Note: Descriptions are shown in the official language in which they were submitted.
2147~61
DES CR I PT I ON
PROSTAGLANDIN El ANALOGUES
Technical Field
The present invention relates to novel prosta-
glandin El analogues.
B ac kg ro u nd A r t
Since prostaglandin (hereinafter referred to
as PG) shows various important physiological actions in
a trace amount, natural PG analogues and a vast number
of derivatives thereof have been studied on synthesis
and biological activities, with attempts to apply these
compounds to pharmaceuticals. In particular, PGEl has
characteristic actions such as platelet aggregation
inhibition, blood pressure lowering and the like and is
already in practical use as a drug for ameliorating pe-
ripheral circulatory disturbance. Therefore, a large
number of PGEl analogues have been investigated. Hith-
erto known PGEl analogues, however, have a drawback ofquick metabolism in living body and consequent short-
term effect. Further, hitherto proposed PGEl analogues
induce diarrhea as a side effect when administered
orally and accordingly have a problem in that they
cannot be administered orally in a sufficiently high
amount to obtain the satisfactory effects.
Meanwhile, 13,14-didehydro-PGEl methyl ester
and 6-hydroxy-13,14-didehydro-PGEl are known as 13,14-
didehydro-PGEl analogues obtained by converting the
double bond between the 13- and 14-positions of PGEl to
a triple bond [Japanese Patent Application Kokai (Laid-
Open) No. 100446/1977 and U.S. Patent No. 4,131,738].
The main object of the present invention is to
provide novel PGEl analogues which have a higher effica-
cy, a more prolonged action and a lower side effect thanhitherto known PGEl analogues.
21~7461
Disclosure of the Inventi on
The present inventors made a study and found
out that PGEl analogues having a triple bond between the
13- and 14-positions, a branch at the 17-position and an
oxygen or sulfur atom at the 3-position in pace of
methylene have excellent and prolonged physiological
activities and a low side effect. The finding has led
to the completion of the present invention.
Thus, the present invention provides a prosta-
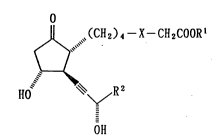
glandin El analogue represented by formula
Jl ~(CH2)4--X--CH2COOR'
15 ~ (I)
'~R 2
OH
(wherein Rl represents a hydrogen atom or a Cl-C6 alkyl
group; and X is an oxygen atom and R represents
~ or / ~ I , or X is a sulfur
atom and R2 represents " " ~'~`- ~ ), and a salt
thereof.
In the present invention, "alkyl group" is a
straight-chain or branched-chain saturated aliphatic
hydrocarbon group. The Cl-C6 alkyl group includes, for
example, methyl, ethyl, n-propyl, isopropyl, n-butyl,
isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl and
n-hexyl groups, etc. Of these, Cl-C4 alkyl groups are
preferable.
In the formula (I), Rl is preferably a hydro-
gen atom or a Cl-C4 alkyl group (a methyl group, in
particular).
2147~61
The compound of formula (I) wherein R1 repre-
sents a hydrogen atom, can be present in the form of a
free acid, or in the form of a salt. Examples of such a
salt include alkali metal salts such as sodium salt,
potassium salt and the like; alkaline earth metal salts
such as calcium salt, magnesium salt and the like; other
metal salts such as aluminum salt and the like; ammonium
salt; salts with organic amines such as trialkylamine
(e.g. triethylamine), pyridine and the like. A pharma-
ceutically acceptable salt is particularly preferable.
The present compound of formula (I) can be
produced, for example, by a process summarized in the
following reaction scheme A.
214~461
React i on Sc heme A
o - C2Hs \
~'-~`N/C21l5 + C2Hs / \\\\ R2,
-
~` (m ) ~R4
First step
o v
~"~R2
OR4
Second step R~IOOC-CH2-X-(Cl~)3Cu(CN)ZnI 2LiCI
chlorotrimethylsilane ( )
(CH3)3SiO -
I (CH2)4-X-CH2COOR "
~"" .
~` Y~\ R2
OR4
Third step Hydrolysis
2147161
Reaction Scheme - A (Cont i nued)
~ ~(CI12)4-X-CHaCOOR
R30~
OR4
Fourth step Deprotection
(CH2) 4-X-CI12COOR"
( I a)
R 2
-
Fifth step Hydrolysis
(CH 2 ) 4 -X-CH 2COOH
~ ( I b)
HO ~/
-
0~
21~7~61
In the above reaction scheme, Rll represents a
Cl-C6 alkyl group; R3 and R4, which may be the same or
different, each represent a protective group for hy-
droxyl group; and X and R have the same definitions as
given above.
The protective group for hydroxyl group can be
a protective group which is removable by an ordinary
reaction for protective group removal, such as hydroly-
sis, hydrocracking and the like and which is ordinarily
used in prostaglandin chemistry. It includes, for
example, tert-butyldimethylsilyl, triethylsilyl,
phenyldimethylsilyl, tetrahydropyranyl,
tetrahydrofuranyl, methoxymethyl, ethoxyethyl and benzyl
groups, etc.
Each of the first to fifth steps is hereinaf-
ter described in more detail.
(First step)
First, a compound of formula (II) known via
the process of Sato et al. [J. Org. Chem., Vol. 53, p.
5590 (1988)] is reacted with about 0.8 - about 2 equiva-
lents of an organoaluminum compound represented by
formula (III) at a temperature of about -10 to about
30C, preferably about 0 to about 10C in an inert
solvent (e.g. benzene, toluene, tetrahydrofuran, diethyl
ether, methylene chloride, n-hexane or the like) to
obtain a compound of formula (IV) stereospecifically.
The organoaluminum compound of formula (III)
used in the above reaction can be produced, for example,
by mixing an acetylene compound represented by formula
NC----C ~R2
OR4
(wherein R2 and R4 have the same definitions as given
above), produced by the process of Sato et al. [Tetrahe-
21471~
dron Lett., Vol. 30, p. 7083 ~1989)] with about 0.8 -
about 1.5 equivalents of an alkyllithium (e.g. n-
butyllithium, tert-butyllithium or the like) at about
-20 to about 30C, preferably about -10 to about 0C,
preferably completing a reaction at about 10 to about
30C, and then adding about 0.8 - about 1.5 equivalents
of diethylaluminum chloride at a temperature of about
-20 to 30C. It is generally preferable that this reac-
tion is conducted in an inert organic solvent (e.g.
benzene, toluene, tetrahydrofuran, diethyl ether, methy-
lene chloride, n-hexane, or the like).
(Second step)
The compound of formula (IV) obtained in the
first step is reacted with about 0.5 - about 4 equiva-
lents of an organocopper compound represented by formula(V) and about 0.5 - about 4 equivalents of
chlorotrimethylsilane in an inert solvent (e.g.
tetrahydrofuran, diethyl ether, methylene chloride,
toluene, n-hexane or the like) at a temperature of about
-78 to about 40C to form a compound of formula (VI).
The organocopper compound of formula (V) can
easily be produced, for example, by a process shown by
the following reaction scheme B.
21~7~6~
Reaction Scheme B
HS-CH2COORII Br-CH2COOR~
(Vm) (X[)
1) HO-(CH2`) 3-OH
AIBN HO-CEI2-CH=CH2 (~)
(lX)
2) NaOCH3/ methanol
J \~/ .
l oHO-(CI12)3-S-CH2COORI I HO-(CH2)3-O-CH2COOR
(X a) (Xb)
1) Methanesulfonic acid chloride/
2) NaI/ acetone triethylamine
\/
I -(CH2)3-X-CH2C()ORI ~ (X m)
Zn/THI~
I Zn-(CI{2)3-X-CH2COOR~ I
Copper cyanide, lithium chloride/THF-
(V)
In the above reaction scheme, R11 and X have
the sàme definitions as given above.
A mercaptoacetic acid ester compound of formu-
la (VIII) is reacted with an alcohol compound of formula
(IX) and 2,2'-azobis(2-methylpropionitrile) (AIBN) to
35 obtain a compound of formula (Xa). Separately, a
br~m~ jc aci,d ~,t~r 9f formula ~XI) is r,eact*~ ,with
2147~61
an alcohol compound of formula (XII) and sodium hydride,
followed by treatment with sodium methoxide in methanol,
to obtain a compound of formula (Xb).
The compound of formula (Xa) and the compound
of formula (Xb) are reacted in the presence of
methanesulfonic acid chloride and triethylamine, fol-
lowed by reaction with sodium iodide in acetone, to
obtain a compound of formula (XIII).
The compound of formula (XIII) is reacted with
about 0.8 - about 5 equivalents of zinc activated with,
for example, 1,2-dibromomethane, chlorotrimethylsilane,
iodine or the like, in an inert solvent (e.g.
tetrahydrofuran, diethyl ether, n-hexane, n-pentane,
dioxane or the like), whereby the compound of formula
( XIII) can be converted into an organozinc compound of
formula (XIV). At this time, heating may be conducted
as necessary. The temperature of heating varies depend-
ing upon the boiling point of the solvent used, but can
be generally about 30 to about 150C, preferably about
40 to about 80C.
The organozinc compound of formula (XIV) is
reacted in the same inert solvent as mentioned above,
containing copper cyanide (about 1 - about 2.5 equiva-
lents) and lithium chloride (about 2 - about 5 equiva-
lents), whereby an organocopper compound of formula (V)can be obtained.
(Third step)
The compound of formula (VI) obtained in the
second step is hydrolyzed by the use of an inorganic
acid (e.g. an aqueous hydrochloric acid solution) or an
organic acid or its amine salt (e.g. p-toluenesulfonic
acid, pyridinium p-toluenesulfonate or the like) in an
organic solvent (e.g. acetone, methanol, ethanol, iso-
propanol, diethyl ether, a mixed solvent thereof or the
like) at a temperature of about 0 to about 40C, whereby
a compound of formula (VII) can be obtained
21~7~61
stereoselectively.
(Fourth step)
The protective group for hydroxyl group, of
the compound of formula (VII) obtained in the third step
is deprotected by a method ordinarily used in prosta-
glandin chemistry, to obtain a compound of formula (I)
of the present invention wherein R is a C1-C6 alkyl
group, i.e. a compound of formula (Ia).
(Fifth step)
The ester moiety (R11) of the compound of
formula (Ia) is hydrolyzed, whereby a compound of formu-
la (I) of the present invention wherein R1 is a hydrogen
atom, i.e. a compound of formula (Ib) can be obtained.
The hydrolysis can be conducted, for example,
by reacting the compound of formula (Ia) with an enzyme
in a buffer such as phosphate buffer, tris-hydrochloride
buffer or the like with an organic solvent (miscible
with water, such as acetone, methanol, ethanol or the
like) used in combination as necessary. The enzyme
usable includes, for example, hydrolases produced by
microorganisms (e.g. enzymes produced by microorganisms
belonging to Candida sp. and Pseudomonas sp.),
hydrolases prepared from animal organs (e.g. enzymes
prepared from pig liver and pig pancreas), etc. Specif-
ic examples of such an enzyme which is commerciallyavailable, are Lipase VII (produced by Sigma Co.; de-
rived from a microorganism of Candida sp.), Lipase AY
(produced by Amano Pharmaceutical Co.; derived from a
microorganism of Candida sp.), Lipase MF (produced by
Amano Pharmaceutical Co.; derived from a microorganism
of Pseudomonas sp.), PLE-A (produced by Amano Pharmaceu-
tical Co.; prepared from pig liver), Esterase (produced
by Sigma Co.; prepared from pig liver), Lipase II (pro-
duced by Sigma Co.; prepared from pig pancreas), Lipo-
protein Lipase (produced by Tokyo Kasei Kogyo Co.;prepared from pig pancreas), etc.
21~7461
1 1
The amount of the enzyme used can appropriate-
l y be determi ned depending upon the potency of the
enzyme used and the amount of the substrate used [the
compound of the formula (Ia)], but is generally about
0.1 to about 20 times (by weight) that of the substrate.
The reaction temperature is about 25 to about 50 C,
p referably about 30 to about 35 C.
Each of the products obtained in the above
steps can be, as necessary, separated from the reaction
mixture and purified by a per se known method, for
example, by chromatography, etc.
The p resent compound of formula (I), as i s
clear f rom the foll owing Test Example, has a strong and
prolonged inhibitory action for platelet aggregation.
Further, the present compound i nduces substantially no
diarrhea (dia rrhea is currently the biggest probl em of
PG) at a dose reliably showing pharmacological actions.
Therefore, the present compound is useful as a drug for
t reating vari ous di seases including periphe ral ci rcula-
tory di sturbance.
Test Example [Test for inhibition of rabbit platelet
aggregation]
New Zealand White-strai n rabbits (g roups of
four rabbits each weighing 2.5-4.0 kg) were tested.
Blood was collected from the common carotid artery of
the rabbits under an ether anesthesia, and mixed with
3.2% sodium citrate in volume ratio of 9: 1. The blood
was centrifuged at 1,100 rpm for 15 minutes to give a
platelet-rich plasma (PRP) as a supernatant.
Blood platelet aggregation was dete rmined
accordi ng to the method of Born [Natu re, Vol. 194, p.
927 (1962)]. To 275 ,u~ of PRP was added 1 ~ of a
solution of various concentrati ons of a test compound
dissolved in ethanol; the mixtu re was stirred at 1,000
rpm at 37C for 3 minutes; then, in this state, 25 ,u~ of
an aggregation-inducing agent ~adenosine di phosphate
2147161
(ADP), final concentration: 5 ~M] was added thereto to
induce platelet aggregation; and by using an
aggregometer, there was determined the maximum aggrega-
tion rate (the maximum change in light transmission
within 5 minutes from the induction of platelet aggrega-
tion).
The aggregation inhibition rate of the test
compound was calculated from the maximum aggregation
rate in the presence test compound relative to the maxi-
mum aggregation rate when ethanol was used instead ofthe solution of the test compound; the IC50 value of the
test compound was determined from the concentration-
response curve. The results are shown in Table 1.
Table 1
Test Compound IC50 ~nM)
Compound 10.60
Compound 20.66
Compound 30.39
Compound 40.26
Compound 50.67
Compound 61.27
Control A42.07
Control B133.6
Control C910.6
Control D1522
Incidentally, in the above table, the
number of each compound corresponds to the number of
each compound shown in Examples described later; and
control A, control B, control C and control D are com-
pounds having the following structures, respectively.
2147~81
~ S-CH2COOH
Control A:
HO \\\
OH
o
~ " O-CI12COOCH3
Control B: ~
1~0~
01
0
~ ~ "~_"S-CH2COOH
Control C~
HO
011
~ O-CI~2COOCH3
Control D:
HO
01{
As described above, the compound of the pres-
ent invention has a strong and prolonged inhibitory
action for pl atelet aggregation . The present compound
can the refore be admi ni stered to mammal s, parti cu 1 arl y
35 humans as a drug for treating various thrombotic diseas-
es incl uding peripheral ci rculatory disturbance.
21~7461
14
In order to use the compound of the present
invention as a drug, the compound can be made into
pharmaceutical preparations suitable for administration,
together with pharmaceutically acceptable adjuvants and
can be administered orally or parenterally (e.g. intra-
venously, intrarectally or intravaginally). As the
preparation for oral administration, there can be used,
for example, solid preparations such as tablets, gran-
ules, capsules and the like; and liquid preparations
such as solution, fat emulsion, liposome suspension and
the like. The present compound, when used in pharmaceu-
tical preparations for oral administration, may also be
made in such pharmaceutical preparations by forming an
inclusion compound with ~ - or ~-cyclodextrin, meth-
ylated cyclodextrin or the like. As the preparation forintravenous administration, there can be used an aqueous
or non-aqueous solution, an emulsion, a suspension, a
solid preparation which is used by dissolving in a
solvent for injection, right before the use, etc. As
the preparation for intrarectal administration, there
can be used suppositories; and as the preparation for
intravaginal administration, there can be used prepara-
tions such as pessary and the like.
As the adjuvants used for making such pharma-
ceutical preparations, there can be cited, for example,
excipients such as crystalline cellulose, lactose, corn
starch, mannitol and the like; lubricants such as magne-
sium stearate, talc and the like; binders such as
hydroxypropyl cellulose, polyvinylpyrrolidone and the
like; disintegrators such as carboxymethyl cellulose
calcium and the like; fluidity improvers such as light
silicic acid anhydride and the like; dissolving agents
such as distilled water for injection, physiological
saline solution, Ringer's solution and the like; preser-
vatives such as methyl p-oxybenzoate, propyl p-
oxybenzoate and the like; emulsifiers such as gum ara-
2147461
bic, lecithin and the like; and surfactants such asTween, Span and the like.
The dose of the present compound can be varied
over a wide range depending upon the age, sex and weight
of patient, the condition of disease, the judgement of
doctor, etc. However, the daily dose for one ordinary
adult is 0.1-100 ~q, and such a daily dose can be admin-
istered in 1-3 portions as necessary.
Best Mode for Carrying Out the Invention
The present invention is hereinafter described
in more detail by referring to Examples.
In the nomenclature of compound, "nor" in the
expression of, for example, "17,18,19,20-tetranor" means
that there is no carbon chain at the positions (in the
above example, there is no carbon chain at the 17-20
positions).
Production Example 1
4-Thia-5-carbomethoxypentylzinc (II) iodide
(1) AIBN (1.93 9, 11.8 mmol) was added to a mix-
ture of methyl thioglycollate (25.0 q, 236 mmol) and
allyl alcohol (19.2 ml, 282 mmol). The mixture was
stirred at 110C for 2 hours. Thereto was added a
saturated aqueous sodium chloride solution (100 ml),
followed by extraction with ethyl acetate (300 ml). The
organic layer was washed with a saturated aqueous sodium
chloride solution, dried over magnesium sulfate, and
filtered. The filtrate was concentrated to obtain
methyl (3-hydroxypropylthio)acetate (38.33 9).
(2) Methyl (3-hydroxypropylthio)acetate (25.68 9)
was dissolved in dimethyl chloride (250 ml). Thereto
were added, at 0C, methanesulfonyl chloride (14.5 ml,
187 mmol) and triethylamine (26.2 ml, 188 mmol). The
mixture was stirred at room temperature for 30 minutes.
Water was added and extraction with dimethyl chloride
was conducted. The organic layer was washed with 3 N
2147461
16
HCl, a saturated aqueous sodium hydrogencarbonate solu-
tion and a saturated aqueous sodium chloride solution,
dried over anhydrous magnesium sulfate, and filtered.
The filtrate was concentrated to obtain methyl (3-
5 methanesulfoxypropylthio)acetate (38.0 q).
(3) Methyl (3-methanesulfoxypropylthio)acetate
(57.2 q, 236 mmol) was dissolved in acetone (600 ml).
Thereto was added sodium iodide (70.7 q, 472 mmol ). The
mixture was stirred at room temperature for 3 days.
10 Water was added to the reaction mixture, followed by
extraction wi th ethyl acetate. The organic layer was
washed with a saturated aqueous sodium hydrogencarbonate
solution and a saturated aqueous sodium chloride solu-
tion, dried over anhydrous magnesium sulfate, and fil-
15 tered. The filtrate was cocentrated. The resultingcrude product was purified by silica gel column chroma-
tography (developing solvent: ethyl acetate / hexane =
1/10) to obtain methyl (3-iodopropylthio)acetate (45.0
9 )
20 l H-NMR (CDCl3, 200MHz) o ppm:
2.02-2.17 (m, 2H), 2.75 (t, J=6.9Hz, 2H), 3.24
(s, 2H), 3.29 (t, J=6.7Hz, 2H), 3.75 (s, 3H)
(4) To a zinc powder (1.93 q, 29.6 mmol ) were
added THF (5 ml) and 1,2-dibromoethane (0.05 ml). The
25 mixture was refluxed with heati ng and stirring. Thereto
were added, with stirring at room temperature,
trimethylsilyl chloride (0.10 ml), methyl
(iodopropylthio)acetate (4.08 q, 14.8 mmol) and THF (10
ml) in this order. The mixture was stirred with heat-
30 i ng, at 40 C for 2.5 hours to obtain a THF soluti on of
the title compound.
Production Example 2
4-Oxo-5-carbomethox ypentylzinc (II) i odide
(1) To sodium hydride (9.6 9, 0.40 mol) was
d ropwise added trimethylene glycol (144.5 ml, 2.0 mol)
at 0 C. The mixture was stirred at room temperature fo r
21~7461
3 hours. Thereto was dropwise added, at 0 C, methyl
bromoacetate (44.4 ml, 0.40 mol ). The mixturè was
stirred at room temeprature for 1.5 hours. The reaction
mixture was subjected to silica gel column chromatogra-
5 phy (developi ng sol vent: ethyl acetate / methanol
19/1) to remove sodium bromide. After concentration,
the concentrate was dissolved i n sodi um methoxide (2.16
9, 0.04 mol) and methanol (200 ml). The mi xture was
s tirred at room temperatu re for 10 mi nutes, then neu-
10 tralized with 6 N hydrochloric acid, and concentrated.The concentrate was purified by silica gel column chro-
matography (developing solvent = ethyl acetate / metha-
nol = 19/1) to obtain methyl 3-hydroxypropyloxyacetate
(36.25 9).
1 H-NMR (CDCl3, 200MHz) o ppm:
1.17-1.93 (m, 2H), 2.19 (br. s, 1H), 3.70 (t,
J=5.5Hz, 2H), 3.76 (s, 3H), 3.81 (t, J=6.OHz,
2H), 4.10 (s, 2H)
(2) Substantially the same procedure as in Produc-
20 tion Example 1 (2) to (4) was repeated to produce a THF
solution of the title compound.
Example
(17R)-1 7,20-Dimethyl-13,1 4-dide hydro-3-oxa-PGE1 methyl
25 ester (compound 1)
(1) In toluene (52 ml ) was dissolved (3S,5R)-3-(t-
butyldimethyl siloxy)-5-methylnon-1-yne (3.49 9). There-
to was added, at 0 C, n-butylli thium (2.5 M, hexane
solution, 4.8 ml). The mixture was stirred at the same
30 temperature for 30 minutes. Thereto was added, at 0C,
diethyl aluminum chl oride (0.94 M, hexane solution, 14.9
ml). The mixture was heated to room temperature and
stirred for 30 minutes.
To the mixture was added, at room temPerature,
35 (4R)-2-(N,N-diethyl amino)methyl -4-(t-
butyldi methyl siloxy)cyclopent-2-en-1-one (0.25 M, ben-
21~7~61
zene solution, 40 ml). The resulting mixtu re wasstirred for 1 5 minutes.
The reaction mixture was poured into a mi xture
of hexane (100 ml), a saturated aqueous ammonium chlo-
5 ride solution (100 ml) and an aqueous hydrochlori c acidsolution (3M, 28 ml ), with stirring. The organic layer
was separated, washed with a saturated aqueous sodium
hydrogencarbonate solution (100 ml), dried and concen-
trated. The resulting residue was purified by silica
10 gel col umn ch romatography (developing solvent: hexane
ethyl acetate = 50/1) to obtain (3R,4R)-2-methylene-3-
[(3'S,5'R)-3'-(t-butyldimethylsiloxy)-5'-methylnon-1'-
ynyl]-4-(t-butyldimethylsiloxy)cyclopentan-1-one (3.13
9 ) -
l H-NMR (CDC13, 300MHz) ~ ppm:
0.03-0.15 (m, 12H), 0.80-0.93 (m, 24H), 1 .06-
1.80 (m, 9H), 2.33 (dd, J=7.4Hz, 17.9Hz, 1H),
2.71 (dd, J=6.4Hz, 17.9Hz, 1H), 3.41-3.56 (m,
1H), 4.20-4.32 (m, 1H), 4.44 (t, J=6.6Hz, 1H),
5.55 (br. s, 1H), 6.14 (br. s, 1H)
I R (neat):
2920, 2850, 2210, 1730, 1630, 1450, 1360,
1240, 1100, 1080, 820, 760 cm l
(2) To the compound obtained in Production Example
2 (0.88 M, tetrahydrofuran solution, 7.10 ml) was added,
at -10 C, copper (I) cyanide-li thium dichloride (1.0 M,
tetrahydrofuran sol ution, 7.85 ml). The mi xture was
stirred at the same temperature for 15 minutes. Thereto
were added, at -78 C, chlorotrimethylsilane (0.72 ml)
and a diethyl ether solution (12.6 ml ) of the compound
obtained in the above (1) (1.55 9). The mixture was
heated to 0 C in about 2 hours, with stirri ng.
To the reaction mixture was added a saturated
aqueous ammonium chloride slution (50 ml), followed by
extraction wi th hexane. The organic layer was washed
with a saturated aqueous sodium chloride solution, dried
2147461
and concentrated. The resulting residue was dissolved
in a mixture of diethyl ether (3.2 ml), isopropanol
(12.8 ml) and pyridinium p-toluenesulfonate (40 mg).
The solution was stirred at room temperature for 15
hours.
To the reaction mixture were added hexane (30
ml) and a saturated aqueous sodium hydrogencarbonate
solution (50 ml), and extraction was conducted. The
organic layer was dried and concentrated. The resulting
residue was subjected to silica gel column chromatogra-
phy (developing solvent: hexane / ethyl acetate = 9/1)
to obtain (17R)-3-oxa-17,20-dimethyl-13,14-didehydro-
PGEl methyl ester 11,15-bis(t-butyldimethylsilyl ether)
(1.17 g).
lH-NMR (CDC13, 200MHz) o ppm:
0.09 (s, 6H), 0.11 (s, 3H), 0.12 (s, 3H),
0.86-0.94 (m, 6H), 0.89 (s, 18H), 1.18-1.85
(m, 15H), 2.04-2.27 (m, 1H), 2.17 (dd,
J=6.9Hz, 17.7Hz, 1H), 2.58-2.76 (m, 2H), 3.52
(t, J=6.5Hz, 2H), 3.76 (s, 3H), 4.07 (s, 2H),
4.22- 4.34 (m, 1H), 4.36-4.45 (m, 1H)
IR (neat):
2955, 2930, 2858, 2234, 1747, 1463, 1439,
1378, 1362, 1254, 1208, 1141, 1094, 1006, 940,
839, 779, 670 cm~l
(3) The compound obtained in the above (2) (1.06
9, 1.70 mmol) was dissolved in acetonitrile (56.5 ml).
Thereto was added 40% hydrofluoric acid (12.8 ml) at
0C. The mixture was stirred for 15 hours with heating
to room temperature. The reaction mixture was poured
into ethyl acetate (100 ml) and a saturated aqueous
sodium hydrogencarbonate solution (300 ml). The aqueous
layer was extracted with ethyl acetate (100 ml). The
resulting organic layer was washed with a saturated
aqeuous sodium hydrogencarbonate solution and a saturat-
ed aqueous sodium chloride solution, dried and concen-
21~7~81
trated. The resulting residue was purified by silicagel column chromatography (developing solvent: hexane /
ethyi acetate = 9/11) to obtain the title compound ~440
mg).
lH-NMR (CDCl3, 300MHz) o ppm:
0.87-0.94 (m, 6H), 1.08-1.88 (m, 15H), 2.20-
2.31 (m, 1H), 2.24 (dd, J=9.OHz, 18.5Hz, 1H),
2.32 (d, J=5.3Hz, 1H), 2.48-2.55 (m, 1H),
2.64-2.71 (m, 1H), 2.75 (ddd, J=1.3Hz, 7.2Hz,
18.5Hz, 1H), 3.51-3.57 (m, 2H), 3.75 ~s, 3H),
4.08 (s, 2H), 4.29-4.38 (m, 1H), 4.43-4.50 (m,
1H)
IR (neat):
3431, 2954, 2930, 2871, 2236, 1746, 1440,
1379, 1285, 1217, 1141, 1072 cm
Example 2
(17R)-17,20-Dimethyl-13,14-didehydro-3-oxa-PGE
(compound 2)
PLE (Sigma Co., 210 units aqueous ammonium
sulfate solution, 190 ~) was dissolved in a phosphate
buffer (pH=8, 17 ml) and acetone (9.6 ml). Thereto was
added the compound obtained in Example 1 (380 mg). The
mixture was stirred at room temperature for 15 hours and
then subjected to salting out with sodium chloride and
extraction with ethyl acetate. The organic layer was
washed with a saturated aqueous sodium chloride solu-
tion, dried over anhydrous magnesium sulfate, and con-
centrated. The resulting crude product was purified by
silica gel column chromatography (developing solvent:
ethyl acetate) to obtain the title compound (330 mg).
H-NMR (CDC13, 300MHz) o ppm:
0.87-0.94 (m, 6H), 1.11-1.89 (m, 15H), 2.24-
2.30 (m, 1H), 2.25 (dd, J=9.1Hz, 18.5Hz, 1H),
2.65-2.79 (m, 1H), 2.76 (ddd, J=1.2Hz, 7.3Hz,
18.5Hz, 1H), 3.53-3.59 (m, 2H), 4.09 (s, 2H),
2i~7461
4.29-4.37 (m, 1H), 4.45-4.50 (m, 1H)
IR (neat):
3402, 2930, 2871, 2238, 1742, 1461, 1377,
1240, 1136, 1050, 677 cm
Example 3
(17S)-20-Isopropylidene-17-methyl-13,14-didehydro-3-
thia-PGEl methyl ester (compound 3)
(1) (3R,4R)-2-Methylene-3-[(3'S,5'S)-3'-(t-
butyldimethylsiloxy)-5',9'-dimethyldec-8'-en-1'-ynyl]-4-
(t-butyldimethylsiloxy)cyclopentan-l-one was obtained
substantially in the same manner as in Example 1 (1),
using (3S,5S)-3-(t-butyldimethylsiloxy)-5,9-dimethyldec-
8-en-1-yne in place of the (3S,5R)-3-(t-
butyldimethylsiloxy)-5-methylnon-1-yne used in Example 1
( 1 ) -
H-NMR (CDCl3, 200MHz) o ppm:
0.10 (s, 6H), 0.11 (s, 3H), 0.13 (s, 3H),
0.78-0.96 (m, 3H), 0.90 (s, 18H), 1.07-2.08
(m, 7H), 1.60 (s, 3H), 1.67 (d, J=0.9Hz, 3H),
2.32 (dd, J=7.4Hz, 18.0Hz, 1H), 2.71 (dd,
J=6.4Hz, 18.OHz, lH), 3.48-3.57 (m, lH), 4.21-
4.34 (m, 1H), 4.38-4.51 (m, 1H), 5.03-5.15 (m,
1H), 5.55 (dd, J=0.7Hz, 2.8Hz, 1H), 6.14 (dd,
J=0.7Hz, 3.1Hz, lH)
(2) ~y using the compound obtained in the above
(1) and conducting substantially the same procedure as
in Example 1 (2), there was obtained (17S)-20-
isopropylidene-17-methyl-13,14-didehydro-3-thia-PGE
methyl ester 11,15-(t-butyldimethylsilyl ether).
H-NMR (CDCl3, 200MHz) o ppm:
0.09 (s, 3H), 0.10 (s, 3H), 0.11 (s, 3H), 0.13
(s, 3H), 0.84-0.92 (m, 3H), 0.89 (s, 9H), 0.90
(s, 9H), 1.05-2.08 (m, 13H), 1.60 (s, 3H),
1.68 (d, J=0.9Hz, 3H), 2.10-2.24 (m, 1H), 2.17
(dd, J=6.5Hz, 18.2Hz, 1H), 2.57-2.74 (m, 4H),
21~7~61
3.21 (s, 2H), 3.74 (s, 3H), 4.23-4.34 (m, lH),
4.36-4.48 (m, lH), 5.03-5.14 (m, lH)
IR (neat):
2954, 2930, 2857, 2235, 1747, 1463, 1437,
1377, 1362, 1279, 1255, 1131, 1099, 1074,
1007, 939, 838, 778, 670 cm l
(3) By using the compound obtained in the above
(2) and conducting substantially the same procedure as
in Example 1 (3), the title compound was obtained.
lH-NMR (CDCl3, 300MHz) o ppm:
0.94 (d, J=6.4Hz, 3H), 1.14-1.28 (m, 13H),
1.61 (s, 3H), 1.68 (d, J=1.1Hz, 3H), 2.13 (d,
J=5.6Hz, 1H), 2.19-2.28 (m, 1H), 2.24 (dd,
J=9.0Hz, 18.5Hz, 1H), 2.50 (d, J=3.4Hz, lH),
2.61-2.69 (m, 3H), 2.76 (ddd, J=1.3Hz, 7.3Hz,
18.5Hz, 1H), 3.22 (s, 2H), 3.74 (s, 3H), 4.28-
4.38 (m, 1H), 4.44-4.52 (m, 1H), 5.06-5.14 (m,
1H)
IR (neat):
3401, 2927, 2858, 2236, 1742, 1438, 1378,
1284, 1153, 1088, 1012 cm
Example 4
(17S)-20-Isopropylidene-17-methyl-13,14-didehydro-3-
thia-PGEl (compound 4)
By using the compound obtained in Example 3
and conducting substantially the same procedure as in
Example 2, the title compound was obtained.
lH-NMR (CDC13, 300MHz) o ppm:
- 30 0.94 (d, J=6.3Hz, 3H), 1.13-2.06 (m, 19H),
2.22-2.31 (m, 1H), 2.25 (dd, J=9.OHz, 18.5Hz,
lH), 2.63-2.73 (m, 3H), 2.76 (ddd, J=1.3Hz,
7.3Hz, 18.5Hz, 1H), 3.23 (s, 2H), 4.27-4.39
(m, 1H), 4.48-4.56 (m, 1H), 5.05-5.14 (m, 1H)
IR (neat):
3392, 2927, 2857, 2238, 1734, 1455, 1379,
2147461
1288, 1157, 1088 cm
Exampl e 5
(17R)-20-Isop ropyl i dene-1 7-methyl -13,14-di dehydro-3-oxa-
5 PGEl methyl ester ( compound 5)
(1) (3R,4R)-2-Methylene-3-[ (3'S,5 'R)-3' -(t-
butyldi methyl si loxy)-5' ,9 '-dimethyldec-8'-en-1 '-ynyl ]-4-
(t-butyldimethylsiloxy)cyclopentan-1-one was obtained
substantially in the same manner as i n Example 1 (1),
using (3S,5R)-3-(t-butyldimethylsiloxy)-5,9-dimethyldec-
8-en-1-yne i n pl ace of the (3S,5R)-3- (t-
butyldimethyl siloxy)-5-methylnon-1-yne used in Example 1
( 1 ) .
l H-NMR (CDCl3, 200MHz) o ppm:
0.10 (s, 6H), 0.11 (s, 3H), 0.14 (s, 3H),
0.75-1.02 (m, 3H), 0.89 (s, 9H), O.90 (s, 9H),
1.07-2.08 (m, 7H), 1.60 (s, 3H), 1.68 (s, 3H),
2.32 (dd, J=7.3Hz, 17.9Hz, 1H), 2.72 (dd,
J=6.5Hz, 17.9Hz, 1H), 3.48-3.57 (m, 1H), 4.21-
4.34 (m, 1H), 4.36-4.51 (m, 1H), 5.03-5.15 (m,
1H), 5.55 (dd, J=0.6Hz, 2.6Hz, 1H), 6.14 (dd,
J = O .6 H z , 3. O H z , 1 H )
(2) By using the compound obtained in the above
(1) and conducting substantially the same procedure as
i n Example 1 (2), there was obtai ned (17R)-20-
i sopropylidene-17-methyl-13,14-didehydro-3-oxa-PGE
methyl ester 11,15- ( t-but yl di me thyl si l yl et her) .
l H-NMR (CDC13, 200MHz) o ppm:
0.09 (s, 6H), 0.11 (s, 3H), 0.13 (s, 3H),
0.87-0.94 (m, 3H), 0.89 (s, 1 8H), 1 .08-2.05
(m, 1 3H), 1 .60 (s, 3H), 1.68 (d, J=1.OHz, 3H),
2.10-2.21 (m, 2H), 2.60-2.73 (m, 2H), 3.52 ~t,
J=6.4Hz, 2H), 3.75 (s, 3H), 4.07 (s, 2H),
4.22-4.34 (m, lH), 4.38-4.51 (m, 1H), 5.07-
5.13 (m, 1H)
2147~61
24
IR (neat):
2954, 2930, 2858, 2235, 1748, 1473, 1463,
1439, 1377, 1362, 1253, 1207, 1141, 1100,
1006, 940, 839, 810, 779, 670 cm l
(3) By using the compound obtained in the above
(2) and conducting substantially the same procedure as
in Example 1 (3), the title compound was obtained.
H-NMR (CDCl3, 300MHz) o ppm:
0.95 (d, J=6.6Hz, 3H), 1.13-2.07 (m, 13H),
1.60 (s, 3H), 1.68 (s, 3H), 2.20-2.31 (m, 1H),
2.24 (dd, J=8.9Hz, 18.5Hz, 1H), 2.30 (d,
J=5.7Hz, 1H), 2.44 (d, J=3.4Hz, 1H), 2.64-2.70
(m, 1H), 2.75 (ddd, J=1.2Hz, 7.2Hz, 18.5Hz,
1H), 3.52-3.56 (m, 2H), 3.75 (s, 3H), 4.08 (s,
2H), 4.31-4.35 (m, 1H), 4.45-4.48 (m, 1H),
5.07-5.12 (m, 1H)
IR (neat):
3430, 2928, 2236, 1746, 1440, 1378, 1285,
1217, 1141, 1082 cm
Example 6
~17R)-20-Isopropylidene-17-methyl-13,14-didehydro-3-oxa-
PGE~ (compound 6)
By using the compound obtained in Example 5
and conducting substantially the same procedure as in
Example 2, the title compound was obtained.
H-NMR (CDCl3, 300MHz) o ppm:
0.94 (d, J=6.6Hz, 3H), 1.15-2.06 (m, 13H),
1.60 (s, 3H), 1.68 (s, 3H), 2.20-2.29 (m, 1H),
2.25 (dd, J=9.1Hz, 18.5Hz, 1H), 2.64-2.80 (m,
1H), 2.76 (ddd, J=1.2Hz, 7.3Hz, 18.5Hz, 1H),
3.56-3.60 (m, 2H), 4.09 (s, 2H), 4.29-4.37 (m,
1H), 4.46-4.51 (m, 1H), 5.07-5.11 (m, 1H)
IR (neat):
3402, 2929, 2237, 1742, 1446, 1376, 1286,
1148, 1087, 1021, 646 cm l