Note: Descriptions are shown in the official language in which they were submitted.
~ W094/08563 2 1 4 ~ 6 3~ PCT/US93/09623
NOVEL CLASS OF PHOSPHOCHOLINE DERIVATIVES
~VING ANTIFUNGAL Ac~ v~
This is a continuation-in-part application of
U.S. patent application Serial No. 07/958,416, filed
October 8, 1993, the entire disclosure of which is
incorporated by reference.
1. Field of the Invention
This invention relates to new classes of
phosphocholine derivatives as well as to various
methods for preparing these compounds -- including
synthetic, enzymatic and extractive using certain
plants. The phosphocholine derivatives of the
invention are non-toxic and exhibit substantial
antifungal activity in slowing fungal growth and in
killing fungi.
2. Backqround of the Invention
The plant species Irlbachia alata has been used
as an anti-infective agent in the Peruvian Amazon
region. The leaves are squeezed and the liquid is
applied to infected skin sores. The same liquid from
the leaves is applied to skin problems and skin fungal
infections. It is utilized to treat vaginal yeast
infections.
Irl bachia alata is one species of 10-12 species
of the plant family Gentianaceae. These species occur
in tropical South America especially in the Amazon and
Negro River basins. The plants in the genus Irlbachia
are generally low herbs characteristically with 3-5
plinerved leaves. The most consistent diagnostic
feature for the genus is the pollen morphology.
A reference to Irlbachia alata and related
species was made in 1775 by the French scientist Fusee
TE SHEE~
2146639
W094/08563 ` " ' PCT/US93/0962
Aublet (Aublet, F. 1775, Histoire des Plantes de la
Guiane Francoise, Didot, Paris). The ethnobotanical
notes from this reference were subsequently compiled
and republished in English. Aublet noted the
following about two species in the genus Irlbac~ia:
Irlbachia alata The entire plant is bitter.
It is used to clear obstructions; I (Aublet) have used
it with good results. The species is called "Bois
creux" (Hollow wood) by the Creoles.
Irlbachia pururascens All parts of this plant
are bitter. It is used as an apertif and to reduce
fever.
3. SummarY of the Invention
We have discovered a class of phosphocholine
derivatives (Class I) having extraordinary antifungal
activity.
Structurally, these compounds are phosphocholine
derivatives (1 or 2-deacyl-phosphatidyl cholines) in
which the 1 or 2-OH-group of the glycerol moiety has
been glycosylated with glucose, galactose, arabinose,
mannose, rhamnose or another sugar. The basic
chemical structure may be drawn as follows:
S~ ~E 8HEET
214663~
W094/0~3 _ 3 _ PCT!US93/09623
R'
R ~ ~
\ ~ C ~ / O \ / / C~ / CH3
CH3
wherein one of R or R' is a sugar moiety and the other
is an acyl or sugar moiety.
The molecular backbone common to all members of
this class of compounds is drawn above. The acyl-
group can be any long-chain fatty acid, while the
sugar unit can be any of the sugars commonly found in
plants, including but not limited to glucose,
galactose, arabinose, mannose, rhamnose, or another
naturally occurring sugar.
We have additionally found a structurally related
class of phosphocholine derivatives of similar or
greater antifungal activity than the above-discussed
class of phosphocholine derivatives (i.e., Class I).
one novel class of phosphocholine derivatives
(Class II) having antifungal activity has the basic
structure shown below:
_ _
A F ~ ~ A
B--\ ' / \D/¦\X--Q--y/¦\D/ \N/--B
O- O' T
where Q is C2 to C30 alkyl, alkenyl, alkynyl, branched
alkyl, branched alkenyl, or branched alkynyl;
Z is oxygen or sulfur; X and Y are independent
oxygen, sulfur, CH2, CF2, or N-RI;
8U~i l l l Ll rE SHE~
wog4/~321~6 63 ~ . PCT/US93/096 ~
A, B, and T are independently alkyl, alkenyl,
alkynyl, branched alkyl, branched alkenyl, or branched
alkynyl radicals of C1 to C20 chain lengths; are
independently or together cycloalkyl or bridged
cycloalkyl radicals of ring size C3 to C20, or
cylcoalkenyl, bridged cycloalkenyl or
cyclo(polyene)radicals of ring size C4 to C20,
cycloalkynyl, bridged cycloalkeynl or
cyclo(polyalkynyl)radicals of ring size C8 to C20;
D is oxygen, sulfur, CH2, CF2, or N-R2;
F is alkyl, alkenyl, alkynyl, branched alkyl,
~ranched alkenyl, branched alkynyl, cycloalkyl,
bridged cycloalkyl, cycloalkenyl or cycloalkynyl
radicals contA;n;ng C1 to C20 carbon atoms;
'5 Rl and R2 are independently hydrogen, alkyl,
alkenyl, alkynyl, branched alkyl, branched alkenyl,
branched alkynyl, cycloalkyl, bridged cycloalkyl,
cycloalkenyl, bridged cycloalkenyl or cycloalkynyl
radicals con~; n; ng C1 to C20 carbon a~oms, or any
protecting group described in the book "Protecting
Groups in Organic Synthesis" by Theodora Greene and
Peter G.M. Wuts.
Another class of phosphocholine derivatives
(Class III) having antifungal activity has the
following structures:
EB BB DD
M ¦ ,or ~^~'M,orM BB
where AA, BB, DD are independent of each other,
~ e~ual to each other, or interchanged as shown above,
the central carbon atom can be either the R and S
S~ 11 LJ1E SHEET
~ WOg4J0~3 21 4 66 ~ 9~ . PCT/US93,09623
-- 5
optical stereoisomer or a mixture of R and S
stereoisomers, and where AA, BB, and CC are defined as
follows:
where AA, is A-J with A being attached to the
carbon atom of the three carbon central unit and J is
defined below;
BB is B-Y, with B being attached to the carbon
atom of the three carbon central unit and Y is defined
below:
DD is
~,,~D~W~E ~G~Q
Il
Z
where W, E, G and Q are defined below;
A is oxygen, sulfur, CH2, CF2 or N-R~;
B is oxygen, sulfur, CH2, CF2 or N-R2;
D is oxygen, sulfur, CH2, CF2 or N-R3;
Y is alkyl, alkenyl, alkynyl, poly(alkenyl),
poly(alkynyl), or poly(alkenoalkynyl) radicals
comprised of C1 to C20 carbon atoms chain lengths, or
alkanoyl, alkenoyl, alkynoyl, poly(alken)oyl,
poly(alkyn)oyl or poly(alkenoalkyn)oyl radicals
comprised of C2 to C20 chain lengths or alkyloxy,
alkenyloxy, alkynyloxy, poly(alkenyl)oxy,
poly(alkynyl)oxy, poly(alkenoalkynyl)oxy radicals
comprised of Cl to C20 carbon atoms;
~JBSmUrE SHE~
2 1 4 6 fi 3 9 ~ t S PCI~/US93/Og62~
J is a furanose or pyranose radical of the type:
L~ ;/Y, , -- !
where X is oxygen, sulfur, CH2, CF2 or N-R4;
F, K, L and M are independently hydrogen,
hydroxyl, protected hydroxyl (as described in the book
"Protecting Groups in Organic Synthesis" by Theodora
Greene and Peter G.M. Wuts), alkyloxy, thiol,
1 alkylthio, arylthio, alkylsulfonyl, arylsulfonyl,
amino, ammonium, alkylamino, alkylammonium,
dialkylamino, dialkylammonium, trialkylamino,
trialkylammonium where the alkyl chain on nitrogen is
comprised of Cl to C20 carbon atoms; or alkyl,
alkenyl, or alkynyl radicals comprised of Cl to C20
carbon atoms.
Z is oxygen or sulfur
E is oxygen, sulfur, CH2 CF2 or N~Rs;
G is alkyl, branched alkyl, cycloalkyl or bridged
cycloalkyl radicals of Cl to C20 chain lengths;
Q is halogen, hydroxyl, protected hydroxyl
utilizing any protecting groups described in the book
"Protecting Groups in Organic Synthesis" by Theodora
Greene and Peter G.M. Wuts, O-arylsulfonyl-, O-
alkylsulfonyl- or O-(perfluoroalkyl)sulfonyloxy,
amino, ammonium, alkylamino, alkylammonium,
dialkylamino, dialkylammoniumj trialkylamino,
trialkylammonium where the alkyl chains on nitrogen
are Cl to C20, or Q=NR~R2R3, where R~, R2, or R3 can
independently or together be a mixture of alkyl groups
Sl.lBSllTU~E SHEET
~ W094/~63 2 1 4 6 6 3 9 PCT/USg3/09623
of C1 to C20 in chain length and a protecting group
described in the book "Protecting Groups in Organic
Synthesis" by Theodora Green and Peter G.M. Wuts, and
Rl can equal R2, R2 can equal R3, or Rl can equal R3
which can equal R3;
Rl, R2, R3, R4 and Rs are independently alkyl,
alkenyl, alkynyl, branched alkyl, branched alkenyl,
branched alkynyl, cycloalkyl, bridged cycloalkyl,
cycloalkenyl or cycloalkynyl radicals of C1 to C20
chain lengths, or any protecting group described in
the book "Protecting Groups in Organic Synthesis" by
Theodora Greene and Peter G.M. Wuts;
where Wl and W2 are P(-OR) (with R being phenyl,
phenylmethyl, or negatively-charged oxygen), S=O,
carbon, or sulfur, provided that if Wl is not P(-OR) W2
is P(-OR) and provided that if J is a furanose or
pyranose radical then Wl is P(-OR).
A preferred subgroup of the above-described
Class III of phosphocholine derivatives have the
20 following structures:
BB OR
\/\ Q
o
BB
R10 1 ORl
Q ~ O~l,O ~ o~l,o
Il 11
O O
SL~ ITE SHEET
-
~1~66~g
W094/0~3 ,- t ' PCT/US93/096 ~
R20 /
AzO ~ 0~0 O
o
R20 \ \ \ \ \ \ \
o- OR1
R2~o~o ¦ o
R~ O
R~
R~ ~ \ O ~ O~¦,O Q
R~ o
where R~ is phenyl or phenylmethyl, hydrogen, or nil;
R2 is hydrogen, phenylmethyl, or any protecting
group described in the book "Protecting Group in
Organic Synthesis" by Theodora Green and Peter G.M.
Wuts which can be cleaved by hydrogenolysis;
AA, BB, and Q are as defined above
where the central carbon atom of the three carbon
unit is either the R optical isomer, the S optical
- isomer, or any mixture of the two optical isomers
thereof;
SUBSTlTUTE SHEET
W094/0~63 ' 21~4 ~ ~ ~ n
~ U~J~ PCT/US93/09623
g
Another preferred subgroup of the above-described
Class III of phosphocholine derivatives have the
following structures:
R2O
R20 _~
~L~R2
R20~ \
~0
~ ORl
R30~ o ¦ ~/\
o
R20 ~ ~/
o o
R O - l R 1R 30 `~"~ OR,
Il 11
O O
SUBSmUTE SHEET
W094/08~3 2 1 ~6 6 3 g ~ ~ PCT/US93/0962 ~
-- 10 --
R20 o~!~o~Q
R2O ~ o ~ ORs .
R20
R2O R9O
R - -T ~ ~ OR1
where Rl is phenyl or phenylmethyl, hydrogen, or
nil; R2 is hydrogen, phenyl methyl or any protecting
group described in the book "Protecting Groups in
organic Synthesis" by Theodora Greene and Peter G.M.
Wuts which can be cleaved by hydrogenolysis;
R3 is hydrogen or a protecting group as described
in the book "Protecting Groups in Organic Synthesis"
by Theodora Greene and Peter G.M. Wuts.;
where the central carbon atom of the three carbon
unit is either the R optical isomer, the S optical
isomer, or any mixture of the two optical isomers
thereof; and Q is defined above.
Still another preferred subgroup of the above-
described Class III of phosphocholine derivatives have
the following structures:
SUts;~ ITE SHEET
~ W094/08563 ~l ~ 6 6 3 ~ PCT/US93/09623
-- 11 --
HO ~ OR1 J ~ Q
O ~d H OR2
where R~ is phenyl or phenylmethyl, hydrogen, or
nil;
R2 is a protecting group as described in the book
"Protecting Groups in Organic Synthesis" by Theodora
Greene and Peter G.M. Wuts, or hydrogen if R1 is not
hydrogen;
and Q is defined above.
We have further found a novel, generally
applicable method for the synthesis of the above
described broad classes of phosphocholine derivatives
(Classes I, II and III).
4. Brief Descri~tion of the Drawinqs
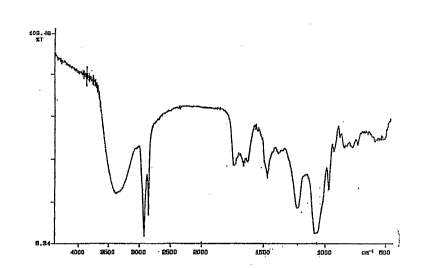
Fig. l is the FTIR spectrum of the composition
comprising a phosphocholine derivative obtained from
Irlbachia alata.
Fig. 2 is the proton NMR spectrum of the
composition comprising a phosphocholine derivative
obtained from Irlbac~ia alata in D20 at 400 mHz.
Fig. 3 is the FAB-/MB mass spectrum of the
composition comprising a phosphocholine derivative
obtained from Irlbachia alata.
5. ~etailed DescriPtion of the Invention
The glysosylated lysolecithins of the invention
can be prepared by synthetic methods or by enzymatic
methods. The phosphocholine derivatives can be
SUBSTITUTE SHEET
~14~3g
W094/0~63 ~ PCT/US93/0962
- 12 -
prepared either by synthetic methods or by methods
entailing extraction from plant materials.
5.1. Chemical SYnthesis of phos~hocholine derivatives
A wide variety of compounds having accessible
alcoholic functionalities can be glycosylated
following the classic Koenigs-Xnorr methodology.
Bochkov, A.F. and Zaikov. G.E., Chemistry of the O-
Glycosidic Bond. Pergamon Press, 1979. As part of the
synthetic route to phosphocholine derivatives with
sugar, all but the anomeric hydroxyl group of the
sugar to be introduced are protected either as esters
or ethers, while the anomeric hydroxyl is being
replaced by a halogen. The aglycon-sugar linkage is
then formed via alcoholysis. Finally, the protective
groups are selectively removed.
In the present invention, benzyl ethers or the
benzilidine moiety are are the preferred protecting
group, since they can be selectively removed by
catalytic hydrogenation, while leaving the sensitive
acyl-glycerol linkage intact. The glycosidation
requires silver, mercury (Helferich modification), or
cadmium salts as catalytic halogen abstractor, in the
presence of a dehydrating agent (Timell, T.E., Can.J.
Chem. 1964, 42, 1456; Dejter-Juszynsky, M. and
Flowers, H.M., Carbohydr. Res. 1973, 30, 287;
Marousek, V., Lucas, T.J., Wheat, P.E., and Schuerch,
C., Carbohydr. Res. 1978, 60, 85), and with or without
auxiliaries such as crown-ethers. (Kn~chel,A. Ger,
R., and Thiem, J. Tetrahedron Letters 197~, 551) More
recent methodology makes use of the halogen-
abstracting power of non-nucleophilic bases such as
diisopropylethylamine and/or of molecular sieves in an
~~ anhydrous media. (Garegg, P.J. and Norberg, T.,
Carbohydr. Res. 1976, 52, 235) The following
SUBSTITUTE SHEET
~ W094/08~3 2 ~ 4 6 6t3 ~ PCT/US93/09623
- 13 -
synthetic scheme is based on the latter reaction
sequence:
OH OBn
5ROJ~ ~p~ ~¢~+
R = Palmitoyl-, Oleyl-, Palmitoleyl-, etc. 1) Et4NE~rtiPr2EtNlMol.Sieves; DCMITHF.
2) H IPd;EtOAclTHF.
0 2
OH OH
~,/
H~
RO~0~ ~ O N~
2 0
2-0-,B-D-Glucopyranosyl Iysolecithin
The synthetic two-step scheme outlined above can be
conducted with commercially available materials.
2,3,4,6-Tetrabenzyl- 2,3-dibenzyl-4,6-benzylidene-
glucose can be converted into the l-bromo- or l-O-
triflate compound by stAn~Ard methodology. Leroux, J.
and Perlin, A.S. Carbohydr. Res. 1976, 47, C8. The
corresponding phosphocholine derivatives are available
through AVANTI POLAR LIPIDS, Inc. All other reagents
are available from ALDRICH. The methodology outlined
above is also applicable to either 1-acyl or 2-acyl
~ (1-acyl detailed above).
SUBSTITUTE StlEET
W094/0~3 2 1 4 6 ~ 3 9 ~ ~ PCT/US93/096 ~
5.2. Enzymatic pre~aration of 1 or 2 qlYcosylated
lysolecithins
As an alternative to the synthetic sequence
outlined above, an in vitro enzymatic glycosidation
simulating the biosynthetic process will produce the
desired compounds in comparable yields. The natural
glycosidation catalysts are glycosyltransferases.
These enzymes operate with uridinediphospho-glycosides
(UDP-sugars) as substrates and ATP as the energy
source. While the enzymes have to be prepared from
fresh plant material, UDP-sugars, ATP, as well as the
respective phosphocholine derivatives are commercially
available. This synthesis has the advantage of being
essentially a one-step process with the high
selectivity and yields expected from an enzymatic
reaction. The following scheme describes the
preparation of a glucoside. Other transferases, not
specific to glucose, could be applied in the
preparation of glycosylated lysolecithins with other
sugars as well:
SUBSmUTE SHE~T
wog4 2~66~9.
3 PCT/US93/09623
- 15 -
H~ O
HO~ 1
HO
S OH p g O ~
R = Palmitoyl-, Oleyl-, Palmitoleyl-, etc.) Uridine-5 -diphospho-glucose
(=UDP-glucose)
1)1-Glucosyl TransferasetATP;Phosphate
Buffer
2) Sephadex LH-20
OH OH
OH /~ /
J~y
~< O
HoO~
~2 O~
RO~ 0 ~ ¦~ 0 ~ t~
2-0-~-D-Glucopyranosyl Iysolecithin
5.3. Total SYnthesis of Phosphocholine Derivatives
A general synthetic method of synthesizing
phosphocholine derivatives of the various structures
described in section 3 is outlined as follows.
An alcohol is phosphorylated or glycosylated.
The product is subsequently deprotected. The
deprotected product is then alkylated or esterified to
produce the phosphocholine derivatives. The general
SIJ~ JTE SHEET
W094/0~63 ~1 4 6 6 3 ~
PCT/US93/0962
- 16 -
scheme for this outlined synthetic method is shown
below.
SUBSTITUTE SHEET
WO 94/08~;63 2 ~ 4 6 6 3 9 PCI/US93/09623
-- 17 --
General Scheme for the Synthesis of Known and Novel Lysolecithins
R2Y~, HO~o R1X,~O
O
HO HO
R2V~OH R1 X~OH
- ~ ~
ZR3 XR1 R2Y ZR3
R2Y ~ ~XR1 R2Y~ ~ ~ZR3 R1X~ ~ ,ZR3R1x~ ~ ~yR2
20
Glycerol Derivative 0 can be either the R or S optical isomer,
racemic, or a mixture of R and S isomers
~ Implies that a number of synthetic transformations
are required
R ~ = Sugar, carbocyclic sugar, functionalized sugar derivative, etc.
R2 = Phosphate or phosphate isostere moiety
R3 = alkyl, alkanoyl, alkenyl, alkenoyl, etc.
X, Y, and Z can be C, O, N, S independently or equal to each other
SUBSTITUTE SHEET
2146~39
W O 94/08563 " ' PC~r/US93/0962
- 18 -
5.4. Methods of Use
The phosphocholine derivative in Classes I, II
and III are all useful in treating fungal infection by
the administration to a warm~blooded animal of a
therapeutically effective a~ount of a phosphocholine
derivative. The pharmaceutical composition comprising
the phosphocholine derivative used for such
administration may also contain pharmaceutically
acceptable excipients and carriers.
Phosphocholine derivatives in Classes I and
II are believed to be novel compositions.
In order to treat a fungal infection, the
antifungal agent of Classes I, II and III may be
administered to a warm-blooded animal intravenously,
intraperitoneally, subcutaneously, intramuscularly,
orally, topically, by aerosol, or combinations
thereof.
The antifungal agent of phosphocholine
derivatives in Class II can be a~ri n; stered
intravenously in a range of about 0.1 to about 10
mg/kg
The fungal agent of Class II can be administered
intraperitoneally in a range of about 0.1 to about 10
mg/kg.
The fungal agent of Class II can be administered
subcutaneously in a range of about 1 to about 20.
The fungal agent of Class II can be administered
intramuscularly in a range of about 1 to about 20.
The fungal agent of Class II can be administered
orally in a range of about 5.0 to about 30 mg/kg.
The fungal agent of Class II can be administered
topically in a range of about.5.0 to about 15% by
weight.
SUt~ 111 lJTE SHEET
~ wo g4/0~3 2 ~ 4 ~ B 3 9 PCT/US93/09623
-- 19 --
The fungal agent of Class II can be administered
by aerosol in a range of about 5.0 to about 30
mg/kg/day.
The above dosage ranges may need to be doubled
for those phosphocholine derivatives in Class I and
III with lower antifungal activity which are identical
or similar to those in table 2 (see below).
6. ~traction of phosphocholine derivatives
from plants
Plants are not known to contain phosphocholine
derivatives.
The general manner of chemical extraction from
the plants can be summarized as follows.
The plant source material, such as the whole
plant, the roots, leaves, stem and/or latex of the
plant, is extracted with water and/or a water miscible
solvent. The preferred solvents are alcohol of 1-3
carbon atoms or acetone. The aqueous extract is
extracted with butanol. The butanol-soluble fraction
is subjected to gel filtration (e.g., over Sephadex),
reversed-phase column chromatography (e.g., C-8), or
gel-permeation chromatography (e.g., divinyl benzene
cross-linked gels) such as PL-GEL or membranes (e.g.,
an Amicon membrane) using water or water and a water
miscible solvent, with or without a buffer, as the
mobile phase. The water miscible solvent is
preferably a 1-3 carbon alcohol, acetone or
acetonitrile.
The useful phosphocholine derivatives containing
compound is the fraction detected by NMR spectroscopy.
A specific member of the class of phosphocholine
- derivatives of the present invention is
SUBSTITUTE SHEET
W094/08563 2 1 4 68 3 ~ PCT/US93/0962~
- 20 -
2-palmitoyl-1-O-glycopyranos~llysolecithin shown
below:
CH~H
Ho
~O 0~o
HO ~ O ~ ~~O
OH ~ I
Z-PaImitoyl-1-0-glucopyranosyllysolecithin
We have found that 2-palmitoyl-1-0-
glucopyranosyllysolecithin is a relatively active
antifungal agent similar in activity to
L-a-Lysophosphatidyl inositol, discussed in Table 2
below.
We have found that one of the most active
antifungal compounds has the following structure.
CH3~ CH2 0 1 0 0 1 0 CH2 CH3
CH3--N~ CH2 11 (CH2)22 11 CH2 N~CH3
1,22-docosan diol bisphosphocholine ester.
SUBSmUTE SHEET
~ W O 94/08563 2 1 4 6~ 3 9~ PC~r/US93/09623
- 21 -
6.1. Extraction
We have isolated by chemical extraction 1,22-
docosan diol bisphosphocholine ester, the active
antifungal compound contained in the plant Irlbachia
S alata. The leaves of Irlbachia alata were milled and
200g of the milled leaves was extracted with lL of
dichloromethane/isopropanol (1:1 v/v) at room
temperature for 24 hours. The extracted material was
separated from the marc (i.e., residual of the plant
after solvent extraction) and ~icc~rded~ The marc was
then extracted with 1.5L of isopropanol/water (1:1
v/v) at room temperature for 24 hours. The marc was
separated from the extract and discarded. The
isopropanol/water (1:1 v/v) soluble extract was
partitioned between water and ethyl acetate. The
ethyl acetate phase was separated and discarded. The
water soluble phase, after extraction with n-butanol,
was then discarded. The n-butanol phase was subjected
to filtration over two Sephadex LH-20 gel columns
using 90% aqueous ethanol (for first filtration) and
20% aqueous acetone (for second filtration) as the
mobile phases. 1,22-docosandiol bisphosphocholine
ester was collected from the early fractions of each
gel filtration.
We believe that several related genera are the
same and/or closely related to the genus Irlbachia,
and would have similar medicinal properties. One
species from a closely related genus, Lisianthus
nigrens is used in Mexico. The leaves are applied as
a poultice to treat fungal infections of the skin,
feet, ankles and hands. A decoction of the root is
also taken orally as a "bitter" and as a febrifuge.
Another species Lisianthus alatus is considered to be
~~ the same as Irlbachia alata. Another species and
genus of interest is Chelonanthus alatus. There are
SUBSTITUTE SHEET
W094/0856~ 1 4 6 6 3 ~ ~ PCT/US93/096
- 22 -
several uses described for Chelonanthus alatus,
including oral decoctions to treat smallpox, fevers
and for gastric disturbances.
S 6.2. S~ectral Characteristics
The isolated phosphocholine derivative fraction
containing 1,22-docosandiol bisphosphocholine ester
has the characteristic IR, proton NMR and FAB- mass
spectra shown in Figs. 1, 2 and 3, respectively.
The IR spectrum has peaks at approximately 1060,
1220, 1475, 1600-1700, 2850, 2950 and 3400 cm~l.
The IH NMR spectrum has major peaks at ~ 1.2, 1.4,
1.7, 3.1, 3.5, 3.7 and 4.3.
The FAB-/MB mass spectrum has major peaks (>40~)
at m/z 657, 612, 587, 586, 555, 493, 491, 475, 403,
277, 233, 201, 194, 179, 168, 165 and 163.
The high resolution mass spectrum (FAB+) has a
molecular ion at 673.4669 amu.
6.3. Total Synthesis of 2-~almitoYl-l-O-
qlucopYranosyllYsolecithin Ex~erimental Section
Gener~l. Tetrahydrofuran (THF) was distilled from
potassium/benzophenone; benzene, triethylamine, and
methylene chloride, N-methylmorpholine, and benzyl
alcohol were distilled from calcium hydride; 2-
bromoethylphosphorodichloridate was prepared according
to the procedure reported by Baumann et al Lipids, 17,
453 (1982) and was freshly distilled prior to use;
trifluromethanesulfonic anhydride was freshly
distilled under inert atmosphere; 0-~-D-
(Glucopyranosyl)trichloroacetimidate was prepared by
the method of Schmidt. (a) R. R. Schmidt, J. Michael,
Angew. Chem. Int. Ed Engl. (1980), 19, 731; (b) R. R.
~~ Schnmidt, J. Michael, TetraAedron Lett. (1984), 25,
821. Anhydrous dimethylformamide (DMF! was obtained
SU~ 11~ ~TE SHEET
W094/08563 2 1 4 ~ 6 3 ~ PCT/US93/09623
from Aldrich. S-(+)-1,2-0-isopropylidene glycerol and
R-(-)-1,2-O-isopropylidene glycerol were obtained from
Lancaster. 2,3,4,6-Tetra-O-benzyl-D-glucopyranose was
obtained from Sigma. Preparative thin layer
chromatography plates was performed on Whatman 2000
TLC silica gel plates. Flash column chromatography
was performed on Whatman 230-400 mesh silica gel using
nitrogen pressure. IH andl3C NMR were provided by using
a Varian 400 MHz spectrometer with chloroform as an
internal reference unless otherwise noted. NMR shifts
were expressed in ppm downfield from internal
tetramethylsilane. Carbon 13 multiplicities as
determined by DEPT experiments are reported in
parentheses following the chemical shift value
according to the following format: (o) for quaternary
carbon, (1) for methine carbon, (2) for methylene
carbon, and (3) for methyl carbons. NMR assignments
were determined on the basis of COSY, HMQC, and HMBC
and DEPT experiments performed on selected
intermediates. NMR coupling constants are reported in
Hertz. Melting points were determined using a Buchi
model 535 melting point apparatus and are uncorrected.
The synthetic routes for the total synthesis of
2-palmitoyl-1-0-glucopyranosyllysolecithin are
outlined in the following diagrams and detailed in the
subsequent discussion that refer to these diagrams.
SU~i I ~ ~ ~ITE SHEEr
214663~
WO 94/08~;63 ~ ~ PCI~/US93/0962
-- 24 --
Scheme 1. Synthesis of the (S),SP-19~01: Preparation of the
Regicisomeric Glycerol Alco~o~s
BnO THF BnO
BnO~O + ~O~............... "" ~ BnO~Lo ~`~""'
BnO~,OH TfO~ o -1 oC BnOB~~
R / 1 68%
60% HOAc ~ +
reflux, 90 mi~ anomer
85%/ 19%
BnO BnO
BnO~LO HO TBDMscl BnO~ LO HO
BnO~O ~OH ~ BnO~,O ~OTBDMS
BnO Imidazole, DMF BnO
2 88% / 3
/palmitic anhydride
Et3N, DMAP
O ~ 97%
O ~ \ ~
2 o BnO~o
BnO~,O`,~OTBDMS
BnO \ TBDMSCI
4\ TBAF 93% \ Imidazole, DMF
~,~OAc, THF
TBAF, THF BnO~
46% BnO~O
B~ /OH
BnO
3 0 BnO~O HO
8nO~O~O ~
6 O TBDMSCI
BnO Imidazole, DM F
BnO~LO 8 ~ 43h, 95%
BnOB~,O~O
7 O
SU~i 111 ~ITE SHE~ET
~ WO 94/08~63 2 1 ~ ~ 6 3 ~ PCI'JUS93/09623
Scheme 2. Synthesis of the (R) SP-19501: Preparation of the
Regioisomeric Glycerol Alcohols,
BnO TH F BnO
BnO~Lo + O~................ "", ~ BnO~O O~
BnO~oH TfO~,o -1 0C BnOB~o~O
S / 8 65-70%
60% HOAc ~ +
reflux, 90 mi~ o~- anomer
81-9
BnO~ BnO
BnO~O HO TBDMSCl BnO~LO
BnO~,O J~ OH ~ BnO~,O ~ OTBDMS
BnO \' `/ Imidazole, DMF BnO \/ V
9 90-95% 10
/mitic anhydride
Et3N, DMAP
O ~/ 90-95%
BnO
BnO~Lo
2 0 Bno~o~oTBDMs ~ TBDMSCI
11 \ TBAF 87-96% \ Imidazole, DMF
~ OAc, THF
46% BnO~O O ~
BnO~,O~OH
BnO 1 2
BnO~LO HO
3o BnO~o~O~
1 3 O TBDMSCI
BnO Imidazole, DMF
- BnO~ TBDMSO " rt, 94%
3 5 B~ V~
14 O
Sl.lts~ 111 ~JTE SHEET
wo 94,o85632 1 4 6 6 3 ~ PCr/US93/0962~
-- 26 --
Scheme 3. Synthesio, cf (S) SP-19501
BnO ll
5BnO~LO
BnO~O ~OH o Cl
~ Br~ ,P~_cl ~ Et3N
2) reagent 5
~3) BnOH, 43%
BnO~
BnO~O\~/ ~p, O Br 15
~CH3)3N
/ BOMB, 55C, 24h, 45%
BnO
BnO~o
BnOB~~`p'O
16 ~\N+(CH3)3Br
H2, 60 ps~ quantitative
MeOH, 24 h
'~ O
BnO ll
BnO~O ~ ~~ ~ \~ r
B~/ ~ `P'-O
'~ ~\
(S) SP-19501 N+(CH3)3
SUBSmUTE SHE,ET
WO 94/08~63 2 1 4 6 ~ 3`~ Pcr/US93/09623
-- 27 --
Scheme 4. Synthesis oi (R) SP-19501
BnO ll
5 BnO~LO
BnO~O OH o C
12 \~) Br~ ~P~_cl ~ Et3N
\ 2) reagent 5
\~3) BnOH, 43%
BnO
BnO~LO ~ \ ~~
BnO~,,O~O~e,OOBn 1 7
--\B
/ (CH3)3N
/ BOMB, 55C, 24h, 21%
O
BnO ll
BnO~Lo
BnO~,O~O~ ,O~
18 ~\N+(CH3)3Br
H2, 60 ps~ quantitative
MeOH, 24 h \
~ O
BnO ll
BnO~O
BnO ~,O~O~ ,O~
(R) SP-l 9501 N+(CH3)3
Su~ JTE SHEET
21466~g
WO 94/08~;63 ~- ~ ' PCr/US93/0962
~ 28 ~
~/ o
o~H BnOH O
098 mol% 500' h~ol% o - \
BrCH2CH20pcl2 N-methylmor~holine~ BrCH2CH20P/ ~
0--RT, 62~i~ 45%
1 9
1 M H3PO4, THF
(C6H5)3CCI, 105 mol% RT, 17h, 80%
O OH i-Pr2EtN, 105 mol% o OH
BrCH2CH2OI / ~/ RT, 40h, 52.1% 1 OH
O2Bln O2On
THF Palmitic anhydride, 110 mol%
15 RT, 3h Et3N, 110 mol%
92.6% DMAP, 20 mol%
o
o~(CH2)14CH3
20 BrCH2CH2OI / ~ HCOOH/THF, 1:1
OBn RT, 2h, 72.7%
22 or
cUso4/c6H6 o
reflux, 2h, 66.2% ~
~ =O (CH2)14CH3
OBn BrCH2CH2OI / ~OH
150 mol ~/ C~ ' 115 mZ1%
CH2CH2 RT, 4h, 22%
B~o dBCH2)14CH3
3s BnO~O O~ ,OCH2CH2Br
17 o
SUBSTITUTE SHEET
~ W094/0~63 2 1 ~6 6 3 9 PCT/US93/09623
- 29 -
(R) 2,3-O-Isopropylidene-l-O-trifluromethylsulfonyl--
glycerol. A nitrogen-purged 250-mL three-necked
roundbottomed flask fitted with a thermometer,
stopper, and septum was charged with S-(+)-1,2-O-
isopropylidene glycerol (1.0 g, 7.6 mmol) dissolved inbenzene (75 ml). Triethylamine (1.25 mL, 9.0 mmol)
was injected into the solution, and the reaction
mixture was chilled until a cloudy solution appeared.
Trifluoromethaneæulfonic anhydride (1.25 mL, 7.6 mmol)
was then added, and the reaction was stirred for 30
minutes with the temperature maintained at 5C. The
solution was then filtered through a bed of silica.
The filtrate was concentrated under reduced pressure
at 30C to give an orange/brown oil (1.84 g, 7.0 mmol)
in 92% yield which was used directly for the next
step.
~2R) tl-0-(2,3,4,,6-Tetra-O-benzyl-~-D-
glu~Gyy~nosyl)-2~3-o-isopropyli~ene] glycerol 1
2,3,4,6-Tetra-O-benzyl-D-glucopyranose (100 g,
0.182 mol) was dissolved in THF (1.4 L) and chilled to
-10C in a nitrogen-purged 3-L three-necked morton
flask fitted with a thermometer, stopper, and
mech~n;cal stirrer. Sodium hydride 60~ in oil (16.1
g, 0.403 mol) was added in 4 increments over 10
minutes, and the solution was stirred for 30 minutes.
(R) 2,3-O-Iso~ylidene-l-O-
trifluoromethylsulfonylglycerol (60.0 g, 0.227 mol)
dissolved in THF (500 mL) was then dropped via an
addition funnel into the reaction mixture over a 30
minute period. The solution was stirred at -10C for
7 hours. Methanol (200 mL) was added dropwise to
- quench excess sodium hydride, the resulting brown
solution was rotary evaporated under reduced pressure
SlJ~S 111 ~ITE SHEET
W094/08563 2 1 4 6 6.3 g ~- PCT/US93/096 ~
- 30 -
and then the residue redlssolved in chloroform (750
mL). The organic la~ye~ was washed with water (2 x 750
mL). The combined aqueous layers were washed with
chloroform (3 x 500 mL). Organic layers were pooled
and rotary evaporated under reduced pressure to give a
white solid which contained both ~ and ~-epimers of
the desired product. The solid was triturated with
diethyl ether to give a white solid of purely ~--
product and a mother liquor which contained ~ and ~-
epimers. The mother liquor was concentrated and flashchromatographed (silica gel, 20% ethyl
acetate/he~ne). Yield of the solid white ~-epimer
product (81 g, 0.123 mol) was 68%, mp 91-91.7C (lit
83-84C);IH-NMR (CDCl3) S7.4-7.29 (m, 18H), 7.20 (m,
2H), 4.96 (d, 2H J=10.8), 4.84 (t, 2H, J=10.8), 4.75
(d, lH, J=10.8), 4.65 (d, lH, J=12.4), 4.6-4.54
(overlapping dd, 2H, J=12H, J=10.4), 4.46 (d, lH,
J=7.2, Hl'), 4.38 (p, lH, H2), 4.12-4.02 (m, 2H, H~"
H2), 3.89 (pseudo t, lH, J=7.2, Hlb), 3.79-3.6 (m, 5H),
3.50 (pseudo t, 2H), 1.46 (s, 3H), 1.40 (s, 3H); 13C-
NMR (CDCl3) S138.529 (0), 138.370 (0), 138.066 (0),
138.013 (O), 128.432, 128.409, 128.129 ,128.015,
127.901, 127.810, 127.734, 127.666, 109.399 (0),
103.824 (Cl'), 84-631 (C3'), 82.120 (C2'), 77.713
(C4'), 75.748 (2), 75.058 (2),.74.891,74.853, 74.315
(2), 73.495 (Cl), 70.317 (2), 68.762 (C6'), 66.896 (C3),
26.880 (3), 25.386(3). Yield of the colorless, oily
~-epimer (23 g, 0.035 mol) was 19~; IH NMR (CDCI3)
S7.4-7.24 (m, 18H), 7.14 (m, 2H), 4.98 (d, lH,
J=10.8), 4.88-4.78 (m, 3H), 4.67 (d, lH, J=12),.4.62
(d, lH, J=11.6), 4.47 (d, 2H J=11.6), 4.37 (t, lH,
J=6.4), 4.07 (pseudo pentet, lH), 3.96 (t, lH, J=8.8),
3.8-3.54 (m, 9H), 1.43 (s, 3H), 1.37 (s, 3H); 13C NMR
(CDCl3) S138.764 (0), 138.203 (0), 138.165 (0), 137.816
(0), 128.440, 128.387, 128.364, 128.030, 127.947,
SU~ 111 ~TE SHEET
W094/0~3 2 1 1~ 6 ~ 9 PCT~US93/09623
- 31 -
127.916, 127.886, 127.696, 127.590, 109.422 (0),
97.482 (Cl'), 81.885 (1), 79.890 (1), 77.508 (1),
75.703 (2), 75.088 (2), 74.535 (1), 73.457 (2), 73.108
(2), 70.279 (1), 69.020 (2), 68.314 (2), 67.040 (2),
26.827 (3), 25.424 (3).
(2R) 1-0-~2,3,4,6-Tetra-O-benzyl-~-D-glucG~y~nosyl)
gly¢erol 2. A 5-L three-necked morton flask fitted
with a mech~nical stirrer, condenser, and stopper was
charged with compound 1 (50 g, 76.2 mmol) in 60%
aqueous acetic acid (2.5 L). The acidic solution was
refluxed for 1.5 hours at 103C and then cooled to
room temperature. Distilled water (1.5 L) was added
to the solution causing precipitation of a white
solid. The acidic solution was extracted with
methylene chloride (4 x 1 L) which was subsequently
neutralized with sodium bicarbonate solution and
concentrated to a white solid. Trituration with
diethyl ether gave white product. The remaining
mother li~uor was flash chromatographed (silica gel,
50% ethyl acetate/hexane~ to give white solid product.
The combined yield (61.9 g, 0.101 mol) was 83 %, mp
101.5-102.4C (lit 76-78C); IH NMR (CDCl3) ~.40-7.26
(m, 18H), 7.19 (t, J=3.5, 2H), 5.0-4.7 (m, 5H), 4.64-
4.5 (m, 3H), 4.46 (d, lH, J=8.0, H~'), 4.0-3.60 (m,
llH, H~'s, H2, H3, H3', H6b', H6~', H~', Hs'~H2~ 2-55 (s,
2H, OH's); 13C NMR (CDC13) 38.529 (0), 138.332 (0),
137.952 (0), 137.740 (0), 128.531, 128.550, 128.478,
128.189, 128.114, 128.091, 127.931, 127.871,
127.749,104.279 (Cl'), 84.654 (C3'), 82.158 (C2'),
77.819 (C41), 75.779 (2), 75.081 (2), 75.028 (2),
74.527 (Csl)~ 73.571 (2), 72.207 (Cl), 71.204 (C2),
68.883, (C6'), 63.353 (C3).
SU~ I ~JTE S~EET
W094/0856~ i 4 6 6 ~ ~ PCT/US93/096
- 32 -
~28) tl-0-(2113,~,C-TQtr~-O-b~nzyl-~-D-
gluc,G~ylO~nosyl)-3-o-t~rt-butyld~imethylsilyl] glycerol
3. In a nitrogen-purged 100-mL round-bottomed flask
fitted with a septum was dissolved!diol 2 (9.0 g, 14.7
mmol), imidazole (2.05 g, 30.2~m~mol), and t-butyl
dimethylsilylchloride (2.28 gq -15.1 mmol) in anhyd DMF
(45 mL). The reaction mixturë was stirred under
nitrogen for 2.5 days, transferred to a 1-L separatory
funnei, and methylene chloride (250 mL) and water (250
mL) were added. The aqueous layer was extracted with
methylene chloride (2 x 250 mL) and then the combined
organic layers were washed with water (2 x 100 mL).
After drying and concentration, purification by flash
chromatography (silica gel, 33% ethyl acetate/hexane)
gave a colorless oil (9.2 g, 12.6 mmol) in 88% yield;
H NMR (CDCl3) ~7.48-7.3 (m, 18H), 7.25-7.21 (m, 2H),
S.00 (d, 2H, J=11.2), 4.89 and 4.88 (overlapping
doublets, 2H, J=10.8, J=10.4), 4.83 (d, lH, J=11.2),
4.67 (d, lH, J-12.4), 4.60 and 4.59 (overlapping
doublets, 2H, J=12.4, J=10.8); 4.50 (d, lH, J=7.6 H~'),
4.06-3.92 (m, 2H), 3.9-3.62 (m, 7H), 3.6-3.52 (m, 2H),
3.04 (s, lH, OH), 0.978 (s, 9H), 0.142 (s, 6H); l3C NMR
(CDCl3) ~188.552 (0), 138.385 (0), 138.005 (0), 137.960
(0), 128.455, 128.440, 128.121, 128.060, 127.931,
127.863, 127.772, 127.734, 127.696, 104.377 (Cl'),
84.692 (C3'), 82.219 (C2'), 77.804 (C4'), 75.771 (2),
75.073 (2), 74.959,(2), 74.717 (C5'), 73.541 (2),
73.078 (2), 71.060 (C2), 68.7~5 (C6'), 63.998 (C3),
25.970 (3), 18.668 (0), -5.299 (3).
(28) t1-0-(2,3,4,6-T6tra-O-benzyl-~-D glucopyranosyl)-
2-O-palmitoyl-3-O-t-butyldimethylsilyl~ glycerol 4. A
nitrogen purged 500-mL round-bottomed flask fitted
~ with a septum was charged with compound 3 (9.3 g, 12.8
mmol) and palmitic anhydride (6.94 g, 14.0 mmol) in
~U~ JTE SHEET
W094/08563 2 1 46 6 3 9 PCT/US93/09623
- 33 -
dry THF (200 mL). Dimethylaminopyridine (316 mg, 2.6
mmol) and triethylamine (2.04 mL, 14.7 mmol) were
added, and the reaction was stirred under nitrogen for
12 h. The mixture was then transferred to a 2-L
s separatory funnel, and diethyl ether (500 mL) and
water (500 mL) were added. The aqueous layer was
filtered through Whatman No. 1 paper and extracted
with diethyl ether (2 x 500 mL). After drying over
magnesium sulfate, the combined organic layers were
concentrated and purifled by flash chromatography
(silica gel, 14% ethyl acetate/hexane) to give a light
yellow oil (12.1 g, 12.5 mmol) in 97% yield; IH NMR
(CDCl3) ~7.40 (br. s, 20H), 5.15 (5, lH), 4.98 (t, 2H),
4.84 (t, 2H), 4.76 (d, lH), 4.67 (d, lH), 4.59 (dd,
2H), 4.52 (d, lH), 4.13(dd, lH), 3.84 (m, 6H), 3.67
(dd, 2H), 3.49 (t, 2H), 2.32 (t, 2H), 1.61 (m, 2H),
1.25 (br. s, 24H), 0.98 (s, 9H), 0.97 (s, 3H), 0.14
(s, 6H). 13C NMR (CDCl3) ~73.280, 138.597, 138.438,
138.127, 138.096, 128.379, 128.356, 128.333, 128.083,
127.977, 127.863, 127.780, 127.605, 127.582, 103.831,
84.556, 81.984, 77.721, 75.695, 75.020, 74.876,
74.603, 73.488, 72.904, 68.754, 67.821, 61.661,
34.428, 33.950, 31.941, 29.717, 29.687, 29.649,
29.619, 29.497, 29.459, 29.384, 29.300, 29.148,
25.826, 24.953, 22.716, 18.268, 14.159, -5.375.
~2R) rl-o-(2~3~6-T~tr~-o-bensyl-~-D-glucG~nosyl!
2-opalmitoyl] glycerol 5.
Procedur~ A. Compound 4 (34.0 g, 35.1 mmol) was
dissolved in THF (1.4 L) in a 3-L three-necked Norton
flask fitted with a mechanical stirrer, thermometer,
and a 500-mL addition funnel. The solution was
chilled to OC, and a solution of tetrabutylammonium
~~ fluoride (TBAF) (520 mL, 1.0 M in THF) which was
buffered to pH=6.5 with acetic acid was added dropwise
SlJtw ~ JTE SHEET
W094/085~ 1 4 ~ ~ 3 g ~ PCT/US9~/096 ~
through the addition funnel. The reaction mixture was
stirred for 11 h at OC, left to sit at -15C for 12
h, and stirred again for 4 h at rt. Water (100 mL)
was added, and the solut'ion was concentrated to 200 mL
of solution. The concentrate was redissolved in
methylene chloride (750 mL) in a 3-L separatory funnel
and washed with water three times (750 mL, 2 x 500
mL). The combined aqueous layers were extracted with
diethyl ether (500 mL). The combined organic layers
were concentrated to give a red oil which was purified
by flash chromatography (silica gel, 33-40% gradient
of ethyl acetate/hexane). A white solid (28.0 g, 32.8
mmol) was obtained in 93% yield. IH NMR (CDCl3) ~7.36
(br. s, 20H), 5.06 (t, lH), 4.96 (dd, 2H), 4.84 (dd,
2H), 4.75 (d, lH) 4.59 (m, 2H), 4.53 (dd, lH), 4.45
(dd, lH), 4.14 (m, 2H), 3.91 (m, 2H), 3.78 (m, 6H),
2.80 (s, lH), 1.64 (m, 2H), 1.27 (br. s, 26H), 0.90
(t, 3H).
Proce~ure B. Compound ~ (500 mg, 0.52 mmol) was
dissolved in THF (20 mL) in a 100-mL three-necked
round-bottomed flask fitted with two stoppers and a
septum. Glacial acetic acid (9.5 mL) was added, and
the solution was chilled to 0C. A solution of TBAF
(5.16 mL, 1.0 M in THF) was syringed into the chilled
solution, and stinting was continued at 0C for 8 h
and then at rt for 25 hours. Methylene chloride (50
mL) was added, and the entire solution was transferred
to a 250-ml separatory funnel where it was neutralized
with lM disodium phosphate solution (2 x 75 mL). The
combined organic layers were rotary evaporated under
reduced pressure and the concentrate was purified by
flash chromatography (silica gel, 25-40% gradient of
ethyl acetate/hexane), yielding a colorless oil (424
- mg, 0.497 mmol, 95~) which later solidified upon
st~n~;ng; IH NMR (CDC13) ~7.36 (br. s, 20H), 5.06 (t,
SUBSmUTE SHEET
~ W094/08~63 2146~ Pcr/us93/o9623
- 35 -
lH), 4.96 (dd, 2H), 4.84 (dd, 2H), 4.75 (d, lH), 4.59
(m, 2H), 4.53 (dd, lH), 4.45 (dd, lH), 4.14 (m, 2H),
3.91 (m, 2H), 3.78 (m, 6H), 2.80 (s, lH), 1.64 (m,
2H), 1.27 (br. s, 26H), 0.90 (t, 3H).
~28) tl-0-~2,3~4~6-T~tr~-O-b~nzyl-~-D-glu~G~y~osyl)-
3-O-palmitoyl] glycerol 6. Compound ~ (3.0 g, 3.1
mmol) was dissolved in THF (120mL) in a 250-mL three-
necked round-bottomed flask fitted with a 60-mL
addition funnel, glass stopper, and septum. TBAF (54
mL, 1.0 M in THF) was added through the addition
funnel over a 15 minute period. Glacial acetic acid
(18 mL) measured in a graduated cylinder was then
poured into the reaction mixture, and the solution was
stirred for 45 minutes. The solution was concentrated
under reduced pressure to approximately 30 mL of
liquid and then redissolved in methylene chloride (150
mL). The organic layer was washed with water (3 x 120
mL) and neutralized with sodium bicarbonate solution
(2 x 150 mL). The combined aqueous layers were
extracted with methylene chloride (100 mL). The
combined organic layers were dried over magnesium
sulfate, filtered, and concentrated. The resulting
dark red concentrate was purified by flash
chromatography (silica gel, 25% ethyl acetate/h~A~e)
to give 6 a colorless oil which corresponded to an
upper TLC spot (1.3 g, 1.52 mmol) in 46% yield. IH NMR
(CDC13) ~7.36 (br. s, 20H), 4.95 (m, 2H), 4.86 (m, 3H),
4.64 (d, lH), 4,58 (m, 2H), 4.47 (d, lH), 4.16 (m,
lH), 3.96 (dd, lH), 3.68 (m, 8H), 2.38 (t, 2H), 1.62
(m, 2H), 1.27 (br. s, 24H), 0.96 (t, 3H). Isolation
of a lower TLC spot gave a white solid (400 mg, 0.469
mmol) in 15% yield which corresponded to compound 5; IH
NMR (CDC13) ~7.36 (br- s, 20H), 5.06, (t, lH), 4.96
(dd, 2H), 4.84 (dd, 2H), 4.75 (d, lH), 4.59 (m, 2H),
SUBSmUTE SHEET
W094/08563 2 1 ~ 6 ~ 3 ~ ~ . PCT/US93/0962~
4.53 (dd, lH), 4.45 (dd, 1H), 4.14 (m, 2H), 3.91 (m,
2H), 3.78 (m, 6H), 2.80(s, lH), 1.64 (m, 2H), 1.27
(br. s, 26H), 0.90 (t, 3H).
Resilation of ~2R) [1-0-(2,3,~,6-Tetra-O-b~nzyl-~-D
glucG~y~anosyl)-2-o-palmitoyl] glycerol 5. In a
nitrogen-purged 50-mL round-bottomed flask fitted with
a septum was placed compound 5 (318 mg, 0.373 mmol)
dissolved in DMF (8 mL). tert-Butyl-dimethylsilyl
chloride (281 mg, 1.86 mmol) and imidazole (254 mg,
3.73 mmol) were added, and the solution was stirred
for 22 h. Methylene chloride (50 mL) was added, and
the reaction mixture was transferred to a 250-mL
separatory funnel. The organic layer was washed with
water (50 ml), and then the aqueous layer was
extracted with methylene chloride (2 x 50 mL). The
pooled methylene chloride layers were washed with
water (2 x 75 mL), dried over magnesium sulfate, and
the filtered. The filtrate was concentrated and
purifled by flash chromatography (silica gel, 14%
ethyl acetate/hexane) to give 4 as a yellow oil (239
mg, 0.247 mmol) in 66% yield.
Re~ilation of (2S) tl-O-(2,3,~,6-TetraDO-benzyl-~
-D-glU~G~ ~08yl)-3-0-palmitoyl~ glycerol 6. In a
nitrogen-purged 25-mL round-bottomed flask fitted with
a septum was placed compound 6 (176 mg, 0.206 mmol)
dissolved in anhyd DMF (5 mL). tert-
Butyldimethylsilyl chloride (155 mg, 1.03 mmol) and
imidazole (140 mg, 2.06 mmol) were added, and the
solution was stirred for 43 h. Methylene chloride (50
mL) was added, and the reaction mixture was
transferred to a 250-mL separatory funnel. The
- organic layer was washed with water (50 mL). The
aqueous layer was extracted with methylene chloride (2
SUB3TITUTE SHEET
W O 94/08563 2 1 ~ 6 6i3 ~ PC~r/US93/09623
- 37 -
x S0 ml). The methylene chloride layers were pooled
methylene and washed with water (2 x 75 mL), dried
over magnesium sulfate, and then filtered. The
filtrate was concentrated and flash chromatographed
(silica gel, 14% ethyl acetate/hexane) to give 7 as a
light yellow oil (190 mg, 0.223 mmol) in 95% yield; lH
NMR (CDC13) ~7.38 (br. s, 20H), 4.99 (dd, 2H), 4.85 (t,
2H), 4.78 (d, lH), 4.68 (d, lH), 4.61 (dd, 2H), 4.48
(d, lH), 4.37 (d, lH), 4.13 (s, 2H), 3.98 (m, lH),
3.77 (m, 2H), 3.67 (m, 3H), 3.52 (m, 2H) 2.35 (t, 2H),
1.67 (m, 2H), 1.31 (br. s, 24H), 0.93 (s, 12H), 0.14
(s, 6H).
(2S) tl-0-(2~3~6-Totra-O-bensyl-~-D-glu~G~y~no~yl)-
2-O-palmitoyl 3-0-(2-bromoethyl)benzylphosphoryl]
glycerol 15.
Procodure A. In a nitrogen-purged 100-mL three-necked
round-bottomed flask fitted with two stoppers and a
septum was dissolved freshly distilled 2-
bromoethylphosophorodichloridate (1.72 g, 7.11 mmol)in diethyl ether (20 mL). The solution was chilled to
0C, and triethylamine (8.15 mL, 58.5 mmol) was
injected into the solution which caused precipitation
of a white solid. A solution of compound 5 (1.0 g,
1.17 mmol) in anhyd diethyl ether (55 ml) was injected
into the chilled reaction mixture, and the ice bath
was removed. The reaction was stirred for 30 minutes
after which benzyl alcohol (1.21 mL, 11.7 mmol) was
injected into the reaction mixture. Stirring was
continued at rt for 5 d. The reaction was then
filtered through a fritted glass funnel, and the
filtrate was concentrated. The orange concentrate was
purified by flash chromatography (0-33% ethyl
- acetate/hexane) to give 15 as a light yellow oil (566
mg, 0.501 mmol) in 43% yield; IH NMR (CDCl3) ~7.38-7.25
SVBSTITUTE SHEET
2146539
WOg4/08563 - PCT/US93/0962
- 38 -
. .
(br. s, 23H), 7.16~;tm, 2H), 5.26 (m, lH), 5.10 (t,
2H), 4.94 (m, 2H), 4.81 (t, 3H), 4.71 (d, lH), 4.61
(d, lH), 4.55 (d, 2H), 4.39 (d, lH), 4.25 (m, 4H),
4.08 (dd, lH), 3.73 (m, 3H), 3.64 (dd, 2H), 3.42 (m,
4H), 2.27 (t, 2H), 1.58 (m, 2H), 1.25 (br. d, 24H),
0.89 (t, 3H).; ~3C NMR (CDCl3) ~173.210, 138.559,
138.362, 138.096, 138.074, 128.667, 128.622, 128.333,
128.318, 128.296, 127.962, 127.878, 127.757, 127.734,
127.696, 127.605, 127.522, 103.862, 84.540, 81.969,
71.652, 75.589, 74.937, 74.906, 74.686, 73.480,
70.469, 70.385, 69.680, 69.619, 68.777, 67.283,
66.099, 66.069, 34.170, 31.887, 29.657, 29.619,
29.596, 29.452, 29.315, 29.239, 29.080, 24,802,
22.647, 14.050.
Proce~ure B. In a nitrogen-purged 100-mL three-necked
roundbottomed flask fitted with a thermometer,
stopper, and septum was dissolved freshly distilled 2-
bromoethylphosphorodichloridate (1.42 g, 5.85 mmol) in
methylene chloride (15 mL). The solution was chilled
to 0C, and compound 5 (1.0 g, 1.17 mmol) and a
solution of N-methylmorphiline (1.28 mL, 11.7 mmol)
dissolved in methylene chloride (35 mL) was injected
into the solution over a 10 minute period. The
reaction mixture was stirred at 0C for 5.5 h at which
point a new TLC spot which co-spotted with secondary
alcohol 6 appeared. Stirring was continued for
another 30 minutes, and benzyl alcohol (1.21 ml, 11.7
mmol) was injected into the reaction. After 6 days of
stirring, the reaction mixture was transferred to a
500-mL separatory funnel, and methylene chloride (150
mL) and water (200 ml) were added. The layers were
separated, and the organic layer was rotary evaporated
under reduced pressure. The resulting oil was flash
- chromatographed (silica gel, 33% ethyl acetate/hexane)
to give 15 as a yellow oil (2 50 mg, 19%); lH NMR
SUBSmUTE SHEET
214663~
W094/0~63 PCT/US93/09623
- 39 -
(CDCl3) ~7.38 8-7.2 5 (br. s, 23H), 7.16 (m, 2H), 5.26
(m, lH), 5.10 (t, 2H), 4.94 (m, 2H), 4.81 (t, 3H),
4.71 (d, lH), 4.61 (d, lH), 4.55 (d, 2H), 4.89 (d,
lH), 4.25 (m, 4H), 4.08 (dd, lH), 3.73 (m, 3H), 3.64
(dd, 2H), 3.42 (m, 4H), 2.27 (t, 2H), 1.58 (m, 2H),
1.25 (br. d, 24H), 0.89 (t, 3H).; l3C NMR (CDC13) ~
173.210, 138.491, 138.286, 137.990, 137.975, 128.720,
128.652, 128.387, 128.364, 128.015, 127.954, 127.878,
127.810, 127.780, 127.727, 127.681, 127.613, 103.854,
84.495, 81.923, 77.781, 77.546, 75.688, 75.020,
74.808, 74.747, 73.473, 70.438, 69.642, 68.633,
67.322, 66.759, 66.129, 34.178, 31.925, 29.702,
29.664, 29.641, 29.490, 29.422, 29.368, 29.285,
29.103, 24.802, 22.700, 14.198.
(28) ~1-0-(2~3~6-Tetra-O-benzyl-~-D-glu~opy~nosyl)-
2-0-palmitoyl-3-O-phosph~tidylcholine] glycerol 16. A
45 mL Parr bomb equipped with a magnetic stirring bar
was charged with a solution of phosphate 15 in toluene
(10 mL). Condensed anhydrous trimethylamine (12 mL)
was added quickly in one portion, and then the vessel
was sealed and heated in an oil bath at 55C for 24 h.
The reaction mixture was concentrated to a viscous oil
and triturated with ethyl ether, upon which a white
precipitate formed. the precipitate was filtered off,
washed with ether, and then the combined ethereal
solutions were concentrated to a glassy solid.
Purification of this residue using preparative TLC
(2000 ~ double elution with 75%,12.5%,12.5% methylene
chloride/reethanol/ hexanes gave inner salt 16 as a
- glassy solid;
t28) ~-D-gluco~nos-l-yl-2-O-p~lmitoyl-3-O-
phosphati~ylcholine] glycerol 8P-19501. A solution of
phosphatidylcholine 16 (200.4 mg, 0.197 mmol) in
SUBSTITUTE SHEET
wo 2146~.39.. ..
94~08~63 ~ PCT/US93/096
- 40 -
i . ,
reagent grade methanol (25 mL) was hydrogenated at 60
psi over 10% Pd/C (40 mg, 20 wt%). After 30 h, the
catalyst was ffltered off through celite and the
methanol wAching were combined and concentrated. The
residue was dissolved in fresh methanol (25 mL) and
resubjected to hydrogenation at 60 psi over 80 mg (40
wt%) of 10%Pd/C. After 48 h, the reaction was still
incomplete. After filtration, washing of the
catalyst, and concentration, the residue was subjected
to hydrogenation using 400 mg (200 wt%) of Pd/C at 60
psi in methanol (25 mL). After 22h, the catalyst was
filtered off through celite and the methanol filtrate
and washings were combined and concentrated to afford
92.8 mg (71.6%) of (8) 8P-19501 as a white solid; IH
NMR (CD30D) ~5.12 (br t, 0.5 H), 4.88 (br m, 4.5 H),
4.25 (br m, 2H), 4.12-3.57(M, 12H), 3.4-3.1 (m
cont~in;ng singlet at 3.18, 12H), 2.3 (m, 2H), 1.55(m,
2H), 1.24 (m, 22H), 0.86 (br t, 3H); l3C NMR (CD30D)
74.93, 104.80, 78.02, 77.93, 75.19, doublet at 71.53
and 71.49, doublet at 70.80 and 70.73, doublet at
67.79 and 67.74, multiplet at 67.50, 62.53, doublet at
60.56 and 60.52, triplet at 54.79, 34.88, 33.15,
30.85, 30.85, 30.66, 30.56, 30.46, 30.26, 26.10 and
26.03, 23.82, 14.56; 3Ip NMR (CD30D) ~1.65.
~2S) 2,3-O-I~opropyiidene-l-0-trifluromethyl~ulfonyl-
glycerol was prepared according to the method
described for the corresponding (R) isomer in 92%
yield and used immediately.
~28) tl-0-~2~3~6-Tetra-O-benzyl-~-D-glu~o~y~nosyl)-
2~3-o-isopropylia~n~] glycerol 8. 2,3,4,6-Tetra-O-
benzyl-D-glucopyranose (65 g, 0.12 mol) was dissolved
in THF (800 mL) and chilled to -10C in a nitrogen-
purged 3-L three-necked morton flask fitted with a
SU I i I UTE SHEEr
~ W094/08563 21.~ 6 ~ 3 D PCT/USg3/~K~3
thermometer, stopper, and mechanical stirrer. Sodium
hydride 60% in oil (33 g, 0.825 mol) was added in 4
increments over 10 minutes, and the solution was
stirred for lh. (S) 2,3-0-Isopropylidene-l-O-
trifluoromethylsulfonylglycerol (0.15 mol) dissolvedin THF (200 mL) was then dropped via an addition
funnel into the reaction mixture over a 20 minute
period at -10 to -15C. The solution was stirred at -
10 to -15C for 6 hours. The reaction mixture was
filtered through a short plug of silica gel and
concentrated to an orange brown oil, 114 g.
Purification of the crude by flash chromatography
using 50% ethyl ether/hexanes gave 39.8 g (67.6%) of
~epimer 8 as a white solid, along with 4 g (5.1 ~) of
a mixture of ~ and ~ epimers; mp of ~ anomer 85.7-
87.2C; IH NMR of ~ epimer (CDC13) ~ 7.4-7.2 (m, 18H),
7.19-7.14 (m, 2H), 4.98-4.92 toverlapping doublets at
4.97 (J=10.8) and 4.94 (J=10.8), 2H], 4.82 (t, 2H, J-
10.8), 4.73 (d, lH, J=10.4), 4.63 (d, IH, J=12.4),
4.58-4.51 [overlapping doublets at 4.55 (J=12) and
4.53 (J=10.8), 2H] 4.45 (dy lH, J=7.2), 4.36 (p, lH,
H2), 4.08 (pseudo triplet, lH), 3.94-3.89 [overlapping
doublets at 3.92 (J=10) and 3.91 (J=9.6), lH], 3.82-
3.57 (m, 6H), 3.47 (pseudo triplet, 2H), 1.44 (s, 3H),
1.38 (s, 3H); ~3C NMR (CDC13) ~ 138.569 (0), 138.384
(0), 138.006 (0), 138.021 (0), 128.341, 128.258,
127.962, 127.856, 127.765, 127.696, 127.620, 127.605,
109.467 (o), 103.869 (Cl'), 84.586 (C3'), 82.075
(C2'), 77.705 (C4'), 75.680 (2), 75.005 (2), 74.815 (2
carbons, C2, C5'), 74.512 (1), 73.457 (Cl), 71.151
(2), 68.785 (C6'), 67.017 (C3), 26.895 (3), 25.393
(3).
- ~2 ) 1-0-~2,3,4,6-Tetra-O-bensyl-~-D-glucopyr~nosyl)--
glycerol 9. A suspension of 8 (20 g, 30.5 mmol) in
SU~S 111 ~TE SHEET
wo g4/08~3 2 1 q ~ ~ ~ 9 ~ PCT/US93/096 ~
- 42 -
60% acetic acid (800)~;was heated to reflux for lh.
Workup was similar`to`that described for the (R) diol
2, providing 18 g (96% yield) of 9 as a white solid,
which was of sufficient purity after trituration with
ether for the subsequent step. Diol 9 could be
recrystallized from ether/hexane, mp 89.6-90.90C; IH
NMR (CDCl3) ~7.38-7.27 (m, 18H), 7.16 (t, J=3.5, 2H),
4.98-4.74 (m, SH), 4.61-4.5 (m, 3H), 4.42 (d, lH,
J=8.0, H~'), 3.89-3.80 (m, 3H, H~'s, H2), 3.72-3.63 (m,
4H, H3 H3', H~'), 3.62-3.44 (m, 4H, H~', H4', Hs~ H2'),
2.59 (s, 2H, OH's); ~3C NMR (CDCl3) ~138.370 (0),
138.119 (0),137.78 (0), 137.69 (0), 128.462, 128.447,
128.432, 128.060, 128.038, 12i.962, 127.894, 127.848,
127.810, 127.704, 104.195 (C~'), 84.616 (C3'), 82.037
(C2'), 77.736 (C4'), 75.733 (2), 75.043 (2, 2 carbons),
74.466 (Cs~)~ 73.480 (2), 72.312 (Cl), 70.772 (C2),
68.731, (C6'), 63.355 (C3).
(2R) tl-0-~2~3~6-T~tra-O-b~nzyl-~-D-glucc~y~nosyl)-
3-O-tert-butyldimQthylsilyl] glycerol 10. In a
nitrogen-purged 100-mL round-bottomed flask fitted
with a septum was dissolved diol 9 (28.0 g, 45 mmol),
imidazole (5.71 g, 90 mmol), and t-butyl
dimethylsilylchloride (6.92 g, 45.3 mmol) in anhyd DMF
(75 mL). The reaction mixture was stirred under
nitrogen ovemight, transferred to a 1-L separatory
funnel, and chloroform (300 mL) and water (300 mL)
were added. The aqueous layer was extracted with
chloroform (2 x 100 mL) and then the combined organic
layers were washed with water (3 x 100 mL). After
= drying (Na2SO4) and concentration, purification by
flash chromatography (silica gel, 50% ethyl
ether/hexanes) gave 10 as a colorless oil (29.5 g) in
90% yield;
SVBSTITUTE SHEET
~ WO ~/U~3 2 1 ~6 3~ PCT/US93/096Z3
~2R) [1-0-(2~3~6-TQtra-O-b~nsyl-~-D-glu~G~ ~nosyl)-
2-opalmitoyl-3-o-t-butyldim~thylsilyl] glycerol 11. A
mixture of 10 (22.2 g, 3 0.4 mmol), palmitic anhydride
(16.5 g, 33.4 mmol), dimethylaminopyridine (741 mg,
6.08 mmol), triethylamine (3.78 g, 5.2 mL, 37.3 mmol)
and anhyd THF (250 mL) was stirred under nitrogen at
rt overnight. The mixture was poured into a 2-L
separatory funnel, diluted with diethyl ether (500 mL)
and water (500 mL), and the layers separated. The
aqueous layer was filtered through Whatman No. 1 paper
and extracted with more diethyl ether (2 x 500 mL).
The combined ether layer was washed with water (3 x
200 mL) and then dried (MgSO4). Following filtration,
purification by flash chromatography (silica gel, 33
ethyl ether/hexane) gave 11 as a light yellow oil,
28.2 g, 96~ yield);
Compound 11 could be carried on to the next
transformation without chromatographic purification.
t2S) [l-o-~2~3~6-Tetra-o-benzyl-~-D-glu~G~ylanosyl)
2-Opalmitoyl] glyc~rol 12. Crude 11 (30.4 mmol based
on 10) was dissolved in THF (100 mL) and the solution
was chilled to OoC. A premixed solution of TBAF (520
mL, 1.0 M in THF) which was buffered to pH=6.37 with
acetic acid was added dropwise via an addition funnel
at 0C for 1 h, and then at -15C overnight. The
reaction mixture was concentrated, water (100 mL) was
added, and the resulting mixture was extracted with
chloroform (3 x 300 mL). The combined chloroform
layer was washed with water (4 x 500 ML), and then the
- combined aqueous layer was backextracted with diethyl
ether (500 mL). After drying the combined organic
layer over Na2S04, concentration gave a red oil which
- was purified by flash chromatography (~0% ethyl
ether/hexane). Evaporation of the product containing
SUBSTITUTE SHEET
2146639
W094/08563 ` PCT/US93/096
fractions afforded 12 as a white solid (24.6 g, 94.6%
yield for two steps);
~2R~ [1-0-(2,3,~,6-T~tra-O~-be~zyl-~-D-gluoG~yl~nosyl)
3-Op~lmitoyl~ glycerol 13. Compound 11 (2.33 g, 2.41
mmol) was dissolved in THF (150 mL) in a 250-mL three-
necked round-bottomed flask fitted with a 60-mL
addition funnel, glass stopper, and septum. After
cooling the solution to OoC, TBAF (24.1 mL, 1.0 M in
THF) was added through the addition funnel over a 5
minute period. Glacial acetiç acid (13.8 mL 241 mmol)
was then poured into the reaction mixture to quench
the reaction, and the resulting solution was stirred
for approximately 30 minutes. The reaction mixture
was poured into a separatory funnel cont~;ning ice
water (500 mL) and methylene chloride (200 mL). The
layers were separated, and aqueous layer was extracted
twice more with methylene chloride (100 mL portions)
and then the combined organic layer was washed with
brine (400 mL). Following dring (MgSO4), flltration,
and then concentration, purification by flash
chromatography using 1/5 EtoAc/ hexanes gave secondary
alcohol 13, 0.96 g (46.8%), as a colorless oil;
Further elution gave 238 mg (11.6%) of primary alcohol
12; Also isolated was a mixture of the two alcohols in
5.3% yield.
Resilation of (28) tl-0-(2,3,~,6-T~tra~O-benzyl-~
-D gluoG~anosyl)-2-O-palmitoyl] glyc~rol 12. The
identity of 12 was established by resilylation of 12
according to the procedure described above for the (R)
isomer, compound 5.
SUBSTITUTE SHEET
~ W094/08~63 2 1 ~ 6 6 3 9 PCT/US93/09623
~2R) tl-O-(2,3,~,6 ~L ~-O-benzyl-~-D-gluoo~.~nosyl)-
2-o-palmitoyl-3-o-(2-bromoethyl)benzylphosphoryl]
glycerol 17. In a nitrogen-purged 1-L three-necked
morton flask fitted with two stoppers and a septum was
dissolved freshly distilled
2-brorhoethylphosphorodichloridate (17.2 g, 71.1 mmol)
in anhyd diethyl ether (500 mL~. The solution was
chilled to 0C and triethylamine (81.5 mL, 0.585 mol)
was injected into the solution, causing precipitation
of a white solid. A solution of 12 (10.0 g, 11.7
mmol) dissolved in diethyl ether (250 mL) was
cannulated into the morton flask, and the solution was
stirred for 1.5 hours. TLC showed disappearance of
12. Benzyl alcohol (12.1 mL, 0.117 mol) was injected
into the reaction mixture, and stirring was continued
at rt for 16 h. The reaction mixture was then filtered
through a fretted glass funnel. Filtrate was then
concentrated and purified by flash chromatography
twice. First chromatography (silica gel, 33% ethyl
20 acetate/hexane) and second chromatography (silica gel,
25% ethyl acetate/hexane) gave 17 as a light oil (5.5
g) in 42% yield;
~28) [1-0-~2,3,4,6-Tetra-O-benzyl-~-D-glucG~l~nosyl)-
2-Op~lmitoyl-3-O-pho~phatidylcholinel glycerol 18. A
45 mL Parr bomb was equipped with a magnetic stir bar
and then charged with a solution of 17 (1.17 g, 1.04
mmol) in benzene (15 mL).
Anhyd trimethylamine (;5 mL, 0.145 mmol) which had
been condensed at -78C was quickly poured into the
reaction vessel, and the bomb was sealed. The
reaction was stirred at 55C in an oil bath for 24
hours behind a blast shield. The bomb vessel was then
- cooled to -78OC, opened, and left in a hood to
evaporate trimethylamine. The remaining solution was
3UBSmUTE SHEET
w094~08s6~1 4 ~ ~ 3 9 - ~ PCT/US93/096 ~
- 46 -
rotary evaporated under reduced ~ressure, and the oily
concentrate was dissolved in;methylene chloride and
purified by preparative TLC ~2000 ~). Double elution
with 75%; 12.5%:12.5% methylene
chloride/methanol/hexane gave inner salt 18 as an
opaque glassy solid (223 mg, 21%). IH-NMR (CDC13) ~.32
(br. s, 20H), 5.21 (m, lH), 4.90 (dd, 2H), 4.82 (m,
2H), 4.64 (m, 2H), 4.50 (t, 2H), 4.42 (d, lH), 4.22
(br. s, 3H), 3.95 (s. 2H), 3.72 (s, 2H), 3.62 (t, 2H),
3.55 (s, IH), 3.40 (m, 4H), 3.10 (s, 9H), 2.19 (m,
2H), 1.47 (m, 2H), 1.20 (br. d, 24H), 0.87 (t, 3H).
3C-NMR (CDC13) ~73.393, 138.453, 138.377, 138.051,
137.998, 128.470, 128.417, 128.356, 128.318, 128.235,
128.053, 128.007, 127.947, 127.856, 127.780, 127.719,
127.636, 127.567, 103.899, 84.472, 81.984, 77.478,
77.394, 77.311, 77.190, 75.672, 74.944, 74.550,
73.336, 68.663, 68.489, 59.158, 59.135, 54.409,
54.349, 34.246, 31.902, 29.710, 29.664, 29.535,
29.353, 29.330, 29.148, 24.7871 22.678, 14.121.
~2R~ D-glu~G~l~nos-1-yl-2-O-p~lmitoyl-3-O-
phosph~ti~ylcholine] glycerol 8P-19501. A solution of
phosphatidylcholine 18 (130 mg, 0.127 mmol) in reagent
grade methanol (25 mL) was hydrogenated at 60 psi over
10% Pd/C (52 mg, 40 wt%). After 23 h, TLC showed an
incomplete reaction. The catalyst was filtered off
through celite and the methanol washings were combined
and concentrated. The residue was dissolved in fresh
methanol (25 mL) and resubjected to hydrogenation at
60 psi over 240 mg (185 wt%) of 10% Pd/C. After 20 h,
the reaction was complete by TLC. The catalyst was
filtered off through celite and the methanol filtrate
and washings were combined and concentrated to afford
- 64.0 mg (76.6%? of (R) 8P-19501 as a white solid; IH
NMR (CDC13) ~ 5.19 (m, lH), 4.97 (s, OH + HDO), 4.34-
SUBSTITUTE SHEET
W094/08~63 2 1 4 6 ~ 3 g PCT/US93/09623
4.26 (br m, 2H), 4.16-3.95 (m, 3H), 3.9-3.6 (m, 6H),
3.42-3.14 (multiplet containing singlet at 3.24, 13H),
2.37 (t, J=7.6, 2H), 1.62 (pseudo t, 2H), 1.31 (m,
24H), 0.92 (t, J=7.2, 3H); l3C NMR (CD30D) 8175.02,
104.88, 78.07, 78.04, 75.04, 72.97, 72.89, 71.S2,
68.56, multiplet at 67.50, doublet at 64.99 and 64.94,
62.65, doublet at 60.52 and 60.48, triplet at 54.74
(J=3.1), 35.14, 33.13, 30.85, 30.69, 30.54, 30.29,
26.01, 23.79, 14.50; 31P NMR (CD30D) ~ 1.35
~2R)1-[Benzyl-~2'bromoethyl)-pho~phproyl]-2,3-
isopropylidene glycerol ~19).
2-Bromoethylphosphodichloridate (20.0 g, 0.08 mol) was
dissolved in CC14 (50 ml) in a nitrogen-purged 0.5 L
three-necked flask fitted with a magnetic stir bar,
thermometer, and a 125-ml addition funnel. The
solution was chilled to 0C, and to this stirred
solution was added dropwise the solution of (S)-form
solketal (10.7 g, 98 mol %) and N-methyl-morpholine
(8.22 g,98 mol %) in CCL4 (25 ml). After 2 hours TLC
showed disappearance of solketal. To the reaction
mixture was added dropwise the solution of benzyl
alcohol (44.6 g, 500 mol %) and N-methylmorpholine
(8.38 g, 100 mol %). The reaction mixture was stirred
under nitrogen for 60 hours at room temperature. TLC
showed the complete reaction. The reaction mixture
was filtered through Shott filter #C, and the solution
was rotary evaporated to volume near 70 ml and
purified by flash chromatography (silica gel, diethyl
ether) to give colorless oil (15.1 g, 0.04 mol) in 45
% yield; ~HNMR (CDCL3) ~ppm: 7.40 (br. s 5 H), 5.2 (d,
2 H), 4.3 (br.s, 3 H), 4.0 (br.s, 3 H), 3.85 (br.s,l
H), 3.2 (s, 2 H), 1.4 (d, 6H); 13C NMR (CDCL3):
- 128.743, 128.682, 1~8.645, 128.114, 128.076, 109.885,
77.364, 77.046, 76.727, 73.920, 73.837, 69.771,
Sl.l~ , JJTE SHEEr
W094/08~63 2 1 ~6 ~ PCT/US93/096
- 48 -
69.710, 67.760, 67.707, 67.654, 66.774, 66.721,
65.955, 29.353, 29.277, 26.-6~3, 25.204; 31p NMR
(CDCL3):-1.108. ~-
~2R)1-[Beuzyl-(2'-bromoethyl)-phosphoroyl] 1-2,3-
~ihydroxy glyc~rol (20). A nitrogen purged 1 L round-
bottomed flask fitted with septum was charged with
compound 19 (19.5 g, 0.048 mol) in dry THF (50 ml) and
the solution of 1 M H3PO4 (800ml) was added. The
reaction mixture was stirred under nitrogen by room
temperature for 15 hours. TLC showed the completness
of the reaction. Then the reaction mixture was
transferred to a 2 L sepapatory funnel. The acidic
layer was extracted with ethyl acetate (7 x 450 ml).
The combined organic extract was washed with water (2
x 850 ml). After drying over sodium sulfate it was
rotary evaporated and dryed in high vacuo for 10 hours
to give a colorless oil (14 g, 0.04 mol %) in 80 %
yield; IH NMR (CDCL3), ~ppm: 7.38 (br.s, 5H), 5.2 (d,
io 2H), 4.25-3.8 (multiplet, 6 H), 3.7-3.25 (br.m 5 H) l3C
NMR (CDCL3): 77.789, 77.774, 77.349, 77.030, 76.712,
70.522, 70. 491, 70.461, 70.431, 70.097, 70.044,
68.898, 68,883, 68.822,
67.085, 67.032, 62.617, 62.496, 42.363, 42.280; 31p NMR
(CDCL3): -0.485 (85 % H3PO4).
t2R)l-tBenzyl-~2~-bromoethyl)-pho~phoroyl-2-hydroxy-3
O-triphenylmethyl glycerol ~21). To a stirred
solution of diol 20 (8.0 g, 21.6 mmol) in DMF (16 nil)
was added diisopropylethylamine (4 ml, 105 mol %)
followed by addition of trityl chloride ( 6.4 g, 105
mol %). After 40 hours at room temperature under
nitrogen the reaction was complete by TLC. The
~ reaction mixture was diluted twice with water and
extracted with diethyl ether (4 x 100 ml). The
SUBSTITUTE SHEET
~ wo g4/08~63 ~ 1 4 6 6 3 ~ PCT/US93/09623
- 49 -
combined extract was dryed over sodium sulfate,
concentrated and purified by flash chromatography
silica gel, ethyl acetate:hexane,1:1) to give 21 as a
light oil 6.9 g (52.1 %); IH NMR (CDCL3) ~.2-7.5 (br.m,
20 h), 5.07 (t, 2H), 4.12-4.26 (m,4H), 3.44 (dd 2H),
2.05 (s, lH), 1.26 (t, lH). 13C NMR (CDCL3): 138.772,
124.017, 123.911, 123.820, 123.342, 123.266, 123-152,
122.462, 122.417, 72.593, 72.274, 71.955, 65.105,
65.090, 65.030, 65.014, 64.954, 64.893, 62.109,
62.056, 58.877, 24.680, 24.604. 31p NMR (CDCL3) -0.158
(S) -
t2R) 1-tBenzyl-t2'-bromoethyl)-phosphoroyl-2-O-
p~lmitoyl-3-0-triphenyl methyl glycerol ~22). To a
stirred solution of compound 21 (6.9 g, 11.3 mmol) in
dry THF (90 ml) was added triethylamine (1.79 ml, 1
lO,mol %), palmitic anhydride (6.13 g, 1 10 mol%) and
dimethylaminopyridine (276 mg, 20 mol%). The reaction
was stirred under nitrogen for 3 hours untill TLC
showed disappearance of the starting material 21. The
reaction mixture was rotary evaporated to a small
volume and purified by flash chromatography (silica
gel, diethyl ether:hexane, 1:3 to elute UV-nonactive
impurities, diethyl ether:hexane,1:1 to elute compound
22). Yield 8.8 g (92.6%0, colorless oil, IHNMR (CDCL3)
7.41-7.22 (m, 20 H), 5.20 (d, lH), 5.04 (t, 2H), 4.23
(m, 4H), 3.58 (s, lH), 3.41 (s, lH), 3.23 (s, 2H),
2.33 (t, 2H), 1.62 (m, 3H), 1.24 (s, 24H), 0.88 (t,
3H). 13C NMR 172.984, 143.407, 143.285, 128.675,
128.607, 128.576, 128.523, 127.985, 127.848, 127.180,
- 127.135, 86.672, 77.319, 77.000, 76.681, 70.901,
70.818, 69.604, 69.581, 66.463, 66.440, 61.828,
34.284, 31.894, 29.672, 29.634, 29.611, 29.437,
~~ 29.346, 29.285, 29.247, 29.232, 29.141, 29.095,
24.832, 22.678, 14.121. 32p NMR (CDCL3)-1.327.
SUBS~ITUTE SHEET
W094~08 ~ 1~ 6 6 3 9~ ~ r PCT/US93/096 ~
- 50 -
~2R)1-rBe~syl(2'- bromoethyl)-phosphoro~-yl-2-O-
palmtoyl-3-hy~roxy glycerol 23. Proce~ure A To a
stirred solution of compound 22 (3.2 g, 3.76 mmol) in
45 ml THF was added 45 ml 96~ formic acid. After 2
hours qt room temperature t~e rèaction was coplete by
TLC. The reaction mixture was diluted twice with
water, neutralized with sodium bicarbonate (3 x 300
ml). The combined extract was washed with water,
dryed over sodium sulfate, rotary evaporated to a
small volume and purified by flash chromatography
(silica gel, ethyl acetat: hexane, 1:3 to. elute less
polar impurities, ethyl acetat:hexane,1:1 to elute
compound 23. Yield 1.65 g (72.7 %), colorless oil.
Procedure B. A nitrogen purged 0.5 L round-bottomed
flask fitted with condenser was charged with compound
22 (1 g, 1.17 mmol) in dry benzene (230 ml) in the
presence of anhydrous CuS04 (17.6 g). The reaction
mixture was stirred at room temperature for 15 hours
and then reflux for 2 hours untill the reaction was
complete by TLC. The CuS04 was filtered off through
Shott filter #C and concentrated in vacuo and purified
by flash chromatography (silica gel, ethyl acetate/
hexane, 1:1) to give a light yellow oil (0.47 g, 0.77
mmol) in 66 % yield. IHNMR (CDCL3): ~7.40 (br. s, 5H),
5.2 (d, 2H), 4.2 (mult, 8H), 2.32 (t, lH), 1.62
(pseudo t, 2H ), 1.31 (m, 24H), 0.88 (t, 3H). 13C NMR
(CDCL3): 130.898, 128.872, 128.789, 128.698, 128.538,
128.S16, 128.114, 127.886, 126.968, 77.326, 77.008,
76.689, 70.097, 70.074, 69.012, 68.951, 68.633,
68,604, 68.572, 68.542, 67.085, 67.047, 67.032,
66.994, 65.272, 64.195, 62.731, 62.716, 34.041,
31.902, 29.672, 29.588, 29.505, 29.444, 29.338,
29.239, 29.103, 24.749, 22.670, 14.113. 31P NMR: -
3.069 (85% H3P04).
SUBST~TUTE SHEET
~ W094/08~63 2 1 4~ ~ 3 ~ PCT/US93/09623
- 51 -
~2R)-1-0-~2,3.~.6-T~tr~-O-b~nzyl-~-D-gluco-pyr~nosyl)-
2'-O-p~lmitoyl-3~-o-[benzyl!2~-bromoethyl)-
phosphoril]-glycQrol ~17). To a stirred solution of
o-(~ -D-glucopyranosyl) trichloroacetimidate (2~) (390
mg, 115 mol %) in dry methylene chloride (3 ml) was
added dropwise a solution of compound 6 (300 mg, 0.49
mmol) and boron trifluoride etherate (70 mg, 100 mol
%) in dry methylene chloride (3 ml). The reaction
mixture was stirred under nitrogen for 2 hours at room
temperature, then more compound 24 (100 mg, 35 mol %)
was added to bring the reaction to the end. After 4h,
the reaction mixture was evaporated to a small volume
and separated by flash chromatography (silica gel,
diethyl ether/hexane, 1:3) to give compound 17 as a
colorless oil (120 mg, 22%), which was identical to
the material described earlier.
6.4. Antifungal Activity
The antifungal activity of the isolated
phosphocholine fraction was determined in vitro by
using three fungal cultures - Candida albicans,
Cryptococcus neoformans and Aspergillus fumigatus.
The method used to determine in vitro antifungal
activity is discussed in McGinnis, M.R., Laboratory
Handbook of Medical Mycology, Academic Press, New
York, London, p661 (1980); and Droughet E., Dupont,
B., Improvisi, L., Vivian, M.A. and Tortorano, A.M.,
"Disc agar diffusion and microplate automatized
tec-hnics for in vitro evaluation of antifungal agents
on yeast and sporulated pathogenic fungi" in In Vitro
and In Vivo Evaluation of Antifungal Agents, Eds.
Iwata, K. and Vanden Bossche, H., Elsevier Science
Publishers, New York, Oxford p303 (1986).
SUBSTITUTE SHEET
21~6~39
W094/08~63 ' ~ PCT/US93/0962
- 52 -
The minimum ihhibitory concentration (MIC) and
the minimum fungicidal concentration (~FC) are
summarized in the table 1 below.
Fungus Culture MIC (uq/ml) MFC fuq/ml)
C. albicans 0.8
C. neoformans < 0.1
A. fumigatus < 0.1 ~0.4-0.8
These results clearly indicate the significant
antifungal activity of the isolated fraction
containing against a variety of fungal cultures.
6.5. Antifunqal Activities of the Phosphocholine
4erivatives Class
A series of related analogs to 2-palmitoyl-1-0-
glucopyranosyllysolecithin obtained commercially from
Avanti Biolipids have also been found to have high
antifungal activities. A summary of the antifungal
screening test is shown in table 2. The analog
compounds were tested for their activity against
C. albicans, C. neoformans, A. fumigatus and T.
rubrum. Partial inhibition of the fungus of between
25 to 75% was measured along with the total inhibition
(MIC) by these anolog compounds. A description of the
partial inhibition measurement can be found in R. L.
Stiller, et al The Journal of Infectious Diseases,
147, No. 6 (1983). The structure of these analog
compounds is as follows.
SUBSTITUTE SHEET
~ W094/0~63 2 ~ 4 6 6 ~ ~ PCT/US93/09623
- 53 -
H
R0 \ CH / \ CH / \ p / \ /
wherein R is the group identified in table 2.
SUBSmUTE SHEEI
214663g
WO 94/08~63 ' 5 4 PCI/US93/096~
o o o
o o
,C E~
., o ` o o
o - o ~ In
Z _ o o ,~
-1 ~ V
_ o o
o o
U --~ ~
o o o
o o o U~
o
o o o o
,,~ o o o ~ U~ U~
V ~ ~ ~ ~ ~ ~1 ~1
:s$
o `t
o o
o o U~ U~
0 z .D --I ~ '
_~ V ,
!~
0
~;
,~ o o o
~ o o o U~
E-~ V ~ ~
O ~ ~ C
.,1 ~ .,~ .~ .,1 ,1
O _ r o O O O
.C o ~ ~ U ~ ~
~: ~ V ~ ~ ~ ~ o ~ _
~ ~ ~ ~ a~ ~ o ~ o J ~ ~
--I O ~ O _, 0 0 ~ V P~ , ~ ; V
r a~ 0 v ~ ~ ~
C ~ ~ C o -- C _I C
E~ s ~ _ O ~' --I ~ ~ ~1 _ --I
l o
j o
~q ~ ~q ~ o ~, ~ rq
a ~ ~ ~ x ~ ~
U~ o U~ o U~
~1 ~I N
SUBSTITIJTE SHEET
21~G639
WO 94/08563 5 5 ~ PCI/US93/09623
o
_I
~o A
a
.
o
o
_I
o
o
,~
o o
,1 o ,1
o
~D ~ ~1
,1 ,1,i
.. , ...
o a~ o
I ~
o ~ o
5 o
.,, ._ . ~ . . .,1
o t~ ~ ~ o
s~ ~ o~ o
O ~
o
o
a o ~ ~ ~ ~'
Ul o U~ o U~
,1 .
SUBSTITUTE SHEET
21~-G3~
W094/08~63 ~ ~ PCT/US93/096
- 56 -
6.6. ToxicitY
The toxicity of the isolated phosphocholine
derivative fraction is low, based~on tests with Hep 2
cells indicating an ID50 of greater than 1000 ug/ml.
The method used in determining cytotoxicity is
discussed in Mosmann, T., "Rapid colorimetric assay
for cellular growth and survival: application to
proliferation and cytotoxicity assays", J. Immun.
Methods, 65, 55-63, 1986.
The isolated fraction having the above-described
in vitro antifungal activity and low toxicity is
expected to similarly exhibit significant in vivo
antifungal activity against fungal infections which
are dermatophytic, systemic, ophthalmic and vaginal.
Other human and animal infections treatable with the
compounds of the present invention include
aspergilliosis, candidiasis, and cryptococcus
infections.
It is expected that the same isolated fraction
would be useful in treating fungal infestation in
plants as well.
It is apparent that many modifications and
variations of this invention may be made without
departing from the spirit and scope thereof. The
specific embodiments described are given by way of
example only and the invention is limited only by the
terms of the appended claims.
A number of references are cited in the present
specification, the entire disclosure of each of which
is incorporated by reference herein, in its entirety.
SUBSTITUTE SHEET