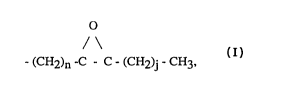Note: Descriptions are shown in the official language in which they were submitted.
O 94/06431 2 1 4 ~; 1 9 ~ PCI/US93/09073
NOVEL EPOXIDE-CO~AINING COMPOUNDS
Technical Field of the Invention
The invention relates to a class of epoxide-containing therapeutic compounds that act
S as drugs on the cellular and bioch~.mir!~l level to modulate cellular responses to noxious,
proinfl~mm~tory stimuli. ~ore sperific~lly, the inventive compounds have at least one
epoxide group on a~side chain bonded to a core moiety.
Bark~-~,u,--l of the Invention
Pentoxifylline (l-(S-oxohexyl)-3,7-dimethyl~nthinP), abbreviated PTX, is a ~nthine
derivative which has seen widespread medical use for the increase of blood flow. PTX is
disclosed in U.S. Patents 3,422,307 and 3,737,433. Metabolites of PTX were snmm~rized in
Davis et al., Applied Environment Microbiol. 48:327, 1984. A metabolite of PTX is 1-(5-
hydroxyhexyl)-3,7-dimethylxanthine, designated M 1. M 1 was also disclosed as increasing
cerebral blood flow in U.S. Patents 4,515,795 and 4,~76,947. In addition, U.S. Patents
4,833,146 and 5,039,666 disclose use of tertiary alcohol analogs of 7~nthine for enhancing
cerebral blood flow.
Furthermore, U.S. Patent 4,636,507 describes an ability of PTX and Ml, to stimulate
chemotaxis in polymorphonuclear leukocytes in response to a stimulator of chemotaxis. PIX
and related tertiary alcohol substituted ~c~nthines inhibit activity of certain cytokines to affect
chemotaxis (U.S. Patent 4,965,271 and U.S. Patent 5,096,906). ~rlminictr~tion of PTX and
GM-CSF decrease tumor necrosis factor (INF) levels in patients undergoing allogeneic bone
marrow tr~ncpl~nt (Bianco et al., Blood 76: Supplement 1 (522A), 1990). Reduction in
assayable levels of TNF was accompanied by reduction in bone marrow transplant-related
complications. However, in norrnal volunteers, TNF levels were higher among PTX
2s recipients. Therefore, elevated levels of TNF are not the primary cause of such
compli~tions.
Therefore, there is a need in the art to discover effective therapeutic compounds that
are safe and effective for human or animal a~lminictration and that m~int:~in cellular
homeostasis in the face of a variety of infl~mm~tory or noxious stimuli. The invention
results from investigations of such compounds.
Summary of the Tnvention
We have found that the compounds described herein can be used to m~int~in
homeostasis of a large variety of target cells in response to a variety of stimuli. In addition,
the inventive compounds and compositions are suitable for normal routes of therapeutic
~ minictration and permit effective dosages to be provided.
The invention is directed to epoxide-containing alkyl side chains bonded to a core
moiety useful in modulating cellular response to external or in sitl~ primary stimuli, as well as
to specific modes of ~llminictration of such compounds in effective amounts.
W O 94/06431 ~ l 4 ~ PC~r/US93/090
The inventive compounds compnse epoxlde-substituted aL~cyl side chain (R) bondedto a core moiety, comprising:
o
/\
Core moiety - (CH2)n -C - C - (CH2)j - CH3,
wherein n is an integer from about 4 to about 16 and j is an integer from about 0 to about 12,
in~luding resolved enantiomers and/or diastereomers, salts, solvates, hydrates and mixtures
thereof. Preferably, n is an integer from about 4 to about 12, more preferably from about 4
to about 10. J is preferably an integer from about 0 to about 3. The aL~yl groups may also be
0 substituted by a hydroxyl, halo or dimethylamino group and/or interrupted by an oxygen
atom, H or aL~yl (1-4C).
The core moiety may be a heterocyclic or a non-heterocyclic moiety. A non-
heterocyclic moiety is, for example, an amino acid (one or two), an hydroxyl group, a
carboxyl group, a sulfoxide group, a sulfonate group, a phosphate group, an amide, an amine,
1S a ketone, a simple ionic functional group, a terminal hydrogen or halogen atom. Exemplary
core moiety amino acids may include one or more of the following: al~ninP, arginine,
~cparagine, aspartic acid, cysteine, glut:~mine, glut~mic acid, glycine, hi~titlinP., isoleucine,
leucine, lysine, methionine, phenyh~l~ninP,, proline, serine, threonine, tryptophan, tyrosine
and valine. The core moiety may preferably be a dipeptide compri.~ing two amino acids
selPcted from the foregoing exemplary list. Exemplary halogen atoms include bromine,
chlorinP., fluorine and iodine.
Exemplary heterocyclic core moieties may be s--hstitllted or uns~lbstitutecl andpreferably may include, but are not limited to, substituted or unsubstituted phth~limitle
homophth~limide, quinazolidinedione, quinazoline, x~nthinP, glut,.rimide, pipçri~line,
piperidone~ ~y- valerolactam, cyclohexane, cyclohPxPne benzene, uracil, thymine, uracil fused
to napth~lP.ne~, ortho-phenol, imi~l~7c)1e amide, pyrrole amide, bPn7~mi~le,
tetrahydrophth~limide. or succinimide. Preferred heterocyclic core moieties may contain at
least one ring nitrogen atom, the R side chain being bonded to a ring nitrogen. Por example,
the heterocyclic moiety may be xanthinç, phthalimide, thymine, alkyl-substitllt~l (Cl-6)
thymine, uracil, alkyl-substituted (Cl-6) uracil, glutarimide, 2,5,4'-trihalobenzol)henone, 1,4-
trihalomethylben7~mide (preferably the halogen groups are selected from chloro, bromo,
iodo and fluoro) and resorcinol. More preferably, the heterocyclic core moiety may be 1,7-
methyl~r~.nthinP, 8-amino-3-methylxanthinP, 7-methylhypox~nthine
dimethyldihydroxypyrazolo[4,3-d]pyrimiclinP7 methylpyrrolo[2,3-d]pyrimirline~ 5- and 6-
suh~stituted uracils, 6-aminouracil, 2,4-dioxohexahydro-1,3,5-triazine, methylbarbituric acid,
isocarbostyril, 1,2,3,4-tetrahydroisoquinolin, 2-hydroxypyridine, 3,3-dimethylflutarimide,
1,3-dihydroxynapthalene, 1,3-cyclopentanedione, 2-pyrrole amide, 3-pyrrole amide, 1-
pyrrole amide and substituted ben7~midçs Preferable compounds of the invention, having
2 1 ~ ~ ~ 9 2 Pcr/US93/09073
a ~nthinP. core moiety, may include, but are not limited to, compounds having a single
epoxide-substit~lt~d alkyl side chain (R) at position 7 of the ~nthint-. nucleus.
The invention includes a method for modulating an immune response or a cellular
response to extçrn~l or in situ primary stimuli comprising ~tlmini.ctering an effective amount
s of an inventive compound. Particularly, on a cellular or biochPmic~l level, the il~ve-llive
compounds have been found to inhibit a specific phospholipid-based pathway that ~mplifi~s a
signal within a cell. This pathway tends to be activated in response to noxious or
infl~mm~tory stimuli. The inventive compounds also decrease prolifPr~tion of tumor cells in
response to an activated oncogene; stimulate hematopoiesis in the presence of agents which
10 inhibit hematopoiesis, such as chemotherapeutic agents; ~u~less the activation of T-cells in
the presence of antigen and the secretion of antibodies by B-cells in the presence of ~ntigen;
suppress the activation of macrophage or endothelial cells by endotoxins, tumor necrosis
factor (TNF), int~rleukin-l (IL-l) or granulocyte macrophage colony stimulating factor
(GM-CSF); enhance the resistance of mesenchymal cells to TNF; inhibit the proliferation of
15 smooth muscle cells, endothelial cells, fibroblasts and other cell types in response to growth
factors, such as platelet derived growth factor (PDGF), PDGF-AA, PDGF-BB, f1broblast
growth factor (FGF), epi-lerm~l growth factor (EGF), etc.; inhibit the activation of T-cells
and viral replic~tion in response to human imm-modçficip~cy virus; inhibit the prolif~r~tio~
of kidney mes~ngi~l cells in response to IL-l; prevent suppression of Steel factor (also called
20 stem cell factor, mast cell growth factor and kit ligand), granulocyte colony stim~ ting
factor (G-CSF),.oncostatin M or interleukin-6 (IL-6) in bone marrow stromal cells in
response to TNF; ~ul~pless expression of adhesion molecules in endothelial cells and suppress
~rlh~sio~ of infl~mm~tion cells to endothelial cells; suppress prolifçr~tion of kidney
mesz.ngi~l cells in response to IL- 1, mip-la, PDGF or FGF; prevent toxicity in kidney
25 glomerular or tubular cells in response to cyclosporin A or amphotericin B; prevent cytotoxic
effects in gastrointestinal or pulmonary epithelial cells in response to a cytotoxic drug or
r~ tion; çnh:~nre the ~ntitllmor effects in tumor cells in response to a nonaLkylating
~ntitumor agent; suppress the production of metalloproteases in synovial cells, other
fibroblasts and a glomerular epithelial cell in response to infl~mm~tory stimuli, such as TNF,
30 IL-l and the like; inhibit production of osteoclast-activating factor (OAP) by osteoclasts in
response to IL-l; inhibit degranulation of mast cells and basophils in response to IgE;
modulate signal transduction of the neurotr~ncmit~r.c epinephrine and acetylcholinP. in neural
palhw..y~ utilizing these transmitters, block activation of platelet activ,ating factor in
infl~mm~tion cells, block release of TNF and IL- 1 in various cell types in response to
35 infl~mm~tclry stimuli, block activation and proliferation of lymphocytes and other cell types
to IL-l and interleukin-2 (IL-2), and the like, including the clinical manifestations of these
cellular and biochçmical events.
In vitro, the inventive compounds: 1) block IL-l signal tr~ncductiQn through
the Type 1 receptor as shown, for example, by preventing IL-l and IL- 1 plus PDGF (platelet
WO 94/06431 2 1 4 ~ 1 9 ~ PCI/US93/0907:~
derived growth factor) induction of proliferation of smooth muscle and kidney mesengial
cells; 2) suppress regulation of adhesion molecules as shown, for example, by blocking
VCAM in endothelial cells of CD18 in neutrophils; 3) inhibit TNF, LPS and IL-1 induced
metalloproteases (an infl~mm~tion model); 4) block LPS, TNF or IL-l induced cellular
s activation (for prevention and tre~tment of septic shock); 5) suppress T cell and B cell
antigen activation by cross-linking CD3 complex; 6) inhibit mast cell activation by IgE; and
7~ suppress m:~lign lnt phenotype in transformed cells and tumor cell lines.
The inventive compounds, inter alia, inhibit signal trancrluction meAi~t~l
through the Type I IL-l receptor, and are therefore considered as IL-1 antagonists. Dinarello
and Wolff, "The Role of Interleukin-1 in Disease," N. Engl. J. Med 328, 106 (Jan. 14, 1993),
descrihe the role of IL-l as "an important rapid and direct determin~nt of ~ ea~e-ll "In
septic shock, for example, IL-l acts directly on the blood vessels to induce vaso~ t:~tion
through the rapid production of platelet activating factor and nitric oxide, whereas in
~qutQimmune disease it acts by stimulating other cells to produce cytokines or en~y.l.cs that
then act on the target tissue." Ibid. The article describes a group of ~lice~çs metli~ted by
IL-1, including many of the foregoing rlice~es
In still another aspect, the invention is directed to a ph~rm~ceutic~l composition
comprising an inventive compound and an effective amount of an agent which reduces the
activity of the enzyme P-450, such as a quinolone, to increase the ph:lnn~cokin~tic half-life
of an inventive compound.
Rrief Description of the Drawin~s
Figure 1 shows a mixed lymphocyte reaction of three inventive compounds CT1605
(N-(5,6-oxidohexyl) glutarimide), CT1808 (N3-(5,6-oxi-lohe~ryl)-Nl-methyluracil), and
CT1906 (N3-(5,6-oxidohexyl) N1-methylthymine). The mixed lymphocyte reaction shows a
~5 proliferative response of PBMC (peripheral blood mononuclear cells) to allogeneic
stimulation determined in a two-way mixed lymphocyte reaction. Each of the inventive
compounds tested was effective (and more potent than PTX although not shown on this
graph) in this immune modulating activity assay procedure.
Figure 2 shows a comparison of three dose levels of CT1808 and CT1906 and no
drug control to inhibit thymocyte proliferation. The thymocytes were obtained from normal
female Balb/C mice and stimulated with Concanavalin A (Con A) and/or interl~ukin-l alpha
(L-la). Drugs were added to the cell cultures two hours before activation with Con A and/or
IL-la. As shown in Figure 2, both drugs inhibited thymocyte proliferation is a dose-
dependent fashion.
Figure 3 shows a comparison of CT1605 and CT1808 on inhibition of B-cell
prolifer~tinn. A Ramos C-cell tumor line was treated with 250 ~LM CT1808 or CT1605 for
one hour prior to stimulation of proliferation with anti-mu antibody or phorbol myristic acid
(PMA, 5 nM). One day later, proliferation was measured with triti~t~ thymidine. Both
CT1605 and CT1808 inhibitPd proliferation in this model.
O 94/06431 ~ 2 Pcr/uS93/09073
Figure 4 shows a compalison of CT1605, CT1808 and CT1906 on PDGF-in~ ced
(platelet derived growth factor) proliferation of human stromal cells. Human stromal cells
were starved in serum-free media for 24 hours and then stimulated with 50 ng/ml of PDGF-
BB. The drugs were added at various in(lir:~ted concentrations one hour prior to PDGF
s stimulation. Tritiated thymidine was added for 24 hrs at the time of PDGF stim~ tion to
measure cellular proliferation. Background counts were approxim~tPly 5% of control levels.
All three drugs inhibited PDGF-indu~ed stimnl~tion in a dose response fashion.
Figure 5 shows the effect of CT1605, CT1808 and CT1906 to inhibit adhesion of
U937 cells to activated human umbilical vein endothelial cells (HUVEC). HUVEC cells
o were activated with 20 ng/ml of TNF for 12 hrs. Drug was added to each culture (except for
controls) one hour prior to adding TNF. U937 cells, preloaded with the fluorescent dye
BCECF, were added to each culture well and then washed. Cell adhesion was det~PrminP~ on
a fluorescence plate reader, showing a decrease in cell adhesion caused by all three drugs in a
dose dependent f~shio~
Figure 6 shows the effects of CT1605, CT1808 and CT1906 to inhibit cell surface
.c;ssion of VCAM in human llmbilic~l vein endothelial cells (HUVEC). The HUVEC
cells were stimulated with 20 ng/ml TNF-oc for 20 hrs and then stained for
immnnofluQrescence using a monoclonal antibody recognizing VCAM, followed by a goat
anti-mouse antibody conjugated to phycoerythrin. The cells were analyzed for antibody
binding using flow cytometry. Figure 6 shows an analysis of mean relative fluOlr.scçl-ce
intensity of 10,000 cells, analyzed by flow cytometry. The mean fluorescerlce levels were
decreased by all three drugs from control levels (TNF tre~tmP-nt no drug).
Detailed Desc~iption of the Invention
The invention is directed to a defined genus of inventive compounds which can
control cellular behavior by a particular phase of a secondary meccpnger pathway system
(Bursten et al., J. Biol. Chen7 266:20732, 1991). The second messenpr~s are lipids or
phospholipids and use the following abbreviations:
PE = phosph~tidyl ethz~nc l~mine.
LPE = lysophosphoethanolamine
PA = phosphatidic acid
LPA = lysophosphatidic acid
DAG = diacylglycerol
LPLD = lysophospholipase-D
LPAAT = lysophosphatidic acid acyl transferase
PAPH = phosph~titlic acid phosphohydrolase
PLA2 = phospholipase A-2.
PLD = phospholipase D
PAA = phospho~r~chidonic acid
PLA-2 = phospholipase A2
WO 94/06431 PCr/US93/0907
~145i92
PC = phosphatidyl choline
"remodeled" PA, cyclic pathway = PAA, LPA, PA and DAG intt~.rm~ tes
s--h~stitutecl with L-saturated, 2-linoleoyl or 1,2-dileolyl/1,2-sn-rlilinol~oyl at the in~lic?~t~l sn-
1 and sn-2 po.citionc.
"Cl~cci~l PI Pathway" = PI, DAG, PA intermetli~tl~c substituted with 1-stearoyl, 2-
~r~hi~lonoyl fatty acyl side chains.
"PLD-generated PA" = PE, PC, LPA, PA and DAG interm~Ai~tPs substituted with,
e.g., 1,2-sn-dioleoyl-, 1-aLkyl, 2-linoleoyl-, and l-aLkyl, 2-docos~heY~neoyl-side chains.
Lysophosph,.ti-lic acid transferase (LPAAT) effects the synthesis of phosph~ti~ acid
(PA) from lysophosphatidic acid (LPA) by incorporation of an acyl group from acyl CoA.
Hydrolysis of the phosphate moiety by PA phosphohydrolase (PAPH) results in the
form~tic-n of DAG. These aspects of the pathway appear to be activated immt~ tely (within
a minute) upon stimulation by a primary stimulus (e.g., a cytokine such as interleukin-l or
TNF) acting at a receptor on a cellular surface. An imme~ te detectable effect is an
elevation of levels of PA and DAG. ~(lminictration of the compounds of the invention
reverse this elevation.
The compounds of the invention, include inhibitors of sukspecies of LPAAT in
PAPH enzymes with substrate specificity for intermerli~t~s with 1,2-~ -nc~t--rated and l-
alkyl, 2-unsaturated subspeci~s One representative example of such an inhihitor (although
not within the genus of inventive compounds) is PTX. PTX blocks PAPH in a specific
activation pathway that does not involve PI but rather derives from a PA that is largely
composed of 1,2-diunsaturated and 1-alkyl,2-unsaturated subspecies. This was shown, for
e~mple, by the demonstration that human mes~ngi:~l cells stimulated with TNF produce
DAG from PI and regenerate PI in the absence and the presence of PTX. In the latter system
there is no evidence to suggest that PA or DAG are derived from sources other than PI. It
should be emphasized that the compounds of the invention affect that subset of PAPH and
LPAAT that relates to substrates with unsaturated fatty acids other than ~r~chi~lon~tt~. in the
sn-2 position, not the housekeeping forms of these enzymes that serve the PI pathway.
Compounds of the Invention
The inventive compounds comprise epoxide-substituted alkyl side chain (R) bondedto a core moiety, comprising:
o
/\
Core moiety - (CH2)n -C - C - (CH2)j - CH3,
wherein n is an integer from about 4 to about 16 and j is an integer from about 0 to about 12,
including resolved enantiomers and/or diastereo~ners, salts, solvates, hydrates and mixtures
thereof. Preferably, n is an integer from about 4 to about 12, more preferably from about 4
to about 10. J is preferably an integer from about 0 to about 3. The alkyl groups may also be
~94/06431 214~1~æ PCr/US93/09073
substituted by a hydroxyl, halo or dimethylamino group and/or interrupted by an oxygen
atom, H or alkyl (1-4C).
The core moiety may be a heterocyclic or a non-heterocyclic moiety. A non-
heterocyclic moiety is, for example, an amino acid (one or two), an hydroxyl group, a
carboxyl group, a sulfoxide group, a sulfonate group, a phosphate group, an amide, an amine,
a ketone, a simple ionic functional group, a terminal hydrogen or halogen atom. Exemplary
core moiety amino acids may include one or more of the following~ ninP, arginine,
~p~r~ginP., aspartic acid, cysteine, ~hlt~minP, glut~mic acid, glycine, hicti-linP, isoleucine,
lP~l~inP, lysine, methionine, phenyl~ nine~ proline, serine, threonine, tryptophan, tyrosine
and valine. The core moiety may preferably be a dipeptide cornpri.~ing two amino acids
selPrtçd from the foregoing exemplary list. Exemplary halogen atoms include, but are not
limited to, bromine, chlorine, fluorine and iodine.
Fy~Pmpl~ry heterocyclic core moieties may be snbstitutp~ or unsubstitut~pd~ and
preferably may include, but are not limited to, substituted or unsubstituted phth~limitle7
homophth~limi~le, quinazolirlinçdione, quinazoline, x~nthinç., glut~rimide, piperi~linP,
piperidc-nP" y- valerol~st~m, cyclohç7c~ne, cyclohP~rPnP~ be--7PnP, uracil, thymine, uracil fused
to napth~lPne, ortho-phenol, imid~7c)1e amide, pyrrole amide, bçn7~mide,
tetrahydrophth~limide or succinimide. Preferred heterocyclic core moieties may contain at
least one ring nitrogen atom, the R side chain being bonded to a ring nitrogen. For ex~mplP,
the heterocyclic moiety may be x~nthine, phth~limide, thymine, alkyl-s-lbstitutP~l (C1-6)
thymine, uracil, aLIcyl-substituted (Cl-6) uracil, glutarimide, 2,5,4'-trihalobenzophPnonP, 1,4-
trih~lomethylbçn~mi(le (preferably the halogen groups are selected from chloro, bromo,
iodo and fluoro) and resorcinol. More preferably, the heterocyclic core moiety may be 1,7-
methyl~r~nthinç, 8-amino-3-methylx~nthinç, 7-methylhypox~nthine,
2s dirnethyldihydroxypyrazolo[4,3-d]pyrimirline, methylpyrrolo[2,3-d]pyrimi-lins, 5- and 6-
substituted uracils, 6-aminouracil, 2,4-dioxohexahydro-1,3,5-triazine, methylb~'Lilulic acid,
isocarbostyril, 1 ,2,3,4-tetrahydroisoquinolin, 2-hydroxypyridine, 3,3-dimethylfl ~ le,
1,3-dihydrokyllapth~lçnç, 1,3-cyclopçnt~n~lione, 2-pyrrole amide, 3-pyrrole amide, 1-
pyrrole amide and substituted ben7~miclçs Preferable compounds of the invention, having
a x~nthine core moiety, may include, but are not limited to, compounds having a single
epoxide-substit--ted alkyl side chain (R) at position 1 of the x~nthine nucleus.The inventive compounds may be provided as enantiomeric or diastereomeric
nli~lur~s or in resolved or partially resolved forms. Standard procedures are used for
resolution of optical isomers. It is contemplated that the different en~ntiome;ic variants (e.g.,
stereoisomers and chiral forms) of the epoxide-cont~ining compounds will have different
drug activities, based upon their differential ability to inhibit PAPH and LPAAT. An optical
isomer s--bst~nti~lly free of the corresponding en~ntiolner and/or diastereomers has at le~st
about 85% relevant optical isomer, preferably at least about 95% relevant optical isomer and
WO 94/û6431 2 1 ~ 2 PCI/US93/0907
especially at least about 99% or higher relevant optical isomer, but most preferably where the
amount of other optical forms is undetectable.
The invention further comprises a pharmaceutical composition comprising one or aplurality of inventive compounds and a pharrn~relltic~lly acceptable carrier or excipient The
s cells to be treated with an inventive compound or inventive pharrn~reutical composition may
either be contacted with the compound of the invention in in vitro culture, in an
extracorporeal tre~tment, or by ~clmini~tering the compound of the invention or
pharma~euti~l composition thereof to a subject whose cells are to be treated.
Illustrative compounds of the invention include both racemic mixture and R and S10 en~ntiom~ors of the following compounds (clesign~tP~ accordingly as R and S) shown in Table
I:
Table I
CT1103 N-(5,6-Oxidohexyl)phth~limitle
CT1105 N-(8,9-Oxidononyl)phth:~limide
CT1109 N-(10,11-Oxidoundecyl)phth~limi~le.
CT1114 N-(10,11-Oxidoundecyl)homophth~limide.
CT1206 1-(5,6-Oxidohexyl)-3-methylbenzoyleneurea
CT1301 N-(5,6-Oxidohexylamido)glutaric acid, methyl ester
CT1409 1-(8,9-Oxidononyl)-3-methyl-7-methylpivaloylx~nthine
CT1410 1-(5,6-Oxidononyl)-3-methyl-7-methylpivaloyl~nthin~.
CT1412 1-(11,10-Oxidoundecyl)-3-methyl-7-methylpivaloylx~nthine
CT1553 1-(7,8-Oxidooctyl)-3,7-dimethylx~nthine
CT1553 1-(7,8-Oxidooctyl)-3,7-dimethylx~nthin~.
CT1555 1-(4,5-Oxidohexyl)-3,7-dimethylx~nthine
CT1560 1-(8,9-Oxidononyl)-3,7-dimethylx:~nthine
CT1565 1-(9,10-Oxidodecyl)-3,7-dimethylx~nthine
CT1569 1-(6,7-trans-O~ri(lononyl)-3,7-dimethylx~nthine
CT1586 1-(6,7-Oxidoheptyl)-3,7-dimethylx~nthine
CT1588 1-(3-(R)-Methyl-7-methyl-(6,7-oxidooctyl)-3,7-dimethylx~nthine.
CT1593 1-(4,5-Oxidopentyl)-3,7-dime~}lylY ~nthine
CT1594 1-(7,8-Oxidoundecyl)-3,7-dimethylx~nthine
CT1605 N-(5,6-Oxidohexyl)glutarimide
CT1606 N-(8,9-Oxidononyl)glutarimide
CT1611 N-(10,110xidoundecyl)glutarimide
CT1618 N-(10,11-Oxidoundecyl)-2-pieridone
CT1619 N-(5,6-Oxidohexyl)piperidine
CT 1804 3-(8,9-Oxidononyl)- 1 -methyluracil
CT1808 3-(5,6-Oxidohexyl)-l-methyluracil
CT 1820 3-(5,6-Oxidoheyl)- 1 -methyldihydrouracil
~ 94/06431 ' 2 1 4 5 1 9 2 PCl/US93/09073
CT1822 3-(10,11-Oxidoundecyl)-l-methyldihydro,uracil
CT1906 3-(5,6-Oxidohexyl)- l-methylthymine
CT1910 3-(8,9-Oxidononyl)- l-methylthymine
CT1932 3-(11,10-Oxidoundecyl)-l-methylthymine
CT2513 1-(3,4-Oxidobutyl)-3,7-dimethylx~nthine
CT2518 1-(11,12-Oxidodecyl)-3,7-dimethyl~nthin~.
-........ CT2~41 1-(9,10-Oxidooctadecyl)-3,7-dimethylxanthine
CT2548 1-(4-Methyl-7,8-oxido-8-methylnonyl)-3,7-dimethylx~nthine.
CT25~2 1-(3,7-dimethyl-2,3,6,7-dioxidooctyl)-3,7-dimethylx~nthine
CT2562 1-(12,13-Oxidotridecyl)-3,7-dimethylx~nthine
CT2563 1-(7,8-cis-Oxidodecyl)-3,7-dimethylx~nthine
CT3503 1-(13,14-Oxidotetradecyl)-3,7-dimethylx~nthine
CT3516 1-(16,17-Oxidoheptadecyl)-3,7-dimethylx~nshint-.
Uses of the Tnvention Compounds ~nd Pharmaceutical Formulations
The compounds of the invention provide a mechanism to m~int~in hom~ost~ci.c in
cells cont~rted by primary stimuli through mitig~ting the effects of these primary stimuli on
the second:lry .cign~ling pathways invoked within secon-ls of the primary stim~ c
S The foregoing in vitro effects give rise to inventive pharm~r~euti~l
compositions comprising an effective amount of at least one of the inventive compounds or a
ph~rmaeeutically acceptable salt, hydrate or solvate thereof and at least one ph~rm:l~e~ltic~lly
acceptable excipient or carrier. Because the inventive compounds, inter alia, inhibit
cellular sign~ling, me~ t.o.~l for example by the IL-1 Type I receptor and are IL-1
antagonists, the inventive pharmaceutical compositions are useful for: 1) protecting and
treating endotoxic shock and sepsis inclucecl by gram positive or negative bacteria; 2)
inhibiting, treating or preventing tumor cell growth, such as cancer; 3) stim~ ting
hematopoiesis inhibited by cytoreductive therapies (e.g., chemotherapy or radiotherapy); 4)
treating or preventing autoimmune ~ice~ces~ such as insulin dependent ~ hetes m~.llitllc
(IDDM), arthritis (including rheumatoid arthritis), multiple scherosis, ~17.h~imers (lice~ce,
glomerular nephritis, Graves tli.ce:~ce, and atheroschlerosis; S) treating or preventing male
pattern b~ltin~ss by stimulation of hair growth through reversal of an apoptotic process; 6)
preventing hair loss caused by cytoreductive therapies; 7) preventing the symptoms of ARDS
(acute respiratory distress syndrome) caused by trauma; 8) treating or preventing ~cthm:l,
infl~mm~tory bowel (lice~ce, acute and myelogenous leuk~.mi~, transplant rejection, psoriasis,
osteoporosis, periodontal lli.ceace, autoimmune thyroiditis, alcoholic hepatitis, premature
labor secondary to uterine infection and even sleep disorders; and 9) preventing synergistic
immunosuppression in GVHD (graft versus host disease).
Excessive or unregulated TNF (tumor necrosis factor) production may play a
2s role in m~.cli~ting or exacerbating a number of li.ce~ces including rhP.-lm~toid arthritis,
rheum~toid spondylitis, osteoarthritis, gouty arthritis and other arthritic conditions; sepsis,
W O 94/06431 2 1 4 ~ 1 9 2 PC~r/US93/090 ~
septie shock, endotoxie shock, gram negative sepsis, toxic shock syndrome, adult respiratory
distress syndrome, cerebral malaria, chronic pulmonary infl~mm:ltory ~lice~ce7 silicosis,
pulmonary sarcoidosis, bone resorption flice~ces, reperfusion injury, graft versus host
rea~tion allograft rejections, fever, myalgias due to infection such as influ~n7~ eachexia
seeondary to infection, AIDS or malignancy, AIDS, other viral infections (e.g., CMV,
inflnPn7~, adenovirus, herpes family), keloid form~tio~) scar tissue form~tiol-, Crohn's
fliceace, uleerative eolitis, or pyresis. The inventive eompounds or ph~rrn~rel~tir~lly
aeeeptable salts thereof ean be used in the manufaeture of a me-lieam-~nt for the prophylaetie
or therapeutie treatmPnt of any disease state in a human or other m:lmm~l, which is
exacerbated or cign~le-l 1) through the specific phospholipid-based mPss~nger pathway that
~mplifies signals within a cell, and 2) by exeessive or unregulated produetion of mPCcPngPr
infl~mm~tory eytokines such as TNF or IL-1. With regard to TNF me~csenger sign~ling,
there are several disease states in which excessive or unregulated monoeyte/macrophage TNF
production exacerbates or causes the tlice~ce These include, for example, neurodegerlP.r~tive
rlice~c-ps such as ~l7~heimers rlice:~ce~ endotoxemia or toxic shock syndrome (Tracey et al.,
Nature 330:662, 1987 and Hinshaw et al., Circ. Shock 30:279, 1990); e~rhpyi~ (Dezube et
al., Lancet 355:662, 1990), and adult respiratory distress syndrome (Miller et al., Lancet
2(8665):712, 1989). The inventive eompounds may be used topieally in the tre~tment of
prophylaxis of topieal disease states me(li:lted or exaeerbated by exeessive TNF or IL 1, sueh
as viral infeetions (herpes or viral eonjunetivitis), psori~ci.c, fungal or yeast infectionc
tringworm, athletes foot, vaginitic, dandruff, etc.) or other d~rm~t~lngie hyperproli~.dlive
disorders. High TNF levels have been implie~ted in acute m~l~ri~ attacks (Grau et al., N.
Engl. J. Med 320:1585, 1989), ehronie pulmonary infl~mm~tory iice~ces such as silieosis
and asbestosis (Piguet et al., Nahlre 344:245, 1990, and Bicso~n~t~P et al., Inflammation
13:329, 1989), and reperfusion injury (Vedder et al., Proc. Natl. Acad. Sci. USA 87:2643,
1990). Therfore, the inventive eompounds may preferably be used to stimulate
hematopoiesis, prevent and treat septic shock, treat acute and chronic infl~mm~tory ~lice~ce~
treat or prevent an autoimmune rliceace~ treat a fungal or yeast infeetion, and stimul~e hair
growth (when applied topieally, as eonfirmed in in vivo results on nude mice).
The inventive compounds are useful as an adjuvant to inhibit toxie side effeets
of drugs. These side effeets include, for example, side effects of: 1) interleukin-2 (IL-2); 2)
eyelosporin A and FK506; and 3) amphotericin B. The inventive compounds also inhibit
antigen-in-luced T cell activation, like eyclosporin or FK506, but, unlike eyclosporin or
FK506, do not: 1) prevent generation of NK and LAK eells; 2) ~up~;ss IL-2 release from T
eells; or 3) suppress IL-8 release.
Metalloproteases mediate tissue damage such as glome'rular rlice~ces of the
kidney, joint destruction in arthritis, and lung destruetion in emphysema, and play a role in
tumor met~ct~ces. Three examples of metalloproteases include a 92 kD type V gel~tin~se
incluced by TNF, IL-1 and PDGF plus bFGF, a 72 kD type IV eollagenase that is usually
~ 94/06431 ~ 2 PCr/US93/09073
constitutive and inr1uced by TNF or IL-l, and a stromelysin/PUMP-l in~uced by TNF and
IL-l. The inventive compounds can inhibit TNF or IL-l induction of the 92 kD type V
gel~tinzl~e induc~ble metalloprotease. Moreover, the inventive compounds can reduce
PUMP-1 activity inrl~lced by 100 U/ml of IL-l. Accordingly, the inventive compounds
5 prevent induction of certain metalloproteases induced by IL-l or TNF and are not involved
with conctit~ltively produced proteases (e.g., 72 kD type IV coll~gçn~ce) involved in normal
tissue remodeling.
For example, the compounds of the invention are used in connection with patientsundergoing bone marrow transplantation (BMT), regardless of whether the BMT is m~tchP-l
10 allogeneic, micm:lt~hPd allogeneic, or autologous. Patients receiving autologous transplants
are aided by treatmPnt with compounds of the invention even though they do not n~ocçssz~. ;ly
need to be z~lmini~tered immuno~.u~lGs~ /e agents, since they do not develop graft-versus-
host disease (GVHD). However, the toxic effect of the chemotherapy or radiation therapy
used in co~nçction with the disease, in response to which the transplantation has been
5 performed, con~titlltPs a negative stimulus with regard to the patients' cells.
In general, all patients undergoing BMT require doses of chemotherapy with or
without total body irradiation that exceed the lethal dose for normal bone marrow recovery.
This provides the rationale for using either stored patient marrow or donor marrow to rescue
the patient. In general, chemotherapy and radiation are delivered to the patient for 7-10
20 consecutive days before the new or stored bone marrow is infused. The day on which the
marrow is given to the patient is referred to as day 0 of the transplant. Previous days on
which the patient received chemo/radiation are ~lPsign~tçd by negative numbers. Subsequent
days are referred to by positive numerals. The median time in which negative responses in
BMT recipients occurs is within the first 100 days after trzln~pl:lnt Therefore, statistically, if
25 p~tientc survive through day 100, their ch:~nces for continued survival are .~ignifirzlntly
enhz~nce-l The inventive compounds are able to increase the percent~gP of p ltientC who
survive. The percentage of f:lt~lities within the first 100 days that is considered acceptable is
15-20% for "good risk" patients and 30-40% for "high risk". These fzt~litiPs are due to the
direct effects of high doses of chemo/radiation. In addition, GVHD contributes to the death
30 rate in allogeneic marrow recipients.
Other indications for which it is useful to ~rlmini~ter the compounds of the invention
include the presence of a tumor burden, a hormone-related disorder, a neurological disorder,
an autoimmune ~lice~e, infl~mm:ltion, restenosis, hypertension, unwanted immune response,
viral infection, nephritis, mucositis, and various allergic responses. Prevention of allergic
35 responses include prevention of acute allergic response and thus moderation or prevention of
rhinorrhea, serious drainage, diffuse tissue edema, and generalized pruritus. Other symptoms
of chronic allergic response include, as well .lS the foregoing, tli77inPss, diarrhea, tissue
hyl~elenlia, and lacrimal swelling with localized lymphocyte infiltration. Allergic reactions
are also associated with leukotriene release and the distal effects thereof, incllltling asthm~tic
1 1
WO 94/06431 2 1 4 5 1 9 2 PCr/US93/0907:--
symptoms including development of airway obstruction, a decrease in FEVl, ch~nges in vital
capacity, and extensive mucus production.
Other suitable subjects for the ;l-lminictration of compounds of the invention, include
patients being :ldmini.ctered toxic agents for the tre~tment of tumors, such as
chemotherapeutic agents or irradiation therapy, as well as tre~tment with biological response
modifiers such as
IL-2 and tumor suppressing cells such as lymphokine activated killer cells (LAK) and tumor-
infiltrating lymphocytes (IIL cells); patients suffering from neoplasias genPr~lly, whether or
not otherwise treated including acute and chronic myelogenous lellkPmi~, hairy cell
lPnkP.mi~, lymphorn~c, megakaryocxtic lel-kemi~ and the like; disease states caused by
b~ctP.ri~l, fungal, protozoal, or viral infection; patients exhibiting unwanted smooth muscle
cell proliferation in the form of, for example, restenosis, such as patients undergoing cardiac
Sulgt;ly; patients who are afflicted with autoimmune tlice~ces~ thus requiring deactivation of
T and B cells, and patients who have neurological disorders.
The compounds of the invention further are able to decrease the çnh~nred levels of a
relevant PA and DAG resulting from stimul ltion of synaptosomes with acetylcholine and/or
epinephrinP. This suggests that the effects of the compounds of the invention are to both
f~nh~n~e the release of inhibitory neural tr~ncmitters such as dopamine, and to modul~te the
distal "slow current" effects of such neurotr~n-cmitters~
Thus, the drugs of the invention are also useful to raise the seizure threshold, to
st~hili7P synapses against neurotoxins such as strichninP, to potentiate the effect of anti-
Parkinson drugs such as L-dopa, to potentiate the effects of soporific compounds, to relieve
motion disorders resulting from a-lminictration of tranquilizers, and to ~liminich or prevent
neuron overfiring associated with progressive neural death following cerebral vascular events
2~ such as stroke. In addition, the compounds of the invention are useful in the tre~tment of
norepinephrine-deficient depression and depressions associated with the release of
endogenous glucocorticoids, to prevent the toxicity to the central nervous system of
deY~mPthasone or methylprednisolone, and to treat chronic pain without ~ iction to the
drug. Further, the compounds of the invention are useful in the tre~tment of children with
1P~rning and attention deficits and generally improve memory in subjects with organic
deficits, in~lutling ~17hPimer's patients.
While dosage values will vary, therapeutic efficacy is achieved when the compounds
of the invention are ~lminictered to a human subject requiring such treatment as an effective
oral, parenteral, or intravenous sublethal dose of about 200 mg to about 5000 mg per day,
depen-ling upon the weight of the patient. A particularly preferred regimen for use in
treating lPukPmi~ is 4-50 mg/kg body weight. It is to be understood, however, that for any
particular subject, specific dosage regimens should be adjusted to the individual's need and to
the professional judgment of the person a~lminict~Pring or supervising the ~lmini~ctration of
the inventive compounds.
12
2 1 4 ~ Pcr/US93/09073
Coadministration With a P-4~() Inhihitor
The co;~-1minictration in vivo of the compounds of the invention along with an
inhibitor of P-450 results in an enhanced effect due to a longer half life of the inventive
compounds. This in vivo effect is due to inhibition of a degradation pathway for the
compounds of the invention. For example, NIH3T3-DSC3 cells can be used to compare
effects of an inventive compound alone or in combination with a P-450 inhibitor by
comparing transformation phenotype control, incubation with an i--ventive compound, and
coincnb~tiQn of an inventive compound with the P-450 enzyme inhibitor.
Compounds that inhibit P-450 include, for example, (mg range daily dosage)
0 ~ro~ olol (20-100), metaprolol (20-100); verapamil (100-400), diltiazem (100-400),
nifedipine (60-100); cimetidine (400-2,400); ciprofloxacin (500-2000), enoxacin (500-
2,000), norfloxacin (500-2000), ofloxacin (500-2,000), pefloxacin (500-2,000); ely~ olllycin
(100-1,000), troleandomycin (100-1,000); ketoconizole (100-2,000), thiabenzadole (100-
1,000); isoniazid (100-1000); mexiletine (100-1,000); and dex~mPth~cone (1-100 mg).
For combination therapy, the compounds of the invention and a P-450 inhibitor can
be ~dminictered individually or in a single composition. A suitable form~ tion will depend
on the nature of the disorder to be treated, the nature of the merlic~ment chosen, and the
jndgment of the attending physician. In general, the inventive compounds are formulated
either for injection or oral a~lminictration, although other modes of a~lministration such as
transmucosal or transdermal routes may be employed. Suitable formulations for these
compounds can be found, for example, in Remington 's Pharrr nce~ticn~ Sciences (latest
edition), Mack Publishing Company. Easton, PA.
Depending on the inventive compound selected, the level of dosage can be
appreciably rliminiched by co~-lminictration of a P-450 inhibitor, such as a quinolone.
Alternatively, a strong synergistic effect may be obtained with such a q-linolone
The invention, illustrated by the following examples, should not be deemed limited
by these examples in any way. In these examples PTX means pentoxifylline.
F.~ ple 1
This example illustrates a synthesis of N-(5,6-Oxidohexyl)phth~limi~le (CT1103). 1-
bromo-5-hexene (6.52 g,40 mmol) was added to a potassium phth~limi~le (7.4 g,40 mmol)
suspension in 50 mL of dimethyl sulfoxide and stirred overnight. After 12 hours of stirring at
room temperature, the reaction was poured into a separatory funnel cont~ining 300 mL of
water and extracted with dichloromethane (5 X 200 mL). The organic extracts werecombined, washed with water (100 mL) and brine (100 mL), dried over anhydrous
m~gne.~illm sulfate and concentrated under reduced pressure. The crude product obtained was
further purified by flash chromatography over silica gel (eluant: 10% acetonelhexane) to
yield 9.2 g (100% yield) of an N-(5-Hexenyl)phth~limide (CT1101).
A solution of N-(5-Hexenyl)ph~h~limide (1.145 g,5 mmol), prepared as described
above, and m-chloroperoxybenzoic acid (2.58 g,7.5 mmol, S0% by wt) in dichloromP,tl lmP.
13
WO 94/06431 2 1 4 ~ 1 9 2 PCr/US93/0907~
(30 mL) was stirred for S hours. The reaction mixture was diluted with 80 mL of
dichlorometh~ne and washed succe~cively with 20% aqueous sodium sulphite solution (20
mL), saturated sodium bicarbonate solution (20 mL), water and brine solutions. The organic
layer was dried over anhydrous m:lgnecillm sulfate and concentrated under reduced pressure.
s The crude product obtained was further purified by flash chromatography over silica gel
(using an eluant of 50% hexane and ethyl acetate) to yield 0.918 g (75% yield) of N-(5,6-
Oxi~loh~xyl)phth~limi~le
F.~ ple 2
This example illustrates a synthesis of N-(9,8-Oxidononyl)phth~limi-le (CT1105). 1-
bromo-8-nonene (8.2 g, 40 mmol) was added to a suspension of pot~c~illm phth:llimi~le (7.4
g, 40 mmol) in 50 mL of dimethyl sulfoxide and stirred overnight. After 12 hours of stirring
at room temperature, the reaction product was poured into a separatory funnel cont~ining 300
mL of water and extracted with dichlorometh~ne (5 X 200 mL). Organic extracts were
combint-d, washed with water (100 mL) and brine (100 mL), dried over anhydrous
m:~gn~cillm sulfate and concentrated under reduced pressure. The crude product obtained
was further purified by flash chromatography over silica gel (using an eluant of 10%
~ceton~/hexane) to yield 7.6 g (70.4 % yield) of N-(8-Nonenyl)phth~limitle (CT1101).
A solution of N-(8-Nonenyl)phth:~limide (1.355 g, 5 mmol), prepared in accordance
with the above process, and m-chloroperoxybenzoic acid (2.58 g, 7.5 mmol, 50% by wt) in
dichloromethane (30 mL) was stirred for 5 hours. The reaction mixture was diluted with 80
mL of dichloromethane and washed successively with 20% aqueous sodium sulphite solution
(20 mL), s~tnr~te-l sodium bicarbonate solution (20 mL), water and brine solutiollc~ The
organic layer was dried over anhydrous m:lgnesium sulfate and concentrated under reduced
pres~u,e. The crude product obtained was further purified by flash chromatography over
silica gel (using an eluant of 50% hexane/ethyl acetate), yielding 1.03 g (70% yield) N-(9,8-
Oxidononyl)phth~limitle
Example 3
This example illustrates a synthesis of N-(10,11-Oxidoundecyl)phth~limi~le
(CT1109). To a suspension of potassium phth~limicle (4.17 g, 22.5 mmol) in
dimethylsulfoxide (30 mL) was added l l-undecenyl bromide (available through MTM) (5.0
g, 21.5 mmol) and the reaction mixture stirred for 16 hours at 60C. The mixture was then
poured into water (80 mL) and extracted with ethyl acetate (3 x 70 mL). The combined
extracted organic portions were washed with water (3 x 100 mL), dried with m~gnPsillm
sulfate and evaporated, resulting in a cream-colored solid. Purification by column
chromatography (using ethyl acetate/hexane) yielded (5.50g, 86%) N-(10-
Undecenyl)phth~limi~le (CT-1107) as a white solid.
A solution of N-(10-Undecenyl)phth~limi(le (4.97 g, 16.6 mmol), prepared as
discribed above, 4-methylmorpholine-N-oxide (8.62 mL, 60% by wt in water, 50.0 mmol)
and potassium osmate dihydrate (58 mg, 0.16 mmol) in 100 mL acetone/water (1:1 by wt)
14
L) 94/06431 2 ~ 2 pcr/us93/09073
was stirred for 16 hours. Water (100 mL) and sodium sulfite (10 g) were added and the
resu~ting solution stirred for an ad-lition:7l hour. The resulting reaction mixture was extracted
with dichlorometh~nf~ (3 x 80 mL) and the organic phase dried using m~gn-~inm sulfate and
evaporated to obtain 3.93 g (71% yield) of N-(10,11-Dihydroxyundecyl)phth~limi~le
(CT1108).
N-(l0,11-Dihydroxyundecyl)phth:~limide (2.35 g, 7.10 mmol) was stirred for 3 hours
with HBr (6.90 mL of a 30% solution in acetic acid, 21.3 mmol). The mixture was then
added over 10 minutes to a solution of water (50 mL), ice (25 g) and NaHCO3 (15 g) and
stirred for an additional 30 min. The resulting reaction product was extracted with
dichlorometh~ne (3 x 70 mL) and the combined organic phase was dried using m~En.ocillm
sulfate and evaporated, yielding a residue of N-(10-acetoxy- 11-bromoundecyl)phth~limi~le
Without further pllri~lc:ltion~ this crude product was treated in methanol (10 mL) with
a solution of sodium methoxide (prepared from sodium -- 0.23 g, 10.0 mmol -- and 10 mL
meth~nol). After 60 minutes the reaction mixture cont~ining treated crude product was
added to water (30 mL) and extracted with dichlorometh~ne (100 mL, 2 x 50 mL). The
extracted organic portions were combined, dried and evaporated, yielding 2.00 g (89% yield)
N-(10,11-Oxidoundecyl)phth~limicle. (CT1109) as a white solid.
F~Y~n1PIe 4
This example illustrates a synthesis of N-(10,11-Oxidoundecyl)homophth~limi-le
(CT1114). A mixture of homophthalic acid (54.0 g; 0.3 mole) and finely powdered urea
(19.82 g; 0.33 mole) were heated to 175-185C until no more ammonia evolves, evidenced
by pH paper. The crude product was refluxed with methanol (500 mL) and
homophth~limide isolated by filteration (29g; 60%). Sodium hydride(95%) (576 mg, 24
mmol) was added to a solution of homophth~limi-le (3.2 g, 20 mmol) in anhydrous
dimethylsulfoxide (75 mL). After 20 minutes of stirring, 1-bromoundec-10-ene (5.6 g, 24
mmol) was added. After 16 hours of stirring at room temperature, the reaction mixture was
poured into a separatory funnel cnnt~ining 500 mL of water and extracted with ethyl acetate
(3 X 100 mL). The organic extracts were combined, washed with water (100 mL) and brine
(100 mL), dried over anhydrous m~gnesitlm sulfate and concentrated under reduced pressure.
The crude product obtained was further purified by flash chromatography over silica gel
using petroleum ether/5% ethyl acetate eluant to yield 2.2 g (35.5% yield) N-(10-
Undecenyl)homophth~limide
A solution of olefin N-(10-Undecenyl)homophth~limide (1.4 g, 4.5 mmol), and m-
chloroperoxybenzoic acid (2.3 ~, 6.7 mmol) (50% by wt) in dichlorom~-th~n~ (50 mL) was
stirred for 5 hours. The reaction mixture was diluted with 80 mL of dichloromethane and
washed successively with 20% aqueous sodium sulphite solution (20 mL), S~t~r?lt~Cl sodium
bicarbonate solution (20 mL), water (50 mL) and brine solutions (50 mL). The organic layer
was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The
crude product obtained was further purified b~ flash chromatography over silica gel using
WO 94/06431 2 1 4 5 1 ~ 2 PCI/US93/0907
75% hexane/ethyl acetate eluant to yield 1.38 g (94% yield) N-(10,11-
Oxidoundecyl)homophth~limide
FY~ e S
This example illustrates a synthesis of 1-(5~6-oxidohexyl)-3-methylbenzoylenplme~
(CT1206). Sodium metal (0.071 g, 3.1 mmol) was dissolved in methanol (3.1 mL) toprepare a 1.0 molar solution of sodium methoxi-lp l-(S-Acetoxy-6-bromohPYyl)-3-
methylbenzoyleneurea (1.17 g, 2.9 mmol) was dissolved into methanol ( 25 mL) and added
to the sodium methoxide solution over 5 minutes. After stirring for 1 hour, 50 mL of water
were added to the solution. The acqueous phase was extracted using three 25 mL aliquots of
dichlorome.th~ne The organic phase was dried over sodium sulfate, filtered and the solvent
removed under vacuum yielding 0.77 g (97% yield) of white, solid 1-(s~6-oyitlohpyyl)-3
methylbenzoyleneurea) .
F.Y~nP1e 6
This example illustrates a synthesis of 3-Methyl-7-methylpivaloyl-1-(8,9-
oxi-lononyl)x~nthine (CT1409). A mixture of 3-methylx~nthin~ (Aldrich, 1.00 g, 6.0 mmol),
sodium hydride (145 mg, 6.0 mmol) and dimethyl sulfoxide (20 mL) was stirred until
homogeneous (0.5 hours). Chloromethylpivalate (865mL, 904 mg, 6.0 mmol) was added
and the reaction stirred for 18 hours. The reaction mixture was poured into water (70 mL)
and then extracted with 25% ethanoVdichlororneth:~ne (4 X 60 mL). The combinP~ organic
extracts were dried over sodium sulfate and evaporated under vacuum to a volume of 40 mL.
This solution was cooled in icewater, whereupon a thick white precipitate was formed. The
solid was filtered off under suction and dried under vacuum to yield 1.43 g (90% yield) 3-
methyl-7-(methylpivaloyl)x:-nthine (CT1404).
A mixture of 3-methyl-7-(methylpivalyl)x~nthine (2.14g, 7.6 mmol) and sodium
hydride (183 mg, 7.6 mmol) in dimethyl sulfoxide (30 mL) were stirred for 15 minut~ps~ after
which period, 9-bromo-1-nonene (1.56 g, 7.6 mmol) was added. After stirring at ~mbit-n~
tem~eldlu~; for 2 days, the reaction mixture was poured into water (50 mL) and extracted
with dichlorometh~ne (3 X 50 mL). The combined organic portions were washed with water
(2 X 20 mL) and saturated aqueous sodium chloride solution (30 mL). The solvent was
removed under vacuum to give a thick oil. Chromatography (silica, ethyl acetate/20%
hexane) of this residue yields 1.46 g (48% yield) of a white, solid, 3-methyl-7-methylpivaloyl-1-(8-nonenyl)x~nlhine (CT1411).
A mixture of 3-methyl-7-methylpivaloyl-1-(8-nonenyl)x~nthinP. (910 mg, 2.3 mmol),
3-chloroperbenzoic acid (1.16 g of a 50% mixture, 3.4 mmol), and sodium bicarbonate (857
3s mg, 10 mmol) in dichloromethane (15 ml) and water (10 mL) was stirred for 18 hours at
~mbient temperature. A saturated aqueous solution of sodium bisulfite was added (15 mL)
and stirring is continued for 30 minutes. The layers are separated and the aqueous layer
eYtractPd with dichloromethane (3 X 30 mL). The combined organic layers were combined
and washed with saturated aqueous sodium bicarbonate solution (20 mL), water (20 mL) and
16
~b 94/06431 2 1 ~ 5 1 9 2 PCr/US93/09073
saturated aqueous sodium chloride solution (20 mL) and then dried over sodium sulfate.
Solvent was evaporated under vacuum. Chromotography of this residue using silica and a
dichlorolneth:~n~/5% ethanol eluant yielded 660 mg (68% yield) 3-methyl-7-methylpivaloyl-
1-(8,9-oxidononyl)~-nthinP (CT1409).
F.x~ ple7
This example i11nctr~tes a synthesis of 1-(8,9-Oxidohexyl)-3-methyl-7-
methylpivaloylx~nthine (CT1410). A mixture of 3-methylx~nthine (Aldrich, 1.00 g, 6.0
mmol), sodium hydride (145 mg, 6.0 mmol) and dimethyl sulfoxide (20 mL) was stirred
until homogeneous (0.5 hours). Chloromethylpivalate (865mL, 904 mg, 6.0 mmol) was
added and the reaction stirred for 18 hours. The reaction mixture was poured into water (70
mL) and then extracted with 25% ethanol/dichloromethane (4 X 60 mL). The combin~d
organic extracts were dried over sodium sulfate and evaporated under vacuum to a volume of
40 mL. This solution was cooled in icewater, whereupon a thick white p-ecipilate formed.
The solid was filtered off under suction and dried under vacuum to yield 1.43 g (90% yield)
3-methyl-7-(methylpivaloyl)x:mthine (CT1404).
Sodium hydride (86 mg, 3.6 mmol) was added to a stirring solution of 3-methyl-7-(methylpivaloyl)x~nthinP CT1404 (1.00 g, 3.6 mmol) in dimethyl sulfoxide (25 mL). After
15 minutes, 6-bromo-1-hexene (589 mg, 3.6 mmol) was added and stirring continued for 72
hours. The reaction mixture was then poured into water (70 mL) and extracted with
dichlorometh~ne (2 X 100 mL) and 20% ethanoVdichloromethane (1 X 100 mL). The
combin~d organic layers were washed with saturated aqueous sodium chloride solution (50
mL) and dried over m~gnecillm sulfate. The solvent was evaporated under vacuum to give a
thick oil. Chromotograpy of the rçsulting thick oil using silica and ethyl acetate yielded 870
mg (67% yield) 1-(5-hexenyl)-3-methyl-7-(methylpivaloyl)x~nthin~. (CT1441)
A mixture of 1-(5-hexenyl)-3-methyl-7-(methylpivaloyl)x~nthin-o. (440 mg, 1.2
mmol) and m-chloroperoxybenzoic acid (828 mg of 50% m-chloropero~ybe~lzoic by wt, 2.4
mmol) in dichloromethane (10 mL) and sodium bicarbonate (807 mg, 9.2 mmol) in water (10
mL) was stirred for 20 hours. Sodium metabisulfite (1.0 g, 5.3 mmol) was added. After 30
mimltes, the reaction mixture was extracted with dichlorometh~ne (3 x 10 mL). The
combinecl organic portions were washed with saturated aqueous sodium bicarbonate solution
(10 mL) and the solvent was evaporated under vacuum. Chromatography of the residue on
silica using 10% pet ether/ethyl acetate eluant yielded 146 mg (32% yield) 1-methyl-7-
(methylpivaloyl)-3-(5,6-oxidohexyl)xanthine (CT1410).
F~nple 8
3~ This example illustrates a synthesis of 1-(11,10-Oxidoun~lecanyl)-3-methyl-7-
methylpivalylx~nthin~ (CT1412). A mixture of 3-methy1x~nthine (Aldrich, 1.00 g, 6.0
mmol), sodium hydride (14~ mg, 6.0 mmol) and dimethyl sulfoxide (20 mL) was stirred
until homogeneous (0.5 hours). Chloromethylpivalate (865mL, 904 mg, 6.0 mmol) was
added and the reaction stirred for 18 hours. The reaction mixture was poured into water (70
WO 94/06431 2 1 4 ~ PCI/US93/090--
mL) and then extracted with 25% ethanolldichloromethane (4 X 60 mL). The combined
organic extracts were dried over sodium sulfate and evaporated under vacuum to a volume of
40 mL. This solution was cooled in icewater, whereupon a thick white precipitate formed.
The solid was filtered off under suction and dried under vacuum to yield 1.43 g (90% yield)
3-methyl-7-(methylpivaloyl)x~nthine (CT1404).
Sodium hydride (76.8 mg, 3.2 mmol) was added to a solution of 3-methyl-7-
pivaloylx~nthine (0.84 g; 3 mmol) and 11-bromoundec-10-ene (0.745 g; 3.2 mmol) in 15 mL
of dimethyl sulfoxide and stirred overnight. After 12hours of stirring at room temp~ lure,
the reaction was poured into a separatory funnel containing 30 mL of water and extracted
with dichloromethane (5 X 50 mL). The organic extracts were combined, washed with water
(30 mL) and saturated aqueous sodium chloride solution (30 mL), dried over anhydrous
m~gn~Sitlm sulfate and concentrated under reduced pressure. The crude product obtained
was further purified by flash chromatography over silica gel using 50% hexane/ethyl acetate
eluant to yield 1.05 g (73.8% yield) 1-(10-un-lecenyl)-7-methylpivaloyl-3-methyLlr~nthine
(CT1403).
A solution of 1-(10-undecenyl)-7-methylpivaloyl-3-methylx~nthin~ (0.60 g, 1.39
mmol), and m-chloroperoxybenzoic acid (0.359 g; 2 mmol, 50% by wt) in dichlorometh~n~
(10 mL) was stirred for S hours. The reaction mixture was diluted with 40 mL of
dichloromethane and washed successively with 20% aqueous sodium sulphite solution(20
mL), saturated sodium bicarbonate solution (20 mL), water and saturated aqueous sodium
chloride solution. The organic layer was dried over anhydrous m~gn~ lm sulfate and
concentrated under reduced pressure. The crude product obtained was further purified by
flash chromatography over silica gel using 50% hexane and ethyl acetate eluant, yielding
0.443g (72% yield) 1 -(11,10-Oxidoundecanyl)-3-methyl-7-methylpivalylx~nthine
Example 9
This example illustrates a synthesis of 1-(11,10-Oxidoun-lec~nyl)-3-methylx~nthinP
(CT1413). A solution of 1-(11,10-Oxidollndec:myl)-3-methyl-7-methylpivalyl~r~nthine
(CT1412), prepared as described above (104 mg; 0.23 mmol), was added to a sollltion of
sodium methc)7ricle (15.1 mg; 0.28 mmol) in 3 mL of methanol and sti'rred for 4 hours. The
reaction mixture was quenched with saturated ammonium chloride solution (5 mL) and
extracted with 20% ethanol/dichloromethane (3x30 mL). The combined organic extract was
dried over anhydrous m~gnesium sulfate and concentrated under reduced ~ft;SSur~. The
crude product obtained was further purified by flash chromatography over silica gel using
10% methanol/ethyl acetate eluant to yield 75 mg (96.7%) of (11,10-Oxidoundec~nyl)-3-
methylxanthine (CT1413).
F.~Tnple 10
This example illustrates a synthesis of 7-(11,10-Oxiclonr1ecyl)-1,3-dimethyl~:lnthin~
(CT-1423). Sodium hydride(95%) (0.575 g, 24 mmol) was added to a solution of
theophylline (3.6 g, 2() mmol) in dimethylsulfoxide (100 mL). After 20 minutes of stirring,
18
2 1 4 ~ Pcr/US93,09073
1-bromoundec-10-ene (4.66 g, 20 mmol) was added and stirred for 12 hours at roomtemperature. The reaction mixture was then poured into a separatory funnel cont~inin~ water
(300 mL) and extracted with dichloromethane (5 X 100 mL). The organie extracts were
combinpd7 washed with water (100 mL) and brine (100 mL), dried ov'er anhydrous
5 m~nPcinm sulfate and concentrated under reduced pressure. The crude product obtained
was further purified by flash chromatography over silica gel using 50% hPY~nP~ethyl acetate
eluant, yielding 6.26 g (94% yield) of 7-(10-undecenyl)-1,3-dimethylx~nthinP, (CT1420).
A solution of 7-(10-undecenyl)-1,3-dimethylx~nthinP. (4.98 g, 15 mmol) and m-
chloroperoxybenzoic acid (7.704 g; 22.5 mmol) (50% by wt) in dichlorometh~nP (100 mL)
10 was stirred for 5 hours. The reaction mixture was diluted with 80 mL of dichlorometh~ne
and washed successively with 20% aqueous sodium sulphite solution (100 mL), saturated
sodium bicarbonate solution (10() mL), water (100 mL) and brine solution (100 mL). The
organic layer was dried over anhydrous m~gnPsjum sulfate and concentrated under reduced
~ssul~. The crude product obtained was further purified by flash chromatography over
silica gel using 50% petroleum ether/ethyl acetate eluant to yield 3.13 g (60% yield) of 7-
(11,10-Oxidondecyl)-1,3-dimethyl~c~nthine
le 11
This example illustrates a synthesis of 7-(11,10-Oxidondecyl)-l-methyl-2,4-
dioxotetrahydropteridine (CT1426). 1-Methyl-4,5-diaminouracil (13.6 g; 59.4 mole) was
suspended in water (150 mL) and converted to its hydrochloride by drop-wise ~ ition of
concentrated hydrochloric acid unitl the solution is strongly acidic. Glyoxan
sodiumbicnlrhit~P (20.4 g; 71.8 mmol) was then added and the reaction mixture rçfln~p~l for
30 minutes. The reaction mixture was cooled to room temperature and the precipitated 1-
methyl-2,4-dioxotetrahydropteridine isolated by filteration, yielding 6.5 g (62%). Sodium
hydride (95%,) (0.575 g, 24 mmol) was added to a solution of 1-methyl-2,4-
dioxotetrahydropteridine (3.56 g, 2() mmol) in dimethylsulfoxide (100 mL). After 20 minl~tPs
of stirring, l-bromoundec-10-ene (4.66 g, 20 mmol) was added and stirred for 12 hours at
room temperature. The reaction mixture was then poured into a separatory funnel cont~ining
water (300 mL) and extracted with ethyl acetate (5 X 100 mL). The organic extracts were
combined, washed with water (100 mL) and brine (100 mL), dried over anhydrous m~gnPcillm
sulfate and concentrated under reduced pressure. The crude product obtained was further
purified by flash chromatography over silica gel using 50% hexane/ethyl acetate eluant to yield
5 g (94% yield) of 3-(10-undecenyl)-1-methyl-2,4-dioxotetrahydl~3pte,idine (CT1421). A
solution of 3-(10-nntlecenyl)-1-methyl-2,4-dioxotetrahydropteridine (3.3 g, 10 mmol) and m-
chloroperoxybenzoic acid (5.13 g; 15 mmol) (50% by wt) in dichlorometh~nP (50 mL) was
stirred for 5 hours. The reaction mixture was diluted with 40 mL of dichlorometh~nP and
washed successively with 2()% aqueous sodium sulphite soluhon (20 mL), saturated sodium
bicarbonate solution (20 mL), water (30 mL) and brine solutions (30 mL). The organic layer
was dried over anhydrous m:-~necium sulfate and concentrated under reduced pressure. The
19
WO 94/06431 2 1 ~ 5 1 9 2 PCr/US93/o907~
crude product obtained was i`urther purifled by flash chromatography over silica gel using 40%
petroleum ether/ethyl acetate eluant to yield 2.61 g (75% yield) of 7-(11,10-Oxi-lontle~yl)-l-
methyl-2,4-dioxotetrahydropteridine.
F.~ rnple 12
s This example illustrates a synthesis of l-Methyl-3-(5,6-oxidohexyl)x~nthinP
(CT1439). A solution of l-methyl-7-(methylpivaloyl)-3-(5,6-oxid~hPxyl)x~nthinP, prepared
as described above, (120 mg, 0.3 mmol) in methanol (5 mL) was treated with a 1 MmPth~nol solution of sodium methoxide (0.33 mL). After 15 minlltes of stirring, water (5
mL) was added and the reaction product was extracted with 25% ethanoWichloromt~th~nP. (4
X 25 mL). The aqueous layer was evaporated under vacuum leaving a solid residue which is
subsequently washed with dichloromethane (4 X 20 mL). The combined organic washings
were evaporated under vacuum to a give a solid, yellowish residue. Chromatography of the
residue using silica and 50% ethyl acetate/meth~nol yields 55 mg (69% yield) of solid 1-
methyl-3-(5,6-oxidohexyl)xanthine.
F~ e 13
This example illustrates a synthesis of 1-(6,7-cis-Oxidononyl)-3,7-dimt;LhyL~anthine
(CT-1509). A mixture of cis-6-nonen-1-ol (TCI, 3.00 g, 21.1 mmol) and ~nPth~nesulfonyl
chloride (1.6 mL, 2.4 g, 21 mmol) in dichloromethane (100 mL) at 0C was treated with
triethylamine (4.4 mL, 3.2 g, 32 mmol). After 1 hour the ice bath was allowed to melt.
After reaching ~mbiçnt temperature, the reaction was poured into a separatory funnel
c~nt~ining water (50 mL) and dichloromethane (50 mL). The layers were se~ala~d and the
aqueous layer washed with dichlor -meth~ne (2 X 50 mL). The combinPcl organic layers
were dried over sodium sulfate, and solvent removed to yield 4.14 g, 18.8 mmol (89% yield)
6-cis-nonene-1-meth~nesulfonate as a yellow oil.
2s Theobromine (3.36 g, 18.8 mmol) and sodium hydride (451 mg, 18.8 mmol) in
dimethylsulfoxide (40 mL) was stined for 40 minlltPs after which time the mesylate (4.14 g,
18.8 mmol) was added. The reaction was stirred at 25C for 3 days, then heated at 80C for
1 hour, and cooled. The reaction mixture was poured into water (100 mL) and ~xlla~;led with
dichlorc meth~ne (3 X 60 mL). The combined organic layers were washed with saturated
aqueous salt solution (2 X 50 mL) and dried over sodium sulfate. The solvent was removed.
Chromotagraphy using silica and ethyl acetate of the residue yields 4.54 g (79% yield) 1-(6-
cis--nonenyl)-3,7-dimethylxanthine (CT1508).
A ~ u~e of 1-(6-cis-nonenyl)-3,7-dimethylx~nthine, prepared as described above, in
dichloromethane (10 mL) was stirred with sodium bicarbonate (2.62 g, 31 mmol) in water
3s (20 mL) and 4-chloroperoxybenzoic acid (2.0 g, or 4.0 g of a 50% mixture) for 15 hours.
.~o~ m sulfite (1.82 g, 14.5 mmol) was added, the mixture added to water (50 mL) and
extracted with dichloromethane (2 x 50 mL). The combined organic layers were washed
with water (40 mL) and saturated aqueous salt solution (40 mL). The.solvent was removed
to yield a yellow oily residue which was subsequently isolated using silica and ethyl acetate
~ 94/06431 2 1 ~ 5 1 9 ~ PC~r/US93/09073
chromatography, yielding 520 mg (42% yield) of a white, solid 1-(6,7-cis-o~citlot onyl)-3,7-
dimethyl~c~nthine.
F.Y~nn~PIe 14
This example illustrates a synthesis of 1-(5,6-Oxidohexyl)-3,7-dimt;lllyk;lnthin~.
s (CT1541). To a mixture of bromohexene (10.7 g, 66 mmol, AldAch) and sodium hydride
(1.58 g, 66 mmol) in dimethylsulfoxide (100 mL) was added theobromine (11.9 g, 66 mmol,
available from Sigma) and stirred for 43 hours. The solution was treated with water (200
mL) and extracted with dichlorometh~ne (3 X 80 mL). The combined extracts were washed
with water (3 X 100 mL), dried over m~gnecium sulfate, and then the solvent evaporated
under vacuum to yield 17 g (65 mmol, 98% yield) of a white powder, l (S-Hexenyl)-3,7-
dimethylxanthine (CT1539).
To a mixture of l-(S-Hexenyl)-3,7-dimethyl~c~nthine (1.07 g, 4.1 mmol) and N-
methylmorpholine-N-oxide (1.44 g, 12.3 mmol) in water (20 mL) and acetone (10 mL) was
added 2.5% ocmillm tetraoxide in t-butanol (6 drops). After stirring for 48 hours, the
lS mixture was treated with 20% aqueous sodium dithionite solution (20 mL). After 2 min~lte
the mixture is extracted with 25% ethanol-dichloromethane solution (3 X 30 mL). The
com~inPcl extracts were dried over m:lgnçcium sulfate and the solvents were evaporated
under vacuum, yielding 750 mg, 2.53 mmol (62% yield) 1-(5,6-dihydroxyhexyl)-3,7-dimethylx~nthine (CT1502) as a white powder.
To 1-(5,6-dihydroxyhexyl)-3,7-dimethylx~nthinP 1.0 g, 3.38mmol) was added 30%
hydrogen bromide-acetic acid (3.4 mL) over 30 seconds and then stirred for 2.5 hours until
all of the solid dissolved. The solution was poured carefully over a mixture of sodium
bicarbonate (12 g) and ice water (S0 mL). After carbon dioxide evolution had sl1bsi-le(l, the
mixture was extracted with dichlorometh~ne (3 X 25 mL). The comhin~l extracts were dried
over m~gn,ocillm sulfate and the solvent evaporated under vacuum, yielding 1.3 g (3.24 mmol,
96%) 1-(S-Acetoxy-6-bromohexyl)-3,7-dimethyl~c~nthinl- as a viscous oil, dissolved in
meth~nol (S mL). A lM sodium methoxide in methanol solution (3.9 mL) was added over 30
secon-lc After stirring for 20 minutes, the solution was reated with water (20 mL) and then
e~ ac~ed with dichlorometh:ln~ (3 X lS mL). The combined extracts were dried over
m~gntocium sulfate and the solvents were evaporated under vacuum to yield 900 mg (3.24
mmol, 100% yield) 1-(5,6-Oxidohexyl)-3,7-dimethylx~nthinP as white crystals.
Example 1
This example illustrates a synthesis of 1-(8,9-Oxidooctyl)-3,7-dime~llyl~ h;l.~
(CT1553). To a suspension of sodium hydride (580 mg, 24.2 mmol ) in dimethylsulfoxide
(100 mL) was added theobromine (3.96 g, 22.0 mmol). After 30 minlltes, 8-bromo-1-octene
(3.96 g, 22 mmol) was added and the reaction stirred 16 hours at 25C. The ~ LLIIe was
then poured into water (200 mL) and extracted with dichloromethane (3 X 50 mL). The
cornbine~l organic portions were washed with brine (50 mL) and dried over sodium sulfate.
WO 94/06431 ~ 1 4 ~ PCr/US93/0907
The solvent was evaporated under vacuum to yield 6.22 g (97% yield) of thick white oil, 1-(7-
octenyl )-3,7-methylx:~nthinP (CT1535) which soli~lifiPd upon standing.
A solution of 1-(7-octenyl)-3,7-dimethylx~nthinP. (1.00 g, 4.5 rnmol), 4-
methylmorpholine-N oxide (553 mg, 4.7 mmol), and a 2.5% solution of q.cminm tetroxide in t-
s butanol (3 drops) in acetone (25 mL) and water (20 mL) was stirred for 4 days. After~dditiQn of a saturated aqueous solution of sodium hydrosulfite (10 mL) and 30 minllte~ of
continued stir~ing~ the reaction mixture was added to water (50 mL) and extracted with 20%
ethanol/dichlorometh~ne (3 X 50 mL). Evaporation of the solvent under vacuum yielded an
off-white residue. The residue was recryst~lli7Pd in eth~nol, yielding 726 mg (63% yield) 1-
(7,8-dihydroxyoctyl)-3,7-dimethyl~:lnthine (CT1538) as a white solid.
A mixture of 1-(7,8-dihydroxyoctyl)-3,7-dimethylx~nthint~, prepared as describedabove, (2.1 lg, 6 mmol) was stirred with a 30% solution of hydrogen bromide in acetic acid
(3.58 mL, 18 mmol) for 90 minutes. The mixture was then added to a flask cont~ining
aqueous sodium bicarbonate solution (4 g in 50 mL) and dichlorometh~ne (30 mL). After 10
15 mimlte.s of vigorous stirring the layers were separated and the aqueous portion was washed
with dichloromPth:~ne (2 X 50 mL). The comhin~d organic portions were dried over sodium
sulfate and the solvent was evaporated under vacuum, yielding 2.51 g (94%) 1-(7-acetoxy-8-
bromooctyl)-3,7-dimethylx~nthine (CT1514) as a yellow oil.
A solution of 1-(7-aoetoxy-8-bromooctyl)-3,7-dimethyl~ .;..r. in meth~nol (10 mL)
was treated with a 1 M solution of sodium methoxide (1.4 mL). After 30 minutPs, the
reaction was added to water (20 mL) and extracted with dichlorometh~n~ (2 X 30 mL).
(~ombin~ci organic portions are dried to give an off-white residue, ~ I .11i7P~l in
dichloromt~th~nP~petroleum ether, yielding 1.42 g (75% yield) 1-(8,9-oxidooctyl)-3,7-
dim~lhylx~thinl- (CT1553).
F. ~nple 1~
This example illustrates a synthesis of 1-(4,5-Oxohexyl)-3,7-dimethyl~ ~nthinP
(CT1555). To a solution of 4-hexen-1-ol (1.22 g, 12.2 mmol) and meth~nPculfonyl chloride
(1.04 mL, 13.4 mmol) in dichloromethane (15 mL), cooled in an ice bath, was added
triethylamine (2.55 mL, 18.3 mmol) dropwise. After 5 minutes, the cooling bath was removed
and the mixture stirred for an additional 45 minutes and then treated with s~tnr~tPd aqueous
sodium bicarbonate solution (25 mL). The layers were separated and the aqueous layer
extracted with dichloromethane (2() mL). The combined organic layers were dried over
m~gnt~cillm sulfate and the solvents evaporated under vacuum, yielding l-mpth~n~sulfon
4-hexene as a viscous oil.
A mixture of theobromine (2.16 g, 12 mmol) and sodium hydride (288 mg, 12
mequivalents) in dimethylsufoxide (1() mL) was stirred for 30 ...i~ Ps and then a solution of
l-meth~nclllfonyloxy-4-hexene in dimethylsulfoxide (10 mL) was added. After 84 hours of
stirring, water (70 mL) is added and the mixture extracted with ether (3 x 50 mL). The
comhinPd extracts were washed wi~h waler (50 mL) and dried over m~gneCium sulfate. The
22
~ 94/06431 ~ 2 1 4 ~ 1 g ~ PCr/US93/09073
solvent was then evaporated under vacuum. The residue was purified by flash
chromatography (22 g silica gel using an ethyl acetate eluant, 500 mL), yielding 2.1 g (67%)
1-(4-hexenyl)-3,7-dimethylx:-nthine To a solution of 1-(4-hexenyl)-3,7-dimethyl~nthinP. (270
mg, 1.03 mmol) in dichloromethane (10 mL) was added saturated aqueous sodium bicarbonate
solution (10 mL), followed by a solution of 50-60% 3-chloroperoxybenzoic acid (889 mg,
2.58 mmol) in dichlorom~-th~nP (5 mL) over 1 minute. After 20 hours of stirring~ 20%
aqueous sodium metabisulfite solution (15 mL) was added over 1 minute. The llli~lUl~ was
then extracted with dichlorometh:-ne (3 x 15 mL). The c~mbinPd extracts were washed with
.C~tllrat~d aqueous sodium bicarbonate solution (3 x 20 mL) and dried over magnPsillm sulfate.
The sovent was evaporated under vacuum. The residue was purified by flash chromatography
using 14 g of silica gel and eluting with ethyl acetate (100 ml) followed by 8% meth~nQl-
dichlorometh:~nP (60 mL), yielding 80 mg (28% yield) of 1-(4,5-oxohexyl)-3,7-
dimethyl~nthine as a white powder.
FY~n~PIe ]7
This example illustrates a synthesis of 1-(8,9-Oxidononyl)-3,7-dimethylY~nthine
(CT1560). A mixture of theobromine (17.64 g, 98 mmol) and sodium hydride (2.35 g, 98
mmol) in dimethylsulfoxide (250 mL) was stirred for 15 minutes. After ~dtlitiorl of 9-bromo-
l-nonene (Alfebro, 20.0 g, 98 mmol) stirring was continued at ambient temperature for 3
days. The reaction mixture was then poured into water (300 mL) and extra~tP~l with
dichlorometh~ne (4 X 200 mL). The combinPd organic layers are washed with s~tllra
aqueous salt solution (2 X 150 mL) and dried over sodium sulfate. The solvent was
evaporated under vacuum to give a thick oil. After cooling a solution of the oil in a minimnm
of dichlorometh~ne and ether, 1-(8-nonenyl)-3,7-dimethyl~r~nthinP- (CT1550) (24.34 g, 77.5
mmol, 99% yield) formed as white crystals .
A solution of 1-(8-nonenyl)-3,7-dimethylx~nthin~ (810 mg, 2.7 mmol), 4-
methylmorpholine-N- oxide (34() mg, 2.9 mmol) and 2.5% oxmium tetroxide in t-butanol (3
drops) in ~cetonP (20 mL) and water (20 mL) was stirred for 24 hours. Saturated aqueous
sodium dithionite solution (S mL) was added. After stirring for 15 minutes, the reaction was
extracted with 25% ethanol-dichlorometh:me (4 X 50 mL). The combined organic portions
were dried over sodium sulfate, and the solvents were evaporated under vacuum. The solid
residue was recrystallized (ethanol-chloroform), yielding 490 mg (54%) 1-(8,9-
dihydroxynonyl)-3,7-dimethyl~nthine (CT1561).
A mixture of 1-(8,9-dihydroxynonyl)-3,7-dimethylx~nfhinP- and 30% hydrogen bromide
in acetic acid (0.8 mL, 3.90 mmol) was stirred for 90 minutes The solution was poured into a
mixture of water (10 mL), sodioum bicarbonate (1.35 g, and dichlorometh~n~. (10 mL). After
10 mimlt~s of vigorous stiring, the layers were separated and the aqueous portion ~n~
with dichlorometh~nP. (3 X 15 mL). The combined organic phases were dried over sodium
sulfate and the solvent evaporated under vacuum, yielding 550 mg (96%) 1-(8-acetoxy-9-
bromononyl)-3,7-dimethyl~c:mthine as a yellow oil. Without further purifi~ ~tion, the oil was
23
WO 94/06431 ~ ; j PCr/US93/0907~
dissoved in methanol (5 mL) and then a 1 M solution of sodium methoxide in meth~n~l (1.4
mL) was added. After 30 minutPs, the reaction mixture was poured into water (30 mL) and
was extracted with dichlorometh~none (3 X 40 mL). The combined organic portions were
dried over sodium sulfate and the solvents evaporated under vacuum. The solid residue was
S ~ lli7Pd (dichlorometh~ne-petroleum ether) to yield 380 mg (91% yield) 1-(8,9-
ononyl)-3,7-dimethylx~nthin~ ~CT1560).
F.Y~n~PIe 18
This example illustrates a synthesis of 1-(9,10-Oxidodecyl)-3,7-dimethylx~nthine(CT1565). To a solution of 9-decene-1-ol (Aldrich, 3.00 g, 19.2 mmol) in dichloromPth~n~
(100 mL) at 0C was added meth~npsulfonyl chlori~le (2.20 g, 1.5 mL, 19.2 mmol), followed
by triethylamine (2.91 g, 28.8 mmol). After stirring for 15 minutes at 0C, the reaction was
allowed to warm to room temperature. After 2 hours, the reaction mixture was poured into
water (100 mL) and extracted with dichloromethane (3 X 60 mL). The combined organic
portions were dried over sodium sulfate and the solvent was evaporated under vacuum
yielding 4.52 g (100%) mesylate as a yellow oil. The mesylate was used without further
pnrifination
To a suspension of sodium hydride (461 mg, 19.2 mmol) in dimethylculfoxi~le (30 mL)
was added theobromine (3.45 g, 19.2 mmol). After 15 mimltP~s, the 9-decenylmesylate (2.25
g, 11 mmol) was added and ~he reaction stirred 18 hours at 25C, then at 100C for 40
minutes- The mixture was then poured into water (100 mL) and extracted with
dichlorometh~nP. (3 X 50 mL). The combined organic portions were washed with saturated
salt solution (60 mL) and dried over m~gnPsillm sulfate. The solvent was evaporated under
vacuum to give a white solid residue. Recryst:-11i7:ltion in ether yields 3.40 g (56% yield) 1-(9-
decenyl)-3,7-dimethylx~nthin~ (CT1563).
A solution of l-(9-decenyl)-3,7-dimethylx~nthinP (3.2 g, 10.1 mmol), 4-
methylmorpholine-N-oxide (1.41 g, 12 mmol) and a 2.5% solution in t-butanol of osmintn
tetroxide (3 drops) in acetone (40 mL) and water (10 mL) was stirred for 24 hours. Following
ad~litiQn of S mL of a saturated solution of sodium dithionite and an ~ r1itio~l 15 minntps of
stirring, the reaction product was extracted with 25% ethanol/dichloromethane (4 X 50 mL).
The combined organic portions were dlied over sodium sulfate. The solvents were evaporated
to a give a white solid residue. Upon recrycl~lli7~tion of the residue in ethanol, 3.30 g (93%
yield) l-(9,10-dihydroxydecyl)-3,7-dimethylx:~nthine (CT1564) were obt~ined.
A mixture of 1-(5,6-dihydroxydecyl)-3,7-dimethylx~nthimP. (2.1 lg, 6 mmol) and a30% solution of hydrogen bromide in acetic acid (3.58 mL, 18 mmol) was stirred for 90
min~lse~ The mixture was then added to a flask cont~ining aqueous sodium bicarbonate
solution (5 g in 40 mL ) and dichloromethane (50 mL). After 10 minutes of vigorous
stirring, the layers were separa~ed and the aqueous portion extracted with dichlorometh:mP. (2
X 50 mL). The combinéd organic phases were dried over sodium sulfate. The solvent was
evaporated, yielding 2.?2 g (lOU%) l-(9-acetoxy-10-bromodecyl)-3,7-dimethylx~nthinP. as a
24
~ 94/06431 2 1 ~ ~ 1 9 2 PCr/US93/09073
yellow oil. Without further purification, the oil was taken up in mPth~nol (30 mL) and
treated with a 1 M solution of sodium methoxide (6 mL). After 30 min~ Ps~ the reaction
mixture was added to water (30 mL) and extracted with dichloromethane (3 X 50 mL). The
organic portions were combined and dried over sodium sulfate to give an off-white solid
residue. Recrystalization in dichlorometh~nP/petroleum ether yields 380 mg (91% yield) 1-
(9,10-oxidodecyl)-3,7-dimethylx~nthine
FY~n~ e 19
This example illustrates a synthesis of 3,7-dimethyl-1-~6,7-trans-~Yirlononyl)Y~nthinP,
(CT- 1569). A mixture of 6-cis-nonen- l-ol (TCI, 990 mg, 7.0 mmol) and thiophenol (60 mg)
was heated at 105- 110C under argon for 4 hours to give 6-nonen-1-ol 872 mg, 88% yield)
with a 4:1 trar~s:cis isomer ratio. Without further purifi~tion, the mixture was stirred with
mPth~nPslllfonyl chloride (694 mg, 6.1 mmol) in dichlorompth~n~p (20 mL) at 0C.Triethylamine (925 mg, 9.2 mg) was added dropwise and stirring continued for 1 hour. The
reaction mixture was added to an aqueous saturated solution of sodium bicarbonate (10 mL)
and the layers were separated. The aqueous layer was eytr~tpd with dichloromPth~nP (2 x 15
mL). The combined organic layers were washed with a 5% sohltion of hydrogen chloride (10
mL), water (10 mL), and an aqueous saturated solution of sodium chlori~le (10 mL) and then
dried over sodium sulfate. The solvent was removed under vacuum to give the mesylate,
which was used in the next step without purification.
A mixture of the mesylate, sodium theobromine (1.21 g, 6.0 mmol) was stirred in
dimethylsulfoxide (10 mL) for 24 hours. The reaction mixture was poured into water (10 mL)
and extracted with dichloromethane (3 X 25 mL). The comkinP(l organic extracts were
washed with water (lS mL) and aqueous saturated salt solution (15 mL). After removing the
solvent under vacuum, the residue was purified by silica/ethyl acetate chromotography,
2s yielding 827 mg (67% yield) 1-(6-trans-noneyl)-3,7-dimethylx~nshinP (CT2512 ), 20% being
cont~min~tPcl with the cis isomer.
1-(6-~ans-noneyl)-3,7-dimethylx:lnthin~ (110 mg, 0.4 mmol), m-chlo,u~,e.l,enzoic acid
(75 mg, 0.4 mmol), and sodium bicarbonate (150 mg, 1.8 mmol) were stirred in
dichlorometh~n~P. (6 ml) and water (5 mL) for S hours at room temperature. A saturated
solution of sodium bisulfite was added (lO mL). The layers were separa~Pd and the aqueous
layer washed with dichlorometh~ne (2 X 20 mL). The organic layers were combinPd and
washed with saturated sodium bicarbonate solution (10 mL), water (10 mL), and brine (15
mL), and dried over sodium sulfate. The solvent was evaporated and the residue f~~ ;.lli7Pd
in ether, yielding 70 mg (54% yield) of 1-(6,7-Oxidoheptyl)-3,7-dimethyL~ P
FY~nple 20
This example illustrates a synthesis of 1-(6,7-Oxidoheptyl)-3,7-dimethylx~nthinP (CT-
1586). To a solution of 6-hepten-1-ol (6.00 g, 52.6 mmol) in dichloromPth~nP. (120 mL) at 0
C was added methanesulfonyl chlnride (6.07 g, 4.0 mL, 53.0 mmol), followed by
triethylamine (7.79 g, 77.() mmol). After stirring for 10 minuS~s at 0C, the reaction was
WO 94/06431 ~ i t ~ 2 PCr/US93/0907--
allowed to warm to 25C and then stirred for 2 hours. The reaction was poured into water
(100 mL) and extracted with dichlorometh mP (2 x 100 mL). The organic portions were
combined, dried over m~gnecium sulfate, and evaporated to give the 7-mPth~nPsulfonyl-1-
heptene as a yellow oil (9.30 g, 93%), which was used without further purifi~ tion
s To a sncpçncinn of sodium theobromine (9.05 g, 50.0 mmol) in dimethylsulfoxide (90
mL) was added 7-mP.th~nPsulfonyl-l-heptene (9.30 g, 48.2 mmol). The reaction was stirred
for 16 hours at 60C. The mixture was then poured into water (100 mL) and eYtrartPd with
ethyl acetate (3 x 100 mL). The organic portions were combimPtl dried, and evaporated to
give an orange solid. Chromatogràphy (silica, ethyl acetate/hexane) yielded 6.50 g (47%
yield) 1-(6-Heptenyl)-3,7-dimethyl~nthine (CT1534) as a white solid.
A solution of 1-4(6-Heptenyl)-3,7-dimethyl~ nthinP (6.00 g, 21.7 mmol), 4-
methylmorpholine-N-oxide (6.82 g,58.0 mmol) and potassium osmate dihydrate (70 mg, 0.19
mmol) in acetone/water 1:2 (120 mL) was stirred for 16 hours. Water (100 mL) and sodium
sulfite (S g) were added and the reaction mixture stirred for 1 hour. The reaction mixture was
extracted with 25% ethanoVdichlorometh~ne (3 x 120 mL), dried over m~gnPcil m sulfate and
the solvent evaporated to obtain a cream solid. Recryst~ ti~ n of the solid from hot
mPth~noVethyl acetate 1:1 yielded 6.45 g (96% yield) 1-(6,7-Dihydroxyheptyl)-3,7-
dimethylx:lnthinP (CT1585) as a white solid.
1-(6,7-Dihydroxyheptyl)-theobromine (4.00 g, 12.9 mmol) was stirred with hydrogen
bromide (12.53 g of a 30% solution in acetic acid, 38.7 mmol) for 2 hours. The mixture was
then added over 10 minutes to water (50 mL), ice (50 g) and sodium bicarbonate (30 g) and
stirred for 30 minutPs. The reaction mixture was extracted with dichlo'romPth~nP- (3 x 100
mL). A comhin~d organic phase was dried over m~gnPcinm sulfate and the solvent was
evaporated to obtain a residue (4.90 g, 91% yield) of 1-(6-acetoxy-7-bromoheptyl)-3,7-
2s dimethyLx~nthin~
Without further puritic:ltion, this crude product was taken up in mPth~nol (10 mL) and
treated with a solution of sodium methoxide (prepared from sodium (0.285 g, 12.4 mmol) and
20 mL methanol). After 60 minutes the reaction was added to water (50 mL) and e~rtr~ctP~
with dichlorometh~nP (3 x 50 mL). The organic portions were combinP~ and dried to give an
off-white solid. Recrystalization in dichlorome,th~nP/petroleum ether yielded 3.30 mg (96%
yield) of 1-(6,7-Oxidoheptyl)-3,7-dimethyl7~:ln~hine.
n~le 21
This example illustrates a synthesis of 1-(3-(R)-methyl-7-methyl-6,7-oxidooctyl)-3,7-
dimethylx~nthine (CT1588R). Sodium hydride(95%) (631 mg, 25 mmol) was added to as~ tion of theobromine (4.14g, 23 mmol) in dimethylsulfoxide (75 mL). After 20 minutes
of stirring~ (R)(-)Citronellyl bromide (5.0 g, 22.8 mmol) was added. After 16 hours of
stirring at room temperature, the reaction was poured into a separatory funnel cont~ining 500
mL of water and extracted with dichloromethane (3 X 100 mL). The organic extracts were
combined, washed with water (10() mL) and brine (100 mL), dried over anhydrous
26
.
214~1~2
~0 g4/06431 PCI/US93/09073
m~g~ sium sulfate and concentrated under reduced pressure. The crude product obtained
was further purified by flash chromatography over silica gel using 30% petroleum ether/ethyl
acetate eluant, yielding 5.9 g (81.5% yield) 1-(3-(R)-methyl-7-methyloct-6-enyl)-3,7-
dimethylx~nthine (CT596R) as a yellowish oil. A solution of 1-(3-(R)-methyl-7-methyloct-
6-enyl)-3,7-dimethyl~c~nthine (318 mg, 1 mmol) and m-chloroperoxybenzoic acid (0.52 g;
1.5 mmol) (50% by wt) in dichlorometh~ne (7 mL) was stirred for 5 hours. The reaction
mixture was diluted with 40 mL of dichlorom~qth~ne and washed successively with 20%
aqueous sodium sulphite solution (10 mL), saturated sodium bicarbonate solution (10 mL),
water and brine solutions. The organic layer was dried over anhydrous m~gn~sillm sulfate
0 and concentr~te.d under reduced pressure. The crude product obtained was further purified
by flash chromatography over silica gel using 5% petroleum ether/ethyl acetate eluant,
yielding 0.253 g (76% yield) of 1-(3-(R)-methyl-7-methyl-6,7-oxidooctyl)-3,7-
dimethylx:-nthint
F.~nple 22
This example illustrates a synthesis of 1-(3-(S)-methyl-7-methyl-6,7-oxidooctyl)-3,7-
dimethyl~nthine (CT1588S). Sodium hydride (95%) (631 mg, 25 mmol) was added to asolution of theobromine (4.14g, 23 mmol) in dimethylsulfoxide (75 mL). After 20 minut~s
of stirring, (S)(-)Citronellyl bromide (5.0 g, 22.8mmol) was added. After 16 hours of
stirring at room temperature, the reaction mixture was poured into a separatory funnel
cont~ining 500 mL water and extracted with dichloromethane (3 X 100 mL). The organic
portions were combinecl, washed with water (100 mL) and brine (100 mL), dried over
anhydrous m~neCillm sulfate and concentrated under reduced pressure. The crude product
obtained was further purified by flash chromatography over silica gel using 30% petroleum
ether/ethyl acetate eluant to yield 5.7 g (80% yield) 1-(3-(S)-methyl-7-methyloct-6-enyl)-3,7-
dimethyl~nthine (CT1596S) as an yellow oil.
A solution of 1-(3-(S)-methyl-7-methyloct-6-enyl)-3,7-dimethyl~nthin~ (636 mg, 2mmol) and m-chloroperoxybenzoic acid (1.04 g; 3 mmol) (50% by wt) in dichlorometh~ne
(15 mL) was stirred for 5 hours. The reaction mixture was diluted with 80 mL of
dichloromethane and washed successively with 20% aqueous sodium sulphite solutio~(20
mL), saturated sodium bicarbonate solution (20 mL), water and brine solutions. The organic
layer was dried over anhydrous m:lgnesium sulfate and concentrated under reduced pressure.
The crude product obtained was further purified by flash chromatography over silica gel
using 5% petroleum ether/ethyl acetate eluant to yield 0.56 g (83% yield) 1-(3-(S)-methyl-7-
methyloct-6-enyl)-3 ,7 -dimethylxanthine.
FY~n~.1e 23
This xample illustrates a synthesis of 1-(4,5-Oxipentyl)-3,7- dimethylx~nthin~
Sodium hydride (95%) (1.38 g, 55 mmol) was added to a solution of theobromine (9.0 g, 50
mmol) in dimethylsulfoxide (3()() mL). After 20 minutes of stirring, 1-bromo-4-pentene(
7.45 g, 50 mmol) was adde(l. After 16 hours of stirring at room temperature, the reaction
27
W O 94/06431 2 1 4 ~ i ~ 2 PC~r/US93/0907 ~
was poured into a separatory funnel containing 1 L of water and extracted with
dichloromethane (5 X 200 mL). The organic extracts were combined, washed with water
(100 mL) and brine (100 mL), dried over anhydrous m~gnecinm sulfate and concentrated
under reduced ples~ulæ The crude product obtained was further purified by flash
5 chromatography over silica gel using 20% petroleum ether/ethyl acetate eluant to yield 9.67 g
(92% yield) 1-(4-pentenyl)-3,7-dimethylx:lnthint~. (CT1575).
A solution of 1-(4-pentenyl)-3,7-dimethylxanthine (2.48 g, 10 mmol), 4-
methylmorpholine-N-oxide (1.49 g, 12.7 mmol) and potassium osmate dihydrate (7.3 mg;
0.02 mmol) in acetone (20 mL) and water (5 mL) was stirred for 6 hours. A solution of 20%
aqueous sodium sulphite (10 ml) was added and stirred for 30 mim1t~s The reaction mixture
was extracted with 25% ethanol/dichloromethane (4 X 250 mL). The combined organic
extracts were dried over anhydrous m~gnesillm sulfate and concentrated under reduced
pressure. The crude product obtained was further purified by flash chromatography over
silica gel using ethyl acetate eluant to yield 2.5 g (88% yield) of 1-(4,5-dihydroxypentyl)-
3,7-dimethylx:lnthin~ (CT1584).
1-(4,5-Dihydroxypentyl)-3,7-dimethylx~nthin~. (2.13 g, 7.6 m,mol) was stirred with
hydrogen bromide (4.74 mL, 6.15 g of a 30% solution in acetic acid, 22.8 mmol) for 90
mimlteS The mixture was then added to a flask cont~ining 50 mL aqueous sodium
bicarbonate solution and 50 mL dichlorometh~ne. After 10 minlltes of vigorous stiTTing, the
layers were separated and the aqueous portion washed with dichlorompth~ne (3 X 50 mL).
The organic portions were combined, dried over m~gn~oS;um sulfate, and the solvent
evaporated to yield 3.4 g (96%) 1-(4-acetoxy-5-bromopentyl)-3,7-dimethylx~nthinP as a
yellow oil. Without further purification, the oil was taken up in methanol (25 mL) and
treated with a solution of sodium methoxide (prepared from 0.2 g, 8.7 mmol sodium, and 25
mL meth~nQl). After 30 min~ltt~s most of the solvent was removed under reduced pressure
and the residue extracted with dichloromethane (3 X 50 mL). The organic portions were
combined and dried over magnesium sulfate to obtain an off-white solid purified by column
chromatography over silica gel using ethyl acetate/(15%) acetone eluant to yield 1.0 g (50%
yield) of 1-(4,5-Oxipentyl)-3,7-dimethylx~nthine
FY~n~PIe 24
This example illustrates a synthesis of 1-(10,11-Oxidonndecanyl)-3,7-
dimethylx~nthint- (CT1594). Sodium hydride(95%) (1.26 g, 50 mmol) was added to asolution of theobromine (7.2g, 40 mmol) in dimethylsulfoxide (300 mL). After 20 mimltes
of stirring, nnclecenylmesylate (7.95 g, 30 mmol) was added and stirred for 12 hours at room
temperature. The reaction was warmed to 70-80C and stirred for 4 hours. The reaction
mixture was then poured into a separatory funnel containing 1 L of water and extracted with
dichlorometh~ne. (5 X 200 mL). The organic extracts are combined, washed with water (100
mL) and brine (100 mL), dried over anhydrous m:~gnecillm sulfate and concentratt-rl under
redllcecl pressure. The crude product obtained was further purified by flash chromatography
28
~ 94/06431 2 1 9L 5 ~ ~ ~J PCI/US93/09073
over silica gel (eluant: 20% hexane/dichloromethane) to yield 4.6 g (46.3% yield) 1-(10-
Undecenyl)-3,7-dimethylxanthine (CT2501).
A solution of 1-(10-~1ndecenyl)-3,7-dimethylx~nthine (4.3 g, 13 mmol), 4-
methylmorpholine-N-oxide (1.942 g, 16.6 mmol) and pot~ccium osmate dihydrate (9.5 mg;
0.026 mmol) in acetone (45 mL) and water (10 mL) was stirred for 6 hours. A solution of
20% aqueous sodium sulphite (12 ml) was added and stirred for 30 minutes. The reaction
mixture was extracted with 25% ethanol/dichloromethane (4 X 100 mL). The combined
organic extracts were dried over anhydrous m~gn~sillm sulfate, concerltrated under reduced
pre~ure and purified by flash chromatography over silica gel using meth~nol
(5%)/dichloromethane eluant tO yield 3.6 g (76% yield) 1-(10,11-dihydro~yu~ ecanyl)-3,7-
dimethyl~nthine (CT1592).
1-(10,11-Dihydroxynn(lecanyl)-3,7-dimethylx~nthine (3.6 g, 10 mmol) was stirred
with hydrogen bromide (6.2 mL, 8.4 g of a 30% solution in acetic acid, 31.1 mmol) for 90
minllteS. The mixture was then added to a flask cnnt~ining 100 mL aqueous sodiumbicarbonate solution and 75 mL dichloromethane. After 10 minutes of vigorous stirring the
layers were separated and the aqueous portion washed with dichlorometh~ne (3 X 75 mL).
The organic portions were combined, dried over m~gnPsjllm sulfate, and evaporated to give
1-(10-acetoxy-11-bromollndecanyl)-3,7-dimethyl~:-nthint~ (3.6 g). Without further
purific~tir-n, the bromoacetate was taken up in methanol (25 mL) and treated with a solution
of sodium methoxide (prepared from 0.28 g, 12.2 mmol sodium, and 25 mL meth~nnl).
After 30 minutes~ most of the solvent was removed under reduced ~JIC5:iUlC and the residue
extracted with dichlorometh~n~ (3 X 75 mL). The organic portions were combined, dried
over m~gnecinm sulfate and concentrated under reduced pressure to obtain an off-white solid
which was purified by column chromatography over silica gel using dichlorometh~ne/(3%)
2s methanol eluant to yield 2.0 g (57% yield) 1-(10,11-Oxidolmdecanyl)-3,7-dimethy~ nthine
FY~nPIe 25
This example illustrates a synthesis of 1-(5,6-Oxidohexyl)glutarimide (CT-1605M).
Sodium hydride (425 mg, 17.7 mmol) was added to a solution of glutarimide (2.00 g, 7.7
mmol) in dimethyl sulfoxide (40 mL). After 20 minutes of stirring, 6-bromo-1-hexene (2.90
g, 17.7 mmol) is added. After 20 hours of stirring, the reaction was poured into a separatory
funnel containing 100 mL water and extracted with dichlormeth~ne (4 X 50 mL). The
organic portions were combined, washed with water (50 mL) and brine (50 mL) and dried to
yield 2.92 g (85% yield) l-(S-Hexenyl)glutarimide (CT1600) as a colorless oil.
1-(5-Hexenyl)glutarimide (630 mg, 3.2 mmol) was dissolved in dichlormethane (10
3s mL) and aqueous sodium bicarbonate (2.20 g, 26 mmol in 10 mL water) was added followed
by m-chloropeloxybenzoic acid (2.5 g of 50% MCPBA by wt, 7.2 mrhol). After 17 hours,
sodium metabisulfite (1.7 g, 9.() mmol) was added. After 30 minlltes, the reaction mixture
was extracted wi~h dichlormethane (3 x 10 mL). Organic portions were combined and
washed with sodium bicarbonate (saturated solution, 10 mL). Purification by
29
WO94/06431 2la5l92 PCr/US93/0907
chromatography on silica using 10% ethanol/dichlormeth:~ne yielded 180 mg (0.9 mmol,
27% yield) 1-(5,6-Oxidohexyl)glutarimide (CT1605).
FY~nPIe 2~
This example illustrates a synthesis of N-(8,9-Oxidononyl)glutarimide (CT1606).
Sodium hydride (1.02 g, 44 mmol) was added to a solution of glutarimide (5.00 g, 44 mmol)
in dimethyl sulfoxide (150 mL). After 20 minutes of stirring, 9-bromo-1-nonene (9.02 g, 44
mmol) was added. After 16 hours of additional stirring at room temperature, the reaction
was poured into a separatory funnel cont~ining 100 mL water and extracted with
dichlormethane (3 X 70 mL). The organic portions were c~mbined, washed with water (2 X
40 mL) and brine (50 mL) and dried to yield 10.09 g (97%) of 1-(8-Nonenyl)gl~ le(CT1604) as a colorless oil.
1-(8-Nonenyl)glutarimide (2.00 g, 8 mmol) was dissolved in rlichlorm~thane (15 mL)
and aqueous sodium bicarbonate (3.20 g, 38 mmol in 20 mL water) was added followed by
m-chloroperoxybenzoic acid (4.5 g of 50% MCPBA by wt, 13 mmol). After 17 hours
sodium metabisulfite was added slowly until no foaming was observed. After 30 minlltes of
stirring, the layers were separated and the aqueous layer extracted with tli~hlQrmeth~n~ (3 x
30 mL). The organic portions were combined and washed with sodium bicarbonate
(s~t~lr~tçd solution, 30 mL), water (20 mL), and brine (20 mL). The residue was purified
using chromotography on silica with ether to yield 756 mg (36% yield) N-(8,9-
Oxidononyl)glutarimide as a colorless liquid.
Fx~-nple 27
This example illustrates a synthesis of N-(11,10-Oxidoundecyl)glutarimide (CT1611).
Sodium hydride (95%) (168 mg, 7 mmol) was added to a solution of ~lu~ ide (565.6 mg,
5 mmol) in dimethyl sulfoxide (15 mL). After 20 minutçs of stirring, 1-bromundec-10-ene
(1.165 g, 5 mmol) was added and stirred for 12 hours at room tçrnperatllre~ The reaction
mixture was then poured into a separatory funnel cont:-ining 100 mL of water and extracted
with dichlormeth~ne (5 X 75 mL). The organic extracts were combined, washed with water
(50 mL) and brine (50 mL), dried over anhydrous m~gn~cium sulfate and concentrated under
reduced pressure. The crude product obtained was further purified by flash chromatography
over silica gel using 2()% ethyl acetate/hexane eluant to yield 0.777g (58.6% yield) N-(10-
Undecenyl)glutarimide (CT 1610).
A solution of N-(10-Undecenyl)glutarimide (0.6 g, 2.24 mmol) and m-
chloroperoxybenzoic acid (1.16 g; 3.37 mmol) (50% by wt) in dichloromçth:-ne (15 mL)
were stirred for 5 hours. The reaction mixture was diluted with 20 mL of dichlorom~thane
and washed successively with 2U% aqueous sodium sulphite solution (10 mL), s~tnrat~d
sodium bicarbonate solution (1() mL), water (10 mL) and brine solutions (10 mL). The
organic layer was dried over anhydrous m;lgn(~Sium sulfate and concentrated under reduced
pressure. The crude product obtaincd was further purified by flash chromatogr~phy over
O 94/06431 2 ~ ~ 5 1 ~ 2 PCI/US93/09073
silica gel using 80% petroleum ether/acetone eluant to yield 0.6 g (94% yield) of N-(11,10-
Oxidoundecyl)glutarimide.
F.Y~nPIe 28
This example illustrates a synthesis of N-(10,11-Oxidoundecyl)-2-piperidone
s (CT1618). A mixture of potassium hydroxide (1.55 g, 25 mmol, pellets ground in mortar
and pestle) and tetrabutylammonium bromide (1.61 g, 5.0 mmol) was stirred in drytetrahydrofuran (10 mL). A solution of d-valerolactam (2.5 g, 25 mmol) and 1-bromo-10-
ece~e (Lancaster, 5.9 g, 25 mmol) in tetrahydrofuran (15 mL) was added by syringe
pump over 1 hour. After stirring for a further 6 hours. Water (60 mL) and dichloromethane
(60 mL) was added to the reaction mixture. The layers were separated and the aqueous layer
was extracted with dichloromethane (2 X 50 mL). The combined organic layers werewashed with water (50 mL) and saturated salt solution (50 mL) and then dried with sodium
sulfate. The residue was further purified by chromatography using silica and 30% h~ n~.,
ethyl acetate to yield 2.88 g (46% yield) of N-(10-Undecenyl)-2-piperi(lQ~e (CT1616) as a
colorless oil.
To a mixture of N-(10-Undecenyl)-2-piperidone (4.00 g, 16 mmol) and a 60 %
aqueous solution of N-methylmorpholine-N-oxide (5 mL, 29 mmol) in water (10 mL) and
acetone (20 mL) was added potassium osmate dihydrate (12 mg, .03 mmol). After stirring
for 3 days, the mixture was treated with sodium dithionite (100 mg). After 30 minl tes,
dichlorometh:~ne (50 mL) and water (30 mL) were added and the organic layer separated.
The aqueous layer was extracted with dichloromethane/10% methanol (2 X 70 mL). The
combined organic extracts were dried over sodium sulfate and the solvents evaporated under
vacuum. The residue was purified by chromatography using silica and ethyl
acetate/methanol in a 0-20% gradient to yield 4.21 g (93%) N-(10,1 1-Dihydroxyundecyl)-2-
2s piperidone (CT1617) as a colorless oil.
To N-(10,11-dihydroxyundecyl)-2-piperidone (4.21 g, 14.8 mmol) was added 30%
hydrogen bromide-acetic acid (8.7 mL) and the mixture was then stirred for 1 hour. The
solution was poured carefully into a mixture of sodium bicarbonate (15 g), ice water (150
mL), and dichloromethane (100 mL). After carbon dioxide evolution has subsi(le~l, the
organic layer was separated and the aqueous layer extracted with dichloromethane (2 X 80
mL). The combined organic layers was dried over sodium sulfate and the solvent evaporated
under vacuum to give 1-(10-acetoxy-11 bromoundecyl)-2-piperidone (4.9 g, 89% yield) as a
viscous oil which dissolves in methanol (10 mL). A 1 M sodium methoxide (15 mL, 15
mmol) in methanol solution was added all at once. After stirring for 1 hour, the solution was
3s treated with water (5() mL) and then extracted with dichloromethane (3 X 50 mL). The
combined extracts were dried over sodium sulfate and the solvent evaporated under vacuum.
The residue was purified by chromatography using ethyl acetate to yield 2.40 g (61% yield)
N-(10,1 1-Oxidoundecyl)-2-piperidone as a colorless oil.
3 1
WO 94/06431 2 1 ~ PCr/US93/0907:~
FY~rnPIe 29
This example illustrates a synthesis of N-(9,10-Oxidodecyl)piperidine (CT1619).
Sodium hydride (95%) (864 mg, 36 mmol) was added to a solution of piperidine (2.554 g, 30
mmol) in dimethyl sulfoxide (75 mL). After 20 minutes of stirring, 1-bromundec-10-ene
s (6.99 g, 5 mmol) was added and stirred for 12 hours at room temperature. The reaction
IIILXIUI~ was then poùred into a separatory funnel containing 100 mL of water and ~Y I ~CIPd
with dichlormeth~ne (5 X 75 mL). The organic extracts were combinPd, washed with water
(50 mL) and brine (50 mL), dried over anhydrous m~gnP,cillm sulfate and concentrated under
reduced ~S~U1~;. The crude product obtained was further purified by flash chromatography
over silica gel using 5% meth~novdichloromethane eluant to yield 3.12 g (44% yield) N-(10-
undecenyl)piperidine (CT 1615) .
A solution of N-(10-undecenyl)piperidine (3.1 g, 13 mmol), 4-methylmorpholine-N-oxide (1.84 g, 15.7 mmol) and potassium osmate dihydrate (13 mg) in acetone (64 mL) and
water (16 mL) were stirred for 6 hours. The reaction was quenched by the ~dtlition of 25 mL
15 of a saturated solution of sodium sulphite and stirred for 15 minutes. The reaction mixture
was then extracted with ethyl acetate (4x 100 mL), the combined organic extract was dried
over anhydrous m~gmPsillm sulphate and concentrated under reduced pressure. The crude
product obtained was further purified by flash chromatography over silica gel using 10%
methanol/ethyl acetate eluant to yield 2.5 g (83.4% yield) N-(11,10-
20 dihydro~yu--decyl)piperidine (CT1622).
A mixture of N-(11,10-dihydroxyundecyl)piperidine (2.0g, 7.4mmol) and a 30%
solution of hydrogen bromide in acetic acid (4.45 mL, 22 mmol) was stirred for 90 min~ltps
The mixture was then added to a flask cont:-ining aqueous sodium bicarbonate solution (15 g
in 40 mL ) and dichloromethane (50 mL). After 10 mimltPs of vigorous stirring, the layers
2s were separated and the aqueous portion extracted with dichloromethane (2 X 50 mL). The
combined organic portions were dried over sodium sulfate. The solvent was evaporated to
give l-(10-acetoxy-11-bromoundecyl)piperidine which was used without further purifi~ation
Bromoacetate was dissolved in methanol (10 mL) and treated with a 1 M solution of sodium
methoxide (8 mL). After 30 minutes, the reaction mixture was added to water (30 mL) and
30 extracted with dichloromethane (3 X 5() mL). The organic extracts were combinP-~l washed
with water (25 mL) and brine solution (25 mL) and then dried over anhydrous m~gnPsillm
sulphate solution and concentrated under reduced pressure. The crude product obtained was
further purified by column chromatography over :llnmin~ (grade-II) using 10%
meth~nol/ethyl acetate eluant to yield 1.5 g (80.6%) N-(9~lo-oxi-loclecyl)pippri-lin
35 FY~Ie ?s0
This example illustrates a synthesis of l-Methyl-3-(8,9-cxidQnonyl)uracil (CT1804).
Sodium hydride (365 mg, 16 mmol) was added to a stirring solution of l-methyluracil (2.00
g, 16 mmol) in dimethyl sulfoxi~e (40 mL). After 15 minut~Ps, 6-bromo-1-nonene (3.26 g,
16 mmol) was added and the mixture stirred for 3 days. The reaction was then poured into
~ 94/06431 2 1 ~ 2 PCr/US93/09073
water (~0 mL) and extracted with dichloromethane (3 X 60 mL). The combmed orgamclayers were washed with water (50 mL), and aqueous saturate~lt solution (30 mL) was
then dried over sodium sulfate. The solvent was evaporated under vacuum to yield 3.72 g
(94%) 1-methyl-3-(8-nonenyl)uracil (CT-1817) as a colorless oil which solidified upon
standing.
A solution of 1-methyl-3-(8,9-nonenyl)uracil (3.72 g, 15 mmol), 4-
methylmorpholine-N oxide (2.1() g, 18 mmol), and potassium osmate (IV) dihydrate (11 mg,
3.0 x 10-5 mmol) in ~cetone (20 mL) and water (10 mL) are stirred for 2 days. After
addition of sodium hydrosulfite (100 mg) to quench the catalyst, the reaction mixture was
extracted with dichlorometh~ne (4 X 50 mL). The comhin~d organic layers were dried over
sodium sulfate and the solvent evaporated under vacuum to give an oily residue.
Cryst~11i7~tion of the residue from ether/dichlorometh:-ne yields 2.66 g (63% yields) 3-(8,9-
dihydroxynonyl)- 1 -methyluracil (CT1818) as white crystals.
A mixture of 3-(8,9-dihydroxynonyl)-1-methyluracil (2.15 g, 7.6 mmol) and a 30%
solutiQn of hydrogen bromide in acetic acid (4.5 mL, 23 mmol) were stirred for 6 hours. The
reaction mixture was added slowly to a mixture of sodium bicarbonate (8.4 g, 0.1 mol), water
(30 mL), and dichloromethane (30 mL). The layers were separated, and the aqueous layer
extracted with dichlorometh:~ne (3 X 40 mL). The combinPd organic layers were washed
with aqueous saturated salt solution (20 mL) and dried over sodium sulfate. The solvent was
removed under vacuum to yield 2.89 g (97% yield) 3-(8-acetoxy-9-bromononyl)-1-
methyluracil (CT1801) as a thick, slightly orange oil.
To a solution of 3-(8-acetoxy-9-bromononyl)-1-methyluracil (2.89 g, 7.4 mmol) inmethanol (10 mL) was added a 1 M meth ~nol solution of sodium mPtho~ e (8 mL). After 3
hours, the reaction mixture was poured into water (30 mL) and extracted with
dichlorom~th~ne (3 X 60 mL). The combined organic layers were washed with water (30
mL) and aqueous saturated salt solution (30 mL), then dried over sodium sulfate. The
solvent was evaporated under vacuum and the residue cryst~lli7P-l in ether to yield 1.61 (82%
yield) l-methyl-3-(8,9-oxidononyl)uracil.
F.Y~n1PIe 31
This example illustrates a synthesis of 3-(5,6-Oxidohexyl)-l-methyluracil (CT1808).
Sodium hydride (86 mg, 3.6 mmol) was added to a stirring solution of 1-methyluracil (500
mg, 4 mmol) in dimethyl sulfoxide (25 mL). After 15 minutes, 6-bromo-1-hexene (647 mg,
4 mmol) was added and the mixture stirred further for 20 hours. The reaction mixture was
then poured into water (50 mL) and extracted with 20% ethanoVdichlorometh~ne (3 X 50
mL). The combined organic layers were washed with aqueous .C~tnrate(l salt solution (20
mL) and dried over sodium sulfate. The solvent was evaporated under v..cuu... to give a
residue which was purified by chromatography with silica and ethyl acetate to yield 598 g
(72% yield) of 3-hexenyl-1-methyluracil (CT1800).
33
WO 94/06431 2 1 ~ 2 PCr/US93/0907:~
A solution of 3-(5-hexenyl)-1-methyluracil (598 mg, 2.9 mmol), 4-
methylmorpholine-N oxide (408 mg, 3.5 mmol), and a 2.5% solution in t-butanol of osmillm
tetroxide (3 drops) in acetone (15 mL) and water (5 mL) was stirred for 3 days. After
ad~litiQn of a saturated solution of sodium hydrosulfite (10 mL) and 15 min~lt~s, the reaction
s mixture was added to water (15 mL) and extracted with 20% ethanol/dichlorom~th~nP (4 X
40 mL). Tbe combined organic layers were dried over sodium sulfate and tbe solvent is
evaporated under vacuum to give 461 mg (66%) 3-(5,6-dihydroxyhexyl)-1-methyluracil
(CT1811) as a colorless oil.
A mixture of 3-(5,6-dihydroxyhexyl)-1-methyluracil (350 mg, 1.4 mmol) and a 30 %solution of hydrogen bromide in acetic acid (0.87 mL, 4.3 mmol) was stirred for 45 minlltes
Tbe mixture was then added to a mixture of sodium bicarbonate (1.6 g), water (10 mL) and
dichlorometh:-ne (20 mL). After 15 minutes of vigorous stirring, the layers were separated
and the aqueous layer extracted with dichloromethane (3 X 40 mL). The combined organic
layers were dried over sodium sulfate, then evaporated under vacuum,to give 3-(5-acetoxy-6-
bromohexyl)-l-methyluracil (500 mg, 100% yield).
Bromo~cet~lte was used in the next step without further pllrific~tion A solution of 3-
(5-acetoxy-6-bromohexyl)-1-methyluracil (360 mg, 1.0 mmol) in meth~nol (5 mL) was
treated with a 1 M methanol solution of sodium methoxide (1.3 mL). After 15 minllt~s~ tbe
reaction solution was poured into water (10 mL) and extracted witb dichlorom~th~nP (3 X 30
mL). The combined organic layers were dried over sodium sulfate and the solvent was
evaporated under vacuum to yield 150 mg (67% yield) 3-(5,6-oxidohexyl)-1-methyluracil as
a colorless oil.
F.~rnple 32
This example illustrates a synthesis of 3-(5,6-oxidohexyl)-2-methyldihydrouracil(CT1820). Sodium hydride (288 mg, 12 mmol) was added to a solution of N-
methylhydrouracil (1.54 g, 12 mmol) and 1-bromo-5-hexene (1.63g, 10 mmol) in 20 mL of
dimethyl sulfoxide at room temperature and stirred for 12 hours. The reaction mixture was
then quenched with water (80 mL) and extracted with dichlorometh:-ne (3xlO0 mL). Tbe
combined organic extract was washed with saturated aqueous salt solution solution (50 mL),
dried over anhydrous m:l~necium sulfate and conrentr~ted under reduced pressure. The
crude product obtained was further purified by flash chromatography over silica gel using
20% ~ceto~/hexane eluant to yield 2.0 g (79%) 3-(5-hexenyl)-1-methylhyd.oul~cil
(CT1812).
A solution of 3-(5-hexenyl)-1-methylhydrouracil (1.5 g, 7.1 mmol), and m-
chloroperoxybenzoic acid (3.68 g, 1().7 mmol) (50% by wt) in dichloromethane (60 mL) was
stirred for 5 hours. The reaction mixture was quenched with 20% aqueous sodium sulphite
solution(75 mL) and extrac~ed with dichloromethane (3xlO0 mL). The combined organic
extract is wasbed successively with saturated aqueous saturated salt solution (50 mL),
s~t~-r;lte-l sodium bicarbonate solution (5() mL), water (50 mL), aqueous saturated salt
34
0 94/06431 2 1 4 5 1 9 ~ PCr/US93/09073
solution (50 mL), dried over anhydrous m;lgn~.cium sulfate and concentrate~ un~er re~uce~
~JI`eS:iUl'e. The crude product obtained was further purified by flash chromatography over
silica gel using 30% acetone/ hexane eluant to yield 1.27 g (78.8 % yiçld) 3-(5,6-
oxi~lohe~yl)-2-methyldihydrouracil (CT1820).
5 Fxample 33
This example illustrates a synthesis of 3-(10,11-Oxidolln~ nyl)- 1-
methylhydrouracil (CT-1822). Sodium hydride (288 mg, 12 mmol) was added to a solution
of N-methylhydrouracil (1.54 g, 12 mmol) and 1-bromo-10-urltlecene (2.33 g, 10 mmol) in
20 mL of dimethyl sulfoxide at room temperature and stirred for 12 hours. The reaction
mixture was then quenched with w~ter (80 mL) and extracted with dichlorom- th~nP (3x100
mL). The combined organic extract was washed with saturated aqueous salt sol~ltion (50
mL), dried over anhydrous m;lgnecium sulfate and concentrated under reduced pressure. The
crude product obtained was further purified by flash chromatography over silica gel using a
20% acetone/hexane eluent to yield 2.04 g (61.8% yield) of 3-(lo-llr~clecenyl)
methylhydrouracil (CT1819).
A solution of 3-(10-undecenyl)-1-methylhydrouracil (0.28 g, 1 mmol), and m-
chloroperoxybenzoic acid (0.517 g, 1.5 mmol) (50% by wt) in dichlorometh~n~ (6 mL) was
stirred for 5 hours. The reaction mixture was diluted with 75 mL of d,ichloromPth~n~ and
washed successively with 20% aqueous sodium sulphite solution(25 mL), saturated NaHCO3
solution (25 mL), water (25 mL), aqueous saturated salt solution (25 mL), dried over
anhydrous m~gn~cillm sulfate and concentrated under reduced 1~1eS~U1e. The crude product
obtained was further purified by flash chromatography over silica gel using 20% acetone/
hexane eluant to yield 0.22 g (74.3 %) 3-(10,11-Oxidonndec~nyl)-l-methylhydroulacil.
FY~rnPIe 34
This example illustrates a synthesis of 3-(5,6-Oxidohexyl)-l-methylthymine
(CT1906). Sodium hydride (343 mg, 14 mmol) was added to a stirring solution of 1-
methylthymine (Sigma, 2.00 g, 14 mmol) in dimethylsulfoxide (30 mL). After 15 minllte
6-bromo-1-hexene (~ ~ncact~r, 2.30 g, 14 mmol) was added and stirring contimle-l for 69
hours. The reaction mixture was then poured into water (100 mL) and extracted with
dichlorometh~ne (4 X SU mL). The combined organic layers were washed with saturated
aqueous salt solution (40 mL) and dlied over sodium sulfate. The solvent was evaporated
under vacuum io give a residue which was cryst:~lli7P-d in dichlorometh~nP/ ethyl ether to
yield 2.80 g (88% yield) 3-(5-hexenyl)-1-methylthymine (CT1905). ~
A solution of 3-(5-hexenyl)-1-methylthymine (2.00 g, 9 mmol), 4-methylmorpholine-
N oxide (1.17 mg, 10 mmol), and a 2.5% sol. in t-butanol of osmillm tetroxide (0.15 mL ) in
acetone (15 mL) and water (10 mL) was stirred for 20 hours. After ~ddition of a s;l~ul~led
solution of sodium hydrosulfite (10 mL) and 15 minutes of stirring, the reaction mixture was
extracted with 20% ethanoVdichloromethane (4 X 40 mL). The combined organic layers
were dried over sodium sulfate and the solvent evaporated under vacuum to a give white
WO 94/06431 2 1 4 ~ :1 9 2 PCr/US93/0907:~
solid residue. The solid was recrystalliæd in ethanol to yield 2.00 g (89%) of 3-(5,6-
dlh~droxyhexyl)-l-methylthymine (CT1907).
A mixture of 3-(5,6-dihydroxyhexyl)-1-methylthymine (1.65 g, 6.4 mmol) and a 30%solution of hydrogen bromide in acetic acid (3.8 mL, 19.3 mmol) in water (5 mL) and
S acetone (10 mL) was stirred for 1.5 hours. The mixture was then added to a flask cont:lining
sodium bicarbonate (6.7 g), water (40 mL) and dichlorometh~ne (50 mL). After 15 minlltes
of vigorous stirring, the layers were separated and the aqueous layer washed with
dichlorometh~ne (2 X 50 mL). The combined organic layers were dried over sodium sulfate.
The solvent was evaporated under vacuum to yield 2.30 g (100%) of 3-(5-acetoxy-6-
bromohexyl)-l-methylthymine as a yellow oil. The bromo~ret~te was used in the next step
without further purific~tion A solution of 3-(5-acetoxy-6-bromohexyl)-1-methylLhy~ e
(2.30 g, 6.4 mmol) in methanol (10 mL) was treated with a 1 M m.oth~nol solution of sodium
methoxide (7 mL). After stirring for 15 minutes, the solution was poured into water (60 mL)
and extracted with 20% ethanoVdichloromethane (2 X 70 mL). The combinP.d organic layers
were dried over sodium sulfate and the solvent evaporated under vacuum to yield 1.30 g
(85%) 3-(5,6-oxidohexyl)-1-methylthymine as a white solid.
F.Y~nlPIe 35
This example illustrates a synthesis of l-Methyl-3-(8,9-oxidononyl)thymine
(CTl910). Sodium hydride (343 mg, 14 mmol) was added to a stirring solution of 1-
methylthymine (2.00 g, 14 mmol) in dimethylsulfoxide (40 mL). After 15 minutes, 9-
bromo-l-nonene (2.93 g, 14 mmol) was added and the mixture stirred for 20 hours. The
reaction was poured into 40 mL water and extracted with dichlorometh~n~. (3 X 50 mL). The
organic layers were combined, washed with water (40 mL), saturated ~queous salt sohltic)n
(20 mL), and dried over sodium sulfate. The solvent was evaporated to yield 2.76 g (73%
yield) 1-Methyl-3-(8-nonenyl)thymine (CT1917) as a colorless oil which solitlifi~d upon
st~ntlin~,
A solution of l-methyl-3-(8-nonenyl)thymine (2.63 g, 9.9 mmol), 4-
methylmorpholine-N oxide (1.39 g, 12 mmol), and potassium osmate tIV) dihydrate (7 mg, 2
x 10-5 mol) in acetone (20 mL) and water (10 mL) was stirred for 18 hours. After addition
of a saturated aqueous solution of sodium hydrosulfite (10 mL) and 15 min~ltes of stirring,
the reaction mixture was extracted with dichloromethane (50 mL) and with
dichlorometh~n~/20% meth;mol (2 X 5() mL). The combined organic layers were washed
with water (15 mL) and saturated aqueous salt solution (15 mL), and then dried over sodium
sulfate. The solvent was evaporated under vacuum to give a white solid residue. The solid
was recryst~lli7~ci in ethanol to yield 2.68 g (91% yield) 3-(8,9-dihydroxynonyl)-1-
me~l,ylLhy",ine (CT1918).
A mixture of 3-(8,9-dihydroxynonyl)-1-methylthymine (2.16 g, 7.6 mmol) and a 30%solution of hydrogen bromide in acetic acid (4.5 mL, 23 mmol ) was stirred for 1 hour. The
reaction was added slowly to a beaker containing sodium bicarbonate (8.4 g, 0.1 mol), ice
36
0 94/06431 2 1 4 !~ 1 9 2 PCI/US93/09073
water (30 mL), and dichloromethane (30 mL). The layers were separated and the aqueous
layer extracted with dichlorometh~ne (2 X 60 mL). The combined organic layers were
washed with water (30 mL), saturated aqueous salt solution (30 mL), and dried over sodium
sulfate. The solvent was removed to yield 2.59 g (85% yield) 3-(8-acetoxy-9-bromononyl)-
s l-methylthymine (CT19Q8) as a thick, sligh~ly or~nge oil.
To a solution of 3-(8-acetoxy-9-bromononyl)-1-methylthymine (2.04 g, 5.1 mmol) in
meth:~nol (15 mL) was added a 1 M solution of sodium methoxide (6 mL). After 3 hours, the
reaction was poured into water (20 mL) and extracted with dichlorometh~ne (3 X 30 mL).
The combined organic layers were washed with water (20 mL) and .s~tllr~sf d aqueous salt
10 solution (20 mL), then dried over sodium sulfate. The solvent was evaporated under vacuum
and the residue crystallized in dichlorometh:lnt -ether to yield 1.09g (76% yield) white
crystals of l-methyl-3-(8,9-oxidononyl)thymine.
F.~ample 3h
This example illustrates a synthesis of 3-(11,10-Oxidoundecyl)-l-melhyllh~ le
(CT1932). Sodium hydride (95%) (168 mg, 7 mmol) was added to a solution of 1-
methylthymine (700.5 mg, 5 mmol) in dimethylsulfoxide (15 mL). After 20 rninlltes of
stirring, 1-bromundec-10-ene (1.165 g, 5 mmol) was added and stirred for 12 hours at room
temperature. The reaction mixture was then poured into a separatory funnel cont~ining lO0
mL of water and extracted with dichloromethane (5 X 75 mL). The organic extracts were
combined, washed with water (50 mL) and saturated aqueous salt solution (50 mL), dried
over anhydrous magnesillm sulfate and concentrated under reduced pressure. The crude
product obtained was further purified by flash chromato~raphy over silica gel using a 20%
ethyl acetate/hexane eluant to yield 1.22 g (83.7% yield) 3-(10-undecenyl)-1-methylthymine
(CT1931).
2s A solution of 3-(1()-nn(lecenyl)-1-methylthymine (2 g, 6.8 mmol), and m-
chloroperoxybenzoic acid (3.5 g, 10.2 mmol) (50% by wt) in dichlororneth~ne (50 mL) was
stirred for S hours. The reaction mixture was diluted with 80 mL of dichlorometh~nt~ and
washed successively with 20% aqueous sodium sulphite solution (30 rnL), s~t~lr~t~d sodium
bicarbonate solution (30 mL), water (3() mL) and brine solutions (30 mL). The organic layer
was dried over anhydrous magnesillm sulfate ;md concentrated under reduced pressure. The
crude product obtained was further puri~ïed by flash chromatography over silica gel using a
70% hexane/ethyl acetate eluant to yield 1.73 g (82% yield) 3-(11,10-Oxidoundecyl)-l-
methylthymine (CT1932).
mple ?s7
3s This example illustrates a synthesis of 1-(3,4-Oxidobutyl)-3,7-dimethylx~nthine
(CT2513). Sodium hydride (95%) (1.26 g, 50 mmol) was added to a solution of thec~romine
(7.2g, 40 mmol) in dimethylsulfoxide (3()0 mL). After 20 minutes of stirring, 4-bromobutene (5.4 g, 4() mmol) was added. After 16 hours of stirring at room temperature,
the reaction was poured into a separa~ory funnel cont:-ining 1 L of water and extracted with
37
.
WO 94/06431 ~ ~ 4 ~ 1 ~ 2 PCr/US93/0907--
dichlorometh lne (S X 200 mL). The organic extracts were combined, washed with water
(100 mL) and brine (100 mL), dried over anhydrous m~gnPcillm sulfate and concentrated
under reduced pressure. The crude product obtained was further purifled by flashchromatography over silica gel using a 20% petroleum ether/ethyl acetate eluant to yield 6.3
s g (67.7% yield) 1-(3-Butenyl)-3,7-dimethylx:-nthinP. (CT2503) as a white solid.
- A solution of 1-(3-Butenyl)-3,7-dimethylx:lnthinP. (5.8 g, 24.8 mmol), 4-
methylmorpholine-N-oxide (3.63 g, 31 mmol) and potassium osmate dihydrate (18.3 mg;
0.05 mmol) in acetone (40 mL) and water (10 mL) was stirred for 6 hours. A solution of
20% aqueous sodium sulphite (20 ml) was added and stirred for 30 minllt~ps. The reaction
~ lure was e~rtr~çt~pd with 25% ethanol/dichloromethane (4 X 250 mL). The combined
organic extracts were dried over anhydrous m~mpsium sulfate and concentrated under
rP~rlllced pressure. The crude product obtained was further purified by flash chromatography
over silica gel using an acetone eluant to yield 4.5 g (67.7% yield) 1-(3,4,-dihydroxybutyl)-
3,7-dimethylx~nthine (CT2509).
1-(3,4-Dihydroxybutyl)-3,7-dimethylxanthine (3.98 g, 14.9 mmol) was stirred withhydrogen bromide (9.2 mL, 12.06 g of a 30% solution in acetic acid, 44.7 mmol) for 90
minutPs. The mixture was then added to a flask cQnt~ining 200 mL aqueous s~ tPd
sodium bicarbonate solution and 75 mL dichloromethane. After 10 minutps of vigorous
stirring, the layers were separated and the aqueous portion washed with dichloromP.th~n~ (3
X 150 mL). The organic portions were co-mbinp~l~ dried (m~gnPsium sulfate), and
evaporated to give 1-(3-acetoxy-4-bromobutyl)-3,7-dimethyl~nthine(s~6 g). Without
further puri~lcation, the bromoacetate was taken up in methanol (50 mL) and treated with a
solntion of sodium methoxide (prepared from 0.414 g, 12.2 mmol sodium, and 25 mLmeth~nol)~ After 30 minutes, most of the solvent was removed under reduced pressure and
the residue extracted with 25% ethanol/dichloromethane (3 X 150 mL). The organic portions
were combinP(l, dried (m~gne~ium sul~;ate) and concentrated under reduced ~r~s~u~c; to give
an off-white solid which is purified by column chromatography over silica gel using an ethyl
acetate eluant to yield 2.2 g (58 % yield) 1-(3,4-Oxidobutyl)-3,7-dimethylx~nthinP
F.~ample 38
This example illustrates a synthesis of 1-(11,12-Oxidododecyl)-3,7-dime~lylx~ .;.... e
(CT2518). To a suspension of m ~gnecium (6.4 g, 265 mmol) and a crystal of iodine in
tetrahydrofuran (40 mL) was added l()-nnclecenyl bromide (12.25 g, 53.0 mmol, available
from MTM) in tetrahydrofuran (30 mL) over 30 minutes and the reaction stirred for a further
30 mimlt~s after the addition was complete. The solution was added via a canula over 5
minnteS to a suspension of paraformaldehyde (1.80 g, 60.0 mmol) in tetrahydrofuran (40
mL) and stirred at 25C for 16 hours. Saturated ammonium chloride (80 mL) is added and
extracted with diethyl ether (2 x 10() mL). The combin~d organic extracts are dried using
m~gneSillm sulfate and evaporated to give a residue which is distilled at 2 mm Hg to yield
6.53 g (67% yield, b.p. 105-1()7C) of l l-dodecenyl alcohol as a clear liquid.
38
0 94/06431 2 1 4 5 1 9 2 PCI/US93/09073
To a solution of 11-dodecen-1-ol (5.5 g, 29.9 mmol) in dichloromethane (70 mL) at 0
C was added methanesulfonyl chloride (3.55 g, 2.40 mL,31.0 mmol) followed by
triethylamine (4.38 g, 46.0 mmol). After stirring for 10 minutes at 0C, the reaction was
allowed to warm to 25C and stilTed for 2 hours. The reaction was poured into water (60
s mL), separated and washed with dichloromethane (50 mL). The organic portions were
combin~d. dried using m~gn~Si~lm sulfate, and evaporated to yield 12-mPth~n~sulfonyl-1-
dode~en~ as a yellow oil which was then used without further purification.
To a suspension of sodium theobromine (6.00 g, 30.0 mmol) in dimethylsulfoxide (60
mL) was added 12-methanesulfonyl-1-dodecene and the reaction stirred for 16 hours at 60C.
The mixture was then poured into water (120 mL) and extracted with diethyl ether (2 x 100
mL). The organic portions were combined, dried using m~gnt~sium sulfate and evaporated to
give a cream solid. Recrystallization from ethyl acetate/hexane 1 :1 yields 6.97 g (67% yield)
l-(ll-dodecenyl)-3,7-dimethylxanthine (CT2516) as a white solid.
A solution of l-(l l-Dodecenyl)-3,7-dimethylx~nthine (4.70 g, 13.6 mmol), 4-
methylmorpholine-N-oxide (4.79 g, 40.7 mmol) and pot~ci-lm osmate dihydrate (52 mg,
0.14 mmol) in acetone/water 1:2 (75 mL) was stirred for 16 hours. Water (50 mL) and
sodium sulfite (5 g) were added and the mixture stirred for 1 hour. The reaction mixture was
extracted with dichloromethane (3 x 100 mL3, dried using m~gnP~illm sulfate and evaporated
to obtain a pale green solid. Recrystallization from hot ethyl acetate yields 4.32 g (84%
yield) 1-(11,12-dihydroxydodecyl)-3,7-dimethylx~nthin~ (CT2517) as a white solid.
1-(11,12-Dihydroxydodecyl)-3,7-dimethylx~nthin~-. (2.50 g, 6.58 mmol) was stirred
with hydrogen bromide (6.39 mL of a 30% solution in acetic acid, 19,73 mmol) for 2 hours.
The mixture was then added over 10 minutes to water (25 mL), ice (30 g) and sodium
hydride CO3 (15 g) and stirred for 30 minutes. The reaction mixture was t;~ c~d with
2s dichlororneth~ne (3 x 50 mL), and the combined organic phases were dried using m~gn~Sil~m
sulfate and evaporated to obtain 3.18 g (99% yield) of 1-(11-acetoxy-12-bromododecyl)-3,7-
dimethylx:7nthine Without further purification, this crude product was taken up in mPth~nol
(10 mL) and treated with a solution of sodium methoxide (prepared from sodium, 0.160 g,
6.90 mmol, and 20 mL methanol). After 60 minutes, the reaction was added to water (30
mL) and extracted with dichloromethane (3 x 50 mL). The organic portions were combined,
dried and evaporated to yield 2.20 g (93% yield) 1-(11,12-oxidododecyl)-3,7-
dimethylx:-nthine as a white solid.
F~Y~n~Ie 39
This example illustrates a synthesis of 1-(9,10-Oxidoctadecyl)-3,7-dimethylx~nthin~
(CT2541). Triphenylphosphine (5.24 g; 20 mmol) was added in portions to a solution of
oleyl alcohol (5.37 g; 20 mmol) and carbontetrabromide (6.63 g; 20 mmol) in 400 mL of
dichlorometh lnt~ and stirred for an hour at room temperature. The solvent was removed
under reduced pressure and ~he residue extracted with hexane (3 X 200 mL). Further
39
WO 94/06431 2 I 4 5 1 9 PCI/US93/0907--
purifil~tion was done by flash chromatography over silica gel using hexane as eluant to yield
5.82 g (88% yield) of l-bromo-9-octadecene.
Sodium hydride (95%) (84 mg, 3.5 mmol) was added to a solution of theobromine
(0.595 g, 3.2 mmol) in dimethylsulfoxide (15 mL). After 20 minllt~s of stirring, 1-bromo-9-
S ct~1ecene(0.995 g, 3 mmol) was added. After 6 hours of stirring at room temperature, the
reaction mixture was warmed to 60C for 3 hours and then poured into a separatory funnel
cont~ining 50 mL of water and extracted with dichlorometh~ne (5 X 40 mL). The organic
extracts were combined, washed with water (50 mL) and brine (50 mL), dried over
anhydrous m~gneSium sulfate and col~centrated under reduced pres~ure. The crude product
obtained was further purified by flash chromatography over silica gel using a 30%
~retonPJpetroleum ether eluant to yield 0.44 g (34 % yield) of )1-(9-oct~-lecenyl)-3,7-
dimethylxanthine (CT2539).
A solution of l-(9-octadecenyl)-3,7-dimethylx~nthin~ (0.15 g, 0.35 mmol) and m-
chloroperoxybenzoic acid (0.15 g, 0.43 mmol) (50% by wt) in dichloromethane (7 mL) was
stirred for 5 hours. The reaction mixture was diluted with 40 mL of dichlorometh~nP and
washed succe~cively with 20% aqueous sodium sulphite solution (10 mL), s~t~lr~ted sodium
bicarbonate solution (10 mL), water and brine solutions. The organic layer was dried over
anhydrous m~gn~cium sulfate and concentrated under reduced pressure. The crude product
obtained was further purified by flash chromatography over silica gel using a 30%
acetone/hexane eluant to yield 0.73 g (48.4% yield) of 1-(9,10-oxidoctadecyl)-3,7-
dimethylx~nthin~.
F.~lnple 40
This example illustrates a synthesis of 1-(4-(S)-Methyl-7,8-oxido-8-methylnonyl)-
3,7-dimethylx~nthine. (CT2548S). To a suspension of m~gneSium (2.74 g, 140 mmol) and a
crystal of iodine in tetrahydrofuran (15 mL) was added (S)-citronellyl bromide (5.0 g, 22.8
mmol) in tetrahydrofuran (10 mL) over 30 minutes and the reaction stirred for a further 30
mimlteS after the addition was complete. The solution was added via a canula over S minutl~.s
to a suspension of paraform~ ehyde (1.80 g, 60.0 mmol) in tetrahydrofuran (15 mL) and
stirred at 25C for 6 hours. Saturated ammonium chloride (40 mL) was added and extracted
with diethyl ether (2 x 30 mL). The combined organic extracts were dried (m~ ps~sulfate) and evaporated to yield 3.25 g (84% yield) 4-(S)-methyl-8-methylnon-7-enyl alcohol
as a clear liquid.
To a solution of 4-(S)-methyl-8-methylnon-7-enyl alcohol (3.25 g, 19.1 mmol) in
dichloromethane (50 mL) at 0C is added methanesulfonyl chloride (2.29 g, 20.0 mmol)
followed by triethylamine (3.()4 g, 30.0 mmol). After stirring for 10 mimltes at 0C, the
reaction was allowed to warm to 25C and stirred for 3 hours. The reaction was poured into
water (50 mL), sep~ ated and washed with dichloromethane (50 mL). The organic portions
were combined, dried using m~gnesium sulfale, and evaporated to give the 1-
~0 94/06431 2 1 ~L 5 1 ~ ~ PCI/US93/09073
mPth~n~sulfonyl-4-(S)-methyl-8-methylnon-7-ene as a yellow oil which is used without
further purification.
To a suspension of sodi~rl theobromine (4.05 g, 20.0 mmol) in dimethylsulfoxide (50
mL) was added 1-methanesulfonyl-4-(S)-methyl-8-methylnon-7-ene and the reaction stirred
s for 16 hours at 60C. The mixture was then poured into water (100 mL) and extracted with
ethyl acetate (100 mL, 2 x 50 mL). The organic portions were combined, dried using
m~gnesillm sulfate, and evaporated to give a residue which was purified by column
chromatography (ethyl acetate/hexane) to yield 1.83 g (30% yield) 1-(4-(S)-Methyl-8-
methylnon-7-enyl)-3,7-dimethylxanthine (CT2536S) as a white solid.
A solution of (0.65 g, 1.96 mmol), 4-methylmorpholine-N-oxide (0.69 g, 5.~7 mmol)
and potassium osmate dihydrate (7 mg, 0.021 mmol) in ~eton~/water 1:2 (12 mL) was
stirred for 16 hours. Water (10 mL) and sodium sulfite (1 g) were added and the reaction
mixture stirred for 1 hour. The reaction mixture was extracted with dichlorom~th~ne (3 x 30
ml), dried using m~gr1esillm sulfate and the solvent evaporated to yield 0.675 g ( 94%) of 1-
(4-(s)-methyl-7~8-dihydroxy-8-methylnonyl)-3~7-dimethylx~nthine (CT2537S) as a colorless
oll.
1-(4-(S)-Methyl-7,8-dihydroxy-8-methylnonyl)-3,7-dimethylx~nthine (0.37 g, 1.00
mmol) was stirred with hydrogen bromide (1.25 mL of a 30% solution in acetic acid, 3.00
mmol) for 4 hours. The mixture was then added over 10 minut~s to water (10 mL), ice (5 g)
and sodium bicarbonate (2 g) and stirred for 30 mimltes. The reaction mixture was t;~LIacled
with dichlornmeth~ne (2 x 15 mL), and the combined organic phases were dried using
m~nPsillm sulfate and the solvent evaporated to yield a residue of 1-(4-(S)-methyl-7-
acetoxy-8-bromo-8-methylnonyl)-3,7-dimethylx~nthine
Without further purification, the crude residue was taken up in methanol (5 mL) and
2s treated with a solution of sodium methoxide (prepared from sodium (0.025 g, 1.09 mmol)
and 5 mL methanol). After 40 minutes, the reaction mixture was added to water (10 mL) and
extracted with dichloromethane (3 x lO mL). The organic portions were combined, dried and
the solvent evaporated to yield 0.32 g ( 92%) 1-(4-(S)-Methyl-7,8-oxido-8-methylnon)-3,7-
dimethyl~ nthin~ as a white solid.
F~ e 41
This example illustrates a synthesis of 1-(4-(R)-Methyl-7,8-oxido-8-methylnon)-3,7-
dimethyl~c~nthine (CT2548R). To a suspension of m:lgn~ lm (2.74 g, 140 mmol) and a
crystal of iodine in tetrahydrofuran (15 mL) was added (R)-citronellyl bromide (5.0 g, 22.8
mmol) in tetrahydrofuran (1() mL) over 30 minutes and the reaction stirred for a further 30
mimlteS after the addition was complete. The solution was added via a canula over 5 minnt~s
to a suspension of paraformaldehyde (1.80 g, 60.0 mmol) in tetrahydrofuran (15 mL) and
stirred at 25C for 6 hours. Saturated ammonium chloride (40 mL) was added and extracted
with diethyl ether (2 x 30 mL). The combined organic extracts were dried (m~gnesillm
41
W O 94/06431 2 i 4 5 ~ 9 2 PC~r/US93/0907 -
sulfate) and the solvent evaporated to yield 3.25 g (84% yield) 4-(R)-methyl-8-methylnonyl-
7-enyl alcohol as a clear liquid.
To a solution of 4-(R)-methyl-8-methylnonyl-7-enyl alcohol (3.25 g, 19.1 mmol) in
dichloromethane (50 mL) at 0C is added methanesulfonyl chloride (2.29 g, 20.0 mmol)
s followed by triethylamine (3.04 g, 30.0 mmol). After stirring for 10 minutes at 0C, the
reaction was allowed to warm to 25C and stirred for 3 hours. The reaction was poured into
water (50 mL), separated and washed with dichloromethane (S0 mL). The organic po,tions
were combin~-l, dried using m~ne~ m sulfate and the solvent evaporated to give the 1-
me~h~n~sulfonyl-4-(R)-methyl-8-methylnon-7-ene as a yellow oil which was used without
further p--rification.
To a suspension of sodium theobromine (4.05 g, 20.0 mmol) in dimethylsulfoxide (S0
mL) was added l-mçth~nesulfonyl-4-(R)-rnethyl-8-methylnon-7-ene and the reaction stirred
for 16 hours at 60C. The mixture was then poured into water (100 mL) and extracted with
ethyl acetate (100 mL, 2 x S0 mL). The organic portions were combined, dried using
m:lgn~cium sulfate and the solvent evaporated to give a residue which was purified by
column chromatography using ethyl acetate/hexane to yield 1.70 g (28% yield) 1-(4-(R)-
Methyl-8-methylnon-7-enyl)-3,7-dimethylxanthine (CT2536R) as a white solid.
A solution of 1-(4-(R)-Methyl-8-methylnon-7-enyl)-3,7-dimethyl~r~nthinto. (0.48 g,
1.44 mmol), 4-methylmorpholine-N-oxide (O.S l g, 4.38 mmol) and potassium osmatedihydrate (S mg, O.OlS mmol) in acetone/water 1:2 (9 mL) was stirred for 16 hours. Water
(10 mL) and sodium sulfite (1 g) were added and the reaction mixture stirred for 1 hour. The
reaction mixture was extracted with dichloromethane (3 x 30 ml) using dried m~gn-~sillm
sulfate and the solvent evaporated to yield O.S l g (97% yield) 1-(4-(R)-Methyl-7,8-
dihydroxy-8-methylnonyl)-3,7-dimethylxanthine (CT2537R) as a colorless oil.
1-(4-(R)-Methyl-7,8-dihydroxy-8-methylnonyl)-3,7-dimethyl~nthine (0.29 g, 0.80
mmol) was stirred with hydrogen hromide (1.00 mL of a 30% solution in acetic acid, 2.40
mmol) for 4 hours. The mixture was then added over 10 minutf~s to water (10 mL), ice (S g)
and sodium bicarbonate (2 g) and stirred for 30 minutes. The reaction mixture was extracted
with dichloromethane (2 x lS mL), the combined organic phases dried using m~n~sillm
sulfate and the solvent evaporated to obtain a residue of 1-(4-(R)-methyl-7-acetoxy-8-bromo-
8-methylnonyl)-3,7 -dimethylxanthine.
Without further purification, this crude residue was taken up in methanol (S mL) and
treated with a solution of sodium methoxide (prepared from sodium (0.020 g, 0.85 mmol)
and S mL methanol). After 4() minutes, the reaction was added to water (10 mL) and
extracted with dichloromethane (3 x 1() mL). The organic portions were cornbin~cl, d;ied and
evaporated to yield 0.24 g (86% yield) of 1-(4-(R)-Methyl-7,8-oxido-~8-methylnonyl)-3,7-
dimethyl~ nthine (CT2548R) as a white solid.
42
~O 94/06431 2 1 4 5 1 9 2 PCr/US93/09073
F.Y~nll~le 42
This example illustrates a synthesis of 1-(3,7-Dimethyl-2,3,6,7-dioxidoctyl)-3,7-
dimethyl~c~nthine (CT2552). Sodium hydride (95%) (0.28 g, 12 mmol) was added to a
solution of theobromine (2.16 g, 12 mmol) in dimethylsulfoxide (50 mL). After 20 mimltes
of stirring, geranyl bromide (2.17 g, 10 mmol) was added. After 6 hours of stirring at room
temperature, the reaction mixture was warmed to 60C for 3 hours and then poured into a
separatory funnel cont~ining 150 mL of water and extracted with dichlorometh~nP (5 X 75
mL). The organic extracts were combined, washed with water (50 mL) and brine (50 mL),
dried over anhydrous m~gnecillm sulfate and concentr~ted under reduced ~res~ure. The
crude product obtained was further purified by flash chromatography over silica gel using
30% acetonPJpetroleum ether eluant to yield 2.1 g (66.5 % yield) 1-(Geranyl)-3,7-
dimethylxanthine (CT2545).
A solution of l-(geranyl)-3,7-dime~hyl~c~nthine. (0.316 g, 1 mmol), and m-
chloroperoxybenzoic acid (1.035 g; 3 mmol) (50% by wt) in dichloromPth~ntq. (15 mL) was
stirred for 6 hours. The reaction mixture was diluted with 80 mL of dichlorometh~np and
washed successively with 20% aqueous sodium sulphite solution (20 mL), s~tnr~tecl sodium
hydride CO3 solution (20 mL), water and brine solutions. The organic layer was dried over
anhydrous m~gnesium sulfate and concentrated under reduced pressure. The crude product
obtained was further purified by flash chromatography over silica gel using a 30%
acetone/hexane eluant to yield 0.25 g (72% yield) 1-(3,7-dimethyl-2,3,6,7-dioxidoctyl)-3,7-
dimethyl~c~nthine.
FY~nPIe 43
This example illustrates a synthesis of 1-(12,13-Oxidotridecyl)-3,7-dimethylx~nthint-
(CT2562). To a suspension of m~gnecium (4.12 g, 172 mmol) and a crystal of iodine in
tetrahydrofuran (40 mL) was added 10-undecenyl bromide (8.00 g, 34.3 mmol) in
tetrahydrofuran (30 mL) over 30 minutes and the reaction stirred for a further 30 minllte~s
after the addition was comple~e. The solution was added via a canula over S minut~s to a
solution of ethylene oxide (2.65 g, 60.0 mmol) in tetrahydrofuran (30 mL) and stirred at 25
C for 16 hours. Saturated ammonium chloride (100 mL) and lM hydrogen chloride (200
mL) were added and extracted with diethyl ether (2 x 200 mL). The combined organic
extracts were dried (m~gnecium sulfate) and evaporated to give a residue which was distilled
at 1.5 mm Hg to afford 12-tridecenyl alcohol as a clear liquid (4.11 g, 61%, b.p. 98-101C).
To a solution of 12-tridecen-1-ol (2.11 g, 10.7 mmol) and carbon tetrabromide (4.37
g, i3.1 mmol) in dichloromelhane (15 mL) at 0C was added triphenyl phosphine (3.45 g,
13.1 mmol) in portions over 5 minutes. After stirling for 1.5 hours at 25C the solvent was
evaporated and the residue ex~rac~ed with hexane (3 x 30 mL), filtering off any solids. The
solvent was evaporated to afford 12-tridecenyl bromide as a clear oil which was used without
further purification.
43
WO 94/06431 ~ ~ PCr/US93/0907~
2145~ 92
To a suspension of sodium theobromine (2.22 g, 11.0 mmol) in dimethylsulfoxide (25
mL) was added 12-tridecenyl bromide and the reaction stirred for 16 hours at 60C. The
mixture was then poured into water 80 mL) and extracted with dichlorometh~nP (3 x 50
mL). The combined organic porti~ns were washed with water (3 x 100 mL), dried using
m~gnP~ m sulfate, and evaporated to give a gummy residue. P-lrific~tion by column
chromatography using an ethyl acetate/hexane eluant yields 1.89 g (50% yield) 1-(12-
Tridecenyl)-3,7-dimethylx~nthine. (CT2555) as a white solid.
A sohltio~ of 1-(12-Tridecenyl)-3,7-dimethylx~nthinP- (1.39 g, 3.86 mmol), 4-
methylmorpholine-N-oxide (1.36 g, 11.6 mmol) and potassium osmate dihydrate (14 mg,
0.040 mmol) in acetone/water 1 :2 (25 mL) was stirred for 16 hours. Water (25 mL) and
sodium sulfite (2 g) were added and the reaction mixture stirred for 1 hour. The reaction
mixturç was extracted with dichloromethane (3 x 50 mL), dried using m~gnP~inm sulfate and
the solvent evaporated to yield 1.25 g (82%) of 1-(12,13-dihydroxytridecyl)-3,7-dimethyl~nthine (CT2556) as a white solid.
1-(12,13-Dihydroxy-tridecyl)-3,7-dimethyl~r~nthinP (1.15 g, 2.92 mmol) was stirred
with hydrogen bromide (2.84 mL of a 30% sohltion in acetic acid, 8.76 mmol) for 2 hours.
The mixture was then added over 10 minutes to water (20 mL), ice (15 g) and sodium
bicarbonate (S g) and stirred for 30 minutes~ The reaction mixture was extracted with
dichlor ~meth~ne (2 x 50 mL), and the combined organic phases were dried using m~nPsillm
sulfate and the solvent evaporated to obtain a residue of 1-(12-acetoxy-13-bromotridecyl)-
3,7-dimethyl~nthine. Without further purification, this crude residue was taken up in
meth~nol (S mL) and treated with a solution of sodium mPthclxide (prepared from sodium,
0.069 g, 3.00 mmol, and 5 mL methanol). After 40 minutes, the reaction mitxure was added
to water (15 mL) and extracted with dichloromethane (3 x 30 mL). The organic portions
were combined, dried and the solvent evaporated to yield 1.00 g (91% yield) 1-(12,13-
Oxidotridecyl)-3~7-dimethy~ nthine (CT2562) as a white solid.
F.Y~n1.PIe 44
This example illustrates a synthesis of 1-(7,8-cis-Oxidodecyl)-3,7-dimethyl~r~nthin~
(CT2563). 7-cis--Decenal (Johnson Matthey Catalog Co., 10.00g, 6.5 mmol) was added
dropwise to a stirring suspension of sodium borohydride (2.45 g, 65 mmol) in ethanol (100
mL) at 0C. The ice bath was allowed to melt and stirring continued for 3 hours.~mmonil-m chloride solution (sat., 60 mL) was added with water (50 mL) and the mixture
was extracted with dichloromethane (3 x 100 mL). The organic extracts were combined and
washed with water (50 mL) and brine (50 mL) and dried (sodium sulfate). E~apul~Lion of
3S solvent gave the alcohol 7-cis--decen-1-ol as a colorless oil (9.19g, 91% yield).
7-cis-Decen-l-ol (5.()() g, 32.1 mmol) and methanesulfonyl chloride (2.5 mL, 3.70 g,
32.3 mmol) in dichlorc)meth:lnp (150 mL) at 0C is treated dropwise with triethylamine (6.7
mL, 4.9 g, 48 mmol), after which time, the ice bath is allowed to melt. After stirring 3 hours
at room temperature, the reaction was poured into a separatory funnel containing saturated
44
~O 94/06431 2 1 ~ ~ I 9 ~ PCr/US93/09073
sodium bicarbonate solution (5() mL) and dichloromethane (50 mL). The layers were
separat~d and the aqueous layer washed with dichlorometh~ne (2 X 50 mL). The organic
layers were combined, washed with hydrogen chloride solution (50 mL, 1%), water (50 mL),
and brine (40 mL), then dried (sodium sulfate). The solvent was removed to yield 6.97 g
s (92%) 7-cis-decene-1-meth~nes--ltonate as a yellow oil. The mesylate was used in the next
step without further purific~tion
1-Sodiotheobromine (6.00 g, 29.7 mmol) and 7-cis-decene-1-mPth~n~s-llfonate (6.97
g, 29.7 mmol) were stirred in dimethylsulfoxide (60 mL) for 15 hours, then at 80C for 3
hours, then cooled. The reaction mixture was poured into water (100 mL) and extracted with
dichlorom~th~ne (3 X 60 mL). The organic layers were combined and washed with water
(60 mL) and brine (50 mL) and dried using sodium sulfate. Addition of ether and petroleum
ether to the residue precipitates a white solid, yielding 3.25 g (32% yield) 1-(7-~is-Decenyl)-
3,7-dimethylxanthine (CT2560).
1-(7-cis-Decenyl)-3,7-dimethyl~r~nthine (0.50 g, 1.6 mmol) in dichloromçth~n~ (25
mL) was added to a solution of sodium bicarbonate (1.30 g, 16 mmol) in water (20 mL). 4-
Chloro-peroxybenzoic acid (326 mg, or 652 mg of a 50% mixture, 1.9 mmol) was added and
the reaction mixture stirred at room temperature for 20 hours. Sodium sulfite (100 mg) was
added to quench residual MCPBA. To the reaction mixture was added dichlorometh~ne (30
mL) and water (20 mL). The organic layer was separated and the aqueous layer washed with
dichlornmeth~n.o (3 x 30 mL). The organic layers were combined and washed with saLulaled
sodium bicarbonate solution (20 mL), and brine (20 mL) and then dried using sodium sulfate.
Removal of solvent results in an oil which solidifies upon standing. The white solid was
washed with ether and dried to yield 460 mg (86% yield) of 1-(7,8-cis-Oxidodecyl)-3,7-
dimethyl~c~n~hine.
2s F.~ n.ple 45
This example illustrates a synthesis of 1-(13,14-Oxidotetradecyl)-3,7-
dimethylx~nthine (CT3503). To a suspension of m~gnesium (1.86 g, 77.2 mmol) and a
crystal of iodine in THF (20 mL) was added 10-nndecenyl bromide (6.00 g, 25.8 mmol) in
THF (14 mL) over 4() minutes and the reaction stirred for a further 30 minlltes after the
addition was complete. The solution was added via a canula over 50 minutes to a suspension
of copper iodide (0.5() g, 2.58 mmol) and 1-bromo-3-chloropropane (3.84 mL, 38.7 mmol) in
THF (20 mL) and stirred at 25C for 16 hours. Sulfuric acid (1.0 M, 50 mL) was added,
extracted with diethyl ether (2 x 60 mL) and the organic solvent dried using m~gnP~ m
sulfate and evaporated. The residue was rli~illed at 0.25 mmHg to obtain 3.06 g (51% yield,
b.p. 98-100C)13-tetradecenyl chlolide as a colourless liquid.
To a suspension of sodium theobromine (1.82 g, 8.68 mmol) in dimethylsulfoxide (20
mL) was added 13-tridecenyl chlolide and the reaction stirred for 48 hours at 50C. The
mixture was then poured into water (6() mL) and extracted with ethyl acetate (3 x 50 mL).
The organic portions were combined, dried using m:~gnt-cillm sulfate and evaporated to give a
W0 94/06431 -; PCr/US93/0907~
2 ~ 2
cream solid. Recrystallization from hot hexane yields 2.38 g (73% yield) 1-(13-
Tetr~decenyl)-3,7-dimethylxanthine as a white solid.
A solution of 1-(13-Tetradecenyl)-3,7-dimethylx~nthine (2.00 g, 5.35 mmol), 4-
methylmorpholine-N-oxide (2.72 mL, 60% wt in water, 15.8 mmol) and potassium osmate
dihydrate (21 mg, 0.05 mmol) in acetone/water 3:1 (80 mL) was stirred for 16 hours. Water
(100 mL) and sodium sulfite (1 g) were added and stirred for 1 hour. The reaction mixture
was extracted with dichloromethane (3 x 100 mL) and the organic phase dried (m~nesjllm
sulfate) and evaporated to yield 2.10 g (96% yield) 1-(13,14-Dihydroxytetradecyl)-3,7-
dimethylx:lnthinP. as a white solid.
1-(13,14-Dihydroxytetradecyl)-3,7-dimethyl~r~nthine (0.80 g, ~.96 mmol) was stirred
with HBr (1.94 mL of a 30% solution in acetic acid, 5.88 mmol) for 2 hours. The mixture
was then added over 10 minut~s to water (20 mL), ice (15 g) and NaHCO3 (3.5 g) and stirred
for 30 minutes. The reaction mixture was extracted with dichloromPth~ne (2 x 50 mL), the
combined organic phases were dried using m~gnecillm sulfate and the solvent was evaporated
to obtain a residue of 1-(13-acetoxy-14-bromotetradecyl)-3,7-dimethylx~nthinP.
Without further purification, this crude residue was taken up in methanol (S mL) and
treated with a solution of sodium mPtho~cide (prepared from sodium (0.069 g, 3.00 mmol)
and S mL meth~nol). After 40 minutes, the reaction was added to water (15 mL) and
extracted with dichloromethane (3 x 30 mL). The organic portions were combinP~l dried and
the solvent evaporated to yield 0.71 g (93% yield) of 1-(13,14-Oxidotetradecyl)-3,7-
dimethyl~nthine as a white solid.
F.~ample 46
This example illustrates a synthesis of 1-(16,17-Oxidoheptadecyl)-3,7-
dimethyl~nthinP (CT-3516). To a suspension of m~gn~sium (3.10 g, 129 mmol) and acrystal of iodine in THF (10 mL) was added 10-nnrlecenyl bromide (6.00 g, 25.8 mmol,
available from MTM) in THF (20 mL) over 40 minutes and the reaction stirred for a further
30 minutes after the addition was complete. The solution was added via a canula over 50
mimltPs to a suspension of copper iodide (0 50 g, 2.58 mmol) and 1-bromo-6-chloro hexane
(6.00 mL, 40.0 mmol) in THF (20 mL) and stirred at 25C for 16 hours. Sulfuric acid (1.0
M, 50 mL) was added, extracted with diethyl ether (2 x 60 mL) and the organic solvent dried
using m:lgnPsium sulfate and the solvent subsequently evaporated. The residue was distilled
at 0.75 mmHg to yield 1.78 g (25% yield, b.p. 130-135C) 16-hep~rlecenyl chloride as a
colourless liquid.
To a suspension of sodium theobromine (2.02 g, 10.0 mmol) in dimethylsulfoxide/
tetrahydrofuran (2:1, 30 mL) was added 16-heptadecenyl chloride and the reaction stirred for
16 hours at 60C. The mixture was poured into water (75 mL) and extracted with ethyl
acetate (3 x 75 mL). The organic portions were combined, dried using m~gnPSinm sulfate,
and the solvent evaporated to give a cream solid. Recryst:111i7~tion from hot hexane yields
2.31 g (~5% yield) 1-(16-Heptadecenyl)-3,7-dimethyl~nthinP- as a white solid.
46
-- -- --
~O 94/06431 2 i ~ ~ 1 9 2 PCr/US93/09073
A solution of 1-(16-Heptadecenyl)-3,7-dimethylx~nthine (1.50 g, 3.60 mmol), 4-
methylmorpholine-N-oxide (1.83 mL, 60% wt in water, 10.6 mmol) and potassium osmate
dihydrate (16 mg, 0.04 mmol) in acetone/water/ tetrahydrofuran (10:7:5, 110 mL) was stirred
for 60 hours. Water (100 mL) and sodium sulfite (1 g) were added and the reaction mixture
stirred for 1 hour. The reaction mixture was extracted with dichloromethane (2 x 100 mL)
and the organic phase dried using nl;lgne~ium sulfate and the solvent evaporated to afford a
cream solid. Recryst~11i7~tion from hot ethyl acetate yields 1.31 g (81% yield) of 1-(16,17-
dihydroxyheptadecyl)-3,7-dimethylx:lntl-in~ (CT3514).
1-(16,17-Dihydroxyheptadecyl)-3,7-dimethylx~nthine (1.10 g, 2.44 mmol) was
stirred with HBr (3.50 mL of a 30% solution in acetic acid, 17.1 mmol) for 4 hours. The
mixture was then added over 10 minutes to water (50 mL),and NaHCO3 (10 g) and stirred
for 30 minlltes. The reaction mixture was extracted with dichlorometh~nt (3 x 30 mL), the
combin~d organic phases were dried using m~gneSium sulfate and the solvent was evaporated
to obtain a residue of 1-(16-acetoxy-17-bromoheptadecyl)-3,7-dimethylx~nthine
Without further purification, 1-(16-acetoxy-17-bromoheptadecyl)-3,7-
dimethylx~nthine was taken up in methanol (S mL) and treated with a solution of sodium
methoxide (prepared from sodium (0.074 g, 3.20 mmol) and 5 mL meth~nol). After 40
minutes, the reaction mixture was added LO water (15 mL) and extracted with
dichloromethane (3 x 30 mL). The organic portions were combined, dried and the solvent
evaporated to 1.00 g (95% yield) 1-(16,17-Oxidoheptadecyl)-3,7-dimethylx~.nthintq as a
white solid.
FY~rnPIe 47
This example illustrates a synthesis of N-(5,6-Oxidohexylamido)glutaric acid, methyl
ester (CT1301). Sodium hydride (425 mg, 17.7 mmol) was added to a solution of
glut~rimide (2.00 g, 7.7 mmol) in dimethyl sulfoxide (40 mL). After 20 minutes of stirring,
6-bromo-1-hexene (2.90 g, 17.7 mmol) was added. After 20 hours of stirring, the reaction
was poured into a separatory funnel cont -ining 100 mL water and extracted with
dichlormeth~.ne (4 X 50 mL). The organic portions were combin~l, washed with water (50
mL) and brine (50 mL) and dlied to yield 2.92 g (85% yield) 1-(5-Hexenyl)~ ;..;...ide
(CT1600) as a colorless oil.
A solution of 1-(5-hexenyl)glutarimide (1.50 g, 7.7 mmol), 4-methylmorpholine-N-oxide (1.08 g, 9.2 mmol) and osmium tetraoxide (3 drops of a 2.5% sol. by wt. in tert-
butanol) in acetone (10 mL) and water (10 mL) was stirred for 69 hours. Following ~ nitio~l
of 10 mL of a saturated solution of sodium dithionite and a further 15 minutes of stirring, the
reaction was extracted with 25% ethanol/dichlormethane (4 X 50 mL). The organic layers
were combined and dried (sodium sulfate) and evaporated to a thick oil which was purified
by chromatography using silica and an ethyl acetate eluant to yield 1.29 g (73% yield) N-
(5,6-Dihydroxyhexyl)glutarimide (CT1603) as a colorless oil.
W O 94/06431 2 1 ~ 2 PC~r/US93/0907
To 1-(5,6-dihydroxyhexyl)glutarimide (1.29 g, 5.6 mmol) was added 30% hydrogen
bromide-acetic acid (3.4 mL) and the resulting mixture stirred until all of the solid had
dissolved (45 minutes). The solution was poured carefully over a mixture of sodium
bicarbonate (6.5 g), ice water (40 mL), and dichlorometh~ne (40 mL); After carbon dioxide
S evolution had subsi(led> the organic layer was separated, and the aqueous layer extracted with
dichlorometh~nP (2 X 30 mL). The combimPd organic layers were dried over m~gnPcil-m
sulfate and the solvent was evaporated under vacuum to yield 1.73 g (92% yield) 1-(5-
Acetoxy-6-bromohexyl)glut~imide as a viscous oil which was dissolved in mPth~nol (5 mL).
A 0.7M sodium methoxide in methanol solution (6 mL) was added all at once. After stirring
for 15 minutps7 the solution was treated with water (20 mL) and then extracted with
dichloromethane (3 X 15 mL). The combined extracts were dried over m~nPsillm sulfate
and the solvents were evaporated under vacuum. The residue was chromatographed (80%
ethyl acetate/20% petroleum ether) to yield 256 mg (18% yield) N-(5,6-
Oxidohexylamido)glutaric acid, methyl ester as a colorless oil.
15 Example 48
This example illustrates the effects of CT154 1 as hair growth stimulant in nude mice.
In a procedure similar to that used in a commercial model for predicting human hair-growing
ability of minoxidil, nu/nu (nude) mice were painted twice daily for 16 days on the right
flank with CT1541 using sterile applicators. Researches h~n-llP~ the ~ice under a laminar
20 flow hood with applicator, wearing face mask and sterile gloves.
After 16 days, one mouse was sacrificed by cervical dislocation and skin biopsies
taken from the treated areas of the shoulder/flank and the non-treated area of the dorsal pelvis
(rump). Specimens were placed in 10% buffered formalin solution.
A microscopic analysis of the skin biopsies confirmed that follicles in the treated
25 areas had hair shafts which sometimes exit to the surface. There were mild accumulations of
mixed infl~mm~tory cells in the dermis. In contrast, hair follicles from untreated skin
biopsies were sm~ller/shorter and less often extend into the subcutis. Hair shafts were rarely
seen. A few mixed infl~mm~tory cells were in the dermis in the untreated areas as well.
The treated sections had more normal appearing hair follicles than the untreated30 sections. In addition, numerous hair shafts were seen exiting follicles in the treated sections.
Six weeks following treatment, a second mouse was euth~n~i7~d and biopsied the
same as previous mouse after lG days.
A microscopic Ç~min:ltion of the skin biopsies taken after six weeks confirmed hair
loss. The biopsies showed slightly more acanthosis (epidermal hyperplasta) in treated areas
3~ and slightly straighter super~lcial portions of follicles with less curling of hair shafts. In
treated sections, the subcutis contained active anagen bulbs located deeply in the subcutis.
The follicles were slightly dila~ed. Scattered mast cells, lymphocytes and a few neutrophils
were present. One section had a few increased cells in the subcutis.
48
~0 94/06431 2 1 ~ ~ 1 9 ~ Pcr/US93/09073
These data show that CT1541, when applied topically, can be used to treat or prevent
balrlness or allopecia.
F.lr~Tnple 49
This example illustrates the effects of CT1105, CT1114, CT1413, CT1439, CT1594,
S CT2518, CT2548R, CT2548S, CT2562 and CT3503 as effective inhibitors of IL-2 in~uced
proliferation of thymocytes. Single cell suspensions of thymus gland cells obtained from 4-6
week old mice were prepared. Two-hundred thousand cells were plated into individual wells
of flat-bottom 96-well plates in RPMI-1640-10% FCS me~ m The invention compoundswere then added to the wells at varying concentrations and the cells incub~ted for 1-2 hours
at 37C.
Following this pre-incubation a mixture of Concanavalin A (ConA) and Interleukin-2
(IL2) was added to the expelimental wells at a final concentration of (0.25ug/ml ConA)/(20
ng/ml IL2). Appropliate positive and negative controls were set up on each plate. Each
variable was set up in quadruplicate.
The plates were incubated for 4 days at 37C in a hllmitlified C2 incubator. On day
4 the wells were pulsed with ltlCi of tritiated thymidine(3H-TdR) and the plates incubated
for an additional 4 hours. The plates were then harvested and the incorporation of 3H-TdR
was determined in a liquid scintillation counter. From the CPM obtained from each
e~creriment~l well an IC-50 for each inventive compound tested was deterrnin~.d Therefore,
a lower reported IC-50 value is indicative of greater inhibition of IL-2-induced prolifer~tion
of thymocytes. Table II illustrates IC-50 values (in ,uM) for inventive compounds inhibiting
proliferation at very low concentrations of compound.
T~ble 1l
Inventive Compound IC-50 (~lM)
CT2562 3.1
CT2518 3.4
CT2548S 4.6
CT2548R 7.5
CT1413 7 7
CT1439 8.0
CT1105 8.5
CT1594 8.5
CT1114 10.0
CT3503 10.0
These data show that ~he illustrated inventive compounds are effective therapeutic
25 agents for the treatment or prevention of autoimune disorders or infl~mm~tory lice~ces
49
.
-
W O 94/06431 2 1 4 ~/ ~ 9 2 PC~r/US93/0907 ~
F.~nn~ple 5()
This example illustrates the effects of CT1114, CT1413, CT1560, CT1565. CT1594,CT2518, CT2548R, CT2548S and CT3503 as effective inhibitors of normal human bone
marrow stromal cells (MSC) proliferation in response to Platelet Derived Growth Factor BB
5 (PDGF B) and IL-loc (50 and 10 ng/ml, respectively). Maximum proliferation occurs when
both PDGF B and IL-lo~ are present, hence a combination of both were used in the following
assay.
MSC were obtained and maintained in exponenti~l growth, released from a growth
plate with trypsin and plated into 96 well tissue culture plates in the presence of Fibroblast
10 Growth Factor (FGF,O for 48 hours. Growth media were removed and the cells were washed
once with serum free media. The cells were then incubated 20-24 hours in serum free media
After incubation, inventive compounds were added to the cells at the a~.prop~iate
con- ensration and then the cells again incubated for 1 hour. PDGF B and IL- la were added
to the cells along with 3H-thymidine and the cells again incubated for an ~d~litiQn~l 24 hours.
After 24 hours of incubation, the cells were then harvested to assess incorporation of
3H-thymidine into DNA (proliferation). From each harvested cell culture, an IC-50 for each
inventive compound tested was determined. Therefore, a lower reported IC-50 value is
indicative of greater activity of the invention compounds in inhibiting MSC proliferation.
Table m illustrates IC-S0 values (in ~LM) for invention compounds inhibiting MSC20proliferation at very low concentrations of compound.
Table m
Inventive Compound IC-50 (~LM)
CT1413 3 7
CT1114 5.1
CT2548S 5.9
CT1560 6.6
CT2548R 8.7
CT1594 14.0
CT3503 14.0
CT2518 16.7
CT1565 17.0
CT1539 18.0
These data illustrate the inventive compounds illustrated are usefule to treat or
prevent rectenosic and atheroscherosis
FY~-nple 51
This example illustrates the effects of CT1605, CT1808 and CT1906 as immune
mod~ tc)r.c in a mixed lymphocyte reaction Figure 1 shows the effects of three inventive
compounds CT16()5 (N-(5,6-oxidohexyl) ~lutarimide), CT1808 (N3-(5,6Oxidohexyl)-N1-
~3 g4/06431 2 l 4 ~ 1 9 2 Pcr/us93/09o73
methyluracil), and CT1906 (N3-(5,6-oxidohexyl) Nl-methylthymine. The mixed
lymphocyte reaction shows a proliferative response of PBMC (peripheral blood mononuclear
cells) to allogeneic stimulation determined in a two-way mixed lymphocyte reaction. Each
of the inventive compounds tested demonstrated activity in this immlln~ mo~ ting activity
assay procedure.
F.~mple 52
This example illustrates a comparison of three dose levels of CT1808 and CT1906
and no drug control to inhibit thymocyte proliferation. The thymocytes were obtained from
normal female Balb/C mice and stimulated with Concanavalin A (Con A) and/or IL-la
(interleukin-l alpha). The thymuses were dissociated and plated into 96-well plates at a
density of 2 x 105 cells/well. Dilutions of Con A and/or IL-la were added to the wells and
the cells were incubated for 4 days at 37C. Drugs were added to the cell cultures two hours
before activation with Con A and/or IL-loc. On day 4, the cells were pulsed with tritiated
thymidine and allowed to incubate an additional 4 hrs. The cells were harvested and
counted. As shown in Figure 2, both drugs inhibited thymocyte proliferation is a dose-
dependent fashion.
F.~rnple 53
This example illustrates a comparison of CT1605 and CT180g for inhibition of B-cell
proliferation. A Ramos C-cell tumor line was treated with 250 ~LM CT1808 or CT1605 for
one hour prior to stimulation of proliferation with anti-mu antibody or PMA (5 nM). One
day later, proliferation was measured with tritiated thymidine. As shown in Figure 3, both
CT1605 and CT1808 inhibited proliferation in this model.
F.xample 54
This example illustrates a comparison of CT1605, CT1808 and CT1906 on PDGF-
inclnced (platelet derived ~rowth factor) proliferation of human stromal cells. Human
stromal cells were starved in serum-free media for 24 hours and then stimulated with 50
ng/ml of PDGF-BB. The drugs were added at various indicated concentrations one hour
prior to PDGF stimulation. Tritiated thymidine was added for 24 hrs at the time of PDGF
stimulation to measure cellular proliferation. Background counts were approximately 5% of
control levels. As shown in Figure 4, all three drugs inhibited PDGF-induced stimulation in
a dose response fashion.
Example 55
This example illustrates a comparis~n of the effects of CT1605, CT1808 and CT1906
to inhibit adhesion of U937 cells to activated human llmbilir -l vein endothelial cells
(HWEC). HUVEC cells (40()()/well, seeded 72 hrs in advance) were activated with 20
ng/ml of TNF for 12 hrs. Drug was added to each culture (except for controls) one hour
prior to adding TNF. U937 cells, preloaded with the ~luorescent dye BCECF, were added to
each culture well and ~hen washed twice wi~h PBS. Cell adhesion was determinecl by
5 1
WO 94/06431 ~ 5 1 ~ ~ PCr/US93/0907
measuring Iluorescence on a fluorescence plate reader. As illustrated in Figure 5, all three
drugs showed a decrease in cell adllesion caused in a dose dependent fashion
F.~ nple 5~
This example illustrates the effects of CT1605, CT1808 and CT1906 to inhibit cell
s surface expression of VCAM in human umbilical vein endothelial cells (HUVEC). The
HUVEC cells were stimulated with 20 ng/ml TNF-a for 20 hrs and then stained for
immlmoflllorescenre using a monoclonal antibody recognizing VCAM, followed by a goat
anti-mouse antibody conjugated to phycoerythrin. The cells were anaiyzed for antibody
binding using flow cytometry. Figure 6 shows an analysis of mean relative fluorescence
intensity of 10,000 cells, analyzed by flow cytometry. The mean fluorescence levels were
decreased by all three drugs from control levels (TNF tre~tme-nt, no drug).