Note: Descriptions are shown in the official language in which they were submitted.
WO 94/08995 2 14 6 9 2 3 PCI~/EP93/02809
- 1 -
HETEROCYCLIC CONDENSED BENZOIC ACID DERIVATIVES AS 5-HT4 RECEPTOR ANTAGONISTS
This invention relates to novel compounds having pharrnacological activity, to
a process for their preparation and to their use as pharmaceuticals.
European Joumal of Pharmacology 146 (1988), 187-188, and
Naunyn-Schmiedeberg's Arch. Pharrnacol. (1989) 340:403-410, describe a non
classical 5-hydroxytryptamine receptor, now designated the 5-HT4 receptor, and that
ICS 205-930, which is also a 5-HT3 receptor antagonist, acts as an antagonist at this
receptor. WO 91/16045 (SmithKline and French Laboratories Limited) describes theuse of cardiac 5-HT4 receptor antagonists in the treatment of atrial arrhythmias and
stroke.
EP-A-501322 (Glaxo Group Limited) describes indole derivatives having
5-HT4 antagonist activity.
WO 93/02677, WO 93/03725, WO 93/05038, WO 93/05040 and
PCT/GB93/00506 (SmithKline Beecham plc) describe compounds having 5-HT4
receptor antagonist activity.
EP-A-234872 (Adria Laboratories Inc.) and EP-A-493041 (Erabomont Inc.),
describe benzobicyclic carboxamides. EP-A-339950 (Rorer International Overseas
Inc.) describes dibenzofurancarboxamides as 5-HT3 receptor antagonists.
WO 92/09284 describes a process for preparing multicyclic oxy-containing ring
system compounds as 5-HT3 receptor antagonists.
It has now been discovered that certain novel compounds also have 5-HT4
receptor antagonist properties.
When used herein, 'treatment' includes prophylaxis as appropriate.
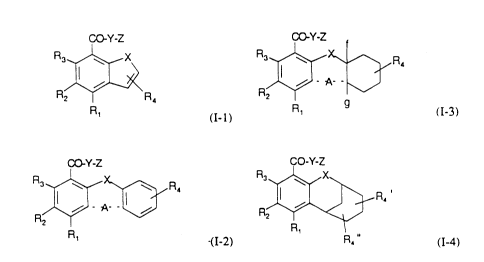
Accordingly, the present invention provides compounds of formula (I),
wherein formula (I) consists of forrnulae (I-1) to (I-4), and pharrnaceutically
acceptable salts thereof, and the use of a compound of formula (I) or a
pharmaceutically acceptable salt thereof:
CO Y-Z
R3~X
R2 f-- R4
R1
(I-l)
wherein
XisOorS;
R I is hydrogen, amino, halo, C 1-6 alkyl, hydroxy or C 1-6 aL~coxy;
WO 94/~63 ~3 2 - PCI~/EP93/02809
R2 is hydrogen, halo, Cl 6 aL~cyl, Cl 6 allcoxy, nitro, amino or C1 6 alkylthio; R3 is hydrogen, halo, Cl 6 aLkyl, C 1-6 alkoxy or amino;
R4 is hydrogen or C 1-6 aL~cyl;
CO Y-Z
$~A~3
R,
(I-2)
wherein
X is O or S;
A represents a single bond, -CH2-, or CO or A is (CH2)a-E-(CH2)b where one of a
and b is 0 and the other is 0 or l and E is O, S or NH;
R 1 is hydrogen, amino, halo, C 1-6 aL~I, hydroxy or C 1-6 alkoxy;
R2 is hydrogen, halo, C 1-6 alkyl, C 1-6 aL~coxy, nitro, amino or C 1-6 alkylthio;
R3 is hydrogen, halo, C 1-6 alkyl, C 1-6 alkoxy or amino;
R4 is hydrogen or C1 6 alkyl;
CO Y-Z
R2~R4
Rl 9
(I-3)
wherein
XisOorS;
A represents a single bond, -CH2-, or CO or A is (CH2)a-E-(CH2)b where one of a
and b is 0 and the other is 0 or l and E is O, S or NH;
20 f and g are both hydrogen or together are a bond;
R1 is hydrogen, amino, halo, C 1-6 allryl, hydroxy or C 1-6 aLkoxy;
R2 is hydrogen, halo, Cl 6 aLkyl, Cl 6 aL~coxy, nitro, amino or Cl 6 alkylthio;
R3 is hydrogen, halo, C 1-6 alkyl, C 1-6 aLcoxy or amino;
R4 is hydrogen or C1 6 alkyl;
CO-Y-Z
R3 ~ X~
ll I ~ R4
R2~
R, R4"
(1-4)
WO 94~08995 2 1 4 6 9 2 3 PCI/~W3/02809
wherein
XisOorS;
R1 is hydrogen, amino, halo, C 1-6 alkyl, hydroxy or C 1-6 alkoxy;
R2 is hydrogen, halo, Cl 6 aLkyl, Cl 6 alkoxy, nitro, amino or Cl 6 alkylthio;
5 R3 is hydrogen, halo, C 1-6 alkyl, C1 6 alkoxy or amino;
R4 and R4 are independently hydrogen or Cl 6 alkyl;
In formulae (I-1) to (1-4) inclusive:
Y is O or NH;
Z is of sub-formula (a), (b) or (c):
(CH2)q
~(CH2)n~_ I--R6
R
(a)
-(CH2) 2~cH2)m
(b)
-(CH2)n3 --N
Rg
(c)
wherein
nl isO, 1,2,30r4;n2isO, 1,2,30r4;n3is2,3,40rS;
qisO,1,20r3;pisO,lor2;misO,lor2;
Rs is hydrogen, C1 12 alkyl, aralkyl or Rs is (CH2)Z-Rlo wherein z is 2 or 3
and Rlo is selected from cyano, hydroxyl, Cl 6 alkoxy, phenoxy,
C(O)CI 6 alkyl, COC6Hs, -CONR1 lR12, NRl lCOR12,
S2NRl lR12 or NR1 1S2R12 wherein Rl 1 and R12 are hydrogen
or C 1-6 alkyl; and
R6, R7 and R8 are independently hydrogen or C 1-6 alkyl; and
Rg is hydrogen or Cl lo alkyl;
or a compound of formula (I) wherein the CO-Y linkage is replaced by a
heterocyclic bioisostere;
in the manufacture of a medicament having 5-HT4 receptor antaoonist activity.
WO 94/0899~ 6 ~ ~ 3 PCr/EP93/02809
Examples of alkyl or alkyl containing groups include C 1, C2, C3, C4, Cs, C6,
C7, Cg, Cg, C 10~ C 11 or C 12 branched, straight chained or cyclic alkyl, as
appropriate. C 1-4 alkyl groups include methyl, ethyl, n- and iso-propyl, n-, iso-, sec-
and tert-butyl. Cyclic alkyl includes cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl, cycloheptyl and cyclooctyl.
Aryl includes phenyl and naphthyl optionally substituted by one or more
substituents selected from halo, Cl 6 alkyl and Cl 6 alkoxy.
Halo includes fluoro, chloro, bromo and iodo.
In formula (I-1):
R1 is preferably hydrogen or amino.
R2 is preferably hydrogen or halo.
R3 is preferably hydrogen or halo.
R4 is often hydrogen.
In formula (I-2):
Rl is preferably hydrogen or amino.
R2 is preferably hydrogen or halo.
R3 is preferably hydrogen or halo.
R4 is often hydrogen.
In formula (I-3):
Rl is preferably hydrogen or amino.
R2 is preferably hydrogen or halo.
R3 is preferably hydrogen or halo.
R4 is often hydrogen.
In formula (I-4):
Rl is preferably hydrogen or amino.
R2 is preferably hydrogen or halo.
R3 is preferably hydrogen or halo.
R4 and R4 are often hydrogen.
A suitable bioisostere for the amide or ester linkage containing Y in formula
(I), is of formula (d):
WO 94/08995 2 I 1 6 9 2 3 PCr/EP93/02809
H--J
~,~U
(d)
wherein
5 the dotted circle represents one or two double bonds in any position in the
5-membered ring; H, J and I independently represent oxygen, sulphur,
nitrogen or carbon, provided that at least one of H, J and I is other than
carbon; U represents nitrogen or carbon.
Suitable examples of (d) are as described for X, Y and Z in EP-A-328200
(Merck Sharp & Dohme Ltd.), such as an oxadiazole moiety.
Y is preferably O or NH.
When Z is of sub-formula (a), nl is preferably 2, 3 or 4 when the azacycle is
attached at the nitrogen atom and n l is preferably 1 when the azacycle is attached at a
carbon atom, such as the 4-position when q is 2.
When Z is of sub-formula (b), n2 is preferably such that the number of carbon
atoms between the ester or amide linkage is from 2 to 4 carbon atoms.
Suitable values forp and m include p = m = l; p = 0, m = 1, p = 1, m = 2,
p=2,m = l.
When Z is of sub-formula (c), n3 is preferably 2, 3 or 4.
R8 and Rg are preferably both aL~yl, especially one of R8 and Rg is C4 or
larger aL~cyl.
Specific values of Z of particular interest are as follows:
\_/
(i)
/~ N /V~
(ii)
`--N~
(iii)
WO 94/0899~, ~ 4 6 9 23 - 6 - PCI~/EP93/02809
~NMe Bu (iv)
~~N~ / (v)
~N~
~J
(vi)
/~ N~
(vii)
The invention also provides novel compounds within formula (I) with side
5 chains (i), (ii), (iii), (iv), (v), (vi) or (vii). In a further aspect, the piperidine ring in
(i), (ii) or (iii) may be replaced by pyrrolidinyl or azetidinyl, and/or the N-substituent
in (i) or (ii) may be replaced by C3 or larger alkyl or optionally substituted benzyl.
In an alternative aspect, the N-substituent in forrnula (i) or (ii) may be
replaced by (CH2)nR4 as defined in formula (I) and in relation to the specific
examples of EP-A-501322.
The pharmaceutically acceptable salts of the compounds of the formula (I)
include acid addition salts with conventional acids such as hydrochloric,
hydrobromic, boric, phosphoric, sulphuric acids and pharmaceutically acceptable
organic acids such as acetic, tartaric, maleic, citric, succinic, benzoic, ascorbic,
15 methanesulphonic, a-keto glutaric, a-glycerophosphoric, and glucose-1-phosphoric
acids.
Examples of pharTn~reu~ic~lly acceptable salts include quaternary derivatives
of the compounds of formula (I) such as the compounds quaternised by compounds
RX-T wherein Rx is C 1-6 alkyl, phenyl-C 1-6 alkyl or Cs 7 cycloalkyl, and T is a
20 radical corresponding to an anion of an acid. Suitable examples of Rx includemethyl, ethyl and n- and ~so-propyl; and benzyl and phenethyl. Suitable examples of
T include halide such as chloride, bromide and iodide.
Examples of pharmaceutically acceptable salts also include internal salts such
as N-oxides.
WO 94/08995 Z I ~ 6 9 2 3 PCI`/EP93/02809
-- The compounds of the formula (I), their pharmaceutically acceptable salts,
(including quaternary derivatives and N-oxides) may also form pharmaceutically
acceptable solvates, such as hydrates, which are included wherever a compound offormula (I) or a salt thereof is herein referred to.
S The compounds of formula (I) wherein CO-Y is an ester or amide linkage are
prepared by conventional coupling of the Z moiety with the appropriate acid.
Suitable methods are as described in GB 2125398A (Sandoz Limited), GB
1593146A, EP-A-36269, EP-A-289170 and WO 92/05174 (Beecham Group p.l.c.).
When CO-Y is replaced by a heterocyclic bioisostere, suitable methods are described
in EP-A-328200 (Merck Sharp & Dohme l.imitPd). Reference is also made to
EP-A-501322 (Glaxo Group T.imitPd).
The invention also comprises a process for preparing the novel compounds of
formula (I) which comprises reacting an appropriate acid derivative with an
appropriate alcoho! or amine. A process comprises reacting an acid derivative
wherein the aromatic substituents are as required in the end compound of formula (I),
or substituents convertible thereto, with an alcohol or amine containing Z or a group
convertible thereto, and thereafter if necessary, converting the benzoic acid
substituents and/or Z, and optionally forming a pharmaceutically acceptable salt.
Suitable examples of conversions in the aromatic substituents include
chlorination of hydrogen to chloro, reduction of nitro to amino, dehydrohalogenation
such as debromination. Any elaboration is, however, usually carried out prior to ester
or amide coupling.
Suitable examples of conversions in the Z containing moiety include
conventional modifications of the N-substituent by substitution and/or deprotection
or, in the case of a 2-, 3- or 4- substituted piperidinyl desired end compound,
reduction of an appropriate pyridyl derivative.
The compounds of the present invention are 5-HT4 receptor antagonists and it
is thus believed may generally be used in the treatment or prophylaxis of
gastrointestinal disorders, cardiovascular disorders and CNS disorders.
They are of potential interest in the tre~tment of irritable bowel syndrome
(IBS), in particular the diarrhoea aspects of IBS, i.e., these compounds block the
ability of 5-HT to stimulate gut motility via activation of enteric neurones. In animal
models of IBS, this can be conveniently measured as a reduction of the rate of
defaecation. They are also of potential use in the treatment of urinary incontinence
which is often associated with IBS.
They may also be of potential use in other gastrointestinal disorders, such as
those associated with upper gut motility, and as antiemetics. In particular, they are of
potential use in the treatment of the nausea and gastric symptoms of gastro-
WO 94/08995 ~14 6 9 2 3 8 - PCr/EP93/0~809
oesophageal reflux disease and dyspepsia. An~i~mPtic activity is determined in
known animal models of cytotoxic-agentlradiation induced emesis.
Specific cardiac 5-HT4 receptor antagonists which prevent atrial fibrillation
and other atrial arrhythmias associated with 5-HT, would also be expected to reduce
5 occurrence of stroke (see A.J. Ec~llm~nn 1990, Naumyn-Schmiedeberg's Arch.
Pharmacol. 342, 619-622, for appropriate animal test method).
Anxiolytic activity is likely to be effected via the hippocampus (Dumuis et al
1988, Mol Pharmacol., 34, 880-887). Activity can be demonstrated in standard
animal models, the social interaction test and the X-maze test.
Migraine sufferers often undergo situations of anxiety and emotional stress
that precede the appearance of he~d~che (Sachs, 1985, Migraine, Pan Books,
London). It has also been observed that during and within 48 hours of a migraineattack, cyclic AMP levels are considerably increased in the cerebrospinal fluid
(Welch e~ al., 1976j Headache 16, 160-167). It is believed that a migraine, including
the prodomal phase and the associated increased levels of cyclic AMP are related to
stimulation of 5-HT4 receptors, and hence that ~dminic~ration of a 5-HT4 antagonist
is of potential benefit in relieving a migraine attack.
Other CNS disorders of interest include schizophrenia, Parkinson's disease and
Huntingdon's chorea.
The invention also provides a pharmaceutical composiLion comprising a
compound of formula (I), or a pharrnaceutically acceptable salt thereof, and a
pharmaceutically acceptable carrier.
Such compositions are prepared by admixture and are usually adapted for
enteral such as oral, nasal or rectal, or parenteral adminisLration, and as such may be
in the form of tablets, capsules, oral liquid preparations, powders, granules, lozenges,
reconstitutable powders, nasal sprays, suppositories, injectable and infusable solutions
or suspensions. Orally ~minicLrable compositions are preferred, since they are more
convenient for general use.
Tablets and capsules for oral a~minictration are usually presented in a unit
dose, and contain conventional excipients such as binding agents, fillers, diluents,
tabletting agents, lubricants, disintegrants, colourants, flavourings, and wetting
agents. The tablets may be coated according to well known methods in the art, for
example with an enteric coating.
Suitable fillers for use include cellulose, mannitol, lactose and other similar
agents. Suitable disintegrants include starch, polyvinylpolypyrrolidone and starch
derivatives such as sodium starch glycollate. Suitable lubricanLs include, for
example, m~gnesium stearate.
WO 94/08995 21 4 6 9 2 3 PCI`/EP93/02809
g
Suitable pharrn~reuti~lly acceptable wetting agents include sodium lauryl
sulphate. Oral liquid preparations may be in the form of, for example, aqueous or
oily suspensions, solutions, emulsions, syrups, or elixirs, or may be presented as a dry
product for reconstitution with water or other suitable vehicle before use. Such liquid
5 preparations may contain conventional additives such as suspending agents, forexample sorbitol, syrup, methyl cellulose, gelatin, hydroxyethylcellulose,
carboxymethylcellulose, aluminium stearate gel or hydrogenated edible fats,
emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous
vehicles (which may include edible oils), for example, almond oil, fractionated
10 coconut oil, oily esters such as esters of glycerine, propylene glycol, or ethyl alcohol;
preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and if
desired conventional flavouring or colouring agents.
Oral liquid preparations are usually in the form of aqueous or oily
suspensions, solutions, emulsions, syrups, or elixirs or are presented as a dry product
15 for reconstitution with water or other suitable vehicle before use. Such liquid
preparations may contain conventional additives such as suspending agents,
emulsifying agents, non-aqueous vehicles (which may include edible oils),
preservatives, and flavouring or colouring agents.
The oral compositions may be prepared by conventional methods of blending,
20 filling or tabletting. Repeated blending operations may be used to distribute the
active agent throughout those compositions employing large quantities of fillers.
Such operations are, of course, conventional in the art.
For parenteral adminictration, fluid unit dose forms are prepared containing a
compound of the present invention and a sterile vehicle. The compound, depending25 on the vehicle and the concentration, can be either suspended or dissolved. Parenteral
solutions are normally prepared by dissolving the compound in a vehicle and filter
sterilisino before filling into a suitable vial or ampoule and sealing. Advantageously,
adjuvants such as a local ~n~esth~tic, preservatives and buffering agents are also
dissolved in the vehicle. To enh~n~e the stability, the composition can be frozen after
30 f1lling into the vial and the water removed under vacuum.
Parenteral suspensions are prepared in substantially the same manner except
that the compound is suspended in the vehicle instead of being dissolved and
sterilised by exposure of ethylene oxide before suspending in the sterile vehicle.
Advantageously, a surfactant or wetting agent is included in the composition to
35 facilitate uniform distribution of the compound of the invention.
W0 94/08995 6 9 ~ 3 1 o - PCI~/EP93~02809
The invention further provides a method of tre~tment or prophylaxis of
irritable bowel syndrome, dyspepsia, atrial arrhythmias and stroke, anxiety and/or
migraine in m~mn-~lc, such as humans, which comprises the ~rlminictration of an
effective amount of a compound of the formula (I) or a pharmaceutically acceptable
salt thereof.
An amount effective to treat the disorders hereinbefore described depends on
the relative efficacies of the compounds of the invention, the nature and severity of
the disorder being treated and the weight of the mamm~l However, a unit dose for a
70kg adult will normally contain 0.05 to lOOOmg for example 0.5 to SOOmg, of thecompound of the invention. Unit doses may be ~dmini.ctered once or more than once
a day, for example, 2, 3 or 4 times a day, more usually 1 to 3 times a day, that is in
the range of applo~ ately 0.0001 to 50mg/kg/day, more usually 0.0002 to 25
mg/kg/day.
No adverse toxicological effects are indicated within the aforementioned
dosage ranges.
The invention also provides a compound of formula (I) or a pharmaceutically
acceptable salt thereof for use as an active therapeutic substance, in particular for use
in the treatment of irritable bowel syndrome, gastro-oesophageal reflux disease,dyspepsia, atrial arrhythmias and stroke, anxiety and/or migraine.
The following Examples illustrates the preparation of compounds of formula
(I), and the following Descriptions relate to the preparation of intermediates. The
compounds of formula (I-1) and interrnediates are prepared in Examples and
Descriptions 1-1, 2-1 etc, the compounds of formula (I-2) are prepared in Examples
and Descriptions 1-2, 2-2 etc and similarly for the compounds of formulae (I-3) to
(I-4).
It will be appreciated that any compound prepared wherein Y is O may be
provided as the corresponding compound wherein Y is NH.
A preferred compound corresponds to any of the compounds prepared in the
Examples, but wherein there is an amino substituent in the 4-position and a chloro
substituent in the S-position of the benzoic acid nucleus depicted in formulae (I-1) to
(I-4) inclusive.
WO 94/08995 2 1 4 6 9 2` 3 Pcr
Description 1-1 (intermedi~e for Example 1-1)
a) Methyl q rr~ ino-3-allyl-S-chloro-2-hydroxyberl7O~te
A mixture of methyl 4-acetylamino-5-chloro salicylate (EP-A-0339950),
(17.0g, 0.070 mol), allylbromide (6.32 ml, 0.073 mol), acetone (350 ml) and
S potassium carbonate (19.35g, 0.140 mol) was heated to reflux with stirring. After
23h, the reaction mixture was allowed to cool, was filtered, and the filtrate evaporated
under reduced pressure and dried in vacuo to give a pale brown solid. The solid was
redissolved in 1,2-dichlorobenzene (300 ml) and was heated to reflux with stirring.
After 24h the reaction mixture was allowed to cool, and was evaporated under
reduced pl~s~ule. The resultant semi-solid brown residue was then purified by silica
gel chromatography (2: 1 Pentane: EtOAc ~ EtOAc as eluant) to give the title
compound (8.24g, 42%) as a yellow solid.
lH NMR (200 MHz, CDC13) ~:
11.10 (s, lH), 7.81 (s, lH), 7.04 (s, lH), 5.88 (m, lH), 5.00 (m, 2H), 3.95 (s, 3H),
3.45 (d, 2H), 2.25 (s, 3H).
b) Methyl-4-acetylamino-5-chloro-2-hydroxy-3-(2-oxoethyl) benzoate
The product from a) (8.23g, 0.029 mol) was dissolved in acetone (300 ml) and
water (60 ml), treated with N-methylmorpholine-N-oxide (6.79g, 0.058 mol),
followed by 4% wt osmium tetroxide in water (1.82 ml, 0.0029 mol) and stirred atroom temperature overnight. After 21 h, 10% sodium bisulphite solution (100 ml)
was added, and the mixture was stirred for 1/2 h, before the acetone was evaporated
under reduced pressure. The reaction mixture was then partitioned between EtOAc
and water. The aqueous layer was then extracted with EtOAc (2X), and the combined
orgarlic layers were dried (Na2SO4) and evaporated under reduced pressure to give
an off white solid, which was dried in vacuo. The solid was then redissolved in
methanol (250 ml) and treated with a solution of sodium periodate (9.41g, 0.044 mol)
in water (60 ml) with stirring. The mixture was then stirred at room temperatureovernight, before the methanol was removed in vacuo. The residue was then
partitioned between EtOAc and water. The aqueous layer was then extracted with
EtOAc, and the combined organic layers were dried (Na2SO4) and evaporated under
reduced pressure to give a dark brown oil. The oil was then purified by the silica-gel
chromatography (EtOAc as eluant) to give the ti~le compoulld as a brown foam
(5.90g, 71%)
lH NMR (200 MHz, CD30D) ~:
8.00 (s, lH), 4.92 (t, lH), 4.10 (s, 3H), 3.12 (d, 2H), 2.33 (s, 3H)
2 ~ 46 9 ~3 PCr/EP93/02809
WO 94/08995
- 12-
c) 2-Acetoxy-7-carbomethoxy-~-chloro-4-diacetvlamino-2,3-
dihydrob~ ofu~a~
The product from b) (5.90g, 0.021 mol) was dissolved in a mixture of acetic
anhydride (55 ml) and pyridine (55 ml), a few crystals of 4-dimethylaminopyridine
were added, and the mixture was stirred at room temperature overnight. After 20h,
the reaction mixture was partitioned between EtOAc and wa~r. The aqueous layer
was then extracted with EtOAc, and the combined organic la~ers were dried
(Na2S04), and evaporated under reduced pressure to ~ive a t rown oil, which was
dried in vacuo. The oil was then purified by silica-gel chromatography (2: 1
Pentane:EtOAc as eluant) to give the ti~le compound as a pale brown oil. (2.87g,37%)
lH NMR (250 MHz, CDC13), ~:
8.00 (s, lH), 7.00 (dd, lH), 3.92 (s, 3H), 3.38 (dd, lH), 3.00 (dd, lH), 2.32 (s, 3H),
2.28 (s, 3H), 2.10 (s, 3H).
d) 7-Carbomethoxy-5-chloro-4-diacetylaminobenzo[b]furan
The product from c) (2.87g, 7.77 mmol) was dissolved in trifluoroacetic acid
(50 ml) and heated to reflux with stirring. After lh, the reaction mixture was
allowed to cool and was evaporated under reduced pressure. The residue was
partitioned between aq. NaHCO3 and dichloromethane. The aqueous layer was then
extracted with dichlorometh~n~ (2x), and the combined organic layers were dried
(Na2SO4), and evaporated under reduced pressure to ~ive a brown oil, which was
purified by silica-gel chromatography (1: 1 Petrol: diethylether as eluant) to give the
title compound as a pale yellow oil (0.765g, 32%).
lH NMR (250 MHz, CDC13), ~:
8.10 (s, lH), 7.81 (d, lH), 6.70 (d, lH), 4.10 (s, 3H), 2.30 (s. 6H).
Description 2-1 (intermediate for Example 2-1)
a) (2-Carboxyphenylthio) Acetic Acid
A solution of thiosalicylic acid (10.Og; 64.9 mmol) in H2O (200 ml)
cont~ining sodium carbonate (34.5g; 0.32 mol) was trealed ~-ith sodium chloroacetate
(7.56g; 64.9 mmol) in H2O (100 ml). The whole was heated at reflux (2 hours),
cooled and acidified to pH2 with c.HCI. The material that crvstallised was collected
by filtration and dried in vacuo to yield the tifle compound as an orange powder(12.5g; 91%)
lH NMR (250MHz; CD3SOCD3) ~:
13.10 - 12.85 (bs, 2H), 7.90 (d, lH), 7.50 (t, lH), 7.35 (d, lH), 7.20 (t, lH), 3.80 (s,
2H)
WO 94/08995 2 ~ ~ PCr/EP93/02809
- 13-
b) Thioindoxyl-7-carboxylic Acid
(2-Carboxyphenylthio)acetic acid (6.5g; 30.66 mmol) was heated at reflux in
thionyl chloride (45 ml) for 1 hour, cooled, evaporated in vacuo and the residueazeotroped with toluene. The residue was redissolved in 1,2-dichlorobenzene (8.0ml) and treated with aluminium chloride (8.18g; 61.3 mmol) portionwise. The whole
was heated to 45-50 C (1 hour) then treated with ice and sodium hydroxide until the
mixture was basic. The aqueous layer was separated, extracted with die~hyl ether and
then acidified with cHCI to pH 1 and left to stand. The precipitate that formed was
collected by filtration and dried in vacuoto yield the ti~le compound as a red powder
(2.45g; 41%)
lH NMR (250MHz; CD3SOCD3) ~:
8.05-7.95 (m, 2H), 7.50 (t, lH), 6.55 (s, lH)
c) Benzothiophene-7-carboxylic Acid
A solution of thioindoxyl-7-carboxylic acid (0.3g; 1.55 mmol) in glacial
acetic acid (Sml) was treated with zinc amalgam (made from zinc dust (1.14g) and the
whole heated at reflux (18h), cooled, filtered through kieselguhr and the filtrate
evaporated in vacuoto yield the ti~le compound and 2,3-dihydrobenzothiophene-7-
carboxylic acid (1: 1) as a red solid (0.152g; 55%).
lH NMR (250 MHz, CD3SOCD3) ~:
8.15 (d, lH), 8.05 (d, lH), 7.85 (d, lH), 7.50 (t, 2H)
Example 1-1 [R1 =NH2,R2=Cl,R3=H,R4=H,X=O,Y=NH,Z=(i)]
(l-Butyl-4-piperidinylmethyl)-4-amino-5-chloro-benzo[b]furan-7-carboxamide
The product from Description I (0.765g, 2.47mmol) was dissolved in a
mixture of 10% sodium hydroxide (15 ml) and ethanol (15 ml). The reaction mixture
was then heated to reflux. After 23 h, the reaction mixture was allowed to cool. The
ethanol was then removed by evaporation under reduced pressure and the aqueous
residue acidifled to pH2 using c. HCl. The resulting grey solid was then filtered off
and dried in vacuo. The solid was then suspended in a mixture of acetonitrile (10 ml)
and DMF (10 ml) and was treated with l,l,carbonyldiimidazole (0.440g, 2.71 mmol)with stirring. After 20h, the reaction mixture was evaporated under reduced pressure
and dried in vacuo. The crude imidazolide was then suspended in dry THF (20 ml)
and N-butyl-4-piperidinylmethylamine (0.461g, 2.71 mmol) (W093/05038) in dry
THF (5 ml) was added. The mixture was then heated to reflux under argon. After 4h, the reaction mixture was allowed to cool and was evaporated under reduced
- pr~ssu,~. The residue was partitioned between EtOAc and 10% NaOH. The aqueous
layer was then extracted with EtOAc, and the combined organic layers were dried
(Na2SO4) and evaporated under reduced pressure to give a brown solid. which was
WO 94/08995 2l 46S ?~3 PCI`/EP93/02809
- 14 -
purified by silica-gel chromatography (20% MeOH/EtOAc as eluant) to give the ti~le
compound as a white foam (0.425g, 46%)
m.pt 88-89 C (from CH2C12/60-80 petrol)
lH NMR (200MHz, CDC13), ~:
S 8.04 (s, lH), 7.64 (d, lH),7.35 (brt, lH), 6.81 (d, lH), 4.71 (s, 2H), 3.43 (t, 2H), 2.97
(d, 2H), 2.33 (t, 3H), 2.05 - 1.60 (m, SH), l.SS - 1.20 (m, SH), 0.91 (t, 3H).
F.Y~mpl~2.1 [Rl =H,R2=H,R3=H,R4=H,X=S,Y=O,Z=(i)]
(1 -Buty1-4-pipe~dinylmethyl)benzothiophene-7~carboxylate
A 1:1 mixture of benzothiophene-7-carboxylic acid and
2,3-dihydrobenzothiophene-7-carboxylic acid (0.262g; 1.46 mmol) was dissolved indry DMF (Sml) and treated with l,l-carbonyldiimidazole (0.161g; 1.61 mmol). The
mixture was stirred (72 hours). N-butyl~-piperidinylmethanol ~WO 93t05038)
(0.275g; 1.61 mmol) was dissolved in dry THF (10 ml) under Ar and treated with
methyllithium (1.18 ml of a l.SM solu~ion in Et2O; 1.77 mmol) then stirred for 15
minutes. This was treated with the solution of imidazolides and the whole stirred (72
hours). Evaporated in vacuo and partitioned H2O/EtOAc. The organic layer was
separated, dried over Na2SO4 and filtered, then the filtrate evaporated in vacuo to an
orange oil. The oil was purified by flash silica-gel chromatography and eluted with
CHC13 ~ 3% MeOH/CHC13 to yield a brown oil which was purified by HPLC
separation to y ield the title compound as a clear gum (0.009g; 2%).
lH NMR (250 MHz; CDC13) ~:
8.15 (d, lH), 8.05 (d, lH), 7.60 (d, lH), 7.30 (d, lH), 7.05 (t, lH), 4.30 (d, 2H),
3.05-2.95 (m, 2H), 2.35 (t, 2H), 2.00-1.75 (m, SH), 1.60-1.25 (m, 6H), 0.90 (t, 3H).
Example 3-1 [R 1 = H, R2 = Cl, R3 = H, R4 = H, X = O, Y = O, Z = (i)]
(1-Butv1-4-piperidinylmethyl)-S-chlorobenzo[b]furan-7-carboxylate
S-Chlorobenzo[b]furan-7-carboxylic acid [US patent 4888353; 33f] (0.5g;
2.54 mmol) was suspended in thionyl chloride (20 ml) and heated at reflux (30
minutP.S) until clear. The solution was evaporated in vacuo and the residue
redissolved in dry THF (10 ml). N-butyl-4-piperidinylmethanol (WO 93/05038)
(0.479g; 2.80 mmol) was dissolved in dry THF (S ml) under Ar and treated with
methyllithium (2.05 ml of a l.SM solution in diethyl ether; 3.08 mmol). The mixture
was stirred at (lS minutes) and treated with the acid chloride solution dropwise. The
solution was stirred (18 hours) evaporated in vacuo and the residue purified by flash
silica gel chromatography wi~h CHC13 ~ 5% EtOH/CHC13 as eluant to yield a pale
yellow oil/solid. This material was triturated with pentane, cooled to -78 C and the
solid that formed collected by filtration and dried in vacuo to yield the ti~le
WO 94/08995 21 4 69 2~
-
compound as a pale yellow solid (0.1 lg; 13%), mp = 39-40 C.
lH NMR (CDC13, 250 MHz) o:
7.90 (d, lH), 7.80 (dd, 2H), 6.80 (d, lH), 4.30 (d, 2H), 3.05 (d, 2H), 2.40 (t, 2H),
2.10-1.80 (m, 5H), 1.65-1.25 (m, 6H), 0.95 (t, 3H)
Example4-1 [Rl=H,R2=Cl,R3=H,R4=H,X=O,Y=NH,Z=(i)]
(l-Butyl-4-piperidinylmethyl)-S-chlorobenzo[b]furan-7-carboxamide
S-chlorobenzo[b]furan-7-carboxylic acid [US patent 4888353; 33f] (0.18g; 0.92
mmol) was suspended in thionyl chloride (2 ml) and heated at reflux (30 minutes)until clear. The mixture was cooled, evaporated in vacuo and the residue azeotroped
with toluene. The residue was redissolved in dry THF (4 ml) and treated with
triethylamine (0.13 ml; 0.92 mmol) and N-butyl4-piperidinylmethylamine
(W093/05038) (0.171g; 1.01 mmol). The solution was stirred at RT (1 hour),
evaporated in vacuo and partitioned H2O/CHC13. The organic phase was dried over
Na2SO4, filtered and evaporated in vacuo to a yellow oil. The oil was purified by
flash silica-gel chromatography with CHC13 ~ 2% MeOH/CHC13 as eluant to yield
the title compound as a pale yellow oil (0.3g; 94%), which was converted to the
oxalate salt, mp = 109-110 C.
lH NMR (250 MHz, CDC13) (free base) ~:
8.10 (d, lH), 7.75 (d, lH), 7.70 (d, lH), 7.55 - 7.45 (m, lH), 6.85 (d, lH), 3.45 (t,
2H), 3.00 (d, 2H), 2.35 (t, 2H), 2.05 - 1.65 (m, 5H), 1.55 - 1.25 (m, 6H), 0.90 (t, 3H).
Description 1-2 (intermediate for Example 1-2)
Dibenzofuran-4-carboxylic acid
A solution of nBuLi (9.7 ml, 1.36 M in hexanes) in hexane (30 ml) was
treated with N,N,N',N'-tetramethylethylene~i~mine (2.0 ml). followed by addition of
dibenzofuran (2g). Stirring was continued at room temperature overni_ht. The
mixture was poured onto solid C2 and diluted with water. The layers were
separated, the aqueous layer acidified to pH2 with 5N HCI and extracted with
dichloromethane. The organic phase was dried (Na2SO4), filtered and concentratedin vacuo to afford title compound as an off-white solid (1.60g).
IH NMR 250 MHz (d6-DMSO)
o 13.34(bs,1H), 8.42(d,1H), 8.21(d,1H), 8.04(d,1H), 7.80(d,1H), 7.36-7.52(m,3H).
W094/08995 7,~ 469~ 16- PCI/EP93/02809
Examplel-2 [R1=H,R2=H,R3=H,R4=H,X=O,Aisasinglebond,Y=O,
z = (i)]
l-Butylpiperidin-4-ylmethyldibenzofuran-4-carboxylate hydrochloride
To a solution of dibenzofuran-4-carboxylic acid (l.OOg) in acetonitrile (50 ml)
was added 1,1-carbonyldimidazo`le (763 mg). Stirring was continued at room
temperature for 2h. The solvent was concentrated in vacuo lo afford crude
imidazolide.
Methyllithium (3.13 ml, 1.5 M in diethyl ether) was added dropwise to a
solution of l-butyl-4-hydroxymethylpiperidine (808 mg) in dry THF (15 ml) at 0C.
Stirring was continued at 0C under a nitrogen atmosphere for 30 min. A solution of
crude imidazolide in dry THF (20 ml) was added to the reaction mixture and stirring
continued at room temperature overnight. Water (2 ml) was added and ~he solvent
concentrated in vacuo. The residue was partitioned between chloroform and water.The organic phase was dried (Na S04), filtered and concentrated under reduced
pressure. The residue was chromatographed on silica using chloroform and ethanolas eluant to afford pure ester. Treatment with ethereal HCI gave the title compound
as a solid (l.OOg).
lH NMR 250 MHz (CDC13) (Free base)
o: 8.12((t,2H), 7.98(d,1H), 7.68(d,1H), 7.50(t,1H), 7.34-7.45(m,2H), 4.32(d,2H),3.02(d,2H), 2.35(t,2H), 1.82-2.08(m,5H), 1.44-1.63(m,4H), 1.26-1.39(m,2H),
0.94(t,3H).
Example2-2 [R1=H,R2=H,R3=H,R4=H,X=O,Ais-CH2-,Y=O,Z=(i)]
(l-Butyl-4-piperidiny1methyl)-9H-xanthene-4-carboxvlate
The title compound is prepared from 9H-xanthene-4-carboxylic acid (P.Yates
et al., Can J. Chem., 1975, 53, 2045 and lithium (1-butylpiperidin-4-yl) methoxide
via the imidazolide.
Example3-2 [R1=H,R2=H,R3=H,R4=H,X=O,Ais-CO-,Y=O,Z=(i)]
(1-Butyl-4-piperidinylmethyl)-9-oxo-9H-xanthene-4-carboxylate
The title compound is prepared from 9-oxo-9H-xanthene-4-carboxylic acid
(S.Akagi et al.,J.Pharm. Soc. Jpn., 1954, 74, 610) (R.Anschutz et al., Ber., 1922, 55,
686) and lithium (1-butylpiperidin-4-yl) methoxide via the imidazolide
Example4-2 [Rl =H,R2=H,R3=H,R4=H,X=O,Ais-NH-,Y=O,Z=(i)]
(l-Butyl-4-piperidinylmethyl)-lOH-phenoxazine-4-carboxylate
The title compound is prepared from lOH -phenoxazine-4-carboxylic acid and
lithium (I-butylpiperidin-4-yl) methoxide via the imidazolide
WO 94/08995 21 4 6 9 2 3 pcr/Eps3/o2sos
-
Example5-2 [Rl=H,R2=H,R3=H,R4=H,X=O,Aisasinglebond,Y=NH,
z = (i)]
(l-Butyl-4-piperidinylmethyl)-1-amino-2-chlorodibenzofuran-4-carboxamide
The ti~le compound was prepared from l-arnino-2-chlorodibenzofuran-4-
carboxylic acid (EP-A-0339950) according to the methodology described in Example- 2-3, and was converted to its oxalate salt.
m.pt 177-178 C
lH NMR (250 MHz, CDC13), (Free base) ~:
8.12 (s, lH), 7.78 (d, lH), 7.54 (d, lH), 7.42 (m, 3H), 4.93 (s, 2H), 3.43 (t, 2H), 2.92
(d, 2H), 2.28 (t, 2H), 1.91-1.65 (m, SH), 1.48-1.35 (m, 6H), 0.87 (t, 3H).
Example 6-2 [Rl = H, R2 = Cl, R3 = NH2, R4 = H, X = O, A is a single bond, Y =
O, Z= (i)]
(1-Butyl-4-piperidinylmethyl)-1-amino-2-chlorodibenzofuran-4-carboxvlate
The title compound was prepared from l-amino-2-chlorodibenzofuran-4-
carboxylic acid (EP-A-0339950) according to the methodology described in Example1-3 and was converted to its oxalate salt.
mpt. 199-200 C
lH NMR (250 MHz, CDC13) (Free base) ~:
8.08 (s, lH), 7.80 (d, lH), 7.70 (d, lH), 7.47 (m, 2H), 5.11 (s, 2H), 4.19 (d, 2H), 3.05
(bd, 2H), 2.39 (t, 2H), 2.12-1.80 (m, 5H), 1.54 (m, 4H), 1.32 (m, 2H), 0.94 (t, 3H).
Example 7-2 ~Rl = H, R2 = Cl, R3 = H, R4 = H, X = O, A is a single bond, Y = O,
Z = (i)]
(l-Butyl-4-piperidinylmethyl)-2-chlorodibenzofuran-4-carboxylate
The title compound was prepared from 2-chlorodibenzofuran-4-carboxylic
acid (EP-A-0339950) according to the methodology described in Example 3-3.
mpt. 80-82 C
lH NMR (250 MHz), CDC13 (Free base) ~:
8.12 (d, lH), 8.05 (d, lH), 7.92 (d, lH), 7.68 (d, lH), 7.53 (t, lH), 7.40 (t, lH), 4.32
(d, lH), 3.03 (d, 2H), 2.38 (t, 2H), 2.10-1.82 (m, 5H), 1.55 (m, 4H), 1.32 (m, 2H),
o.gO (t, 3H).
W094/08995 ?,~69~3 - 18- PCI/EP93/02809
Examplel-3 [R1=H,R2=H,R3=H,R4=H,X=O,Aisasinglebond,f,g=H,
y=o,Z=(i)]
(l-Butyl-4-piperidinylmetlhyl)-1-amino-2-chloro-Sa,6,7,8,9,9a-
tetrahydrodibenzofuran~-carboxylate
S l-Amino-2-chloro-5a,6,7,8,9,9a-hexahydrodibenzofuran-4-carboxylic acid
(EP-A-0339950) (0.267g, 0.998 mmol) was suspended in acetonitrile and treated with
bis-carbonyldiimidazole (0.178g, 1.098 mmol) with stirring. After 4 h, the reaction
mixture was evaporated under reduced pressure and dried in vacuo to give the crude
imidazolide as a white solid. Meanwhile, a solution of l-butyl-4-piperidinemethanol
(WO 93/103725) (0.171g, 0.998 mmol) in dry THF (8 ml) was treated with 1.5M
methyllithium in Et2O (0.665 ml, 0.998 mmol) with stirring under argon. After 0.25
h, a suspension of the crude imidazolide in dry THF (5 ml) was added slowly. After
24 h, the reaction mixture was evaporated under reduced pressure and partitionedbetween EtOAc and water. The aqueous layer was then extracted with EtOAc and thecombined organic layers were dried (Na2SO4) and evaporated under reduced pressure
to give a yellow oil, which was purified by silica-gel chromatography (5%
MeOH/CH2C12 as eluant) to give the tifle compollnd as a pale yellow oil (0.082g,20%), which was converted to its oxalate salt m.pt. 105-107C.
1H NMR (200 MHz, CDC13) (free base) ~:
7.68 (s, lH), 4.70 (m, lH), 4.35 (s, 2H), 4.12 (d, 2H), 3.13 (bd, 2H), 3.00 (m, lH),
2.55-1.45 (m, 17H), 1.43-1.15 (m, 4H), 0.93 (t, 3H).
Example 2-3 ~Rl = H, R2 = Cl, R3 = NH2, R4 = H, X = O, A is a single bond,
f, g, together are a bond, Y = NH, Z = (i)]
(1-Butvl-4-piperidinvlmethyl)-1-amino-2-chloro-Sa,6,7,8,~,9a-
tetrahydrodibenzofuran-4-carboxamide
l-Amino-2-chloro-5a,6,7,8,9,9a-hexahydrodibenzofuran-4-carboxylic acid
(EP-A-339950) (0.292g, 1.092 mmol) was suspended in acetonitrile (20 ml) and
treated with bis-carbonyldiimidazole (0.186g, 1.46 mmol) with sti~ring. After 20h,
the reaction mixture was evaporated under reduced pressure and dried in vacuo togive the crude imidazolide as a white solid. The imidazolide was then redissolved in
dry THF (10 ml) and (1-butyl-4-piperidinyl) methylamine (WO 93/05038) (0.204 g,
1.201 mmol) in dry THF (2 ml) was added under Ar. The mixture was then heated
under reflux. After 8 h, the reaction mixture was allowed to cool, and was evaporated
under reduced pressure. The residue was then partitioned between CH2CI2 and aq.
NaHCO3. The aqueous layer was then extracted with CH~CI2 (lX), and the
combined organic layers were dried (Na2SO4), and evaporated under reduced
pressure to give a colourless oil, which was purified by chromatography (10%
WO 94/08995 2~ 1 g 6 g 2 3 PCI`/EP93/02809
- 19-
.._
MeOH/CH2C12 as eluant) to give the title compound as a colourless oil (0.161 g,
35%) that was converted to its oxalate salt.
m. pt 214-215 C
lH NMR (250 MHz CDC13) (free base) o:
7.82 (s, lH), 7.54 (t, lH), 4.72 (m, lH), 4.29 (s, 2H), 3.32 (t, 2H), 3.03 (m, 3H), 2.32
(m, 3H), 2.12-1.15 (m, 18H), 0.91 (t, 3H)
F.Y~mple33 [Rl=H,R2=Cl,R3=H,R4=H,X=O,Aisasinglebond,f,g=H,
y=o,z=(i)]
(1-Butyl-4-piperidinylmethyl)-2-chloro-cis-~a,6,7,8,9,9a-
hexahydrodibenzofuran-4-carboxylate
2-Chloro-cis-5a,6,7,8,9,9a-hexahydrodibenzofuran-4-carboxylic acid
(EP-A-0339950), (O. lOOg, 0.396 mmol) was suspended in thionyl chloride (5 ml) and
heated to reflux with stirring. After Ih, the reaction mixture was allowed to cool, and
was evaporated under reduced pressure to give a pale brown oil, which was dried in
vacuo in give the crude acid chloride. Meanwhile a solution of 1-butyl-4-
piperidinylmethanol (0.075g, 0.436 mmol) in dry THF (3 ml) under argon was treated
with 1.6M n-butyllithium (0.272 ml, 0.436 mmol). After 0.25h, a solution of the
crude acid chloride in dry THF (5 ml) was added, and the resultant mixture stirred at
room temperature overnight. The reaction mixture was then evaporated under
reduced pressure and purifled by silica-gel chromatography (2% MeOH/CH2C12 as
eluant) to give the ti~le compound (0.071g, 44%) as a colourless oil, which was
converted to its oxalate salt.
m.pt 154-155 C
lH NMR (250 MHz, CDC13) (free base) ~:
7.70 (d, lH), 7.20 (d, lH), 4.85 (m, lH), 4.18 (d, 2H), 3.25 (m, lH), 3.05 (d, 2H),
2.43 (t, 2H), 2.13-1.70 (m, 8H), 1.65-1.25 (m, llH), 0.95 (t, 3H).
Example4-3 [Rl =H,R2=Cl,R3=H,R4=H,X=O,Aisasinglebond,
f, g, together are a bond, Y = O, Z = (i)]
(l-Butyl-4-piperidinylmethyl)-2-Chloro-6,7,8,9-tetrahydrodibenzofuran-4-
carboxylate
The ~i~le compollnd was prepared from 2-chloro-6,7,8,9-
tetrahydrodibenzofuran-4-carboxylic acid (EP-A-0339950) according to the
methodology described in Example 3-3 and was converted to its oxalate salt.
m.pt 188-190 C
wo 94/oW5 ~69~3 20 - PCI/EP93/02809
lH NMR (200 MHz, CDC13) (free base) ~:
7.78 (d, lH), 7.53 (d, lH), 4.28 (d, 2H), 3.03 (d, 2H), 2.80 (l, 2H), 2.58 (t, 2H), 2.10-
1.7S (m, 9H), 1.52-1.25 (m, 6H), 0.90 (t, 3H).
Examplel-4 rX=O,R1=H,R2=Cl,R3,R4,R4=H,Y=O,Z=(i)]
10-(1-Butylpiperidin-4-ylmethyl)-8-chloro-3,4,5,6-tetrahydro-2,6-methano-2H-1-
benzoY ~inel~rboxylate
The title compound is prepared from 8-chloro-3,4,5,6-tetrahydro-2,6-methano-2H- 1-
benzoxacin-10-carboxylic acid (R.D. Youssefyeh et al., J.Med.Chem. 1992, 35, 903)
and lithium (1-butylpiperidin-4-yl) methoxide via the imidazolide.
5-HT4 RECEPTOR ANTAGONIST ACTIVITY
1) Guinea pig colon
Male guinea-pigs, weighing 250-400g are used. Longitudinal muscle-
myenteric plexus preparations, approximately 3cm long, are obtained from the distal
colon region. These are suspended under a 0.5g load in isolated tissue baths
containing Krebs solution bubbled with 5% CO2 in 2 and maintained at 37C. In
all experiments, the Krebs solution also contains methiothepin 10-7M and granisetron
10-6M to block effects at 5-HT1, 5-HT2 and 5-HT3 receptors.
After construction of a simple concentration-response curve with 5-HT, using
30s contact times and a 15min dosing cycle, a concentration of 5-HT is selected so as
to obtain a contraction of the muscle approximately 40-70% maximum(10~9M
approx). The tissue is then alternately dosed every 15min with this concentration of
5-HT and then with an approximately equi-effective concentration of the nicotinereceptor stimulant, dimethylphenylpiperazinium (DMPP). After obtaining consistent
responses to both 5-HT and DMPP, increasing concentrations of a putative 5-HT4
receptor antagonist are then added to the bathing solution. The effects of this
compound are then determined as a percentage reduction of the contractions evoked
by 5-HT or by DMPP. From this data, pICso values are determined, being defined as
the -log concentration of antagonist which reduces the contraction by 50%. A
compound which reduces the response to 5-HT but not to DMPP is believed to act as
a 5-HT4 receptor antagonist.
The compounds generally had a pICso of at least 7.