Note: Descriptions are shown in the official language in which they were submitted.
~ W094/07~07 2 1 4 S ~ 8 I PCT/US93/08168
METHOD FOR ANTAGONIZING INOSITOL 1,4,5-TRISPHOSPHATE
BACKGROUND OF THE INVENTION
This application relates to a series of diamino
benzenedisulfonic acid oligomers that have demonstrated an
affinity for the receptor sites of inositol 1,4,5-
triphosphate (IP3) and are, therefore useful in diminishing
the bioactivity of IP3, especially with regard to its
effect on the release of intracellular calcium ions.
DESCRIPTION OF THE PRIOR ART
The diamino benzenedisulfonic acid oligomers
demonstrating utility as IP3 antagonists according to this
invention are described in detail in the European Patent
Application published January 22, 1992 under Publication
No. 0467185 A2. In that publication, the oligomers of the
present invention were described as having utility in the
diagnosis and/or treatment of AIDS and AIDS related
complex.
SUMMARY OF THE INVENTION
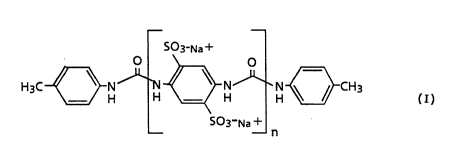
The invention herein disclosed relates to a method of
inhibiting the activity of inositol 1,4,5-triphosphate
(IP3) by occupying the receptor sites specific to IP3 with a
compound of the formula:
W094/07507 PCT/US93/08168
2~ 8 l -2-
S03~Na +
H3C~N~I ~NJ~N ~CH3
S03~Na +
-- --n
wherein n is a whole number selected from the range of 5 to
20 inclusive and the pharmaceutically acceptable salts
thereof.
.,
DETAILED DESCRIPTION OF THE INVENTION
Inositol 1,4,5-triphosphate (IP3) is a naturally
occurring and active component of animal physiology. It is
formed intracellularly upon the activation of cell-surface
receptors linked to the enzyme phospholipase C. Once
generated in sufficient quantities, IP3 acts to stimulate
the release of calcium ions from storage organelles within
the cell. In this role IP3 is characterized as a "second
messenger". Depending upon the type of cell, the calcium
released by IP3 ~unctions to stimulate a variety of
physiologic processes such as smooth muscle contraction,
histamine secretion and the hyperpolarization of nerve
cells. Any compound or agent that can promote or interfere
with the function of IP3, will promote or interfere with the
generation of calcium ions and thereby elicit predictable
pharmacological effects.
The process by which IP3 releases calcium ions begins
with the binding of IP3 to a specific receptor protein
located on an intracellular calcium storage compartment
located typically on the endoplasmic reticulum. This
receptor protein has been cloned and has been shown to form
a calcium "channel" with unique structural properties when
bound to IP3. Therefore, when IP3 binds with its receptor,
a calcium channel is opened causing the release of calcium
stored in the cell's endoplasmic reticulum. In turn, the
W094/07507 2 1 4 5 6 8 1 PCT/US93/08168
released calcium will elicit the appropriate cellular
response.
Heretofore, the only verified potent antagonist of the
5 IP3 receptor was heparin, a complex glycosaminoglycan. The
diamino benzenedisulfonic acid oligomers of this invention
also appear to antagonize the effects of IP3 by competing
for the receptor site. In most cases, these compounds are
more effective than heparin and demonstrate fewer secondary
effects. In addition to providing utility as laboratory
"tools" in evaluating the therapeutic potential of other IP3
receptor antagonists, the oligomers of this invention would
also be administered to modulate IP3-induced calcium release
and have a salutary effect on any number of disorders that
are caused or exacerbated by an inordinately productive IP3
second messenger pathway.
EXPERIMENTALS
Measurement of IP3 Bindinq
Cerebella from male Sprague-Dawley rats (200 g) were
homogenized in 30 volumes of ice-cold buffer A (50 mM Tris,
1 mM dithiothreitol, 1 mM EDTA, pH 7.7 with HCl) with a
polytron (setting 9 for 10 seconds). The tissue is then
washed twice by centrifugation (20,000 x g, 15 minutes;
Sorvall 28-S., SS-34 rotor) and resuspended in 30 volumes
of ice-cold buffer A.
For the binding assays, 1.5 ml eppendorf tubes
containing 50 ~1 of test compound (made up as a lOx stock
in water) or water, 50 ~1 [3H] IP3 (17Ci/mmol; Dupont-NEN;
usually made as a 25 nM (lOx) stock solution in buffer),
and 350 ~1 of buffer B (buffer A with pH adjusted to 8.4)
. on ice. Tubes for non-specific binding also contained 50
~1 of non-radioactive IP3 (100 ~M stock (lOx); final
concentration 10 ~M), with an appropriate reduction in the
volume of buffer B. Reactions were initiated by the
addition of 50 ~1 tissue to make the final volume 500 ~1,
W094/07507 PCT/US93/08168
214~681 ~
--4--
followed by vortex mixing. Samples were incubated on ice
for 10 minutes and then were centrifuged (14,000 x g) in a
microfuge (Eppendorf model 5415) for 5 minutes followed by
aspiration of the supernatant fraction. The tissue pellets
were solubilized overnight in 100 ~1 of Protosol (Dupont-
NEN). After solubilization, 73 ~1 of glacial acetic acid
were added to decrease chemiluminescence, and the mixture
was transferred to scintillation vials. To these vials was
added 7 ml of Ecoscint-A (National Diagnostics) and the
radioactivity determined by li~uid scintillation
spectrophotometry.
Specific binding was defined as the difference between
total binding (radioactivity in the absence of test
compound and cold IP3) and non-specific binding
(radioactivity in the absence of test compound but in the
presence of cold IP3). This number was taken as 100%
specific binding. Data points obtained with the test
compounds were fit by a computer program (GraphPad-InPlot)
to determine their inhibitory potency. The inhibitory
potencies of the test compounds were expressed as the
concentration of compound that produces 50~ inhibition of
specific binding (the ICso value).
The binding data are presented in Table 1 and
demonstrate that compounds within the scope of the present
invention effectively compete for [3H] IP3 binding sites in
rat cerebellar membranes. The compound identified as MDL
102,869 was the most potent competitor for binding with an
IC50 of 50 nM, whereas low molecular weight heparin (5100
MW) had an IC50 of 74 nM. MDL 102,869 is the compound
according to the claimed invention wherein n = 15.
The potency for binding also seems to correlate with
the ability to antagonize IP3-induced calcium ion release.
Thus, 1 and 3 ~M of MDL 102,869 inhibited calcium ion
release by 42 and 100~, respectively (see Fig. 1), whereas
WO 94/07507 2 1 4 5 ~ 8 1 PCI/US93/08168
10 llM of heparin inhibited release by 9o%. MDL 101,828,
which had an IC50 binding of 104 nM, inhibited IP3-induced
calcium ion release by 72% at 3 micro moles. MDL 101,828
is the compound according to the claimed invention wherein
n = 9.
TABLE 1
BindingInhibition of Ins(1,4,5) P3-
CompoundICso (nM)Induced Ca2+-release
Heparin (low MW)74 9o% at 10 ~M
MDL 101,828 104 42% at 3 llM
MDL 102,869 50 42% at 1 llM
100% at 3 ~M
The tracing of Fig. 1, dramatically illustrates the IP3
inhibition data set forth in the third column of Table 1.
S03~Na +
O ~ O
~H H 4~H N ~CH3
S03~Na +
--n
The y-axis represents the concentration of free calcium
ions in arbitrary units. The tracing shows that two
successive additions of 0.1 llM of IP3 stimulated similar
amounts of calcium ion release from cerebellar microsomes.
The addition of 1 ~lM of MDL 102,869 stimulated a small
increase of calcium ion for unknown reasons. In the
presence of 102,869, however, calcium ion release
stimulated by 0.1 ~lM of IP3 was inhibited by 42%. This
inhibition was overcome by the addition of 1 llM of IP3,
consistent with competitive antagonism by MDL 102,869.