93/07830
~",
2: L 4 ~
- 121 -
Spectra: `
MS m/z 787 (MI); 509 (MI-Ph3PO); 262 (Ph3P).
IR 2965 (CH2Cl~): 3600 (OH); 1740 (carhonyls, 1740 cm~
lH NMR (300 MHz, CDC13): ~ selected absorbances 1.15
(d, J=6 Hz, CH3CEO-); 2.78 (dd, J-2 ~ 10 Hz, H6); 4.87
(s, CH2OH); 5.74-6.00 (m two -CH2-CH=CH2).
ST~P B:
Yield: 49%
Spectra: ;
IR(CH2C12): 1780 (~-lactam); 1740 ~ 1715 (carbonate
and ester (cm-l). ;1
1~ NMR (300 MHz, CDC13): ~ 1.49 (d, J=6 Hz, CH3CHO-);
1.85 (t, J=6 Hz, OH); 3.13(dd, J=10 and 18 Hz, Hla);
3.32 (dd, J=9 ~ 18 Hz, Hlb); 3.43 (dd, J=3 & 8 Hz,
H6); 4.59-4.74 (m's, two CH2CH=CH2); 4.88 (d, J=6 Hz,
C~2OH); 5.13-5.40 (m, two CH2CH=C~2); 5.76-6.01 (m, ;~
two CH2C~=CH2); 7.24-7.55 (m's, phenyl and thienyl
protons)-
`
OH
~ H H
~ N-CH
C~2- W ~P-`~
W~ 94/05669 PCr/US93/07830
~, . ,~; . i
21~70~
- -- 122 --
.,
Yield: 21%
Spectra:
W (~2) ~a~ = 265 m~; ~sh = 303 m~ (N~20
quenchable); ~ - 8,800.
1~ NMR (300 MEz, D20)~ 6 ~d, J=6 ~z, C~3CEO-);
2.98 (dd, J=10 & 18 ~Z. ~la); 3 34 (dd, J=8 & 18 Hz
~lb); 3.42 (dd, J=2.5 & 6 Hz, H6); 3.79 (s, NCE3?;
4.20 (m's, H5 & Hl,); 5.52 (s, thiophene-CH2-N);
7.11-7.60 (phenyl, thiophene and two imidazole
10 protons); 8.72 (s, N=C~-N of imida~ole). -~
. PREPARA2ION OF INTERMEDIATES:
ST~P Q: `~
:~
~, .,
I3r Br fi
~ ' ~53~ ~3
I3r CHO (diforrrylation~
1 2 .;~
, -''.
To a solution of l (2.86 g, 9 mmol) in TEF
(30 m}) with stirring at -78 under N2, 1.6 M ~uLi
(5.8 ml, 9.3 mmol) was added dropwise ~ia an addition
funnel. After a few mins for the addition and 5 min
additional stirring, DMF (0.9 ml, 116 mmol);was
added, and the reaction was allowed to warm to
ambient temperature. Stirring was continued ~or 3
h. The yellow solution was then poured into brine
~: . (200 ml) and Et20 (100 ml), shakeni and separated.
The aqueo~s layer was again e~tracted with Et20, and
~ the combined organic layers were washed with 1
:.`~ brine:1~20 (100 ml), dried (~gS04), filtered and
~ ~: ;
, "~
WO~qJOSfi69 PCT/US93/07830 ~,
2 1 4 2 7 u ~
- 123 - ! `
concentrated in ~3~Q to a yellow liquid with a tan
precipitate. ~exane (a few ml) was added, and the ¦.
residue was slurried and filtered. The insoluble
portion was washed 2x with hexane (few ml), and the
solid dried in ~~Q to give ~ (496 mg, 19% yield).
The he~ane-soluble filtrate was re-concentrated in ~ s
vacuo (2.38 g) and chromatographed on 60 g of ~akers
Si gel (60-200 M ~z) packed in hexane. The material
was applied to the column in 1:2 CH2CI2/hexane and
eluted with the same solvent system (300 ml) after
which 10~/o Et20 in he~ane was used to elute 2.
Approximately 814 mg of 2 was eluted, but 630 mg of
that required further purification on 1000 ~ Si Gel ;
GF plates (eluting and extracted with CH2C12) to
provide a total of purified 2 ~713 mg, 30% yield)
Data for 2:
MS: m/z 266/268 (MI)
lH NMR ~300 ~ ~z, CDCl3): ~ 7.06 (dd, J=4 and 6, H~);
7.32 (dd, J=0.5 and 6 Hz, Ha); 7.33 (dd, J=0.5 and 4
Hz, B~,); 7.82, 7.90 and 7.96 (3 m's, 3 phenyl H's); ~ `
9.9~ (s, C~0) "
. .
Data for 3:
MS: m/æ 294/296 (MI)
H NM~ (300 M~z, CDC13): ~ 7.43 & 7.73 (2 d's, J=4
~æ, ~ ~ E~,): 7.95, 7.97 and 8.01 (3 m's, 3 phenyl
~'s); 9.87 & 9.94 (2s's, 2 C~O's). I :
~ . .
3~ ~ :
~: i
wos~/0~669 PCT/US93/07~30
21 ~ 2 rl ~ '~
- 124 -
':
;. .
Br
2 . ~ ~ :
OH
To a solution of 2 (707 mg, 2.7 mmol) in
MeO~(26 ml) with stirring at 0O was added Na~H4 (125
mg, 3.3 mmol), and after some initial foaming, ~-
stirring was continued at 0 for 35 min. The . -
reaction miæture was concentrated to a small ~olume :
of yellow oil under a N2 stream. Et2O (30 ml) and
brine ~30 ml) were added, and the rection mixture was
~haken in a separatory funnel~ After phase ;~
separation, the aqueous layer was again extracted
with ether~ The combined organic layers were
backwashed with brine, dried (MgSQ4), ~lltered and ~.
concentrated in Y~~Q to ~i~e the crude ~lcohol (735 ` ~;
mg~ as an of~-white solid~ Preparative TLC o.f 304 mg
of this substance o~ 4-1000 ~ Si Gel GF plates ;- -~:
(eluting with 5% EA/CH2C12 and extracting with 10% .
MeOH/CX2C12) provided purified alcohol 4 (278 mg). ;~
H NMR (30 M Hz, CDC13): ~ 472 (s, C~20H); 7.~6 (m, ~3
4"'- H of thiophene); 7~30 (m, 3"1 ~ 51~ 's of :
thiophene);; 7.42, 7.50 ~ 7.66 (3 br m~s, phenyl ~'s)~
. ' . .
. ", . " . .
~'094/0~6$9 PCTt~S93/0783~
.. ~", " .
2 ~
- - 12~ -
EXAMPLE 19
HO H H ,r3 ~ . ~
s ~,~-S ' ~
CO~ \~N~\N-CH3
1 o , . . .;,
SILYLATION OF 2-(3'-BR0~0-5~-HYDROX~METHYL)P~ENYLTeIO- ~ :
P~ENE
Conditions: 1) TBDMSiCl; ~t3N; DMF/C~2C1
2) overnight `
~ield: 80%
Spectra: .~
MS m/z 382, 384 (MI); 325, 327 (MI-t-butyl); 251, 253 :;
~MI-(CH3)2t-BuSiO~
1~ NMR (300 MHz, CDC13): ~ 0.12 (s~ t-butyl); 0.96
(s, Si(C~3)2); 4.73 (s, CH20); 7.06-7.62 (thiophene
phenyl protons).
PR~P~RATION QF YLI~E_E~E~ ~`
,, ~,.
~3 1 ~
osi-t
30 Conditions: 1) Mg; BrCH2CH2Br/TEF; rfl~; ~ hours 2,. """ .2) Pyridylthioester; 0; 2 hours ~ ;
: Yiel~: 33% ~ ~
~' ';:'".
:: .
~ .:
~V094/0~669 PCT/US93/07830 ~
21 0 ~
~ - 126 - I :
,
Spectra:
lH NMR (300 M~z, CDC13): ~ ~mixture of correct
product and diaddition of Grignard) 0.12 & 0.14 (2s,
~-butyls); 0.96 & 0.97 (2s; Si(C~3)2); 1.15 (d, CH3).
~:
DESILYLATION TO GARBINOL YLIDE KETONE :~
Conditions: 1) aq. ~Cl/MeO~
2) 0; 1 hour ~ .;
3) prep. TLC in 1:1 ~tOAC:CH2C12 to `: ;
remove diaddition product
Yield: 74% o~ càrbinol
Spectra: .
MS m/z: 787 (MIj; 509 (MI-Ph3PO); 262 (Ph3P)
1~ MMR (300 M~z, CDC13): ~ 1.16 (d, CH3); 5~75~6rO1
(m, CH2=C~-C~2~
CXCLIZATION OF YLI~ CARBIN~L TO CAR~APENEM C ~ INOk `l :
Conditions: Benzene; 80; overnight
~` Yield: 83%
:~ 20 ~Spectra:~
MS m/z: 509 (MI); 339 (~-lactam cleavage).
IR: 1780 (~-iactam C=O); 1745 ~ 1720 (C=O's) cm~l . ;;
1~ MMR (300 MHz, CDC13): ~ 1.49 ~d, CH3); 1.84 (t,
~ 0~), 3.42 (dd, ~); 4.30 ~m, ~5); 5.75-6.00 (m,
: 25 CE~=C~-CH2-); 7.06-7.56 (thiophene and phenyl
~: protonæ).
. QU~T~RNIZATION OF.CARBINO~
Conditions: 1) 2.5 eq. 1-methylimidazole/CH2C12
: 2) 1.1 eq. triflic anhydri:de
3~ 0-;~30 min
Yield: 94%
no data; used immedlately
wo94/n~66s PCT/US93/07830 ~
~ I; 2 ~ ~1 2 7 ~
.~ .
- 127 - i ~;
DEALLYLATION
Conditions: PPh3; Pd(PPh3)4 ~i
~ ~ .
CO2K
~/~ ''` '
CO2H
, ~", .
1:1 C~2C12:EtOAc; 2 hours
Yield: 20%
.
Spectra: ~:
W (H2Q): ~max = 293 m~
lH NMR (300 M~z, 2:1 D20:CD3CN): (no internal
standard - DOH at 4.80); ~ 1.55 (d, C~3C~O~-); 4.1
(s, N-CH3); 5.63 (s, thiophene-C~2); 4.03 (s, N=C~-N).
~ EXAMPLE ~0
STEP A :
.Ar-13r=Br
~ /
Conditions:
Al: 1) Mg/THF; 3 hrs./R.T. .
3 Yield: 2) 0"; 15 min; THF; pyridylthioester ,~
';
' ~:
wos4~0s669 PCT/~S93/07830
i , . . .
'~ 1 4 27 (3 5~
~ - 128 -
Conditions:
A2: CH30HIH2SO4; 0; 1.25 hrs
Yield: 81% ~ -.
: `
ST~
Conditions .- `
Bl: Xylene; 130; 1.5 hrs.
Yield: 83% .
Conditions .
10 B2: Tri~lic anhydride; C~2Cl2; .~ ~
`: ` .
H O .. ..
N - C O ~/; ~
N ~ `
,,'~
B2 0 15 min
: 20 ~Q~
IR: 1775; 1740 ..
NMR: H6: 3.43~3.52; dd; J = 3 & 8 ~z
~5: 4.24-4.38; ddd; J = 3, 9 ~ 10
C~2N+: 5.78 (s); Aromatic ~'s: 7.2-9.65 !
:
; ;~
W 0 94/0~669 PC-r/US93/07830 ~ ~ :
2142'~ ù3 ` I
.. . j
: - ].29 - I .
~,` `"' '~, .
STEP C
Conditions: PPh3; Pd~PPh3)4
~ ; .. :
C ~ K :
~ ` . .
C ~ H `~
",.. .
`., .:
CH2C12; 0; 4 hrs ;~;
Yield: 19%
15 W 298 :
ext 2397
EXAMPLE 31 , . .
~ m~ r~ A `~
Ar-Br= Br
~c .
Conditions A: 1) MgtT~ 3 hr./R.T.
2) 0; 15 min T~F pyridylthioester
Yield of A: 67% : -~
Conditions A2: C~3~ 2$4
3 09; 1. 25 hr
Yield of A2: ~1%
~., ~ . .
; ~ .
WOg~t/()~fi69 PCT/US93/07830 ~ .
2il~æ7~
- 130 -
~P B
Conditions:
B1: Xylene; 130; 1.5 hrs.
Yield ~3% ~ -
Conditions:
B2: Triflic anhydride; CH2C12;
:. ,. . ~ :.
H O ~ .
N ~ N - CO
0; 15 min. -~
1 5 ,' " ` "~:','
Conditions: PPh3; Pd(PPh3)4: ~ ,;
~,~ ~
C0011
CE12C12; 0 ; , ,~
~ ext 1107
3~
~ '~'';~.` ''`f
, ,,, , ~
,. .
~ .
- ....~ ;.,
: . - ~
~vo~l/0~69 PCT/US93/07830
,.~ .
2 1 4 ~ 7 0 ~
~ - 131 -
~,
EXAMPLE 32
f : '
~r - Br = Br : -
S ~ ,C~
~P .9
Conditions:
10 Al: 1) Mg/THF; 3 hr.s/R.T.
2) 0~; 15 min; T~F; pyridylthioester
Yield: 67%
Conditions:
A2: C~30H/H2S04; 0; 1.25 hrs.
~ield: 81%
~1æ~ '
Conditions:
Bl: Xylene; 130; 1.5 hrs.
Yield: 83% ~
Conditions: ;
B2: Triflic anhydride; CH2C12; :~
~5 ~SMe :~
N~:
0; 15 min.
;
~ ,'`
::
'~
~09~/0~669 PClt'US93/07830 ~^ ~
2 1 9~ ~ r~
- 132 -
Spectra~
IR: 1780; 1745; 1715 ,~ ;
NMR: SCH3: 2.02(s); ;
H6: 3.42-3.52; dd; J - 3 & 8 ~z
H5: 4.24-4.40; ddd; J = 3, 9 & 9 Hz
SC~2: 3.88 (s);
NCH2: 6.12(s);
Aromatic H's: 7.22-9.02 ~;
,
.
1 0 ~P
Conditions: PPh3; Pd(PPh3)4;
!i . '
~ ,~
` COOK
i..:`.
~; ~.. ,
COOH
C~2C12 : c`::
Yield: 14%
W : 293 ;~
ext ` 3847
, .
. .
.`,, ; I , - !:`
. i~
.
.
~ir ~ r~ ~ ;