Note: Descriptions are shown in the official language in which they were submitted.
21435~8
LD6 9
I~IB~TO~S OF FA~æSY~ PROTEIN T~NSFER~SP!
This invention relates to compounds that
inhibit farnesyl-protein transferase and Ras
protein farnesylation, thereby making them useful
as anti-cancer agents. The compounds are also
useful in the treatment of diseases, other than
cancer, associated with signal transduction
pathways operating through Ras and those associated
with CAAX-cont~i n i ng proteins other than Ras that
are also post-translationally modified by the
enzyme farnesyl protein transferase. The compounds
lS may also act as inhibitors of other prenyl
transferases, and thus be effective in the
treatment of diseases associated with other prenyl
modifications of proteins.
7o
The mammalian ras gene family comprises three
genes, H-ras, K-ras and N-ras. The Ras proteins
are a family of GTP-binding and hydrolyzing
proteins that regulate cell growth and
2~ differentiation. Overproduction of normal Ras
proteins or mutations that inhibit their GTPase
activity can lead to uncon~rolled cell di.vision.
-
2143588
LD69
-- 2
The transforming activity of ~as is dependenton localization of the protein to plasma membranes.
This mem~rane b;n~i n~ occurs via a series of post-
translational modifications of the cytosolic Ras
proteins. The first and m~ tory step in this
sequence of events is the farnesylation of these
proteins. The reaction is catalyzed by the enzyme
farnesyl protein transferase (FPT), and farnesyl
pyrophosphate (~PP) serves as the farnesyl group
donor in this reaction. The Ras C-terminus
contains a sequence motif termed a UCys-Aaa1-Aaa2-
Xaa~ box (CAAX box~, wherein Cys is cysteine, Aaa
is an aliphatic amino acid, and Xaa is a serine or
methionine. Farnesylation occurs on the cysteinyl
residue of the CAAX box (Cys-186), there~y
attaching the prenyl group on the protein via a
thio-ether linkage.
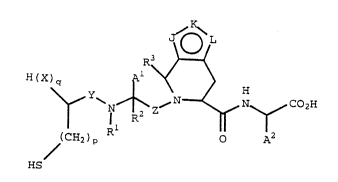
In accordance with the present invention, a
compound of the formula
I
J ~ L
H(X)q ~ ~ H
\~ Y~ ~ N--~ N~ CO2H
HS
its enantiomers and diastereomers, and
pharmaceutically acceptable salts, prodrugs and
so~vates thereof inhibit S-farnesyl protein
transferase, which is an enzyme Lnvolved in Ras
_
21435~8
_ 3 _ LD69
oncogene function. In formula I and throughout
this specification, unless otherwise specified, the
above symbols are defined as follows:
Al and A2 are each independently H, alkyl,
substituted alkyl, phenyl or substituted phenyl;
Y and Z are each independently -CH2- or
--C (O) --;
Rl and R2 are each independently H or alkyl;
Rl and Al taken together may be ~(CH2)m-;
R3 is H, alkyl or phenyl;
J, K and L are each independently, N, NR4, O,
S or CR5 with the provisos that only one of the
groups J, K and L can be O or S, one or two o~ the
groups J, K and L may be N or NR4, and at least one
~ the groups J or L must be N, NR4, 0 or S to form
a used five-membered heteroring;
the bond between J and K or K and L may also
form one side of a phenyl ring fused to the fused
five-membered heteroring;
X is O or NR6;
R4 is H, alkyl or phenylalkyl;
R5 is H or alkyl;
R6 is H, alkyl, phenyL, phenylalkyl,
substituted phenyl, (substituted phenyl)alkyl or~ -C~o)R7;
R7 is H, alkyl, phenyl or substituted phenyl;
m is 3 or 4;
p is 0, l or 2; and
q is 0 or l, with the proviso that when p is0 0, q is also 0.
-
2143588
_ 4 _ LD69
Listed below are definitions of various termsused to descri~e this invention. These definitions
apply to the terms as they are used throughout this
speci~ication, unless otherwise limited in specific
instances, either individually or as part of a
larger group.
The terms ~alkyl~ and "alk-" refer to straight
or branched chain unsubstituted hydrocarbon groups
of 1 to 7 carbon atoms. The expression '~lower
alkyl~ refers to unsubstituted alkyl groups of 1 to
4 carbon atoms.
The term "substituted alkyl~ refers to an
alkyl group substituted by, for example, one to
four substituents such as halo, hydroxy, alkoxy,
alkanoyl, alkanoyloxy, amino. alkylamino,
dialkyl~mino, alkanoyl~mino, thiol, alkylthio,
alkylthiono, alkylsulfonyl, sul~onamido, nitro,
cyano, carboxy, carbamyl, N-hydroxycarbamyl.
alkoxycarbonyl, phenyl, substituted phe~yl,
guanidino, indolyl, imidazolyl, furyl, thienyl,
thiazolyl, pyrrolidyl, pyridyl, pyrimidyl and the
like.
The term ~halogenu or ~halo~ refers to
fluorine, chlorine, bromine and iodine.
The term ~alkoxy~ refers to alkyl-O-.
The term ~alkanoyl" refers to alkyl-C(O)-.
The term ~alkanoyloxyu refers to
alkyl-C(O)-O-.
The terms ~alkylamino~ and n dialkylamino~
refer to (alkyl)NH- and (alkvl)2N-, respectively.
The term ~alkanoylamino~ refers to
alkyl-C(O)-NH-.
2143588
LD69
-- 5
The term ~alkylthio~ refers to alkyl-S-.
The term ~'alkylthiono~ refers to alkyl-S(0)-.
The term ~alkylsulfonyl~ refers to
alkyl-S(O)2--
S The term ~carbamyl~ refers to -C(0)NH2.
The term "alkoxycarbonyl" refers t~
alkyl-O-C(O)-.
The term ~substituted phenylU refers to a
phenyl group substituted by, for example, one to
four substituents such as alkyl, halo, ~ydroxy,
alkoxy, alkanoyl, alkanoyloxy, amino, alkyl~mi no,
dialkyl ~mi no, alkanoyl ~mi no, thiol, alkylthio,
nitro, cyano, carboxy, carbamyl, alkoxycarbonyl,
alkylthiono, alkylsulfonyl, sulfonamido and the
like.
The compounds of formula I form salts which
are also within the scope of this invention.
Pharmaceutically acceptable (i.e., non-toxic,
physiologically acceptable) salts are preferred,
although other salts are also useful, e.g., in
isolating or purifying the compounds of this
invention.
The compounds of formula I may form salts with
alkali metals such as sodium, potassium and
Iithium, with alkaline earth metals such as calcium
and magnesium, with organic bases such as
dicyclohexylamine, tributylamine, pyridine and
amino acids such as arginine, lysine and the like.
Such salts may be obtained by exchanging the
car~oxylic acid protons in compound I with the
desired ion in a medium in which the salt
precipitates or in an a~ueous medium followed by
e~aporation.
~ 2 ~ 8 8
LD69
-- 6
When compound I comprises a basic moiety, such
as amino or substituted amino, it may form salts
with a variety of organic and inorganic acids.
Such salts include those ~ormed with hydrogen
ch~oride, hydrogen bromide, meth~nesulfonic acid,
sulfuric acid, acetic acid, trifluoroacetic acid,
maleic acid, benzenesulfonic acid, ~oluenesulfonic
acid and various others (e.g., nitrates,
phosphates, borates, tartrates, citrates,
succinates, benzoates, ascorbates, salicylates and
the like). Such salts may be formed by reacting
compound I in an equivalen~ amount of the acid in a
medium in which the salt precipitates or in an
a~ueous medium followed by evaporation.
In addition, zwitterions ("inner salts~) may
be formed.
A compound of the formula I may also have
prodrug forms. Any compound that will be converted
Ln v vo to provide the bioactive agent (i.e., the
compound of formula I) ls a prodrug within the
scope and spirit of the invention. For exampie,
compound I may be in the form o~ a prodrug having
the formula
\0/
H ( X ~ q Al ~--) H
\~ ~N Z~f N~CO~R9
/ CH~)p 0 A2
R9S
wherein R8 is:
214~a~8
LD69
-- 7
lower alkyl, such as methyl, ethyl and the
like;
substituted lower alkyl, such as 2-(N-
morpholine)ethyl and the like;
lower aralkyl, such as benzyl, biphenylmethyl
and the like;
(acyloxy)alkyl, such as (pivalyloxy)methyl, 1-
(propanoyloxy)-2-methyl-1-propyl and the like;
(aminoacyloxy)aroyloxyalkyl, such as ~la-
glycyloxybenzoyloxymethyl and the like;
(aminoalkoxy)aroyloxyalkyl, such as ~aLa-2-
[(N-morpholine)ethoxy]benzoyloxyme~hyl and the
like;
substituted amides, such as N,N-di(2-
hydroxyethyl)acetamido, 4-methylpiperazine-1-
acetyl, 4-(2-hydroxyethyl)piperazine-1-acetyl and
the like; or
a dioxolanemethyl, such as (5-methyl-2-oxo-
1,3-dioxolan-4-yl)methyl and the like;
and Rg is:
k~noyl, aroyl, acyloxyalkyl, acylaminoalkyl,
alkylthio, phenylalkylthio,
J O L
R8 ~
~(X~q A1 ~ ) H
Y~ ~N~ N~CO~R8
(CH2)p O A
~S
2143~88
- 8 - LD69
H2N~J~
- OH
~CH2
and the like. ~Of course, one of R8 and R9 may be
H while the other one of R8 and R9 is as described
above.)
Additionally, for a compound of formula I
where X is NR6, R6 and R9 may be ~oined to form a
thiazolidine ring. (See, e.g., H. T . Nagasawa, et
al., J. Me~. Chem., 27, 591 (1984)).
Further, for a compound of formula I where A2
is substituted alkyl of the formula -(C~2)WOH
(where w is 2 or 3), A2 may be joined with the
car~oxyl group to form a lactone ring which can be
opened L~ vivo to give a compound of formula I
where A2 is -(CH2)WOH (again where w is 2 or 3).
various forms of prodrugs are well known in
the art. For examples of such prodrug derivatives,
see:
a) Decian of Pro~rllas, edited by H. Bundgaard,
(Elsevier, lg85) and Metho~s in Fnzvmol oov, Vol .
~2, p. 309-396, edlted by K. Widder, et al.
(Academic Press, 19853;
b) A Texthook of ~7rua Desicrn an~ DeveloDment,
edited by Krogsgaard-Larsen and H. Bundgaard,
Chapter 5, ~Design and Application of Prodrugs,~l by
~. Bundgaard, p. 113-191 (1991~;
c) H. Bundgaard, A~v~nce~ Drua Del iverv Reviews,
~, 1-38 (1992);
d) H. Bundgaard, et al ., Jol~rn~l of
Ph~r~ceutic~l Sciences, 77, 285 (1988); and
2143588
- LD69
g
.
e) N. Kakeya, et al., Chem Ph~rm B~ , 692
(1984).
It should further be understood tha~ solvates
(e.g., hydrates) of the compounds o~ formuia I are
also within the scope of the present in~ention.
Methods of sol~ation are generally known in the
art.
For compounds of the formula I, the following
moieties are preferred:
A1 and A2 are each independently H or
D-, L- or DL- -CH3, -cH(cH3)2~ -cH2cx(cH3)2~
-CH(CH3)CH2CH3, -C(CH3)3, -CH2OH, -CH2CH20H,
-CH2~3
-CH2CH2CH2OH, -CH(OH)CH3,
{:H2~ N~l
~H2 ~ OH ~ -C~2 CH2 C~2 CH2 MH2
~2 CH2 CH2 NH~ NH2
N~
-CH2C(O)OH, -CH2CH2C(O)OH, -cH2c(o)NH2
-CH2CH3,
-CH2CH2C(O)NR1OR11 where R10 and Rll are each,
independently, H, alkyl, phenyl or phenylalkyl, or
R10 and R11 taken together are (-CH2)t- where t is
an integer from 2 to 6, -CH2CH2OCH3,
-CH2CH2C(O)NHOH, -CH2SH, -cH2cH2sH~ -cH2cH2s(o)2NH2
or -CH2CH2SCH3.
The following moieties are particuiarly
preferred:
~ 21~3S88
LD69
-- 10 -
Al is L- -CH3, -cH(cH3)2~ -cH2cH(cH3)2/
-C(CH3)3, ~ , -CH(CH3 )CH2CH3, -CH2OH or
-CH(OH)CH3;
A2 is L- -CH2CH2SCH3, -CH2CH20H, -CH2CH2CH20H,
S -CH2CH2C(O)NRlO~ll where R10 and Rll are each,
independently, H, alkyl, phenyl or phenylalkyl,-or
RlO and Rll taken together are (-CH2)t- where t is
an integer from 2 to 6, or -CH2CH20CH3;
the fused five-membered (substituted)
heteroring is ~ , ~ , ~ ,
s~1 o,~ HN~1 N~ NH
, , or
X is NR6;
Rl, R2, R3, R~, R6 and R7 are H; and
p is 1.
The compounds of formula I are inhibitors of
S-farnesyl protein transferase. ~hey are thus
useful in the treatment of a variety of cancers,
including (but not Limited to) the following:
- carcinoma, including that of the bladder,
breast, colon, kidney, liver, lung, ovary,
pancreas, stomach, cervix, thyroid and skin;
- hematopoietic tumors of lymphoid lineage,
including acute lymphocytic leukemia, B-cell
lymphoma and Bur~etts lymphoma;
_
2~3~88
LD69
- hematopoietic tumors of myeloid lineage,
including acute and chronic myelogenous leukemias
and promyelocytic leukemia;
- tumors of mesenchymal origin, including
fibrosarcoma and rhabdomyosarcoma; and
- other tumors, including melanoma, sPminom~,
tetratocarcinoma, neuroblastoma and glioma.
The compounds of formula I are especially
useful in treatment of tumors having a high
incidence of Ras involvement, such as colon, lung,
and pancreatic tumors. By the a~mi n i ~tration of a
composition having one (or a combination) of the
compounds of this invention, development of tumors
in a m~m~Alian host is reduced.
Compounds of formula I may also be useful in
the treatment of diseases other than cancer that
may be associated with signal transduction pathways
operating through Ras, e.g., neuro-fibromatosis.
Compounds of formula I may also be useful in
the treatment of diseases associated with CAAX-
cont~ining proteins other than Ras (e.g., nuclear
lamins and transducin) that are also post-
translationally modified by the enzyme farnesyl
protein transferase.
Compounds of formula I may also act as
inhibitors of other prenyl transferases (e.g.,
geranylgeranyl transferase), and thus be effective
in the treatment of diseases associated with other
prenyl modifications (e.g., geranylgeranylation~ of
proteins (e.g., the rap, rab, rac and rho gene
products and the like). For example, they may find
use as drugs against Hepacitis delta virus (HDV)
infections, as suggested by the recent finding thac
geranylgeranylation of the large isoform of the
~ 2143588
LD69
- 12 -
delta antigen of HDV is a requirement for
productive viral infection [J. S. Glenn, et al.,
Science, 25~, 1331 (1992)].
The compounds of this invention may also be
useful in combination with known anti-cancer and
cytotoxic agents. If formulated as a fixed dose,
such combination products employ the compounds of
this invention within the dosage range described
below and the other pharmaceutically active agent
within its approved dosage range. Compounds of
formula I may be used sequentially with known
anticancer or cytotoxic agents when a combination
formulation is inappropriate.
The compounds of this invention may be
formulated with a pharmaceutical vehicle or diluent
for oral, intravenous or subcutaneous
;ni~tration. The pharmaceutical composition can
be formulated in a classical m~nnPr using solid or
liquid vehicles, diluents and additives appropriate
to the desired mode of A~mi ni stration. Orally, the
compounds can be administered in the form of
tablets, capsules, granules, powders and the l_ke.
These compounds may be a~minictered in a dosage
range of about 0.05 tO 50 mg/kg~day, preferably
less than 50 mg/kg/day, in a single dose or in 2 to
4 divided doses.
A compound of formula III below, wl~erein prol
is an amine protecting group (e.g., t-butyloxy-
carbonyl (Boc), benzyloxycarbonyl (Cbz) and the
like):
2143~88
LD6 9
-- 13 --
III
J~j~jL
R3_~ >
Pro l~ N~ OH
can be coupled with a carboxylic acid protected
derivative o~ an amino acid of formula rv:
IV
H~N~ H~CO,Pro~
to form a compound of formula V below, wAerein pro2
is a carboxylic acid protecting group (e.g., alkyl,
benzyl, p-methoxybenzyl and ~he like):
V
~ N ~ N~_~CO,Pro~
Pro n T
O A-
For additional examples of amino and
carboxylic acid protecting groups (as well as means
of formation and eventual deprotection), see T.W.
Greene and P.G.M. wuts, ~Protective Groups in
Organic Synthesis~, Second Edition, John Wiley &
Sons, New York, '99l.
A variety of coupling agents may be used for
this coupling, including l-(3-dimethylaminopropyl)-
3-ethylcarbodiimide hydrochlorlde (E~CI~ ~ith l-
-
~14353~
LD69
- 14 -
hydroxybenzotria~oie ( HOBT ), dicyclohexyl-
carbodiimide (DCC) with ~OBT, benzotriazol-1-
yloxytris(dimethylamino)phosphonium hexafluoro-
phosphate (BOP) with or without ~OBT,
carbonyldiimidazole (CDI), bis(2-oxo-3-
oxazolidinyl)phosphinic chloride (BOP chloride),
isopropylchloroformate (IPCF) and the like.
Compounds of the formula IV are known in the
art. See, for example, R. M. Williams, I'Synthesis
of Optically Active a a-Amino Acids," Pergamon
Press, Oxford, 1989.
Compounds of formula II~ can be prepared by
methods known in the art. For example, for
compounds of formula III wherein 1) J is -CH-, K is
-CH- and L is S, see J. P. Maffrand, U.S. Patent No.
4,147,787 issued April 3, 1979; 2) J is S, K is -CH-
and L is -CH-, also see J. P. Maffrand, U.S. Patent
No. 4,147,787 issued April 3, 1979; 3) J is S and
the bond between K and L form one side of a fused
phenyl ring, see, e.g., H. Xawakubo, et al., J. Me~.
Chem., ~, 3110 (1990); ~ J is NR4, and the bond
between K and L form one side of a fused phenyl
ring, see D. G. Harvev, ~t ai., J. Chem Soc., i53
(1941) and A. srossi, et al., ~. Med. Chem. 16, 418,
(1973); 5) J and L are NR4, and K is CR5, see M.
Cain, et al . , Heterocv~les, 19, 1003 (1982); and 6)
J is O, and the bond between K and L form one side
of a fused phenyl ring, see S. ueki, e~ al ., U. S .
Patent ~Jo. 5,126,448 issued June 30, 1992.
A compound of formula v can be treated with a
suitable N-deprotecting agent to proviàe the
corresponding free amine of formula VI:
~ 2143588
- 15 - LD69
VI
J ~ L
N~CO~Pro-
O A
(The prol and pro2 protecting groups are chosen so
that prol can be selectively remo~ed in the
presence of Pro2).
An amine of formula VI can be coupled with a
suitable amine-protected amino acid of formula VII
(wherein Pro3 is an amine protecting group~:
VII Al
P ' OH
R ¦ ¦
R~ O
using an approprla~e coupling reagent (e.g., 30P-
Cl) to form a compouna of formula VIII:
VIII
J ~ L
AL ~ > H
Pro ~ ~ N ~ N ~ C02Pro
(Compounds of formula VII are known in ~he art.
See, for exampLe, R.M. '.~illiams, ~Synthesis of
-
21435~8
L~69
- 16 -
Optically Active a-Amino Acids,~ Pergamon Press,
Oxford, 1989).
Compounds of formula VIII can also be prepared
by coupling a C-terminal protected amino acid of
the formula IX (wherein Pro4 is a carboxylic acid
protecting group):
IX
\~ O~L
R~
N ~ OPro4
with an amine-protected amino acid of formula VII
above to provide a compound of formula X:
J O L
A~ ~
Pro ~ N ~ ~ ~ OPro4
IL R2 ll
R o o
The Pro~ protecting group o~ compound x can be
selectively removed by me~hods known in the art to
provide a compound o~ formula XI below:
~ 21~3~8
LD69
- 17 -
XI
Pro~ ~ OH
(The Pro3 and Pro4 protecting groups are chosen so
that Pro4 can be selectively removed in the
presence of Pro3).
Coupling of a compound of formula XI with an
amino ester of formula IV would then pro~ide
compound VIII.
A compound of formula VIII can be selectively
N-deprotected by methods known in the art to
pro~ide an amine of formula XII:
XII
2,.1R~
H~ ~ N ~ N ~ CO2Pro
(The pro2 and Pro3 protecting groups are chosen so
that Pro3 can be selectively removed in the
presence of Pro2).
Coupling of an amine of formula XII with an
acid of formula XIII (wherein X is optionally
protected and Pro5 is a thiol protecting group such
as trityl):
~ 2143588
LD69
-- 18 --
XIII
H (X~ l~
`f OH
( CH2 ) p
ProsS
with a suitable coupling agent provides a compound
5 of formula xrv
XIV
H(X~
~cyl~ R o O
Pro5S
3eprotection o~ the pro2 and ProS groups of
XIV, and removal of the optionai protecting group
on X, then provides a compound of formula I where Y
and Z are -C(O)-. (See T.W. Greene and P.G.M.
wuts, ~Protective Groups in Organic Synthesis",
John Wiley & Sons, New York, l99l, for exemplary
protectins groups for X, their formation and
removal).
Alternatively, removal of the nitrogen
protecting group (Pro3) from a compound of formula
X gives an amine of formula XV:
2~13S88
LD69
-- 19 --
XV
R O O
which can then be coupled with an acid of formula
XIII to pro~ide a compound of formula XVI wherein X
is optionally protected:
XVI
J O L
H(X~
~ I ~ ~
( CH2 ) p O
Pro S
Selective deprotection o~ the Pro~ protecting
group of XVI provides an acid of formula XVII:
XVII
.~3
H(XJ~ >
(CH2)p R O O
Pro'S
2143588
LD69
- 20 -
which can be coupled with an amine of formula IV to
pro~ide a compound of formula XIV. Deprotection of
the pro2 protecting group of XIV, and removal of
the optional protecting group on X, then also
provides a compound of formula I where Y and Z are
--C ~O) --.
Further, a compound of formula I where Y and
Z are -C(O)- can be prepared by automated solid
phase peptide synthesis using methods that are well
known in the art. See, for example:
a) M. Bodansky and A. Bodansky, ~The Practice
of Peptide Synthesis~, springer-Verlag,
Berlin/Heidelberg/New York/Tokyo, 1984i and
b) J. M. Stewart and J. D. Young, "Solid Phase
Peptide Synthesis~, Pierce C~emical Co.,
Rockport, Illinois, 1984.
In another process, an amino acid of formula
VII (wherein Pro3 is a suitable amino protecting
group) can be con~erted to the N-methoxy-N-
methylamide of formula XVIII by methods known inthe art:
XVIII
Al fH3
Pro ~ N~ ~ C~ ~
R2 11
Rl o
A compound of formula XVIII can be reduced tO
an aldehyde of formula XIX by methods known in the
art. (See, e.g., Fehrentz, et al ., Svnthe~is, ~76
(1983)):
- - -
21435~8
LD69
XIX
Al
Pro3~ ~CI~ ~ H
R' O
Alternatively, a compound of formula XIX can
be prepared by reduction of a compound of formula
VII with a reducing agent such as borane, followed
by oxidation of the resulting alcohol using, for
examp7e, the Swern method of oxidation. (See,
e.g., Luly, et al., J. Or~. Ch~m., 52, 1487 (1987);
or Stanfield, et al., J. Or~. Chem~ 4~, 4797
(1981)). In yet another alternati~e, a compound of
formula XIX can be prepared by reduction of an
ester of a compound of formula VII with a reducing
agent such as diisobu~ylaluminum hydride. See,
e.g., Rich, et al ., ~ . Ora . Chem., 43, 3624 ( 1978).
An N-protected aldehyde of formula XIX can be
reductiveLy aminated with an amine of formula VI to
form a compound of formula xX using methods ~nown
in the art (e.g., sorch, et al., ~. ~. Ch~. coc ,
~, 2897 (1971)):
XX'
ALR~
L~H
~ 2~3588
LD~9
- 22 -
A compound of formula XX can be treated with a
suitable N-deprotecting agent to provide the
corresponding free amine of formula XXI:
XXI
",i ~
R H O
(The pro2 and Pro3 protecting groups are chosen so
that Pro3 can be selectively removed in the
presence of Pro2).
A compound of formula XXI can be coupled with
an acid of formula XXII (wherein X is optionally
protected) using an appropriate coupling reagent tO
form a compound of formula XXII:
XXII
~(L
Pross
Deprotection of the pro2 group of the compound of
formula XXII, and removal of the optional
protecting group on X, provides a compound of
formula I where Y is -CtO~-, Z is -CH2-.
21~3~8
- 23 - LD69
An acid of formula XIII (wherein X is
optionally protected) can also be converted to the
N-methoxy-N-methylamide of formula XXIII by methods
known in the art (e.g., Fehrentz, et al .,
Svnth~sis, 676 (1983)):
XXIII
~C~
C N- O
/ \
I CH3 CH3
(CH~)p
Pro S
A compound of formula XXIII can be selectively
reduced to an aldehyde of formula xxrv by methods
known in the art (e.g., r ehrentz, et al .,
Svnthesis, 676 (1983)):
XXIV
H(X)~
C~
I H
(C~,)p
Pro~S
An aldehyàe of formula XXIV can be reductiveiy
aminated with an amine of formula XII to form a
compound of formula XXV using methods known in the
art (e.g., ~orch, et al ., J . ~m . Chem. Soc., ~,
2897 (1971)):
~ 3 5 8 8
LD69
- 2~ -
XXV
J ~ L
H(X) H H ~1 ~ ~ H
N ~ C02Pro2
/ CH2)p O A~
Pro~S
Deprotection of the Pro2, and removal of the
optional protecting group on X, provides a compound
of formula I where Y is -CH2-, Z is -C(O)-.
Alternatively, reductive amination o~ a
compound of formula XXVI where pro6 is a carboxyLic
acid protecting group:
XXVI
.;~,1
H~ ~ oPro~
1~n
R' O
with a compound o f f ormula XXIV provides a compound
of formula XXVII:
XXVII
H(X)~ V
OPro~
I 1~ R
(CH2)p 0
/
Pro'S
Selective deprotection of the pro6 group provides a
compound o f f ormula XXVIII:
~ 21~3588
LD69
- 25 -
XXVIII
H(X) ~.H
( CH2 ) p
Pro5S
which, on coupling with a compound of formula VI
(using, for example, BOP-Cl) provides a compound of
S formula XX~.
By combining the appropriate steps above, one
skilled in the art can also prepare a compound of
formula I where both Y and Z are -CH2--
Side-chain protecting groups may be used in
these processes with amino acids ha~ing reactive
functionalities, such as hydroxyl, carboxyl, amino,
mercapto, guanidino, ; mi ~olyl, indolyl and the
like. The particular protecting groups used for
any amino acid residues depend upon the sidechains
to be protected and are generally known in the art.
Exemplary sidechain protecting groups incLude
acetyl, benzoyi, benzyl, t-butyl and the ike for
hydroxyl; cyclohexyl, benzyl, methyl, ethyl, t-
butyl and the like for carboxyl; benzyl, 4-
methylbenzyl, 4-methoxybenzyl, acetyl,
acetamidomethyl, triphenylmethyl (trityl) and the
like for mercapto; t-butoxycarbonyl (Boc),
benzyloxylcarbonyl (Cbz), ~-~(9~-Fluoren-~-
ylmethoxy)carbonyl] (Fmoc~, phthaloyl (Pht), p-
toluenesul~onyl (Tos), trifluoroacetyl, 2-
(trimethylsilyl)ethoxycarbonyl (Teoc) and the like
for amino; 2,4-dinitrophenyl, benzyloxymethyl, Tos,
Boc, trityl and the like for imidazolyl; formyl,
Cbz, Teoc, 2,2,2-trichloroethyl carbamate (TROC)
2143588
LD69
- 26 -
and the like for indolyl; and tosyl, nitro, bis(1-
adamantyloxycarbonyl) and the like for guanidino.
Side-chain protecting groups may be removed,
if desired, by, for example, treatment with one or
more deprotecting agents in an inert solvent or
solvent mixture. For examples of protec~ing groups
and suitable deprotecting agents, see M. Bodansky
and A. Bodansky, ~The Practice of Peptide
SynthesisU, springer-Verlag~ Inc. ~1984); and T. W.
Greene and P.G.M. Wuts, "Protective Groups in
Organic Synthesis~, Second Edition, John Wiley
Sons, New York, 1991.
The invention will now be further described by
the following working examples, which are preferred
embodiments of the invention. All temperatures are
in degrees Celsius (C) unless otherwise indicated.
Compounds exemplified herein, which comprise a
basic moiety such as an amine or substituted amine,
may exist as a salt of an organic or inorganic
acid. This information is not necessarily
explicitly described in all the examples, but would
be understood by those skilled in the arc. These
examples are illustrative rather than limiting.
~ ~143~88
LD69
- 27 -
~mn le
N-[[5-[N-[(R)-2-Amino-3-mercaptopropyl]-L-valyl]-
4,S,6,7-tetrahydrothieno~3,2-cipyridin-6-yl]-
c~rhonvll-r,-methionine, trifllloro~cetAte (1:1) sAlt
A. N2-~(1,1-Dime~hylethoxy)car~onyl]-N-
methoxy-N-methyl-S-(triphenylmethyl)-
r.-cvStein~mi ~le
N-Butyloxycarbonyl-S-trityl-L-Cysteine (20.0
g, 43.14 mmol), 1-ethyl-3-(3-dimethyl~mi~opropyL)
car~odiimide-~Cl (8.3 g, ~3.1 mmol) and 1-
hydroxybenzotriazole hydrate (5.8 g, 43.1 mmol)
were dissolved in dimethyl~ormamide (100 mL). N,O-
Dimethylhydroxylamine hydrochloride (4.91 g, 50.6
mmol) and N,N-diiso~oy~lethylamine (6.13 g, 8.3
mL, 47.5 mmol) were then added. The reaction
mixture was stirred ~or 5 hours at room
temperature, poured into water (50 mL) and
2~ extracted with e~hyl acetate (3 x 200 mL). The
combined organic layers were washed with water (3 x
500 mL) and brine (200 mL), dried magnesium
sulfate, concentrated and chromatographed (silica
gel, eluting with 40% ethyl acetate, 60% hexane).
Fractions containing the proàuct were collected and
concentrated to yield Compound A as a white solid
(19.95 g, 92%)
B. N-[(1,1-Dimethylethoxy)car~onyl]-S-
(trinhenYImethvl)-r.-cysteinAl
Lithium aluminum hydride in tetrahydro~uran
(lM, 59.3 mL, 59.3 mmol) was added dropwise to a
solution of Compound A (25 g, ~9.4 mmol) in diethyi
ether (500 mL) at -lSC, while keeping the
2143588
.
LD69
- 28 -
temperature below -14C. The reaction was stirred
at -15C for 30 minutes, quenched slowly with
potassium hydrogen sulfate (134.4 g, 99.8 mmol) in
water (400 mL), warmed to room temperature, and
stirred for 1 hour. The organic layer was
separated and washed sequentially with 10%
potassium hydrogen sulfate (1 x 200 mL), lN
hydrochloric acid (1 x 200 mL) and 10% sodium
bicarbonate (1 x 300 mL). The organic layer was
dried (magnesium sulfate), filtered and
concentrated under ~acuum to a~ford the aldehyde
Compound B (20 g, 90%), which was used withou~
further purification.
mp: 48-54C
TLC: Rf : 0.80 (1/1 hexane/ethyl acetate,
visualization by W)
MS: (M-H)- 446
IR: (KBr), 1713 cm~1
[a]D = + 16.0, ( c = 1.19 methanol)
C. (R)-N-[2-[(1,1-Dimethylethoxy)carbonyl]-3-
[(triphenyimethyl)thio3propyl]-1-valine,
meth~] ester
Acetic acid (2.6 mL, 44.74 mmol) was added
eo a solution of Compound B 120 g, 44.74 mmoL) and
L-valine methyl ester HCl (9.0 g, 53.7 mmol) in
methanoi (50 mL). The mixture was stirred at room
temperature for 30 minutes. Sodium
cyanoborohydride (2.8 g, 44.74 mmol) in
tetrahydrofuran (50 mL) was added dropwise to the
mixture over 30 minutes, and the resulting mixture
was stirred at room temperature for 2 hours. The
reaction was quenched with sodium bicarbonate (3.75
g, 44.74 mmol) in water (20 mL) and concentrated
~ , _ , .
2143588
LD69
- 29 -
under vacuum. The residue was dissolved in 10%
sodium bicarbonate (10 m~) and extracted with
dichloromethane (3 x 100 mL). The combined organic
extracts were dried (magnesium sulfate), filtered
and concentrated under vacuum. The residue was
purified by flash chromatography (eluting with 9:1
hexane/acetone) to afford Compound C (17.7 g, 70%).
D. (R)-N-[2-[(1,1-Dimethylethoxy)carbonyl~-3-
o r (tri~hen~lmethvl)th;ol~ro~vll-1-v~line
A solution of lithium hydroxide (lN, 7.2 mL,
7.2 mmoi) and Compound C (2.0 g, 3.6 mmol~ in 2/2/1
tetrahydrofuran/dioxane/methanol (10 mL) was
stirred at room temperature for 16 hours. The
mixture was concentrated under vacuum, and diluted
with water (50 mL) and lN hydrochloric acid (11.1
mL). The aqueous mixture was extracted with
dichloromethane (3 x 50 mL). The combined organic
extracts were dried (magnesium sulfate), filtered,
concentrated under vacuum, and used without further
purification to afford Compound D (2.08 g, 100%) as
a white soiid.
mp: 155-i66C
TLC: Rf = 0.45 (9/1/0.05 chloroform~methanol/acetic
acid, visualization by W)
MS: (M+H)+ 549
E. 5-[(1,1-Dimethylethoxy)carbonyl~-4,5,5,7-
tetrahyarothieno[3,2-cipyridine-5-carboxylic
~ci~
To a suspension of 6-carboxy-4,5,6,7-
tetrahydro-thieno[3,2-c]pyridine (0.9 g, 4.9 mmol;
for preparation, see J. P. Maffrand, U.S. Patent
No. 4,147,,87 issued April 3, 179) in dioxane (7
.
~ 2~3S~8
LD69
- 30 -
mL) at 0C was added 1~ sodium hydroxide (5 mL, 10
mmol) and water (5 mL). To the resulting clear
solution was aàded soc-anhydride (1.53 g, 7.0 mmol)
and the cold bath was removed. ~fter 1 hour, the
reaction mixture was concentrated to hal~ the
~olume, and ethyl acetate (15 mL) and lN
hydrochloric acid (8 mL) were added with vigorou;s
stirring. The aqueous layer was separated and
extracted with ethyl acetate (10 mL). The organic
layers were combined, dried (magnesium sulfate),
filtered and concentrated in vacuo to afford
Compound E (i.52 g, 110% of theoretical).
~S (FAB), (M+H)+ = 284'
lS F. N-[tS-[(1,1-Dimethylethoxy)carbon~l]-
4,5,6,~-tetrahydrothieno~3,2-c~pyridin-6-
vllc~rhon~ll-r-methionine methvl ester
To a solution of Compound E (1.52 g) in
dichloromethane (lS mL) at O~C under argon was
added sequentially benzotriazol-1-yloxytris-
(dimethylamino)phosphonium hexafluorophospnate
(2.21 g, ~.0 mmol), N-methylmorpholine (2.1 mL, 1
mmol) and the hydrochloride salt of L-me~hionine
methyl ester (1.O g, S mmol). The reaction was
allowed to warm to room temperature. ~fter 16
hours, the reaction mixture was washed successively
with lN hydrochloric acid, saturated sodium
bicarbonate and brine (lS mL each). Each aqueous
layer was extracted separately with chloroform (10
mL). The organic layers were com~ined, dried
(magnesium sulfate), iltered and concentrated.
The residue was purified by flash silica gel column
chromatography eluting with 30% ethyl acetate in
hexanes to afford Compound F (1.15 g).
2143~88
LD69
- 31 -
MS (CI), (M+H)+ = 429+
TLC : Rf 0.46 (50%, ethyl acetate in hexanes,
visualized by W and ceric ammon. moiybdate)
HPLC: YMC, S3, C18 (6 x 150 mm) column, 220 nm, 1.5
S mL/min: 0-90% aqueous methanol with 0.2% phosphoric
acid, gradient over 30 minutes : Retention time :
28.2 and 28.3 minutes (two diastereomers)
G. N-[[5-[N-[(R)-2-[(1,1-Dimethylethoxy)-
carbonyl]-3-[(triphenylmethyl)thio]propyl]-
L-valyl]-4,5,6,7-tetrahydrothieno[3,2-
c~pyridin-6-yl]carbonyl]-~-methionine,
methvl ester
Anhydrous hydrochloric acid (4M in dioxane,
1~ 3 mL, 12 mmol) and triethylsilane (0.4 mL, 2.5
mmol) were added to Compound F (1.1 g, 2 57 mmol)
at room temperature under argon. After stirring
1.5 hours, the volatiles were removed in vacuo and
the residue was triturated with ether and dried in
vacuo (1.0 g).
To the resulting amine hydrochloride (0.8 g)
in dichloromethane (8 ~L) at 0C under argon was
added the acid Compound D (1.0 g, 1.82 m~ol) and
dimethylformamide (1 mL) to obtain a homogeneous
2~ solution. soP-cl (0.48 g, 1.9 mmol) and N,N-
diisopropylethylamine (0.73 mL, 4.1 mmol) were
added and the reaction mixture was stirred
overnight at 5C. The reaction mixture was diluted
with ethyl acetate (15 mL~ and washed sequentially
with lN hydrogen chloride, saturated sodium
bicar~onate and brine (10 mL eacA). The organic
layer was dried (magnesium sulfate), filtered and
concentrated in vacuo . The r~sidue was purified by
flash silica gel column c~romatography eluting with
,
~ 2143~8
LD69
- 32 -
step gradient of 30%. 40% and 50~ ethyl acetate in
hexanes to afford Compound G (240 mg, 15~), as a
white foam.
In a separate, small scale experiment,
S dimethyiformamide was not used as co-solvent. In
this case, the yield of Compound G was 40%.
MS: (FAB) ~M+H)+ = 859+
TLC : Rf 0.51 (1:1, ethyl acetate in hexanes,
visualized by W and ceric ammon. molybdate, shows
two diastereomers when spotted very dilute).
HPLC: YMC, S3, C18 (6 x lS0 mm) column, 220 nm, ~.5
mL/min: 0-90% aqueous methanoi with Q.2% phosphoric
acid, gradient over 30 minutes : Retention time :
30.9 minu~es.
1~
H. N-[[5-[N-[tR)-2-Amino-3-mercaptopropyl]-~-
~alyl]-4,5,6,7-tetrahydrothieno[3,2-c]-
pyridin-6-yl]carbonyi]-L-methionine,
triflllQro~cet~e (1:1) s~lt
~0 To a solution of compound 5 (0.22 g, 0.26
mmoi) in methanol (1 mL) and tetrahydrofuran (2 mL)
at room tempera~ure was added a~ueous lN lithium
hydroxide (0.3 mL, a . 3 mmol). ~ter 8 hours, 1~
hydrogen chloride (l mL~ was added and the mixture
was ex~racted with ethyl acetate (5 mL). The
organic layer was dried (magnesium sulfa~e),
filtered and concentrated in ~acuo to afford a
white soiid (240 mg; MS (M+H)+ = 845+).
To a solution of the above solid (230 mg) ~n
dichloromethane (1.2 mL) at room tempera~ure under
argon was added triethylsilane (0.08 mL, 0.48 mmol)
and trifluoroacetic acid (1 mL). After 1.5 hours,
the volatiles were removed in vacuo and the residue
was dissolved in deoxygena~ed 40% aqueous methanol.
21~3588
LD69
- 33 -
The precipitate formed was filtered through nylon
0.45 ~ filter and the filtrate was purified by
RPHPLC (YMC, S10, C18, 30 x 500 mm column) eluting
with a linear gradient of ~2%-66% aqueous methanol
cont~i~ing 0.1% trifluoroacetic acid over 1 hour.
Appropriate fractions were collected and
concentrated in vacuo to remo~e methanol. The
r~aining solution was lyophili~ed to obtain the
title compound (150 mg, 80%), as a white
lyophilate.
m.p. 64-66C
MS:(FAB); (M+H)' = 502'
HPLC: YMC, S3, C18 (6 x 150 mm) column, 220 nm, 1.5
mL/min: 10-90% a~ueous methznol with 0.2%
phosphoric acid, gradient over 30 minutes :
Retention time : 16.0 minutes
[a~D = -16.2 (c= 0.6, methanol)
Elemental analysis for C21H34N4O452. 1.2 ~2 2.05
trifluoroacetic acid
~ E
Calculated: 39.77 5.11 7.39 12.69
~ound: 39.72 5.15 7.47 15.46
21~35~8
LD69
-- 34 --
~x;lmn le 2
(S~,R*)-N-[[2-[N-(2-Amino-3-merCaptOy ~yl)-L-valyl]-
2,3,4,9-tetrahydro-lH-pyrido~3,4-b]indol-3-
vllc~rhonvll-rl-methionine~ trifllloro~cet~te (1:2) s~lt
A. (S)-1,3,4,9-Tetrahydro-2H-pyrido[3, 4 -
b]indole-2,3-dicar~oxylic acid, 2-(1,1-
~imethvlethvl) e~ter
To a solution o~ (s)-2~3~4~9-Tetrahydro-l~-
pyrido[ 3, 4 -b]indole-3-car~oxylic acid (2.4 g, 11.1
mmol) in lithium hydroxide (lN, 11.1 mL,
deoxygenated) was ~e~ tetrahydro~uran (10 mLJ and
Boc-anhydride (2.g g, 13.3 mmol). After 30
minutes, a small amount of precipitate formed.
Water and tetrahydrofuran (10 mL each) were added
and stirred vigorously. After 2.5 hours, lN
hydrochloric acid (12 mL) was added and the mixture
- was extracted with ethyl acetate (20 mL). The
organic layer was dried (magnesium sul~ate) and
concentrated in vaCuo to afford Compound ~ (3.4 g,
97~).
~S (CI): (M+H)~ = 317.
s. (R~)-N-[[2-[(1,1-Dimethylethoxy)carbonyl]-
2,3,4,9-tetrahydro-lH-pyrido[3,4-b]indol-3-
vllc~rhon~ll-r-methionine meth~l ester
To a solution of Compound A (1.6 g, 5.0
mmol) in dimethylformamide (lS mL) at room
temperature under argon were added L-Methionine,
methyl ester, hydrochloride (1.0 g, S.0 mmol) and
diisopropylethyl~mine (2.65 mL, 15 mmol). The
mixture was stirred overnight (16 hours). The
reaction mixture became dark. The reaction mixture
-
21~3~88
LD69
was partitioned between ethyl acetate and lN
hydrochloric acid (25 mL each). The organic layer
was separated, washed with saturated sodium
bicarbonate and 10% lithium chloride (20 mL each),
dried (magnesium sulfate) ~nd concentrated in
vacuo. The residue was purified by silica gel
co~umn chromatography eluting with step gradient o~
25, 35 and 50% ethyl acetate in hexanes to afford
Compound B (980 mg, ~2%).
MS (CI): (M+H)~= 461.
TLC: Rf 0.27 (1:1 ethyl aceta~e:hexanes, visualized
by U.V. Ceric ammon. molybdate)
C. (R~)-N-[[2,3,4,9-~etrahydro-lH-pyrido[3,4-
b~indol-3-yl]-carbonyl]-L-methionine, methyl
ester. trifll~oro~cet~te s~lt
To a solution of Compound B (0.2 g, 0.43
mmol) in dichloromethane (1 mL) at room temperature
under argon were added dimethyl sulfide (0.4 mL),
trifluoroacetic acid (0.1 mL) and triethylsilane
(0.04 mL, 0.25 mmol). .~fter 3 hours,
trifluoroacetic acid (0.25 mL) was added. .~fter an
additional 2 hours, .he voiatiles were removed n
vacuo and the residue was -riturated with ether (3
x 2 mL) and dried in vacuo to afford a reddish oil
Compound C (220 mg, 100%).
Al~ernatively, Compound C was also prepared
by treating the same amount of Compound B with 98%
formic acid overnight (16 hours) and exchanging the
formate salt with TsOH salt tO afford Compound C
(219 mg, 100 %).
~14~588
LD69
- 36 -
D. (S*,R*)-N-[[2-[N-[2-[[(1,1-Dimethyl
ethoxy~carbonyl]amino]-3-~(triphenylmethyl)
thio]propyl]-~-valyl]-2,3,4,9-tetrahydro-
lH-pyrido[3,4-b]indol-3-yl]carbonyl]-L-
methi oni ne, meth~l e~t~r
Compound D was prepared using the method
described in Compound G o~ Example 1 ~except
dimethylformamide was not used.] Thus, Compound 2C
(210 mg, 0.~ mmol) and (S~)-N-[2-[[(1,1-
Dimethylethoxy~carbonyl]amino~-3-
[(triphenylmethyl)thioipropyl]-T-valine (219 mg,
0.~ mmol) were converted to compound D (82 mg,
23%).
MS (FAB): (M~H)' = 8g2.
TLC: R~ 0.21 (1:1 ethyl acetate:hexanes, visualized
by U.V., Ceric ammon. molybdate).
Compound D was also prepared on the same
scale using TsOH salt (81 mg, 23%). Both the
batches were combined and used in the next step.
E. (S~,R*)-N-~[2-[N-(2-Amino-3-mercaptopropyl)-L-
~alyl]-2,3,4,9-te~rahydro-lH-pyrido[3,4-b3indol-
3-yl]carbonyl]-L-methionine, trifluoroacetate
~ ) s~lt
The title compound was prepared using ~he
two step procedure described in Example lH ~except
that in the first step sodium hydroxide was used in
place of lithium hydroxide and the reaction mixture
was stirred for 1.5 hours and in the second step
1.2 equivalents instead of 2.0 equivalents of
triethylsilane was used.] Thus, Compound D (160
mg, 0.18 mmol) was converted to the title compound
(60 mg, 42%).
MS (FAB): (M~H)+ = 536.
2~3~8
LD69
- 37 -
m.p. 110-115C.
[a]D = + 18.9~ (c = 0.23, methanol).
IR (KBr): 2974, 1676, 1437, 1204 cm~l.
HPLC: YMC, S3, C18 (6 x 150 mm3 column, 220 ~m, 1.5
mL/minute: 10-90% a~ueous methanol with 0.2%
phosphoric acid, linear gradient o~er 30 minutes.
Retention time : 19.9 minutes.
Elemental analysis for C25H37N5O4S2~ 1.3 H2O .
2.1 TFA
C H N
Calculated 43.92 5.26 8.77
Found 44.12 5.04 8.42
lH NMR (CDCl3, with 5% CD30D, 270 MHz): d 7.49 (lH,
d, J=7.8 Hz~, 7.33 (lH, d, 7.8 Hz), 7.16-7.09 (2 H,
1~ m), 5.15 (lH, d, J-16 Hz~, 5.04 (lH, d, J= 4 Hz),
4.6 (lH, d, J=16 Hz~, 4.35 (lH, m), 4.09 (lH, d, J=
S Hz), 3.3-2.82 (7H, m), 2.3-1.8 (5H, m), 1.95 and
1.68 (3H, tWO S), 1.18-1.03 (6H, m).