Note: Descriptions are shown in the official language in which they were submitted.
2~.47~~8
'O 94/10175 PCT/US93/09629
-1-
TOPOISOMERASE II INHIBITORS
AND THERAPEUTIC USES THEREFOR
Backctround of the Invention
Enzymes categorized under the rubric of "DNA
topoisomerase" control the topology of DNA over the
course of conformational and topological changes which
occur during many cellular processes. For example, DNA
topoisomerases are involved in DNA replication, RNA
transcription and :recombination.
Two kinds of DNA topoisomerases are recognized
generally. Type T and type II enzymes catalyze
topological changes in DNA by transiently breaking one
stand or two strands of the DNA helix, respectively. The
relaxation of superhelical DNA is a characteristic
reaction catalyzed by a topoisomerase I ("top 1"), while
a topoisomerase IT ("top 2") catalyzes the passing of two
DNA segments i;n a manner leading to such
topoisomerization reactions of DNA as
supercoiling/relaxation, knotting/unknotting and
catenation/decantenation.
DNA topoisomerase II has been implicated as the
chemotherapeutic target for a diverse group of antitumor
agents, including epipodophyllotoxins, anthracyclines,
acridines, anthracenediones and ellipticines. See
Macdonald et a1 . , in DNA TOPOISOMERASES IN CANCER 199-214
(Oxford University Press 1991) (hereafter "Macdonald
(1991) ") . Under the influence of such drugs, top 2 is
believed to cleave DNA and form a concomitant covalent
association with the broken strands) of duplex DNA. The
formation of such "c:leavable complexes" of drug, DNA and
top 2 enzyme has been attributed to the stabilization by
the drug of a covalE~nt, DNA-bound catalytic intermediate
in the cleavage-resealing sequence of the enzyme. Id.
New inhibitors of top 2 have been identified after
they were noted fox- their antitumor properties. Some,
such as amonafide, genistein and the terpenoides, act in
CA 02147608 2005-07-11
~~47oos
-2_
the manner of the above.-mentioned drugs, by trapping
cleavabla Complexes. Antitumor compounds like marbarone
and fostriecin, by contrast, inhibit top 2 activity
without stabilizing cleavable complexes. -
while attempting to elucidate mechanistic issues in
this field, including the nature of binding sites) fox
top 2 inhibitors ir. the ternary complex, Macdonald ( 1991)
formulated a composite l0.odsl by superi,z~posing structural
subunits of top 2 inhibitors from the give classes of
to compounds mentioned previously, namely,
epipodophyllatoxins, anthracyclines, acridines,
anthracenediones arid elliptici,nes. The composite, three-
domain pharmaaophore encompassed, inter alia, a family of
hybrid structures which incorporated, respectively,
substructural elements from each class of top 2
inhibitors. The °unitied pharmacophore" model was not
refined sufficiently, however, to allow fwr a priori
predictions of any certainty regarding the activity, if
any, of actual molecules deemed within the ambit of the
2o composite.
Hulkenbarg et al., 8P 357,122 discloses beta-
carbolines that 2~re rtported as having good cytostatic
pzoperties. Hulkenberg do net describe the effects the
compounds described therein have on tubulin inhibition or
topoisomerase II catalytic activity. Lateurtre et al.,
Cancer Research, Vol 52(16), 4478-83 (1992) discloses
azatoxin as exhibiting topoisomerase II inhibition
activity, and is described in detail below.
Snmmarv Of the T,nV~nt3on
It is an object of the present invention, therefore,
to provide top 2 inhibitors which display properties that
not only distinguish theM from known inhibitor compounds
but also recommend them for therapeutic uses in
anticancer and antiviral contexts,
In accomplishing tris object and others there has
been provided, in accordance ~rith one aspect of fiche
present invention, co~apounds that are represented by
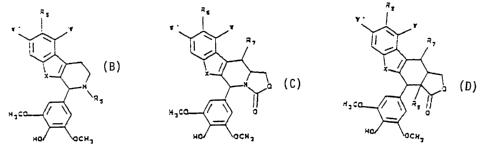
the following formulae (A) - (D):
Ar~~~o~~ s~~
L~2~
Image
'~~ WO 94/10175 _3_ ~ ~ ~~ ~ ~ ~ PCT/US93/09629
wherein
(i) Y represents the formula
i
R~ R3
R2
R1 and R3 are the same or different and denote,
respectively, hydrogen or methoxy,
and
R2 denotes hydroxyl;
and
(ii) R4 denotes hydrogen, hydroxyl, alkyl ether or
hydroxyalkyl ether;
In a preferred embodiment, R1 and R3 both denote methoxy
and R4 denotes hydrogen;
Rs
(B) w
ERs
H3t
a
wherein
(i) X denotes NH, S or O;
(ii) R5 denotes COOCH3, COCH3, COCHZOH;
(iii) R6 denotes F, Cl, Br, CN, OH, H or NH2; and
(iv) W and W' are the same or different and denote,
respectively, H or F;
CA 02147608 2005-07-11
2~.476U~
WO 94/10175 _4 _ PCT/US93/096?~
(C) Rs
w
H3
wherein
(i) X is the same as above;
(ii) W and W' are the same as
above;
(iii) R6 is the same as above; and
(iv) R~ denotes H, OH, the formula
R
-NH
wherein R denotes H, OH F, Br, C1, NO2, NH2, CN, OCH3,
or COZCHZCH3;
the formula
-T- ( CHZ ) a Z
wherein
T denotes NH or O;
Z denotes NHZ, OH, N (CH;) 2, or N ( CH2CH2C1 ) 2;
n is 2, 3 or 4; and
R~ may be derivatized with a 4,6 o-protected sugar;
R6
(D) ~,
H3
CA 02147608 2005-07-11
wherein
(i) X is the same as above;
(ii) W and W' are the same as above;
5 (iii) R4 is the same as above;
(iv) R~ is the same as above; and
(v) R4 denotes H or OH.
The aforesaid compounds are usefull to inhibit topoisomerase II catalytic
activity.
Advantageously, the invention relates to a compound of formula (B):
Rfi
(B)
R5
in which W', W, X, RS and Rb are as defined hereinabove. Preferably, R6 may be
F or
Br.
Advantageously, the invention relates to a compound of formula (C):
RF
Vv
(C)
in which W', W, X, R~ and R~ are as defined hereinabove. Preferably, R~ may be
H;
CA 02147608 2004-05-04
-Sa-
and/or R7 may be selected from the group consisting of
-NH~
and -NH(CH2)ZN(CH3)2.
Advantageously, the invention relates to a compound of formula (D):
R6
w
R
Kg 11
~ 3 C °---~~ o
H0~ ~OCN
3
in which W', W, X, R6, R7 and R8 are as defined hereinabove. Preferably, R~
may be
H; and/or R7 may be - ~ ~
-NN
The term "4,6 o-protected sugar" includes etoposide analogs such as o
glucosyl sugars protected with a conventional protecting group such as a
methyl
acetal or a thiophene acetal group.
It has surprisingly been found that compounds of formula (C), preferably
when R6 is H, R~ is
-N i-1
and R is F, have increased topoisomerase II catalytic activity when compared
to
compounds of formula (A). The activity is increased by a factor of at least 3,
and
preferably by at least 5, when compared to compounds of formula (A). The
compounds of formula (A) are not claimed in the present invention. Those
skilled in
the art readily recognize that the dosage amount required for compounds having
such
an increase in activity are respectively decreased to maintain the same or a
similar
effect.
m
CA 02147608 2004-05-04
Sb-
In accordance with another aspect of the present invention, a pharmaceutical
composition is provided that comprises a tumor-affecting amount of a compound
represented by formula (B), (C) or (D), and a physiologically compatible
carrier for
that compound. In one preferred embodiment, the pharmaceutical composition is
an
injectable, or infusible preparation.
In accordance with yet another aspect of the present invention, a use and a
method is provided for treating cancer in a mammal, which method comprises the
step
of bringing a pharmaceutical composition as described above into contact with
cancerous tissue in a mammal suffering from
y. r . . W v ~ cr n - ~.. W G. W .. .a... . ~.
2~47~pg
- 6 -
a tumor, such that neoplastic development in the
cancerous tissue is retarded or arrested. Thus,
preferred modes of administration in the context of such
a method are those that maximize contact between -_
cancerous tissues and the active agent of the
pharmaceutical composition.
other objects, features and advantages of thA
present invention will become apparent from the following
detailed description. It should be understood, however,
that the detailed description and the specific examples,
while indicating preferred embodiments of the inventions,
are given by way of illustration only, since various
changes and modifications within the spirit and scope of
the invention Will become apparent to those skilled in
the art rrom this detailed description.
Brief Description ~ha Dra,~inas
Figure s is a graphical representation of results
generated by testing the inhibitory activity of azatoxin
against panel of tumor cell lines which is a standard of
the National Cancer Institute Developmental Therapeutics
Program (GI5a: "50% azowth inhibition"; TGI: ~~tumor
growth inhibition~~; LCSQ: ~~50% lethal concentration's ] .
Figure 2 depicts, at left, the structural formula of
a less preferred compound, azatoxin and, at right, a
stereochemical superimposition of the top 2 poison
demethyldesoxy--podophyllotoxin (DIP; dashed lines) and
azatoxin (solid lines).
t d a cri t o t a fsr ed Efibodiments
A new class of top 2 poisens has been discovered
which, dsapite certain structural similarities to
demethylepipodophyllotoxins and other known top 2
inhibitors, are distinguishable in terms of DNA cleavage
pattern and structure-activity relationships which could
not have been predicted from any structural
superiraposition of known anthracycl;ne, acridine',
epipodophyllotoxin and antriracenedione structures.
Archetypical of the new class of inhibitors i.s the
compound azatoxin and derivatives thereof, SR,llas-3-one-
~,?~~F~~;DtD S~tEcT
tc~ v . v uw : tr.y-:w t:wrtt.v t . ~+- 1 1 -a'~ ~ __. ~ m ~ -uy, wtW ;3'.i--
+~.a ti:3 '':35:J4-~b5 : tt 1()
- 6A -
1H,6H,-5,4,11,11a-tetrahydra-5-(3,5-dimethoxy-4-
AMENDED ShE~T
'YO 94/10175 PCT/US93/09629
-7-
hydroxyphenyl)-oxazolo[3',4':1,6]pyrido[3,4-b]indole,
which is represented by formula (A) above when R1 and R3
are methoxy, R2 is hydroxyl and R4 is hydrogen.
Compared to other top 2 inhibitors, azatoxin induces
the largest number of top 2 cleavage sites both in SV40
and c-myc DNA, and is very active in inducing protein
linked DNA breaks in cells. Accordingly, azatoxin and
other pharmacophores of the present invention should be
useful as reagent: in the context of mapping top 2 sites
in chromatin.
Azatoxin also displays a pattern of differential
growth inhibition against human tumor cell lines which is
indicative of an antitumor drug action reminiscent of
that of top 2 poisons like the nonintercalative
epipodophyllotoxins, VP-16 (etoposide) and VM-26
(teniposide). See: Yang et al., Cancer Res. 45: 5872-76
(1985), and Liu (1989), supra, at 361-63. More
specifically, azatoxin evidenced significant inhibitory
activity when screened against a panel of sixty human
tumor cells lines representing leukemia and melanoma, as
well as cancers of the lung, colon, kidney, ovary and
central nervous system. Pursuant to the convention of
Paull et al., J. Nat'3 Cancer Inst. 81: 1088-92 (1989),
Figure 1 depicts these results, i.n terms of parameters
conventional to cancer research, by graphs which are
centered at the arithmetic mean of the logarithm of each
parameter. See also Monks et al., J. Nat'1 Cancer Inst.
83: 757-66 (1991), and Boyd, Principles & Practice of
Oncology 3: 1-12 (:1989) .
Since a right-extending bar in such a "means graph"
indicates a sensitivity by the cell line to the test
substance, Figure 1 evidences the cytotoxicity of
aratoxin to cells associated with disseminated cancers
(leukemias) as well as several consolidated cancers (non-
small cell lung and colon) . It also has been discovered,
using a conventional in vitro tubulin polymerization
assay, that azatoxin is a potent tubulin inhibitor in the
manner of several compounds, such as vineristine,
WO 94/10175
-8 - PCT/US93/096?~'~
vinblastine and taxol, that are very active in cancer
chemotherapy. Thus, azatoxin effectively prevents
tubulin polymerization in vitro at concentrations in the
range of 1 to 10 ~M.
Compounds of the present invention can be synthesized
via a modified Pi.ctet-Spengler reaction. More
specifically, the inventive compounds are obtained by
reacting a corresponding pendent-group dimethylacetal
with a precursor carbamate in the presence of a catalytic
amount of para-toluenesulfonic acid.
Because the azatoxin skeleton is synthetically
accessible, a large number of derivatives are readily
prepared and screened for top 2 inhibitory activity in
accordance with the present invention. Such screening can
be effected by means of an in vitro assay which employs
purified top 2 and 3~P-end-labelled DNA. In such an
assay, top 2 inhibition by a test substance results in
DNA fragmentation which can be detected by agarose gel
electrophoresis, as described by Fesen and Pommier, J.
Biol. Chem. 19: 11354-59 (1989); by the filter-binding
assay described by Pommier et al., Biochem. 24: 6410-16
(1985); or by the sodium dodecylsulfate precipitation
assay employed by Nelson et al., Proc. Nat'1 Acad. Sci.
USA 81: 1361-65 (1984).
Although a myriad of azatoxin derivatives may be
synthesized, the structure-activity relationships
illuminated in this description indicate that those
derivatives falling within the present invention will
conform to certain guidelines of molecular design. Thus,
with reference to formula (A) and Figure 1, there should
be a conservation of the relative spatial orientation
between the polycyclic ring system and the pendant ring
(Y) in azatoxin. The R/S stereochemistry of the
polycyclic ring system also should be carried over from
azatoxin. Methoxy groups at the 3' and 5' positions (R1
and R3, respectively) enhance top 2 inhibition, while a
4' hydroxyl group (R2) is essential for inhibitory
activity. In contrast to R2, greater flexibility is
_ 2~ ~ 7~0~
~~ WO 94/10175 PCT/US93/09629
-9-
realized at the 11R position (R4) , where substitutions of
the preferred hydrogen can be a hydroxyl group, an alkyl
ether group or a hydroxyalkyl ether group such as
ethoxyethyl and hydroxypropyl.
The present invention also contemplates the use of
compounds according to formulae (A), (B), (C) and (D) in
a pharmaceutical composition which further comprises a
physiological compatible carrier for the compound. The
phrase "physiological compatible carrier" here denotes a
solid, liquid or gaseous material that can be used as a
vehicle for administering a formula (A), (B), (C), or (D)
compound as a medicament because the material is inert or
otherwise medically acceptable, as well as compatible
with the active agent,. in a particular context of
administration. In addition to a suitable excipient, a
physiologically compatible carrier can contain
conventional additives such as diluents, adjuvants,
antioxidants and other preservatives, solubilizing
agents, and the like.
The preferred routes for administering a
pharmaceutical composition of the present invention are
those that maximize introduction of the active agent into
the immediate region of the tumor under treatment. It is
advantageous, therefore, that the pharmaceutical
composition should be an injectable or infusible
preparation, the formulation of which would typically
require initial solubilization in a nonaqueous solvent,
such as dimethyl sulfaxide (DMSO) , that is employed in
the field to accommodate hydrophobic anticancer agents.
Similarly, intraarterial administration is preferred for
therapy when the tumor is supplied by a known artery,
while intrathecal administration can be used for tumors
located in the brain.
Intradermal and intracavitary administration is
feasible with tumors that are restricted to areas close
to a particular skin region and particular portion of the
body cavity, respectively. By the same token, an active
agent of the present invention can be administered via
WO 94/10175 ~ ~ ~ ~ ~ ~ PCT/US93/0962'
-10-
application to a mucosal lining (sublingually, for
example), when the tumor is accessible through the
lining, or via inhalation or insufflation with a gaseous
carrier, when the tumor is accessible to the respiratory
tract. It is possible, furthermore, to administer an
active agent of the present invention in a topical
preparation applied to a superficial tumor or, more
generally, to a lesion associated with a viral infection
against which compounds of the present invention prove
effective.
Alternatively, an inventive pharmaceutical
preparation can be in a form suitable for oral
administration (cachet, tablet, hard- and soft-gelatin
capsule, etc.). This route takes advantage of the
targeting afforded by the particular sensitivity of
proliferating, neoplastic cells to the cytoxic effects of
the inventive compounds.
A pharmaceutical composition of the present invention
also can take the form of a solid dosage preparation
(implant) for introduction surgically into the tumor or
its immediate vicinity. A so-called "implantation
tablet" would be made up primarily of the active
substance and, hence, could be absorbed completely. On
the other hand, an implant featuring a non-absorbable
skeleton (plastic matrix) or coating would effect
controlled release of the inventive compounds upon
implantation and then would be removed from the tissues
after ceasing to exert an antitumor influence.
A pharmaceutical composition within the present
invention preferably contains an inventive compound in an
amount that itself is tumor-inhibiting or, in the context
an infusion regimen, that permits accumulation in the
tumor of a cytotoxic level of the active agent. Since an
inventive compound typically inhibits both topoisomerase
II activity and tubulin polymerization, it is possible to
realize a therapeutic spectrum combining aspects both of
a top 2 poison like doxorubicin and a tubulin inhibitor
like vincristine. See GOODMAN AND GILLMAN'S THE
"V0 94/10175 -11- PCT/US93/09629
PHARMACOLOGICAL BASIS OF THERAPEUTICS (7th ed. 1985), at
pages 1279-85. Symptoms of clinical toxicity associated
with these two types of anticancer agents also may be
observed and, if so, will require remedial
countermeasures which are conventional to the field of
cancer therapy.
From these considerations it will be apparent that
the optimum dosage of the inventive compound will vary
with the particular case. The relevant pharmacokinetics
can be determined routinely in the clinical context,
which may be therapeutic or prophylactic. "Therapeutic
treatment" in this context means treatment intended to
kill a maximum number of neoplastic cells, while a
"prophylactic treatment",is one aimed at retarding or
preventing re-establishment of a proliferating neoplastic
population and, hence, a relapse in the disease, once
remission has been achieved. It is anticipated that a
typical dosage regimen will be similar to that of
etoposide (VP-16), i.e., on the order of 100 mg/m2 per
day.
Without further elaboration, it is believed that
those skilled in the art, informed by the preceding
description, can utilize the present invention fully.
Accordingly, the following examples are presented for
purposes of illustration only. The materials and methods
employed in the examples are described below:
Chemicals, and enzymes: Azatoxin and its
derivatives, as well as the demethyldesoxypodo
phyllotoxin (DMDP) and demethylepipodophyllotoxin (DMEP),
were synthesized by conventional methods. The compounds
m-AMSA and mitoxantrone were obtained from the Drug
Synthesis and Chemistry Branch, National Cancer
Institute, Bethesda, MD. The compounds VP-16 and VM-26
were obtained from Bristol-Myers Company (Wallingford,
CT) . Drug stock solutions were made in dimethylsulfoxide
at 10 mM. Further dilutions were made in distilled
water. Various other reagents, including simian virus 40
(SV40) and c-myc human DNA inserts in plasmid pBR322,
2~4760~
WO 94/10175 PCT/US93/0962'
-12-
restriction endonucleases, DNA topoisomerase I, T4
polynucleotide kinase, calf intestine phosphatase,
agarose and polyacrylamide/bis, were purchased from
Bethesda Research Laboratories (Gaithersburg, MD), from
the American Type Culture Collection (Rockville, MD) or
from New England Biolabs (Beverly, MA) . [Gamma 32P]ATP
was purchased from New England Nuclear Research Products
(Boston, MA).
DNA topoisomerase II was purified from mouse leukemia
L1210 cell nuclei as described, for example, by Pommier
et a1. (1985), supra, and was stored at -70°C in 40%
(v/v) glycerol, 0.35 M NaCl, 5 mM MgCl2, 1 mM KHZP04, 0.2
mM dithiothreitol and 0.1 mM phenylmethanesulfonyl
fluoride (pH 6.4). The purified enzyme yielded a single
170 kDa band after silver staining of SDS-polyacrylamide
gels, in accordance with the description of Pommier et
al., Cancer Res. 46: 3075-81 (1986).
Preparation of end-labeled DNA fragments: DNA
fragments were 5' end-labeled as described, for example,
by Fesen and Pommier (1989), supra. Briefly, native DNA
was first linearized with a restriction enzyme, then the
5'-DNA was first linearized with a restriction enzyme,
then the 5'-DNA termini were dephosphorylated with calf
alkaline phosphatase and labeled with [gamma-3zP]ATP using
T4 polynucleotide kinase. For double-strand breaks
assays using HL-60 nuclear extract, SV40 DNA was digested
with BclI endonuclease and labeled at both DNA termini.
For sequencing experiments, SV40 and c-myc DNAs were
first 5'-end labeled at the XhoII and XbaI restriction
sites, respectively. Then, in order to generate uniquely
5'-end-labeled fragments, labeled DNA was subjected to a
second enzyme digestion, PfIMI for SV40, and EcoRI plus
HindIII for c-myc DNA. The resulting DNA fragments were
separated by agarose gel electrophoresis and isolated by
electroelution. Purification by phenol-chloroform
extraction and ethanol precipitation were included
between each step and at the end of the labeling
~-~ ~ 760
4~V0 94/10175 -13- PCT/US93/09629
procedures, pursuant to Pommier et al., J. Molec. Biol.
222: 909-24 (1991).
Topoisomerase II-induced DNA cleavage reactions: DNA
fragments were equilibrated with or without drug in 10 mM
Tris-HC1, pH 7.5, 50 mM KC1, 5 mM MgCl2 0.1 mM EDTA, 1 mM
ATP and 15 ~cg/ml bovine serum albumin for 5 minutes
before addition of purified topoisomerase II (40 to 70
ng) or HL-60 nuclear extract in 20 ~1 final reaction
volume. Reactions were stopped by adding sodium dodecyl
sulfate (SDS) to a final concentration of 1% and
proteinase K to 400 ~.g/ml, followed by incubation for 1
hour at 40°C.
For agarose gel analysis, 3 u1 (10 X) loading buffer
(0.3% bromophenol blue, 16% Ficoll, 10 mM Na2HP04) was
added to each sample which was then heated at 65°C for 1
2 minutes before loading into an agarose gel made in (1X)
TBE (89 mM Tris, 89 mM boric acid, 2 mM EDTA, pH8), in
accordance with Fesen & Pommier (1989), supra. Agarose
gel electrophoresis was conducted overnight at 2 V/cm.
The quantification of drug-induced DNA double-strand
breaks in the presence of HL-60 nuclear extract was
effected as described .in the next paragraph.
Radioactive gels were counted in betascope 603 blot
analyzer. For each lane radioactivity then was measured
in the DNA cleavage products (C) (size between 600
5243bp), and in the total DNA present in the lane with a
size superior to 600 by (T). Drug-induced cleavage was
expressed as:
C/T - C./T.
Percent DNA cleaved = 100 x
1 - C./T.
where C. and T. are the counts for cleaved and total DNA,
respectively, in presence of nuclear extract without
drug.
For DNA sequence analysis, samples were precipitated
with ethanol and resuspended in 2.5 ~,l loading buffer
(80% formamide, 10 mM NaOH, 1 mM EDTA, 0.1% xylene cyanol
CA 02147608 2004-05-04
-14-
and 0.1% bromophenol blue). Samples were heated to 90°C and immediately
loaded into
DNA sequencing gels (6% polyacrylamide; 19:1, acrylamide:bis) containing 7 M
urea in
(1X) TBE buffer. Electrophoresis was at 2500 V (60 W) for 4 hours. Gels were
dried on
3M paper sheets and autoradiographed with Kodak* XAR-2 film. See Pommier et
al.
(1991), supra.
EXAMPLE 1. SYNTHESIS OF AZATOXIN AND OTHER COMPOUNDS
1H NMR spectra were taken on a General Electric QE300 Spectrometer at 300
MHz. Mass spectra were recorded on a Finnegan MAT4 615 GC/MS/DS instrument
using chemical or electron impact ionization techniques. Elemental analyses
were
determined by Atlantic Microlab Inc. (Norcross, GA). Melting points were
determined
on a Thomas-Hoover UNI-MELT* apparatus and are uncorrected.
Thin-layer chromatography was performed using E. Merck glass plates pre-
coated with silica gel 60 F-254 and visualized with a phosphomolybdic
acidlethanol
solution. Woelm silica 32-63 was employed for column chromatography which was
carried out using.a modified shortlflash column technique.
Tetrahydrofuran was distilled from sodium benzophenone immediately prior to
use. Dichloromethane was distilled from CaH2 immediately before use. All
chemicals
were purchased from Aldrich Chemical Company except for D and L-tryptophan
methyl
ester-HCL, which was purchased from Sigma Chemical Company.
All reactions were carried under an argon atmosphere.
48- (1H-Indol-3-ylmethyl)-2-oxazolidone (Compound 1A):
Sodium (1.56 g, 68.1 mmol) was dissolved in absolute ethanol (150 ml) and 1-
trptophanol (12.94 g, 68.02 mmol) in ethanol (100 ml) and diethyl carbonate
(8.83 g, 74.8
mmol) were added. The solution was heated at reflux for 5 hours and was
concentrated
after cooling. Saturated NH3C1 (100 ml) and CHZC12 (200 ml) were added, and
after
mixing well the layers were separated. The organic layer
*Trademark
YO 94/10175 -15- PCT/US93/09629
was washed with CHZC12 (2 X 100m1) and the organic
fractions were combined, dried over Na2S04, and
concentrated. Recrystallization from MeOH/H20 yielded
11.66 g (79%) of a white solid: mp 155oC; 1NMR (CDCL3)
8.17(sb, 1H) 7.57(d, J=7.94 Hz, 1H), 7.40(d, J=8.06 Hz,
1H) , 7 .19 (m, 2H) , 7 . 08 (d, J=2 . 2 Hz, 1H) , 5. 22 (sb, 1H) ,
4.50(m, 1H), 4.21(m, 2H), 3.04(m, 2H). [g]~°D +8.4 (c
9.4, MeOH) . Anal. (C~ZH12N202) C, H, N.
4R-(1H-Indol-3-ylmethyl)-2-osazolidone:
This compound was prepared in a manner analogous to
the preparation of Compound la. [a]~'D -8.3 (c 1.25,
MeOH) , Anal. (C12H,2N202) C, H, N.
General Method for the Preparation of Dimethvl Acetals
To a solution of the aromatic aldehydes (1 g) in
trimethyl orthoformate (7 ml) a catalytic amount of p-
TSOH (40 mg) was added and the reaction was followed to
completion by TLC. The solvent was removed under reduced
pressure and the remaining oil was dissolved in CHC12 and
filtered through a plug of silica. The solvent was again
removed under reduced pressure and the remaining oil was
stored in a desiccator until use.
Preparation of 5,lla-trans-aza toxins:
Method A
To a solution of Compound la (2 mmol) and the
corresponding aldehyde (2 mmol) in a CHZCIz/MeOH (9:1)
solution (6 ml) was added concentrated HZS04 (4 mmol).
The reaction was followed to completion by TLC (20%
acetone in CHCL3). The solution was added to sat NaHC03,
the layers were separated, and the aqueous layer was
washed with CHZC12 (3X). The combined organic fractions
were dried over Na2S04, filtered and concentrated. The
product was purified by flash chromatography (acetone-
CHZC12) .
WO 94/10175 PCT/US93/096"
-16-
Method B
To a solution of the corresponding dimethyl acetal
( 2 . 2 mmol ) and Compound 1a ( 2 mol ) in CHC13 ( 8 ml ) , p-
toluenesulfonic acid (.2 mmol) was added . The reaction
was followed to completion by TLC. If the reaction
proceeded too slowly the reaction was heated at reflux.
Saturated NaHC03 was added. The layers were separated,
and the aqueous layer was washed with CH2C13 (2 x 20 ML).
The organic layers were combined, dried over Na2S04,
filtered and concentrated. The product was purified by
f lash chromatography ( acetone-CHZC12) .
Method C
To a solution of the corresponding dimethyl acetal
(3 mmol) and Compound la ~(2 mmol) in anhydrous THF
(8 ml), anhydrous TFA (10 mmol) was added and the
solution was heated at reflux. The reaction was followed
to completion by TLC. After cooling, the solution was
added to saturated NaHC03, the layers were separated, and
the aqueous layer was washed with CHZC12 (3 X 20 ml) . The
combined organic fractions were dried over Na2S04,
filtered and concentrated. The product was purified by
flash chromatography (acetone-CHZC12).
5R,11x8-3-One-5,4,11,i1a-tetrahydro-5-(3,5-dimethouy-4-
hydro$yphenyl)-1H,6H-oxazolo[3',4':1,6]pyrido[3,4-
b]indole (Compound 1)
This compound was prepared as described in method C.
Purification by flash chromatography (12% acetone in
CHC13, Rf=.28) produced a white solid in 91% yield: mp
(decomposed slowly around 175°C) ; 1NMR (CD3CN) 7.94 (sb,
1H), 7.51(d, J-7.91 Hz, 1H), 7.30(d, J=7.57 Hz, 1H),
7.09(m, 2H), 6.59(s, 2H), 6.27(s, 1H), 5.88(d, J=1.7 Hz,
1H), 4.54(dd app t, J=8.3 Hz, 1H), 4.33(m, 1H), 4.21(dd,
J=8.5 Hz, 4.7 Hz, 1H), 3.75(s, 6H), 3.16,(dd, J=15 Hz,
4.6 Hz, 1H), 2.76(ddd, J=15 Hz, 10.38 Hz, 1.73 Hz, J=1.73
Hz, 1H) , [a]ZZD=-139.6. Anal. (CZ~,HZO,Nz,05) C, H, N.
~'VO 94/10175 -17- PCT/US93/09629
58,iiaR-3-One-5,4,11,i1a-tetrahydro-5-(3,5-dimethogy-4-
hydrogyphenyl)-1H,6H-oxazolo[3,4,:1,6]pyrido[3,4-b]indole
This campound was prepared in a manner analogous to
the preparation of Compound 1. [a]nD=+139.4. Anal.
(Cz~~Hzo~Nz.Os) O~ H~ N.
5R,11a8-3-One-5,4,ii,iia-tetrahydro-5-(3-methosy-4-
hydrouyphenyl)-iH,6H-oxazolo[3',4':1.6]pyrido[3,4-
b]indole (Compound 2)
This compound was prepared as described in method A.
Purification by flash chromatography (15% acetone in
CHC13, Rr=.30) gave a white solid in 40% yield: 'NMR
(CD3CN) 8.94(s, 1H), 7.5~1(d, J=7.59 Hz, 1H), 7.29(d,
J=7.95 Hz, 1H), 7.09(m, 2H), 6.91(d, J=1.7 Hz, 1H,),
6.79(d, J=8.08 Hz, 1H), 6.74(dd, J=8.08 Hz, 1.7 Hz 1H),
6.55(sb, 1H), 5.90(d, J=1.5 Hz, 1H), 4.52(dd app t,
J=7.94 Hz, 1H), 5.27(m.lH), 4.20(dd, J=8.21 Hz, 4.83 Hz,
1H), 3.78(s, 3H), 3.16(dd, J=14.93 Hz, 8.48 Hz, 1H),
3.77(ddd, J=14.91 Hz, 10.07 Hz, 1.59 Hz, 1H). Anal.
(CzoH~aN20a) C. H~ N.
SR,ilaB-3-one-5,4,ii,ila-tetrahydro-5-(4-hydrogyphenyl)-
iH,6H-ogazolo[3~,4':1,6]
pyrido[3,4-b]indole (Compound 3)
This compound was prepared as described in method C.
Purification by flash chromatography (20% acetone in
CHC13, Rf=.28) followed by recrystallization from CH3CN
gave a white solid in 89 % yield: ~NMR (db-DMSO) 9.47 (s,
1H), 7.43(d, J=7.56 Hz, 1H), 7.24(d, J=7.91 Hz, 1H),
6.99(m, 4H), 6.70(d, J=8.6 Hz, 2H), 5.81(s, 1H), 4.49(dd
app t, J=8.03 Hz, 1H), 4.14(m, 2H), 3.10(dd, J=12.14 Hz,
4.69 Hz, 1H), 2.68(dd, J=14.3 Hz, 10.6 Hz, 1H). Anal.
(Cl9.Hi6~Nz.03/CH3CN) C, H, N.
SR,ila8-3-one-5,4,i1,ila,tetrahydro-5-(3,4,5-
trimethoxyphenyl)-iH,6H-ouazolo[3',4':1,6]pyrido[3,4-
b]indole (Compound 4)
WO 94/10175 ~ ~ ~ ~ ~ ~ -18- PCT/US93/096~~
This compound was prepared as described in method C.
Purification by flash chromatography (7% acetone in
CHC13, Rf=.30) yielded a white solid in 91% yield: mp
203°C, ~NMR (CD3CN) 8.95(s, 1H), 7.51(d, J=7.64 Hz, 1H),
7 . 31 (d, J=7. 93 Hz, 1H) , 7 . 09 (m, 2H) , 6. 61 (s, 2H) , 5. 89 (s,
1H) , 4 . 57 (dd app t, J=8. 24 Hz, 1H) , 4 . 35 (m, 1H) , 4.23 (dd,
J=8.42 Hz, 4.68 Hz, 1H), 3.74(s, 6H), 3.70(s, 3H),
3.17(dd, J=15.02 Hz, 4.48, Hz, 1H), 2.77(dd, J=14.85 Hz,
10.41 Hz, 1H).
Methyl-3-(1-benzenesulfonyl-indol-2-yl)-2-amino-
propanoate (Compound Sa)
This compound was prepared, according to the method
of Schollkopf et al., Angew, Chem. Int. Ed. Engl. 18: 863
(1979), using (1-benzenesulfonyl)-2-chloromethyl-indole
(Compound X) and 2,5-diethoxy-3,6-tetrahydropyrazine
(Compound Y) as starting materials. 'H NMR(CDC13)
8,19(d,lH), 7,79(d, 2H), 7,55-7,18(m, 6H), 6.51(s,lH),
4,19(q, 2H), 4,00(dd, 1H), 3.55(dd, 1H), 1.18(t, 3H).
3-(1-benzenesulfonyl-indol-2-yl)-2-aminopropanol
(Compound 8b)
To a well-stirred solution of 0.44g (4.1 eq.) NaBH4
in 20 mL 75 % ethanol, 0. 85g (8a) in 10 mL 75% ethanol was
added and the solution heated at reflux 15 hours. TLC
showed no starting material. The solution was allowed to
cool and then it was diluted with 20 mL water and the
ethanol was removed by rotary evaporation. The residue
was extracted with ethyl acetate (4 X 20mL), dried over
sodium sulfate, and concentrated to yield 0.56g (56%) of
Compound 8b as a clear oil which was used without further
purification. 'H NMR (CDC13) 8.18(d, 1H), 7.71(d, 2H),
7.60-7.19(m, 6H), 6.50(s, 1H), 3.70(m, 1H), 3.50(dd, 1H),
3.45(dd, 1H), 3.25(dd, 1H), 2.90(dd, 1H).
iH-Indol-2-ylmethyl-2-oxazolidone (Compound 8c)
To a solution of 0.07g (1.5 eq) Na dissolved in 5 mL
absolute ethanol, 0.67g of Compound 8b in 10 mL absolute
ethanol and 0.36g (1.5 eq) diethyl carbonate were added
~~~'O 94/10175
PCT/US93/09629
-19-
and the solution was heated at reflux overnight (16
hours). TLC showed a higher Rf spot(80:20) ethyl
acetate: hexane, Rt=0.35). After cooling the ethanol was
evaporated by rotary evaporation and the residue was
diluted with 15 mL saturated ammonium chloride and
extracted with CHZC12 (3 X 20 mL). Purification by flash
chromatography using an 80:20 ethyl acetate: hexane
mixture as eluent yielded 0.22g (71%) of Compound 8c as
a tan solid. 'H NMR(CDC13) 8.85(s, 1H), 7.55(d, 1H),
7.30(d, 1H), 7.15(m, 2H), 6.45(s, 1H), 6.20(g, 1H),
4.30(m, 1H), 4.00(m, 2H), 2.82(d, 2H).
S,iia-traps-3-one-5,,4,11,i1a-tetrahydro-5-(3,5-dimethouy-
d-hydrogyphenyl-1H, lOH-ouazolo[3',4':1,6]pyrido[4,3-
b]indole (Compound 8)
To a solution of 0.7g (1.5 eq) syringealdehyde
dimethyl acetal and a few grains p-TSOH in 2 mL CHZC12 was
added 0.04 g of Compound 8c. The solution was stirred
for 2 hours. TLC showed no starting material. The
solution was concentrated and purified by flash
chromatography using a 15% acetone in chloroform solution
as eluent to yield 0.05g( 69%) of Compound 8 as a beige
solid. 'H NMR(CDCN) 9.28(bs, 1H), 7.40(d, 1H), 7.15(t,
1H), 7.00(d, 1H), 6.92(t, 1H), 6.58(s, 2H), 6.22(bs, 1H),
5.98(s, 1H), 4.47(t, 1H), 4.31(m, 1H), 4.16(q, 1h),
3.69(s, 6H), 3.11(dd, 1H), 2.91(ddd, 1H).
Raaemic methyl-3-(acenapth-1-yl)-2-aminopropanoate
(Compound 7a)
This compound was synthesized according to the method
of Schollkopf et al., Liebigs Ann. Chem. 1987: 393-97,
using acenapthylene-1-carboxaldehyde (Compound Z) and
2,5-diethoxy-3,6-tetrahydropyrazine (Compound Y) as
starting materials. The final product was synthesized in
an identical manner as described for Compound 8. Due to
the substitution in the 4-position, however, the
intermediates existed as an inseparable mixture of
-2 0- PCT/US93/096"
WO 94/10175
diastereomers and 'H NMR analysis proved impossible,
except for identification of important groups.
S,ila-trans-3-one-5, 4, 11, ila-tetrahydro-5-(3,5
dimethosy-4-hydroxyphenyl)-iH, 10H-ouazol [3', 4' :1,6]
pyrido[1, 2-b] acenapthylene (Compound 7)
0.01 mL BP3oEtz was added to a solution of 0.03g of
Compound 7d and 0.01 mL triethyl silane in 1mL CH2C12 at -
78 °C. The solution was warmed slowly to room temperature
over a period of 2 hours. TLC showed no starting
material. 5 mL Hz0 was added, the layers were separated,
and the aqueous layers were extracted with 2 X 5 mL
portions of CHZC12, dried and concentrated. The residue
was purified by flash chromatography using a 15% acetone
in chloroform solution as eluent to yield 19.4 mg (70%)
of Compound 7. 'H NMR(CDC13) 7.81(d, 1H), 7.71(d, 1H),
7 . 65-7 . 51 (m, 2H) , 7. 38 (t, 1H" ) , 7 .11 (d, 1H) , 6. 72 (s, 2H) ,
6.14(d, 1H), 5.52(s, 1H), 4.58(t, 1H), 4.25(m, 2H),
3.77(s, 6H), 3.24(dd, 1H), 2.90(ddd, 1H).
iR,3R-1-(3-5-dimethoxy-4-hydroayphenyl)-3
methosycarbonyl-1,2,3,4-tetrahydro-~-carboline(Compound
9A)
To a solution of 1-tryptophan methyl ester
hydrochloride (9.52 g, 37.4 mmol) in CHC13 (150 ml) was
added 14% ammonium hydroxide (30 ml). The biphasic
mixture was allowed to stir for 1 hour. The layers were
separated and the aqueous layer was extracted with CHC13
(2 X 100 ml). The organic layers were combined, dried
over Na2S04, and concentrated to yield a yellow oil. The
oil was dissolved in benzene (200 ml). Syringaldehyde
(6.818, 37.4 mmol) and NaZS04 (10 g) were added, and the
solution was allowed to stir for 60 hours. A white
precipitate formed. The mixture was again concentrated,
and anhydrous CHZCIz (150 ml) and anhydrous TFA (5.76 ml,
74.8 mmol) were added at 0°C. The solution was allowed
to stir at 0°C for 12 hours. The mixture was again
2.~~'~~0~
"~O 94/10175 -21- PCT/US93/09629
concentrated, and the remaining solid was added to a
biphasic mixture of saturated NaHC03 and ether. The
mixture was allowed to stir for 1.5 hours and the white
solid that formed was collected in a sintered glass
funnel. The solid was washed with water, dried in a
vacuum oven, and recrystallized from CH3CN/water to
produce 13.34 g (93%) of a white solid: Anal.
(C21iH22iN2i~3) Ci Hr N.
18,38-1-(3,5-dimethoay-4-hydrosyphenyl)-3-
methouycarbonyl-1,2,3,4-tetrahydro-~-carboline
This compound was prepared in a manner analogous to
the preparation of Compound 9A. Anal . (c21, Hue, N2, OS) C,
H, N.
iR,3R-1-(3,5-dimethoxy-4-hydro8yphenyl)-3-hydrosymethyl-
1,2,3,4-tetrahydro-~-carboline (Compound 98)
To a solution of Compound 9a (2.01 g, 5.31 mmol) in
1:1 dioxane/water (20 ml) NaBH~ (1.00 g, 26.6 mmol) was
added, and the solution was allowed to stir at room
temperature for 3 hours. The solvent was removed under
reduced pressure and the remaining solid was redissolved.
The product was precipitated by the addition of NaCl, and
collected by filtration and dried in a vacuum dessicator
to yield 1.43 g (76~) of a white solid which was used
without further purification.
18,38-i-(3,5-dimethouy-4-hydroxyphenyl)-3-hydrosymethyl-
1,2,3,4-tetrahydro-f~-carboline
This compound was prepared and analyzed in a manner
analogous to the preparation of Compound 9B.
WO 94/10175 PCT/US93/096'
-22-
SR,iiaR-3-One-5,4,ii,iia-tetrahydro-5-(3,5-dimethosy-4-
hydrosyphenyl)-iH,6H-osazolo[3'4':1,6']pyrido[3,4-
b]indole (Compound 9)
To a suspension of the amino-alcohol (1.31 g 3.70
mmol) in THF (10 ml), carbonyl diimidazole (1.79 g, 11.1
mmol) was added and the suspension was allowed to stir
for 5 hours. The suspension was concentrated and 10%
NaOH was added. After stirring for an additional 3
hours, the solution was carefully acidified to pH 6 by
the addition of concentrated HC1 and the resulting
mixture was extracted with EtOAc (3 X 50 ml), dried over
Na2S04 and concentrated. ~NMR (CDC13) 7.58 (s, 1H) , 7. 51 (d,
J=8. 37 Hz, 1H) , 7.19 (m, 3H) , 6. 57 (s, 2H) , 5. 52 (s, 1H) ,
5.24 (s, 1H) , 4. 63 (t, J=6. 65 Hz, 1H) , 4.22 (m, 2H) , 3.83 (s,
OH), 3.22(m, 1H), 2.92(ddd, J=16.4 Hz, J=10.1 Hz, J=1.8
Hz, 1H) . Anal (C21, HZO, N2, OS) C, H, N.
58,i1a8-3-One-5,4,ii,ila-tetrahydro-5-(3,5-dimethouy-4-
hydroxyphenyl)-iH, GH-oxazolo[3'4':1,6]pyrido[3,4-
b]indole (Compound i0)
This compound was prepared in a manner analogous to
the preparation of Compound 9 . Anal . (CZ" H2o, NZ, OS) C, H,
N.
SR,iiR,iiaB-3-One-ii-methosy-5,4,ii,iia-tetrahydro-5
(3,5-dimethoxy-4-hydrogyphenyl)-iH,6H
oaazolo[3'4':1,6]pyrido[3,4-b]indole (Compound 1C)
To a solution of syringaldehyde dimethyl acetal (.50
g) and catalytic p-toluenesulfonic acid in anhydrous
CHzCl2/MeOH 9 : 1 ( 8 ml ) , Compound 1B was added at 0 ° C in
small portions. After stirring for 4 hours the
precipitate that formed was collected by filtering the
reaction mixture through a sintered glass funnel, and was
dried in a vacuum desiccator to yield the pure product in
47% yield. ~NMR (db-DMSO) 8.42(sb, 1H), 7.65(d, 1H),
7.31(d, 1H), 7.06(m, 2H), 6.48(s, 2H), 5.85(s, 1H),
~'O 94/10175 ~ ~ ~ ~ ~ -23- PCT/US93/09629
4.63(d, J=1.8 Hz, 1H), 4.43(m, 3H), 3.64(s, 6H), 3.28(s,
3H).
SR,iiR,ilaB-3-One-ii-hydrosy-5,4,ii,iia-tetrahydro-5
(3,5-dimethouy-4-hydrosyphenyl)-iH,6H
Oaazolo[3'4:1,6]pyrido[3,4-b]indole (Compound 6)
To a suspension of Compound Ic (1 mmol) in water (8
ml ) KIOH ( 2 mmol ) was added, and the solution was brought
to reflux and immediately cooled. The solution was then
acidified to ph 7 with 10% HC1, and the precipitate that
formed was collected by filtration in a sintered glass
funnel and dried in a vacuum dessicator.
5R,liR,ilaB-3-One-ii-methoxy-5,4,ii,iia-tetrahydro-5-
(3,5-dimethosy-4-hydrouyphenyl)-iH,6H-
oxazolo[3'4~:1,6]pyrido[3,4-b]indole (Compound 1C)
To a solution of syringaldehyde dimethyl acetal (.50
g) and catalytic p-toluenesulfonic acid in anhydrous
CHZC12/MeOH 9 :1 ( 8 ml ) , Compound 1B was added at 0 ° C in
small portions. After stirring for 4 hours the
precipitate that formed was collected by filtering the
reaction mixture through a sintered glass funnel, and was
dried in a vacuum desiccator to yield the pure product in
47% yield. ~NMR (db-DMSO) 8.42(sb, 1H), 7.65(d, 1H),
7.31(d, 1H), 7.06(m, 2H), 6.48(s, 2H), 5.85(s, 1H),
4.63(d, J=1.8 Hz, 1H), 4.43(m, 3H), 3.64(s, 6H), 3.28(s,
3H).
SR,iiR,iiaB-3-One-1i-methouy-5,4,ii,ila-tetrahydro-5-
(3,5-dimethouy-4-O-carbomethoxyphenyl)-iH,6H-
osazolo[3',4':1,6]pyrido[3,4-b]indole (Compound 1D)
Compound 1C (1.96g, 4.77 mmol) was suspended in 10
ml of CH2C12 and 6.7 ml (47.7mmo1) TEA. In the
suspension, 3.69 ml (47.7 mmol) of methyl chloroformate
was added dropwise at 0 °C, the solution was diluted with
CHZC12 and washed with water. The organic fraction was
dried over Na2S04, filtered and concentrated.
Purification by flash chromatography eluting with 8%
PCT/US93/0962'
WO 94/10175
-24-
acetone in CHC13 yielded 2.09 g (94%) of a white solid.
An analytical sample was obtained by recrystallization
from ethyl acetate.
5R,11R,liaS-3-One-11-hydrouy-5,4,ii,ila-tetrahydro-5
(3,5-dimethouy-4-O-carbomethouyphenyl)-iH,6H
ouazolo[3',4':1,6]pyrido[3,4-b]indole (Compound 1E)
To a solution of 1.88 g (7.00 mmol) of compound 1D
in a 9:1 dioxane/water solution (30 ml), 130 mg (0.70
mmol) of paratoluenesulfonic acid (p-TSOH) was added.
The solution was followed to completeion by TLC and
diluted with CHC13. The resulting mixture was washed
with saturated NaHC03, dried over NaZS04, filtered and
concentrated. Purification by flash chromatography
eluting with 20% acetone in CHC13 yielded 1.21 g (66%) of
a white solid.
5R,11R,i1a8-3-One-11-(4-fluoroanilino)-5,4,ii,ila-
tetrahydro-5-(3,5-dimetho$y-4-O-carbomethosy)-iH,6H-
osazolo[3',4':1,6]pyrido[3,4-b]indole (Compound 1F)
To a solution of 0.33 g (0.73 mmol) of Compound 1E
in anhydrous dioxane (4 ml) , 350 ~1 (2.5 mmol) of TEA and
160 ~1 (2.1 mmol) of acetyl chloride were added. After
stirring for 15 minutes, the solvent and unreacted acetyl
chloride were removed under reduced pressure. The
reaction vessel was recharged with dioxane, (diethylene
dioxide DDO), 1.4 mmol of 4-fluoro aniline and 1.4 mmol
TEA. The reaction mixture then was heated at 50°C and
stirred for 6 hours. To this solution, CHC13 was added
and the resulting solution was washed with water, dried
over Na2S04, and concentrated. Purification by flash
chromatography eluting with 10% acetone in CHC13 yielded
62 mg (16%) of a white solid.
5R,ilR,liaB-3-One-li-(4-fluoroanilino)-5,4,ii,ila-
tetrahydro-5-(3,5-dimethosy-4-hydrouyphenyl)-1H,6H-
oxazolo[3',4':1,6]pyrido[3,4-b]indole (Compound 11)
214'~~~&
''O 94/10175 PC'T/US93/09629
-25-
To a solution of sodium methoxide (90 ~mol) in 1 ml
methanol, 16.5 mg (30 ~,mol) of Compound 1F was added and
the reaction was followed to completion by TLC.
Saturated NH4C1 (200 ~1) was added followed by addition
of CHZC12. The layers were separated and the organic
layer was dried over Na2S04, filtered and concentrated.
Purification by flash chromatography eluting with 20%
acetone in CHC13 yielded 10.8 mg (73%) of a white solid.
Representative syntheses for the aforementioned compounds
are depicted below:
Compound 1A:
C02Me
H
~NH3C1
\ \ NaBH4 -
N~ EtOH/H20
H H
(known
(Et0)2C=0
EtONa, EtOH
0
~~ 0
\ H
N
H
( 1A~
WO 94/10175 2, ~ ~~ ~ ~ ~ PCT/US93/0962''
-26-
Compounds 1-5:
~o
ArCH[OMe~2
CH2C12, p-TSOH
H Ar
H
C1
C 1A]
Wherein Ar can be Y, as defined above.
Compound 6:
O Ac0 O
0 0
H
DDD ~ ~ ~ H ARCH[OAAe)2
/ H HOAC/THF / N CH2C12, AIEOH
H -79'C H p-TSOH
C 1A) ( 18]
OH
04a
KOH
H
H ~ H2p, a - 0
0
/
/ Na OMe
AIeO OMe
OH
OH
(6)
C ~C)
°'~O 94/10175 PCT/US93/09629
-27-
Compounds X, Y and Z have the following structures:
OEt
CHO
CI
\ \N I
I ~ N~--~
SOzPh OEt
CX~ ~Y~ CZ~
WO 94/10175 PCT/US93/096:'
Q -28-
Compound 7:
OMEAI
OMEM
COZEt NaBH ~ ~ OH CO~H30)ZC=0
C H ONe
75% EtOH ' ~ ~ J NH2 2 5
NH2 4 a
[ 7n] C 7b)
OAIEIA OIAe
O
H P'TSOH I ~ ~0
Syringaldehyde
0 dlmethyl acetal
O
[7c) 1A6 / OMe
OH
[ 7d)
OMe
~0 Et3St H
BF3'OEtz
0
/
Me0 ~ home /
Me0 OMe
OH
OH
7d)
'V0 94/ I 0175 ~ _
_2 9 _ PCT/US93/09629
Compound 8:
SOzOH S020H
\ Na BH
~ N EtONe/EtOH
/ ~ C02Ne EtOH/H 0 ~ / ~ [Et0]zC=0
NHZ NH2 OH
( BA] [ 88)
Mo0 OMe
H
N
HN ~e0 home
0 OH Me
p p-TSOH, CHZC~
(BC]
(8]
Compound 9
cozue
~NH3C 1 ~Me
\ ~ 1) NH3, CHC1~
/ N 2]SyrlnOe aldehyde, NatSO~
H 3) TFA
[9A]
NeBH~ Carbonyl
dllmldazole
dfoXene/ THF
H?0
(98)
[8)
WO 94/10175 'Z ~ ~ _3 ~_ PCT/US93/096'
Compound 11:
0 ACO 0
0 ~O
H DDO ~ \ ~ H A~CH(OMe]2
/ N HOAc/THF / CHpCl2, AIeOH
H -78'C H p-TSOH
[1A) (1B]
ONe
OMe
.O
0
CICOOMe H ~ p-TSOH
H ~ O~C 0 CHC13
0 \
/ , Me OMe
Me0 OM8
OCOZMe
OH
( 1 D]
(1C]
'O 94/10175 ~ ~ ~ ~ -31-
PCT/US93/09629
F
OH NH
0 TEA, CH3COC1 ~N ~0
DDO, C HqFNH H
6 2 p
\ ~ \
/ /
Me0 OMe Me0 OMe
OCOZMe OC02Me
(1E] [1F]
F
NH
D
CH30H
i
NH4C1
GHZG12
Me0 OMe
OH
[ 11]
EXAMPLE 2. INDOCTION OF DNA DOOBLE-STRAND BREARS BY
AZATO%IN IN THE PRESENCE OF HL-60 NOCLEAR E%TRACT
Drug-induced DNA double-strand breaks were measured
first in SV40 DNA in the presence of HL-60 nuclear
extracts. SV40 DNA was chosen because it is a natural
substrate of top 2 and is cleaved at many sites by other
WO 94/10175 ~ ~, ~ ~ ~ ~ -32- PCT/US93/096'
top 2 inhibitors. See, for example, Fesen & Pommier
(1991), supra. The smallest azatoxin concentration that
produced detectable cleavage was 5 to 10 ~M. Above 10
~M, cleavage occurred at many sites and was proportional
to the logarithm of azatoxin concentration. The potency
of azatoxin was comparable to that of VP-16 and, as in
the case of VP-16, azatoxin-induced DNA cleavage was not
suppressed at high drug concentrations (up to 1 mM),
consistent with azatoxin's not intercalating into DNA
(see below).
EXAMPLE 3. SEQUENCING OF TOPOISOMERASE II CLEAVAGE SITES
BY AZAT08IN
Induction of top 2 cleavage by azatoxin was tested
directly with the use of purified murine leukemia top 2.
Since the SV40 nuclear matrix-associated region has been
shown to be cleaved preferentially by top 2, see Pommier
et a1. (1991), supra, this region was chosen for
analysis. Sites of cleavage were also determined by DNA
sequencing in the 5' flank of c-myc first intron.
Azatoxin induced many cleavage sites both in the SV40 and
the c-myc DNA fragments. In general, azatoxin induced
more cleavage sites than mitoxantrone, m-AMSA, VM-26 or
VP-16. Thus, azatoxin was shown to be a potent top 2
inhibitor, with a cleavage pattern differing from those
induced by other top 2 inhibitors.
The cleavage pattern of azatoxin also was compared
to that of epipodophyllotoxin derivatives the structures
of which seem quite similar (for drug structures and
abbreviations, see table below). The compound 4'-
demethyl-4-desoxypodophyllotoxin (DMDP), with a structure
most similar to azatoxin, induced less cleavage that
azatoxin and at distinct cleavages sites. The ~-4-
hydroxy derivative of DMDP, 4'-demethylepipodophyllotoxin
(DMEP) , was at least as potent as VP-16, and its cleavage
patterns, while similar to that of VP-16 with some local
differences, was different from that of azatoxin.
i !i
CA 02147608 2004-05-04
-33-
EXAMPLE 4. EFFECTS ON TOPOISOMERASE ACTIVITY
Two different assays were conducted to illuminate the nature of azatoxin's
effects
vis-a-vis DNA relaxation. To study the inhibition of top 2 catalytic activity,
topoisomerase reactions were performed with 0.4 ~,g native SV40 DNA in 30 w1
reaction
buffer for 30 minutes at 37°C and stopped by adding SDS to a final
concentration of 1%
and proteinase K to 400 ~g/ml, followed by incubation for 1 hour at
42°C, essentially as
described by Fesen & Pommier (1989), supra. Agarose gel electrophoresis vas
performed in 1 % gels made in Tris-Acetate-EDTA (TAE) buffer (40 mN Tris-
Acetate,
pH 7.6, 10 mM NaZEDTA). Gels were run at 2 V/cm overnight, washed in water and
then stained with 1 ~,M ethidium bromide for 45 minutes. After an additional
45 minutes
destaining in 1 mM MgZS04 the DNA was visualized under UV light and
photographed
with a Polaroid* type 57 film.
To assess DNA unwinding, see Pommier et al., Nucleic Acids Res. 15: 6713-31
(1987), the DNA was relaxed first by treatment for 15 minutes with top 1 (20
units), after
which the test agent was added. These steps were carried out at 37°C.
DNA-agent-top 1
mixtures were incubated for an additional 30 minutes and then stopped as
described
above. Samples were then subjected to agarose gel electrophoresis as described
above.
From the assay data it was determined that azatoxin inhibits top 2-mediated
relaxation of native SV40 DNA. At the same time, azatoxin was observed to
produce a
substantial amount of linear DNA without significant increased of nicked DNA.
The DNA unwinding assay, with excess topoisomerase I and relaxed SV40 DNA,
was employed to assess azatoxin intercalation in accordance with Pommier et
al., Nucleic
Acids Res. 15: 6713-31 (1987). In fact, azatoxin did not induce detectable DNA
unwinding even at drug concentrations as high as 1 mM. This was also the case
for the 2
azatoxin isomers, 8 and 10, and for the
*Trademark
WO 94/10175 ~ ~ ~ ~ ~ ~ -34- PCT/US93/096,"
demethylepipodophyllotoxins, DMDP and DMEP. Similar
results were obtained with supercoiled DNA. The lack of
unwinding by azatoxin strongly indicated that the drug
does not intercalate into DNA.
EXAMPLE 5. EFFECTS OF AZATO$IN BTRDCTORAL MODIFICATIONS
ON TOPOISOMERASE II INHIBITION
Three isomers and six azatoxin derivatives, the
synthesis of which is described in Example 1, were tested
for drug-induced cleavage efficiency in the presence
either of HL-60 extract or of purified top 2. The
compounds and test results are set out in the table
below.
'O 94/10175 ~ ~ ~~ _35_ PCT/US93/09629
R4
R~
1'1a \ 1~a
\ \
1= I T 0 2=_ I 0
s ~ / s
H 0
Y Y
R4
~~n
a= / \ I s ~0
r~.~~~~0
Y
Y= I \
R~ R3
R4
R2
'1 1 n ,
1=_ ~ \ I ~ .0
s
0
Y
~~ .'~ Gag
WO 94/10175 PCT/US93/096.
-36-
r s~~n~mia~y Topoi.oounx
a
i
hibi
i
on
n
t
COMPOUND STRUCTURER1 ~ ~ R4 5 lla
AZATOXINS
Autooin I OCH3 OH OCH3 H R S +++
2 I H OH OCH3 H R S
3 1 H OH H H R S O
4 1 OCH3 OCH3 OCH3 H R S O
1 H NHS02CH2H H R S O
6 1 OCH3 OH OCH3 OH R S +
7 4 OCH3 OH OCH3 H R S O
8 3 OCH3 OH OCH3 H R S O
9 1 OCH3 OH OCH3 H R R O
1p 1 OCH3 OH OCH3 H S S O
EPIpODOPHYLLOTOXINS
DMDP 2 OCH3 OH OCH3 H R S +
DMEP 2 OCH3 OH OCH3 OH R S +++
VP-16 2 OCH3 OH OCH3 a R S +++
VM-26 2 OCH3 OH OCH3 6 R S ++++
a t: H b
H H
0 0
OH OH
Azato$in Isomers
The three azatoxin isomers (compounds 8_-10 in the
table) were found not to be active as top 2 inhibitors in
DNA cleavage assays. The finding that the two
diastereoisomers (9 and 10) were inactive demonstrated
that a strict stereochemical relationship between the
polycyclic ring system and the pendant aromatic ring must
exist for activity. The inactivity of isoazatoxin (8_)
was surprising and indicated the great sensitivity of the
binding site for these agents to minor structural
modification. Thus, azatoxin and isoazatoxin (8_) differ
only in the orientation of the tetrahydrooxazolopyrido
ring fusion into the indole ring; this change in
.1~V0 94/10175 -3,~- PCT/US93/09629
orientation imparts (1) only a subtle differential
"curve" to the tetracyclic nucleus of the molecule,
without altering the spatial relationship between the
indole and phenyl ring systems, and (2) a change in
orientation of the nitrogen indole.
Azatouin Derivatives
The results from testing the six azatoxin derivatives
for top 2 inhibition also are set out in table. Two of
derivatives were modified on the polycyclic ring system
and the others were modified on the pendant ring.
Hydroxylation at position 11 (R4) of the azatoxin
polycyclic ring system yielded a compound (6) that was
structurally similar to DMEP and that displayed
measurable but not strong t,op 2 activity. This 4-hydroxy
substitution differentiates azatoxin from the 4'-
demethylpodophyllotoxin framework, since hydroxyl
substitution at this site significantly decreases top 2
activity of azatoxin (_1 versus 6) and increases activity
of the podophyllotoxins (DMDP, DMEP). Differing from
azatoxin by its polycyclic ring system, Compound 7 is
inactive, again indicating that the structure of the
polycyclic ring system is critical for azatoxin activity.
Modification to the pendant (Y) ring gave the
following results. Monodemethoxylation (2_) reduced top
2 activity by a factor 10, while didemethoxylation (3)
abolished the top 2 activity. Position 4' (R2) was also
crucial as methylation of the hydroxyl residue (4_)
abolished top 2 inhibition. These results are not
inconsistent with those obtained for demethyl-
epipodophyllotoxins. See, for example, Sinha et al.,
Eur. J. Cancer 26: 590-93 (1990). Notably, compound 5_,
in which the azatoxin pendant ring had been placed by
that of AMSA was inactive. These results differentiate
the azatoxin family of top 2 inhibitors from the m-AMSA
family of inhibitors.