Note: Descriptions are shown in the official language in which they were submitted.
2159660
-- 1 --
PHENYLALANINE-GLYCINE DERIVATIVES PROCESS
FOR PREPARATION THEREOF AND PHARMACEUTICAL
COMPOSITION CONTAINING SAID DERIVATIVES
BACKGROUND OF THE I~v~NllON
This application is a divisional application of copending
Canadian Application Serial No. 2,091,316, filed March 9, 1993.
1. Field of the Invention
The present invention relates to a novel conjugate of a
phenylalanine derivative with an antitumor substance, a process
for preparation thereof, and a pharmaceutical composition, in
particular, an antitumor agent, containing it. More particu-
larly, the present invention relates to a conjugate of L-
phenylalanine-glycine with an antitumor substance, a process for
preparation thereof, and a pharmaceutical composition containing
it.
2. Description of the Related Art
Hitherto, chemotherapeutic agents have proved efficacy in
treating tumors, but many problems still remain. For example,
chemotherapeutic agents not only have effects on tumor cells, but
also affect the host cells and exhibit cell toxicity. Therefore,
the agents cannot be administered for a long time to physically
weakened patients and thus it was difficult to secure a suffi-
cient therapeutic effect. The mechanism of action of chemothera-
peutic agents is based on the inhibition of biosynthesis (in
particular, that of nucleic acids) in the cells and inhibition
of the metabolism necessary to maintain cell life. Strictly
speaking, it means that the chemotherapeutic agents were not
based on specificity to the tumor. Namely, these agents had
toxicity to the normal cells of the host suffering from the tumor
as well. It was desired to develop antitumor agents with an
improved selectivity to the tumor cells and an improved method
for concentrating conventional antitumor agents into the tumor
cells.
In the meanwhile, 5-fluorouracil (5-FU) as it is does not
2159660
- la -
exhibit antitumor activity, but exhibits antitumor activity when
bonded with the pentose phosphate in the cells to form fluoro-
deoxyuridine-5'-monophosphate (FdUMP), fluorouridine-5'-
triphosphate (FUTP) or the like. Namely, FdUMP inhibits the
thymidylate synthetase activity to inhibit the synthesis of DNA.
FUTP is taken up in an RNA and causes critical damage to the RNA,
thereby inhibiting the production of cell proteins. Therefore,
if
21ss66a
the pentose phosphate in the tumor cells could be increased
selectively, selective chemotherapy to tumor cells would become
possible by 5-FU
Further, pyruvate kinase is the rate-determining enzyme in the
anaerobic glycolysis which relates to the production of pentose
phosphate in the cells, and includes L-type, M1-type, and M2-type
isoenzymes. Tumors contain almost only M2-type isoenzyme. It was
known that the M2-type isoenzyme is selectively inhibited by a low
concentration of L-phenylalanine. Therefore, it was expected that
L-phenylalanine could cause inhibition specific to the pyruvate
kinase activity in the tumor cells and enhance the production of
pentose phosphate only in the tumor cells. Thus, Lee disclosed and
ascertained a method of enhancing the activity of 5-FU to inhibit
the tumor in combination with L-phenylalanine [Med. J. Kagoshima
Univ., Vol.37, No. 3-4, 285-308, 1985].
In the method of Lee, however, L-phenylalanine was mixed in a
laboratory chew and ingested. 5-FU was separately administered.
Therefore, L-phenylalanine and 5-FU were conveyed separately to the
lesion. The present inventors engaged in various studies with the
object of conveying L-phenylalanine and 5-FU to the target lesion
in a bonded form and separating them quickly at the target site.
As a result, the inventors found that the above object can be
achieved by bonding 5-FU with L-phenylalanine-1-acetoxyglycine, and
that there is a similar effect in antitumor substances other than
5-FU. The present invention is based on these findings.
SUMMARY OF THE INVENTION
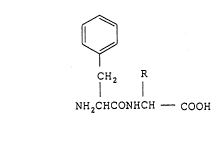
Accordingly, the present invention relates to a phenylalanine-
glycine derivative of the general formula (I)
fH2 1 (I)
NH2cHcoNHcH - COOH
2159660
wherein R represents a residue of an antitumor substance, a salt
thereof or an ester thereof (hereinafter optionally referred to
as the "the present conjugate"), a process for preparation of the
present conjugate, and a pharmaceutical composition containing
the present conjugate.
In accordance with an embodiment of the present invention
there is provided a carbobenzoxyphenylalanine amide of the
formula (IV):
c~l ~IV)
CH20CONHCHCONH2
.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
The glycine moiety contained in the present conjugate may
be any residue of the L- or D-glycine, or a mixture of LD-
glycine. Further, the phenylalanine moiety is preferably residue
of L-phenylalanine, but may also be residue of D-phenylalanine
or a mixture of LD-phenylalanine. The present conjugate may be
non-toxic salts or esters which the amino acids in the glycine
and/or phenylalanine residues can form.
The non-toxic salts may be acid-addition salts or metal
complexes. The metal complexes are complexes with, for example,
zinc, iron, calcium, magnesium, or aluminum. As the acid-
addition salts, there may be mentioned hydrochloride, hydro-
bromide, sulfate, phosphate, tannate, oxalate, fumarate,glauconite, alginate, maleate, acetate, trifluoroacetate,
citrate, benzoate, succinate, malate, ascorbate, tartrate, or the
like. Further, the salts may be carbonates, for example, salts
with alkali metals tsodium, potassium salts, etc.), salts with
alkaline earth metals (calcium, magnesium salts, etc.), or
2I5966~
- 3a -
ammonium salts.
The esters may be any esters conventionally used for amino
acids, such as aryl or alkyl esters. In particular, there may
be mentioned straight-chain or branched alkyl esters having 1 to
4 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-
butyl, or isobutyl ester.
The antitumor substance residue R in the present conjugate
may be a residual group of an alkylating antitumor substance, an
antimetabolic antitumor substance, an antibiotic antitumor sub-
stance, or the like. As the antitumor substances, there may be
mentioned, for example, 5-fluorouracil, 5-amino-7-hydroxy-lH-v-
triazolo(4,5-d)pyrimidine, 4-amino-N1~-methylpteroyl-glutamic
acid, 4-aminopteroyl-glutamic acid, 6-mercaptopurine, 5-[bis(2-
chloroethyl)amino]-uracil, mitomycin C, bleomycin, daunorubicin,
doxorubicin, p-[bis(2-chloroethyl)amino]-L-phenylalanine or ester
21~966D
thereof, N,N-bis(2-chloroethyl)-N1, O-propylene-phosphate ester
diamine, 4-[p-(bis~2-chloroethyl)amino)phenyl]-butylic acid or
es~er thereof.
The present conjugate may be prepared by the steps (a) and (b)
as follows:
(a) The step comprising introducing an antitumor substance residue
R into a compound of the general formula (IIIb):
CHz OCOCH3 (IIIb)
~ CH20CONHCHCONHCHCOR
wherein R2 represents a benzyloxy group or an alkoxy group with 1
to 4 carbon atoms, to obtain a compound of the general formula
(II):
CH2 R (II)
~ CH20CONHCHCONHCHCOR2
wherein R and R2 have the same meanings as above.
(b) The step comprising removing group(s) for protecting amino
group(s) from the compound of the general formula (II) to obtain
the compound of the general formula (I) or ester thereof.
In the step (a), the compound of the general formula (IIIb)
and the antitumor substance are reacted in the presence of an
2159660
organic solvent (for example, dimethylformamide) and preferably a
base at -10 to 50CC, preferably a~ 10 to 30~C, for 10 to 120
minutes, preferably for 15 to 60 minutes, to thereby obtain the
compound of the general formula (II). As the base, triethylamine,
pyridine or the like may be used. After the reaction is completed,
the reaction product is used in the next step without or with
purification by recrystallization, distillation, extraction,
precipitation, washing, column separation, concentration,
lyophilization, or the like. In the step (b), a solution of the
compound of the general formula (II) in an organic solvent is added
to an alcohol solution of palladium carbon and treated at -10 to
120~C, preferably at -5 to 100~C, for 10 to 300 minutes, preferably
for 15 to 240 minutes. After cooling, the filtrate is concentrated
to obtain the crystal. Further, the crystal was recrystallized or
washed with a solvent to obtain a compound of the general formula
(I) with a high purity. If necessary, the D-form and L-form of the
compound of the general formula (I) or ester thereof are resolved
from each other by, for example, chromatography. The compound of
the general formula (I) may be converted to the corresponding salt
or ester before used as an antitumor agent.
Examples of the present conjugate will be shown hereinafter:
(1) L-phenylalanyl-2-(5-fluorouracil-1-yl)-D, L-glycine
(2) L-phenylalanyl-2-(5-fluorouracil-1-yl)-D, L-glycineamide
(3) L-phenylalanyl-2-(5-fluorouracil-1-yl)-D, L-glycinemethylester
(4) L-phenylalanyl-2-(5-fluorouracil-1-yl)-D, L-glycineethylester
(5) L-phenylalanyl-2-(5-fluorouracil-1-yl)-D, L-glycinepropylester
(6) L-phenylalanyl-2-[5-(bis(2-chloroethyl)-amino)uracil-1-yl]-D,L-
glycine ,-
(7) L-phenylalanyl-2-[5-(bis(2-chloroethyl)amino)-uracil-1-yl]-D,L-
glycinemethylester
(8) L-phenylalanyl-2-[5-(bis(2-chloroethyl)amino)-uracil-1-yl]-D,L-
glycineethylester
(9) L-phenylalanyl-2-[p-(bis(2-chloroethyl)amino)-L-
phenylalaninemethylester-2-yl]-D,L-glycineethylester
(10) L-phenylalanyl-2-(5-fluorouracil-1-yl)-L-glycine
(11) L-phenylalanyl-2-(5-fluorouracil-1-yl)-D-glycine
The compound of the general formula (IIIb) may be prepared,
for example, by the following steps (c) to (e):
21~9660
(c) The step comprising reacting a salt or a compound of the
fonmula (v):
[~
(v)
fH2
H2NCHCONH2
with carbobenzoxychloride to obtain a compound of the formula (IV):
CH2 (Iv)
~ CH20CONHCHCONH~
More particularly, L-phenylalanine amide hydrochloride and
sodium hydrogencarbonate are qissolved in water and then
carbobenzoxychloride and an organic solvent are added at 0 to 30~C.
The mixture is agitated and reacted, for 1 to 24 hours, preferably
for 2 to 8 hours. Further carbobenzoxychloride and sodium
hydrogencarbonate may be added thereto. After the reaction is
completed, a white crystal is taken out and recrystallized from
ethyl acetate to obtain the compound of the formula (IV).
(d) The step comprising reacting the compound of formula (IV) with
a glyoxylic acid compound of the general formula (VI):
O O
ll ll (VI)
H - C - C - R2
2ls966~
wherein R2 has the same meaning as above, to obtain a compound of
~he general formula (IIIa):
c~, OH (lIIa)
~ CH OCONHCHCONHCHCOR
wherein R2 has the same meaning as above.
More particularly, the compound of the formula (IV) is
dissolved or suspended in an organic solvent. The glyoxylic acid
of the general formula (VI) is added to the mixture while agitating
at 0 to 30~C and reacted for 50 to 240 hours. The product is
concentrated under reduced pressure to obtain a white crystal of
the general formula (IIIa).
(e) The step comprising reacting the compound of the general
formula (IIIa) with acetic anhydride to thereby obtain the compound
of the general formula (IIIb).
More particularly, the compound of the general formula (IIIa)
is agitated for 1 to 48 hours at 0 to 30~C in acetic anhydride and
pyridine and then the reaction mixture is extracted with ethyl
acetate. The resulting solution is washed with hydrochloric acid,
water, sodium hydrogencarbonate aqueous solution, distilled water,
saturated saline solution, or the like. The organic layer is then
concentrated to obtain an oily substance. The oily substance is
purified by silica gel chromatography, and recrystallized to obtain
the compound of the general formula (IIIb) as a white crystal.
In comparison with the case that an antitumor agent is used
singly, or the case that an antitumor agent is used in the form of
a mixture with phenylalanine, the antitumor activity is enhanced by
the present conjugate containing said antitumor agent. Further, in
comparison with the case that the antitumor agent is used singly,
the acute toxicity of the present conjugate containing said
antitumor agent is reduced.
2159660
The present conjugate can be formulated to, for example,
syrups, injections, ointments, tablets, or the like. The present
conjugate may be contained in the formulation in an amount of 0.1
to 99.5 % by weight, preferably in an amount of 1 to 90 % by
weight. The formulation of the present conjugate may be
administered orally or parenterally. A dose varies with the method
of administration, the extent of the treatment, and also the kind
of the antitumor substance contained therein. Generally speaking,
however, the dose of the present conjugate is in the range of 100
to 1000 mg/kg/day orally, while in the range of 5 to 500 mg/kg/day
parenterally, which is divided into 1 to 4 dosages in a day.
As above, the novel present conjugate of the phenylalanine-
glycine derivative and the antitumor substance exhibits a superior
antitumor activity compared with the case that an antitumor
substance is administered singly, or the case that the antitumor
substance is administered in the form of a mixture with
phenylalanine.
Examples
The present invention now will be further illustrated by, but
is by no means limited to, the following examples. The
physicochemical data described in the following Examples were
obtained by the following methods:
(1) Elemental Analysis
A Yanagimoto MT3-type automatic elemental analyzer was used
and the decomposition gas was detected with a thermal conductivity
type detector (TCD).
(2) Optical Rotation
A Nihon Bunko automatic polarimeter DIP-360 was used to
measure the optical rotation for a solution of the present
conjugate to determine the [~]D-
(3) NMR
A Nihon Denshi JNM-GSX500 was used.
(4) Infrared Absorption Spectrum
A Nihon Bunko A-202 apparatus was used to measure the infrared
absorption spectrum by the KBr tablet method and to determine ~max-
(5) Thin Layer Chromatography
The Rf value was determined by a hexane-ethyl acetate system,
2t59660
g
butanol-acetic acid-pyridine-distilled water system, or 5 %
methanol-dichloro~ethane system.
(6) Melting Point
The melting point was measured by a Yanagimoto micro melting
point detector (DSC).
Exam~le 1
(1) Pre~aration of carbobenzoxv-L-~henvlalanineamide (IV)
~J ~3 CH20COCl
CH2 CH2
HClH7NCHCONH2 NaHCO3 ~ CH20CONHCHCONH2
(v)
(IV)
I~phenylalaI~ine amide hydrochloride (V) (1.00 g, 5.0 mmmol) and
1.09 9 of sodium hydrogencarbonate (13.0 mmol) were dissolved in 40
ml of distilled water. The solution was agitated at room
temperature while adding 0.72 9 of carbobenzoxychloride (4.2 mmol)
and 25 ml of dichloromethane. After 2 hours, a further 0.72 9 of
carbobenzoxychloride (4.2 mmol~ and 0.55 9 of sodium
hydrogencarbonate (6.5 mmol) were added. When agitation was
continued, a white crystal was precipitated in the dichloromethane
layer. To the crystal layer, dichloromethane was further added to
completely dissolve them, and the dichloromethane was washed with a
saturated sodium hydrogencarbonate solution. The dichloromethane
layer was dried over anhydrous sodium sulfate, then concentrated
under reduced pressure, and recrystallized with dichloromethane-
hexane, whereby 1.46 9 of a white compound (IV) was obtained. The
physicochemical data of the product was as follows:
Yield: 99.0 %, Melting point: 160.1 to 162.1~C
Elemental analysis (%)
Found: C, 68.32; H, 5.93; N, 9.38
2159660
1~
Calculated (for C17H1gN2O3): C, 68.44; H, 6.08; N, 9.39
~a]D: -5.55~ (c 1.0, CH30H)
H-NMR (CDCl3): ~3.05 (dd, lH, J=13.8Hz, 7.3Hz) phenylalanyl CH2,
~3.13 (dd, lH, J=13.8Hz, 7.3Hz) phenylalanyl CH2, ~4.30 (m, lH)
NCHCO, ~5.09 (s, 2H) Ph-CH2O, ~5.35 (br, 2H) CON_2, ~5.66 (br, lH)
CON_2
IR(KBr)~max: 3425m, 3210s, 1690m, 1660s (amide group)
Rf: 0.42
5 % methanol-dichloromethane (phosphomolybdic acid reagent W +).
(2) Pre~aration of N-carbobenzoxv-L-~henvlalanvl-D,L-2-
hvdroxyalvcinebenzvlester (IIIa-1)
CHOCOOCH, ~ ¢~
( IV) CH2 OH
CH20CONI{C~ICONHCHCOOCH
( I I I a - 1 )
To a suspension of 2.81 9 of carbobenzoxyphenylalanine amide
(IV) -(9.4 mmol) in dichloromethane, 1.93 9 of benzyl glyoxylic acid
(11.8 mmol) was added while agitating at room temperature and
reacted for about 160 hours. The resulting suspension was
concentrated under reduced pressure to obtain a crystal. The
product was filtered with suction while washed with
dichloromethane-hexane and then dried to give 3.78 9 of the
compound (IIIa-1).
Yield: 86.8 %, Melting point: 120.6 to 127.7~C
Elemental analysis (~)
Found: C, 67.00; H, 5.66; N, 6.77
Calculated (for C26H26N2O6): C, 67.52; H, 5.67; N, 6.06
[a]D: -13.0~ (c 1.0, CH30H)
H-NMR (CDCl3): ~3.06 (m, 2H) phenylalanyl CH2, ~3.86 (d, 0.5H) OH,
~3.97 (d, 0.5H) OH, ~4.43 (m, lH) Ph-CH2-CH, ~5.06 (m, 2H) Z group
CH2, ~5.20 (m, 2H) benzylester CH2, ~5.49 (t, 2H) glycyl CH, ~7.1
2159660
to 7.4 (m, 15H) aromatic ring
IR(KBr)vmax: 3420m (NH), 3310s (NH), 1750m (COO), 1695m,
1660s (CONH)
Rf: 0.45
5 % methanol-dichloromethane (phosphomolybdic acid reagent W+).
(3) PreDaration of N-carbobenzoxv-L-Dhenvlalanvl-D,L-2-
acetoxvalvcinebenzYlester (IIIb-1)
~ ~2 OCOCH
(IIIa-1) , ~ CH2OCONHCHCONHCHCOOCH
(CH3CO)2O
(IIIb-1)
N-carbobenzoxy-L-phenylalanine-D,L-2-hydroxyglycinebenzylester
(IIIa-1) (4.82 9, 10.4 mmol) was dissolved at room temperature in
50.0 ml of acetic a~hydride and 36.5 ml of pyridine and agitated
for about 24 hours. After the end of the reaction had been
confirmed by thin layer chromatography, the reaction mixture was
extracted with 100 ml of ethyl acetate and the organic layer was
washed three times with 100 ml of distilled water, then twice with
100 ml of saturated saline solution. The washed organic layer was
dried over anhydrous sodium sulfate and concentrated under reduced
pressure to obtain 5.64 9 of a yellow oily substance. The
resulting oily substance was purified by silica gel chromatography
using n-hexane/ethyl acetate (2/1) and recrystallized from ethyl
acetate-n-hexane to obtain 1.31 9 of the compound (IIIb-1).
Yield: 34.0 ~, Melting point: 112.5 to 114.0~C
Elemental analysis (%)
Found: C, 66.63; H, 5.52; N, 5.68
Calculated (for C2gH2gN2O7): C, 66.66; H, 5.59; N, 5.55
[~]D: -7-0~ (c 1.0, CH3OH)
1H-NMR (CDCl3): ~2.05 (s, 3H) acetyl CH3, 83.07 (m, 2H)
phenylalanyl C_2, 84.46 (m, lH) phenylalanyl N-CHCO, ~5.06 (m, 2H)
2I59660
12
Z group CH2, ~5.19 (m, 2H) benzylester CH2, ~6.37 (d, lH) glycyl
CH, ~7.1 to 7.4 (m, 15H) aromatic ring
IR(~CBr)~max: 3300s (NH), 3060w, 3030w, 2973w, 1770s, 1740s, 1695s,
1665s
Rf: 0.42
Hexane: Ethyl acetate = 2:1 (phosphomolybdic acid reagent W+).
(4) Pre~aration of N-carbobenzoxv-L-~henvlalanvl-2-(5-fluorouracil-
1-vl-D,L-qlvcinebenzvlester (II-1)
H N
O N
(IIIb-1) ~ ~ O f
(II-1) HN I F
N-carbobenzoxy-L-phenylalanyl-D,L-2-acetoxyglycinebenzylester
(IIIb-1) (0.25 9, 0.5 mmol) and 73.5 mg of 5-fluorouracil (0.57
mmol) were dissolved in 1.0 ml of dimethylformamide at room
temperature and 0.51 9 of triethylamine (5.0 mmol) was added and
the mixture was agitated for about 20 minutes. After the end of
the reaction had been confirmed by thin layer chromatography, the
reaction mixture was concentrated under reduced pressure and
extracted with 50 ml of ethyl acetate. The organic layer was
washed with 20 ml of distilled water and 40 ml of saturated saline
solution. The washed layer was dried over anhydrous sodium
sulfate, concentrated under reduced pressure, and recrystallized
from ethyl acetate-n-hexane to obtain 0.16 9 of the compound tII-
1) .
Yield: 53.1 %, Melting point: 170.3 to 175.0~C
Elemental analysis (%)
215966~ .
13
Found: C, 62.68; H, 4.68; N, 9.76
Calculated (for C30H27N~C7F): C, 62.71; H, 4.74; N, S.75
[a]D: -7.0~ (c 1.0, CH3OH)
1H-NMR (CDCl3): ~2.8 to 3.0 (m, 1.5H) diastereomeric phenylalanyl
CH2, ~3.12 (dd, 0.5H, J=13.8, 5.9Hz), ~4.67 (d, 0.5H, J=7.3Hz)
phenylalanyl CHCO, ~4.73 (d, 0.5H, J=6.4Hz) phenylalanyl CHCO, ~4.9
to 5.2 (m, 4H) benzyl CH2, ~5.51 (d, 0.5H), ~5.57 (d, 0.5H), ~5.80
(d, 0.5H) glycyl CH, ~5.84 (d, 0.5H) glycyl CH, ~7.0 to 7.3 (m,
15H) aromatic ring, ~7.50 (d, 0.5H, J=5.5Hz) uracil 6H, ~7.60 (d,
0.5H, J=5.5Hz) uracil 6H, ~9.19 (br, 0.5H) uracil 3NH, ~9.66 (br,
0.5H) uracil 3NH
IR (KBr)~max: 3405m, 3300s, 3160m, 3140m, 1760s, 1700s, 1660s.
(5) Preparation of L-PhenvlalanYl-2-(5-fluorouracil-1-vl)-D,L-
qlvcine (I-1~ -
Pd/C fH2
(II-1) ~ H2O-L-NH2CHCONHCIHCOOH (I-1)
~ ~I
HN ~ F
Methanol was added dropwise to 246.5 mg of 10 % palladium
carbon at 0~C. Further, there was added thereto dropwise at 0~C a
solution of 0.29 9 of N-carbobenzoxy-L-phenylalanyl-2-(5-
fluorouracil-1-yl)-D,L-glycinebenzylester (II-1) (0.5 mmol) in
methanol and cyclohexene added. The solution was heated under
reflux at 70 to 80~C for about 20 minutes. After the end of the
reaction had been confirmed by thin layer chromatography, the
reaction liquid was diluted with methanol and filtered through a
fluted filter paper. The filtrate was concentrated under reduced
pressure to obtain a white crystal. The crystal was washed with
~ 2159660
14
ethyl acetate to obtain 0.16 9 of the compound (I-1).
Y:eld: 89.6 %, Melting point: 197.3 to 201.3~C
Elemental analysis
Found: C, 48.8; H, 4.5; N, 14.8
Calculated (for C1sH1sOsN4F-H2O): C, 48.9; H, 4.7; N, 15.2
[~]D: 11-9~ (c 2.0, CH3OH)
1H-NMR (D2O): ~2.93 to 2.97 (dd, 2H) diastereomeric phenylalanine
CH2, ~3.20 to 3.29 (dd, 2H) diastereomeric phenylalanine CH2, ~4.26
to 4.33 (m, 2H), ~5.79 (s, lH) glycyl C_, ~5.90 (s, lH) glycyl CH,
~7.0 to 7.3 (m, 10H) aromatic ring, ~7.55 (d, lH, J=6Hz) uracil 6_,
~7.85 (d, lH, J=6Hz) uracil 6H
IR (KBr)~max: 3400m, 3170m, 3030m, 2800m, 1690s, 1500m, 1370s,
1240s
Rf: 0.62, 0.57
Butanol: Acetic acid: Pyridine: Distilled water = 4:1:1:2
(ninhydrin reagent W+).
(6) The procedures of Examples 1 (4) and (5) were repeated, except
that 5-[bis(2-chloroethyl)amino]uracil was used instead of 5-
fluorouracil, to obtain L-phenylalanyl-2-[5-(bis(2-
chloroethyl)amino)uracil-1-yl]-D,L-glycine at a yield of 45 %. The
results of the elemental analysis were as follows:
Found: C, 48.01; H, 4.73; N, 15.02
Calculated (for C19H23OsNsCl2): C, 48.32; H, 4.91; N, 14.83.
Exam~le 2
(1) Preparation of N-carbobenzoxy-L-phenylalanyl-D,L-2-
hydroxyglycineethylester (IIIa-2)
(IV) CH2C12 fH2 OH
+ ~' ~ CH20CONHCHCONHCHCOOCH2CH3
HOCOOCH2CH 3
(IIIa-2)
, 2159660
To a suspension of 0.94 9 of carbobenzoxyphenylalanine~ amide
(IV) (3.1 mmcl) prepared from Ex~mpie 1(1) in dichloromethane, 0.40
g of ethyl glyoxylate (3.9 mmol) was added while agitating at room
temperature, and then the suspension was agitated for about 160
hours. The suspension was concentrated under reduced pressure to
obtain a white crystal. The crystal was filtered with suction
while washing with dichloromethane-hexane and then dried to obtain
3.78 9 of the compound (IIIa-2).
Yield: 77.6 %, Melting point: 143.7 to 148.7~C
Elemental analysis (%)
Found: C, 61.60; H, 5.72; N, 7.02
Calculated (for C21H24N2O6): C, 62.99; H, 6.04; N, 7.00
[~]D: 6.85~ (c 1, DMSO)
1H-MMR (d6-DMSO): ~1.19 (t, 3H, J=6.91) ethylester CH3, ~2.74 (m,
lH) phenylalanyl CH2, ~2.98 (m, lH) phenylalanyl CH2, ~4.13 (m, 2H)
ethylester CH2, ~4.31 (m, lH) phenylalanyl CH, ~4.93 (d, 2H,
J=3.33) Z group CH2, ~5.49 (t, 2H) glycyl CH, ~6.60 (d, 0.5H,
J=6.67) O_, ~6.68 (d, 0.5H, J=6.67) OH, ~7.19 to 7.34 (m, 10H)
aromatic ring
IR(KBr)umax: 3300s, 3060m, 3030m, 2950w, 1747s, 1685s, 1658s, 1530s
Rf: 0.32
5 % methanol-dichloromethane (phosphomolybdic acid reagent W+).
(2) PreParation of N-carbobenzoxv-L-Phenvlalanvl-D,L-2-
acetoxvql~cineethvlester (IIIb-2)
CH2 OCOCH3
( CH3C0 ) 20 ~
(IIIa-2) > ~ CH2OCONHCHCONHCHCOOCH2CH3
~ (IIIb-2)
N
N-carbobenzoxy-L-phenylalanine-D,L-2-hydroxyglycineethylester
(IIIa-2) (2.65 9, 6.6 mmol) was dissolved in 31.8 ml of acetic
anhydride and 23.2 ml of pyridine at room temperature and agitated
2159660
16
for about 3 hours. After the end of the reaction had been
corfirmed by thin layer chromatsgraphy, the reac~ on mixture was
extracted with 70 ml of ethyl acetate and the organic layer was
washed three times with 70 ml of distilled water and then twice
with 70 ml of saturated saline solution. The washed organic layer
was dried over anhydrous sodium sulfate, then concentrated under
reduced pressure to obtain 2.31 9 of a yellow oily substance. The
oily substance was purified by silica gel chromatography (n-
hexane/ethyl acetate = 2/1), and recrystallized (ethyl acetate-
hexane) to obtain 1.18 9 of the compound (IIIb-2).
Yield: 40.3 %, Melting point: 141.5 to 147.0~C
Elemental analysis (%)
Found: C, 62.13; H, 5.65; N, 6.32
Calculated (for C23H26N2O7): C, 62.43; H, 5.92; N, 6.33
[~]D: 23-55~ (c 1, CHCl3)
lH-MMR (CDCl3): ~1.27 (t, 3H, J=7.10) ethylester CH3, ~2.08 (s, lH)
acetyl CH, ~3.11 (m, 2H) phenylalanyl CH2, ~4.22 (m, 2H) ethylester
CH2, ~4.49 (br, lH) phenylalanyl CHCO, ~5.09 (s, 2H) Z group CH2,
~5.23 (br, lH) phenylalanyl CON , ~6.32 (d, lH, J=9.16) glycyl CH,
~7.16 to 7.37 (m, 10H) aromatic ring
IR (~3r)Umax: 3300s, 3050w, 3030w, 2960m, 1735s, 1690m, 1660s,
1540m
Rf: 0.54
Hexane: Ethyl acetate = 1:1 (phosphomolybdic acid reagent W+).
(3) PreParation of N-carbobenzoxv-L-phenYlalanvl-2-(5-fluorouracil-
1-Y1) -D,L-alvcineethYlester (II-2)
HN
O N
CH2
(IIIb-2J ~ ~ CH2OCONHCHCONHfHCOOCHzCH3
N ~
(II-2) HN ~ F
o
~ 17 2 1 5 9 6 6 ~
N-carbobenzoxy-L-phenylalanyl-D,L-2-acetoxyglycineethylester
(IIIb-2) (0.47 9, 1.1 mmol) and 0.15 9 of 5-fluorouracil (1.20
mmol) were dissolved in 2.2 ml of dimethylformamide at room
temperature and then 1.06 9 of triethylamine (10.5 mmol) was added
thereto. The mixture was agitated at room temperature for about 20
minutes. After the end of the reaction had been confirmed by thin
layer chromatography, the reaction mixture was concentrated under
reduced pressure and the residue was extracted with 100 ml of ethyl
acetate. The extracted organic layer was washed with 40 ml of
distilled water and 80 ml of saturated saline solution. The washed
organic layer was dried over anhydrous sodium sulfate, concentrated
under reduced pressure, and recrystallized (ethyl acetate-hexane)
to obtain 0.54 9 of the compound (II-2).
Yield: 90.7 %, Melting point: 98.0 to 102.3~C
Elemental analysis (%)
Found: C, 57.98; H, 4.94; N, 10.56
Calculated (for C2sH2sN4O7F): C, 58.59; H, 4.92; N, 10.93
[~]D: -10.i20 (c 2, CHOH)
1H-NMR (CDCl3): ~1.19 (t, 1.5H, J=7.11) ethylester CH3, ~1.25 (t,
1.5H, J=7.11) ethylester CH3, ~3.01 (m, 1.5H) phenylalanyl CH2,
~3.15 (dd, 0.5H, J=13.75, 5.96Hz) phenylalanyl CH2, ~4.24 (m, 2H)
ethylester CH2, ~4.67 (d, 0.5H, J=7.3Hz) phenylalanyl C_CO, ~4.73
(d, 0.SH, J=6.4Hz) phenylalanyl C_CO, ~5.05 (m, 2H) Z group CH2,
~5.63 (d, 0.5H, J=8.25Hz) phenylalanyl CON_, ~5.70 (d, 0.5H,
J=8.25Hz) phenylalanyl CONH, ~5.78 (d, 0.SH, J=7.33Hz) glycyl CH,
~5.83 (d, 0.5H, J=7.33 Hz) glycyl C_, ~7.05 to 7.31 (m, 10H)
aromatic ring, ~7.56 (d, 0.5H, J=5.04Hz) uracil CH, ~7.65 (d, 0.5H,
J=5.04Hz) uracil C_, ~9.70 (br, 0.5H) uracil NH, ~10.0 (br, 0.5H)
uracil NH
IR (KBr)vmax: 3300s, 3050m, 3020m, 1750s, 1700s, 1660s, 1510m
Rf: 0.26
Hexane; Ethyl acetate = 1:1 (phosphomolybdic acid reagent W+).
(4) Preparation of L-Phenvlalanvl-2-(5-fluorouracil-1-vl)-D,L-
qlvcineethvlester (I-2)
215966~
18
CH2
Pd/C
,. 2 HC 1 /NH2CHCONHCHCOOCH2CH3
(II 2) H2 0
(I-2) HN ~
A O . lN hydrochloric acid-methanol solution was added dropwise
to 98.6 mg of 10 % palladium carbon at 0~C. Further, a O.lN
hydrochloric acid-methanol solution (total volume of 4.7 ml) of
0.10 9 (0.2 mmol) of N-carbobenzoxy-L-phenylalanyl-2-(5-
fluorouracil-1-yl)-D,L-glycineethylester (II-2) was added thereto
dropwise and then the mixture was agitated under an H2 gas stream
at room temperature for 3 hours. After the end of the reaction had
been conflrmed by thin layer chromatography, the reaction mixture
was diluted with methanol and filtered through a fluted filter
paper. The filtrate was concentrated under reduced pressure to
obtain a white crystal. The crystal was washed with diethylether
and filtered to obtain 65.4 9 of the compound (I-2).
Yield: 72.4 %, Melting point: 174.0 to 185.3~C
Elemental analysis (%)
Found: C, 45.72; H, 4.66; N, 12.41
Calculated (for C17H1gOsN4F-2HCl): C, 4S.25; H, 4.69; N, 12.41
[a]D: 27.03~ ~c 1, CH3OH)
1H-NMR (D2O): ~1.31 (t, 1.5H, J=7.10Hz) ethylester CH3, ~1.36 (t,
1.5H, J=7.10Hz) ethylester CH3, ~3.12 (dd, 0.5H, J=13.29, 10.54Hz)
phenylalanyl CH2, ~3.38 tm, lH) phenylalanyl CH2, ~3.47 ldd, 0.5H,
J=13.29, S.SOHz) phenylalanyl CH2, ~4.37 (m, 2H) ethylester CH2,
~4.47 (m, lH) phenylalanyl CHCO, ~6 36 ~d, lH, J=7.33Hz) glycyl CH,
~7.26 to 7.54 (m, SH) aromatic ring, ~7.90 (d, O.SH, J=S.SHz)
uracil CH, ~8.05 (d, 0.5H, J=5.5Hz) uracil CH
IR(KBr)umax: 3420m, 3180m, 3020s, 1705s, 1530m, 1495m, 1470m
2159660
Rf: 0.24
5 % methanol-dichloromethane (phosphomolybdic acid reagent W+3.
Ex~m~le 3
(1~ Pre~aration of N-carbobenzoxv-L-~henvlalanvl- 2-~-(bis( 2-
chloroeth~l)amino)-L-~henvlalaninemethvlester-2-vll-D,L-
alvcineethvlester (II-3)
NH2
ClCH2CH2 > N ~ CH2CHCOOCH3
C
(IIIb-2) CH2
CH20CONHCHCONHCHCOOCH2CH3
NH
ClCH2CH2 > N ~ CH2CHCOOCH3
(II-3)
The compound (IIIb-2) obtained from Example 2(2) was reacted
with melphalan methylester [p-(bis(2-chloroethylamino)-L-
phenylalaninemethylester] in the manner same as that in Example
2(3) to obtain the compound (II-3). The melphalan methylester was
treated with a sodium hydrogen carbonate solution (2. 2 equivalents)
to remove the hydrochloride and extracted with dichloromethane.
Yield: 84.3 ~, Melting point: 112.3 to 115. 3~C
Elemental analysis (%)
Found: C, 60.04; H, 6.00; N, 7.93
Calculated (for C3sH42O7N4Cl2): C, S9.91; H, 6.03; N, 7.98
[~]D: 19.1~ (c 1, CHCl3)
lH-MMR (CDCl3): ~1.26 (t, 3H, J=7.10Hz) ethylester CH3, ~2.73 (dd,
0.5H, J=13.75, 7.79Hz) phenylalanine CH2, ~2.91 (dd, O.5H, J=13.98,
5.73Hz) phenylalanine CH2, ~3.05 (m, 2H)- melphalan Ph-CH2, ~3.57 to
3.62 (m, 4H) melphalan CH2-CH2, ~3.65 to 3.70 (m, 4H) melphalan Cl-
~15966a
-
CH2, ~3.69 (s, 3H) melphalan CH3, ~4.14 (m, 2H) ethylester CH2,
~4.36 (m, lH) melphalan CH-CO, ~5.07 (m, 2H) Z group CH2, ~5.18 (m,
lH) melphalan NH, ~6.59 (d, lH, J=8.71Hz) glycyl CH, ~6.96 to 7.31
(m, 14H) aromatic ring
IR(KBr)umax: 3300s, 3070m, 3040m, 2950m, 1730s, 1645s, 1610m, 1520s
Rf: 0.50, 0.56
Hexane: Ethyl acetate = 1:1 (phosphomolybdic acid reagent W+).
(2) Pre~aration of L-~henvlalanvl-2-~-(bis(2-chloroethvl)amino)-L-
~henvlalaninemethvlester-2-vll-D,L-alvcineethvlester (I-3)
H2 Cti
(II-3) ~ 3HC1/NH2CHCONHCHCOOCH2CH3
Pd/C
NH
ClCH2CH ~ N C ~ CH2CHCOOCH3
(I-3)
The compound (II-3) obtained from Example 3(1) was reacted
with palladium carbon under an H2 stream in the manner same as that
in Example 2(4), to obtain the compound (I-3).
Yield: 89.7 ~, Melting point: 102.3 to 108.0~C
Elemental analysis (%)
Found: C, 47.33; H, 5.97; N, 8.16
Calculated (for C27H36OsN4C1-3HC1): C, 47.90; H, 5.80; N, 8.28
~~]D: 36-5~ (c 1, CH3OH)
1H-NMR (D2O): ~1.25 (t, 3H, J=7.10Hz) ethylester CH3, ~3.17 (m, 2H)
phenylalanyl CH2, ~3.27 (m, 2H) melphalan Ph-CH2, ~3.70 to 3.76 (m,
4H) melphalan CH2-C~2-, ~3.86 to 3.90 (m, 4H) melphalan Cl-CH2,
~3.89 (s, 3H) melphalan CH3, ~4.37 (m, 2H) ethyl ester C~ 4.55
(m, lH) melphalan CH-CO, ~7.35 to 7.71 (m, 9H) aromatic ring
IR(KBr)umax: 3410s, 3150m, 2950s, 1740s, 1705s, 1610m
2159660
Rf: 0.38, 0.44
5 ~ methanol-dichloromethane (phosphomolybdic acid reagent WTj.
Exam~le 4: Acute Toxicitv
(1) The conditions in the feeding chamber were arranged to a
temperature of 23+1~C, a relative humidity of 55+5 %, a ventilation
frequency of 20 times per hour, and a lighting term of 12 hours.
Wistar rats (male; five-week old; 120 to 141 9) were used. The
rats were placed in metallic cages having wire walls in the front
and bottom thereof, at the rate of 5 rats per one cage. They were
allowed food (MF: Oriental Yeast) and water ad libitum.
(2) Method of Administration
The conjugate (I-1) of the present invention obtained from
Example 1(5) was suspended in a 1.5 % methylcellulose aqueous
solution. The suspension was a~mi n; stered by force. The amount of
the administration was 1 ml per 100 9 body weight of ~n;m~ls forced
to fast for 18 hours, that is, 250 mg/kg was administered.
Symptoms of addiction and survival were observed at every one-hour
up to 8 hours after the administration, then twice a day until 14
days after administration. No death was observed.
ExamPle 5: Antitumor Activitv
The concentration of the solid tumor cells (Sarcoma-180) was
aseptically adjusted to 1 X 106/0.2 ml with a medium [prepared by
filtering 10 % bovine fetal serum-added MEM (Eagles' Minimum
Essential Medium) for sterilization, and stored at 4~C] and
subcutaneously implanted at the axillas of ICR mice (5 weeks old;
females; one group consisting of 10 mice). From the next day, the
samples (0.15 % physiological saline solution) shown in Table 1
were administered intraperitoneally 10 times every other day. On
the 22nd day, the tumors were excised and the inhibition rate (IR)
was obtained from the weight of the tumors. The results are shown
in Table 1. The inhibition rate (IR) (~) was calculated by the
following formula:
IR (%) = ~ - (T/C~ X 100
wherein T is the average tumor weight of the treated group, whereas
C is the average tumor weight of the control group.
2159660
22
Table 1
Group Amount Average tumor IR (~)
(mg/kg) weight+SD
Control - 3.438+1.481
S-Fu 15 1.467+1.437 57.3
MP 10 1.358+1.369 60.5
5-CAU 10 1.574+1.285 54.2
Phe~5-Fu 15+15 1.385+1.297 59.7
Phe+MP 15+10 1.407+1.295 59.1
Phe+5-CAU 15+10 1.550+1.375 54.9
Phe-5-Fu 30 0.770+0.397 77.6
Phe'-5-Fu 30 0 808+0.389 76.5
Phe-MP 20 0.943+0.587 72.6
Phe-5-CAU 20 0.854+0.664 75.2
5-Fu: 5-Fu alone
MP: *Melphalan alone
5-CAU: 5-[bis(2-chloroethyl)amino]uracil alone
Phe+5-Fu: Mixture of L-phenylalanine and 5-Fu
Phe+MP: Mixture of L-phenylalanine and MP
Phe+5-CAU: Mixture of L-phenylalanine and 5-CAU
Phe-5-Fu: Present Conjugate (I-l) prepared from Example 1
Phe'-5-Fu: Present Conjugate (I-2) prepared from Example 2
Phe-MP: Present Conjugate (I-3) prepared from Example 3
Phe-5-CAU: Present Conjugate prepared from Example 1(6).
Example 6 Pre~aration of Iniection
A 500 mg amount of the compound (I-l) of the present invention
obtained from Example 1(5) was dissolved in 50 ml of ethanol to
prepare an injection.
Although the present invention has been described with
reference to specific embodiments, various changes and
modifications obvious to those skilled in the art are deemed to be
within the spirit, scope, and concept of the invention.
*Trade Mark